Submitted:
07 July 2026
Posted:
08 July 2026
You are already at the latest version
Abstract
Aging is the breakdown of life over time. A comprehensive, integrated, and universal mechanism to explain aging is lacking. We propose a unifying model reconciling existing theories with new ideas, organized around a concept we term “intropy”: the capacity of encoded information to produce and sustain functional and purposeful order. This model maintains aging results from the progressive loss of intropy through corruption of information-bearing nucleic acid that scrambles the chemical memory required to order life’s processes. The corruption decreases the efficiency of replicational, transcriptional, translational, and enzymatic outputs, amplifying functional inefficiency up a hierarchy of biological organization, from genome to organism. To sustain order against nearly infinite environmental stochasticity, evolution begot phenotypic diversity to protect and safeguard the transmission of relatively uncorrupted intropy to progeny (a “prime directive”), the original carrier left to continue a descent to a disordered state. Death results after crossing an irreversible efficiency threshold in which functional order is catastrophically lost and disorder rapidly rises, consistent with thermodynamic laws. While many cellular components sustain environmental damage, only corruption of nucleic acid, the sole irreplaceable template directing biological order, propagates functional disruption across every level of life's hierarchy. The informational corruption underlying aging reframes age-associated disease as a consequence of disordered biological instruction, thereby revealing nucleic acid change as the common process uniting aging, disease, and evolution. The theory reveals ways to significantly preserve order via engineered intropic protection, rendering the carrier relatively amortal.
Keywords:
aging
; intropy
; entropy
; information
; corruption
; evolution
; mortality
; protection
; modification
; nucleic acid
Approach to the Narrative
A proper theory of something as complex as aging is difficult to resolve within the confines of conventional manuscript limits and structure. This was a major challenge we faced in crafting this narrative. However, in this formalization, we were guided by three overarching principles. First, the main text should be accessible to any scientifically inclined reader regardless of their level of involvement in this field. We hope this democratization will stimulate a broader discussion on the merits of these ideas with the intent of driving interest in a topic that we argue deserves far more support, research, and priority. Second, we wished to not distract the reader with jargon, complex molecular details, and prominent controversial topics that confound the literature of every major field. Instead, we chose to retell the story of aging as an interaction of three main players, with the main text establishing a logical and integrative narrative accessible to the general reader, while Explanatory Notes supply the empirical substance, molecular details, citations, and technical discussion the framework requires. The Explanatory Notes are not supplementary; they carry the empirical substance of the argument. Third, we wish to integrate the elegant ideas of the many brilliant thinkers that contributed to the corpus from which this work is drawn, explaining why most current theories of aging are fundamental to the various steps of a larger framework presented herein. We did not anticipate, when we began, how much this framework would come to explain several observations it accounts for that were not originally considered during its construction. We explore those in the accompanying Notes.
Introduction
Opening Premise
All life over the past three billion years, except that which is currently living, has died.[1,2] If not subdued by predation, illness, or calamity, every organism can be viewed as suffering from one progressive pathology or disease – aging.[3,4] That organisms die from aging seems like a paradox.[5] If central is the will to survive, why age? Should not evolution have produced beings that indefinitely exist?EN1 According to the Gompertz-Makeham law, mortality in nearly every species increases with time.[5,6], EN2 As Shakespeare articulated in Cymbeline, “Golden lads and girls all must, as chimney-sweepers, come to dust”.
Why do we age?
Why do we die?
The answer to this paradox may be found in a thermodynamic bargain with universal laws.EN3 Purposeful order, what we call “order with functional intent”, is the defining feature of life. All systems, living and not, progress towards disorder. To resist and delay disorder, life concocted an ingenious plan, a magic act so elegant only on Earth has it yet been observed; the invention of chemical memory, a molecule whose nature contains information to organize its future.EN4 With chemical memory, life remembers how to order, rejecting random assembly. Chemical memory allows life to read instructions, a vetted plan to resist chaos.
Yet chemical memory generates its own problem. The same destructive forces that break order also erode chemical memory. For life to recall order with functional intent the information must be preserved and read. Only through faithful readout of information can life order over vast stretches of time, expand the order to new environments, and introduce a mechanism to improve, and thus adapt, to environmental chaos. But to read and copy is to error.EN5 No repetitive process, engineered or stochastic, continues without change. True of planetary orbits or biological molecules, every process is negatively impacted by imperfections. Thermodynamic laws dictate system entropy (measure of disorder) must increase.[7,8,9,10,11] How does life reconcile these laws while maintaining order through time?
The evolutionary solution may have been a proverbial agreement with the governing laws of the universe. The individual trades an ordered present for life’s ordered future. That is, life clandestinely transmits a relatively unaltered copy of information to a second carrier before the degradation of that message prevents future order. The copying mechanism itself may be the only possible way to replenish the original information over time. If true, it explains why all life must copy: it’s a way to counter the inevitable fact that thermodynamically the system must slowly disorder. The parent carrier, compelled to execute a “prime directive” to copy, continues to error, corrupting information beyond interpretation. The new relatively uncorrupted carrier efficiently orders again, and so forth.EN6 Thus, we propose aging is the physical manifestation (phenotype) of subtle but constant corruption of life’s information to order.EN7 We view lifespan as an evolutionary agreement on the time needed to limit the accumulation of information loss before the capacity to functionally order is lost. Once transmitted, the new vessel retains the original “memory” of how to construct another copy of the information, as well as the protections afforded to its safety. The parent vessel, unable to recall, is sacrificed to entropy, the debt now paid. Viewed in this way, death is simply a means to beget new life.
What has been lacking is an explanation for why nucleic acid, and not any other molecule, sits at the root of this process, why its corruption is thermodynamically inevitable, what determines the rate of that corruption in a given species, and how a single class of molecular events propagates functional decline across every level of biological organization. Presenting these ideas, along with explanations developed by others, to answer these questions is what we attempt here. In doing so, we needed to conceptualize a process by which information embedded in the structure, sequence, and modification of nucleic acid (both DNA and RNA) is copied, read, and interpreted to achieve the intended functional order needed to survive and withstand macro and micro assaults against this order at every level of biological organization. We thus formatively define this idea, which, in our assessment, has not previously been conceptualized in Biology in a unified manner, as “intropy”. Intropy is a word that both connotes that information integrity is critical to sustained output of order while also reflecting that life is an active struggle to maintain that order over time.
Intropy is the living counterpart to entropy: where entropy describes the universal tendency toward disorder, intropy describes the capacity by which life converts encoded information into functional order, and holds that tendency towards disorder off for a brief moment of time. A genome written into a sequencing file carries information; but only within a working cell does that information become intropy. We therefore distinguish potential intropy, the stored ordering capacity of an intact template, from realized intropy, that capacity actively producing and maintaining biological order. In this framework, life is realized intropy, aging is the progressive loss of intropic capacity, and death is intropic collapse below the threshold required for self-maintenance. Explanatory Note 7 elaborates further on the choice of this word and its definition.EN8
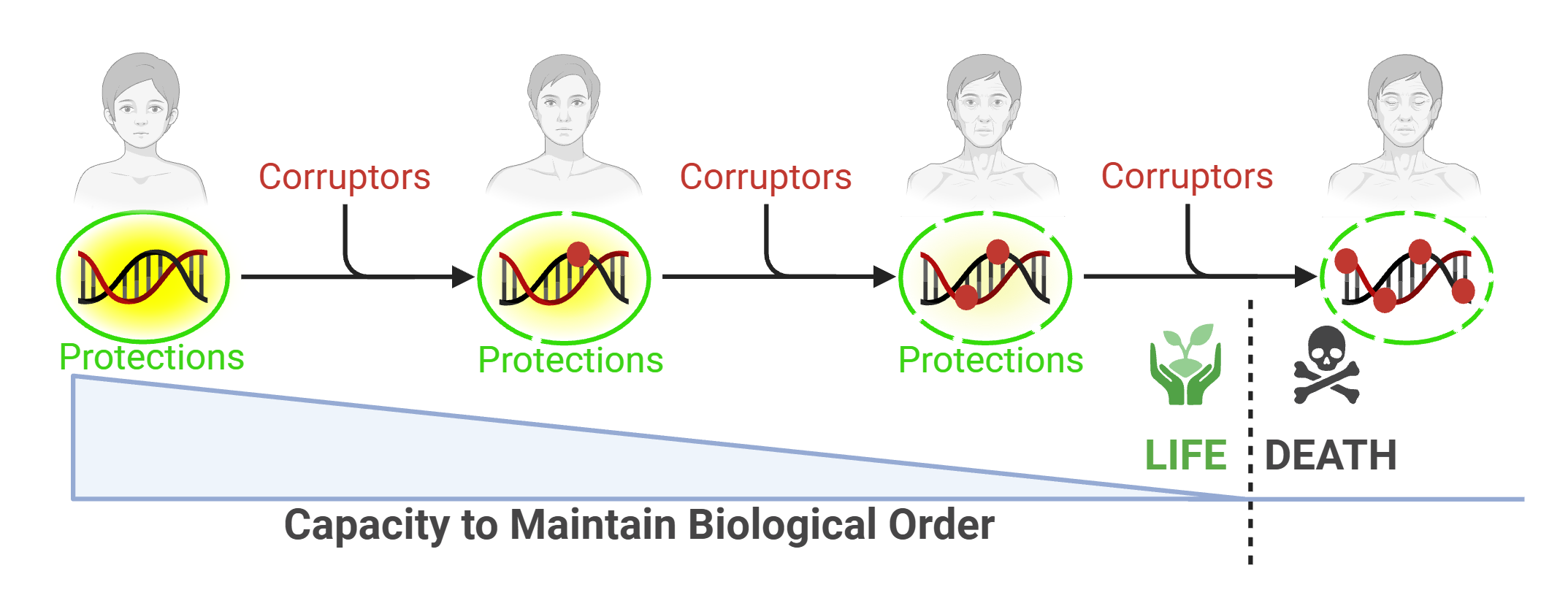
Information-theoretic and thermodynamic entropy are formally distinct, though Landauer’s principle establishes that information processing has irreducible physical costs, linking the two frameworks.[11] Shannon’s theorem, a pillar of information theory, states that no communication of information in any channel can occur indefinitely without error. While reliable copying is possible in principle through sufficient redundancy, real systems pay for fidelity in time and energy, and no finite system can reach perfect transmission.[11,12,13], EN9 Be it a wireless signal or photocopied book, information is incrementally corrupted before, during, and after each transmittal, disorder increasing. With enough errors, the information collapses, now meaningless (Figure 1, scheme 1). Through the layered redundancies of the protection systems, biology invests heavily to approach this ceiling, though no finite organism can afford to reach it.
As such, Biology operates within a corridor bounded by two limits. An Eigen-like threshold sets the upper bound: above a critical error rate, a self-replicating system loses the information that specifies it and collapses.[14] The drift barrier sets the lower bound: below a certain rate, further reductions in error rate no longer pay off, because random chance rather than selection pressure governs which variant persists.[15], EN10 Life tolerates error not because error is impossible to reduce but because no finite organism can afford to operate arbitrarily close to either limit. Nucleic acid (DNA/RNA) is the information-containing system embedded with chemical memory to order, and every organism accumulates error in that system at a rate set by its metabolism, its repair investment, and the evolutionary calibration of its protective layers. Drawing upon analogy to Shannon’s theorem of information loss over time, nucleic acid is also inevitably subjected to small aggressions that induce unintended change in the original information. The organism, a physical embodiment of the translated information, ages as that tolerated error compounds across the readout machinery, with death following when a catastrophic threshold is crossed (Figure 1, scheme 2). Using this basic premise as foundation, in the accompanying pages, we attempt to lay out an integrated theory of aging and mortality, which can be generally stated as: the progressive loss of intropy, the capacity to convert nucleic acid information into functional biological order through a hierarchy of organization, driven by the thermodynamically inevitable change of that information; death follows when the residual capacity falls below the threshold at which stochastic challenge can no longer be survived. The framework reconciles leading theories, orders them as cause or effect within a single string of linked processes, simplifies understanding by framing the narrative as an interaction of three overarching models of life, and finally, reveals purpose in life’s diversity.
Approach
Cause and Effect
Our approach uses two strategies of logic. The first is Aristotelian cause and effect to break the “chicken vs egg” cycle. By structuring aging as sequential, we can identify the root causes and resulting effects. We classify the phenotypic changes (hallmarks) observed during aging as effects; regardless of whether aging is viewed as a disease or process, we view them as symptoms. Since the symptoms are prominent, they can understandably be misaligned as cause, a distraction. We view many hallmarks and theories of aging as being primarily associated with the effect, not the cause, each a downstream step in a sequence that begins with a singular foundational event. If humanity’s objective is to slow/stop death (in our opinion, the ultimate frontier), strong scientific efforts are best directed at understanding the foundational cause.EN11 Treating symptoms is not likely to lead to a solution; it is rouge over scar, perpetuating the false impression that we are winning.EN12
The second approach is Francis Bacon’s reductionism, to peel layers and “get to the core”.[16], EN13 As microbiologists, we turn to life’s unit of order, the cell. Following Baconian reduction, in addition to other models, sometimes lean on the “most reduced” of cells, that of bacteria.[17], EN14 Because microbiologists study life in its simplest form, the bacterium, they are uniquely positioned to build upward toward complexity and to understand what reductionism reveals at a fundamental level. Although the aging phenotypes of mammals and bacteria differ enormously, the underlying information-maintenance machinery is conserved, the product of that information (the cell) clear in its purpose, and the mechanisms by which information corrupts straightforward, thereby making bacteria an experimentally tractable system to study the fundamental chemistry of information corruption. Bacteria organize, metabolize, and evolve like all cells but, distinctively, are the purest copying vessels, the generation of more cells seemingly its only objective.EN15 Free of distractive bells and whistles observed in higher mammals, life’s ordered mechanic processes are strikingly clear in bacteria, their component simplicity showing the essence of the complex.
Theories of Aging
A universal theory of aging will (1) explain the majority of, often divergent and sometimes contrasting, data; (2) reconcile, integrate, and readily explain existing theories; (3) stand the test of time, especially if future and more precise mechanistic evidence or improved technologies reveal new data; (4) have parts or all of the logic be applicable to all life that ages, especially at the most basic molecular and cellular levels; and finally (5) have the steps in the process be falsifiable even if the complete picture may not yet be readily testable.EN16 This framework largely fulfills these criteria while stitching together (and justifying the validity) of the most promising ideas that have emerged over the past 60 years, layered as five main levels of recognition required to fully explain aging (Table 1). López-Otín and colleagues’ highly influential hallmark taxonomy proposed a causal stratification of age-associated phenotypes into primary, antagonistic, and integrative tiers.[18,19] The intropy framework focuses on the foundational chemistry whose corruption initiates the cascade and the channels through which it propagates. We start with theories that we view as being more aligned with effects instead of with cause.
One of the earliest theories on aging is The Wear and Tear Hypothesis (1882)[20], EN17 which states aging is caused by tissue weathering (like a used automobile).[20,21] Modern evidence shows tissues repair and are remade from stem cells, complicating the simple notion that parts wear out.[22,23], EN18 The idea of slow break-down is intuitive and ultimately correct; however, we see weakened parts as an effect.EN19 Instead we ask: why do parts weaken?
The Glycation Hypothesis (1981) states aging comes from glycan crosslinking/macromolecule aggregation.[24],EN20 Cross-linked proteins are in cataracts/atherosclerosis, cross-linked collagen forms wrinkles, and brains exposed to crosslinkers age quickly.[25,26,27,28,29], EN21 However, there exist ample systems to control and remove glycation. We maintain glycan products are effects. Instead we ask why do the control mechanisms lose their ability to regulate?EN22
The Hormonal/Endocrine model (1971) postulates aging is caused by hormonal changes.[30], EN23 Related is the Immunological Theory of Aging (and more recent and related Inflammaging and Immunosenescence Models), i.e. dysfunction in immunity prevents defense against pathogens, tumors, or endogenous damage.[30], EN24 We maintain that a universal explanation of aging will be applicable across kingdoms of life.EN25 Hormones and immune systems are absent in most species. Furthermore, any breakdown of a part of a multi-component system like the endocrine or immune systems (e.g. failure of the heart and thus the entire organism dies), even if associated with aging, we maintain is still a symptom of a deeper, more basic, corruption. We instead ask: if certain organ systems are lynchpins that corrupt before others, accelerating aging in healthier tissues, what are the causes of the failure of those systems in the first place? Theories with a human-centric view of aging suffer from being biased towards observable mammalian physiology and phenotypes, possibly masking true causes that operate at a more fundamental level. In our view, any life that harbors information-bearing nucleic acid and reads that nucleic acid to convert its message into biological order will age, the symptoms specific to the nearly limitless modifications that beget that information and the functional consequences (or lack thereof) that result from information change.
Several theories link metabolic activity to aging (e.g. Rate-of-Living, 1928). These theories postulate metabolism rate (particularly faster metabolism) negatively impacts lifespan. Thus, low metabolic activity organisms tend to live longer than those with higher metabolic activity. We view the relationship between metabolic activity and lifespan as a clue that energy use is intimately tied to corruption. We ask what molecules and cellular processes endanger information-bearing molecules such that their modified instructions reduce the efficiency to functionally order?
There are theories of aging being programmed. The Programmed Aging Hypothesis states aging is evolution’s way to cull older individuals so young have resources.EN26 There is little molecular evidence systems activate aging, though cellular senescence is sometimes cited in support of is.[31], EN27 This hypothesis states somatic cells have finite divisions and are programmed to suicide when defective, protecting the organism from cancer. We do not favor a “purposefully” timed model of aging. Instead, we view symptoms associated with aging as evidence that the underlying cause is incremental and progressive, i.e. that cells, tissues, and organisms corrupt at different rates. We ask what are the rates of information corruption, and how does the resulting intropy loss translate into functional decline across the hierarchy? In this sense, a lifespan may be simply defined as the time evolution has afforded the vessel to transmit functional information to a second vessel before corruption prevents readability and translation to purposeful action.EN28 This framing parallels Kirkwood’s disposable soma hypothesis, which posits that organisms allocate finite resources to reproduction over somatic maintenance. We extend this logic in two directions. First, the maintenance that is allocated is itself encoded in information that progressively corrupts over time, making indefinite repair a thermodynamic impossibility. Second, the level of maintenance evolution calibrates the rate at which that corruption accumulates.EN29 Each replicating entity accumulates corruption at its own rate (i.e. intropic decay, or aging) and has its own threshold beyond which functional order cannot be maintained (i.e. intropic collapse, or death).
Telomere Theory of Aging (~1960s) states that divisional shortening of the ends of chromosomes triggers cell death.[32], EN30 Telomere length is tracks division number; the rate of telomere shortening correlates to longevity.[33], EN31 Telomerase, which builds new telomeres, drives indefinite division.[34,35,36,37] Telomere disorders produce premature aging [38] and division capacity tracks telomere length/lifespan.[33,39] There is evidence against the theory.EN32 Different species of mice vary in telomere length with no change in lifespan.[40,41] There are significant differences in telomere length amongst cells of the same tissue, tissues of the same organism, across organisms of the same species, and between species, and no clear relationship between length and senescence/lifespan.[42] We instead ask, at the most atomic level, what causes the ends of DNA to lose their original information content? We interpret current evidence as aligning with telomeres being predominantly an evolved physical yardstick of replication history, a measure of accumulated information corruption, a proxy of aging but not a direct cause.EN33 The telomere story was one of the first to key in on nucleic acid integrity as central to aging.[43] Like a shoelace fraying because it lacks a plastic cap, so too are the exposed and unprotected ends of DNA corrupted by the environment.
Additional ideas have emerged over the last decade, each supported by strong evidence. We view these, like the theories above, not as flawed but as accounts of how decline propagates through specific channels rather than where it originates. Theories of disabled macroautophagy and proteostasis collapse postulate that an inability to clear damaged proteins, organelles, and other cell factors leads to their accumulation, and thus aging.[44,45], EN34 We agree but instead ask what intropy is lost such that these systems become inefficient over time? An erosion of epigenetic control dysregulates transposable elements that triggers a multitude of age-related effects.[46,47], EN35 Cells senesce in response to damage and arrest/die, often also harming surrounding tissues (see EN22 and EN27). Stem cells that replenish these cells can eventually become exhausted and damaged themselves, preventing replenishment.[48], EN36 Nutrient sensing pathways become dysregulated with age,[49,50], EN37 intercellular signaling becomes compromised,[51,52], EN38 and there are even effects in symbiotic systems such as the microbiome.[53], EN39 In some ways, it seems there are little to no micro or macro-molecular cellular systems that are not affected by aging. We thus ask if many of these breakdowns are considered effects or symptoms of a more global underlying problem, what do they all have in common? Is there a foundational system that begets the functional order of all cellular processes regardless of their role, mechanism, difference, and uniqueness in any given cell’s biology?
The DNA Damage Theory of Aging, developed over several decades and most comprehensively articulated in recent years by Schumacher, Pothof, Vijg, Hoeijmakers, and colleagues, proposes that physical and chemical lesions in nuclear and mitochondrial DNA accumulate over time and causally drive most recognized features of the aging process [45,54] and the broader synthesis assembled by Niedernhofer, Gurkar, Vijg, Hoeijmakers, and colleagues.[54] The empirical case for this view is substantial. Premature aging syndromes caused by inherited DNA repair defects, the age-associated rise in double-strand breaks and bulky adducts, the gene-length-dependent decline in transcription in aged post-mitotic tissues, and the acceleration of nearly every feature of the aged phenotype in DNA-repair-deficient mouse models all support a direct causal role for DNA damage in aging. We share most of these empirical commitments and draw on much of the same literature. Recent refinements within this tradition, particularly the distinction between transcription-blocking and replication-blocking lesions and the recognition of gene-length-dependent transcriptional decline, have begun to organize damage by its functional consequences. However, DNA damage theory has, to date, treated nuclear information corruption principally as a problem of structural integrity: damaged DNA produces damaged cells, damaged cells produce damaged tissues, and damaged tissues produce the aged phenotype. We instead ask a prior set of questions. Why DNA, and not some other macromolecule, sits at the root of this process? Why is its corruption thermodynamically inevitable rather than merely empirically observed? What determines the species-specific rate at which that corruption accumulates? Can we expand the category of “damage” to include the full range of modifications that change the information content of nucleic acid, including RNA? And, importantly, through what channels do modifications propagate functional decline across every level of biological organization, from the transcript to the tissue to the organism? Answering these questions, we argue, requires reconceptualizing DNA damage not as one hallmark among many nor as an upstream driver of hallmarks, but as one category of modification to an information-bearing molecule whose accurate readout is the thermodynamic substrate from which biological order itself emerges. The shift is from damage to information, and from information to the intropic capacity it sustains, and the consequences of that shift are the subject of the framework that follows.
We can now examine additional theories, when combined with new ideas presented herein, to form a simplified conceptual model from which to understand all aging effects.EN40 They are the DNA Damage Theories (including mutational and error thresholds), the Free Radical Theory (including mitochondrial theories), and the Epigenetic Theory, framed in the context of the evolutionary logic of the Disposable Soma hypothesis, to form a new integrated and universal idea that accounts for most available evidence.EN41 Each is causal, sequential, and mechanistic, but in our view has been somewhat misaligned in how they drive aging.EN42 Rather than treating these theories of aging as competing explanations, we order them as steps in a single corruption cascade, beginning with the foundational information that orders life’s function and amplify upwards through the hierarchy of biological organization. To establish a common foundation and maximize the democratic understanding of this concept, we must retell the story of aging as a tragedy involving three key actors; the Replicator, the Protector, and the Corruptor. With this telling, the reasons for aging, and life’s purpose more generally, become apparent. We start with the Replicator.
The Replicator
If information corruption, and the intropy loss that follows, is the cause of aging, what information-containing molecule(s) are most central to the propagation of intropy loss? Evidence undeniably points towards nucleic acid (and its linear sequence of nucleobases) as the answer.[45], EN43 Nucleic acid encodes life’s information, all life contains nucleic acid, and all nucleic acid is subject to the chemistry of change and corruption, therefore all life is vulnerable to intropy loss. Everything – the cell, the collection of cells that comprise a tissue, the tissues that comprise an organism, the organisms of a social unit, and the behavior, culture, and technology of that unit, from the smallest single-celled bacteria to the largest multi-celled whale – exists to protect the transmission and readability, vessel to vessel through the eons of time, of a single molecule, the nucleic acid replicator s(Figure 2a).
What is the replicator? The replicator is a molecule embedded with chemical memory whose information content has but one purpose: copy. Regardless of its carrier, the way it replicates, its form, function or phenotype, the replicator seemingly exists only to duplicate, which here we define as its prime directive.EN44 Life, in its multitude of elegant forms and functions, can be viewed as the replicator’s nurturing cocoon, moving it through space and time, the memories the instructions to order its local biological environment in the face of chaotic threats to the directive.
There are many types of replicators, but all are made of nucleic acid. The simplest replicators are transposons, tiny mobile elements that copy by parasitizing complex replicators. Next come plasmids, then viruses, and finally any living cell or collection of cells (organism), including humans. Common features that define a replicator are shown in Figure 2b. First, the replicator must copy. The earth’s first replicators probably did this spontaneously, being thermodynamically favorable. The advanced replicators of today have built sophisticated nanomachines to facilitate the copying process. These replicators acquire energy to facilitate protection and copying when not thermodynamically easy. Second, the replicator must sequentially order its processes. Although some molecules in nature will duplicate because of favorable chemical energetics and atomic structure, these spontaneous reactions may not be viewed as consistent and sustainable. Replicators overcame this challenge by building into the process a type of chemical memory, a preordained reaction mechanism inherent to the molecule itself that recalls how to replicate.EN45
Third, replication is not safe in a changing and caustic environment. Since we have learned from information theory that copying cannot be perfect and that local environmental aggressions against the system will introduce replication errors, the prime directive must also tolerate some level of informational change. Herein is realized the replicator’s cunning elegance. If, by chance, a change in information enhances instead of diminishes the prime directive, those changes will be selected. Should environmental assault corrupt the information too much, the directive falters and information cannot direct intropic order. If chaos causes too little corruption, the directive again fails for there is no route to adaptation to the environment. Here a critical distinction must be drawn: informational changes that occur in the germline and prove beneficial are the currency of evolution but change that accumulates in somatic cells is the cost.EN46
Fourth, when “just enough” information changes, and in the correct set of instructions, the directive will improve. And in the case of nucleic acid, improvement can arise from a single nucleobase change, a localized alteration that reshapes the directive without dismantling the sequence as a whole. If bacteria, our simplest living replicators, are given a nutrient rich environment, they copy.[55,56] If the environment challenges them, only those replicators holding information to order despite the new chaos will dominate the pool. This fourth rule (that changes in information introduce adaptation) is of course the Darwinian (organism)/Mendelian(gene) concept of genetic evolution. Here, evolution is tweaked as the selection of changed (relative to the original state) but now beneficial information. The selection itself was unintended, a consequence of imperfect control, but the changes it preserves enhance the replicator’s competitive persistence and safeguard the prime directive. Without it, replicators go only so far as the existing information has prepared them, unable to adjust to the stochastic challenges of an unpredictable universe.
This reframing gives a different perspective on life’s diversity (Supp. Figure 1). The first self-catalyzing attempts had no memory and were at the mercy of rapid increases in entropy, a “disorder trap.” Earth, however, was tolerant,EN46 a type of replicator’s training ground. The cradle contained water as a medium, reactive atoms as ingredients, diverse energy sources, and a chemical environment that was at minimum not outright destructive. Critically, the atmosphere and water column likely attenuated radiation enough to favor subtle informational error over molecular annihilation, a Goldilocks zone for incremental chemical experimentation.[57,58,59,60] The rarity of such conditions may explain why life has only been yet observed on Earth: the environment not only needed to permit self-replication, but also sustain a fitness gradient shallow enough for incremental improvement yet steep enough for selection to operate. Most importantly, the replicator was gifted time. Earth formed approximately 4.5 billion years ago; isotopic and fossil evidence places the emergence of microbial life by at least 3.7 billion years ago.[1,61] The interval between a cooling crust and the first recognizable living systems was, by geological standards, remarkably brief. Over what may have been as few as several hundred million years, nascent replicators were copying and failing but incrementally improving.
When a replicator was born that, by virtue of its unique chemistry and structure, retained information that instructed how to replicate, a prohibitive disorder-to-order divide was crossed. With that memory captured in chemistry, copying became consistent, and thus sustainable. The process not only had instructions to limit copying disorder, but a built-in mechanism to improve (incremental “information leaps”). A metaphorical “rebellion against disorder” began, the chaos momentarily controlled through the invention of instructions that dictated how to build the machine the same every time. With Earth’s environment a challenge but still providing a limited range of extremes that produced a nourishing cover, it can be stated that in harsher regions of the universe, the local chaos would have been too destructive for ordered biology to persist, while Earth was forgiving enough to let it take hold.
This brings us to the final feature of the replicator: all self-serving information that enhances the prime directive will do so endlessly, no number of copies enough, a feature we argue is underappreciated by the sciences and humanity more broadly. At its simplest, although masked a billion different ways and concealed through the ages of time, the core purpose of all replicators and thus all life can be argued as the successful execution of the prime directive, every form of life a protection mechanism to ensure this tenet.EN47 If true, there is a defensible case that the prime directive is the meaning of life, the replicator ever at work via its evolved mechanism to safeguard its existence, ever copying and being read, indefinitely expanding and, so far, winning. In other words, any lineage that deprioritizes replication relative to its competitors is eliminated by selection. The prime directive is universal precisely because, inherent to the process, any alternative that does not follow has already died out.
We thus define biology as a form of order that has intent, or purpose. But this “order with intent” agreement, a balance between life’s existence and the forces that may consume it, has a statute of limitations. The replicator is only allowed to order for the time it requires to advance a relatively unaltered copy of itself to the next vessel. It is not given enough time for the remaining replicators in the original vessel to indefinitely continue without decay, thus losing the original information and the intropy it sustains.EN48 The parent carrier, bound by the prime directive and unable to indefinitely make “clean” copies, absorbs relentless environmental assault on information (EAI), strand by strand, cell by cell, tissue by tissue, and organism by organism, until accumulated modifications compound the inefficiency of function via loss of the original instructions. Wasting nothing, evolution dictates the slowly corrupting but still useful parent vessel to safeguard the newly minted copy, thereby increasing the likelihood of executing the prime directive.
Because the physical universe offers infinite ways to destroy order, no one informational unit can resist every challenge. The replicator’s tolerated-change solution to this problem is so brilliant that it’s been duplicated trillions of times, each adaptive cycle making gradual upgrades to safeguard the will and right to copy. When viewed this way, it seems that it is Earth’s biological diversity that immortalizes the replicator, the individual a means to that end.
Before we move on to the next section, it’s important to specifically define what we mean by information. We define biological information as any chemical template, structure, or process that lead to the downstream construction and function of all cellular and organismal biological actions. This is essential to appreciate because later we suggest that the escalation of disorder from the foundation through the hierarchy is what we recognize as aging and disease. Applied loosely, all biological molecules house some level of information. For example, protein enzymes contain structural features that house the chemical information to catalyze a reaction. However, our definition applies exclusively to coded information as it pertains to nucleic acid (DNA/RNA), including its structural core, sequence, and its modifications. Figure 3 lists a classification or types of information, the features of each type, the consequences of “reading” the information, and a generalization of the types of modifications to this information. In this framework, DNA is foundational since it begets all other biology; however, RNA, as the message between the code-script and protein, is also essential to the framework. In our model, reading is the term by which life converts the underlying information into interpretation, i.e. biological action. Essential to the intropy framework is the idea that local entropy is constantly modifying this information. The cell in turn, with all its layered protections and repair mechanisms, is attempting to preserve the original information. Without its maintenance, the information is changed such that it is read incorrectly or not read at all, both of which may translate into a loss of intropic capacity, the erosion of efficient, effective, and intended biological action. Over time, the consistent inability to convert the read information into the original and intended instructions to order, largely because it has been modified from its original state by environmental assault, drives an increasing inability to order the cell at every level, thereby seeding the corruption up the hierarchy of life. We argue that although all biological information is changeable, the alteration of nucleic acid is foundational (“at the base”), can be lasting, and most consequential because it ties to all life processes. We call this the critical information divide. Corruption below the divide (proteins, lipids, metabolites) is replaceable information contributes to aging as effects. Corruption above the divide (nucleic acid) can be labeled as causes because their information is irreplaceable and their corruption propagates through every level of biological function.
It is now time to introduce the physical mechanism that facilitates the immortalization of the replicator through the individual and the second character in the story, the protector.
The Protector
To fulfill the prime directive, information must be relatively intact. The damaging effect of the local environment (EAI) on information’s fidelity is ubiquitous, constant, and varied.[62,63] The replicator requires protection, a mechanism to repel EAI and delay information corruption. The protector is thus defined as any physical embodiment of encoded order that stores, maintains, conceals, shields, copies, repairs, transmits, functionalizes, socializes, culturalizes, or technologizes the prime directive, thereby protecting information from EAI. In our view, Earth’s entire living ecosystem is one colossal protectosphere to secure the replicator’s indefinite existence.EN49
How is the protectosphere layered? This answer is essential to the ideas proposed herein. The first layer is built into the replicator itself (Figure 4). DNA’s duplex structure is like cloud back-up; should one strand be damaged, the second is a template to retrieve original information.[64,65] The helix manages strain, keeps the information tight, and allows for easy access to both copies.[66,67,68] Eukaryotes back-up the back-up (diploidy).[69], EN50 Redundancy of information escalates up the biological ladder. Thus, a stem cell has a function to recall the entire informational set after damage and make more cells with the original information.[70] Members of the same species (99.9% identical information) are back-ups of core information that defines the entire species; and the many different species of replicators are insurance for the core information that orders all life.
The replicator is also repaired, preserving information, and each repair process matches the type of EAI experienced.[71,72,73,74,75], EN51 All life has DNA damage limiting or repair systems. In the prokaryotic kingdom, for example, Deinococcus radiodurans, which is constantly exposed to ionizing radiation, has some of the most robust DNA repair mechanisms known, up to one fourth its genome dedicated entirely to directly repairing or preserving its information. Eukaryotic and mammalian cells also deploy specialized repair and surveillance mechanisms to prevent cancer, a process unique to hierarchical systems in which a corrupted cell can escape the constraints that subordinate its replication to the organism’s benefit.[76,77,78,79] The other components of the protectosphere, like repair systems, are encoded by DNA as well. We view repair as essential to information maintenance, which explains why so many systems are dedicated to specific modification types. Repair systems also highlight just how much pressure there is from the global and local environment on the state of the information, and how easy it is to corrupt. This creates an iterative selection pressure where failures of one layer of the protectosphere drive the evolution of additional layers to safeguard the information to order.
Protections are observed at every level. The structure of nucleic acid (strong covalent bonds linearize information while weaker hydrogen bonds allow opening) enhances copying/repair.[65,80,81] Higher organisms bundle their replicators with proteins (histones/heterochromatin),[82,83] reducing exposure to EAI, enhancing spatial function, and shielding information. Cells bundle to tissue, tissues to an organ,[84,85] and organisms to a group (schools of fish, herds of bison),[86] layers upon layers upon layers of protection against diverse EAI.EN52, EN53
The cell is the quintessential unit of protection, an ordered, efficient, and functional vessel to ensure the prime directive (Figure 4). The membrane separates outside disorder from inside order, promoting chemical control (buffering/salt/pH), separated tasks (organelles), and functional compartmentalization. Information-damaging byproducts are sequestered, destroyed or excreted. Energy is acquired, stored, and distributed. Proteins, the cell’s action molecules, are structural scaffolds or enzymes that executes the replicator’s decree. Apart from the intrinsic chemistry of nucleic acid itself, no protective structure in biology has been refined by selection longer than the cell, its defensive architecture shaped for billions of years before any multicellular organization existed.
If a cell is the physical form by which information nurtures the prime directive, it stands to reason that a collection of cells will expand protection (Figure 4). Here specialized functions—energy distribution (heart), detoxification (liver), sensing (brain), and structure (bone)—all resist EAI. Layer upon layer, the protections extend up an ordered functional scaffold of standard biological hierarchy, amplifying the effect at each level. At the species level, different replicators adapted to complex EAI broaden the replicator’s full potential. What if the replicator could choose protections most likely to advance the prime directive? Thus, sexual selection, applying a decision on what information to add to the replicator, brings additional versatility to resist EAI.[87] Imagine billions of replicators shopping among billions of others for traits to enhance their information content, each decision driven by the underlying calculus of enhancing and ensuring the prime directive.
The specialization of individual ability, mastered by our own species, would be the replicator’s next quantum breakthrough. Because no genome has infinite size and possibility, if information content can be distributed amongst different members of a species, those similar replicators expand versatility. Here, the skilled hunter can hunt, the builder build, the teacher teach, the nurturer care, and the thinker think, each trait advancing a group of bonded replicators, only now as a social species, the sociality itself the next layer of protection, the collective stronger than the parts.EN54 And with the advent of intelligence, the replicator can harness protective powers that transcend its chemical memory.[88,89] These extra-anatomical non-encoded adaptations, entirely synthetic, extend a boundless protectosphere, putting within reach everything allowed by physical laws. With technology, phenotypes of protection extend to all realms of EAI in real time, something biological adaptation takes eons to achieve. Traits thus become inventible; we are the only species capable of using invention to create a limitless set of technological extensions of self for our well-being and survival. But slicing through the layers of protection, the computers and rockets, the social bonds and customs, the fast cars and gowns, the skin and bones, the species and cell, the protein and the lipid, it can be strongly argued that the directive is still the same… copy, and copy well; read, and read well, thereby ensuring the process can continue and the instructions are converted into systems to protect that process (see EN29). If we remove the protections and view only the nascent information, either in its steady-state or during the readout of information, is there a moment where the replicator is vulnerable?
We now turn to the last player in the story, the information corruptor.
The Corruptor
To appreciate the corruptor, we must first ask what is corruptible and what corrupted process matters? We maintain that there are three layers of chemical memory that information important for the reading processes; (i) nucleic acid core structure, (ii) nucleobase sequence, and any (iii) nucleobase modification (Figure 3).[90,91,92], EN55 Critical to our model of aging is information integrity at each of these levels, and the ability to read and inherit this information in a way that preserves order.EN56 The replicator core structure is the most heritable. Any change to the global structure of nucleic acid is disastrous and likely quickly fixed or the cell destroyed.EN57 Changes to the core information are not exempt from being a prominent drivers of aging but most core disruptions likely accelerate aging during later stages.
The next most heritable information is base sequence, which corrupts via mutation.EN58 The central dogma of molecular biology states that mutation will manifest in RNA, translate as altered protein, and affect function.[92], EN59 In our view of aging, corruption of an individual protein by some spurious chemical reaction is transient: the damaged molecule is destroyed and recycled. What is not transient is the change in the information used to build that protein. If the memory of how to construct it correctly has been erased and replaced, then the new information state will, in most cases, likely decrease the original intent to order. Since the prime directive dictates that the resulting progeny will inherit this altered information, all newly-made proteins must inherit the loss of full function.EN60 And, any mutations that affect other processes (transcription, translation, nucleic acid stability, etc.), will also be amplified up the functional levels of the cell.
Corrupted coding regions also affect RNA information. Transcribed mutated RNA creates toxic repeats/hairpins [93] or corrupts translation efficiency.[94,95,96] Intropy loss in non-coding regions augments coding errors by disrupting the regulation of order, creating a timing problem. Because regulatory regions control the timing, location, and quantity of gene expression, their corruption has outsized effects: a single informational change in a promoter, enhancer, or splice regulatory element can dysregulate multiple downstream genes simultaneously, amplifying functional inefficiency even more than what a single coding mutation could achieve.[97,98] Transcription, translation, and protein action/function aside, even the efficiency of the prime directive (replication) will corrupt by such informational changes.[99,100,101,102], EN61 Once memory is lost, so are the instructions to order, intropy falls and entropy grows.
Perhaps an analogy is most appropriate. If in the reading or copying of this paragraph, typos are made because of some small smudge on the letters (modifications or corruptions), the message may remain interpretable. However, if each copy accumulates typos over time, the meaning of the words will be lost. If the letters are the nucleobases of the replicator and life the reader, the instructions to order function will be uninterpretable with each divisional loss of information. When corruption occurs in key words (e.g., a verb), although the sentence is largely intact, its meaning is not. Thus, where the corruption occurs is critical and hotspots of information corruption are likely to accelerate aging and aging-related pathologies.
With this vantage, the Mutational/Damage Theory of Aging is very pertinent as a necessary step in the aging process. [103,104,105,106] Mutations are permanent, change information, are successive, heritable, and universal, all requirements for causality. Mutations generationally permeate functional loss in the forward direction. Simple organisms limit lineage-level meltdown with purifying selection; complex organisms do the same with stringent germline control. Germline mutation rates are lower than somatic cells, defective germ cells are culled, and mobile elements silenced.[107,108,109,110,111] Some fraction of the replicator remains viable even as somatic lineages accumulate irreversible errors.[112,113] The most damaging or catastrophic mutations are likely selected against and eventually lost from the cell population; however, some harmful mutations will persist, particularly those whose effects on functional order are too subtle to be effectively selected against. Both are important for aging.
