
Submitted:
04 September 2018
Posted:
04 September 2018
You are already at the latest version
Abstract
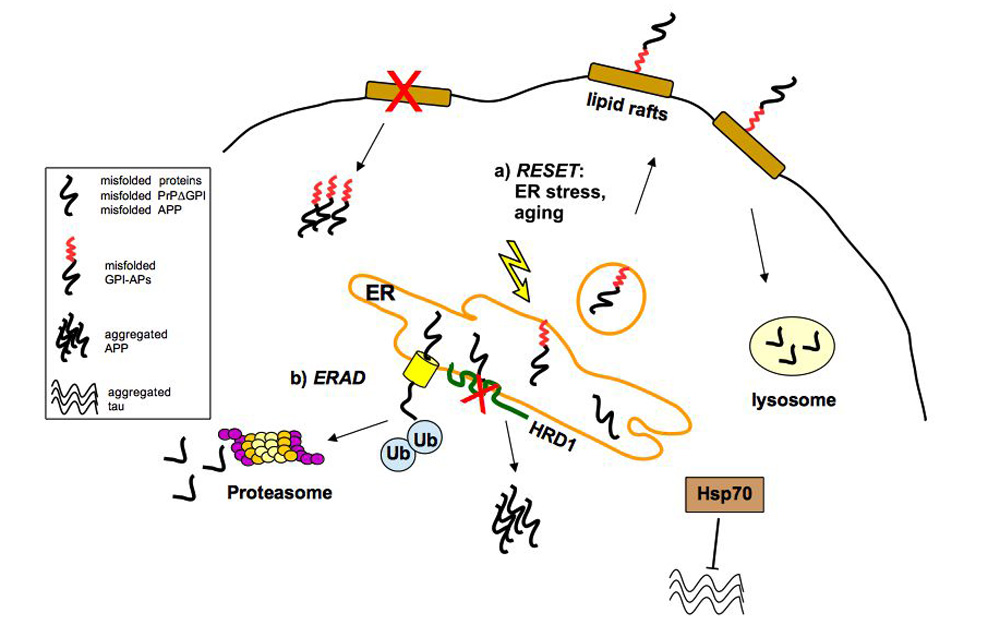
The misfolding and aggregation of proteins is the neuropathological hallmark of numerous diseases including Alzheimer’s disease, Parkinson’s disease, and prion diseases. It is believed that misfolded and abnormal -sheets forms of wild-type proteins are the vectors of these diseases by acting as seeds for the aggregation of endogenous proteins. Cellular prion protein (PrPC) is a glycosyl-phosphatidyl-inositol (GPI) anchored glycoprotein which is able to misfold to a pathogenic isoform PrPSc, the causative agent of prion diseases which present as sporadic, dominantly inherited and transmissible infectious disorders. Increasing evidence highlight the importance of prion-like seeding as a mechanism for pathological spread in Alzheimer’s disease and tauophaty, as well as other neurodegenerative disorders. Here, we report the latest findings on the mechanisms controlling protein folding, focusing on the ER quality control of GPI-anchored proteins and describe the “prion-like” properties of amyloid- and tau assemblies. Furthermore, we highlight the importance of pathogenic assemblies interactions with protein and lipid membrane components and their implications in both prion and Alzheimer’s diseases.
Keywords:
A
; aggregation
; amyloid
; APP
; misfolding
; prion protein
; prion-like
; PrP
; seeds
; tau
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



