
Submitted:
05 April 2018
Posted:
06 April 2018
Read the latest preprint version here
Abstract
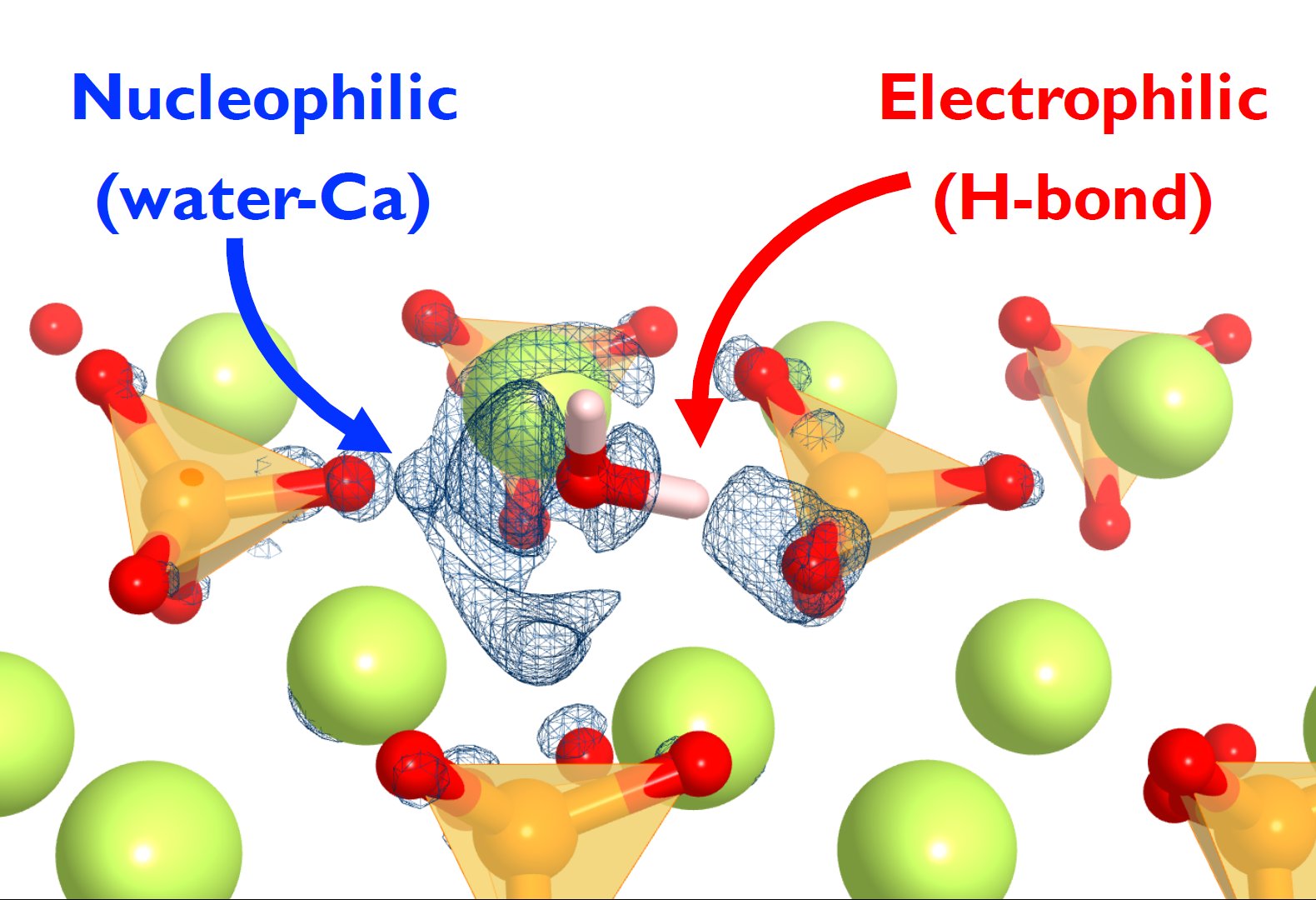
β-dicalcium silicate (β-Ca2SiO4, or β-C2S in cement chemistry notation) is one of the most important minerals in cement. An improvement of its hydration speed would be the key point for developing environmentally friendly cements with lower energy consumption and CO2 emissions. However, there is a lack of fundamental understanding on the water/β-C2S surface interactions. Therefore, in this work we aim to understand the water adsorption and dissociation mechanism on the β-C2S (100) surface using density functional theory (DFT) calculations. Our results indicate that thermodynamically favorable water adsorption process takes place in several surface sites, with a broad range of adsorption energies (-0.78 to -1.24 eV), depending on the particular water conformation and surface site. To clarify the key factor governing the adsorption, the electronic properties of water at the surface sites were analyzed. The partial density of states (DOS), charge analysis, and electron density difference analyses suggest a dual interaction of water with β-C2S (100) surface: a nucleophilic interaction of the water oxygen lone pair with surface calcium atoms, and an electrophilic interaction (hydrogen bond) of one water hydrogen with surface oxygen atoms, being the first one the stronger interaction. Hence, we suggest that β-C2S hydration could be enhanced by introducing chemical or structural changes that increase both the electronegative/positive character of the surface.
Keywords:
Belite
; Hydration
; Density Functional Theory
; Water Adsorption
; Calcium Silicate
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