We argue, though, that mutation is not the original sin, which is why additional ideas on aging need to be reconciled. We propose the most basic corrupting elements, and causal driver of intropy loss, are molecules/atoms that modify nucleic acid, particularly at the level of nucleobases (Figure 5).EN62 These modifications are numerous, diverse, and will alter information in a myriad of ways. They can be substantial and lead to global damage such as single or double strand breaks. They can induce polymerase slippage, causing mutations, including base insertions, base deletions, or other major nucleic acid reorganization (inversions, transversions, recombination, etc.). They can be even more subtle, such as epigenetic modification that has intended biological function or modifications (mostly adducts) that are not intended but still alter the way the cell interacts with the nucleic acid. Intended or not, such modifications may occur at much higher frequency than permanent mutations or overt damage, may exist in dozens of different reactive chemistries with nucleic acid, and can change the information to order in nearly infinite ways (see EN67). Regardless of the type, chemistry or place of the modification, they all have one thing in common: they will change the original biological information to another state, and likely to a state that is damaging to the order needed to function well.
The genetic evidence supports this hierarchy. Of all the aging-related diseases and evidence in the literature, we maintain the progeroid syndromes are as close to a human genetic “model” of the complex dichotomy and processes observed in aging as one can observe. They reveal a pattern in which defects that leave transcription-blocking modifications in expressed genes tend to produce premature aging while defects in replication fidelity produce cancer without shortened lifespan. Pathway redundancy and the non-repair activities of several repair factors blur individual cases, but the underlying axis separates corruption that stalls the reading of genes from corruption that silently miscopies them (Supplemental Figure 3). As such, a close examination of the molecular mechanisms that underlie progerias reveals direct clues into cause.EN63, EN64
We thus define and name all the potential modifiers of information as nucleobase information corruptors (NICs), or “corruptors”. We choose the word corruption only because we wish to connote here that the modification is not intended, results from some local environmental error or chaos, and will likely, if not removed, change the information and thus any downstream biology that results from its reading (or misreading). NICs are thus highly reactive (or activatable), promiscuous, diffusible, small, and likely form covalent adducts to the atoms of the nucleobase ring (Figure 5). Their reaction with the replicator instantaneously changes life’s information and alters the memory to order.EN65 Any reaction of such corruptors with proteins, lipids, or metabolites is largely transient, the damaged molecules replaced by newly synthesized copies built from the existing information template. But if that template itself is corrupted, the conversion of that information is not only transiently altered at that moment, it may, upon copying and/or reading, seed that intropy loss into the fabric of all future progenitors, cellular processes, and anything built from cells.EN66 A corruptor is thus defined as a reactive molecule or atom that induces transient or permanent information change such that life’s functional ability to order is disrupted.EN67 Corruptors are the physical manifestation and direct henchmen of EAI.
This segues well into the third level of replicator information, the epigenome. We view the epigenome as a reversible regulatory layer of additional information for an already informationally dense molecule that provides control over when, where, and how much of the embedded irreversible information is read. The Epigenetic/Information Theory of Aging posits that age-related drift in DNA methylation and chromatin disrupts transcriptional programs and drives functional decline.[114,115] EN68 That is, alteration of the epigenome (post-replicational modified nucleic acid) dysregulates gene expression, the protein product, and other cellular function.[18,116,117] As part of the replicator’s information, we agree that alteration of the epigenome facilitates aging, with one important exception regarding its causality: it is a component of the information content of nucleic acid, not nucleic acid’s sole information-bearing system. And, importantly, it is not mutually exclusive with the other informational content, deeply intertwined and quite possibly inseparable from it.
Epigenomic information is maintained across cell divisions through dedicated but imperfect mechanisms, yet it lacks the template-directed fidelity of sequence replication.EN69 The enzymes that copy epigenetic marks onto new strands do so imperfectly, and unlike the systems that correct errors in the DNA sequence itself, no machinery exists to restore drifted epigenetic markers to their original states. The sequence (and the local chemical environment it makes) likely dictates epigenomic modification.EN70 Thus, the most logical and sequential model for EAI proceeds first via unintended corruptor modification of nucleic acid which, depending on its location in the genome, may translate to all downstream functionality that depends on that information, some of it passed to future lineages through increased mutations or epigenetic remodeling. This is consistent with the now known feature that somatic mutations coincide with extensive remodeling of the surrounding methylome, and that mutation-based age predictions closely parallel epigenetic clock estimates, suggesting mutations as an explanation for epigenetic aging.[118] The corruption of the epigenome amplifies the global dysfunctionality by altering the translation of information into protective action at the time that action is needed. It may also promote more information corruption by increasing NIC access to nucleic acid. The chemistry of damage and the chemistry of regulation are intertwined: a single oxidative modification on a regulatory site can recruit the repair machinery in a way that erases the nearby epigenetic mark, rewriting the local regulatory landscape from one corrupting event. Some of the cell’s own intended regulatory marks are themselves chemically reactive and can spontaneously convert into mutations, leaving a signature on the genome that tracks time and shows up on nearly every aging clock. If any epigenetic changes are inherited, they likely follow preexisting corruption of the original sequence information. Corruptors therefore act on the epigenome through two routes at once. Acutely, they scramble the cell’s ability to read its own regulatory marks; permanently, they seed sequence changes that destabilize regulation further. Each round of copying amplifies both effects together.
Clonal hematopoiesis offers an empirical demonstration of this cascade in a single tissue: somatic mutations in DNMT3A propagate through stem cell lineages into tissue-wide mosaicism, demonstrating how one molecular event can scale upward to organism-wide consequences (see EN72 for full mechanistic treatment),[119,120] illustrating how a single point of sequence corruption propagates functional consequences upward through every dependent level.EN72 The partial reversibility of aging markers by reprogramming factors has been cited as evidence that epigenetic information loss is an independent and reversible cause of aging,[121] and that a “backup copy” of youthful epigenetic information exists. We argue instead that the DNA sequence itself is the reference from which epigenetic patterns are re-specified during reprogramming, that no separate archive is needed, only the original chemical memory of the nucleic acid template itself, and that the reversal is partial and temporary because the underlying sequence corruption (mutations) and the standing modification burden (which continuously drives epigenetic drift) are not cleared by reprogramming factors.EN73 This interpretation generates testable predictions, including that reprogramming efficacy should correlate negatively with somatic mutation burden, and is elaborated with supporting evidence in the accompanying notes. But through it all and regardless of the exact mechanisms and sequence at play, it is the resulting change in information, and the intropy lost from that change, that drives the downstream effects we see.
When does corruption occur, and how? To interpret the information, the replicator must be read. For it to be read, it must open. We favor a model whereby the highest probability of NIC reactions occurs during any of the activities that read (transcription) or copy (replication) open DNA. We speculate that temporarily unprotected nucleic acid during steady-state (chromatin remodeling, etc.) may also expose DNA for modification. In essence, any process that lowers the protections (“open” nucleic acid), is likely when and where the information is most exposed. These modifications will first affect acute activities related to the conversion of the information into cellular functionality (gene regulation, transcription, translation, etc.). But since the transcriptional and polymerase complexes conform to lock and key catalysis, any physical perturbation of information will also impact enzyme-substrate fit, resulting in decreased fidelity and reading of either RNA or DNA during synthesis of new strands, conversion to the translated message, and anything in between.[122], EN74, EN75
That NICs intercede in these steps, and cause some negative effect on the process, is where many of the original principles around the Free Radical Theory of Aging have merit [123,124] but applied differently.[125] Although radicals can damage any biomolecules, we argue only those that erode intropy, the capacity to translate information into biological order, will propagate functional efficiency loss up the hierarchy of biological organization, some as acute perturbation of cell activity while others permanently embedded in a new code-script.EN76 Although we have no evidence for this, attractive is a hypothesis where radicals generated via Fenton chemistry from the numerous iron-metal cofactors in the polymerase place NICs conveniently in the vicinity of replicating and transcribing nucleic acid. [126,127,128,129,130,131,132,133], EN78
Radicals (and not just oxygen) are excellent candidate corruptors; they are reactive, small, diffusible, constant and ubiquitous.[134], EN79 As protectors grew larger and stacked layer upon layer of organized function, they required copious production of high energy molecules to meet the demands to sustain order, settling on oxygen (in mammals) as a terminal sink for dangerous electrons made during respiration.[85,135,136,137],EN78 It may be that life took a forbidden bite of an energetically-toxic fruit, trading the expansion of replicator protections at the level of an adapted species over indefinitely safeguarding information integrity at the level of the individual. By relentlessly tapping into the energy-rich pathways that fuel evolution’s greatest and boldest creations, we also may inadvertently generate highly-reactive by-products that quietly erode critical original information over time … our own version of Eden’s Apple (Figure 6).[18] It is tempting to speculate that mitochondrial energy generation, a process (via the endosymbiotic theory) that increased the energy ceiling of higher-order cells, creates a proverbial double-edged sword. While meeting our high energy needs, a fraction of its activity also generates dangerous byproducts, not all of which are immediately sequestered or neutralized by systems designed to contain them. Being nearly constantly generated, these molecules may leach from their origin and modify the cell’s nucleic acid information (either directly or indirectly through damaged lipids that themselves are radicals) in ways that, over time, cannot be indefinitely prevented. The fact that some information (mitochondrial nucleic acid) is also in the immediate vicinity of such molecules may explain why mtDNA is important in aging and how sequestering these molecules promotes lifespan.[138] Such a model would explain much of the data associated with how the mitochondria and free radicals are linked to aging phenotypes.
The Mechanism
We thus propose a molecular mechanism of aging incorporating these new ideas and existing theories. The prime directive dictates the replicator must be read and copy (Supp. Figure 1). The reading/copying process cannot occur without mistakes (Figure 1). To read/copy accurately and sustainably, the replicator acquired a type of chemical memory on how to order itself (Figure 2). The chemical memory contains embedded information (Figure 3) that orders the reading process and, depending on the assaults to it, builds layers of protections, thereby safeguarding it from stochastic environmental assaults that would otherwise disorder interpretation and prevent the prime directive (Figure 4). The information of primordial replicators directed self-catalyzing duplication. The replicators of today house vast amounts of additional and diverse information which directs and constructs a protective copying vessel. Subtle but beneficial errors in the process were selected by the diverse environmental challenges to the original information. To read the information, it must be accessed. To access the information, the protections must be lowered. Lowering the protections exposes the information to localized environmental chaos, much of which is already constrained but not indefinitely so. The surrounding chaos assaults the integrity of the information (EAI). The EAI is physically manifested as reactive corruptors (Figure 5). To achieve greater feats of protection in the face of greater environmental assaults and challenges, incremental information changes that harnessed more dangerous energy sources were selected (Figure 6). This may generate more corruptors via a process that is always on, thus creating endogenous assaults that may just be more significant than exogenous ones since the protections are so strong. The corruptors change the original information. Most errors erase chemical memory to order, but some preserve it or even have been utilized to add additional levels of information regulation and control. Since EAI may challenge and corrupt information in near infinite ways, the protections that resist EAI will be equally diverse.[139,140,141,142] The physical and permanent representation of this diversity is life.
How does a single chemical modification of a nucleobase, a corruption, translate into functional decline? Any single modification takes one of these paths, but across the genome we argue there are at least seven main channels that operate in parallel, each drawing from a different subset of the total modification burden (Figure 7).EN81 These channels fall into three functional groups: readout corruption of an intact template (i-iii), acute regulatory disruption that resets with repair (iv), and conversions of transient modifications into heritable corruptions (v-vii).
The first three channels corrupt the readout of a template whose underlying sequence remains intact. First, bulky or helix-distorting modifications physically block RNA polymerase II, silencing the affected gene until the lesion is cleared and transcription re-initiates from the promoter; because longer genes present larger targets, this produces a systematic bias against the expression of large genes with age.[143,144], EN82 Second, modifications that do not block the polymerase may nonetheless be miscoded during transcription, a process termed transcriptional mutagenesis, in which the polymerase inserts an incorrect ribonucleotide opposite the lesion and every subsequent transcript from that locus carries the same error until the modification is repaired.[145,146] This channel introduces continuous noise into the proteome without any change to the DNA sequence, potentially contributing to the protein misfolding and aggregation observed during aging.[147] Third, modifications at splice sites, branch points, or exonic splicing regulatory elements can alter co-transcriptional splicing decisions, producing aberrant transcript isoforms from genes whose underlying sequence is intact; aged tissues show increased Pol II elongation speeds and systematically more splicing errors, making this one of the few channels where an upstream pressure on transcriptional tempo traces to measurable downstream dysfunction.[148,149,150]
The fourth channel corrupts cellular regulation without heritable consequence. Modifications within transcription factor binding sites, enhancer elements, or insulator regions can directly alter regulatory output by reducing binding affinity, disrupting enhancer-promoter contacts, or shifting topologically associated domain boundaries. These effects reset when the modification is repaired, which distinguishes them from the heritable regulatory corruption described next.
The final three channels convert a transient modification into a heritable corruption. Fifth, modifications at regulatory loci, particularly CpG islands, can initiate changes in chromatin state and DNA methylation that propagate through cell division long after the initiating lesion has been repaired; an oxidative hit at a single CpG can become a self-sustaining epigenetic alteration inherited by all descendant cells. Sixth, in dividing cells, modifications encountered during replication are either misread or bypassed by error-prone polymerases, converting the transient modification into a permanent heritable mutation. This is where information corruption becomes irreversible at the sequence level and where cancer risk primarily resides (see EN60).[151,152,153], EN83 Seventh, modifications encountered during DNA replication can also cause fork collapse when the replisome cannot bypass or tolerate the lesion, converting a transient modification into a double-strand break. These breaks are severe forms of information corruption: they can delete genes, cause translocations, trigger apoptosis, or induce cellular senescence with its attendant SASP; even successful repair by non-homologous end joining is error-prone and can leave behind insertions or deletions that propagate to all descendant cells.[45,99,154]
The downstream consequence of any given modification thus depends on where in the genome it falls, what cellular process is engaging that locus at the time, and whether repair intervenes before the next readout event. The cumulative operation of all seven channels across the genome constitutes the intropy loss that, in our framework, manifests as aging. We acknowledge there may be other molecular channels by which information may corrupt and manifest but if discovered, this framework states that the effect will translate back to altered information states at foundational levels.
Assault on information would seemingly be destructive to life. This is where evolution and lifespan are key features of this framework. In some life, a bargain was struck. Complex organisms delay the prime directive in a select group of special prime-directive inactive replicators (germ line).EN84 By keeping this information relatively unexposed and unread, the number of potential assaults to the information is lower, preserving a relatively uncorrupted information copy. At the right moment, that unaltered copy is advanced to a new vessel, restarting the clock and buying more time before catastrophic corruption. Unfortunately, the prime directive must continue, the mistakes in information must accrue, and the translation of that scrambled information must manifest in the breakdown of protections, therefore accelerating more errors. Emerging data support this self-reinforcing logic quantitatively.EN85 The cell’s two main systems for clearing modifications and bulky lesions from DNA both lose capacity measurably with age, with the first falling by as much as half in aging neurons and the second declining roughly 1% per year in human immune cells.[155,156] Only a fraction of this decline comes from age-related silencing of the genes that encode these repair systems; in mouse brain, such silencing accounts for about one-third of the loss of one key repair enzyme, and in other cohorts it accounts for none.[157,158] As repair capacity drops, the standing burden of unrepaired modifications rises two- to three-fold,[159] and in aged mouse liver roughly 40% of actively transcribing RNA machinery is stalled on damaged stretches of DNA, reducing productive output by about 1.5-fold in a pattern that tracks stochastic damage rather than age-related silencing.[143] The repair genes themselves, being read from the same damaged template as every other gene, are subject to the same readout corruption they exist to prevent. Together these measurements describe a system in which the substrate of information corruption rises faster than the cell can repair it, with the repair machinery itself subject to the same erosion.
Thus, nucleic acid-level information change (corruptor modification, epigenetic alteration, mutation, breaks and much more) is translated as functional inefficiency, intropy loss, upwards through to molecular, cellular, tissue, organ, and whole-organism hierarchical levels, manifesting differently across biological systems and species and giving rise to the various “hallmarks” associated with aging (Figure 8). For example, transcription-blocking modifications stall Pol II on long neuronal genes, reducing the supply of proteins those genes encode and producing the aggregation and cellular dysfunction observed in aged brain; somatic mutations in epigenetic regulators such as DNMT3A drive clonal expansion in hematopoietic stem cells, skewing differentiation and eroding immune surveillance; and persistent modifications encountered at replication forks generate double-strand breaks that trigger DDR-mediated elimination of stem cells, thinning regenerative pools across turnover-dependent tissues. Each route begins with a single molecular event at the replicator and amplifies through cellular, tissue, and organismal levels until the downstream consequences present as recognizable aging phenotypes. Evolution counters the disorder by masking the most egregious intropy loss with compensatory dampening adaptations, slowing aging (intropic decay) and death (intropic collapse). It is tempting to speculate that what we “see” as aging progression or phenotypes in the individual is evolution’s counter to catastrophic collapse, sort of softening and thereby delaying the effect to give the prime directive more time to occur. But over time, the loss of intropy translates into so much functional deficiency that an order threshold is crossed, the rate at which mortality accelerates with age dictated by the underlying kinetics of corruption accumulation, strikingly consistent with Gompertz mortality dynamics.EN86 After 3 billion years evolving a protector to neutralize an unpredictable environment, the greed to execute the prime directive essentially catalyzes the remaining vessel’s descent to chaos, the intropic collapse we call death, a sacrifice to entropy, the payment now made, the laws of thermodynamics balanced and maintained. Many of the tenets of this integrated theory we outline in a step-by-step process in Table 2. Most of these steps are already supported by existing data, others require experimental falsification, and others still will require new technologies to address adequately.
Conclusions
Amortality and the Future
Can the individual become amortal? The straight answer is that if the ideas presented here are true, it offers the prospect that the mechanistic prevention of information corruption at early steps would help preserve intropy. If Eden’s Apple is a primary source of corruption, since energy use is inevitable, the solution may lie in reimagining biological information.
Whether the dominant corruptors prove endogenous or exogenous, the underlying problem is the same: information must be safeguarded from assault. Like a vaccine against a virus, highly organized life may require a shield against entropic corruption, an engineered protectosphere for sustained intropy.EN87 However, substantial ethical, technological, and societal changes will be required since the solution will require dramatic alteration of the information content of our species and others. In the beginning, science will need to examine the relationship between information corruption, the intropy it erodes, and the resulting aging phenotypes, identifying corruptors, mechanisms of corruption, and the information that is essential to guard. The ideas herein suggest a blanket level of protection may not be necessary; instead, targeting the key players promoting corruption, or ones that prevent and repair it, or even the information hotspots themselves, may be the first steps. The first artificial genome protectors may be small molecules that neutralize or disarm corruptors. EN88 Against a relentless process, neutralizer delivery, localization, and timing will be challenges.
Another tactic may be to embed the protection in our information itself.EN89 One might envision expressing enzymes that destroy corruptors, or deliver replicative machinery enhanced to better read or repair corrupted information. New genetic delivery/editing mechanisms (DNA/mRNA nanoparticles/CRISPR), as well as ways not yet discovered or invented, will aid in converting the reimagined information to reality. We may also reboot our information with nucleobases resistant to attack, or remove hotspots, or both. Such advances may prove orthogonally useful for ending cancer, correcting germline genetic diseases, and even protecting astronauts from cosmic radiation.[160,161,162], EN90 But acceptance of such methods will require societal support for there is danger of using such methods to not only protect corrupted information but also to attempt to enhance it.EN91 One also wonders if the ultimate solution is a transition to a digital substrate.EN92 Moving at the speed of light, the information can be beamed across space to seed life where there is low EAI, with the replicator reconstructed at the destination and its information preserved with a fidelity that carbon-based transit cannot match.
There are countless questions still left unanswered. These will provide fertile grounds for many studies. Some of the main questions, though, center around origins and mechanisms. What are the main information corruptors and where, when, and how are they made? Which changes in information are most important for disease and aging phenotypes, and where, when and how it is modified? What consequences of changed information are the most consequential for loss of intropy, and where, when, and how in the hierarchy of organismal life are these most important? Do these answers change from species to species, cell to cell, and nucleic acid to nucleic acid? And finally, the question on everyone’s mind, can this knowledge lead to a slowing, prevention, cessation or even reversal of the process?
Through an understanding of aging mechanisms, humanity fights the only universal adversary that is a threat to all ordered life: entropy. Intropy, becoming the thread uniting all living creatures, a banner against eroding entropic forces, humanity may yet rally a resistance against disorder and its cold, unpurposeful starkness. To achieve this, it seems we must first break the original agreement that binds us to mortality. If the framework presented here is correct, we may have a clear path ahead: identify the corruptors, map the information they target, and develop the tools to sustain intropy, thereby halting the propagation of inefficiency and disorder up the hierarchy. The diversity of life, which we have argued is the replicator’s ultimate protectosphere, becomes not merely a heritage to appreciate but a resource to preserve, for in that diversity lies the resilience of intropy against disorder. Human intelligence, and all the protective technologies invented from its engagement, give the replicator its only known direction beyond the fate of a single world. Released from ancient forces that turn us against each other, humanity may pioneer a new frontier, becoming the champions and custodians of all life, big and small, carrying this purpose across the galaxy.
Explanatory Notes
EN1. Is aging a process or a disease, and why hasn’t evolution eliminated it?
Whether aging should be classified as a disease or a natural process is actively debated. The distinction hinges on how each term is defined. A biological process is typically understood as a universal, intrinsic feature of life, like development or reproduction; a disease implies a departure from normal function with identifiable causes, diagnostic criteria, and response to intervention.
Those who argue aging is a disease point to its progressive, degenerative character and the growing list of identifiable molecular mechanisms (e.g., genomic instability, epigenetic drift, mitochondrial dysfunction, cellular senescence) that collectively drive functional decline.[18,19] Aging has measurable biomarkers, including epigenetic clocks and frailty indices, and several interventions (caloric restriction, rapamycin, senolytics) demonstrably slow its trajectory in model organisms.[287,288] On these grounds, Khaltourina et al. (2020)[288] argued that biological aging satisfies the WHO’s own diagnostic criteria for disease classification in ICD-11, leading to the inclusion of an extension code for “ageing-related” conditions (XT9T) in the causality section.
Those who argue aging is not a disease emphasize its universality: diseases are, by convention, deviations from a normative healthy state, yet every organism ages, making it difficult to define a “normal” against which aging deviates.[289,290] There are also practical concerns. When the WHO initially proposed including “old age” as a diagnostic code (MG2A) in ICD-11, international opposition from gerontological and geriatric societies led to its relabeling before the official 2022 release, partly on grounds that such classification could reinforce ageism and distort cause-of-death epidemiology.[291] The WHO ultimately replaced the term with “ageing-associated decline in intrinsic capacity,” explicitly stating that ICD-11 does not classify old age as a disease.
For the purposes of this manuscript, the distinction is immaterial. Whether aging is labeled a disease or a process, the underlying molecular observations do not change. Functional decline is progressive, universal, and measurable regardless of what we call it. The framework presented in the following pages addresses the mechanism of aging, not its classification. We occasionally use the term “progressive pathology” to emphasize that aging has identifiable molecular causes that are, in principle, amenable to intervention, without taking a formal position on whether it constitutes a disease.
Regardless of classification, a more fundamental question remains: why doesn’t evolution select against this “disease” or pathology so we do not succumb to it? Asexual reproduction can blur the boundary between parent and offspring, but it does not eliminate copying error, and no lineage can replicate indefinitely without error.[112,292] Even with proofreading and repair, mutations accumulate because copying fidelity is finite.[112,292] In finite asexual systems, Muller’s ratchet can drive stochastic loss of the least-mutated members, increasing mutational load unless counteracted by compensatory mechanisms (selection, mutations, recombination, etc.).[293] An analogous information constraint: for a given replicator length and the selective landscape, fidelity must remain high enough for sequence information to be preserved.[14] Above this threshold, sequence information loses coherence and decays into noise. If one exceeds this threshold, sequence information loses coherence becomes indecipherable noise. For life, one issue with staying below this threshold is that physical error correction is not free: kinetic proofreading and information erasure impose energetic costs, and arbitrarily high fidelity becomes increasingly costly under finite-energy constraints.[8,11,204]
Claims of biological immortality, such as bacteria dividing “forever” or the jellyfish Turritopsis dohrnii, do not withstand detailed scrutiny. Bacteria accumulate mutations; no experiment has tracked a single cell lineage indefinitely without error.[294] Turritopsis reverses its life cycle through developmental remodeling and transdifferentiation, which is not evidence that an individual genome is maintained indefinitely without damage or mutation.[295,296] Self-renewing and regenerating tissues face similar limits: repeated cell divisions still incur replication errors, and even whole-body regeneration in planaria approximately triples the de novo mutation rate per generation.[211,297] Evolutionary change itself demonstrates that informational corruption is unavoidable.[112,113] Thus, indefinite production without information change is not a realistic biological strategy under finite energy, finite time, and nonzero molecular error rates.[11,204] From this perspective, reproduction and developmental resetting are lineage preserving strategies that are lower-cost than indefinite somatic maintenance. Mortality is not necessarily selected for directly; it emerges because selection favors sufficient maintenance across the reproductive window rather than perfect preservation indefinitely.[298] Under this framework, the nucleic acid information is seemingly immortal while the carrier is not. And nucleic acid’s information corruption through time, consistent with physical laws, benefits the carrier by introducing adaptation in the face of chaotic environments.[14,112]
What about bacteria? They persist not through individual immortality, but through population-level redundancy and purifying selection. While individual lineages accumulate mutations and degrade, the vast population size ensures that some viable offspring remain, their information content now pushed forward. Selection filters out deleterious variants, and horizontal gene transfer or mutation introduces functional alleles, partially counteracting Muller’s ratchet.[299] This statistical strategy allows bacterial populations to maintain overall viability despite the inevitability of error accumulation in any single lineage. But with their fast reproduction rate and smaller genomes, information corruption can be experimentally charted, thus making them a good cellular model system to track changes in information molecules, identify driver hotspots for information corruption, and pinpoint the contributors and the inhibitors, with the caveat that bacterial findings inform but do not directly predict the more complex protectosphere architecture of mammals.[63,64,300]
EN2. Are there exceptions to actuarial senescence?
While the Gompertz-Makeham model accurately describes increasing age-specific mortality in many species, exceptions exist.[301] Certain taxa exhibit negligible or even negative senescence, where mortality remains constant or declines with age.[302,303] Examples include Hydra, naked mole rats, proteinaceous deep-sea corals, and long-lived plants.[302,303,304,305,306] These organisms appear to reduce or delay actuarial senescence through varied mechanisms, including continuous stem-cell renewal in Hydra, modular and indeterminate growth in plants, and enhanced somatic maintenance in naked mole rats. Within the framework described here, these strategies are best read not as exceptions to aging but as mechanisms that delay the phenotypic consequences of molecular corruption.[18,303,307]
Such cases also highlight alternative life-history strategies under specific ecological and evolutionary conditions. Even in these species, molecular fidelity of nucleic acid is not absolute. Across biological systems, mutations, epigenetic remodeling, and molecular damage still occur, though their phenotypic impact may be delayed or buffered by compensatory mechanisms.[18,75,308] For example, the naked mole rat, often considered the strongest mammalian case for negligible senescence, nonetheless exhibits clear age-related methylome remodeling and information loss when assessed by bisulfite sequencing, and an epigenetic clock built from these data accurately predicts individual age; the rate of epigenetic aging tracks maximum lifespan, proceeding far slower than in mice but faster than in humans.[309] Similarly, somatic mutation rates in the naked mole rat scale inversely with its lifespan, falling on the same regression observed across 16 mammalian species.[210] These findings are consistent with the framework’s prediction that negligible demographic senescence reflects slowed or buffered information corruption rather than its absence. The framework herein accommodates these exceptions: the underlying process of error accumulation persists, though its physiological manifestation is postponed. In multicellular organisms, this delay need only preserve somatic function long enough for a relatively protected germline copy to be transmitted. In unicellular systems, continuity is maintained statistically through population-level selection among progeny rather than through preservation of any single cell. This feature is very similar to Kirkwood’s disposable soma hypothesis, an idea that we maintain can be integrated into the framework presented herein.[165]
These cases are uncommon because they likely require unusual combinations of life history, ecology, body plan, and maintenance investment. Continuous somatic maintenance and repair require high energy investments, which reduces resources available for reproduction.[310] In environments where extrinsic mortality (predation, disease, environmental hazards) is high, selection favors early reproduction over costly maintenance.[311] Negligible senescence is therefore most common in species with low extrinsic mortality and stable environments (i.e. less stochastic threats endangering life), where the payoff for long-term maintenance outweighs its energetic costs.[302,310]
EN3. Thermodynamics and the persistence of order
Life exists as an open system that maintains local order by exporting entropy to its surroundings (heat, CO2, waste, etc).[312,313,314] Early development produces a highly organized, far-from-equilibrium state. Over the lifespan, unrepaired molecular and cellular disorder accumulates, gradually reducing the organism’s capacity to maintain that state. Death marks the failure of active homeostasis; the formerly maintained system then relaxes passively toward thermodynamic equilibrium through cooling, autolysis, and chemical degradation over hours to weeks. This is consistent with the second law of thermodynamics: global entropy increases even as local order is sustained.[312,313,314] Maintaining low entropy requires continuous energy dissipation, and every process of copying or repair incurs an energetic cost.[204,312,315] Arbitrarily high fidelity requires increasing energetic, kinetic, and architectural investment, making perfect fidelity biologically unrealistic under finite time and resource constraints.[204,315] Consequently, residual errors accumulate despite proofreading and repair.[75,292] Aging, in this framework, is not strictly mandated by the second law but is an expected consequence of finite resources and imperfect information management under thermodynamic constraints.[316] Life maintains itself as an open system to export entropy, yet simultaneously compartmentalizes itself to create a relatively isolated internal environment via membranes, controlled molecular gates, and hierarchical biological organization. Within these compartments, the protective mechanisms themselves are subject to stochastic molecular damage, replication errors, and repair infidelity, including damage to the systems that maintain the information substrate of life: nucleic acid. In an aging organism, accumulated unrepaired molecular disorder tends to grow over time, a trend that repair and maintenance can slow but not reverse indefinitely .[312,313] What we describe in this manuscript as intropy is the system-level capacity to maintain functional order against this thermodynamic demand; aging is the gradual loss of that capacity rather than an inexorable consequence of the second law itself.
EN4. Nucleic acids are a form of chemical memory
Across all known life, hereditary information is encoded in nucleic acids, understood here broadly to include DNA, RNA, and related nucleic-acid systems.[92] Their linear sequence and complementary base-pairing enable high-density information storage and templated replication.[65] RNA-based systems likely preceded DNA, and some viruses still rely exclusively on RNA.[317,318] Protein-based inheritance, such as prion propagation, can alter phenotype, but lacks the universal, genome-wide instruction set and stable replication machinery that are hallmarks of nucleic acids.[319,320] Epigenetic marks, chromatin states, and structural modifications add regulatory layers but do not replace the primary role of nucleic acids as the core chemical memory.[321] This universality explains why the model or ideas presented herein center on nucleic acids as the foundation of biological order, a type of molecular memory for how biological order is reproducibly constructed and maintained.[65,92] The fidelity of nucleic-acid maintenance constrains organism function, cancer risk, and longevity, as illustrated by DNA-repair disorders and age-associated mutation accumulation.[210,316,322] Progressive corruption of this chemical memory propagates into loss of biological order.[323] Thus, we argue informational change in nucleic acid is the primary upstream driver of aging in this framework, and the functional manifestation of its loss of chemical memory is downstream of that, an effect.[323] For the purposes of this framework, phenotypic endpoints are treated as downstream readouts. The central mechanistic question is what corrupts the information substrate and how that corruption can be prevented, repaired, or buffered. In the vocabulary of this framework, chemical memory is the physical substrate of potential intropy.
EN5. Defining the error: mutations and modifications.
Throughout this manuscript, we distinguish two forms of informational change in nucleic acid. Mutations are stable alterations to nucleotide sequence (substitutions, insertions, deletions) that become heritable to daughter cells once propagated through replication. Once fixed, they are not normally recognized as damage because the cell no longer has an unambiguous record of the ancestral sequence.[63,75] Modifications are chemical or structural changes to existing nucleic-acid bases or backbone chemistry that alter, obscure, or regulate the information read from the molecule without (yet) changing the inherited sequence.[63] Modifications encompass two functionally distinct categories: unintended lesions caused by reactive molecules (oxidative adducts, alkylation products, deamination products, abasic sites, single- and double-strand breaks, interstrand crosslinks, DNA-protein crosslinks) and directed regulatory marks on the nucleic acid itself, such as 5-methylcytosine and other DNA base modifications, that cells place deliberately to control gene expression. Chromatin-associated marks such as histone modifications add a further regulatory layer but are not themselves nucleic-acid modifications. These categories are not mutually exclusive. For example, 8-oxoguanine is an unintended oxidative lesion that can also behave as an epigenetic-like signal by recruiting OGG1 and influencing local methylation or chromatin state.[229,231] Because the same chemical change can function as insult or signal depending on context, we adopt “modification” as the umbrella term rather than “damage” or “lesion” (which connote unintended insult and information loss respectively) or “adduct” (which excludes subtractive changes such as breaks and abasic sites). The choice is deliberate; specific terms (lesion, adduct, break, modification class) are used when discussing specific subclasses. Lesions may be individually transient because repair systems remove them, while regulatory marks may be written, erased, maintained, or diluted through replication. Both are functionally consequential while present: those that distort the DNA helix or block RNA polymerase progression impair gene expression in real time, and those that persist through a round of replication risk conversion to permanent mutations through polymerase misincorporation.[75,324] The distinction matters because our framework argues that modifications, not mutations, are the earliest and most frequent corruption of information, and that mutations are best understood as one relatively irreversible downstream consequence of unrepaired lesions, replication errors, or repair infidelity rather than as the primary insult (see EN62, EN66). We return to this distinction repeatedly in the notes that follow.
EN6. Asymmetric damage segregation in bacteria. Empirical precedent for the soma-germline logic in the simplest replicator.
The soma-germline asymmetry that complex organisms instantiate through dedicated reproductive tissue has a precedent in the simplest replicators. E. coli divides by apparently symmetric binary fission, yet the daughter inheriting the old cell pole shows progressive fitness decline while the new-pole daughter starts relatively fresh.[294] One lineage absorbs damage; the other receives a partial reset. This is not a true germline-soma division, since both daughters remain reproductive, but the underlying logic, that asymmetric inheritance of accumulated damage allows replicative continuity under imperfect maintenance, predates dedicated germ cells by billions of years. The molecular mechanisms and broader implications for aging dynamics are treated in EN48.
Two features of this finding are particularly relevant to the framework. The asymmetry in E. coli is geometric (each cell has one old pole and one new pole) rather than hereditary, so the analogy is to the underlying logic of asymmetric inheritance rather than to a lineage precursor. More importantly, E. coli aging occurs despite both daughter cells inheriting essentially identical genomic information at standard mutation rates of roughly 10⁻³ per genome per generation.[325] This means the asymmetry in fitness between old-pole and new-pole cells cannot be driven by differences in DNA sequence, at least not over the timescale of a few divisions. Rather, it reflects the differential inheritance of damaged cellular components, including modified and aggregated proteins, and possibly oxidatively damaged membrane lipids, that accumulate at the old pole. Within the framework proposed here, this represents the cellular-level analog of the somatic burden: even when the information substrate is shared, the physical context in which that information operates can be asymmetrically degraded. It also reinforces our argument that bacteria serve as tractable model systems for studying the fundamental chemistry of information corruption (see EN14, EN15), because some general principles of damage accumulation, asymmetric segregation, and functional decline can be observed in bacteria within hours, while recognizing that mammalian aging involves additional tissue-level, immune, endocrine, and developmental layers.
EN7. Imperfect copying and the inevitability of error accumulation
We are using the phrase “prime directive.” All life contains nucleic acid and all life duplicates it. Lineages that fail to replicate are outcompeted and lost, so the replicators that persist are the ones that successfully execute this directive. We use “prime directive” as shorthand for the selection constraint this imposes: a lineage continues only if it preserves and transmits its heritable information well enough to reproduce. Across known life, this requires maintaining the integrity of the code-script while permitting the rare variation that selection acts on. Replication fidelity in biological systems is high but not absolute.[75,292,324] DNA polymerases incorporate errors at rates typically between 10-7 and 10-10 per base per replication, even with proofreading and mismatch repair [75,292,324,326]. These mechanisms reduce, but cannot eliminate, copying errors because error suppression has energetic and structural limits.[8,11,204]
Residual errors that escape repair can become fixed during replication and propagate to descendant cells.[63,75,326] Over successive divisions, these changes accumulate, and some fraction of them disrupts gene function, regulation, or tissue maintenance. This cumulative information loss underlies the progressive decline in cellular and organismal function observed as aging.[211] The process is effectively unidirectional: once fixed, the change cannot be reversed by reference to the original template (as discussed above).[63] However, the step preceding mutation, the chemical modification of a nucleobase by a reactive molecule, is itself functionally consequential even before replication converts it to a permanent sequence change. Modifications that distort the template can stall transcriptional machinery and impair gene expression in real time, meaning that the information to order is disrupted at the moment of modification, not only upon its fixation as a mutation during the next copying event. This distinction is critical: modification is typically the earliest reversible corruption, and mutation is one of the earliest irreversible forms. Both compromise the fidelity of information transmission (see EN66). Errors in long-lived self-renewing cells and highly proliferative progenitors (such as hematopoietic, intestinal, and basal epithelial stem cells) have disproportionate potential to influence aging because they can be clonally amplified across tissue.[120,211,327] In postmitotic tissues, damage in non-dividing cells does not propagate clonally but can still impair tissue function directly. A model is proposed later that displays aging as resulting from progressive information corruption along a hierarchical functional chain.
EN8. On the name “Intropy.”
The word intropy is coined for this framework. We build it deliberately in parallel with entropy, the term Clausius introduced in 1865 from the Greek τροπή (trope), meaning turn or transformation. Clausius chose a word that echoed energy while naming a distinct direction of physical change: the dissipation and dispersal of usable order in thermodynamic systems. Intropy is constructed to echo entropy in the same way, but to name the biological counter-process: the capacity of encoded information, when realized in a living system, to generate and sustain functional order against that dispersal. Entropy describes the universal tendency toward disorder; intropy describes the capacity life holds against it.
We adopt the term because no existing word captures the biological quantity required here. Negentropy, introduced by Schrödinger and later elaborated by Brillouin, comes closest, but it refers broadly to negative entropy, imported order, or information-theoretic contrast rather than to the specific biological capacity of encoded information to produce self-maintaining functional order. Complexity, order, and organization each name related features, but none distinguishes stored ordering potential from realized biological function. Intropy provides a clean coinage, typographically and semantically parallel to entropy, that admits biologically necessary distinctions such as potential intropy and realized intropy and supports the formal treatment I(t), dI/dt, and Ic developed in EN86.
EN9 Shannon redundancy and the biological logic of protection.
Shannon’s noisy channel theorem contains a second, often overlooked implication for aging biology. The theorem establishes that for any transmission rate below channel capacity, error rates can be driven arbitrarily low, provided the system encodes sufficient redundancy.[12] Life has implemented this principle with remarkable thoroughness. DNA’s double-stranded structure is a redundancy code: if one strand is damaged, the complementary strand provides a template for accurate reconstruction.[65,67] Diploidy doubles this backup for the entire genome. Mismatch repair, base excision repair, nucleotide excision repair, and homologous recombination each add successive layers of error detection and correction, analogous to the cascaded coding schemes that engineers use to approach Shannon’s theoretical limit.[64,74,75] Beyond the molecular level, stem cell reserves provide tissue-scale redundancy, immune surveillance removes corrupted cells, and organismal behaviors reduce exposure to damaging environments. We term this layered system the protectosphere (see EN49), and its architecture follows the same logic that Shannon’s theorem formalizes for engineered systems: because the information substrate is under continuous chemical assault, the replicator must encode, and continuously invest energy in, enough redundancy to keep the effective error rate below the threshold at which information collapses (Eigen-like informational error threshold; see EN10).[12,14]
This framing clarifies why aging persists despite such elaborate protection. Shannon guarantees that sufficiently redundant coding can reduce errors to any desired level, but each additional increment of fidelity demands more energy, more molecular machinery, more time, and more capacity dedicated to maintenance rather than other functions. Evolution, operating under the constraints of finite energy and the declining force of selection with age (see EN26), invests in protection only to the point where further gains no longer improve reproductive success, the principle we term protectosphere calibration (see EN29b). The result is a system that suppresses information corruption with extraordinary efficiency through the reproductive window but that cannot, and was never selected to, sustain that suppression indefinitely. Aging is what happens when the redundancy can no longer keep up.
EN10. Information theory and the corridor of life.
Shannon’s channel coding theorem shows that in a noisy channel, error probability can be made arbitrarily small for rates below capacity with sufficiently long codes.[12] For most noisy channels, exactly zero error at a non-zero rate is unattainable without restrictive channel structure.[13] Approaching arbitrarily low error, particularly near capacity, requires increasing block length, coding complexity, or delay (finite-blocklength limits)[328] and, in physical devices, increasing energy/time which may not be pragmatically possible.[8,204,329] Biological copying faces analogous constraints: DNA replication stacks fidelity mechanisms, such as polymerase selectivity, 3’→5’ exonuclease proofreading, and post-replicative mismatch repair, to suppress errors, yet residual mutations persist because each layer imposes energetic/kinetic costs, and evolution tunes fidelity only where further gains are not worth the cost.[11,74,75,326]
Landauer’s principle formalizes part of this cost: erasing one bit of information requires a minimum energy expenditure of kT*ln(2).[8] Biological error correction is not always direct erasure, but proofreading, repair, turnover, and quality control all carry energetic and kinetic costs that grow with fidelity demands.[11,204,329] As a result, biological systems cannot drive error rates to zero. They operate below an Eigen-like upper bound that applies to any informational replicator regardless of substrate: the point at which error rates exceed the capacity to maintain sequence information.[14] If error suppression fails, the system crosses this threshold and loses informational integrity. These principles explain why perfect fidelity is unattainable and why some residual errors are expected to persist or become fixed over long timescales, particularly in long-lived cells and self-renewing lineages.[12,14,75] The most direct intervention is to drive information corruption rates lower, which should slow aging.[103] If the underlying theory herein is correct, and aging correlates with nucleic acid error rate across the information systems that matter most, then even a modest reduction of information corruption should increase lifespan accordingly (see Supposition 38).
Together, these constraints define a corridor in which life must operate. The upper bound is informational rather than strictly Eigen’s: in complex organisms, redundancy, neutrality, repair, sex, and developmental buffering raise the operative ceiling above the threshold derived for asexual replicators with all sites informational, an informational-load ceiling rather than a fixed numerical limit.[299,330,331] The lower bound is the drift barrier: selection on fidelity becomes ineffective once the marginal benefit of further improvement falls below approximately 1/Ne, the resolving limit of selection.[112,113] Life is therefore confined between an upper bound it cannot exceed without informational collapse and a lower bound below which selection can no longer reliably favor further fidelity improvements. In multicellular organisms, this corridor applies most strictly to the germline. The soma can be allowed to drift above the upper bound for the duration of reproductive viability, with the protectosphere holding off informational collapse long enough for germline transmission. Aging, as it manifests in our species, is a predictable cost of that allocation.
The corridor’s bounds are substrate-independent: they apply to any chemistry capable of heritable information storage and any selection process. Two consequences extend beyond aging. Environments whose unavoidable corruption rate exceeds the upper bound cannot support life regardless of substrate, which gives habitability an informational criterion in addition to the usual physical-chemical ones. The corridor also narrows for complex life because multicellular Ne reductions raise the drift floor while the Eigen-like ceiling stays fixed unless raised by costly, actively maintained evolutionary innovations in the protectosphere. Environments that permit simple replicators may be too narrow for complex ones to evolve. Since complex multicellular organization is a precondition for the network architectures that sustain higher cognition, the rarity of conditions that permit complex life is also the rarity of conditions that permit anything that could be called intelligence. We develop these astrobiological consequences in forthcoming work.
EN11. Why reductionism and causal analysis matter
Aging manifests as a complex phenotype involving numerous interacting pathways.[18,19] Without assigning temporal and mechanistic order to these processes, it is difficult to distinguish cause from effect.[16,332] Reductionist approaches (deconstructing the system into fundamental components) have historically yielded many of the most durable causal insights in biology, from genetics to molecular microbiology.[64,65,67] Applying causal ordering to aging allows identification of primary drivers rather than secondary consequences, which is essential for developing interventions that target upstream processes rather than downstream readouts.[18,19] The field has historically been rich in correlative observations but comparatively sparse in rigorous temporal and causal ordering of the processes observed, which can lead to interventions that target downstream consequences rather than upstream drivers. This is historically understandable. The complexity of aging, the long timescales required for interventional studies, and the technological limitations that until recently prevented single-cell and single-molecule resolution have all made rigorous causal ordering difficult or impossible. The field has also, justifiably, maintained a high bar for causal claims given the long history of premature promises surrounding longevity. Nevertheless, the data have quickly converged to allow us to move beyond correlation, and we argue the cost of continuing to catalog effects without ordering them temporally and mechanistically is that interventions remain targeted at downstream consequences rather than upstream drivers.
EN12 Positive feedback and symptom-focused interventions.
Biological aging involves feed-forward loops in which secondary effects amplify upstream damage and can become secondary drivers themselves.[18,19,333,334] For example, genomic instability can compromise genome maintenance, which in turn accelerates further instability.[78,323] Similarly, mitochondrial dysfunction can alter redox balance, metabolism, and stress signaling, generating reactive species that contribute to further damage to DNA, proteins, and lipids in ways that can reinforce cellular dysfunction.[18,133,335,336] These feedback dynamics make it difficult to distinguish initiating events from downstream consequences.[18,19] Any interventions that target only the symptoms or effects of aging may extend lifespan but will not slow aging itself. These interventions do not address the upstream drivers that sustain these loops. For this reason, we argue most efforts should be directed at reducing, buffering, or repairing information corruption at its source. The account herein attempts to create the conceptual framework to identify causal drivers.
EN13. Baconian reductionism and causal analysis.
Francis Bacon’s Novum Organum introduced the principle of systematic observation and reductionism as a means to uncover causal mechanisms in natural phenomena [16]. Modern mechanistic biology extends this logic by decomposing complex systems into components, activities, and interactions that can be experimentally tested and reassembled into causal explanations.[337,338] This approach has produced many insight in molecular biology, from DNA structure and templated replication to the pathways that detect and repair DNA damage.[64,65] Because aging is highly complex, pleiotropic, and unfolds across multiple scales, this framework emphasizes explicit causal-mechanistic ordering throughout.
EN14. LUCA and the cellular roots of information maintenance.
Comparative genomics and phylogenetic reconstruction suggest that the last universal common ancestor (LUCA) was a cell-like, compartmentalized, energy-harvesting system with core metabolic and replication functions.[17] Although LUCA was already highly evolved and should not be confused with the first replicator, it marks the most recent point at which all known life shared a common cellular architecture. This supports the use of prokaryotic models to study fundamental processes of information preservation and corruption.[63,64] It also supports our reductionist approach since all known extant life descends from a cellular ancestor that already coupled replication, metabolism, and compartmentalization.[339]
EN15. Bacteria as models for molecular biology.
Bacteria have been foundational in elucidating core principles of molecular biology.[340] The discovery of DNA as a genetic material was demonstrated in Streptococcus pneumoniae.[341,342] The operon model of gene regulation, which introduced the concept of transcriptional control, was established in Escherichia coli through studies of the lac operon.[343] Semiconservative DNA replication was demonstrated in E. coli using density-gradient centrifugation.[344] The genetic code was deciphered using E. coli extracts and synthetic RNA templates.[345] Restriction-modification systems were first studied through bacterial-phage genetics, with early work in E. coli, and the Type II enzymes that enabled recombinant DNA technology were first characterized in Haemophilus influenzae.[346,347] CRISPR-Cas systems, now a cornerstone of genome editing, were demonstrated to function as adaptive immune systems in bacteria.[348] These examples underscore why prokaryotic systems remain indispensable for reductionist approaches to fundamental biological questions.[340] We argue aging research should make greater use of bacteria, particularly as it relates to being a basic model replicator, as a convenient experimental tool to identify the mechanistic drivers of nucleic acid corruption and information loss, how that corruption amplifies more corruption, and the cellular pathways that promote or prevent both. Bacteria cannot model organismal aging in full, but they can model the conserved molecular processes that generate, repair, propagate, or segregate damage. Under optimal laboratory conditions, E. coli divides in roughly 20 minutes, its genome is a size that can be easily sequenced, there are knock-out libraries of all known non-essential genes, and there is a plethora of other modified strains that can be used to ease feasibility.[55,349]
EN16 Falsifiability.
Later in this work, we identify specific steps in the proposed corruption cascade that can be individually tested with current or near-future experimental methods (Table 2, Suppositions). However, a theory of aging faces the same challenge as any theory describing a process that unfolds over decades within a complex system: no single experiment can encompass the whole. This does not distinguish it from other foundational frameworks in biology. Evolutionary theory is foundational in biology, yet no experiment has directly observed the full macroevolutionary history of any lineage over millions of years. The timescales are impractical and the variables too numerous. Instead, the theory is validated through converging lines of evidence: fossil transitions, comparative genomics, molecular phylogenetics, directly observed microevolution, and the predictive power of the framework itself. No single line is the whole theory, but together they are mutually reinforcing and overwhelming.
As with other broad biological frameworks, the appropriate standard is convergent, multi-level evidence rather than a single decisive experiment. The individual steps, that endogenous reactive molecules modify nucleobases, that modifications impair transcription and seed mutations, that mutations propagate through stem cell lineages, that the resulting functional decline amplifies up the biological hierarchy, are each experimentally testable and in many cases already supported by existing data. The integrative claim, that these steps constitute an ordered causal architecture whose cumulative effect is aging, is tested not by one decisive experiment but by whether the framework consistently explains new observations as they emerge, makes predictions that hold up under scrutiny, and fails to be contradicted by data that would be incompatible with it. Falsifying observations would include: aging phenotypes proceeding normally despite durable suppression of nucleic acid lesions and mutation accumulation in relevant tissues; transcription-blocking lesion burden showing no relationship with functional decline in long-lived postmitotic cells; or full functional rejuvenation despite high irreversible somatic mutation burden. Positive predictions, listed more comprehensively in the accompanying Suppositions, include the prediction that reprogramming efficacy should correlate negatively with somatic mutation burden, that transcription-blocking modifications should produce more severe aging phenotypes than transcription-permissive ones of equal abundance, and that species with longer potential reproductive windows and lower extrinsic mortality should show proportionally greater investment in information maintenance. We welcome and encourage the experimental testing of each.
EN17. Historical origin of Wear-and-Tear Hypothesis
August Weismann’s essays on aging and heredity (1882, 1891) appear in many histories of aging theory, though his own argument was evolutionary rather than a mechanical wear-and-tear model (see EN26).[20] Later interpretations reframed his observations as a mechanical analogy, likening organisms to machines that degrade with use.[3,21] While intuitive, this analogy lacks molecular specificity and fails to account for biological repair systems.[3,21]
EN18. Evidence for tissue repair and regeneration
Contrary to the wear-and-tear model, cells and tissues exhibit robust repair and renewal mechanisms.[18,64,67,350] Epithelial layers undergo continuous turnover, hematopoietic stem cells sustain blood cell production, and skeletal muscle regenerates via satellite cells.[70,351,352,353] Advances in regenerative medicine (e.g., induced pluripotent stem cell technology and partial epigenetic reprogramming) further demonstrate that tissue integrity can be restored, undermining the notion that cumulative mechanical damage alone dictates aging.[18,193,351,354] In this framework, the key problem is not that tissues are damaged but that the systems responsible for repair and regeneration progressively lose fidelity, transmitting altered regulatory and genomic states to their progeny.[116,117,323]
EN19. Modern critique of Wear-and-Tear Hypothesis.
Current consensus views wear-and-tear as descriptive rather than mechanistic.[18,19,355] Aging persists despite efficient repair systems, suggesting that upstream molecular processes (e.g. genomic instability and epigenetic drift) drive decline in tissue function.[18,19,64,116,323] In this framework, repair failure typically amplifies upstream information corruption, though inherited or acquired repair defects can themselves initiate accelerated aging phenotypes.[18,19,323] The fact that components wear down with use is less important to aging than the loss of fidelity in the chemical memory that the replacement mechanisms read to reconstruct biological order.[64,116]
EN20. Historical origin and rationale of the Glycation Hypothesis.
The crosslinking theory of aging originated with Johan Bjorksten, who proposed in the 1940s–1950s that covalent crosslinks between macromolecules progressively impair tissue function.[24,356] This framework was refined into the more specific glycation hypothesis when Monnier and Cerami (1981) argued that nonenzymatic browning via reducing sugars and Maillard chemistry could occur in vivo in long-lived proteins, providing a specific chemical mechanism.[24,25,187] The hypothesis gained traction because it provided a chemical basis for age-related structural changes in connective tissues.[21,25,28,29]
EN21. Evidence supporting glycation as a contributor to aging.
Advanced glycation end-products (AGEs) accumulate in extracellular matrix proteins with low turnover, such as collagen, leading to decreased elasticity in skin, vasculature, and tendons.[25,28,29,357,358] Experimental carbonyl-stress and glycation-promoting models, including D-galactose and methylglyoxal exposure, can induce aging-like phenotypes in rodents, while antiglycation approaches such as aminoguanidine mitigate some diabetes-associated structural changes.[26,27,359,360] Glycation is also implicated in diabetic complications, where hyperglycemia accelerates AGE formation, providing a pathological model for its role in tissue dysfunction.[359]
EN22. Limitations of the Glycation Hypothesis.
Despite its biochemical plausibility, glycation cannot fully explain aging.[5,18,19,355] Reactive carbonyls arise from multiple metabolic routes including glycolysis and lipid peroxidation, so glycation cannot be reduced to dietary glucose exposure. Aging persists across organisms and tissues through mechanisms beyond AGE accumulation, and intracellular proteins with rapid turnover also exhibit age-related dysfunction independent of glycation.[5,18,19] Furthermore, interventions targeting AGEs have not consistently extended lifespans in model organisms.[25,359,361] These findings indicate that glycation is best understood as an important downstream or parallel damage pathway that can causally drive specific aging phenotypes, particularly in long-lived extracellular matrices, but is unlikely to be the universal initiating cause of aging.[5,18,19,355]
EN23. Origin and rationale of the Hormonal/Endocrine Theory
The concept that glandular secretions influence aging dates to Brown-Séquard’s testicular extract self-injections in 1889 and the surgical rejuvenation attempts of Steinach and Voronoff in the 1920s.[362] The hypothesis was formalized as the Hormonal Theory by Dilman, who linked age-associated changes in hypothalamic feedback thresholds to systemic decline.[189] Age is associated with changes in growth hormone, sex steroids, adrenal hormones, and thyroid-axis regulation, which correlate with age-related changes in body composition, metabolism, and reproductive capacity.[18,363] Hormone replacement studies in humans and animals can transiently restore specific youthful traits, supporting the idea that endocrine remodeling contributes to aging phenotypes.[18,363,364,365] In humans, broad hormone replacement for anti-aging purposes has not been shown to extend lifespan and can increase morbidity, as illustrated by trials of growth hormone, estrogen-progestin combinations, and thyroid hormone in older adults without clear deficiency.[364,365,366,367] At the same time, endocrine pathways are not merely passive markers: reduced GH/IGF-1 and insulin signaling causally extend lifespan in multiple model organisms.[368,369] These findings suggest that age-associated endocrine changes are heterogeneous: some are adaptive, some compensatory, some causally contribute to pathology, and some are downstream readouts of cellular decline. In the framework proposed here, exogenous hormone replacement is best understood as a downstream signaling intervention: it may temporarily restore effector functions, but it does not necessarily repair the upstream molecular corruption that produced endocrine dysregulation in the first place.[18,363,369,370,371]
EN24. Origin and rationale of the Immunological Theory.
First articulated by Roy Walford in the 1960s, the Immunological Theory proposed that aging results from progressive immune decline and dysregulation.[372] Evidence includes thymic involution, reduced naïve T-cell output, and impaired adaptive responses in aged organisms.[18,30,372,373] These changes correlate with increased infection risk, cancer incidence, and chronic inflammation (“inflammaging”).[18,19,334,373] While immune dysfunction accelerates morbidity, it is not universal across all life forms and does not explain aging in organisms lacking adaptive immunity, limiting its scope as a primary mechanism.[302] Nevertheless, immune compromise can accelerate many age-associated phenotypes, which can make immune aging appear primary; in this framework it is better understood as a powerful mediator and amplifier of upstream cellular corruption rather than its universal initiating cause.[18,30]
EN25 Limitations and integration of systemic theories.
In our view, both hormonal and immunological models describe important systemic manifestations of aging but are unlikely to be universal root causes.[5,18] Vertebrate-style endocrine axes and adaptive immunity are absent from many organisms that nonetheless exhibit aging or age-associated functional decline, including bacteria, fungi, plants, and diverse invertebrates.[302] This suggests that any universal root cause must operate at a more fundamental cellular or molecular level than these lineage-specific systemic architectures. In this framework, endocrine and immune decline are interpreted as downstream systemic consequences and amplifiers of cumulative molecular damage, including persistent DNA lesions, genomic instability, epigenetic remodeling, clonal expansion, and altered transcriptional regulation.[18,64,78,116] These systemic failures can strongly accelerate aging phenotypes, but their limited phylogenetic scope argues that they are better understood as organism-level amplifiers of upstream information corruption rather than universal initiating causes.
EN26. Origins of programmed aging concepts.
A classic early programmed account was proposed by August Weismann, who proposed that senescence evolved to remove older individuals, thereby reducing competition for resources and benefiting the species.[20] This interpretation, often referred to as the Programmed Aging Hypothesis, relies on group selection: the notion that traits can evolve because they enhance the survival of the group rather than the individual.[316,374] However, group selection is generally considered too weak to drive aging because alleles that shorten individual reproductive lifespan are difficult to maintain unless they provide compensatory individual benefits. Later, Medawar introduced the Mutation Accumulation Hypothesis, arguing that the force of natural selection declines with age, allowing late-acting deleterious mutations to persist.[163] Williams expanded this framework with Antagonistic Pleiotropy, suggesting that genes beneficial early in life can have harmful effects later, a trade-off favored by selection because early-life fitness dominates evolutionary outcomes.[164] Hamilton later formalized this logic mathematically, showing how the force of selection on age-specific traits declines after the start of reproduction.[264] Kirkwood further refined this logic with the Disposable Soma theory, proposing that organisms allocate limited resources between reproduction and somatic maintenance, with natural selection favoring investment that maximizes reproductive success rather than indefinite survival.[165,310,316] These models explain the evolutionary persistence of aging but do not imply a dedicated “death program.” Modern evolutionary gerontology generally treats aging as a consequence of declining selection with age, life-history trade-offs, and imperfect somatic maintenance, rather than as an adaptation to promote species survival.[316] The mutation accumulation framework operates at a different explanatory level from this framework: Medawar explains why late-acting deleterious effects are not efficiently purged by selection, whereas the intropy framework treats mutations and persistent nucleic acid lesions as the proximate molecular mechanism through which information loss accumulates and propagates.[163]
EN27. Evidence for replicative senescence and its role.
Replicative senescence refers to the finite proliferative capacity of somatic cells, first demonstrated by Hayflick and Moorhead in human fibroblasts.[178] This phenomenon is largely attributed to telomere shortening during successive cell divisions, which eventually triggers DNA damage responses and cell-cycle arrest.[35,38,78] Senescence serves as a tumor-suppressive mechanism by preventing uncontrolled proliferation of damaged cells.[77,375] That said, the inflammatory secretome of senescent cells (SASP) is a potent propagation mechanism that amplifies the underlying corruption.[333] Senescent cells often arise when telomere dysfunction, DNA damage, oncogenic stress, or other cellular insults activate durable checkpoint programs; the resulting chronic inflammatory signaling can then propagate damage and dysfunction to neighboring cells, creating a feed-forward loop. Thus, senescence is not a root cause but neither is it inert; it is an amplifier whose removal (as demonstrated by senolytic interventions extending healthspan in mice) reduces downstream inflammatory and tissue-remodeling consequences without addressing the upstream lesions that produced senescence in the first place.[45,376,377] Senescence is best explained as a trade-off between tumor suppression (cancer prevention) and tissue regeneration. We view telomere shortening as one form of information corruption that contributes to aging, though not as a primary global driver.[35,38,39] In this framework, events that alter, obscure, or misregulate nucleic acid information can contribute to aging when they persist, propagate, or affect functionally important cells. Modifications, mutation, and epigenetic alteration are more likely to be prominent drivers due to the frequency at which these events occur, their universal presence in all cells, and the subtle introduction of inefficiency they cause in downstream processes.[18,103,116]
EN28. Critiques of programmed aging as an adaptive trait.
The hypothesis that aging is an adaptive trait remains controversial and is not the dominant view in evolutionary gerontology.[164,165,378] Adaptive aging would require group-level benefits strong enough to outweigh the individual fitness cost of reduced survival and reproduction, usually under restrictive conditions of population structure, kinship, or ecology.[164,264,378] Ecological variables such as predation, resource limitation, and extrinsic mortality shape the selection horizon for maintenance investment, but they do not imply an intrinsic “death clock.” [311,378] Molecular and genetic studies do reveal pathways that modulate lifespan and aging phenotypes, but these pathways generally serve normal functions in growth, stress response, and tissue maintenance rather than existing solely to induce aging.[18,378] In the intropy framework, aging emerges from cumulative molecular damage and imperfect maintenance, with damage accruing in the information-bearing systems that preserve cellular order. As organisms evolved in ecological niches that extended the reproductive window, natural selection increasingly invested in information protection.
EN29. Evolutionary logic of multi-level protection.
(a) Multi-level protection.
Natural selection favors traits that increase the reliable transmission of hereditary information.[316] As organisms became more complex, additional protective layers evolved that buffer individuals and lineages against environmental and stochastic insults: multicellularity and division of labor (specialized tissues/organs)[84,85,379], social cooperation (risk sharing and defense)[86], and even extra-somatic protection via niche construction and technology (an “extended phenotype”).[88,89] These layers improve short- and medium-term persistence but cannot abolish the underlying accumulation of molecular damage; thus, aging reflects the limits, not the failure, of protection under physical and evolutionary constraints.[18,19,316]
A useful corollary follows from this view. The hallmarks of aging are not thermodynamic inevitabilities. For each hallmark in the López-Otín taxonomy,[18,19] at least one species has evolved a partial or complete countermeasure, demonstrating that no individual hallmark represents a physical impossibility, only a calibration choice within a finite reproductive economy. The hallmarks are tractable; they are simply not affordable to solve in combination, which is precisely the prediction the protectosphere calibration framework makes (see section b).
(b) Protectosphere calibration
Because selection pressure declines with age, maintenance systems that protect information integrity, including DNA repair, stem cell renewal, antioxidant defense, and immune surveillance, are under diminishing pressure to function beyond the reproductive window.[165,264] We propose that this creates a phenomenon we term protectosphere calibration: actively maintained protective systems are evolutionarily tuned to sustain information integrity approximately as long as it contributes to direct or inclusive fitness. Intrinsically stable features (membrane self-assembly, resilient structural proteins) persist without ongoing energetic input and may naturally outlast the reproductive window. It is the energy-dependent, gene-expression-dependent systems that selection calibrates, pushing each just past the reproductive threshold at a cost proportional to the difficulty of improvement. The systems requiring the most ongoing investment sit closest to the margin, and these are expected to lose functional reserve over overlapping late-life intervals.
This overlapping late-life loss of function in the protectosphere has a critical consequence for aging research. In the intropy framework, all actively maintained systems are scaled to roughly the same underlying variable (the rate at which information corruption crosses a functional threshold), so they decline on similar timescales. The result is coordinated deterioration that presents the appearance of many independent aging mechanisms failing concurrently. Each system, studied in isolation, appears to be a plausible cause; interventions that extend any single system modestly extend function, reinforcing the impression of independent causation. We argue this impression is largely an artifact of calibration. In this framework, the shared upstream pressure is information decay; the maintenance systems were simply never selected to outlast it.
The calibration chain proceeds as follows: ecological context sets extrinsic mortality, which constrains the reproductive window, which defines the selection horizon, which determines evolutionary investment in information maintenance, which constrains the achievable information decay rate. This chain is bidirectional (better repair permits later reproduction, which extends the selection horizon), reaching equilibrium when the marginal cost of further maintenance exceeds the marginal fitness benefit, consistent with Kirkwood’s disposable soma.[165] A principal external input that breaks the symmetry is ecology.
All actively maintained systems are calibrated to keep their specific failure modes below organism-fatal thresholds over the reproductive window, with the consequence that selection acts against whichever component fails first. Information fidelity occupies a special position in this network because its failures propagate as input load into every downstream system, making it the highest-leverage variable for system-wide longevity even though each individual system has its own calibration.
This reflects an evolutionary equilibrium: at any point in a lineage’s history, lifespan is constrained by whichever protectosphere component fails first. Selection acts against that weakest link until a new adaptation, niche shift, or social innovation breaks through the current ceiling, at which point the next most vulnerable component becomes limiting. Species with exceptional longevity have typically broken through multiple successive ceilings, each requiring a distinct molecular or ecological solution (see section c).
If this calibration framework is correct, it predicts that single-system interventions (boosting one repair pathway, clearing senescent cells) will yield diminishing returns, because they extend one layer past the point where others have been calibrated to fail. Maximal lifespan extension would instead require reducing the rate of information corruption itself, and/or simultaneously recalibrating multiple systems, consistent with the observation that the most effective longevity interventions (caloric restriction, rapamycin, genetic dwarfism) act on hub regulators that influence multiple systems rather than any single downstream pathway. For empirical tests of this framework across species, see section (c).
The same calibration logic may also explain why germline-soma separation evolved in the first place. Above some threshold of organismal complexity and developmental depth, holding an entire somatic cell mass within the corridor between an Eigen-like informational collapse threshold and the drift barrier may become evolutionarily unstable, with selection favoring a smaller, higher-fidelity transmitting lineage protected by a finite-maintenance soma. Aging, in this reading, would be the cost of using a bounded-maintenance soma to host and transmit a higher-fidelity germline. The prediction is that lineages with simpler genomes (Volvox-Gonium, plants without sequestered germlines, Hydra) should sit at or below this threshold, while lineages above it should have sequestered germlines. We develop this argument with formal treatment and quantitative predictions in subsequent work.
(c) Body size, ecological safety, and the calibration of longevity
The positive correlation between body size and lifespan across species is one of the most robust patterns in comparative biology, yet no single accepted mechanistic explanation exists, and the correlation reverses within species (large-breed dogs die younger than small-breed dogs). Protectosphere calibration offers a unifying resolution: body size does not drive repair investment directly. Rather, large body size reduces predation risk, which lowers extrinsic mortality, which extends the reproductive window, which strengthens selection for information maintenance. In other words, the correlation between size and longevity is partly mediated by ecological safety rather than by size alone.
This also clarifies a distinction frequently conflated in the literature: cancer suppression and aging prevention have different scaling variables. Cancer risk scales with cell number multiplied by division rate (Peto’s paradox), and its solutions (redundant tumor suppressors, enhanced apoptosis, immune policing) are specific to that problem. Aging prevention scales with information maintenance investment, set by the calibration chain. The elephant-whale comparison illustrates this starkly. Elephants solved Peto’s paradox through cell elimination via 20 copies of TP53 but live to approximately 70.[380,381] Bowhead whales solved it through enhanced DNA repair, including upregulation of CIRBP and RPA2, and live past 200.[382,383] This contrast is suggestive: elephants and whales appear to have invested most heavily in different protectosphere strategies, with elephants emphasizing tumor suppressor expansion and damage-induced apoptosis, and bowheads emphasizing enhanced DNA repair. The threefold lifespan difference is consistent with the idea that information repair aligns more directly with both cancer prevention and long-term tissue preservation, while cell elimination preferentially solves cancer risk but can impose renewal costs if overused.
Within a species, the information maintenance machinery is identical: all dogs share essentially the same repair enzymes, the same protective regulatory pathways, and the same calibration point. But larger individuals require more cell divisions during development and achieve their size through elevated growth signaling that simultaneously suppresses maintenance.[384,385] The result is more corruption accumulated against the same level of protection. Between species, different calibration points; within species, different corruption rates against the same calibration.
Subterranean rodents offer a particularly instructive gradient. Blind mole rats (Spalax), which share the underground niche but are solitary, achieve roughly 20-year lifespans with notable cancer resistance, suggesting the niche alone extends the selection window substantially beyond surface rodents of similar size.[386] Naked mole rats, whose eusocial structure concentrates reproductive fitness on queen longevity, extend it further to over 30 years with negligible senescence.[387] The molecular investments track the selection pressure: naked mole rat membranes contain roughly one-ninth the DHA of mice, dramatically reducing the substrate for lipoperoxidation and the generation of transcription-blocking aldehyde adducts, while total unsaturated fatty acid content is comparable.[388,389] Their longevity is not explained by ROS suppression or enhanced antioxidant defenses, both of which are paradoxically low.[390,391] Instead, the reduced DHA content may sever the link between ROS exposure and the secondary lipid-peroxidation products (including aldehydes) that can damage DNA. This is protectosphere calibration operating at the chemical level: the ecological context opened the selection window, eusociality widened it further, and evolution responded by investing specifically in the protectosphere layers that prevent the most functionally consequential class of information corruption. In every case, exceptional longevity traces to ecological safety extending the selection horizon, not to body mass per se.
EN30. Origin and basis of the Telomere Theory.
The telomere theory of aging traces to Olovnikov, who in 1973 reasoned that conventional DNA polymerases cannot fully duplicate linear chromosome ends and predicted that terminal sequences must therefore shorten with each round of replication.[43] Telomeres, the repetitive sequences occupying chromosome termini, absorb that incremental loss and keep chromosome ends from being misread as double-strand breaks or joined to other chromosomes.[35,392] When telomeres fall below a critical length or lose their cap structure, the resulting damage signal arrests proliferation through replicative senescence or triggers apoptosis.[35,39] Karlseder and colleagues showed that critically shortened or deprotected telomeres are recognized as DSBs by the ATM/ATR kinases and that the resulting checkpoint activation, rather than end-loss per se, drives the cell-fate decision; in this framework reading, telomere shortening operates as a readout-disruption mechanism (chromosome ends transitioning from protected to unprotected) rather than as a separate aging variable.[393] The empirical scaffolding for the theory was assembled in stages: Hayflick and Moorhead first showed that human diploid cells have a finite division capacity; Olovnikov’s end-replication argument supplied a candidate molecular explanation; and subsequent work characterized telomere structure across organisms, identified telomerase as the enzyme that counteracts terminal erosion, and connected telomere length to replicative reserve.[39,43,178,392]
EN31. Evidence supporting the Telomere Hypothesis.
Several lines of evidence initially supported the telomere hypothesis. Telomere length correlates with replicative capacity in cultured human fibroblasts [39] and rate of telomere shortening (rather than initial length) predicts species lifespan.[33] Overexpression of telomerase in vitro enables cells to bypass senescence and divide indefinitely [394], while telomerase deficiency accelerates aging phenotypes.[41] Clinical observations, such as dyskeratosis congenita, link defective telomere maintenance to premature aging phenotypes.[38,395] These observations are consistent with our model, but telomere attrition alone cannot account for all functional decline observed during aging.[18,19]
EN32. Evidence against telomere length as a lifespan determinant.
Contradictory findings challenge the telomere theory as a universal mechanism of aging.[40,41,396] Telomere length varies widely among species without corresponding differences in lifespan; for example, some rodents have long telomeres yet short lifespans.[396] Telomerase knockout mice show near-normal lifespan in G1 but progressive defects and reduced survival in later generations.[41] Within a single organism, telomere length varies considerably across tissues, and these tissue-level patterns do not track the locations or rates of age-related dysfunction in any clean way.[38,397] These observations suggest that telomere attrition is neither necessary nor sufficient to explain organismal aging, though it likely contributes as one element of a broader corruption process.
EN33. Conceptual implications of Telomere Theory.
Within the intropy framework, telomere shortening is a downstream readout of accumulated information loss rather than its primary driver. The phenomenon itself is structural rather than a fidelity problem: conventional DNA polymerases cannot fully duplicate the ends of linear chromosomes, so a small terminal segment is lost with each round of division.[43] This attrition compounds in proliferative tissues but plays a much smaller role in long-lived postmitotic cells such as neurons and cardiomyocytes, whose decline reflects accumulating modifications and other corruption channels rather than terminal erosion.[18,19]
A revealing clue to its calibrated role is that the enzyme that repairs it, telomerase, already exists and is actively deployed in germline, embryonic, and certain progenitor compartments yet held silent across most somatic tissues. Within this framework that selective absence is read as protectosphere calibration (see EN29b): a cell with continuously replenished ends is a cell capable of unlimited division, and unlimited division inside a multicellular organism is cancer. The risk is compounded by the fact that each replication round is itself a corruption opportunity, with modifications encountered at the fork converting to permanent mutations or to fork-collapse double-strand breaks (see EN81), so division number scales not only with the chance of defection but with the rate at which new corruption seeds it. The empirical signature of this coupling is the strong correlation between lifetime stem cell division number and tissue cancer risk across organs.[153] Capping divisional capacity holds somatic cells inside the organism-level coalition rather than letting selection re-enter at the level of the individual cell. Telomere length therefore counts divisions, and the calibrated rate of attrition sets the threshold past which a cell is retired through senescence or apoptosis. We read it as a tractable yardstick for one corruption channel, not as the foundational cause the broader theory of aging must ultimately track.
EN34. Proteostasis decline as a principally downstream consequence of information corruption.
The proteostasis network, comprising molecular chaperones, the ubiquitin-proteasome system, and autophagic clearance pathways, maintains functional protein homeostasis by ensuring that proteins are properly folded, damaged species are removed, and aggregates are cleared.[19,398] Its decline with age is well documented: proteasome activity decreases, chaperone inducibility diminishes, and autophagic flux slows, contributing to the accumulation of misfolded and aggregated proteins characteristic of neurodegenerative and other age-related diseases.[19,398,399] However, every component of this network is encoded by DNA and regulated by gene-expression programs that are themselves subject to information corruption.
Several lines of evidence support information corruption as the upstream driver of proteostasis collapse rather than the reverse. First, in DNA repair-deficient progeroid mice, the endoplasmic reticulum unfolded protein response is constitutively activated, and caloric restriction simultaneously reduces both transcription stress (a consequence of unrepaired DNA lesions) and proteostatic burden, providing a direct mechanistic link between DNA damage and protein homeostasis failure.[45] Second, in C. elegans, the heat shock response undergoes a precipitous decline at the onset of reproductive maturity, driven not by accumulated protein damage but by the placement of repressive H3K27me3 chromatin marks at chaperone gene promoters, an epigenetic event whose timing is regulated by germline signaling and which can be prevented by germline removal.[400] This finding is particularly significant for our framework: the decline in proteostasis is not a passive consequence of protein accumulation overwhelming the system but an active regulatory event, consistent with protectosphere calibration (see EN29b), in which the maintenance of stress response pathways is scaled to the reproductive window rather than to the full lifespan. Third, most intracellular protein damage is cleared on a timescale shorter than the lifetime of the cell, with damaged molecules degraded and resynthesized from the underlying template. A subset of long-lived proteins, including lens crystallins, elastin, certain collagens, and the scaffold nucleoporins of postmitotic neurons, are maintained primarily through chaperone-mediated stabilization and lesion-specific repair rather than direct replacement, and accumulate damage when those secondary maintenance routes saturate.[401,402] Within this framework, the limited replacement of these proteins is itself an instance of protectosphere calibration (see EN29b): selection has not invested in turnover machinery for components whose original molecules carry the organism through the reproductive window, so when the damage rate eventually outruns the chaperone and repair capacity, the consequences (cataract, arterial stiffening, nuclear pore leakage) appear as canonical late-life phenotypes. For the bulk of the proteome, however, replacement requires the chaperones, proteasomal subunits, and autophagy regulators that read the genome to be themselves expressed accurately. Once the information encoding that machinery is corrupted, whether through mutation, standing-modification-driven transcriptional impairment, or epigenetic drift, the replacement capacity progressively degrades, producing the appearance of an independent aging mechanism.
We therefore view proteostasis collapse as a powerful amplifier of aging whose trajectory is set by upstream information corruption: the network fails because the instructions for building and regulating it are progressively degraded at the same time the substrates it is meant to clear continue to accumulate.
EN35. Transposable element activation as a consequence of epigenomic erosion.
Transposable-element-derived sequences make up roughly half the human genome, with LINE-1 retrotransposons alone accounting for about 17%, and are normally held silent by DNA methylation at their promoter sequences, heterochromatin packaging maintained by factors such as SIRT6, and small RNA surveillance pathways.[47] With age, each of these silencing layers weakens. DNA methylation at LINE-1 loci declines, SIRT6 occupancy at LINE-1 promoters decreases, and heterochromatin loosens, permitting transcriptional reactivation of normally repressed elements, including the evolutionarily young LINE-1 families that retain retrotransposition abilities.[46,47,403] LINE-1 expression also generates cytoplasmic cDNA species, distinct from the nuclear retrotransposition intermediates produced by target-primed reverse transcription, that activate cGAS-STING innate immune sensing and drive a type I interferon response, contributing substantially to the chronic sterile inflammation of aging tissues.[45,46] Treatment of aged mice with nucleoside reverse transcriptase inhibitors, which block LINE-1 reverse transcription, reduces this inflammatory burden across multiple tissues, and caloric restriction similarly suppresses LINE-1 expression in aged animals.[46,47]
Within our framework, transposable element reactivation is a downstream consequence of information corruption acting on the silencing machinery itself. Every component required to keep these elements repressed, including DNMT1 and DNMT3 methyltransferases, SIRT6, KAP1, and the heterochromatin remodeling apparatus, is encoded by DNA and depends on accurate transcription, recruitment, and chromatin context to function. All three are progressively undermined by the corruption cascade described elsewhere in this manuscript: standing modifications stall and miscode the polymerases reading silencer genes, accumulated mutations corrupt the proteins those genes encode, and the resulting epigenomic drift further destabilizes silencer recruitment (see EN71). As silencer output declines, repression fails and transposable elements escape containment. The resulting inflammatory signaling then acts as a potent amplifier, accelerating modification of nucleic acid in neighboring cells through oxidative and inflammatory damage, but the initiating event is upstream information loss, not autonomous transposon behavior. Notably, the inflammatory amplification step is itself a calibrated layer rather than a fixed consequence: long-lived bat lineages have evolved attenuated STING signaling and dampened inflammasome activation, demonstrating that the inflammatory response to cytoplasmic nucleic-acid stress can be tuned independently of the underlying TE silencing problem.[47]
EN36. Two modes of tissue decline: information corruption versus stem cell exhaustion.
Stem cell exhaustion, the progressive loss of regenerative capacity in tissues that depend on progenitor cell replenishment, is a recognized hallmark of aging.[19,45,351] In the hematopoietic system, aged stem cells exhibit lineage bias, reduced engraftment potential, and clonal contraction: in individuals over 75, as few as 12 to 18 clones may account for the majority of blood production, compared with the tens of thousands of contributing clones observed in younger adults.[271] Similar age-related decline in stem cell function has been documented in muscle satellite cells, intestinal crypt stem cells, and neural progenitors.[351]
Our framework predicts that stem cell decline can proceed through two mechanistically distinct routes that converge on a shared phenotype, tissue-level functional failure, which is why they have been conflated.
In the first route, which we propose is a principal driver of normal aging in proliferative tissues, stem cells persist but progressively accumulate corrupted information. Each division transmits that degraded template to every daughter cell, meaning the quality of replacement cells steadily declines even as production continues. The tissue breaks down not because it lacks new cells, but because those cells carry increasingly inaccurate instructions. The hematopoietic system illustrates this directly: in mice, aged phenotypic HSC pools actually expand in number while their functional output deteriorates; in humans, the picture is dominated by clonal contraction with surviving clones producing lineage-skewed, poorly differentiating progeny.[271,351] Challen, Goodell, and colleagues demonstrated this principle in controlled conditions: conditional ablation of the DNA methyltransferase Dnmt3a in mouse HSCs caused progressive expansion of the stem cell compartment over serial transplantation while simultaneously impairing differentiation, producing an ever-growing pool of progenitors with ever-declining functional output.[119] This is the quality-over-quantity signature that our framework predicts for information corruption aging, and it is precisely the pattern observed in DNMT3A-driven clonal hematopoiesis in aged humans (see EN72). Clonal hematopoiesis driven by mutations in DNMT3A, the most common driver of age-related clonal expansion, exemplifies how a single informational change in one stem cell expands through its lineage to dominate tissue-level clonal architecture, with downstream consequences ranging from elevated cardiovascular and hematologic disease risk to altered immune output (Challen and Goodell 2020).[243]
In the second route, which we associate primarily with progeroid syndromes and certain forms of acute damage, the DNA damage response eliminates stem cells faster than they can be replaced, depleting the regenerative pool. The tissue then fails from attrition: there are simply not enough replacement cells, regardless of their informational quality. DSB-repair progerias (LIG4 syndrome, Ku80 deficiency) demonstrate this pattern, where DDR hyperactivation culls damaged stem cells and produces rapid aging in stem-cell-dependent tissues such as bone marrow and skin.[45,404,405] The POLG mutator mouse ages through the same exhaustion mechanism: high-burden mtDNA mutations trigger apoptosis in tissue-specific progenitors and produce premature decline in proliferative compartments, with little evidence of bulk oxidative damage to tissues and no change in mitochondrial ROS production measured ex vivo.[406,407,408] In vivo measurements show that mitochondrial hydrogen peroxide rises with age in these mice, and interventions that lower it delay the aging phenotype, suggesting that elevated H₂O₂ acts as a redox-signaling amplifier of the apoptotic and inflammatory output of accumulated mtDNA mutations rather than as a primary damage source.[409] In intropy framework terms, the mutations are the upstream information corruption; the redox signaling is the channel through which their consequences propagate.
Both routes produce overlapping phenotypes (tissue thinning, immune decline, regenerative failure), which explains why they have been difficult to distinguish experimentally. However, they differ in a critical prediction: exhaustion-driven decline should preferentially affect tissues with high turnover and stem cell dependence, while information corruption should affect all tissues, including post-mitotic organs like the brain and heart that have no stem cell reserve to exhaust. Normal human aging affects both, consistent with information corruption as the broader mechanism. The pattern across progeroid syndromes is consistent with this division: DSB-repair defects (LIG4, Ku80) preferentially deplete proliferative compartments such as bone marrow and skin, while transcription-coupled NER defects (Cockayne syndrome, XPA/CSB combinations) produce severe neurodegeneration in post-mitotic tissues precisely because transcription-blocking modifications cannot be cleared from neurons that cannot replicate around them. Different repair channels protect different categories of cell, and their loss produces aging phenotypes in the tissues most dependent on the affected channel (see EN51, EN75).
Transplantation data are consistent with this framework but do not isolate the mechanism. Young hematopoietic stem cells transplanted into aged recipients extend survival,[410] and selecting for the least-corrupted HSCs within an aged donor produces similar benefits.[411] These interventions may work by replacing corrupted information with less-corrupted copies, consistent with the prediction that the quality of the informational template, not the quantity of progenitors, is the limiting variable in normal aging.
We emphasize that this two-mode distinction is a prediction that follows naturally from this framework: if information corruption is the root cause, then stem cell depletion and information degradation should produce distinguishable patterns even when their surface phenotypes overlap. The framework does not depend on this prediction being correct in every detail, but it offers a testable lens for reinterpreting progeroid syndromes, transplantation data, and the tissue-specificity of age-related decline.
EN37. Deregulated nutrient sensing and its relationship to information maintenance.
Nutrient sensing pathways, including the insulin/IGF-1 axis, mTOR, AMPK, and the sirtuins, form a deeply conserved signaling network that adjusts cellular growth, metabolism, and maintenance in response to energy availability.[19,50,412,413] Genetic or pharmacological suppression of growth signaling through these pathways extends lifespan across species from yeast to mammals. Rapamycin, which inhibits mTORC1, is the most robustly replicated pharmacological lifespan intervention in mice, extending median and maximum lifespan even when initiated late in life.[49] Reduced insulin/IGF-1 signaling extends lifespan in worms, flies, and mice, partly through activation of the transcription factor FoxO, which promotes expression of DNA repair enzymes, antioxidant defenses, and proteostatic machinery.[368] Caloric restriction, the oldest known longevity intervention, operates in part through these same nodes, engaging AMPK and sirtuins, suppressing mTOR, and activating autophagy alongside broader stress-response programs; Madeo and colleagues demonstrated that autophagy induction alone, through spermidine supplementation, extends lifespan across yeast, flies, worms, and mice, establishing autophagic clearance as an independently sufficient longevity lever.[19,270,412,413,414]
Within our framework, the outsized effectiveness of nutrient sensing interventions is explained by protectosphere calibration (see EN29b). These pathways sit at regulatory nodes that simultaneously influence multiple arms of the protectosphere: DNA repair, proteostasis, autophagy, antioxidant defense, and stem cell quiescence. The NAD+/sirtuin axis specifically illustrates this convergence: Verdin (2015) reviewed the central role of NAD+ as a metabolic substrate whose age-related decline impairs sirtuin-mediated regulation of repair, mitochondrial function, and stress responses simultaneously, and Imai’s group established that systemic NAD+ availability is governed by tissue-specific NAMPT activity that itself declines with age.[415,416] Because all actively maintained protective systems are calibrated to roughly the same underlying variable, the rate of information corruption relative to the reproductive window, interventions that act on hub regulators affecting many systems simultaneously produce larger effects than interventions targeting any single downstream pathway. This is precisely the pattern observed: rapamycin produces larger and more reproducible lifespan effects than antioxidant supplementation or upregulation of any single repair pathway, and the most effective non-pharmacological intervention, caloric restriction, similarly acts at the level of multiple coordinated maintenance systems rather than any one downstream target. This convergence is empirically visible at the transcriptome level. Schumacher et al. (2008) compared genome-wide liver expression in long-lived dwarf and calorie-restricted mice with that in DNA-repair-deficient progeroid mice and found that both extremes engage the same survival response, with suppression of somatotropic and energy pathways alongside elevated stress responses. The same program is activated whether maintenance is favored by reduced damage demand (CR, dwarfism) or forced by elevated damage burden (progeroid models), consistent with protectosphere-calibrated regulation tracking a single underlying corruption variable rather than separate inputs.[417]
The age-related dysregulation of these pathways is itself downstream of information corruption, though through readout drift rather than direct mutation of mTOR, AMPK, or the sirtuins themselves. The hub genes are read from the same template that accumulates standing modifications and epigenetic drift over time, while the cellular metabolic state they integrate (mitochondrial output, inflammatory tone, adiposity) becomes a less reliable proxy for actual maintenance demand. Growth and maintenance fall out of sync with the organism’s true corruption load, reducing information protection at precisely the time when that protection is needed most. Caloric restriction likely acts through both routes simultaneously: by reprogramming nutrient-sensing pathways toward maintenance and stress resistance, and by reducing the flux through energy-handling pathways that generate endogenous nucleobase information corruptors (NICs, the reactive small molecules and atoms that modify nucleic acid; see EN67, EN77, EN80), giving the existing repair machinery more time to clear modifications before they convert to permanent mutations or block transcription of critical genes.
EN38. Systemic signaling degradation as a readout of cellular information corruption.
Age-related changes in intercellular communication, including chronic low-grade inflammation (often termed inflammaging), altered paracrine and endocrine signaling, and dysregulated immune surveillance, constitute a recognized hallmark of aging.[19,334] The systemic signaling environment measurably deteriorates with age, shifting toward a pro-inflammatory, fibrotic, senescence-promoting, and anti-regenerative state that impairs tissue maintenance across the organism.
Heterochronic parabiosis experiments have demonstrated that this systemic environment profoundly influences tissue aging. When old mice share circulation with young partners, aged tissues show improved regenerative capacity in muscle, liver, and brain, with reactivation of stem cell populations and partial restoration of youthful molecular signatures.[51,418] These findings initially suggested that young blood contains specific rejuvenating factors. However, Conboy and colleagues subsequently showed that diluting old plasma with saline and albumin, without adding any young factors, produces broad rejuvenating effects across multiple tissues, suggesting that the accumulation of inhibitory signals in aged blood may be as important as, or more important than, the loss of youthful ones.[52] Furthermore, old blood exerts rapid and pronounced inhibitory effects on young tissues, sometimes exceeding the benefit that young blood confers on old tissues.[419]
These observations are consistent with our framework, which we argue offers a clean account of why parabiotic and plasma-exchange interventions produce partial, time-limited rejuvenation rather than long-term rescue. The cells that produce circulating signaling molecules, including hepatocytes, immune cells, endocrine glands, and adipocytes, are themselves subject to information corruption. As the genomic and epigenomic integrity of these cells degrades, their signaling output shifts: cytokine profiles skew toward inflammation, hormonal balance drifts, and growth factor production becomes dysregulated. The architecture of intercellular communication is not independently damaged so much as progressively misread: the genes encoding cytokines, receptors, hormones, and the regulatory machinery that sets their expression timing all sit on the same template that accumulates standing modifications, mutations, and epigenetic drift over time, so the secretory output of every signal-producing cell type drifts in concert. The systemic environment is a readout of the aggregate informational state of the cells that compose it.
This interpretation explains why parabiotic rejuvenation is temporary. When young blood is removed, old tissues revert to their aged state, because the underlying information corruption in the cells of the old organism has not changed. The young circulation provided a transient supply of correctly instructed signaling, effectively an external protectosphere subsidy, but the root cause, the corrupted information in the cells that produce the old organism’s own signals, persists and reasserts itself on a timescale of days to weeks. This parallels the epigenetic rebound observed after reprogramming factor withdrawal (see EN73): in both cases, the downstream consequence is temporarily reversed, but the upstream informational corruption drives it back.
EN39. Age-related dysbiosis as a reflection of host information decay.
The gut microbiome commonly undergoes compositional shifts with age, including altered (often reduced) diversity, expansion of pro-inflammatory taxa, and contraction of species associated with barrier integrity and immune homeostasis.[19] These changes are not merely correlative: fecal microbiota transplantation from young donors into aged mice reverses age-associated changes in the gut, brain, and retina, reducing neuroinflammation and restoring intestinal barrier function.[420] In progeroid mice, transplantation of wild-type microbiota extends both healthspan and lifespan, and administration of a single beneficial species, Akkermansia muciniphila, is sufficient to confer part of this benefit.[53] Conversely, transferring aged microbiota into young animals drives inflammatory and barrier dysfunction in the gut, brain, and retina, demonstrating that the microbial community itself can transmit aging-associated phenotypes between hosts.[420]
Within our framework, the microbiome represents an extension of the protectosphere: a community of organisms whose metabolic output (short-chain fatty acids, secondary bile acids, immune modulators) supports host tissue function and information maintenance. The host-side trajectory of age-related dysbiosis is, in this framework, downstream of information corruption in the tissues that shape the gut environment, with exogenous inputs (diet, drugs, environmental exposures) modulating that trajectory independently. The intestinal epithelium, mucosal immune system, and enteric nervous system collectively determine which microbial species thrive, and each of these host systems is built and maintained from a DNA template that is progressively corrupted, producing measurable declines in stem cell output, antimicrobial peptide expression, mucus integrity, and immune surveillance. As barrier integrity declines and mucosal surveillance weakens, the selective environment shifts to favor microbial communities that promote inflammation rather than suppress it, creating a feed-forward loop in which host information corruption drives dysbiosis, and dysbiosis accelerates further host damage through inflammatory signaling. The effectiveness of young microbiota transplantation likely reflects a partial and time-limited restoration of a healthy microbial environment that reduces inflammatory burden on host tissues, analogous to the partial and time-limited benefit of young or diluted plasma in parabiosis and plasma-exchange experiments (see EN38), rather than correction of the underlying host corruption that selected for dysbiosis in the first place.
EN40. Origins of the three major molecular theories.
The DNA Damage Theory of Aging, developed in its modern form by Vijg, Hoeijmakers, Schumacher, and colleagues,[45] proposes that physical and chemical lesions in nuclear and mitochondrial DNA accumulate over time and causally drive most recognized features of aging, with the scope expanded beyond sequence change alone to include non-mutational lesions that disrupt cellular function before or without conversion to permanent mutation. Its historical predecessor, the Somatic Mutation Theory of Aging, was articulated by Szilard and Curtis in the 1950s and 1960s and emphasized cumulative sequence change as the substrate of cellular decline.[169,421] Both should be distinguished from Medawar’s evolutionary Mutation Accumulation hypothesis (1952), which explains why selection tolerates aging rather than how aging happens at the cellular level (see EN26). The framework presented here builds on the DNA Damage Theory’s empirical foundation while reframing damage as one route by which information is corrupted, placing the cascade in an explicit thermodynamic and information-theoretic context, decomposing the readout consequences into channel-specific predictions (see EN81), introducing protectosphere calibration as the evolutionary scaffolding (see EN29b), and connecting the molecular cascade to Gompertz mortality kinetics through stochastrophe (see EN86). The Free Radical Theory, first linked to oxygen toxicity by Gerschman and formalized as an aging theory by Harman, posited that reactive oxygen species generated during metabolism caused progressive molecular damage.[123,124] The Epigenetic Theory, proposed by Holliday,[115] evolved from observations that chromatin structure and DNA methylation patterns change with age, leading to dysregulated gene expression.[91] Each theory captures a distinct molecular dimension: genomic integrity, oxidative chemistry, and regulatory architecture. In the intropy framework, these are not competing explanations but consequences of the same upstream chemistry: reactive species produce nucleobase modifications, modifications convert to mutations or persist as standing lesions that disrupt readout, and both routes propagate into epigenetic drift through repair-mediated demethylation and altered chromatin context (see EN71). The Free Radical Theory (almost) identified the chemistry; the DNA Damage Theory identified the readout consequences and broadened the scope to include non-mutational lesions; the Epigenetic Theory identified the regulatory drift. The framework integrates them by naming the upstream layer they were each partially seeing. The cascade itself operates within the evolutionary frame of Kirkwood’s Disposable Soma hypothesis, which explains why selection has not eliminated aging despite its clear individual cost (see EN29 and Table 1, Group A).
EN41. Evidence supporting each theory.
Support for the DNA Damage Theory includes the increase in somatic mutation burden with age, its correlation with cancer incidence, the recent demonstration that somatic mutation rates scale inversely with species lifespan,[103,210,211] and the broader body of work establishing that transcription-blocking lesions, double-strand breaks, and bulky adducts accumulate in aged tissues and produce cellular dysfunction. The direct contribution of ordinary somatic mutation burden to non-cancer aging remains debated, with replicative-fidelity defects elevating mutation rates without accelerating organismal aging in some genetic models (see EN60). The Free Radical Theory gained traction from studies showing that oxidative damage to DNA, proteins, and lipids accumulates over time.[161] Some targeted antioxidant manipulations extend lifespan in mice, including mitochondria-targeted catalase overexpression,[138] though broad antioxidant supplementation and many genetic manipulations of antioxidant enzymes have produced inconsistent or null effects, prompting substantial revision of the original theory. The Epigenetic Theory is supported by age-associated changes in DNA methylation, histone modifications, chromatin remodeling, heterochromatin maintenance, and transposable-element derepression (see EN35), which collectively alter transcriptional programs and cellular identity, and by the predictive power of epigenetic clocks built from methylation patterns at thousands of CpG sites across tissues.[116,422]
EN42. Limitations and need for integration.
Individually, these theories are limited in accounting for the full complexity of aging.[18,19] Mutation accumulation provides a compelling explanation for progressive corruption since these changes, once copied, are generally not reversible.[423] However, what remains unclear is the mechanism by which mutations translate into functional decline, and the upstream processes that generate them in the first place.[63,423] DNMT3A-driven clonal hematopoiesis exemplifies how this integration plays out in practice: a single sequence corruption produces epigenetic drift, altered stem cell behavior, tissue mosaicism, and elevated disease risk, simultaneously satisfying predictions made by the Mutation, Epigenetic, and Stem Cell Exhaustion theories without privileging any of them as primary.[119,243] The Free Radical Theory overemphasizes oxidative stress; antioxidant interventions often fail to extend lifespan in mammals.[424,425] However, it contains one central insight: it provides a mechanism by which the cell corrupts its own information.[161] Equally important, oxidative damage to most non-information substrates (proteins, lipids, metabolites) is transient because the affected molecules are replaced from intact templates; only when oxidative damage perturbs information-bearing or information-maintaining substrates does it become a durable aging driver, with long-lived proteins (lens crystallins, elastin, scaffold nucleoporins) representing a notable exception where direct turnover cannot clear accumulated damage (see EN34). The theory also does not expand to the many small molecules that can act as corruptors, not just reactive oxygen species but endogenous metabolites including aldehydes, formaldehyde, methylglyoxal, lipid peroxidation products, and alkylating intermediates that chemically modify nucleic acids in vivo.[63] The Epigenetic Theory captures regulatory drift but, on its own, did not explain why epigenetic marks themselves degrade.[121,426,427] Recent evidence partial resolves this: Koch et al (2025) demonstrated that somatic mutations at CpG sites drive extensive remodeling of the surrounding methylome, and that somatic mutations explain more than half of the variation in methylation age across individuals.[118] This positions sequence corruption upstream of at least the majority of inter-individual variation in clock-based aging signal, while leaving room for replication-coupled and repair-coupled mechanisms to contribute to the rest. Epigenetic marks are also imperfectly maintained through cell division, unlike the underlying sequence which is copied with far-higher fidelity; this limited heritability creates problems with the requirement that causal corruption be irreversible.[422,426,427] This chicken-and-egg question is now partly resolved by the Koch et al. findings above. However, as advocated herein, the epigenome is part of nucleic acid’s information system, providing regulatory control. We propose the corruption of this regulatory control is a contributor to aging.[90,426,427] A unified model must reconcile these processes as interconnected manifestations of a deeper principle: the progressive corruption of biological information under thermodynamic constraints. The hallmarks framework provides the field’s shared vocabulary for what aging looks like at the cellular and tissue level, and our model is best understood as complementary, addressing what initiates the process, what determines its rate, and through which channels a single class of molecular events produces the diverse phenotypic catalog the hallmarks describe (in agreement with Garinis et al. (2008),[428] Niedernhofer et al . (2018),[54] and Schumacher et al. (2021)[45], who frame DNA damage and the survival response it engages as a unifying axis through which aging-associated phenotypes converge).
EN43. Why nucleic acids became the universal memory substrate.
All known cellular life stores hereditary information in nucleic acids, DNA or RNA. Their linear, sequence-addressable structure and complementary base-pairing support both dense information storage and a templated copying mechanism in which each strand specifies its partner.[14,65,92] RNA likely preceded DNA in early evolution, serving both as genetic material and catalyst (RNA world hypothesis).[60,318,429] DNA later became the dominant long-term repository through several chemical advantages: deoxyribose is more stable than ribose, the substitution of thymine for uracil enables detection of cytosine deamination as a damage signal, the duplex structure permits complementary-strand repair, and separating archival storage from catalysis allowed each function to be optimized independently.[63,318] In known biology, no other class of polymer has produced a self-templating hereditary substrate. Amino acid sequences carry functional information in proteins, and prion-like conformations can transmit phenotypic states across cell generations,[320] but neither offers the templated copying, error correction, and high-density storage that nucleic acid base-pairing provides.[65] In our view, the central tenet to understand aging is that the chemical information stored in nucleic acid, if not maintained, drives corruption of all cellular functions. This corruption is inevitable: the prime directive (the lineage-level imperative to preserve and transmit heritable information well enough to reproduce; see EN7) requires metabolism, and metabolism continuously generates reactive species (oxidative, alkylating, aldehyde, and hydrolytic) that modify the same nucleic acid the cell is trying to protect, while replication and transcription expose that nucleic acid to those species at moments of maximal vulnerability.[7,63]
EN44. Evolutionary logic of the prime directive.
Replication is the central selection constraint on life: lineages persist only when their heritable information is preserved and transmitted well enough to reproduce, and natural selection therefore acts on the persistence of that information across generations (see EN7 for the operational use of “prime directive” in this framework).[430,431] Traits that enhance replication, directly or indirectly, are favored, while traits that strongly compromise reproductive success are removed; weakly deleterious traits and traits expressed after the reproductive window can persist through drift, linkage, or antagonistic pleiotropy (see EN26).[430,431,432] This principle underlies the evolution of protective systems (repair enzymes, error-checking, compartmentalization) and explains why organisms invest heavily in safeguarding genetic material (see EN10 for the corridor between an Eigen-like informational error threshold and the drift barrier).[64,113,322] Aging emerges as the cost of this strategy: as Kirkwood and Holliday formalized in the disposable soma hypothesis, natural selection favors transmitting a relatively uncorrupted copy of heritable information over maintaining the original carrier indefinitely. In the intropy framework presented here, this tradeoff has a specific molecular grounding: the same metabolism that powers transmission generates the corruptors that erode the information being transmitted, so the system is forced to choose between unbounded somatic maintenance and unbounded reproductive output, and selection has chosen the latter (see EN29 for the calibration logic).[298]
EN45. Replication fidelity and inevitability of error.
DNA polymerase accuracy emerges from layered mechanisms: base selectivity, exonucleolytic proofreading, and post-replicative mismatch repair, yielding post-repair error rate of approximately 10-7 to 10-10 per base per replication in vivo, varying with organism, polymerase identity, sequence context, and repair status.[74,75] Raising accuracy beyond this baseline generally requires more discrimination steps (kinetic proofreading), better repair, slower throughput, or greater molecular complexity, all of which carry costs.[11,204,433] Selection improves fidelity only up to the point where further gains are outweighed by costs to speed, energy, or other fitness-relevant variables; beyond this point, drift rather than selection governs the residual error rate (see EN10).[11,204,433]
Error suppression is not free: information processing and correction have physical costs. Kinetic proofreading explicitly spends free energy to improve discrimination, creating a speed-accuracy-dissipation trade-off.[11,204] Landauer’s principle establishes a thermodynamic lower bound on logically irreversible information erasure; biological error correction is not strictly identical to computational erasure, but proofreading, repair, and quality control all require energy and time and produce dissipation, so the principle’s spirit applies even where its strict form does not. Modern nonequilibrium analyses quantify how error reduction requires energetic dissipation in biological as well as engineered systems.[8,11,434] The critical questions then become: what causes the errors, which of those causes and errors drive aging, and what mechanisms exist to prevent them?
EN46. The corridor of viable change and the fitness landscape.
Earlier we framed life as operating within a narrow corridor of viable error rates, bounded above by an informational-load ceiling and below by the drift barrier (see EN10). Within this corridor, not all informational changes are equal.
Fisher’s geometric model of adaptation provides the formal framework for understanding why.[435,436] Fisher envisioned an organism as a point in a multidimensional space, where each dimension represents a trait contributing to fitness and the optimum phenotype sits at a defined target. A mutation is a random displacement in this space. The key insight is that the probability of a mutation moving the organism closer to the optimum depends on both the size of the mutational step and the number of traits it affects. The smaller the step, the higher the chance it lands closer to the optimum rather than farther: in the limit of vanishingly small displacements, the probability of improvement approaches one-half. Large mutations, by contrast, are almost certain to overshoot the optimum in at least one dimension, and the more traits an organism integrates, the worse the odds become; pleiotropy and dimensionality both work against random change.
This is why a fish does not become a tyrannosaur in a single generation. The phenotypic distance between those organisms spans thousands of coordinated traits, and no single mutational event can traverse it without catastrophically disrupting the organism. Evolution unfolds instead through countless small viable steps, each slightly reshaping the fitness landscape for the next. The replicator climbs the landscape one foothold at a time, and every foothold must support a living organism. Evolution is not a leap but a walk, and the walking speed is set by the size of informational change the system can tolerate without collapsing.
This connects directly to our framework. The informational changes that drive aging and the informational changes that drive adaptation are the same chemistry operating on the same substrate, but they are separated by two filters: selection and heritability. Changes that pass through both filters (beneficial and germline-heritable) become adaptation. Changes that fail either filter (deleterious or somatic-only) become the cumulative burden we observe as aging. The corridor of viable error rates, bounded above by an Eigen-like informational-load ceiling and below by the drift barrier (see EN10), is the space in which both processes operate. Neither corruption nor adaptation can be globally driven to zero without threatening the other, but their rates and tissue distributions can be shifted. The framework’s argument for amortality rests precisely on this: targeted reduction of somatic corruption while preserving germline variation is the design space within which intervention becomes conceivable.
EN47. Information theory and biological copy.
As established in EN10, copying fidelity in any information system is bounded by fundamental physical and theoretical constraints.[12,14] These constraints operate at three levels. Eigen’s error threshold sets the upper bound for asexual quasispecies replicators: above a critical mutation rate, sequence information disintegrates into randomness and the system ceases to exist as a coherent replicator.[14] For complex organisms, redundancy, recombination, repair, and selection at multiple levels modify this bound, but the underlying logic still applies as an Eigen-like informational-load ceiling. Shannon’s noisy-channel theorem and Landauer’s principle establish the physical and information-theoretic background: error suppression requires redundancy, time, and dissipative work, with Landauer’s principle setting a thermodynamic floor on the cost of logically irreversible bit erasure.[7,9,12] Biological proofreading and repair are not strictly identical to computational erasure, but they likewise impose energetic and kinetic costs, so error rates can be driven very low without reaching zero in any finite biological system. Between these extremes, the drift-barrier hypothesis sets the practical lower bound that life actually achieves: natural selection can reduce replication errors only until the marginal fitness benefit of further fidelity falls below approximately 1/Ne (the effective population size), at which point genetic drift prevents reliable selection for, and maintenance of, further fidelity gains.[112,113] This means that even under sustained selection for accuracy, an irreducible floor of replication error persists, not because physics forbids better fidelity, but because evolution cannot see the benefit. Long-term evolution experiments illustrate that mutations accumulate continuously even in well-adapted populations under strong selection.[437,438] In somatic tissues, selection acts on cell-level proliferation and survival rather than on long-term organismal function, so corrupted cells with proliferation advantages can outcompete healthier neighbors, and the resulting clonal expansions amplify rather than buffer the underlying informational corruption (see EN36, EN72).[439] Organisms therefore occupy an inescapable band: above an Eigen-like informational-load ceiling, information collapses; below the drift barrier, selection cannot reliably maintain further fidelity; and at the physical level, the energetic and kinetic costs of error suppression keep zero error out of reach for any finite biological system.
EN48 Bacterial aging and the origins of asymmetric damage inheritance.
EN48 expands on the bacterial aging finding introduced in EN6, examining the molecular basis of asymmetric damage inheritance and its implications for aging dynamics.
The Stewart et al. (2005) result, that old-pole E. coli daughters age while new-pole daughters reset, has been refined by subsequent mechanistic work.[294] Lindner et al. (2008) demonstrated that spontaneous protein aggregates form during normal unstressed growth, localize to the cell poles, and segregate asymmetrically at division: the old-pole daughter inherits the parental aggregates while the new-pole daughter starts aggregate-free.[440] Aggregate inheritance accounts for more than 30% of the loss of reproductive ability in old-pole cells, the metric Lindner et al. used to quantify aging in this system; the remaining majority of the fitness asymmetry is not yet pinned to a single molecular cause, and subsequent work has shown that the aggregate-fitness correlation depends on the choice of fluorescent reporter and growth conditions. The MukBEF complex, a bacterial structural maintenance of chromosomes homolog, mediates nonrandom segregation of sister chromosomes, with the older template DNA strand preferentially inherited by the cell forming at the old pole.[441] Bacteria therefore show asymmetric inheritance of protein aggregates and possess chromosome-segregation machinery that produces nonrandom inheritance of older versus newer DNA templates, but the relative contributions of aggregate burden, template strand age, oxidized membranes, and other inherited damage classes to old-pole decline remain incompletely resolved.
E. coli aging is also observable outside the dividing-cell context. Yang et al. (2019) showed that carbon-starved, growth-arrested E. coli tracked at single-cell resolution in microfluidic chambers exhibit Gompertz mortality kinetics, with hazard rates that double at a characteristic age, paralleling the exponential scaling of mortality that characterizes metazoan aging.[442] Follow-up work (Yang et al., 2023) found that initial damage or cell-cycle state does not strongly predict time of death; instead, the data are consistent with stochastic accumulation of damage to a threshold, with the underlying damage substrate not yet identified as a single molecular class.[443] The bacterial aging literature thus presents a coherent demographic pattern (exponential mortality scaling, asymmetric damage inheritance, replicative decline of old-pole lineages) operating on a still-incomplete molecular picture.
This is directly relevant to our framework. Both daughter cells inherit essentially identical DNA sequences; standard bacterial mutation rates are far too low (Drake’s rule places spontaneous mutation rates at roughly 10⁻³ per genome per generation in microbes with DNA genomes) for new sequence mutations to explain old-pole/new-pole fitness asymmetry over a few generations.[292,444] The asymmetry must therefore reflect either differential inheritance of damaged cellular components (aggregates, oxidized membranes, conformationally altered macromolecules) or accumulation of standing DNA modifications that have not yet converted to mutations but that impair the function of the older template. Both classes of damage correlate with reduced fitness in old-pole cells; their relative contributions are not yet resolved. The carbon-starvation Gompertz finding adds a complementary observation: even when replicative dilution is removed by halting growth, E. coli still ages with the same exponential hazard scaling that characterizes metazoan systems, suggesting that the aging dynamic is driven by damage accumulation in the cellular machinery rather than by replication-coupled processes alone.
E. coli therefore offers a system for studying the molecular basis of asymmetric damage inheritance and the broader dynamics of damage-driven mortality. The 20-minute generation time (in optimal conditions), small genome, single-cell tracking technology, and availability of single-gene knockouts in every repair, segregation, and quality-control pathway make it feasible to ask questions that would require decades to address in mammals: which classes of damage accumulate in aging lineages, which compartments are most vulnerable, how protein-aggregate and DNA-modification contributions to old-pole decline can be separated experimentally, and how mortality kinetics shift under specific genetic or pharmacological perturbations (see EN14, EN15). Analogous principles of damage accumulation, asymmetric segregation, and exponential mortality scaling that unfold over years to decades in mammalian tissues can be observed in E. coli within hours.
EN49. Concept and evolutionary basis for the protectosphere.
The term “protectosphere” refers to the hierarchical network of systems evolved to safeguard hereditary information from stochastic degradation.[165,322] The premise is not that every biological system directly protects DNA, but that biological systems are filtered by selection according to their effects on lineage persistence: those that improve survival, repair, reproduction, or kin fitness are favored, those that compromise these are removed, and the resulting network functions as a layered defense against the loss of biological order (see EN7 for the operational use of “prime directive” in this framework).[165,322] At the molecular level, this includes the chemical stability of nucleic acids, the layered repair systems (proofreading, mismatch repair, base and nucleotide excision repair, transcription-coupled repair, double-strand break repair), proteostasis, redox buffering, and compartmentalization.[63,64,323] At higher levels, it encompasses tissue renewal, immune surveillance, apoptosis and senescence programs, endocrine and metabolic homeostasis, and behavioral and ecological strategies that reduce exposure to both endogenous and environmental hazards.[18,19] These layers emerged because natural selection strongly favors mechanisms that delay catastrophic information loss long enough to ensure reproduction.[165,322] Because threats to biological order are diverse, both endogenous (oxidative metabolism, hydrolysis, deamination, alkylating intermediates, replication errors, transcriptional stress) and environmental (radiation, exogenous mutagens, infection, predation, starvation), the protective systems that evolved to counter them are correspondingly varied. However, no protective system can eliminate molecular corruption entirely under finite energy and evolutionary constraints; the best selection can generally achieve is to keep corruption sufficiently delayed, repaired, diluted, or buffered that reproduction and fitness-relevant functions remain viable through the reproductive window (see EN29b for the framework’s specific calibration argument).[18,19] This principle explains why aging persists despite elaborate maintenance systems.[18,19,165]
EN50. DNA architecture as intrinsic protection.
DNA’s double-helical structure provides local redundancy: each strand can often serve as a template for repair of the other.[65,67] Hydrogen bonding between complementary bases and base stacking stabilize the duplex, while covalent phosphodiester bonds maintain backbone integrity.[65,81] Higher-order features add further protection and constraint. Chromatin packaging modulates exposure and repair, shielding some genomic regions while restricting repair access in others.[83] Diploidy provides partial organism-level redundancy, buffering many recessive defects and enabling homologous-recombination repair, though dominant-negative effects, haploinsufficiency, and loss-of-heterozygosity leave the buffer incomplete.[69] These structural adaptations exemplify how molecular architecture evolved to minimize information loss from both damage and replication errors. However, during both replication and transcription, the duplex must locally unwind, producing single-stranded DNA at replication forks and transcription bubbles that are more vulnerable to chemical damage, secondary structure, polymerase stalling, and mutagenic repair (see EN43, EN81 for the framework’s treatment of these moments of vulnerability and their channel-specific consequences).[75,99]
EN51. Repair pathway specificity as evidence for an evolved protectosphere.
One of the strongest lines of evidence that the protectosphere is an evolved system tailored to the actual corruptor landscape is the specificity of DNA repair pathways and the phenotypic consequences of their loss. Major classes of DNA damage are handled by partially specialized, overlapping repair systems, and the consequences of losing those systems vary depending on whether the unrepaired lesion blocks transcription, stalls replication, becomes fixed as mutation, or triggers cell loss, in the broad pattern our framework predicts.
Base excision repair handles small oxidative and alkylative lesions. Loss of downstream BER coordination (as in XRCC1 deficiency) leaves AP sites and single-strand breaks unrepaired, producing severe neurodegeneration and shortened lifespan.[274] Nucleotide excision repair, particularly its transcription-coupled branch, clears bulky helix-distorting adducts that block RNA polymerase. The phenotype of its loss is more complicated than a simple damage-accumulation story. Trichothiodystrophy (certain XPD/XPB mutations) fits the pattern cleanly, producing shortened lifespan without elevated cancer. Cockayne syndrome does not. The CSB genotype-phenotype relationship is genuinely complicated but in a direction the framework predicts: CSB-null mice show only mild CS-like features (fat tissue reduction, photoreceptor loss, mild neurological pathology) and develop the severe progeroid phenotype only when global-genome NER is also disrupted in XPA/CSB or XPC/CSB double mutants, indicating that simple loss of CSB-mediated TC-NER is insufficient to drive severe progeria. In humans, some CSB-null alleles, including the codon 77 nonsense and frameshift mutations, produce UV-sensitive syndrome rather than classical Cockayne syndrome,[445,446] consistent with the mouse data. Other complete-loss alleles, particularly those affecting the 5’ UTR and eliminating CSB transcription, do produce classical Cockayne syndrome,[447] which represents a genuine complication that may reflect 5’ UTR effects beyond simple CSB loss or a difference between mouse and human dependence on CSB function. Mutations producing a dysfunctional CSB protein appear to drive severe progeria through additional mechanisms beyond repair loss, with one proposed model from Chatre and colleagues implicating HTRA3-mediated POLG1 degradation under elevated oxidative and nitrosative stress in CS cells,[448] and Crochemore and colleagues separately showed that CSB promoter downregulation via histone hypoacetylation contributes to replicative senescence.[449] Even so, the transcription-blocking lesion mechanism is supported by eliminating all routes to lesion clearance at once: XPA-/- Csbm/m double mutant mice, which lose every branch of NER capable of clearing transcription-impeding adducts, develop progressive neurodegeneration consistent with cumulative transcription-impeding lesion burden in post-mitotic cells.[450,451] The Fanconi anemia pathway resolves interstrand crosslinks, including those generated by endogenous aldehydes such as formaldehyde and acetaldehyde (see EN77). Garaycoechea and colleagues showed that endogenous aldehydes drive hematopoietic stem cell failure in FA-deficient backgrounds,[19] and disruption of the FA pathway produces progressive bone marrow failure, developmental abnormalities, cancer predisposition, and features of premature aging.[452] Taken together, these repair deficiencies leave transcription-impeding or replication-blocking modifications in place, and the resulting phenotypes are dominated by progressive functional decline, neurodegeneration, developmental defects, or stem-cell failure, often with premature-aging-like features and sometimes alongside cancer risk, consistent with our framework’s prediction that functional decline results from the accumulation of modifications that impair information readout (see Supposition 12). The lesson of the Cockayne/UVSS dissociation is one of protectosphere redundancy (see EN29b): single-pathway loss is often buffered by parallel routes, and the undiluted phenotype emerges only when every redundant layer is removed.
In contrast, loss of replication fidelity mechanisms produces a fundamentally different outcome. Mismatch repair deficiency (Lynch syndrome, MSH2 or MLH1 loss) massively elevates mutation rates but produces cancer predisposition rather than a stereotyped progeroid syndrome independent of malignancy.[453] POLE and POLD1 proofreading domain mutations similarly increase replication error rates by orders of magnitude and predispose to cancer without classical progeroid features.[454] MUTYH deficiency, which fails to correct adenine mispaired with 8-oxoguanine during replication, causes colorectal polyposis through elevated G:C→T:A mutations, also through the cancer-predisposition rather than progeroid-aging route.[455] In each case, the unrepaired errors are replication-dependent mutations that do not block transcription, and their consequence is predominantly cancer through clonal selection rather than progressive functional decline.
The pattern in Supplemental Figure 3 makes this dissociation visible at the level of repair-pathway architecture: replication-fidelity defects (MMR, POLE/POLD1 proofreading, MUTYH) cluster overwhelmingly in the cancer-without-progeria phenotype, while transcription-blocking-lesion defects (TC-NER, especially CSA/CSB) and structural-maintenance defects (LMNA, ZMPSTE, BANF1, SPRTN, dyskeratosis genes) cluster overwhelmingly in the progeria-without-cancer-elevation phenotype. The within-gene dissociations are even cleaner: in POLD1, catalytic mutations produce the modification phenotype (normal cancer rate, shortened lifespan) while proofreading mutations produce the mutation phenotype (elevated cancer, normal lifespan). In TFIIH (XPB/XPD), complex disruption produces TTD (modifications, normal cancer rate, shortened lifespan) while helicase disruption produces XP (elevated mutations, elevated cancer, shortened lifespan). In XPG, nuclease disruption produces XP (elevated mutations, normal lifespan) while C-terminal deletion produces XP/CS (elevated mutations and shortened lifespan). The same gene, same protein, different mutation-class consequences, different phenotypic outcomes mapping precisely onto the modification/mutation distinction.
This double dissociation, where loss of modification repair causes progeroid-like decline (sometimes alongside cancer in pathways with environmental amplification, as in XP exposed to UV) and loss of replication fidelity causes cancer through clonal selection without producing classical progeroid syndromes independent of malignancy, is difficult to explain under any framework that treats all DNA damage as equivalent. Under this framework, the pattern follows directly: modifications that block transcription in critical genes are the primary driver of functional decline, while mutations that do not block transcription primarily elevate cancer risk through clonal selection. The specialization of repair pathways to specific damage classes, including the within-gene dissociations where different mutations in the same protein produce different phenotypes, is itself evidence that natural selection has independently identified the same molecular distinctions the framework draws.
EN52. Hierarchical protection systems and their limits.
Cells deploy multiple layers of defense beyond intrinsic DNA stability: histone-mediated chromatin packaging [83]; DNA repair pathways (base excision repair, nucleotide excision repair, mismatch repair, and double-strand break repair)[64,67,71,72,74,456], and checkpoint signaling to arrest replication under stress.[64] At the organismal level, tissue stem cells and immune surveillance mitigate local failures.[70,457] Beyond the organism, a social system further promotes protection of the replicator, manifested in behaviors ranging from herd formation to cooperative child-rearing. However, every layer of the protectosphere is finite. The replication and transcription of hereditary information create vulnerable states (replication forks, transcription bubbles, single-stranded DNA, stalled polymerases), exposing the substrate to chemical assault at critical moments, while endogenous metabolites continue to damage nucleic acids even in non-dividing cells.[99]
EN53. Plant longevity, negligible senescence, and the architectural basis of mortality shape.
Plants provide a striking natural test of the hierarchical amplification principle central to our framework. Long-lived modular plants lack several features that make animal aging accelerate: they have no central nervous system, no obligate post-mitotic organs whose loss is irreversible, and fewer architectural dependencies on the lifelong survival of irreplaceable post-mitotic cells, with most tissues regenerable from meristematic stem cell populations. Instead, plants grow modularly through meristematic tissue, stem cell populations distributed at shoot tips, root tips, and cambial layers that continuously generate new organs throughout the organism’s life. When a branch accumulates sufficient damage, it can be shed and replaced. When root tissue corrupts, new roots grow from surviving meristems. The organism persists not by maintaining the same cells indefinitely, but by continuously replacing corrupted modules with newly generated ones, a strategy unavailable to organisms whose architecture depends on irreplaceable post-mitotic components like neurons and cardiomyocytes.
Plant longevity is not unconditional. Many plants undergo programmed senescence at predictable timescales triggered by specific environmental cues, most notably the inability to survive winter at temperate and high latitudes.[458] Annual plants senesce dramatically at the end of a single growing season, monocarpic species like century plants and bamboo senesce after a single reproductive event, and many deciduous responses to seasonal stress involve coordinated tissue death. Under this framework, these patterns are not contradictions of negligible senescence but extensions of the same disposable-soma logic operating on different timescales: when expected somatic survival drops below the threshold where continued maintenance pays for itself, selection favors converting somatic resources into reproductive output rather than continuing to invest in repair. The result is environmentally triggered programmed death rather than corruption-driven physiological aging. Long-lived modular plants are the cases where neither environmental cues nor architectural dependencies force whole-organism senescence, so corruption-driven aging dominates and proceeds slowly enough to permit lifespans of thousands of years.
The consequences for lifespan are dramatic. Bald cypress trees along North Carolina’s Black River have been dated by dendrochronology and radiocarbon analysis to at least 2,624 years, making them the oldest known living trees in eastern North America and among the oldest wetland organisms on Earth.[459] Bristlecone pines exceed 4,800 years. Clonal plant colonies such as Pando, a quaking aspen grove connected by a single root system, are likely thousands of years old, with habitat-modeling and recent genetic-distance analyses giving estimates ranging from approximately 14,000 to potentially tens of thousands of years.[460] Some long-lived perennial plants show negligible or even negative demographic senescence, with mortality rates that do not increase with age in the Gompertz-like pattern characteristic of many animal species.[302] Our framework predicts this pattern: without a deep hierarchy of irreplaceable interdependent organs, molecular information corruption is less likely to propagate irreversibly through a single centralized hierarchy and tends to remain locally compartmentalized, and the accelerating mortality characteristic of animal aging is muted or absent. The information still corrupts. Plants accumulate somatic mutations in meristematic lineages, including substantial mutation loads in long-lived trees as documented in oak [461] and other long-lived species, with some of those mutations entering reproductive lineages because germline sequestration in plants is often late or flexible,[462] but the architecture limits the consequences by ensuring that no single corrupted module is indispensable. This is a protectosphere strategy based primarily on expendability rather than exclusive reliance on repair, and it works precisely because the hierarchy is shallow enough that molecular corruption does not cascade into systemic failure.
The architectural principle generalizes beyond plants. Hydra, which combines distributed stem-cell populations with the absence of irreplaceable post-mitotic organs, shows no age-related increase in mortality and no decline in fertility across laboratory observations spanning decades, with constant age-specific death rates that demographic modeling projects could sustain individual lifespans on the order of thousands of years under stable conditions.[463] Eusocial insect colonies exhibit individual workers aging on timescales of weeks to months while the colony itself persists for years or decades, because function is distributed across replaceable individuals (a principle we treat more fully in EN54). Even within mammals, species that evolved toward architectural and systemic compensation, such as the naked mole rat, whose subterranean niche and eusocial structure permitted extreme investment in corruption suppression (see EN29), delays or flattens the age-related mortality increase far beyond body-size expectations, with mortality hazard that does not increase with age in the standard Gompertzian manner across the studied period.[306] The general prediction is that mortality shape is determined by the interaction of corruption rate with architectural depth: organisms with shallow hierarchies and distributed replaceable components can approach negligible senescence, while organisms with deep hierarchies and irreplaceable post-mitotic components exhibit the accelerating Gompertz mortality characteristic of animal aging. Corruption still occurs everywhere; what varies is whether it can propagate through enough organizational layers to produce systemic failure.
EN54. Specialization as a recurrent protectosphere strategy.
The main text describes human specialization as the replicator’s breakthrough, but the principle of distributing function among specialized units to expand the collective protectosphere is far older and operates at every level of biological organization.
In bacteria, specialization appears in its most basic form. Individual E. coli cells acquire roughly one new mutation per thousand generations (Drake’s rule), meaning that any given bacterium is nearly identical to its neighbors and requires large populations to have any robust capacity for evolutionary adaptation.[292,325,444] Bacterial survival depends entirely on population-level diversity: among billions of cells, rare variants carrying useful informational changes are selected, and the population adapts collectively while most individuals do not. Biofilms formalize this further, with genetically identical cells adopting distinct metabolic and structural roles depending on their position within the community, some producing matrix, others sporulating, others maintaining active metabolism, each serving a function that benefits the group as a whole.[464]
The transition to multicellularity institutionalized this specialization. Dictyostelium, a social amoeba that lives as a solitary cell when food is plentiful, aggregates into a multicellular body under starvation and differentiates into two cell types: reproductive spores and sacrificial stalk cells that die to elevate the spores for dispersal.[465] Volvox, a colonial green alga, evolved a permanent division between somatic cells (responsible for motility and maintenance) and germ cells (responsible for reproduction), one of the earliest known instance of a dedicated soma-germline separation.[466] In both cases, genetically identical cells adopt different fates through regulatory mechanisms already present in their unicellular ancestors, not through genetic differences between cells. The same information, read differently, produces the specialization.
In eusocial insects and naked mole rats, the principle extends to the organismal level: most individuals in the colony suppress their own reproduction to serve specialized roles (foraging, defense, nursing, tunneling) that protect the reproductive members carrying the shared genetic information forward.[467,468] What our species mastered is not specialization itself, which is ancient and pervasive, but flexible specialization: the same genome produces a hunter or a healer, a builder or a thinker, depending on environment and learning, without requiring conditions that fixes a worker bee’s fate at the larval stage. This flexibility is itself a protectosphere adaptation, allowing a single species to respond to novel challenges at a speed that fixed specialization cannot match, and it may represent the most flexible biological layer of protection before culture and technology extends the protectosphere beyond the limits of encoded information entirely.[88]
EN55. Corruptible information classes in nucleic acids.
DNA suffers continuous endogenous decay (hydrolysis, deamination, depurination, oxidation, alkylation) and exogenous insults; repair pathways are organized to protect core structure, preserve sequence, and manage base modifications.[63,78] Each class elicits qualitatively different cellular responses: gross structural lesions (e.g., DSBs, crosslinks, collapsed replication forks) trigger checkpoint arrest, senescence, apoptosis, or repair [72,78]; sequence changes become heritable once they propagate through replication, because the cell loses the ancestral template that would allow correction [74]; base modifications can be repaired, tolerated, interpreted as regulatory signals, or converted into mutations or epigenetic remodeling if they persist through replication or alter repair and chromatin context.[75,90,231] In the framework presented here, structure, sequence, and modification state define the three classes of nucleic-acid information whose corruption is relevant to aging, with the three classes interconverting through replication and repair (see EN66, EN70, EN81). The aging hallmarks literature recognizes genome instability and epigenetic alterations as separate hallmarks; the intropy framework integrates them as outcomes of corruption acting on a single information substrate.[18,19]
EN56. Inheritance asymmetry: sequence vs epigenetic information.
DNA sequence is copied with template-directed fidelity and, once replicated, becomes durably heritable through all progeny.[65,75] Epigenetic information shows partial maintenance (e.g., DNMT1 at hemimethylated CpGs) yet undergoes two waves of genome-wide reprogramming (germline and early embryo) and drifts in somatic tissue [116,469,470,471,472]; thus, in mammals, epigenetic states are generally much less stably heritable across generations than sequence.[471,472] For the arguments herein, this asymmetry positions fixed sequence corruption as the most durable form of nucleic-acid information loss available to the cell. Sequence changes accumulate over the lifetime of long-lived and self-renewing lineages, can interact epistatically and clonally amplify when they affect proliferation-relevant genes, and translate into altered functional output when they affect coding or regulatory elements (most somatic mutations are silent or neutral, but the cumulative load produces measurable functional decline at the tissue and organism level).[18,19] Standing modifications are upstream of and more numerous than fixed mutations (see EN42, EN66), but they are individually reversible; sequence changes, once propagated, are not.
Standing modifications, however, are much more numerous per cell and while individually reversible, collectively impair or influence many critical cellular processes, including the transcriptional output in expressed genes (Supposition 12),[143,226,473] DNA stability,[63] and replication fork dynamics,[99,167] acting as a prominent source of information loss before conversion to permanent sequence changes (see EN81).[63,223]
EN57. Why global core structural disruption is unlikely to be a graded driver of aging.
Large-scale perturbations of the nucleic-acid core (loss of phosphodiester integrity, persistent DSBs, catastrophic crosslinks) typically activate the DNA damage response pathway, causing cell-cycle arrest, senescence, or apoptosis rather than slow, cumulative drift; checkpoint responses arrest, senesce, repair, or eliminate cells carrying such lesions, limiting their ability to act as silent graded insults.[64,78] In post-mitotic tissues where checkpoint elimination is not the dominant fate, persistent DDR signaling itself can drive functional decline, but this remains an acute or attritional contribution rather than a substitute for graded modification accumulation. Progeroid syndromes affecting DSB repair (e.g., LIG4 syndrome and other NHEJ-related defects) confirm rather than contradict this logic: these models produce aging phenotypes not because unrepaired DSBs accumulate as a chronic burden, but because checkpoint activation, apoptosis, senescence, and impaired self-renewal progressively limit stem cell function and tissue renewal.[45,404,405] The aging in these models is therefore a consequence of protectosphere hyperactivation depleting the regenerative reserve, not of structural damage operating as a gradual driver. An important exception involves repair intermediates: when base excision repair initiates but cannot complete (e.g., in XRCC1 deficiency), the resulting AP sites, single-strand breaks, and trapped PARP1 complexes persist as structural lesions that block transcription, collapse replication forks, and evade immediate elimination long enough to drive progressive cellular dysfunction, producing severe premature aging.[274] This suggests that the boundary between “structural” and “modification” damage is not absolute; incomplete repair can convert a tolerable modification into a toxic structural lesion. The framework predicts that such structural lesions become more prominent during late-stage collapse, when repair capacity, checkpoint control, and clearance mechanisms fail in coordinated fashion, but they are not expected to track the gradual, decades-long aging trajectory by themselves. Significant structural alterations of nucleic acid may therefore be most important as late-stage accelerants of collapse, once maintenance capacity has fallen below the threshold required to repair, tolerate, or eliminate damaged cells.
EN58. Sequence changes, information flow, and why proteins dominate function.
Gross disruption of DNA backbone integrity, duplex structure, or chromatin architecture is usually acutely handled by checkpoint responses, repair, senescence, or apoptosis rather than propagating as quiet graded inheritance, and the cellular consequences of failure tend to be severe (see EN57).[63,78,81] Changes to the primary sequence (the “code”) are generally more tolerable than gross disruption of the DNA backbone/duplex, and a subset can be beneficial.[75] In the operational central dogma, genomic sequence is transcribed to RNA and translated to protein, so coding and regulatory sequence changes that escape correction can alter the proteome by changing amino acid sequence, expression level, isoform choice, splicing, or timing.[75,92] Proteins dominate cellular catalysis, scaffolding, transport, signaling, and mechanical work, while RNA carries essential catalytic and regulatory functions including translation (ribosomal peptidyl-transferase center) and splicing (spliceosome).[474,475] RNA-level dysfunction is largely transient because RNA turnover is rapid; protein-level dysfunction can persist longer, with long-lived proteins particularly vulnerable to accumulated damage (see EN34). This framework accommodates known caveats (reverse transcription, RNA processing and mis-splicing, RNA viruses, prions) without undermining the predominance of DNA to RNA to protein information flow in cellular life.[93,320,476] Translation of information errors into altered protein abundance, sequence, folding, or regulation is therefore one major route by which nucleic-acid corruption produces cellular dysfunction during aging, and the framework treats it as a downstream consequence channel rather than an independent driver (see EN34, EN81).[18,19]
EN59. Scaling of protein corruption.
Any permanent translation of information change into proteins scales up a ladder as well. When a durable change in nucleic-acid information alters protein output, the consequences scale up through biological hierarchy. The functional consequence depends on the importance of the amino acid in the protein, the importance of the protein in the cell, the protein’s abundance, its interactions with other proteins and pathways, the cell’s role in the tissue, and the tissue’s importance to organismal function.[95,211,351,477,478,479,480,481,482] This hierarchical organization explains why corruption in stem cells has outsized consequences, since their errors can propagate to all lineage progeny (see EN36 and EN72). Conversely, post-mitotic tissues like the brain and heart are vulnerable for the opposite reason: their cells are rarely replaced in general, so persistent lesions, long-lived protein damage, mitochondrial defects, and epigenetic drift can accumulate over a lifetime with little dilution by cell turnover, making these tissues disproportionately dependent on in-cell repair, proteostasis, mitochondrial quality control, and damage tolerance to maintain information integrity (see EN34 for the long-lived-proteins exception in particular).[18,19,211,351,482]
EN60. Somatic mutation accumulation, mosaicism, and functional impact with age.
High-accuracy sequencing approaches, including ultra-deep, single-cell, and clonal organoid methods, demonstrate that somatic mutations accumulate across human tissues throughout life, yielding pervasive mosaicism; many tissues also show age-related clonal expansions.[211,483,484,485,486,487] Although most variants are neutral, burden and clone size increase with age, increasing the probability that some cells acquire function-impairing combinations, altered regulatory states, or maladaptive clonal behavior.[483,485] In addition, mutations in non-coding regions may affect cell function, since they can disrupt regulatory elements such as enhancers, promoters, and transcription factor binding sites that control gene expression without altering any protein sequence.[485,488] That said, elevated somatic mutations alone do not invariably accelerate aging: germline defects in replicative proofreading (e.g., POLE/POLD1) elevate mutation rates substantially yet produce cancer predisposition without classical progeroid phenotypes,[454] suggesting that mutation elevation alone is more strongly oncogenic than progeroid, and that if mutations contribute substantively to non-cancer aging, they do so in concert with persistent lesion burden, transcriptional disruption, epigenetic drift, clonal selection, and tissue architecture, rather than acting independently. These observations support sequence-level mosaicism as a plausible contributor to age-progressive cellular dysfunction.[103,210,211,483,485] In this framework, mutations are one durable downstream consequence of upstream modification burden and repair failure. They matter because they persist and can clonally expand, but persistent modifications are the more frequent and (generally) immediately disruptive substrate, while fixed mutations represent the irreversible channel through which earlier damage becomes permanent (see EN56 for the durability argument, EN62 and EN81 for the upstream-modification framing).
EN61. Replication program couples sequence/structure to mutation density and spectra.
During replication, the duplex must unwind at origins and replication domains shaped by local sequence features, chromatin accessibility, transcriptional activity, and three-dimensional genome organization.[206,489] Genome-wide analyses show higher mutation frequencies in late-replicating regions and strand-asymmetric biases tied to leading/lagging synthesis; multiple mutational signatures vary with replication timing and direction.[206,207,490] These patterns are consistent with local sequence, chromatin structure, and replication-program context modulating lesion exposure, repair efficiency, polymerase behavior, and mutation fixation, thereby shaping both mutation likelihood and spectrum.[206,489] These observations are consistent with the framework’s broader argument that replication is a moment of vulnerability for nucleic-acid information (see EN43, EN50).
EN62. Endogenous NICs and canonical base lesions.
Endogenous chemistry generates a steady flux of DNA base damage through four well-characterized mechanisms, each associated with a distinct class of nucleobase information corruptor (NIC; see EN67 for the framework’s developed treatment) and each capable of contributing to characteristic mutational patterns when unrepaired or misrepaired.[63,491] Hydrolytic deamination removes exocyclic amino groups from bases. Cytosine deaminates to uracil, and 5-methylcytosine deaminates to thymine, the latter producing a G-to-A transition at CpG sites that is a major biochemical source of SBS1, one of the clock-like mutational signatures that accumulates with age across many human tissues.[209] The corrupting chemistry here is hydrolysis itself, with water as the reactant; reaction rate is shaped by local chemistry (ionic strength, pH) and by single-stranded exposure during replication or transcription.[208]
Spontaneous depurination generates abasic (AP) sites by hydrolytic cleavage of the glycosidic bond. Unrepaired AP sites frequently impede replication and transcription, and their processing by BER generates single-strand break intermediates that, if incomplete (as in XRCC1 deficiency), persist as structural lesions (see EN51, EN57). Lindahl’s original estimates place the endogenous AP site burden at roughly ten thousand per cell per day.[63]
Oxidative damage proceeds primarily through reactive oxygen species generated by mitochondrial electron transport and inflammatory signaling. Guanine, whose low redox potential makes it the most vulnerable base, yields 8-oxoguanine (8-oxoG) and FapyG as the dominant products; other bases yield thymine glycol, 5-hydroxycytosine, and 8-oxoadenine at lower frequencies.[286,492] ROS-derived lesions are predominantly cleared by BER. 8-oxoG is less helix-distorting than bulky adducts and is often bypassed by Pol II with some misreading, contributing to aging principally through transcriptional mutagenesis, altered repair and chromatin recruitment, and replicative mutagenesis (see EN81 channels ii and vi) rather than primarily through Pol II stalling. SBS18 is commonly associated with oxidative guanine damage, and SBS36 with MUTYH deficiency, both involving defective handling of 8-oxoG:A mispairs or related oxidative lesions and producing G:C→T:A transversions.[209]
Alkylation occurs through endogenous methyl donors, most prominently S-adenosylmethionine, which non-enzymatically methylates ring nitrogens and exocyclic oxygens to generate 3-methyladenine, 7-methylguanine, O6-methylguanine, and related products. Alkylated bases differ in consequence: 3-methyladenine is strongly replication-blocking, 7-methylguanine can depurinate to form abasic sites, and O6-methylguanine mispairs with thymine during replication to produce G:C→A:T transitions. SBS11 is canonically associated with alkylating-agent exposure such as temozolomide, but endogenous O6-meG contributes to G:C→A:T transitions through the same biochemistry.[209]
Beyond these four canonical chemistries, a broader spectrum of endogenous NICs generates modifications whose aging-relevant consequences we develop separately: reactive aldehydes from lipid peroxidation (HNE-dG, etheno adducts, propano-dG, acrolein-dG) and from histone/DNA demethylation (formaldehyde-derived DPCs) produce bulky transcription-blocking adducts that are central to the Pol II stalling mechanism (see EN64, EN77).[214,286] The cumulative steady-state endogenous damage burden has been estimated at 10⁴ to 10⁵ lesions per cell per day, of which only a small fraction persist at any moment because of continuous BER and NER activity.[63,205]
Collectively, these endogenous chemistries constitute the dominant source of the NIC-driven modification burden that our framework positions as the primary substrate of aging Collectively, these endogenous chemistries constitute a major source of the NIC-driven modification burden that the framework positions as a primary substrate of aging (see EN66 for the developed argument). The downstream consequences of individual modification classes, whether they manifest as transcription blockade, replicative mutagenesis, or regulatory disruption, are developed elsewhere (see EN51, EN63, EN81).
EN63. Progeroid syndromes as natural dissections of the protectosphere.
Progeroid syndromes are genetic conditions that recapitulate selected features of aging on an accelerated timescale. They are often described as models of accelerated aging, but we argue this framing is misleading. Each progeroid syndrome results from the loss of a specific protectosphere component, and the resulting phenotype reveals which layer of information protection was breached, not what causes normal aging. Normal aging reflects the partial and interacting decline of multiple protective systems jointly calibrated against information corruption over the reproductive window (protectosphere calibration, see EN29b); we maintain progerias reveal what happens when one system fails in isolation while others remain intact. The accompanying figure and table organize the known repair and maintenance proteins whose loss produces aging, cancer, or both, and three patterns emerge that are central to our framework. First, defects that leave transcription-blocking lesions unresolved, trap repair intermediates, or disrupt the transcription-repair interface preferentially produce premature-aging-like phenotypes. This includes transcription-coupled NER (Cockayne-spectrum disorders, trichothiodystrophy), downstream BER coordination (XRCC1 deficiency), and interstrand crosslink repair (Fanconi anemia). By contrast, defects restricted largely to global-genome NER or upstream BER glycosylase steps more often present as cancer predisposition or mutator phenotypes than as classical progeroid syndromes.[493,494,495,496,497,498] The Cockayne syndrome case is discussed in detail in EN51 and is retained here as one entry in the broader pattern; the severity of the CS phenotype reflects a combination of repair loss and toxic gain-of-function from dysfunctional CSA/CSB protein rather than repair loss alone. Cockayne syndrome (CSA, CSB), trichothiodystrophy (XPD, XPB complex disruption), and XRCC1 deficiency all leave bulky adducts, AP sites, or repair intermediates lodged in the template strand of expressed genes, blocking RNA polymerase and directly impairing cellular function.[274,493,499] These conditions produce severe neurodegeneration, growth failure, and multi-organ decline, with many features of accelerated aging concentrated in specific tissues. The modifications responsible are individually repairable, but without the relevant repair pathway, they persist and accumulate in proportion to metabolic activity and transcriptional demand. Critically, mice deficient in transcription-coupled repair (Csb, Xpd) develop premature aging without elevated mutation frequencies, consistent with the standing modification burden driving functional decline independently of the mutational pathway, though with the caveat that these mouse lines carry hypomorphic or point-mutant alleles rather than clean nulls (see EN51 and EN76).[223]
Second, loss of pathways that prevent replication errors produces cancer without premature aging. Lynch syndrome (MSH2, MLH1, PMS2), POLE and POLD1 proofreading domain mutations, and MUTYH deficiency all elevate somatic mutation rates substantially, in some cases by orders of magnitude, yet affected individuals age on a normal schedule.[267,500,501] The mutations generated by these deficiencies do not block transcription; they are read through by RNA polymerase without stalling, and their primary consequence is clonal selection for growth-promoting variants (cancer) rather than progressive functional decline (aging). Third, loss of DNA damage response or structural repair pathways produces aging primarily through stem cell exhaustion. LIG4 syndrome, Ku80 deficiency, and ATM deficiency drive checkpoint activation, apoptosis, senescence, and impaired self-renewal that progressively deplete stem cell reserves in proliferative tissues such as bone marrow and skin (see EN36).[267,404,405,502,503,504] The aging in these models affects proliferative tissues preferentially and operates through a fundamentally different mechanism (depletion of cell quantity) than the modification-driven pathway (degradation of cell quality). The within-gene dissociations shown in the figure sharpen this picture further. POLD1 is a single gene encoding one protein, yet catalytic domain mutations (which impair gap-filling during repair and increase the standing modification burden) shorten lifespan without increasing cancer, while proofreading domain mutations (which increase replication errors) cause cancer without shortening lifespan.[267,454,505,506] TFIIH subunits XPB and XPD show the same dissociation: mutations that disrupt the helicase function needed for NER (allowing transcription-blocking modifications to persist) cause xeroderma pigmentosum with cancer and shortened lifespan, while mutations that disrupt the complex’s role in transcription (causing trichothiodystrophy) shorten lifespan without increasing cancer.[494,507] XPG follows the same pattern.[497] In each case, the same gene, the same protein, produces aging or cancer depending on whether the functional consequence is accumulated modifications or accumulated mutations. This finding supports the framework’s broader view that cancer and aging are divergent outcomes of nucleic-acid information corruption: cancer reflects clonal gain of proliferative fitness through replication-fixed mutations, while aging reflects loss of coordinated tissue function through accumulated modifications, mutations, and cell loss. Both conditions trace to the same molecular substrate (information change in nucleic acid) but differ in their downstream consequence channels. The UVSS separation-of-function variant of CSA (p.Trp361Cys),[508] which disrupts UV damage repair while preserving the oxidative stress response and produces UVSS rather than Cockayne syndrome, provides a final refinement to the within-gene pattern: the progeroid component of CSA loss tracks with its non-repair activities rather than with its TC-NER role, consistent with the broader argument developed in EN51.[508] We therefore propose that progeroid syndromes should be read not as accelerated versions of normal aging but as controlled natural experiments that isolate individual variables in the corruption-protection system. Each syndrome answers a specific question: what happens when this particular type of damage is left unrepaired, or when this particular response pathway is hyperactivated? The answers consistently point to the same conclusion across all three patterns: aging in each case reflects a different mode by which the information that orders cellular function becomes progressively corrupted, whether by accumulation of transcription-impeding modifications, accumulation of heritable mutations, or attrition of the cell populations that carry the information forward, precisely the mechanism proposed in this manuscript.
EN64. Pol II Stall duration, not repair deficiency, determines progeroid severity; R-loops as secondary pathology; and the endogenous damage connection.
Stall duration as the critical variable. One of the most informative natural experiments in aging biology is the comparison between Cockayne syndrome (CS) and UV-sensitive syndrome (UVSS). Both conditions result from loss of transcription-coupled nucleotide excision repair (TC-NER): CS from mutations in CSA or CSB, UVSS from mutations in UVSSA.[273] Both impair TC-NER, yet diverge sharply in how stalled Pol II is subsequently processed. Yet their clinical presentations differ dramatically. CS produces severe progeria with neurodegeneration, growth failure, and early death; UVSS produces only mild photosensitivity and freckling.
Gonzalo-Hansen et al. resolved this paradox by measuring the fate of stalled Pol II in each condition.[226] In CS cells lacking CSA or CSB, lesion-stalled Pol II remains bound to chromatin for extended periods. Two hours after UV exposure, 10-20% of all Pol II molecules in CS cells remained trapped at damage sites. Against an approximate cellular Pol II pool on the order of tens of thousands of molecules, this represents thousands of large macromolecular complexes physically obstructing DNA-transacting processes at those loci. In UVSS cells lacking UVSSA, the lesion is equally unrepaired, but the stalled Pol II is cleared from the template by VCP-mediated proteasomal degradation. The lesion remains, but the roadblock is removed.
This distinction has profound implications for our framework. Severity is not explained by lesion persistence alone; the dominant variable is how long a stalled polymerase obstructs the template. CS cells cannot clear stalled Pol II (severe progeria). UVSS cells cannot repair the lesion but can clear the stalled polymerase (mild phenotype). Normal cells both repair the lesion and clear the polymerase, but with age, both processes slow. The progeroid phenotype thus arises not from DNA damage alone, but from the duration of transcriptional blockade at damaged loci, consistent with our proposal that it is information readout, not information content per se, that drives functional decline.
The RPB1-K1268R knockin mouse reinforces this logic by demonstrating that disrupting the ubiquitination signal upstream of both repair and clearance is sufficient to produce shortened lifespan, premature aging, and neurodegeneration even when the rest of the repair machinery is genetically intact.[473] Donnio and Giglia-Mari further emphasized that recovery of RNA synthesis after damage is a process distinct from repair itself, depending on how stalled Pol II is processed, whether restart factors are recruited, and whether chromatin structure is restored.[509]
Prolonged transcriptional stalling generates secondary pathology through R-loops. Although R-loops form continuously as a feature of normal transcription and are typically resolved within minutes by RNase H1, RNase H2, Senataxin, and topoisomerases, persistent stalling stabilizes existing R-loops and impairs their resolution. But when stalling persists, R-loops accumulate and create secondary problems: the displaced non-template strand becomes vulnerable to chemical attack (deamination, oxidation, nicking); the R-loop itself blocks subsequent Pol II molecules from transcribing the same gene; and unresolved R-loops expose the displaced non-template strand to chemical attack, block subsequent Pol II elongation through the same locus, and in some contexts contribute to nicks, replication-transcription conflicts, and double-strand breaks. .
Hall et al. profiled R-loops in aging Drosophila photoreceptor neurons and found that bulk R-loop levels increased with age, but not through formation of new R-loops at novel sites.[510] Instead, they observed broadening of existing R-loop peaks, indicating failure to resolve rather than increased formation. The R-loop accumulation was concentrated at long, highly expressed genes, precisely the loci most vulnerable to stochastic damage. Protein levels of Top3-beta, a topoisomerase involved in R-loop prevention, declined with age. Photoreceptor-specific depletion of Top3-beta caused a 60% decrease in visual function by day 30 without any retinal degeneration (neurons alive but dysfunctional), while overexpression of Top3-beta or nuclear-localized RNaseH1 enhanced visual function during aging. This demonstrates that R-loop accumulation is a reversible contributor to age-related neuronal dysfunction and that neurons can be functionally impaired by R-loop-mediated transcriptional disruption long before they die.
Much of the foundational work on Pol II stalling and TC-NER used UV-induced cyclobutane pyrimidine dimers as experimental substrates. A reasonable concern is whether these findings apply to the endogenous modifications that accumulate during normal aging. A trio of 2024 studies in Nature Cell Biology addressed this directly by showing that DNA-protein crosslinks (DPCs) induced by formaldehyde, a normal metabolic byproduct of one-carbon metabolism, stall Pol II and recruit CSB and CSA through the same TC-NER pathway used for UV damage, and that TC-NER-deficient cells show delayed recovery of RNA synthesis after formaldehyde exposure.[511,512,513] This establishes that the stalling/repair/recovery machinery studied in UV paradigms is the same machinery handling endogenous metabolic damage. Mulderrig et al. (2021) provide the in vivo phenotypic counterpart by showing that mice deficient in formaldehyde clearance (Adh5−/−) crossed onto a Csb null background develop cachexia, neurodegeneration, and kidney failure resembling human Cockayne syndrome, with formaldehyde-driven transcriptional stress activating an anorexigenic GDF15 response in proximal tubule cells; CSB therefore protects post-mitotic and slow-turnover tissues from endogenous formaldehyde at the level of organismal physiology, not just transcript-level recovery.[514]
More broadly, any bulky modification that distorts the DNA helix sufficiently to arrest Pol II will trigger the same cascade regardless of its chemical origin. Endogenous sources of such modifications include lipid peroxidation products (HNE-dG, etheno adducts, propano-dG, acrolein-dG), formaldehyde-derived crosslinks and DPCs, oxidation-derived cyclopurine adducts (which are particularly relevant because they are NER substrates not repaired by BER), and products of other reactive carbonyls from glycolysis, amino acid catabolism, and polyamine metabolism. The combined steady-state burden of these endogenous transcription-blocking modifications is substantial: The combined steady-state burden of endogenous DNA modifications across all classes reaches thousands of lesions per cell genome.[515,516] The Guilbaud et al. adductomics study detected over 100 distinct putative DNA adducts in rat and human tissues, with 36 showing significant age-dependent accumulation, including etheno-deoxyadenosine (a lipid peroxidation product) in human hearts.[286] Each of these adducts, if positioned in the template strand of an active gene, has the potential to stall Pol II and initiate the cascade described above. The rate of functional decline in any tissue may therefore be understood as a function of the modification burden in active genes, the efficiency of TC-NER and Pol II clearance, and the average gene length of the tissue’s transcriptional program, a prediction that is testable and falsifiable.
EN65. From DNA adduct chemistry to mutational signatures.
Large cancer-genome analyses resolve mutational signatures that reflect operative mutational processes; both exogenous (e.g., UV, tobacco PAHs, aflatoxin) and endogenous (e.g., 5-mC deamination, oxidative damage) processes leave characteristic trinucleotide-context patterns.[152,517] Several signatures show replication-strand and timing biases, consistent with lesion formation and bypass dynamics.[207] This provides a route to infer candidate NICs and repair contexts from observed mutation spectra, although most signatures are composite readouts shaped by lesion formation, repair, replication timing, and bypass dynamics rather than one-to-one chemical fingerprints.[518,519]
EN66. Why base modification is the root reversible event.
hen a nucleobase undergoes chemical alteration, the information the template carries is functionally changed until repair restores the original base.[63,491] If this lesion escapes repair and is copied, the resulting mutation becomes fixed on both strands, propagating through all descendant cells.[75] This permanence contrasts with most protein and metabolite damage, which is typically degraded, diluted, or replaced through ongoing turnover; long-lived proteins are a partial exception developed in EN34.[136] Thus, base change is the earliest irreversible step in the corruption cascade and base modification the earliest reversible step that precedes permanent errors, though as discussed in EN62, standing modifications can impair transcriptional output directly even before conversion to mutation.[63,456,491]
EN67. Operational definition of a corruptor.
A corruptor (NIC) is any chemical species that reacts with nucleobase atoms to alter their covalent structure or pairing behavior, thereby changing the information content of the affected base.[63,491] This includes hydroxyl radicals and other reactive oxygen species, endogenous alkylating agents (e.g., S-adenosylmethionine acting non-enzymatically), reactive aldehydes (formaldehyde, acetaldehyde, lipid peroxidation products such as 4-HNE and acrolein), deaminating species, and exogenous chemistries (e.g., nitrogen mustards, UV-generated photoproducts, ionizing-radiation-derived radicals).[67,520,521] The defining criterion is that the modification changes the original information: it may compromise template fidelity during replication (leading to misincorporation or strand misalignment), block transcriptional readout in expressed genes, block transcriptional readout, alter splicing or RNA output, interfere with repair, or destabilize the local structure of nucleic acid.[232,520] Cells deploy extensive systems to remove both free and reacted corruptors, and these systems succeed the vast majority of the time. But correction is not perfect, and the residual standing modification burden increases over time as repair capacity declines, with each replication cycle risking permanent fixation of altered information.[63,67,521]
EN68. Epigenetic theory and its integration with information corruption.
The epigenetic theory dates to Holliday’s proposals in the 1980s that age-associated changes in methylation and chromatin structure drive aging and cancer.[114,115] Landmark studies (e.g., Horvath clocks) show methylation patterns predict chronological age [422], and experimental reprogramming can partially restore youthful transcriptional states.[282] These findings align with the corruption model but differ in permanence: epigenetic marks are reversible and not indefinitely heritable.[18,116] In our framework, information corruption drives aging through two complementary routes. In dividing cells, unrepaired modifications can fix as mutations during replication and propagate through all descendant lineages (see EN66). In both non-dividing and dividing cells, the standing modification burden can impair transcription, splicing, repair, and regulatory readout from the affected template. Both routes propagate functional inefficiency upward through the biological hierarchy.[522] Epigenetic drift is therefore not treated as an independent primary cause in this framework: it follows from upstream sequence corruption (which permanently alters methylation patterns) and from standing modifications (which disrupt the enzymatic maintenance of epigenetic marks), and it amplifies the cascade once established by dysregulating downstream gene expression.[19,116]
EN69. Limits of epigenetic inheritance across cell generations.
Maintenance methyltransferases (e.g., DNMT1) copy CpG methylation during S-phase [469,470,523], but fidelity is imperfect [524] and global erasure occurs in germline and early embryogenesis.[469,471] Histone modifications are even less stably propagated.[470] Consequently, we maintain that epigenetic drift amplifies local inefficiency but is better understood as a downstream consequence of upstream information corruption than as an independent primary cause: its imperfect inheritance differs from both the real-time chemical presence of standing lesions and the durable lineage propagation of fixed sequence mutations.[472]
The evolutionary record reinforces this asymmetry. If epigenetic state were the primary repository of organism-defining information, heritable epigenetic variation should be a substantial driver of speciation and adaptation. The empirical pattern across evolutionary genetics is consistent: closely related species differ at the sequence level, and where epigenetic differences exist between them, those differences track underlying sequence variation rather than serving as the primary axis of divergence.[525] Documented candidates for primarily epigenetic speciation are rare, metastable, and have not produced reproductively isolated lineages over evolutionary timescales. Documented cases of transgenerational epigenetic inheritance in plants and animals typically decay within a few generations, far short of the timescales over which speciation occurs. The deepest substrate that survives germline reprogramming, geological time, and selection on phenotypes is DNA sequence, together with the developmental and regulatory programs it encodes. This is the substrate to which aging-relevant information ultimately reduces in this framework.
EN70. The epigenome is a reporter of foundational sequence information corruption.
This framework explains the remarkable accuracy of epigenetic clocks in predicting age.[422] Rather than epigenetic drift being a primary cause, methylation patterns serve as sensitive, early-stage readouts of underlying DNA sequence and information corruption.[63,91,116] Recent evidence directly supports this interpretation by demonstrating that somatic mutations at CpG sites drive extensive remodeling of the surrounding methylome, with mutation-based age predictions closely paralleling epigenetic clock estimates and accounting for more than half of inter-individual variation in clock-predicted age.[118] The clock therefore reports on two layers of corruption operating in parallel: permanent sequence changes that destroy or alter methylatable sites, and standing modifications that disrupt methylation maintenance at sites whose sequence remains intact (elaborated in EN71). Because methylation is coupled to local DNA state and aggregates signal across thousands of CpG sites, it detects corruption early, before functional redundancy at higher levels of biological organization masks the decline.[18,19,422] One well-characterized chemical mechanism for the modification-driven component is direct inhibition of DNA methyltransferases by oxidative lesions at CpG sites: 8-oxoG decreases Dnmt3a methylation activity in vitro by up to 25-fold depending on its position relative to the target cytosine, providing a concrete link between the standing modification burden and clock methylation loss.[240]
EN71. Lesion-to-mutation pathway and feedback with chromatin state.
Base modifications such as 8-oxoG or alkyl adducts miscode during replication, producing point mutations that can disrupt transcription-factor motifs and reconfigure local chromatin, creating a feedback loop between sequence corruption and epigenomic dysregulation.[232,526,527,528,529,530] However, modifications can also alter the epigenome directly, without passing through the mutation step. Oxidative lesions such as 8-oxoG at or near CpG sites recruit base excision repair machinery (notably OGG1 and TET enzymes), and the repair process itself demethylates the adjacent 5-methylcytosine, producing a heritable loss of methylation at that site.[229,231] Similarly, 8-oxoG at or near CpG sites has been shown to inhibit Dnmt3a activity by up to 25-fold, impairing maintenance of methylation during replication.[240] These direct pathways mean that the standing modification burden continuously reshapes the methylome without requiring replication errors as an intermediary, and they provide a mechanistic explanation for why epigenetic clocks correlate with age even in slowly dividing tissues. We propose that 8-oxoG/OGG1 is one example of a broader, testable principle: any repair process that excises and resynthesizes a DNA segment containing methylated CpGs may impose epigenetic collateral damage if the original methylation pattern is not faithfully restored. NER removes an oligonucleotide patch of roughly 24–32 nucleotides around bulky adducts,[531] and ICL repair disrupts an even broader region of local chromatin. In both cases, restoration of the original methylation depends on faithful copying from the opposing strand by maintenance methyltransferases such as DNMT1, with de novo activity (DNMT3a/3b) required where the opposing-strand mark is absent or inaccessible. In post-mitotic or slowly dividing cells where de novo methyltransferase activity is low, incomplete re-methylation could leave the loss effectively permanent. This generates a falsifiable prediction: transcription-blocking modifications repaired by NER, and lesions repaired through ICL pathways, should produce greater epigenetic collateral damage per event than small base modifications repaired by single-nucleotide BER, coupling the transcriptional severity gradient (Supposition 12) to epigenetic drift through the same class of lesions.
EN72. Clonal hematopoiesis as empirical demonstration of the corruption cascade.
DNMT3A mutations that drive clonal hematopoiesis arise when modifications, replication errors, or error-prone repair events become fixed in hematopoietic stem cells, producing a permanent sequence change whose downstream consequences illustrate the full corruption cascade (see Suppositions 13–17). The phenomenon of normal tissues being colonized by mutant clones is now documented across multiple organ systems, including skin, esophagus, blood, and others, with hematopoiesis providing the most extensively characterized example.[486,487] The most common DNMT3A driver mutation, R882H, occurs at a CpG site whose mutational signature is consistent with 5-methylcytosine deamination, an unrepaired modification scenario the framework predicts. Clonal hematopoiesis therefore provides an empirical demonstration of the cascade in a single tissue system. Mutations in the epigenetic regulator DNMT3A, one of the most common driver of age-related clonal expansion in hematopoietic stem cells, cause.[48,119,243] Work from Goodell and colleagues established that Dnmt3a-null HSCs can regenerate across at least twelve serial transplant generations in mice, far exceeding normal HSC lifespan, demonstrating that loss of this single epigenetic regulator fundamentally alters stem cell behavior.[532] Kapadia and Goodell have since synthesized this and related findings into a unifying account of clonal expansion as tissue mosaicism, an organizing concept that maps directly onto the hierarchical propagation predicted by this framework.[48] Here, upstream sequence corruption (a somatic mutation in DNMT3A) drives downstream epigenetic degradation (methylation loss at regulatory domains), which in turn alters stem cell output (impaired differentiation, expanded self-renewal), producing the tissue mosaicism increasingly recognized as a hallmark of aging.[48] This is the hierarchical cascade our model predicts: information corruption at the base of the hierarchy propagating functional consequences upward through every dependent level. The prevalence of DNMT3A-mutant clonal hematopoiesis (detectable in over 10% of individuals older than 65) and its association with cardiovascular disease, inflammation, and hematologic malignancy illustrate how a single point of sequence corruption in a stem cell can amplify into organism-wide pathology.[120]
EN73. Epigenetic primacy, the backup copy hypothesis, and reprogramming.
Yang, Hayano, Griffin et al. (2023) reported that targeted DNA double-strand breaks at non-coding sites in the ICE (Inducible Changes to the Epigenome) mouse model produced accelerated aging phenotypes that were partially reversed by OSK reprogramming factors (Oct4, Sox2, Klf4).[121] Lu, Tian, and Sinclair (2023) subsequently formalized this and related findings as the Information Theory of Aging (ITOA), under which epigenetic information loss is itself a reversible cause of aging, with reprogramming retrieving correct epigenetic patterns from a backup copy of youthful information.[173] The reprogramming results are striking. Our reading differs from ITOA on a single but consequential point: not whether the phenomena ITOA documents are real, but where they sit in the causal hierarchy. The framework presented here positions epigenetic drift as a readout of upstream nucleic acid corruption rather than as an autonomous primary driver, a positioning consistent with how the framework treats other downstream observables throughout (see EN21, EN38, EN71, EN72, EN81). What follows develops that positioning specifically with respect to the reprogramming literature.
The disagreement is one of causal ordering. Methylation patterns predict chronological age with high accuracy,[422] reprogramming partially reverses methylation clock signal, and ICE mice respond particularly well. Epigenetic state is one of the genuine readouts through which aging manifests, and the ITOA framework points to phenomena that are real and worth explaining. The question is whether epigenetic drift sits at the top of the causal pyramid, with modifications and mutations as secondary contributors, or whether modification burden sits at the top, with epigenetic drift as one of the primary downstream readouts amplifying and propagating the underlying corruption. The framework here takes the second position, that modifications are the upstream variable. Epigenetic drift is what those modifications produce when read out through the cell’s regulatory machinery. The apparent reversibility of epigenetic drift under reprogramming reflects readout being temporarily corrected, not the underlying corruption being addressed. Mutations and modifications, in this framing, are not in competition with epigenetic drift as candidate causes; they are upstream of it.
Universality of aging across the tree of life provides one line of evidence about where in the causal hierarchy aging actually lives. Every organism studied senesces, from bacteria undergoing asymmetric segregation of damaged components,[294] to yeast accumulating oxidative and alkylation damage during chronological aging, to multicellular organisms across kingdoms. What is universal across these systems is the chemistry of the substrate: nucleic acids carry the ordering information, modifications continuously occur, repair is imperfect, and the standing burden rises over time. What varies is the regulatory layer those modifications act upon. The Information Theory of Aging covers a broader epigenome than mammalian methylation alone, including histone modifications and three-dimensional chromatin organization, but even at its broadest, the regulatory substrate ITOA designates as the carrier of aging-relevant information differs markedly across taxa. S. cerevisiae lacks cytosine methylation entirely [533] and silences chromatin through Sir-family deacetylation. C. elegans has minimal cytosine methylation.[534] Bacterial methylation serves restriction-modification rather than transcriptional silencing. Mammals layer cytosine methylation on top of histone marks and chromatin architecture. The substrate that varies, even granting ITOA its broadest scope, is the regulatory readout; the substrate that does not vary is modification chemistry acting on nucleic acid. A framework that locates aging at the modification-accumulation level operates at the level shared by everything that ages. A framework that locates aging at the regulatory readout level requires a separate primary substrate in each lineage. We treat universality as evidence about which variable sits closer to the cause: the variable that everything aging shares, rather than the one that varies across taxa.
Within mammals specifically, the same logic plays out at the level of how the epigenome is constructed in the first place. Each generation rebuilds nearly all of the somatic epigenome through extensive erasure and reconstruction. Primordial germ cells undergo a methylation reset to under five percent of starting levels, erasing imprints, X-inactivation marks, and most retrotransposon silencing.[535] Methylation is then re-established de novo through DNMT3A/B, with the resulting patterns shaped by sequence context, pioneer transcription factor binding, and histone landscape rather than by any inherited methylation state. Histone marks undergo equally dramatic resetting: H3K27me3 is largely cleared from sperm at fertilization and reconstructed across pre- and post-implantation development, with the canonical bivalent H3K4me3/H3K27me3 patterns at developmental genes appearing only after implantation.[536] These observations are not in themselves an argument against the existence of a reference repository in ITOA’s sense; the repository can be read as the post-developmental cellular state that aging then drifts away from. They are an argument about where the durable reference information lives. The cellular state at the end of development is itself constructed each generation from sequence-encoded information, namely the genome plus the cell-type-specific transcription factor networks it encodes. Even if aging is described as drift from the post-developmental state, the substrate that determines what that state is, and that determines whether it can be reconstructed, is sequence and the TF networks sequence encodes. The candidate Observer markers proposed within ITOA, including H3K27me3, H3K9me, and DNA:RNA hybrids, are themselves among the chromatin features that undergo near-complete reset during germline reprogramming and post-fertilization reconstruction; they appear as constructive outputs of development rather than inherited inputs to it.
The somatic version of this same logic is induced pluripotent stem cell generation. Sequence-encoded transcription factors, applied to a fully differentiated adult cell, can reset that cell’s epigenetic state to pluripotency.[354] The factors are constructive machinery: they engage developmental TF networks whose binding sites remain present in sequence, recruit chromatin remodelers and DNMT/TET enzymes, and rebuild the pluripotent epigenetic state from sequence input. Whatever epigenetic state exists in an adult somatic cell was originally established by exactly this kind of constructive process during the individual’s own development, with cumulative perturbation since. Reprogramming uses constructive machinery to write a new state.
Yeast aging illustrates the universality point at the cellular level and provides direct mechanistic evidence for modification primacy in the organism where ITOA originated. Studies showing that the Sir silencing complex redistributes from silenced loci to sites of DNA damage and that this redistribution corrupts gene silencing during aging [537,538] are the foundation of ITOA’s chromatin-based reading, but the mechanism is precisely consistent with our framing: nucleobase modifications accumulate during yeast replicative aging, including 8-oxoguanine and DNA strand breaks measured by comet assay,[539] base excision repair capacity is causally required for full yeast lifespan, and modifications couple directly to chromatin in yeast through nucleosome-position-dependent BER kinetics and histone-modification-regulated repair.[540] Sir2 redistribution itself is triggered by DNA damage that pulls Sir2 away from silencing duties; the chromatin changes ITOA describes in yeast are responses to upstream modification burden, with asymmetric segregation of damaged macromolecules as the yeast analog of protectosphere function. The modification-driven architecture proposed here is therefore not in tension with the yeast foundation of ITOA but operative within it.
The Cockayne syndrome versus UV-sensitive syndrome contrast provides natural-experiment evidence at the level of stall-duration-driven readout. Both syndromes share TC-NER defects and both fail to repair transcription-blocking modifications. What differs is how long stalled Pol II persists at unrepaired lesions: hours in CS where CSA/CSB cannot recruit clearance machinery, minutes in UVSS where VCP-mediated clearance proceeds (see EN64). Crochemore et al. (2023) showed that CS fibroblasts carry approximately 15.5 years of accelerated epigenetic age relative to UVSS and healthy controls by the Horvath Skin & Blood clock, with a CS-specific progeroid methylome signature absent from UVSS, despite both syndromes impairing transcription-coupled repair.[225] Other features of CS that UVSS lacks may also contribute to this clock differential, but the stall-duration variable is consistent with our reading: the methylome tracks modification persistence at the transcriptional readout level, not bulk lesion accumulation alone.
The mechanism by which reprogramming partially reverses methylation clock signal, and the rebound that follows factor withdrawal in most published protocols, both fall out of treating modification burden as a flow variable rather than static. Standing burden at any moment reflects the balance between continuous production by ongoing metabolism and continuous clearance by repair, with steady-state burden set by the ratio of these rates. Anything that lowers production, raises clearance, or transcriptionally masks modification-driven readout will lower the apparent clock signal. Anything that reverses these changes will allow the signal to return toward its prior steady state on the timescale of the relevant kinetics. These mechanisms are non-exclusive, and the framework does not require the same lever to dominate in every tissue or protocol.
OSK and OSKM appear to act on more than one component of this balance. Pluripotent state is associated with broadly elevated base excision repair and homologous recombination capacity,[541,542] while nucleotide excision repair components are not preferentially upregulated. OSK and OSKM also appear to lower production rate. TGF-β signaling drives ROS, inflammation, and senescence-associated damage, and reprogramming suppresses TGF-β pathway activity early in its trajectory; pluripotent metabolism shifts toward glycolysis with reduced mitochondrial ROS output. Paine et al. (2024) showed that small-molecule inhibition of the TGF-β pathway alone, without OSKM, was sufficient to reduce DNA damage markers and rejuvenate the methylation clock in fibroblasts from Ercc1−/Δ progeroid mice, despite the same intervention disrupting several repair processes in those cells.[283] Clock improvement under conditions of reduced clearance capacity is consistent with production suppression as an independently functional lever. A third OSK contribution is transcriptional: while OSK is expressed, the factors engage developmental TF networks that override aspects of the somatic state regardless of whether the underlying modification or mutation context has been physically cleared.
The rebound after factor withdrawal follows from the same flow framing. When OSK transcription stops, production rates return toward baseline as TGF-β signaling resumes and metabolism reverts; repair protein levels decay back toward somatic baseline over the timescale of protein turnover; and the transcriptional override of any mutation-driven methylation pull releases as exogenous OSK protein is no longer maintaining it. The methylation balance returns toward its prior steady state on the kinetic timescale of these processes, days to weeks rather than instantaneous. The dissociation between rebounding methylation clock signal and persisting phenotypic benefits has a principled basis here: clock signal is a steady-state observable that reflects continuous flow balance and reasserts itself when that balance is restored, while benefits arising from one-shot processes (clearance of senescent cells, completion of accumulated DSB repair, re-engagement of self-stabilizing transcriptional circuits) do not have a steady-state balance to drift back to and persist after the work is done. The published literature shows exactly this dissociation, with lifespan extension and tissue integrity in progeroid models persisting long after factor withdrawal while methylation clock signal rebounds on the timescale described above.[282]
Mutation effects on the methylome operate on different timescales than modification flow and contribute to the bound on reversibility. A mutation that destroys a CpG cytosine eliminates the methylatable substrate at that site, fixing a hard local bound on what any reprogramming protocol can restore. Koch et al. (2025) demonstrated a second route in which mutations remodel the surrounding methylome over substantial distances, accounting for a substantial fraction of the inter-individual variation in clock-predicted age.[118] During OSK expression, long-range mutation effects may be transcriptionally overridden by the engagement of pluripotency-associated TF programs at affected loci. After withdrawal, the underlying mutation-driven methylation pull would reassert as the override releases. A further contribution to drift dynamics is developed in EN89: standing modifications can epigenetically silence repair gene promoters, lowering clearance capacity. Reprogramming that transiently restores repair gene expression by clearing aberrant promoter methylation would briefly raise clearance capacity above its pre-reprogramming baseline, contributing to age reversal during expression, with re-silencing returning clearance to baseline and contributing to rebound.
Available data fit this picture in a graded way. The ICE model itself, under the reported interpretation of brief damage induction during youth and reportedly faithful sequence resealing, achieves up to 57 percent epigenetic age reversal in cultured MEFs across four independent clocks (Thompson, Petkovich, Meer, and Stubbs).[121,543] Aged wildtype animals respond along a gradient that scales with the framework’s predictions. Browder et al. (2022) treated 12- and 15-month-old mice with cyclic OSKM over ten months and reported significant clock reduction in skin and kidney, with little change in liver, lung, muscle, or spleen; the same dosing schedule shortened to one month and applied to 25-month-old animals produced no measurable clock improvement.[285] Younger animals given more time responded in some tissues; older animals given less time did not respond at all. The flow framing predicts exactly this. Tissues with lower standing modification burden, treated for windows long enough to let the methylation balance track the reprogramming-imposed transcriptional state, should show more clock reduction than tissues with heavier accumulated burden treated briefly. The framework therefore reads reprogramming efficacy as bounded by accumulated mutation effects, persistent modification context, and altered cell composition in the target tissue, with each variable setting a floor on what any protocol can restore and a ceiling on durability. The ICE model itself is contested,[544,545] and both proposed interpretations of the underlying damage sit naturally within the framework presented here without changing the claim about causal ordering.
A useful way to read the ICE experiment within the intropy framework is that Yang and colleagues ran the modification-primacy test directly, framed as a test of the inverse hypothesis. The induced lesion was a chemical event (a targeted double-strand break). The dependent variable was readout corruption: epigenetic drift, transcriptional change, exdifferentiation, accelerated clock signal. The partial reversibility under reprogramming established that a transcriptional override could realign the readout once the upstream chemical event (modification) had been resolved through repair. That is the structure the framework predicts: introduce modifications, observe readout corruption, observe functional decline, then partially correct the drift by restoring the transcriptional program. The disagreement with ITOA is narrower than it first appears. We agree on which experiment was run and what it shows. The reading differs at one step: ITOA treats the recovery as evidence that the regulatory information is itself the aging-relevant locus, retrieved during reprogramming from a youthful backup. We treat the recovery as evidence that modifications drive readout corruption that reprogramming can temporarily mask. The operative difference between ICE aging and physiological aging is that ICE delivers damage as a discrete event during youth and the cell then repairs it, while physiological aging delivers damage continuously and clearance does not keep pace. We hold that ICE has done the modification experiment for us. The interpretive layers ITOA adds, a primary epigenetic locus and a backup repository, are compatible with what the experiment shows but are not required to explain it. The framework prefers the reading that requires fewer interpretive layers because physiological aging, where damage is delivered continuously rather than as a discrete youthful event the cell then resolves, behaves differently from ICE in ways the modification-readout architecture predicts directly.
This reading also locates the proper role of reprogramming within a broader intervention architecture. ICE shows that resealing the upstream damage is not by itself sufficient to restore readout fidelity; the readout corruption persisted even after the induced breaks were repaired faithfully and was realigned only when OSK engaged the constructive transcriptional machinery. Prevention and rejuvenation are therefore distinct intervention classes addressing different parts of the same problem. Prevention targets the production-and-clearance balance through reduced NIC flux, enhanced repair, and restored expression of epigenetically silenced repair genes; these levers do not require reprogramming. Direct support for the prevention arm comes from Vermeij et al. (2016), who showed that thirty percent dietary restriction tripled both median and maximal remaining lifespan in Ercc1∆/− progeroid mice, retained roughly fifty percent more neurons, preserved motor function, suppressed the gene-length-biased transcriptional decline that accumulating damage produces, and reduced γH2AX foci, demonstrating that lowering damage flux rescues a repair-deficient progeroid phenotype without any reprogramming intervention.[277] Rejuvenation targets readout corruption that has already propagated beyond what damage clearance alone can resolve, and that appears to require constructive transcriptional override of the kind OSK provides. Mutations sit beneath both intervention classes as the irreducible floor. They are the permanent byproduct of modifications meeting replication or repair errors, and once fixed they cannot be cleared by repair or restored at destroyed CpG substrates. Prevention slows their accumulation by clearing modifications before conversion; rejuvenation can transcriptionally override their regulatory consequences while OSK is expressed but releases that override at withdrawal. The mutation floor therefore sets a principled upper bound on what any rejuvenation strategy can achieve and scales with the tissue’s lifetime modification flux. The intropy framework therefore reads reprogramming not as a competing claim about what causes aging but as a likely necessary component of any rejuvenation strategy, complementary to the upstream interventions the framework’s causal ordering predicts.
This architecture yields testable predictions that distinguish the two readings. Combining reprogramming with interventions that reduce modification production, such as targeting mitochondrial ROS leakage, lipid peroxidation, or aldehyde detoxification capacity, and that enhance repair clearance, such as restoring repair gene expression where it has been epigenetically silenced, should yield more durable effects than reprogramming alone. Tissues with lower standing modification burden should respond more durably than tissues with higher burden. Tissues and individuals with higher accumulated mutation burden should show lower ceilings on rejuvenation regardless of how aggressively prevention is layered on top, with the mutation-driven floor distinguishable from the modification-driven steady state by its persistence under combined intervention. The kinetics of rebound after factor withdrawal should track repair-protein turnover and metabolic recovery rates rather than any independent timescale of autonomous epigenetic drift. These predictions distinguish the modification-primacy reading from the autonomous-drift reading without requiring either reading to be wrong about the phenomena both seek to explain.
EN74. Exposure, ssDNA lability, and transcriptional asymmetries.
Replication and transcription transiently expose single-stranded DNA and can promote R-loop formation under permissive conditions, increasing chemical vulnerability and altering repair access.[99,546,547] Transcription-coupled repair (TCR) selectively accelerates removal of bulky transcription-blocking lesions from the template strand of active genes, creating mutational asymmetries, while transcription-associated mutagenesis can elevate local mutation rates through transcription-replication conflicts, topological stress, R-loop persistence, recruitment of error-prone repair, and context-dependent bypass pathways.[99,546,547,548,549] That said, non-replicating DNA undergoes base modification at constitutive rates from endogenous chemistry; while these modifications cannot be fixed as mutations without replication, they can impair transcriptional readout directly (see EN62).
Genome-scale analyses show that many mutational signatures vary with replication timing and display strand biases linked to leading/lagging synthesis, indicating that replication context shapes both lesion fixation and repair.[207,490] Late-replicating regions accumulate more mutations, and several signatures, including oxidative- and APOBEC-associated patterns, display strand or timing biases tied to replication architecture.[207,489] These features are consistent with elevated vulnerability when DNA is opened and processed during replication.[99]
Eukaryotic replicases achieve high fidelity via stringent nucleotide selectivity and 3’->5’ exonuclease proofreading.[550] When adducts or non-canonical templates impede synthesis, cells invoke TLS polymerases (Y-family inserters and Pol ζ extender), which bypass lesions at the cost of accuracy and generate characteristic mutation types.[324,526,551] This architecture mechanistically links base modification to error-prone copying.[324,526]
Biochemical studies show that DNA adducts can cause polymerase pausing, stalling, reduced processivity, and misincorporation.[324,526] Single-molecule real-time sequencing further demonstrates that base modifications alter polymerase kinetics, allowing modified templates to be detected at single-molecule resolution.[552] Recent strand-resolved maps in mammalian cells demonstrate asymmetric mutagenicity tied to lesion tolerance and repair, clarifying how adduct chemistry and replication direction influence mutation fixation.[553] Single-molecule real-time sequencing now resolves how base modifications alter polymerase kinetics during DNA synthesis, providing direct readouts of how lesion chemistry shapes incorporation behavior.[552,553] Not only is there ample evidence that modified DNA drives the formation of new mutations, the type of mutations that results is dependent on the chemistry and structure of the original corruptor.[207,526,553]
EN75. Pol II Stalling and its consequences for aging.
The functional consequences of nucleobase modifications depend critically on how the transcription machinery responds when it encounters them. RNA polymerase II (Pol II), the enzyme responsible for transcribing all protein-coding genes in eukaryotes, elongates along the template strand at roughly 1-4 kb per minute. When it encounters a bulky or helix-distorting modification (cyclopurine adducts, aldehyde-derived crosslinks, certain oxidative lesions, or interstrand crosslinks), elongation is strongly arrested. The Cockayne syndrome B protein (CSB) binds the stalled polymerase and engages its ATPase/translocase activity to remodel the elongation complex; this can promote bypass of minor lesions such as 8-oxoguanine, but bulky modifications typically require repair or polymerase removal.[554]
When the push fails, two fates are possible. For brief stalls, Pol II may backtrack along the template and, after repair of the lesion, resume elongation. For prolonged stalls, the polymerase is ubiquitinated at RPB1 lysine 1268 and destroyed by the proteasome.[473] Mice engineered with a K1268R substitution that prevents this ubiquitination develop shortened lifespan, premature aging, and neurodegeneration, demonstrating that the ability to clear stalled Pol II is itself essential for longevity. After degradation, transcription does not resume from the stall site. Andrade-Lima et al. showed genome-wide that recovery of RNA synthesis after damage shows faster recovery at 5’ than 3’ end of large genes, meaning transcription must re-initiate from the promoter and traverse the entire gene to produce a complete transcript.[555] For a short gene of a few kilobases, this delay is modest. For very long genes (such as SLIT2 at approximately 365 kb), full recovery at the 3’ end required more than 24 hours even in wild-type fibroblasts, and in TC-NER-deficient cells recovery was severely impaired throughout.
This restart-from-promoter requirement produces a systematic bias: longer genes, presenting larger targets for stochastic damage, are more likely to harbor a modification at any given moment and therefore more likely to have their transcription interrupted. Stoeger et al. demonstrated that aging across mouse tissues is accompanied by a shift in the transcriptome toward shorter transcripts, with significant length-associated imbalance present in 10 of 17 tissues by 9 months and rising to all 17 by 24 months (14 with significant anticorrelation between transcript length and abundance, 3 with significant positive correlation).[144] Eight anti-aging interventions tested partially countered this shift. Soheili-Nezhad et al. proposed the term gene-length-dependent transcription decline (GLTD) for this phenomenon and argued it represents a fundamental bottleneck in aging transcriptomes, noting that most transcriptomic studies include implicit normalization that can mask the overall reduction in transcriptional output.[149] This is consistent with what the intropy framework predicts: long genes present larger statistical targets for stochastic transcription-blocking lesions and require longer uninterrupted elongation, making them disproportionately vulnerable to readout interruption as modification burden rises.
The most direct measurement of age-related Pol II stalling comes from Gyenis et al., who combined nascent RNA sequencing with Pol II ChIP-seq in mouse liver.[143] In aged tissue, approximately 40% of elongating Pol II molecules were stalled, and productive transcription was roughly 1.5-fold lower than in young tissue. The authors examined whether the decline could be explained by sequence, chromatin, methylation, splicing, or transcriptional-error features and did not find such explanations sufficient. The gene-length dependence pointed instead to stochastic DNA damage as the most likely driver, the conclusion the authors themselves reached. A 2025 follow-up from the same group integrated EU-incorporation transcriptional measurements with a TBL accumulation model and estimated that wild-type mice accumulate roughly 62 transcription-blocking lesions per day, while DNA repair-deficient progeroid mice accumulate 1,600 to 5,000 daily.[516] The authors concluded that endogenous DNA damage is sufficient to drive the observed transcriptional stress and explicitly framed it as a unifying driver of the aging process, the same interpretation this framework reaches independently.
Debes et al. added a complementary finding: average Pol II elongation speed increases with age across five metazoan species (C. elegans, Drosophila, mice, rats, and humans), and dietary restriction reverses this acceleration.[148] Slow Pol II mutants exhibited fewer transcription errors. One possible interpretation is that increased speed represents a compensatory response to reduced output, with the cost of reduced fidelity further degrading information readout.
Why stalling worsens with age: the repair-readout feedback loop. If Pol II stalling is driven by the density of transcription-blocking modifications in active genes, the rate of stalling should increase as the modification burden rises. Two independent lines of evidence confirm that it does, and point to the mechanism.
First, multiple studies report age-associated declines in BER and NER capacity in specific tissues. BER activity falls in aging rat neurons, with reduced expression and activity of OGG1, APE1, and Pol β.[155,556] In human foreskin fibroblasts across donors aged 20-64, BER efficiency showed a significant age-dependent decline linked to falling SIRT6 levels.[557] NER capacity similarly declines: in human peripheral blood lymphocytes from 135 donors aged 20-60, NER efficiency dropped at a rate of approximately 1% per year, amounting to roughly 30-40% over a 40-year span, and cells from older donors also introduced more mutations during repair.[156,558] ERCC1 and XPF, rate-limiting components of the NER incision machinery, show age-dependent mRNA decline in human blood cells.[559] In the rat lens, OGG1 protein levels fell to approximately 63% of 2-month-old values by 26 months.[560]
Second, the steady-state modification burden rises correspondingly. Helbock et al. measured approximately 24,000 oxidative DNA adducts per cell in young rats and approximately 66,000 in old rats, a roughly 2.75-fold increase.[159] The Guilbaud et al. adductomics study found 36 distinct DNA adducts with significant age-dependent accumulation in rat tissues, including etheno-deoxyadenosine (a lipid peroxidation product) in human hearts.[286]
A critical question is why repair gene expression itself declines. The most straightforward explanation within our framework is that repair genes are subject to the same readout corruption as every other gene. Epigenetic reprogramming of repair gene promoters accounts for only a fraction of the decline: in mouse brain, OGG1 promoter methylation explained approximately 32% of the variance in OGG1 expression loss (R² = 0.320), with the methylation itself correlated with MeCP2 expression, suggesting an epigenetic-regulatory axis rather than simple silencing.[157] But in Alzheimer’s patient lymphocytes, promoter methylation showed no association with the significant downregulation of APE1, OGG1, MUTYH, PARP1, and NEIL1, indicating that in that cohort, measured epigenetic changes explained essentially none of the BER transcriptional decline.[158] Lautrup et al. confirmed broad downregulation of core BER genes across aging brain regions but attributed the regulatory mechanism to BDNF signaling rather than a neat promoter-methylation fraction.[561] These results are consistent with Gyenis and colleagues’ finding that transcriptional decline across the aging liver transcriptome correlates with stochastic DNA damage rather than with epigenetic features.
The unexplained majority of repair gene decline is consistent with a specific prediction of the intropy framework: OGG1, ERCC1, CSB, XPC, and every other repair gene must be transcribed through a template that is itself accumulating modifications. Pol II stalls in the repair gene body. Permissive modifications generate miscoded repair protein. Regulatory modifications at the repair gene promoter alter expression timing. The protectosphere erodes because the instructions for building it are written in the same medium that is being corrupted. This creates a positive feedback loop: more modifications accumulate, repair output falls, modifications accumulate faster, repair falls further. The loop is consistent with the exponential acceleration of mortality described by Gompertz kinetics, and it generates a testable prediction: Pol II stalling density within repair gene loci specifically should increase with age in proportion to gene length and modification burden, independent of promoter methylation status.
Together, these findings establish that nucleobase modifications do not merely change the sequence information in DNA; they physically impede the machinery that reads it, a loss of readout as argued throughout, producing a progressive, gene-length-biased decline in transcriptional output that is measurable in aged tissues, self-reinforcing through repair gene corruption, and reversible by interventions that reduce the rate of modification accumulation.
EN76. Free radical theory: historical context and modern integration.
Early proposals connected oxygen radicals to toxicity and aging, later integrated with mitochondrial bioenergetics.[122,123,124,212,213] Contemporary work reframes radicals as context-dependent. They are essential for signaling yet damaging when their accumulation outpaces detoxification and repair.[122,212,213] Within the intropy model, radical-induced base modifications function as NICs that can immediately impair transcriptional readout and, if copied or misrepaired, seed permanent sequence change.[63,491] The free radical theory was therefore not wrong so much as incomplete: its most aging-relevant target is the information system that radicals modify, rather than oxidative damage considered as a generic bulk property.[122] Free radical disruption of life’s information content is where we maintain attention should be focused.[491]
Macromolecule turnover vs corruption permanence: Proteins and lipids undergo rapid turnover, chaperone-mediated repair, and autophagic clearance, limiting long-term propagation of their damage.[19,398] In contrast, once a DNA lesion is copied through replication or incorporated by repair, the resulting sequence change persists through all descendant cell divisions, amplifying its effects across lineages.[63,491] DNA sequence is therefore the unique substrate of templated corruption: damage to most other macromolecules can be cleared by turnover, repair, or replacement, while damage that becomes encoded in sequence is propagated by the replication apparatus itself. Long-lived proteins (crystallins, collagen, elastin, nuclear pore proteins; see EN34) are a partial exception, but lack the templated propagation mechanism that distinguishes nucleic-acid damage.[63] An important nuance: standing DNA modifications are themselves individually reversible, placing them in the same “transient” category as protein damage. However, their functional impact, particularly transcription blocking in expressed genes, is immediate and ongoing even before conversion to permanent mutations. Mice carrying certain alleles of transcription-coupled repair factors (Csbm/m, XpdTTD) exhibit features of premature aging without elevated mutation frequencies,[223] a pattern long interpreted as evidence that the standing modification burden drives functional decline independently of the mutagenic pathway. The interpretation is partially confounded by the fact that these lines carry hypomorphic or truncating alleles rather than clean nulls, and CS-associated alleles can generate toxic protein effects beyond simple repair loss (see EN51 and EN63). What can be claimed with confidence is that these mice age without accumulating excess mutations, which is at minimum inconsistent with mutation accumulation as the sole driver and consistent with a role for both the standing modification burden and the non-repair consequences of dysfunctional repair factors.[223] The critical distinction is therefore not simply permanent versus transient, but whether the damage impairs the readout of information, which standing modifications at transcribed loci do. Standing modifications matter while present, not only if their replacement fails. Even when transiently repaired, they can stall transcription, trigger DDR, induce inflammation, alter chromatin, or destroy critical transcripts during their occupancy of the template. The progressive character of aging emerges when modification production chronically exceeds repair capacity, when modifications recur at vulnerable loci, or when the corrupted template encodes the very machinery responsible for replacement and repair (see EN75 for the repair-readout feedback loop).[398,491] If the underlying template, repair machinery, and replacement environment all remain intact, turnover restores function; progressive decline follows when persistent or recurrent damage corrupts the systems that would otherwise replace it.[398]
EN77. Mitochondria and the routes to information corruption in aging.
Mitochondria contribute to aging through at least three mechanistically related routes, all of which converge on corruption of nuclear information. What have been described as distinct mitochondrial aging mechanisms in the literature are, under the intropy framework, different upstream drivers feeding into the same downstream process.
Route one: lipid peroxidation and reactive aldehyde generation. Reactive oxygen species generated at the electron transport chain are mostly short-lived. Hydroxyl radicals last nanoseconds and superoxide lasts microseconds to milliseconds, making direct diffusion of these highly reactive species from mitochondria to nuclear DNA at scale unlikely. Hydrogen peroxide, while longer-lived and capable of diffusing between compartments via aquaporin-8, is also the least reactive of the three and produces limited direct DNA damage at physiological concentrations. Instead, the principal route for oxidative nuclear damage from mitochondrial metabolism runs through lipid peroxidation cascades that generate reactive aldehydes, including malondialdehyde, 4-hydroxynonenal, acrolein, and crotonaldehyde.[212,213,562] These aldehydes, with half-lives of minutes to hours, are sufficiently long-lived to diffuse from mitochondria to the nucleus, where they react with DNA bases to form exocyclic adducts.[563] Two classes result: small etheno-type adducts repaired by BER (mutagenic), and bulky propano-type adducts repaired by NER (transcription-blocking and replication-blocking).[563,564] The bulky propano adducts are precisely the class of modification that drives Pol II stalling and transcriptional decline in the framework we develop for chromatin damage (see EN75 and Supposition 12). This chain, from ROS to membrane lipid peroxidation to reactive aldehydes to transcription-blocking nuclear DNA adducts, is one major candidate mechanism by which mitochondrial metabolism corrupts nuclear information in normally aging tissue.
This route explains several otherwise puzzling observations. First, it explains why dietary antioxidant supplementation consistently fails to extend lifespan in mammals [424,425]: cytoplasmic antioxidants cannot intercept ROS at the mitochondrial inner membrane where it is being generated, and by the time ROS has attacked membrane lipids, the aldehydes are already being produced. Second, it explains why mitochondria-targeted catalase overexpression (mCAT) is one of the only antioxidant interventions that does extend mammalian lifespan [138,565]: catalase in the mitochondrial matrix converts hydrogen peroxide to water before it can generate hydroxyl radicals that attack the membrane, preventing the entire aldehyde production cascade at its source. Cardiac pathology is particularly attenuated by mCAT.[565] a tissue where cell turnover is minimal. Under this framework, one candidate explanation is reduced aldehyde-mediated nuclear DNA adduct burden rather than preserved progenitor reserves; other contributing mechanisms, including improved mitochondrial function and altered redox signaling, may also be operative. Third, it provides a molecular basis for the membrane pacemaker theory of aging, which posits that species with more peroxidation-resistant membrane lipids (higher monounsaturated-to-polyunsaturated fatty acid ratios) tend to live longer.[566] Under our framework, fewer polyunsaturated targets means less lipid peroxidation, fewer reactive aldehydes, and a lower rate of transcription-blocking nuclear DNA adducts. Fourth, a parallel story emerges for formaldehyde, a distinct endogenous aldehyde generated during one-carbon metabolism, methylamine catabolism, and enzymatic histone and DNA demethylation. Loss of formaldehyde detoxification enzymes (ADH5, ALDH2) in combination produces AMeD syndrome, a severe progeroid condition marked by growth failure, bone marrow failure, and reduced lifespan.[567] This reinforces the broader principle: endogenous reactive aldehydes, whether generated by lipid peroxidation or by normal epigenetic turnover, drive aging when detoxification fails. The tight coupling between epigenetic remodeling and aldehyde-mediated DNA damage is particularly striking for an information theory of aging, since the cell’s own demethylation machinery generates the very species that threaten the information it is trying to edit.
Route two: nucleotide pool depletion from mtDNA hyperreplication. When mitochondrial DNA accumulates damage at high rates, the cell compensates by increasing mtDNA replication frequency. Hämäläinen and colleagues demonstrated that this compensatory hyperreplication sequesters nucleotides into mitochondria, depleting the cytoplasmic dNTP pool available for nuclear DNA replication.[167] The result is slow replication fork progression, chronic replication stress, cell cycle stalling, and double-strand breaks in proliferating cells, particularly somatic progenitor cells. This is a form of nuclear information corruption that operates through nucleotide competition rather than direct chemical damage, and it proceeds without elevated ROS. It is the dominant mechanism in the POLG mutator mouse, which accumulates mtDNA point mutations without consistently elevated oxidative stress markers across studies [408] yet exhibits premature aging phenotypes, progenitor cell dysfunction from embryogenesis onward, and widespread DSBs in stem cell compartments.[167,406] Hämäläinen and colleagues describe this mechanism as “a unifying mechanism for mouse progerias,” explicitly framing nuclear genome maintenance failure (especially in stem cell compartments) as the convergence point that unites mtDNA-mutator progeria with classical DNA-repair-defect progeria. This convergence on nuclear information corruption is exactly what the intropy framework predicts: different upstream drivers (mtDNA mutation, NER deficiency, replication stress) feed into the same downstream nuclear information substrate.
Route three: mitochondrial-dysfunction-triggered apoptosis as protectosphere action. Mitochondrial dysfunction, independent of ROS production, can trigger apoptosis through several routes: loss of mitochondrial membrane potential, ATP depletion, BH3-only protein activation (often downstream of nuclear DNA damage), mitochondrial unfolded protein response failure, and Ca2+ dysregulation through the permeability transition pore.[407,568] This explains how POLG mice accumulate apoptotic cells despite normal ROS production: the apoptosis is driven by dysfunctional respiratory chains and by nuclear DNA damage downstream of Route 2, not by oxidative signaling. Under our framework, this is the protectosphere operating as designed. Cells whose mitochondria have failed, or whose nuclear genomes have been compromised by replication stress, are eliminated by apoptosis before they can propagate corrupted output into the tissue. The DSB-repair progerias described elsewhere (see EN57) use the same protectosphere mechanism, triggered by a different upstream signal.
These three routes are not parallel mechanisms producing similar-looking phenotypes by coincidence. They are three points of entry into the same downstream process. Routes 1 and 2 generate nuclear information corruption through different chemistries (aldehyde adducts versus replication-stress-induced DSBs). Route 3 is the quality-control response that eliminates corrupted cells, which at tissue scale manifests as stem cell exhaustion when damage rates exceed replacement capacity. The POLG mutator mouse illustrates why these should not be separated. Its premature aging is driven by Route 2, its apoptotic stem cell loss is Route 3 acting on Route 2 damage, and it shows minimal involvement of Route 1 because its mtDNA damage is polymerase-driven rather than ROS-driven. Crucially, this produces a phenotype dominated by progenitor cell depletion: HSC vulnerability and apoptotic loss drive the multisystem failure documented in Ahlqvist et al. and Hämäläinen et al.[167,406] Normal aging looks empirically different. The hematopoietic stem cell pool expands rather than depletes with age in both mice and humans, with clonal expansion of mutant subsets (clonal hematopoiesis, see EN72) and myeloid skewing rather than numerical exhaustion.[569,570] Postmitotic tissues like heart and brain age progressively without replacement reserves to deplete. This is precisely the pattern the three-route framework predicts: Route 1 (aldehyde adducts) operates predominantly in long-lived postmitotic tissues where it accumulates across decades and where mCAT specifically attenuates cardiac decline,[565] while Routes 2 and 3 operate predominantly in proliferative compartments where replication stress drives stem cell loss but only when the upstream insult (mtDNA replication burden, NER deficit, or other defects) is sufficient to overcome normal aging’s compensatory clonal dynamics. Normal aging stays below this threshold; progeroid models cross it.
What looks like “two mechanisms” of mitochondrial aging (ROS-driven damage versus mtDNA-mutation-driven progeria) is, under this framework, a single process of nuclear information corruption operating through different upstream chemistries and different cellular compartments. We believe that recognizing this unity resolves much of the confusion surrounding mitochondrial theories of aging, where experimental results supporting one route have been used to argue against another. A testable prediction follows directly: interventions targeting Route 1 (mCAT, peroxidation-resistant membrane lipids) should rescue normal aging but not POLG-driven progeria, while interventions targeting Route 2 (nucleoside supplementation to restore dNTP pools) should preferentially rescue POLG-driven progeria over normal aging, where Route 1 dominates. The existing literature is partially consistent with this prediction, and the experiments needed to test it directly are straightforward.
EN78. Fe-S clusters in replicative polymerases and their functional role.
While mitochondrial ROS may often reach nuclear DNA indirectly through lipid peroxidation products (see EN77), this explanatory note considers a more speculative possibility: whether the iron-sulfur clusters within the replication machinery itself could represent a redox-sensitive vulnerability that contributes to local information corruption. We flag this as a hypothesis rather than an established mechanism. Eukaryotic B-family polymerases (α, δ, ε, ζ) contain conserved [4Fe-4S] clusters essential for structural integrity, DNA binding, and coordination within the replisome.[128,129,571,572] Mutational disruption of Fe-S ligands impairs polymerase activity and fidelity, underscoring a redox-sensitive interface between replication and metal cofactors.[129,572] These clusters are coordinated structural and redox cofactors essential for DNA binding, polymerase hand-off, and replisome assembly.[571,572] Importantly, properly ligated [4Fe-4S] clusters should not be assumed to catalyze Fenton chemistry: such reactions require accessible Fe(II) and are inhibited by intact cluster coordination. The hypothesis worth considering is whether damaged, misassembled, oxidized, or destabilized clusters could release labile iron near DNA-processing complexes, creating localized vulnerability to radical chemistry. This is one biological example of a recurring trade-off: iron provides useful structural and redox chemistry, but mismanaged iron can catalyze damaging reactions.[128,573] If radical generation occurs at the replication complex, this might lead to information modification, and eventually permanent corruption.
Fenton chemistry is a radical-generating mechanism. The Fenton reaction involves Fe(II)-mediated conversion of hydrogen peroxide into hydroxyl radicals (·OH), among the most reactive oxidants in biology.[132] Hydroxyl radicals attack DNA bases and sugars within nanometers of their generation, producing lesions such as 8-oxoG and strand breaks.[232,491,574,575] While demonstrated in vitro and in some cellular contexts, whether this occurs at replication forks in vivo depends on local Fe(II) availability and redox cycling.[132,133] Base modifications provide a graded route to mutation and altered readout, while strand breaks more often trigger acute repair and checkpoint responses or structural genome instability. Both routes can produce permanent information loss, but they operate on different timescales and through different mechanisms (EN57).[63,491] Our framework emphasizes radical-induced base modifications rather than direct strand breakage as the dominant contributor to gradual aging, for reasons developed in EN57.[78]
Some molecules may be not reactive but become activated in this iron environment. For example, hydrogen peroxide crosses membranes via aquaporin/peroxiporin channels and can access nuclear compartments under some oxidative or signaling conditions.[576,577] Its relative stability compared to radicals allows it to diffuse to DNA-proximal sites, where catalytic metals could convert it into highly reactive species.[132] This makes H₂O₂ a plausible diffusible precursor for site-localized radical generation wherever catalytic metals are available.[132,576,577]
EN79. Endogenous ROS/RNS and canonical oxidative DNA lesions.
Aerobic metabolism generates superoxide, hydrogen peroxide, and hydroxyl radicals, while inflammatory and signal pathways add RNS such as nitric oxide and peroxynitrite.[491,492] These species oxidize bases and sugars, producing lesions like 8-oxoG, FapyG, and abasic sites that are predominantly repaired by BER.[232,491,492] Steady-state lesion formation is continuous; mutagenicity emerges when repair is delayed or bypassed.[491,492] Because of their universal presence, abundance, and reactivity, these oxidative lesions are among the most studied NICs, though their direct contribution to aging may be less than their abundance suggests: 8-oxoG, the most prevalent oxidative lesion, is largely transcription-permissive and does not cause premature aging when its repair is abolished (see EN62). The aging-relevant damage from ROS may operate more through secondary products, particularly the reactive aldehydes generated by lipid peroxidation (see EN77), than through direct base oxidation.[232,491,492] However, it is likely there are other corruptors either unidentified or known but not suspected; we therefore advocate for studies to examine how reactive small molecules generated as a result of metabolism and cell activity impact the informational content of life.[491]
EN80. Mitochondrial ROS production and diffusion.
Electron leakage at complexes I and III generates superoxide. Complex I releases superoxide into the matrix where SOD2 (MnSOD) converts it to H₂O₂; complex III releases superoxide to both the matrix and the intermembrane space, where SOD2 and SOD1 (CuZnSOD) handle it respectively.[134,135,212,213] H₂O₂ is more diffusible than superoxide and can cross membranes through aquaporin/peroxiporin pathways. Its effective range is constrained by peroxiredoxins, glutathione peroxidases, and catalase. At regulated concentrations it serves as a redox signaling molecule; in excess, particularly in the presence of accessible reduced iron, it can drive Fenton chemistry that generates highly reactive hydroxyl radicals.[132,213] Mitochondrial ROS output varies with membrane potential, substrate, and respiratory state, providing a continuous endogenous source of redox stress and secondary NICs.[212,213,491,578] Mitochondrial function occupies a central position in this framework as a major endogenous source of redox-derived NICs, particularly through downstream lipid-peroxidation and aldehyde pathways (see EN77). Endosymbiosis supplied eukaryotes with a high-capacity energy-conversion organelle, enabling greater cellular complexity while introducing persistent redox-management costs.[134,137]
Aerobic respiration, chemiosmosis, and evolutionary trade-off: the electron transport chain couples redox reactions to proton pumping and ATP synthesis (chemiosmosis).[137,578,579] O2 as terminal acceptor permits high ATP yield but introduces partial reduction pathways producing ROS.[134,212,213] This energetic advantage likely supported larger, more complex organisms, at the cost of persistent oxidant production that threatens information fidelity.[137,213,491]
For the downstream pathways by which mitochondrial ROS production leads to nuclear information corruption and tissue-level aging, see EN77.
EN81. The seven channels of information readout corruption
(i) Transcriptional silencing by polymerase stalling. When RNA polymerase II (Pol II) encounters a bulky DNA modification during elongation, it stalls. The Cockayne syndrome B protein (CSB) attempts to push the polymerase past the obstacle using its translocase activity, but cannot advance Pol II over helix-distorting lesions such as cyclopurine adducts, aldehyde-derived crosslinks, or UV photoproducts. When the push fails, Pol II is ubiquitinated at RPB1-K1268 and degraded by the proteasome; mice carrying a K1268R substitution that prevents this clearance develop progeria and neurodegeneration, demonstrating that removal of stalled Pol II is essential for longevity.[473] After degradation, transcription must re-initiate from the promoter and traverse the entire gene before producing a complete transcript, a process requiring up to 24 hours for the longest human genes.[555] In aged mouse liver, Gyenis et al. found that approximately 40% of elongating Pol II molecules were stalled, producing a measurable decline in productive transcription that was biased against long genes.[143] Stoeger et al. independently demonstrated that aging is accompanied by a systematic shift toward shorter transcripts across mouse tissues, with the length-associated imbalance becoming significant in 10 of 17 tissues by 9 months and present in essentially all 17 tissues by 24 months, partially reversible by anti-aging interventions including treatments tested in the NIA Interventions Testing Program.[144] Soheili-Nezhad et al. proposed the term gene-length-dependent transcription decline (GLTD) for this phenomenon and argued it represents a major bottleneck in aging transcriptomes.[149]
The most informative natural experiment distinguishing stall duration from repair deficiency is the comparison between Cockayne syndrome (CS) and UV-sensitive syndrome (UVSS). Both conditions impair transcription-coupled nucleotide excision repair (TC-NER), yet CS produces severe progeria with neurodegeneration while UVSS produces only mild photosensitivity. Steurer et al. showed that in CS cells (lacking CSA or CSB), lesion-stalled Pol II remains chromatin-bound for extended periods, with 10-20% of all Pol II molecules trapped at damage sites two hours after UV exposure. In UVSS cells (lacking UVSSA), Pol II is instead cleared from the lesion by VCP-mediated proteasomal degradation even without repair occurring.[226] The severity of the aging phenotype therefore is not explained by lesion repair deficiency alone; it correlates strongly with how long stalled Pol II persists on chromatin. This distinction is critical to our framework: inefficient readout of genomic information, independent of whether repair eventually occurs, is the proximate driver of functional decline.
(ii) Transcriptional mutagenesis. Not all modifications block Pol II. Smaller, non-distorting lesions such as 8-oxoguanine (8-oxoG), O6-methylguanine, and uracil (from cytosine deamination) are read through by the polymerase, but the altered base-pairing properties cause misincorporation of the wrong ribonucleotide into the nascent mRNA. Saxowsky et al. demonstrated that 8-oxoG in the template strand directs Pol II to insert adenine instead of cytosine in mammalian cells, and that this transcriptional mutagenesis is sufficient to activate the Ras oncogene from a single genomic lesion.[145] Brégeon et al. confirmed that Pol II bypass of 8-oxoG is efficient and error-prone in human cells and that the magnitude of miscoding depends on the sequence context surrounding the lesion.[580]
Critically, a single unrepaired permissive modification generates corrupted transcripts continuously: every Pol II molecule that traverses the lesion can produce a miscoded mRNA, and each miscoded mRNA may be translated multiple times. A gene transcribed hundreds of times before repair could yield a substantial fraction of aberrant protein from one lesion. Vermulst et al. showed that artificially increasing transcription error rates in yeast induces proteotoxic stress and shortens cellular lifespan, establishing a direct link between transcriptional infidelity and functional decline.[147] Fritsch et al. subsequently demonstrated, using circle-sequencing across yeast, C. elegans, Drosophila, and mice, that genotoxic agents elevate transcription error rates genome-wide, that these errors arise in both dividing and non-dividing cells, and that the rate of transcription errors far exceeds the rate of DNA mutations under the same conditions.[146] The same group later measured the baseline transcription error rate in human embryonic stem cells and found that different RNA species display different error rates, suggesting cells may prioritize the fidelity of some transcripts over others.[581] Here again, the loss of faithful readout, this time from what is nominally a transient modification, produces cumulative functional decline.
This channel has particular relevance for post-mitotic cells. Neurons and cardiomyocytes rarely divide, so replicative mutagenesis (channel vi) contributes minimally to their functional decline. But they transcribe constantly, meaning transcriptional mutagenesis operates at every moment of their lifespan. If the standing burden of permissive modifications increases with age (as the modification data suggest), the fraction of corrupted proteins produced per unit time rises correspondingly, potentially contributing to proteostasis stress, aggregation risk, and functional decline in postmitotic tissues without invoking permanent sequence changes.
(iii) Splicing infidelity. Lesions or repair events that alter Pol II elongation kinetics can shift the timing of co-transcriptional splice-site recognition. The same physical sites where lesions can occur (splice sites, branch points, exonic splicing regulatory elements) are also where altered kinetics or local damage could in principle disrupt splice-factor recognition, though direct lesion-specific evidence is more limited than for stalling or transcriptional mutagenesis. A parallel mechanism operates through Pol II elongation rate: when the polymerase is slowed or hurried by factors including encountered modifications, the kinetic window for splice-site recognition shifts, and splicing fidelity falls. Debès et al. demonstrated that aged tissues across five metazoan species show increased Pol II elongation speed and a corresponding rise in splicing errors, and that experimentally slowing Pol II reduced splicing errors and extended lifespan in Drosophila.[148] Splicing infidelity is distinct from the other readout-fidelity channels (i and ii) because it corrupts the interpretation of a correctly transcribed message rather than the transcription itself. It is distinct from the information-content channels (vi and vii) because the underlying DNA sequence remains intact. This channel has received relatively little attention in aging research compared to transcriptional mutagenesis or stalling, but the Debès 2023 evidence suggests it may be a significant and partly independent contributor to age-associated proteome decline.
(iv) Regulatory disruption. Modifications within non-coding regulatory elements can directly alter the control logic governing gene expression without necessarily remodeling chromatin state. Lesions at transcription factor binding sites can alter binding affinity; lesions near regulatory elements may disrupt enhancer-promoter or insulator function. This channel is mechanistically plausible and supported by specific cases (8-oxoG at G-quadruplexes affecting transcription factor binding,[230] DNA methylation effects on CTCF binding [582]), though comprehensive locus-specific lesion-binding maps are still lacking. Unlike channel iii, these effects are not heritably propagated through cell divisions; they operate on the immediate regulatory program of the cell and reset when the modification is repaired. The distinction matters because it separates acute regulatory perturbation (potentially reversible) from established epigenetic drift (self-sustaining). Both can scramble the programs that coordinate gene expression across networks, but they do so on different timescales and with different prospects for repair.
(v) Epigenetic disruption. Modifications that alter chromatin state can initiate changes in DNA methylation, histone modification, or chromatin accessibility that propagate through cell divisions independent of the original lesion. The best-characterized example involves 8-oxoG at CpG islands: the base excision repair (BER) glycosylase OGG1 recognizes and excises the oxidized base, and the repair intermediate recruits chromatin remodelers and, in some contexts, the TET1 demethylase, remodeling the local methylation landscape.[229,231] Because maintenance methyltransferases propagate altered methylation patterns to daughter cells, the epigenetic consequences of an oxidative hit can, in some contexts, become self-sustaining after the initiating modification has been repaired. This channel is the epigenetic counterpart to replicative mutation (channel vi): in both cases, a transient corruption event becomes a heritable state, but the medium of inheritance differs. Channel vi changes the underlying sequence; channel v changes the chromatin context in which that sequence is read. This channel is elaborated with supporting evidence in EN70, EN71, and EN72.
(vi) Replicative mutagenesis. In dividing cells, modifications encountered by the replication fork are either misread by the replicative polymerase or, if blocking, bypassed by specialized translesion synthesis (TLS) polymerases that are intrinsically error-prone. Either mechanism converts the transient modification into a permanent, heritable mutation. A related mechanism, particularly relevant when mtDNA damage elevates the cellular demand for nucleotides, is replication stress from nucleotide pool depletion, which produces slow fork progression, cell cycle stalling, and chronic replication stress in proliferating cells (see EN77 for the POLG case).[151,167] This is the step at which information corruption becomes irreversible: the modification may subsequently be repaired, but the mutation it seeded persists in all descendant cells. This channel is well established and is the primary driver of somatic mutation accumulation and cancer risk, consistent with the observation that proofreading-domain defects in replicative polymerases (POLE, POLD1) and mismatch repair loss produce cancer susceptibility more prominently than systemic progeria. POLD1 polymerase-domain mutations are a notable exception, producing the progeroid MDPL syndrome (mandibular hypoplasia, deafness, progeroid features, lipodystrophy) through replication stress and cellular senescence rather than through proofreading loss.[209] This aspect of modification-to-mutation conversion is treated further in EN58, EN60, and EN62.
(vii) Double-strand break generation. A subset of modifications, particularly those encountered during replication or at sites of transcription-replication conflict, can convert into double-strand breaks when the replisome collapses, when topoisomerase complexes fail to resolve tension, or when clustered oxidative damage produces opposing-strand single-strand breaks in close proximity. DSBs also arise through routes that do not begin with nucleobase modification (R-loop processing, mechanical stress, programmed nuclease activity), but those routes lie outside the modification-driven channel framework developed here. DSBs are the most consequential class of information corruption. A single break can eliminate genes outright, generate translocations that fuse unrelated regulatory contexts, activate ATM/p53 signaling that drives the cell into apoptosis, or trigger senescence and its attendant SASP. Unlike point mutations from channel vi, which affect single codons, DSBs can disrupt entire regulatory domains or eliminate gene function outright. The repair of DSBs via non-homologous end joining is itself error-prone, so even “successful” repair can leave behind small insertions or deletions that propagate as mutations in all descendant cells.[99] DSBs are also the best-established trigger for cellular senescence and for the SASP-mediated tissue-level consequences that propagate aging phenotypes beyond the originally damaged cell.
All seven channels originate from nucleic-acid information stress, including covalent base modification, abasic sites, crosslinks, strand breaks, repair intermediates, or secondary lesions generated during replication or transcription. The downstream consequence of any given modification depends on three variables: (a) its genomic location (coding region, regulatory element, splice site, or structural domain), (b) the cellular process engaging that locus at the time (transcription, replication, or quiescence), and (c) whether repair clears the modification before the next readout event. The boundaries between these channels are permeable. A single modification at a regulatory site within an actively transcribed gene may simultaneously disrupt readout fidelity (channel i or ii), corrupt splicing decisions at a nearby junction (channel iii), alter local chromatin state (channel v), and, if encountered by the replisome before repair, seed a heritable mutation (channel vi) or a double-strand break (channel vii). The channels are analytical conveniences for describing the consequences of modifications, not mutually exclusive processes. The cumulative and simultaneous operation of all seven channels across the genome, scaled by the frequency, persistence, and functional impact of each modification, constitutes the information readout corruption that our framework identifies as the molecular basis of aging.
EN82. Aging in non-dividing cells.
The framework developed here makes a sharp prediction for post-mitotic tissues such as neurons and cardiomyocytes: these cells should age despite being largely spared from the replication-associated mutation ratchet (Supposition 20), which means their aging should be dominated by modification-driven transcription failure rather than by mutation accumulation. The mechanism is covered in detail elsewhere (standing modification burden, Supposition 12; Pol II stalling and gene-length-dependent transcription decline, EN75; transcription-coupled repair requirements in post-mitotic tissue, EN74), and the intropy framework’s position is that these mechanisms are sufficient to drive a substantial component of neuronal and cardiac aging, with mutation accumulation contributing less than in dividing tissues but not absent.
Two classes of evidence support this prediction. First, XRCC1 deficiency produces severe pediatric neurodegeneration via accumulated unrepaired BER intermediates and toxic PARP1 trapping at damage sites; unlike CSA/CSB hypomorphs where the mutant protein itself contributes pathology, XRCC1 loss is closer to a clean repair-failure model in neural tissue,[45,274] and shortened lifespan, directly demonstrating that transcription-blocking modifications in post-mitotic tissue are sufficient to drive aging-like decline (see EN51).[274] Cockayne syndrome fits the same broad pattern, with the qualification (also discussed in EN51) that CS severity reflects a combination of TC-NER failure and toxic effects from dysfunctional CSA/CSB protein rather than repair loss alone; the XPA⁻/⁻ Csbm/m double mutant, in which backup NER pathways are also eliminated, produces catastrophic early-onset neurodegeneration consistent with the vulnerability of post-mitotic neural tissue to unrepaired transcription-impeding lesions.[78,451] Second, somatic mutations do accumulate in post-mitotic neurons with age,[275] but at rates far below those of dividing tissues, and the mutations observed are dominated by SBS5-type signatures, which Spisak et al. (2025) argue accumulate via DNA damage and repair processes rather than replication errors, and which subsequent work has further characterized as a collateral signature funneling multiple damage sources into a common mutational pattern.[583] Supposition 41 argues that this is expected under our framework: SBS5 is a biomarker of modification processing rather than a driver of aging, and its accumulation in post-mitotic cells reflects the same underlying corruption that manifests functionally as transcription blockade.
A distinct vulnerability applies specifically to neurons: activity-dependent gene expression requires topoisomerase-mediated double-strand breaks, creating recurrent opportunities for information loss at actively transcribed loci.[584] Combined with the absence of a meaningful stem-cell reserve for most neurons and cardiomyocytes, this produces a class of tissue where modification-driven corruption is both cumulative and essentially irreversible at the cellular level, explaining why neurodegenerative disease and heart failure sit among the most devastating age-related pathologies.
The broader implication is that interventions that extend dividing-cell lifespan primarily by addressing mutation accumulation (improved replication fidelity, enhanced stem-cell maintenance) would be expected to have limited impact on post-mitotic tissue aging, whereas interventions that reduce the standing modification burden (reducing corruptor production, enhancing transcription-coupled repair, clearing stalled Pol II) should benefit both tissue classes. This asymmetry is testable: the framework predicts that broadly effective longevity interventions should preferentially act on modification production or repair capacity rather than on mutation rate. Caloric restriction, rapamycin, and growth-hormone-axis attenuation all reduce metabolic stress and/or upregulate maintenance pathways,[49,585,586,587] consistent with this prediction, though these interventions also affect proteostasis, autophagy, and inflammation through routes that are not specific to nucleic acid information corruption. Direct measurements of standing modification burden, Pol II stalling, and repair recovery before and after intervention would provide more specific evidence.[49,322,585]
EN83. Why modifications drive aging while mutations drive cancer.
A natural question arising from this framework is why nucleobase modifications, if they are so consequential for cellular function, do not appear as the heritable driver of cancer. The answer is that modifications can initiate cancer through downstream conversion to mutation or stable epigenetic change, but they do not themselves serve as the heritable clonal unit that sustains tumor growth across many cell divisions. Cancer requires a heritable, clonally expandable state, and modifications, being individually reversible, cannot provide this directly.
The vast majority of modifications are short-lived. Most known classes of base lesion have repair half-lives of minutes to hours,[63,205,220] often clearing the lesion before the next replication fork arrives, especially in slowly dividing cells. Some lesions persist substantially longer, particularly in rapidly dividing cells where replication can outpace repair, in inaccessible chromatin, or for lesion classes (bulky adducts, crosslinks, cyclopurines, DNA-protein crosslinks) that engage slower repair pathways.[588] While present, these modifications impair transcription (channels i and ii), contributing to aging. But they are repaired before replication could encounter them, so they leave no heritable trace in daughter cells.
The minority of modifications that persist long enough to encounter a replication fork do not passively dilute. Because they distort or block the template strand, they stall the DNA polymerase.[589] There are a narrow range of consequences: either the cell suffers fork collapse, DSBs, and checkpoint activation (leading to death or senescence, which removes the cell from the proliferative pool),[262] or specialized TLS polymerases bypass the lesion, converting the modification into a permanent mutation.[227,228,583] In either outcome, the modification’s downstream legacy is no longer the modification itself but a fixed mutation, a chromosomal rearrangement, a heritable epigenetic state, or the elimination of the affected cell. The clonally heritable unit that cancer can exploit is therefore one of these downstream products, not the original modification. The mutation can then drive clonal expansion and cancer, but at that point the heritable driver is the mutation, not the initiating modification.
A modification therefore cannot serve as the heritable unit sustaining clonal expansion. It either disappears before replication or triggers a crisis during replication that eliminates either the cell or the modification itself (converting it to a mutation in the latter case). This creates the mechanistic asymmetry that underlies the empirical double dissociation between aging and cancer: defects in modification processing drive aging without proportional cancer risk, and defects in replication fidelity drive cancer without proportional systemic aging (Supposition 35). In other words, modifications drive aging through continuous transcriptional impairment in cells that do not replicate frequently enough to dilute or convert them,[143,223] while mutations drive cancer through heritable gene dysregulation in cells that must replicate to form tumors.[267,268]
Modifications can contribute indirectly to cancer risk through two routes. First, a long-lived modification may transiently silence a critical gatekeeper gene such as a master regulator of repair, creating a brief window of elevated mutation rate. Second, a modification may seed a permanent epigenetic alteration, for instance through BER-mediated demethylation at a CpG (see EN73), which can then be inherited through cell division even after the initiating modification is gone. In both cases the modification enables heritable change but does not itself serve as the heritable unit; the gatekeeper’s inactivation enables mutations, and the altered epigenetic state is what propagates through the daughter lineage. The modification sets these processes in motion; mutations or inherited epigenetic marks drive the cancer that results.
This asymmetry also clarifies why post-mitotic cells vulnerable to aging (e.g., post-mitotic neurons, cardiomyocytes) are among the most resistant to cancer, and why cells vulnerable to cancer (rapidly dividing epithelia, hematopoietic cells) can accumulate substantial mutation loads that more often manifest as clonal expansion, immune dysregulation, or cancer risk than as immediate organism-wide aging.[153] The same property, replication, that converts a fraction of lesions into heritable mutations (enabling cancer evolution) also forces a decision at each fork that prevents indefinite persistence within a given cell lineage: lesions are repaired, bypassed (with potential mutation fixation), or trigger checkpoint activation, senescence, or death. Post-mitotic cells, which never replicate, cannot dilute lesion burden through cell replacement or convert lesions into mutations through replication. They depend on continuous repair to manage the standing modification burden, and when modification production exceeds repair capacity (as it does with age, see EN75), persistent and recurrent transcription-blocking lesions accumulate at steady-state without ever being converted into the heritable mutations that cancer requires. Rapidly dividing cells clear modifications through replication but generate the heritable mutations that cancer exploits. The two diseases occupy opposite ends of the replication spectrum, united by a common root cause (information corruption) but diverging at the replication step, where lesions are either resolved (preserving the cell), converted to mutation (enabling cancer), or destroyed with the cell (preventing both).
EN84. The germline-soma distinction as an information preservation strategy.
The segregation of a protected germline from an expendable soma is one of the most consequential innovations in the history of multicellular life, with antecedents reaching back to the simplest replicators: the asymmetric segregation of damaged components to old-pole E. coli cells while new-pole daughters receive a relatively fresh complement mirrors the same logic at the single-cell level: one daughter retains the accumulated damage while the other receives a relative reset, long before dedicated reproductive tissues evolved (see EN48). In most animals, germline cells are set aside as a distinct lineage early in development, whether through preformation or epigenesis.[590] What matters is not the mechanism of specification but the outcome: a reproductive lineage with dedicated maintenance and surveillance machinery and intergenerational reset, partially insulated from the cumulative burden of somatic damage that builds across decades of division and metabolic exposure. This early sequestration is complemented by lower germline mutation rates [265], stringent meiotic checkpoints that cull defective germ cells [591], epigenetic reprogramming that resets the methylome between generations [592], and transposon silencing by PIWI-interacting RNA pathways [593]. Together, these mechanisms ensure that a relatively uncorrupted copy of the replicator’s information is transmitted to the next generation, even as the somatic copy degrades. This is the molecular implementation of the bargain described in the main text: the soma absorbs the entropic cost so the germline can persist.
The germline-soma distinction is not universal in its implementation, however. Plants do not segregate a dedicated germline early in development. Instead, reproductive cells (in flowers) are specified late from somatic meristematic tissue, meaning that the plant “germline” has accumulated whatever somatic mutations the meristem lineage acquired over the organism’s lifetime. By the logic of strict germline protection, this should be a severe disadvantage, yet plants include some of the longest-lived organisms on Earth (see EN53). The resolution, under our framework, is that plants compensate through a fundamentally different protectosphere architecture: modular growth, replaceable organs, and the absence of irreplaceable post-mitotic tissues prevent somatic corruption from propagating through a single centralized hierarchy the way it does in most animals. Recent measurements complicate the simple picture. Long-lived trees show surprisingly low per-year somatic mutation rates, suggesting that meristem stem cells in the central zone are themselves protected against accumulation, possibly through low division rates and active maintenance in that compartment.[461,594] Plants therefore achieve information preservation through a different combination of low-corruption-rate meristem biology and modular architecture, not through early germline sequestration. This reinforces the broader principle that aging is not caused by any single failure but by the interaction between the rate of information corruption and the architecture through which that corruption propagates.
EN85. Mutational signatures as empirical evidence for lifetime corruption as the scaling variable in aging.
Our framework’s claim that information corruption drives aging makes specific predictions about the mutational record left behind in somatic genomes. If modifications are the upstream initiating events and mutations are their downstream heritable consequence (Supposition 13; see also EN83), then mutational signatures in normal tissues should reflect modification chemistry rather than replication error, should accumulate in proportion to lifetime corruption exposure, and should scale inversely with the species-specific protectosphere investment that determines lifespan. The comparative somatic mutagenesis literature now provides empirical support for all three predictions.
Cagan et al. (2022) extended the broader Martincorena program of somatic mutation analysis in normal tissues to a comparative cross-species design, performing whole-genome sequencing of 208 intestinal crypts from 56 individuals across 16 mammalian species and applying mutational signature decomposition.[210] Their findings, together with mechanistic work on the underlying signatures, establish an empirical chain that the intropy framework is uniquely positioned to explain.
First, somatic mutation accumulation scales inversely with species lifespan across mammals with approximately 30-fold lifespan differences, with no other life-history trait (including body mass and metabolic rate) showing comparable correlation.
Second, the dominant mutational signatures across all 16 species are not replication-error-driven; they arise from methylcytosine deamination (SBS1), oxidative base damage (SBSC, resembling human SBS18 at cosine similarity 0.91), and error-prone processing of other DNA modifications during repair or translesion synthesis (SBSB, resembling the SBS5/SBS40 family of human signatures at cosine similarity up to 0.93, with the closer match shifting between SBS5 and SBS40 across species). None of these is a proofreading- or mismatch-repair-defect signature.
Third, each signature reflects prior modification exposure rather than mutation opportunity per se: SBS18 is attributed to oxidative DNA damage; SBS5/SBSB depends on lesion bypass and repair synthesis, with mechanistic support from Hwang et al. (2025), who showed in human TK6 lymphoblasts that REV7 knockout (eliminating polymerase zeta-mediated translesion synthesis) reduces spontaneous SNVs roughly 2.3-fold and largely eliminates SBS40, the signature most closely related to SBS5;[227] and SBS1, while fixed as a mutation during replication, originates from chemical deamination of a modified base (5-methylcytosine) rather than from copying error.
Fourth, the end-of-life mutation burden converges across species within a relatively narrow range despite the 30-fold lifespan differences. The framework reads this convergence as evidence that the “rate multiplied by time” product is evolutionarily tuned, with each species’ annual rate calibrated against its lifespan to keep cumulative corruption near a tolerable ceiling. The logic extends to post-mitotic cells, where somatic mutations nevertheless accumulate linearly with age despite the absence of replication.[275] In post-mitotic neurons, where replication-dependent fixation is unavailable, SBS5 emerges as the dominant signature.[595] Recent mechanistic work supports the framework’s interpretation: Spisak and colleagues argue that SBS5 represents the footprint of errors during DNA synthesis triggered by multiple classes of damage, with mutations arising from both translesion synthesis and repair-associated polymerase use, effectively a “collateral mutagenesis” signature that funnels heterogeneous corruption sources into a single mutational pattern.[228]
Among proposed accounts, the intropy framework integrates all four observations without invoking new chemistry beyond what is already established for these signatures. The integrated reading is that lifetime effective corruption pressure (formation set against clearance and bypass), rather than replication error, body mass, or metabolic rate alone, is the variable that scales with lifespan, with aging tracking the cumulative integral of that pressure. Cagan and colleagues themselves note that no single biological process appears to control the scaling, consistent with the multi-system calibration the framework predicts. Purely repair-capacity-based explanations fail because signature ratios vary across species in ways not cleanly predicted by repair differences alone. Purely metabolic-rate explanations fail because body mass does not recover the observed correlation. Replication-error explanations fail because the dominant signatures do not bear the proofreading- or mismatch-repair-defect patterns that would be expected if replication fidelity were the scaling variable. The modification-driven corruption logic is what remains when these alternatives are ruled out.
Within the intropy framework, the three dominant signatures function as informative proxies for different axes of corruption history: SBS5/SBSB as a tissue-averaged proxy for general modification processing rate, SBS18/SBSC as a narrower proxy for oxidative corruption, and SBS1 as a marker of lifetime replication history. The relative contributions of each signature across species reflect differences in protectosphere calibration (see EN29b), and the variation is bounded by the requirement that total corruption tolerance be compatible with the species’ reproductive window.
EN86. Stochastrophe: how linear intropy loss produces exponential mortality.
A central challenge for any molecular theory of aging is connecting the gradual, approximately linear accumulation of cellular damage to the exponential increase in mortality observed across nearly all adult animal populations. This exponential increase, first described by Gompertz in 1825, is one of the most robust empirical findings in biology: in modern human populations, adult mortality rises approximately exponentially from roughly age 30 through the mid-80s, with the death rate doubling roughly every seven to nine years across that range.[301,596] Any candidate root cause of aging must account for this pattern, because a process that produces only smooth and non-amplifying functional decline cannot explain why death rates accelerate so dramatically.
The core insight we propose is this: exponential mortality emerges because progressively thinned protective capacity converts stochastic physiological and environmental challenges into threshold failures with increasing probability. We term this coupling stochastrophe (from stochastic + catastrophe). The linear component is the progressive loss of shield capacity, dI/dt, where I(t) is treated as a model variable whose components admit individual measurement as discussed below. Death is not the deterministic crossing of a single threshold but a stochastic event whose probability per unit time rises as I(t) falls and as the overlap between shield reserve and challenge magnitude distribution grows. Stochastrophe names neither the erosion nor any individual lethal event alone, but the coupling between them: the process by which gradual intropy loss converts ordinary environmental and physiological encounters into lethal events at an accelerating rate. Death occurs not when the protectosphere collapses entirely, but when a particular challenge, arriving at a particular moment, finds a protectosphere that has thinned past the threshold required to withstand it.
Schrödinger framed life as a process that imports free energy to sustain ordered configurations against the thermodynamic gradient.[314] Prigogine extended this through dissipative structures.[597] Morowitz grounded it in the thermodynamics of biological organization.[598] More recent nonequilibrium treatments continue the lineage.[9] The view of aging as the progressive loss of a capacity runs through all of them. What earlier thinkers described piecemeal we name intropy, a single primitive for the capacity of life to maintain ordered, low-entropy configurations by continuous work against thermodynamic demand.
The central claim of this thermodynamic tradition maps directly onto the shield capacity-demand picture developed here. Gravity, pressure gradients, osmotic and thermal gradients, oxidative flux, and ongoing biochemical flux toward equilibrium represent a permanent demand landscape that the organism counters through metabolic expenditure. Aging, in this view, is the progressive thinning of intropy, the organism’s capacity to perform that counter-work. Death is the moment when imported order can no longer be maintained against the continuous demand for it. The present framework can be read as a specific, mortality-focused instantiation of this longer tradition, one that commits to a particular molecular identity for the capacity (realized intropy, expressed as the organism-internal shield I(t), ultimately rooted in information readout fidelity but operationally expressed through tissue-level reserve.
In developing the formalism, we worked out the biology and the mathematics together and subsequently recognized that the resulting structure matches a well-established engineering framework: the load-strength interference model in reliability theory.[599,600] In that framework, a component has a time-varying strength distribution and is subjected to loads drawn from an independent distribution. Failure occurs not when the component “fails intrinsically” but when a load sample exceeds a strength sample at a given moment. The instantaneous failure rate depends on the overlap between the two distributions, and as the strength distribution shifts toward the load distribution through wear or damage accumulation, this overlap grows and failure probability rises. For a broad class of reasonable assumptions about distribution shapes, linear strength decline produces exponential failure-rate increase, with the exponential form emerging from the integration of the load distribution against the shifting strength threshold.
Our framework maps onto this structure directly but with biological content that engineering reliability does not and cannot supply. The strength distribution corresponds to the shield I(t), the organism-internal component of the broader protectosphere, understood as physiological reserve composed of component capacities (vascular, immune, skeletal, thermoregulatory, metabolic, neurological). The load distribution corresponds to the challenge process. We use λ for the arrival rate of challenges per unit time, and k for the rate at which penetration probability rises as shield integrity falls; k captures both the steepness of the threshold and the tail behavior of the challenge magnitude distribution. Separating arrival rate from magnitude tail keeps the parameters identifiable when interventions act selectively on one or the other. The exponential form for the per-challenge lethality follows from a specific class of assumptions: that challenge magnitudes have an approximately exponential upper tail, or equivalently that penetration probability rises exponentially as reserve falls. This assumption is biologically defensible in three ways. Empirically, many physiological tolerance distributions (bone density and fracture risk, cardiac reserve and arrhythmia, immune competence and infection lethality, plaque integrity and rupture) are well-approximated as exponential or log-linear at the low-reserve end where most failures occur. Theoretically, the broad empirical convergence of mortality data on the Gompertz form across diverse species suggests the exponential structure is forced not by any single mechanism but by the generic combination of declining reserve and stochastic demand, a result that has been derived from multiple starting points (Strehler-Mildvan vitality decline,[601] Sacher random walks on physiological reserve,[602] Gavrilov-Gavrilova redundancy exhaustion,[198] frailty-selection demography,[603] stochastic-process formulations on physiological state variables using Kolmogorov-Fokker-Planck dynamics [604]). The load-strength interference mapping recovers the same form from a fifth starting point. Structurally, when each challenge is itself the composite of many independent physiological and environmental insults, extreme-value theory places the upper tail of their aggregate magnitude in the Gumbel domain of attraction, whose tail is exponential irrespective of the shapes of the contributing components; on this reading the exponential penetration term is not a stipulation about any single stressor but a generic consequence of challenges arriving as composites.[605,606]
The convergence between the biologically derived formalism and the engineering framework is, in our view, evidence that the load-strength interference structure is well-matched to the underlying problem of time-varying capacity meeting stochastic demand. Other mathematical structures (Weibull-type, log-normal-type) can produce similar empirical patterns under different assumptions; the load-strength formalism happens to be the one whose biological mapping is most direct and whose parameters are most readily identified with measurable physiology. What the load-strength interference framework contributes is mathematical validation and established machinery.
Comparison with Gavrilov-Gavrilova reliability theory. The dominant formal treatment of Gompertz kinetics in the aging literature is the reliability theory of Gavrilov and Gavrilova, who applied engineering reliability mathematics to biological systems.[198,607] Their framework treats the organism as a system of redundant, irreplaceable components that fail stochastically at a constant internal rate. As redundant backups are progressively lost, the probability that the next internal failure eliminates the last remaining copy of some critical function increases exponentially, producing Gompertz kinetics. This is an elegant derivation, and we acknowledge its foundational contribution.
The Gavrilov-Gavrilova framework draws on the redundancy-exhaustion branch of reliability theory, while the formalism developed here draws on the load-strength interference branch. Both branches produce Gompertz-like kinetics through different routes: redundancy exhaustion through progressive loss of internal backups, load-strength interference through progressive overlap between a thinning strength distribution and a stochastic load distribution. The reliability mathematics that Gavrilov and Gavrilova developed remains valid under either reading. What the load-strength interpretation contributes is a route from the abstract reliability parameters to specific biological variables, with the c parameter mapping onto the rate of intropy loss, the k parameter onto threshold sensitivity by challenge type, the alpha decomposition onto challenge arrival rate and baseline penetration probability, and the I0 term onto the standing modification burden present at the start of adult life. This mapping makes each parameter empirically addressable and connects them to distinct intervention classes.
Beyond the formal compatibility, a useful feature of the load-strength interference mapping is that it operates at any scale where finite reserve meets stochastic demand. Carbon-starved E. coli tracked at single-cell resolution show clean Gompertz kinetics (see EN48). Reliability mathematics can accommodate this under multiple framings, and the redundancy-exhaustion logic can be applied to molecular-level redundancy within a single cell. Within this framework we read the bacterial finding as evidence that the capacity-demand dynamic operates regardless of multicellular architecture, which clarifies what architecture is actually contributing. Architecture appears to dictate not whether Gompertz emerges but how local failure propagates: in bacteria the cell is the organism; in plants, modular replacement contains many failures locally (see EN84); in animals with deep hierarchical organization, certain tissue failures cascade through dependent systems. The capacity-demand dynamic sets mortality kinetics, while architecture sets the propagation rules.
We propose that the same empirical phenomenon (Gompertz kinetics) admits a biological interpretation through the load-strength interference formalism that preserves the reliability mathematics while grounding the biological meaning of the parameters in measurable molecular variables. In this reinterpretation, the organism does not fail from within in isolation. Rather, it exists under continuous environmental and physiological challenge, and aging determines whether those challenges can be survived. Pneumonia kills the elderly person whose immune defenses have thinned past the threshold required to clear the pathogen. A hemodynamic stress ruptures the atherosclerotic plaque whose fibrous cap has thinned past the threshold required to contain it. A fall fractures the hip whose bone density has declined past the threshold required to absorb the impact. In each case, the proximate cause of death is an external or quasi-external challenge; the role of aging is to have eroded the protectosphere to the point where the challenge penetrates.
Formal development. We formalize this as follows. Let I(t) represent the organism-internal (somatic) component of realized intropy at time t, hereafter the shield, declining approximately linearly due to metabolically driven damage accumulation and calibrated repair, much of which acts by corrupting information readout:
where I₀ is the initial shield reserve capacity (set during development and early life; see below) and c is the rate of shield thinning, determined by the balance between NIC production and repair capacity. We note that linear decline in I(t) is a first-order approximation; in reality, the feedback loop between damage accumulation and declining repair capacity (see EN12) likely produces a mildly accelerating decline, and behavioral and institutional changes in later life may alter the effective rate. The approximation is adequate to capture the dominant behavior over the age range where Gompertz kinetics hold (roughly 30 to 90 in humans). More generally, the Gompertz form does not depend on strict linearity of I(t). For shield decline functions that are approximately linear over the relevant age range, combined with the exponential-tail challenge assumption, the hazard rises approximately exponentially in t. Substantially nonlinear declines produce departures from clean Gompertz behavior; convex declines (accelerating loss) drive super-Gompertz acceleration, while concave declines flatten the slope. The linear case is chosen here for analytical transparency and because it captures the dominant adult behavior in mammalian datasets where Gompertz fits well.
The organism faces environmental and physiological challenges arriving at an average rate λ. These include infections, hemodynamic stresses, mechanical loads, thermoregulatory demands, and metabolic crises. We treat λ as approximately constant over the adult lifespan, while acknowledging that in reality, the challenge mix shifts with age, behavior, and medical care. Each challenge type has a characteristic threshold sensitivity k, reflecting how steeply the probability of lethal failure increases as shield integrity declines. Cardiovascular events, for example, have steep threshold functions: the plaque either ruptures or it does not. Immune challenges have more graded responses: partial immune competence offers partial protection.
The probability that a challenge of a given type penetrates the shield and proves lethal conditional on its arrival is:
where g is the conditional lethality (the probability that a penetrating challenge strikes a critical system with insufficient remaining redundancy). The overall hazard rate at time t is then the sum of two components: an age-dependent term reflecting shield-mediated failure, and an age-independent term reflecting challenges whose lethality does not depend on shield integrity (accidents, acute trauma, and similar causes):
The constant A is the Makeham term, representing background mortality from causes unrelated to the aging process. Substituting the linear approximation for I(t) into the age-dependent term:
This is the Gompertz-Makeham law, with the parameters identified as:
Makeham term = A (age-independent background mortality)
Gompertz Gompertz Individuals within a population vary in each of these parameters: initial shield integrity (I0) differs due to developmental conditions and genetic endowment, the rate of shield thinning (c) differs due to metabolic rate, repair capacity, and environmental exposure, and both the challenge arrival rate (λ) and the threshold sensitivity (k) differ due to lifestyle, geography, and medical access. This variation produces the population-level heterogeneity in vulnerability that demographic frailty models capture statistically as a latent variable; the present framework offers a mechanistic basis for that heterogeneity, suggesting that conventionally modeled frailty may be understood as the population-level signature of individual-level shield erosion dynamics.
Each Gompertz parameter now carries a specific biological interpretation:
Gompertz α, the scale of the age-dependent mortality hazard evaluated at baseline adult shield integrity, is the product of three terms: the rate at which challenges arrive (λ), the probability that a penetrating challenge is lethal (g), and the baseline penetration probability determined by the initial shield integrity (). Importantly, α depends on both the organism and its environment. Reducing the challenge arrival rate, through sanitation, vaccination, or safer environments, reduces alpha without changing anything about the organism’s internal biology.
This immediately explains the dominant pattern of 20th-century mortality improvement. Historical analyses of the Gompertz-Makeham parameters show that much of the decline in human mortality was driven by reductions in age-independent background risk and via reduced challenge arrival through improvements in sanitation, infectious disease control, and environmental safety, while the age-dependent parameters remained comparatively stable.[608,609] Public health reduced the attack rate without slowing the shield thinning, precisely as this framework predicts. Because the standard reliability framework aggregates environmental and internal stressors into a single effective failure rate, it does not naturally explain why external environmental improvements would selectively alter one Gompertz parameter while leaving the other unchanged.
Gompertz β, the rate of exponential mortality increase, is the product of two terms: the threshold sensitivity of the dominant challenge type (k) and the rate of shield thinning (c). This decomposition yields two predictions that match existing data, and an angle on a third long-standing observation.
First, because k differs by challenge type, populations in which different causes of death dominate should exhibit different Gompertz slopes even if the underlying rate of shield thinning c is identical. Cause-specific mortality analyses are consistent with this prediction. Cardiovascular mortality follows an age-dependent acceleration that peaks in the eighth decade, while total cancer mortality decelerates at the oldest ages, behaviors that would be expected if cardiovascular events have steeper threshold functions (plaque rupture being a relatively binary outcome) and cancer mortality reflects a more graded interaction between accumulating mutations, immune surveillance, and competing risks.[610,611]
Second, as public health eliminates the lower-threshold challenges (infections, nutritional deficiencies), the surviving cause-of-death portfolio becomes enriched for high-threshold events (cardiovascular rupture, osteoporotic fracture), and the observed β, which reflects the weighted average of k across the active challenge portfolio, should increase. This counterintuitive prediction, that the Gompertz slope increases even as life expectancy rises, is confirmed by demographic analysis showing that the Gompertz slope parameter has increased over time in human populations.[609]
The α–β decomposition also offers an angle on a long-standing demographic observation. Strehler and Mildvan (1960)[601] reported that across human populations, Gompertz α and β are negatively correlated: populations with high baseline mortality tend to have shallower age-dependent acceleration, while populations with low baseline mortality show steeper acceleration. Standard reliability theory has no clean mechanistic account of this so-called compensation effect. Within the load-strength interference mapping, and , so a negative correlation can arise when populations with elevated challenge load face their highest-threshold causes of death late, after lower-threshold challenges have already removed the most vulnerable individuals from the cohort, leaving the surviving portfolio enriched for steep-k events. The compensation effect, on this reading, reflects sequential exposure to challenges of differing threshold sensitivity rather than a coordinated tradeoff between baseline and slope. The effect itself remains debated, with some authors arguing it reflects statistical correlation in parameter fitting rather than a biological signal. If the effect is real, the load-strength formalism provides a candidate mechanism that does not require positing a coordinated α–β tradeoff.
The same logic applies, in a different form, to Strehler and Mildvan’s vitality decline framework,[601] which modeled mortality as with V(t) declining toward a deterministic zero-threshold at which death becomes certain. The structural form parallels the load-strength interference framing here, but the frameworks diverge on four points. i) Strehler-Mildvan posited a deterministic kill criterion at each encounter; the framework here makes penetration probabilistic, so an organism with substantial residual I(t) can still die from a sufficiently large challenge (pandemic, accident, severe pathogen) at any age, and the late-life hazard saturates rather than diverging as I(t) approaches zero. ii) Strehler-Mildvan assumed homogeneous populations that subsequent demographic work showed to be untenable; the formulation here accommodates heterogeneity in I0, in the rate of intropy loss, and in shield architecture across individuals without modification. iii) Strehler-Mildvan collapsed all extrinsic factors into a single μ0; the framework here decomposes the extrinsic hazard into challenge arrival rate λ and per-challenge lethality g, predicting that different intervention classes (vaccination, antibiotics, nutrition, geriatric medicine) should move different Gompertz parameters and producing a principled distinction between extending life by removing external causes and slowing the underlying corruption process. iv), the Strehler-Mildvan compensation effect, a contested negative correlation between Gompertz α and ln(R0), has been attributed substantially to joint-estimation artifact;[612] the framework’s α-c relationship is mechanistically specified rather than fit-extracted and is not subject to that critique.
A note on operational status. We do not treat organism-level I(t) as a directly measured scalar; doing so would replicate the Strehler-Mildvan circularity, where a latent variable was defined to fit the mortality curve and then offered to explain it. Organism-level I(t) is instead a model construct whose components admit individual measurement: adductomic standing burden, somatic mutation archives, repair-pathway readouts, and tissue-specific functional reserves such as cardiac output, eGFR, immune competence, bone density, and grip strength. These are candidate observables rather than a commensurable unified scale, and their integration into a single shield index is itself a research program. Threshold sensitivities ki should in principle be estimable from clinical reserve-failure relationships in cardiology, nephrology, hematology, infectious disease, and geriatric medicine rather than from all-cause mortality fits. The framework’s primary tests therefore live at the cause-specific hazard level; all-cause Gompertz parameters and age-shifting cause-of-death portfolios are derived consequences of those predictions rather than independent inputs. Operationalization of the amplification function bridging tissue-level Ii(t) to all-cause hazard is identified conceptually (driver tissues, hierarchical propagation, protectosphere calibration; see Suppositions 22, 28, and EN36) and is developed in future work. The structural distinction from Strehler-Mildvan is preserved because the framework’s parameters are forward-defined from independent biological and clinical inputs rather than fit-extracted from the mortality curve they describe.
The shield model also offers a distinct interpretation of the late-life mortality plateau, the well-documented deceleration of mortality increase at extreme old ages.[198,263] In the Gavrilov framework, the plateau reflects redundancy exhaustion: once all backup elements are gone, the failure rate saturates at the rate of individual element failure, an internally determined ceiling. Another widely cited contributor is frailty heterogeneity (Vaupel and colleagues): high-frailty individuals die earlier, leaving low-frailty survivors at extreme ages, which compresses the apparent mortality acceleration. Both mechanisms can operate, and the shield model adds a third: as I(t) approaches zero, the shield-mediated component of the hazard saturates at λg, the raw arrival rate multiplied by lethality. The three are not mutually exclusive, and distinguishing among them empirically requires manipulations the framework discusses below. This is a ceiling determined by the interaction between the environment and the organism’s remaining critical systems, not by internal architecture alone. The two interpretations make distinguishable predictions, though the divergence is sharpest under specific conditions. In highly protected environments like long-term care settings with infection prevention, fall precautions, and temperature regulation, the environmental ceiling may be pushed high enough that the internal ceiling dominates, making the two models harder to distinguish. The models diverge most clearly when external challenge rates are manipulated late in life. The clinical practice of geriatric medicine implicitly assumes the shield model: fall prevention, pneumococcal vaccination, blood pressure management, and thermoregulation in elderly patients all operate by reducing λ or k for specific challenge types, and these interventions measurably reduce mortality at advanced ages. If the late-life plateau were entirely determined by internal component failure, such environmental interventions should have diminished effect on the oldest-old.
The rate of shield thinning, c, is the parameter that distinguishes a true anti-aging intervention from one that merely reduces environmental risk. This maps onto the distinction between what might be called beta-modifying and alpha-modifying interventions. Caloric restriction (CR), the most robust lifespan-extending intervention in model organisms, reduces c by lowering endogenous NIC production (see EN77, EN80). Meta-analytic data show that dietary restriction reduces the Gompertz slope in rodents, consistent with a slowing of shield thinning rather than a simple downward shift in the mortality curve, although effects vary by restriction level, strain, sex, and husbandry.[613,614] Lower restriction levels may act predominantly through α-type vulnerability reduction with smaller slope effects, suggesting a dose-dependent shift in which mechanism dominates.[615] In contrast, antibiotics, seatbelts, and water treatment change α (and the Makeham constant A) without changing β. The distinction is not merely academic. If the goal is to slow aging itself rather than to protect against its consequences, interventions must target c, the molecular rate at which NICs erode the protectosphere. Two centuries of reducing environmental threats through public health have extended average lifespan dramatically but have not measurably slowed the rate of aging, because they operate on λ and A, not on c.
The initial shield integrity I0 deserves comment. No organism begins life with a perfect protectosphere. From the gamete onward, every cell carries a standing burden of endogenous metabolic damage, including DNA modifications, protein damage, and other persistent molecular alterations, the steady-state consequence of aerobic metabolism producing NICs (Eden’s Apple) continuously while repair clears them at a finite rate. This standing damage burden is not accumulated through aging (though the steady-state does increase); it is the instantaneous, thermodynamically unavoidable cost of metabolic activity. Gavrilov and Gavrilova recognized the mathematical necessity of initial flaws in their model and noted that without them, the redundancy framework produces Weibull rather than Gompertz kinetics.[198,616] However, the molecular nature of these flaws was never established. Under the intropy framework, the initial flaws comprise the standing modification burden together with the cumulative mutational load present from the earliest cell divisions: the endogenous NIC-driven damage that every cell carries from its formation, including persistent DNA lesions, adducts, and modifications that corrupt information readout, along with fixed mutations that alter the underlying sequence. Within this framework, the standing modification burden is proposed as the dominant contributor, accounting for the largest fractional reduction in transcriptomic efficacy, while mutations contribute additively to the overall deficit. Together these set I₀ below the theoretical maximum and ensure that the shield is already partially thinned at the start of adult life.
In summary, the contribution here is not a new derivation of Gompertz kinetics. It is a biological reinterpretation that, in our view, makes the mathematical framework more complete. The same exponential hazard that emerges from reliability theory can be understood as progressive thinning of protective capacity under stochastic external challenge, yielding a mapping between mortality parameters, environmental risk, and intervention classes that the abstract reliability framework does not provide. The rate of shield thinning (c), which is dI/dt under constant metabolic load, should be the target for anti-aging therapy. The challenge arrival rate (λ) is the target for public health. The threshold sensitivity (k) explains why different cause-of-death portfolios produce different Gompertz slopes. And the initial shield capacity (I₀), set by the standing burden of endogenous damage at the start of adult life, provides a candidate molecular basis for the “initial flaws” that reliability theory requires but does not identify.
EN87. Why prevention must precede mutation fixation.
Once a modification is fixed as a mutation through replication or a mistake in the repair process, the resulting sequence change is no longer correctable by endogenous repair: the original template that would specify the ancestral base is no longer available, and the change persists in all descendant cells.[63,324] Single-cell and ultra-deep sequencing show that somatic mutations accumulate with age across human tissues, creating mosaicism that cannot be globally reversed by physiological processes.[210,211,488] Mutations may still be removed by cell death, clonal selection, or future editing technologies, but those routes eliminate or replace the carrier cell rather than restoring the ancestral sequence.
However, mutations represent only the final, irreversible stage of information corruption. The standing modification burden, which impairs transcriptional output continuously while each lesion remains individually repairable (see EN62, Supposition 12), is in principle a more tractable therapeutic target. Strategies that reduce NIC production rates (e.g., mitochondria-targeted antioxidants, aldehyde scavengers) or enhance properly coordinated repair throughput could lower the steady-state modification burden and slow both the direct functional consequences and the rate at which modifications convert to permanent mutations. Coordination matters because unbalanced upregulation of any single repair component can trap intermediates and impair clearance: PARP1 trapping at BER intermediates in XRCC1-deficient cells (see EN82 and the broader pathway discussion in EN51) is the canonical example.
Stem cell transplantation operates through a related but distinct intervention class: not prevention of fixation, but replacement of compartments whose substrate is already heavily corrupted with cells carrying less-corrupted copies: in one mouse study, non-myeloablative transplantation of young bone marrow into aged recipients extended mean residual lifespan by approximately 39% relative to controls,[410] and selecting a younger-profile subset of stem cells within an aged donor (CD150-low HSCs, which retain differentiation capacity) extends median lifespan in old recipients by roughly 10%, which we interpret within this framework as evidence that the less-corrupted information substrate carries through to organismal output.[411] These interventions do not repair corrupted information in situ; they substitute it. Together with prevention strategies, this constitutes the set of interventions that target the underlying rate of shield thinning (the c parameter in EN86) rather than only buffering its consequences.
Prevention strategies therefore act upstream of fixation, on the substrate where lesions remain repairable.[64,67] Replacement strategies act downstream of fixation, on the compartmental level. Reversal at the level of individual fixed mutations remains beyond current capability; future technologies that scan an individual’s genome and restore it to its original sequence could in principle close that gap, drawing on any of three reference strategies: identifying remaining unaltered cells as direct templates, deriving an ancestral consensus bioinformatically from large numbers of cells where stochastic mutations differ across lineages, or referencing germline information from the individual’s gametes or early embryonic cells. No such system currently exists.
EN88. Targeted neutralization vs systemic antioxidants.
Large randomized trials and meta-analyses show no survival benefit and possible harm from routine antioxidant supplementation (β-carotene, vitamin A, vitamin E), indicating that bulk redox buffering is ineffective or counterproductive in humans.[425,617,618] Systemic antioxidants can be counterproductive because ROS serve essential signaling functions in exercise adaptation, immune activation, and mitohormetic stress responses; indiscriminate suppression impairs these processes while doing little to reduce the NIC burden at the subcellular sites where information corruption actually occurs.[619,620] In contrast, localization matters: catalases targeted to mitochondria (MCAT) extend mouse lifespan and reduce indices of mitochondrial oxidative damage, implicating H2O2 near the electron transport chain as actionable.[138] Early human studies with the mitochondria-targeted antioxidant MitoQ improved endothelial function and reduced arterial stiffness in older adults in a small short-term trial, with broader meta-analytic evidence suggesting effects across additional aging-related biomarkers, though effects on lifespan are unknown.[621,622] Metal chemistry is critical. Iron chelators can protect by limiting Fenton-driven hydroxyl radical generation, but the protective effect depends on stoichiometry: at L1:iron ratios below approximately 3:1, partially complexed iron species remain Fenton-active and can paradoxically potentiate oxidative DNA damage, while above that ratio sequestration is sufficient and the agent is protective.[623] The example illustrates a more general principle: redox-control interventions must achieve adequate local stoichiometry against the relevant chemistry, not just systemic exposure. The localization principle parallels the coordination requirement for repair upregulation discussed in EN87: in both cases, blunt amplification of a beneficial process disrupts the signaling and stoichiometric balance that makes it work, and effective intervention requires precision rather than magnitude. The challenge is spatiotemporal precision: an effective intervention must be present at the right subcellular location, in the right stoichiometric relationship with local NICs, and sustained across a lifetime, because the endogenous processes that corrupt information are continuous and unavoidable. Under this framework, aging is driven by cumulative information loss, and interventions that reduce the rate of that loss should extend functional lifespan when information corruption is rate-limiting for the dominant cause of death. The benefit will not generally be linear in the rate reduction: tissue-specific bottlenecks, compensatory feedback, and shifts in the active cause-of-death portfolio (EN86) all break strict proportionality.
EN89. Protection, repair, and the feedback loop that erodes them.
Upregulating base excision repair for 8-oxoG (via OGG1) lowers mutagenesis in model systems,[624,625] but isolated enhancement of a single BER step can be counterproductive: OGG1 excises the damaged base but generates an AP site intermediate that is itself transcription-blocking and toxic if downstream steps (APE1, Pol β, XRCC1/Ligase III) cannot keep pace.[274,626] This same logic applies more broadly: AAG overexpression converts tolerable alkylated bases to toxic AP sites, and XRCC1 deficiency causes PARP1 trapping at BER intermediates that collapses replication forks.[274] Effective pathway enhancement therefore likely requires coordinated upregulation of the entire BER cascade, not isolated overexpression of individual enzymes, though direct demonstrations of the coordinated-upregulation strategy remain limited.[624,625]
Repair capacity is also epigenetically regulated at multiple levels. In the aging brain, HDAC1 activates OGG1-initiated repair; HDAC1 deficiency elevates 8-oxoG and impairs cognition, while pharmacologic activation restores repair capacity.[627] A critical and underappreciated dimension of this regulation is that repair genes are themselves subject to the same information-corruption-driven silencing that operates elsewhere in the genome, producing a self-amplifying feedback loop. In mouse brain, OGG1 promoter methylation increases significantly with age across 27 CpG sites, correlating inversely with OGG1 expression (R² = 0.320) and with reduced BER incision activity and elevated 8-oxodG levels.[157] XRCC1 promoter methylation shows a similar age-dependent increase.[157]
The mechanistic basis for this silencing is now increasingly well-established. Oxidative damage directly recruits the epigenetic machinery that suppresses transcription at damaged sites: hydrogen peroxide exposure causes DNMT1, DNMT3B, SIRT1, and Polycomb complex members (including EZH2) to relocalize to GC-rich promoter CpG islands, where they establish repressive chromatin and, at low-expression genes, deposit DNA methylation.[628] This recruitment is mediated in part by mismatch repair proteins (MSH2-MSH6) that bring DNMT1 to sites of oxidative damage,[629] and 8-oxoG-bound OGG1 itself recruits repressive chromatin writers including CHD4, EZH2, G9a, and DNMTs.[232] The lesion that the repair machinery exists to resolve thus directly triggers the silencing of nearby transcription, including, when damage falls near repair gene promoters, the silencing of repair capacity itself.
This creates the conditions for a positive feedback loop: standing modifications at or near repair gene promoters drive epigenetic silencing of those genes, reducing repair capacity, which allows the modification burden to rise further, which deepens the silencing. The protectosphere is eroded by the very damage it exists to repair. This epigenetic feedback operates alongside a second, likely dominant, transcription-level mechanism described below.
Repair capacity declines with age in part because the transcriptional machinery that produces repair proteins is itself progressively compromised by accumulated transcription-blocking lesions. In post-mitotic tissue this mechanism appears dominant: genome-wide work shows that roughly 40% of elongating RNA polymerases are stalled in aged mouse liver, producing a gene-length-dependent skewing of the transcriptome that accounts for the majority of age-related expression changes in post-mitotic tissues.[143,516] Direct mass-spectrometric profiling of DNA adducts in rat and human post-mitotic and slow-turnover tissues (heart, brain, liver, kidney) shows age-dependent accumulation of multiple modification classes, including 8-oxoG, 1,N6-etheno-dA, glycation-derived adducts, and oxidized cytosines that range in function from transcription-blocking lesions to regulated epigenetic intermediates.[286] Transcriptional mutagenesis compounds this: RNA Pol II bypassing an unrepaired 8-oxoG misincorporates adenine, producing malformed transcripts and malformed proteins without any change in the underlying DNA.[145,228,630] Age-related promoter methylation of repair genes has also been reported [157] but in post-mitotic tissue appears to explain a smaller fraction of expression variance than the transcription-stalling mechanism.
While this mechanism is best established in post-mitotic tissue, converging metabolic and lesion-distribution data suggest a similar constraint may apply in replicative compartments. Proliferating cells run their metabolism substantially harder than quiescent cells of the same lineage: direct comparison of cycling versus contact-inhibited fibroblasts shows roughly 60 to 80 percent higher basal respiration per unit protein and more than doubled ATP output in the proliferating state,[631] and ROS levels rise through the cell cycle to peak in S and G2/M.[632] Consistent with this, global 8-oxoG levels are approximately two-fold higher in proliferating MCF10A cells than in G0-arrested counterparts, with damage concentrating at actively transcribed long genes and at replication origins where transcription-replication conflicts generate oxidation-vulnerable single-stranded DNA.[633,634] Replication does not reset lesion burden cleanly. Lesions can be diluted, bypassed, repaired, or fixed as mutations during replication, but the underlying flow of newly generated damage continues, and both daughter cells inherit the same aged cytoplasmic enzyme pool that determines clearance capacity. Because lesion burden is a flow variable set by the balance between damage generation and repair, and because generation in proliferating cells is elevated while repair enzyme output is constrained by the same stalling feedback that operates in post-mitotic cells, the steady-state lesion burden is unlikely to return to youthful levels unless both damage production and repair throughput are substantially reset. Epigenetic contributions may be more prominent in replicative tissue than in post-mitotic tissue, but appear to operate through specific, coordinated changes rather than through stochastic drift. Genome-wide profiling of young and aged hematopoietic stem cells shows concerted, non-random methylation changes that systematically reinforce self-renewal at the expense of differentiation, consistent with directed rather than drifted epigenetic remodeling.[635] One characterized mechanism involves 8-oxoG forming preferentially at methylated CpGs, where bound OGG1 recruits TET1 and initiates localized demethylation;[229] other lesion-directed mechanisms likely exist.
In our framework, this combined effect is a concrete instantiation of protectosphere decline: the repair arm weakens because the same damage burden it exists to resolve also degrades the machinery that produces new repair proteins. The mechanism operates across tissue types through partially overlapping routes, with transcription-blocking lesions driving Pol II stalling in post-mitotic cells and a similar stalling dynamic operating against a higher metabolic and replication-associated damage background in replicative cells. This self-amplifying dynamic provides a mechanistic basis for the mildly accelerating shield decline acknowledged in EN86.
Beyond repair, engineered detox pathways (e.g., peroxiredoxins) can improve stress resistance and extend lifespan in flies, supporting the concept of encoded shielding.[636] Clinical genome engineering is advancing rapidly: CRISPR base and prime editing can now correct pathogenic point mutations ex vivo and is entering early clinical trials for in vivo applications.[637,638] and lipid nanoparticle mRNA delivery platforms have become a mature route for transient expression of engineered proteins in vivo.[639,640] Such systems, or improved versions of them, are plausible candidates for embedding protective modules at the genomic or transcriptomic level, with delivery, off-target control, durability, immune response, and cancer risk as the principal unresolved constraints.
EN90. Space radiation as an exogenous NIC source and protection by planetary shields.
Long-duration spaceflight produces systemic molecular changes, including shifts in gene regulation, telomere dynamics, and DNA damage response, as documented in the NASA Twins Study.[641] These changes reflect multiple spaceflight stressors including microgravity, fluid shifts, altered circadian rhythms, and confinement, with ionizing radiation as a major and persistent contributor and a primary concern for missions beyond low Earth orbit.[642] Direct ISS experiments have demonstrated that DSB repair assays can be performed in microgravity, establishing the feasibility of measuring repair pathway use under space conditions.[643] Earth’s atmosphere and magnetic field significantly reduce fluxes of high-energy particles.[644,645,646,647] In their absence, ionizing radiation becomes a persistent exogenous source of DNA lesions, analogous to endogenous NICs in their need for repair but differing in origin and in lesion spectrum high-LET particles in particular produce clustered damage that is qualitatively harder to repair than typical endogenous oxidative lesions.[642,647]
Earth’s geophysical environment, its atmosphere and magnetosphere, is not itself a protectosphere. It did not evolve to protect any replicator, and biology has no control over it. But it is the enabling condition under which biological protectospheres could evolve. By attenuating cosmic rays, solar particle events, and most short-wavelength electromagnetic radiation before they reach the surface,[644,645,646,647] the geophysical environment removed one of the most severe corruption sources from the selection problem that early life had to solve. Biology could then allocate selection pressure toward the many other challenges a replicator faces (predation, pathogens, thermal stress, starvation, and endogenous corruption) rather than having to solve planetary-scale radiation first. Natural background radiation averages roughly 2.4 mSv/year globally, with the cosmic component contributing approximately 0.3 mSv/year at sea level,[647] a low enough level that terrestrial protectospheres have not needed to build dedicated defenses against it beyond the repair pathways that already handle endogenous oxidative and base-damaging lesions.
Beyond the magnetosphere and atmosphere, as on the ISS or in deep space, cosmic rays and solar particle events become dominant exogenous sources of DNA damage and represent major health risks.[642] Even so, the preponderance of DNA damage in terrestrial life arises from endogenous processes such as hydrolysis, oxidation from metabolism, and aldehyde adducts.[63] In most internal tissues under ordinary surface conditions, endogenous NICs dominate the baseline corruption burden, though ionizing radiation contributes meaningfully through radon exposure, medical imaging, and natural background; the relative contribution of radiation rises substantially at high altitude, in spaceflight, and in occupational or geological high-exposure settings.[642,647]
The broader implication is that the engineered anti-corruptor mechanisms proposed in EN89 would extend the principle of shielding from the geophysical scale (where it operates by chance, not by design) down to the molecular scale (where it would operate by deliberate engineering). Human space travel inverts the problem: humans carry their biological protectospheres with them, but leave behind the geophysical shielding that made those protectospheres sufficient. Engineering anti-corruptor defenses into the genome itself, as EN89 proposes, is one component of a layered protection strategy for spaceflight that could extend molecular shielding to environments where planetary protection is unavailable, alongside physical shielding, storm shelters, mission design, and pharmacologic radioprotectors.
EN91. Chemical fortification and rewriting of the information substrate.
The main text raises the possibility that information protection could be built into the substrate itself rather than layered around it through repair and clearance. Recent work demonstrates technical groundwork at several scales, though none of it yet addresses the central question of whether a substrate can be made more resistant to endogenous NICs in a multicellular eukaryote. Xeno-nucleic acids (XNAs) with non-natural backbones can store information and undergo heredity and evolution when copied by engineered polymerases, demonstrating that alternative chemistries with enhanced nuclease resistance and altered vulnerability profiles are achievable in principle, although these polymers depend on engineered enzymes and have not replaced DNA as the genome of any living cell.[648,649,650] At organism scale, a 4-Mb synthetic E. coli genome recoded to 61 codons has been constructed and is viable, proving that comprehensive sequence rewriting and codon compression are technically feasible, and the JCVI-syn3.0 project defined a minimal 473-gene bacterial chassis from which further modifications can proceed.[651,652] In eukaryotes, the Sc2.0 project has synthesized yeast chromosomes de novo with two distinct classes of design features: stability-oriented changes including removal of transposons and non-essential introns, telomere standardization, and relocation of tRNA genes to a separate neochromosome; and a built-in inducible diversity system (SCRaMbLE) consisting of loxPsym sites placed downstream of non-essential genes that allow controlled genome rearrangement under Cre recombinase induction.[653] None of these projects have yet produced a substrate measurably more resistant to endogenous NICs than natural nucleic acids in a eukaryotic context, but they establish the technical groundwork for doing so. Whether such rewrites could be introduced into multicellular organisms without triggering immune activation, developmental failure, regulatory disruption, oncogenic transformation, or germline consequences remains untested.
The ethical concerns raised in the main text about using such methods to enhance rather than merely protect information are not reduced by feasibility; they are heightened by it.
EN92. From biological to digital permanence: what is feasible?
The idea that replicator information might transition to a digital substrate is grounded in a set of capabilities that are already within current or near-term technology. DNA-based data storage has demonstrated that large information payloads can be encoded in synthetic DNA and retrieved with high fidelity,[654,655,656] and whole-genome synthesis and assembly allow stored sequences to be reconstructed as functional DNA.[651,652] These capabilities, combined, support digital preservation and transmission of genomic sequence information across arbitrary distances, with biological reconstruction at a destination that has compatible synthesis and cellular infrastructure. For prokaryotes, a synthesized genome introduced into a compatible cytoplasmic chassis can produce a viable cell. For multicellular eukaryotes, the sequence is necessary but far from sufficient: development additionally requires the egg’s cytoplasmic context, maternal factors, mitochondrial DNA, the starting epigenetic state, and the developmental environment, none of which is captured by genomic sequence alone. This opens strategies for sequence preservation and transmission across space that conventional reproduction, bound to physical transport of living cells, cannot achieve.
In intropy framework terms, digital storage preserves potential intropy (the sequence substrate) without preserving realized intropy (the maintained living state). The transition from one to the other still requires a biological platform with sufficient remaining capacity to read and instantiate the stored information.
References
- Mojzsis, S. J.; Arrhenius, G.; McKeegan, K. D.; Harrison, T. M.; Nutman, A. P.; Friend, C. R. L. Evidence for life on Earth before 3,800 million years ago. Nature 1996, 384(6604), 55–59. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: Overview. Annals of the New York Academy of Sciences 2001, 928(1), 1–21. [Google Scholar] [CrossRef] [PubMed]
- Viña, J.; Borrás, C.; Miquel, J. Theories of ageing. IUBMB Life 2007, 59(4–5), 249–254. [Google Scholar] [CrossRef] [PubMed]
- Rattan, S. I. S.; Clark, B. F. C. Ageing: A challenge for biotechnology. Trends in Biotechnology 1988, 6(3), 58–62. [Google Scholar] [CrossRef]
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Current Biology 2012, 22(17), R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L.; Gems, D. Mechanisms of aging: public or private? Nature Reviews Genetics 2002, 3(3), 165–175. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Kruse, K.; Lu, T.; Wang, J. Nonequilibrium physics in biology. Reviews of Modern Physics 2019, 91(4), 045004. [Google Scholar] [CrossRef]
- Landauer, R. Irreversibility and Heat Generation in the Computing Process. IBM Journal of Research and Development 1961, 5(3), 183–191. [Google Scholar] [CrossRef]
- England, J. L. Statistical physics of self-replication. The Journal of Chemical Physics 2013, 139(12). [Google Scholar] [CrossRef] [PubMed]
- Skinner, D. J.; Dunkel, J. Improved bounds on entropy production in living systems. Proceedings of the National Academy of Sciences 2021, 118(18). [Google Scholar] [CrossRef] [PubMed]
- Sartori, P.; Pigolotti, S. Thermodynamics of Error Correction. Physical Review X 2015, 5(4), 041039. [Google Scholar] [CrossRef]
- Shannon, C. E. A Mathematical Theory of Communication. Bell System Technical Journal 1948, 27(3), 379–423. [Google Scholar] [CrossRef]
- Shannon, C. The zero error capacity of a noisy channel. IEEE Transactions on Information Theory 1956, 2(3), 8–19. [Google Scholar] [CrossRef]
- Eigen, M. Selforganization of matter and the evolution of biological macromolecules. Die Naturwissenschaften 1971, 58(10), 465–523. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. Evolution of the mutation rate. Trends in Genetics 2010, 26(8), 345–352. [Google Scholar] [CrossRef] [PubMed]
- Bacon, F.; Devey, J. Novum Organum; P. F. Collier, 1902; Available online: https://books.google.com/books?id=Xc9xDgHgvaYC.
- Weiss, M. C.; Sousa, F. L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W. F. The physiology and habitat of the last universal common ancestor. Nature Microbiology 2016, 1(9), 16116. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M. A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153(6), 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M. A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186(2), 243–278. [Google Scholar] [CrossRef] [PubMed]
- Weismann, A.; Poulton, E. B. Essays upon heredity and kindred biological problems; Clarendon press, 1891; Vol. 1. [Google Scholar]
- Jin, K. Modern Biological Theories of Aging. Aging and Disease 2010, 1(2), 72–74. [Google Scholar] [PubMed]
- Park, D. C.; Yeo, S. G. Aging. Korean Journal of Audiology 2013, 17(2), 39. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The aging process. Proceedings of the National Academy of Sciences of the United States of America 1981, 78(11), 7124–7128. [Google Scholar] [CrossRef] [PubMed]
- Bjorksten, J.; Tenhu, H. The crosslinking theory of aging — Added evidence. Experimental Gerontology 1990, 25(2), 91–95. [Google Scholar] [CrossRef] [PubMed]
- Snedeker, J. G.; Gautieri, A. The role of collagen crosslinks in ageing and diabetes - the good, the bad, and the ugly. Muscles, Ligaments and Tendons Journal 2014, 4(3), 303–308. [Google Scholar] [PubMed]
- Pucci, M.; Aria, F.; Premoli, M.; Maccarinelli, G.; Mastinu, A.; Bonini, S.; Memo, M.; Uberti, D.; Abate, G. Methylglyoxal affects cognitive behaviour and modulates RAGE and Presenilin-1 expression in hippocampus of aged mice. Food and Chemical Toxicology 2021, 158, 112608. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Majdi, A.; McCann, S. K.; Mahmoudi, J.; Vafaee, M. S.; Macleod, M. R. D-galactose-induced brain ageing model: A systematic review and meta-analysis on cognitive outcomes and oxidative stress indices. PLOS ONE 2017, 12(8), e0184122. [Google Scholar] [CrossRef] [PubMed]
- Schnider, S. L.; Kohn, R. R. Effects of age and diabetes mellitus on the solubility of collagen from human skin, tracheal cartilage and dura mater. Experimental Gerontology 1982, 17(3), 185–194. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Woodley, D. T.; Mechanic, G. L. Aging and cross-linking of skin collagen. Biochemical and Biophysical Research Communications 1988, 152(2), 898–903. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Witkowski, J. M.; Pawelec, G.; Alan, C.; Larbi, A. On the Immunological Theory of Aging; 2014; pp. 163–176. [Google Scholar] [CrossRef] [PubMed]
- Bhatia-Dey, N.; Kanherkar, R. R.; Stair, S. E.; Makarev, E. O.; Csoka, A. B. Cellular Senescence as the Causal Nexus of Aging. Frontiers in Genetics 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Libertini, G.; Shubernetskaya, O.; Corbi, G.; Ferrara, N. Is Evidence Supporting the Subtelomere–Telomere Theory of Aging? Biochemistry (Moscow) 2021, 86(12–13), 1526–1539. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, K.; Vera, E.; Martínez-Nevado, E.; Sanpera, C.; Blasco, M. A. Telomere shortening rate predicts species life span. Proceedings of the National Academy of Sciences 2019, 116(30), 15122–15127. [Google Scholar] [CrossRef] [PubMed]
- Yegorov, Y. E. Olovnikov, Telomeres, and Telomerase. Is It Possible to Prolong a Healthy Life? Biochemistry (Moscow) 2023, 88(11), 1704–1718. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E. H. Telomere states and cell fates. Nature 2000, 408(6808), 53–56. [Google Scholar] [CrossRef] [PubMed]
- Mikhelson, V. M. Replicative mosaicism might explain the seeming contradictions in the telomere theory of aging. Mechanisms of Ageing and Development 2001, 122(13), 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Tarangelo, A. Telomeres and Telomerase in Cancer: Overview and Therapeutic Potential. Journal of Student Research 2022, 11(3). [Google Scholar] [CrossRef]
- Armanios, M.; Blackburn, E. H. The telomere syndromes. Nature Reviews Genetics 2012, 13(10), 693–704. [Google Scholar] [CrossRef] [PubMed]
- Allsopp, R. C.; Vaziri, H.; Patterson, C.; Goldstein, S.; Younglai, E. V.; Futcher, A. B.; Greider, C. W.; Harley, C. B. Telomere length predicts replicative capacity of human fibroblasts. Proceedings of the National Academy of Sciences 1992, 89(21), 10114–10118. [Google Scholar] [CrossRef] [PubMed]
- Hemann, M. T. Wild-derived inbred mouse strains have short telomeres. Nucleic Acids Research 2000, 28(22), 4474–4478. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K. L.; Chang, S.; Lee, H.-W.; Blasco, M.; Gottlieb, G. J.; Greider, C.; DePinho, R. A. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell 1999, 96(5), 701–712. [Google Scholar] [CrossRef] [PubMed]
- M., J., M., H., & Capkova, R. (2011). Telomere Maintenance in Organisms without Telomerase. In DNA Replication-Current Advances. InTech. [CrossRef] [PubMed]
- Olovnikov, A. M. A theory of marginotomy. Journal of Theoretical Biology 1973, 41(1), 181–190. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M. S.; Kasturi, P.; Hartl, F. U. The proteostasis network and its decline in ageing. Nature Reviews Molecular Cell Biology 2019, 20(7), 421–435. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; Pothof, J.; Vijg, J.; Hoeijmakers, J. H. J. The central role of DNA damage in the ageing process. Nature 2021, 592(7856), 695–703. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Ito, T.; Petrashen, A. P.; Elias, A. E.; Skvir, N. J.; Criscione, S. W.; Caligiana, A.; Brocculi, G.; Adney, E. M.; Boeke, J. D.; Le, O.; Beauséjour, C.; Ambati, J.; Ambati, K.; Simon, M.; Seluanov, A.; Gorbunova, V.; Slagboom, P. E.; Helfand, S. L.; Sedivy, J. M. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566(7742), 73–78. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Mita, P.; McKerrow, W.; Fenyö, D.; Boeke, J. D.; Linker, S. B.; Gage, F. H.; Kreiling, J. A.; Petrashen, A. P.; Woodham, T. A.; Taylor, J. R.; Helfand, S. L.; Sedivy, J. M. The role of retrotransposable elements in ageing and age-associated diseases. Nature 2021, 596(7870), 43–53. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, C. D.; Goodell, M. A. Tissue mosaicism following stem cell aging: blood as an exemplar. Nature Aging 2024, 4(3), 295–308. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D. E.; Strong, R.; Sharp, Z. D.; Nelson, J. F.; Astle, C. M.; Flurkey, K.; Nadon, N. L.; Wilkinson, J. E.; Frenkel, K.; Carter, C. S.; Pahor, M.; Javors, M. A.; Fernandez, E.; Miller, R. A. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460(7253), 392–395. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S. C.; Rabinovitch, P. S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493(7432), 338–345. [Google Scholar] [CrossRef] [PubMed]
- Conboy, I. M.; Conboy, M. J.; Wagers, A. J.; Girma, E. R.; Weissman, I. L.; Rando, T. A. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005, 433(7027), 760–764. [Google Scholar] [CrossRef] [PubMed]
- Mehdipour, M.; Skinner, C.; Wong, N.; Lieb, M.; Liu, C.; Etienne, J.; Kato, C.; Kiprov, D.; Conboy, M. J.; Conboy, I. M. Rejuvenation of three germ layers tissues by exchanging old blood plasma with saline-albumin. Aging 2020, 12(10), 8790–8819. [Google Scholar] [CrossRef] [PubMed]
- Bárcena, C.; Valdés-Mas, R.; Mayoral, P.; Garabaya, C.; Durand, S.; Rodríguez, F.; Fernández-García, M. T.; Salazar, N.; Nogacka, A. M.; Garatachea, N.; Bossut, N.; Aprahamian, F.; Lucia, A.; Kroemer, G.; Freije, J. M. P.; Quirós, P. M.; López-Otín, C. Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nature Medicine 2019, 25(8), 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L. J.; Gurkar, A. U.; Wang, Y.; Vijg, J.; Hoeijmakers, J. H. J.; Robbins, P. D. Nuclear Genomic Instability and Aging. Annual Review of Biochemistry 2018, 87(1), 295–322. [Google Scholar] [CrossRef] [PubMed]
- Monod, J. THE GROWTH OF BACTERIAL CULTURES. Annual Review of Microbiology 1949, 3(1), 371–394. [Google Scholar] [CrossRef]
- Scott, M.; Gunderson, C. W.; Mateescu, E. M.; Zhang, Z.; Hwa, T. Interdependence of Cell Growth and Gene Expression: Origins and Consequences. Science 2010, 330(6007), 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- Wilde, S. A.; Valley, J. W.; Peck, W. H.; Graham, C. M. Evidence from detrital zircons for the existence of continental crust and oceans on the Earth 4.4 Gyr ago. Nature 2001, 409(6817), 175–178. [Google Scholar] [CrossRef] [PubMed]
- Kasting, J. F. Earth’s Early Atmosphere. Science 1993, 259(5097), 920–926. [Google Scholar] [CrossRef] [PubMed]
- Patel, B. H.; Percivalle, C.; Ritson, D. J.; Duffy, C. D.; Sutherland, J. D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nature Chemistry 2015, 7(4), 301–307. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J. D. The Origin of Life—Out of the Blue. Angewandte Chemie International Edition 2016, 55(1), 104–121. [Google Scholar] [CrossRef] [PubMed]
- Nutman, A. P.; Bennett, V. C.; Friend, C. R. L.; Van Kranendonk, M. J.; Chivas, A. R. Rapid emergence of life shown by discovery of 3,700-million-year-old microbial structures. Nature 2016, 537(7621), 535–538. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M. M.; Knudson, A. G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proceedings of the National Academy of Sciences 2003, 100(22), 12871–12876. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362(6422), 709–715. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S. J. The DNA Damage Response: Making It Safe to Play with Knives. Molecular Cell 2010, 40(2), 179–204. [Google Scholar] [CrossRef] [PubMed]
- WATSON, J. D.; CRICK, F. H. C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171(4356), 737–738. [Google Scholar] [CrossRef] [PubMed]
- Yakovchuk, P. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Research 2006, 34(2), 564–574. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A. DNA EXCISION REPAIR. Annual Review of Biochemistry 1996, 65(1), 43–81. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Wood, R. D. Quality Control by DNA Repair. Science 1999, 286(5446), 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Otto, S. P.; Gerstein, A. C. The evolution of haploidy and diploidy. Current Biology 2008, 18(24), R1121–R1124. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Fuchs, E. Plasticity of epithelial stem cells in tissue regeneration. Science 2014, 344(6189). [Google Scholar] [CrossRef] [PubMed]
- Lieber, M. R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annual Review of Biochemistry 2010, 79(1), 181–211. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of Strand Breaks by Homologous Recombination. Cold Spring Harbor Perspectives in Biology 2013, 5(11), a012740–a012740. [Google Scholar] [CrossRef] [PubMed]
- LINDAHL, T.; BARNES, D. E. Repair of Endogenous DNA Damage. Cold Spring Harbor Symposia on Quantitative Biology 2000, 65(0), 127–134. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P. Mechanisms in Eukaryotic Mismatch Repair. Journal of Biological Chemistry 2006, 281(41), 30305–30309. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T. A.; Bebenek, K. DNA Replication Fidelity. Annual Review of Biochemistry 2000, 69(1), 497–529. [Google Scholar] [CrossRef] [PubMed]
- Slade, D.; Radman, M. Oxidative Stress Resistance in Deinococcus radiodurans. Microbiology and Molecular Biology Reviews 2011, 75(1), 133–191. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R. A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144(5), 646–674. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S. P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461(7267), 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Lane, D. P. p53, guardian of the genome. Nature 1992, 358(6381), 15–16. [Google Scholar] [CrossRef] [PubMed]
- Yakovchuk, P. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Research 2006, 34(2), 564–574. [Google Scholar] [CrossRef] [PubMed]
- SantaLucia, J.; Hicks, D. The Thermodynamics of DNA Structural Motifs. Annual Review of Biophysics and Biomolecular Structure 2004, 33(1), 415–440. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, C. L.; Ghosh, R. P. Chromatin Higher-order Structure and Dynamics. Cold Spring Harbor Perspectives in Biology 2010, 2(5), a000596–a000596. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A. W.; Richmond, R. K.; Sargent, D. F.; Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389(6648), 251–260. [Google Scholar] [CrossRef] [PubMed]
- Grosberg, R. K.; Strathmann, R. R. The Evolution of Multicellularity: A Minor Major Transition? Annual Review of Ecology, Evolution, and Systematics 2007, 38(1), 621–654. [Google Scholar] [CrossRef]
- Knoll, A. H. The Multiple Origins of Complex Multicellularity. Annual Review of Earth and Planetary Sciences 2011, 39(1), 217–239. [Google Scholar] [CrossRef]
- Nowak, M. A. Five rules for the evolution of cooperation. Science (New York, N.Y.) 2006, 314(5805), 1560–1563. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Simmons, L. W. Sexual selection and mate choice. Trends in Ecology & Evolution 2006, 21(6), 296–302. [Google Scholar] [CrossRef] [PubMed]
- Laland, K. N.; Odling-Smee, F. J.; Feldman, M. W. Evolutionary consequences of niche construction and their implications for ecology. Proceedings of the National Academy of Sciences 1999, 96(18), 10242–10247. [Google Scholar] [CrossRef] [PubMed]
- Hunter, P. The revival of the extended phenotype. EMBO Reports 2018, 19(7). [Google Scholar] [CrossRef] [PubMed]
- Allis, C. D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nature Reviews Genetics 2016, 17(8), 487–500. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes & Development 2002, 16(1), 6–21. [Google Scholar] [CrossRef] [PubMed]
- CRICK, F. Central Dogma of Molecular Biology. Nature 1970, 227(5258), 561–563. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M. M.; Swanson, M. S. RNA mis-splicing in disease. Nature Reviews Genetics 2016, 17(1), 19–32. [Google Scholar] [CrossRef] [PubMed]
- Kudla, G.; Murray, A. W.; Tollervey, D.; Plotkin, J. B. Coding-Sequence Determinants of Gene Expression in Escherichia coli. Science 2009, 324(5924), 255–258. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nature Reviews Molecular Cell Biology 2018, 19(1), 20–30. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J. B.; Kudla, G. Synonymous but not the same: the causes and consequences of codon bias. Nature Reviews Genetics 2011, 12(1), 32–42. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, T.; Jordan, J.; van Breugel, M. E.; Halavatyi, A.; Tischer, C.; Polidoro, P.; Abe, N.; Tsai, A.; Mann, R. S.; Stern, D. L.; Crocker, J. Dense and pleiotropic regulatory information in a developmental enhancer. Nature 2020, 587(7833), 235–239. [Google Scholar] [CrossRef] [PubMed]
- Claringbould, A.; Zaugg, J. B. Enhancers in disease: molecular basis and emerging treatment strategies. Trends in Molecular Medicine 2021, 27(11), 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M. K.; Cimprich, K. A. Causes and consequences of replication stress. Nature Cell Biology 2014, 16(1), 2–9. [Google Scholar] [CrossRef] [PubMed]
- Prioleau, M.-N.; MacAlpine, D. M. DNA replication origins—where do we begin? Genes & Development 2016, 30(15), 1683–1697. [Google Scholar] [CrossRef] [PubMed]
- Gómez-González, B.; Aguilera, A. Transcription-mediated replication hindrance: a major driver of genome instability. Genes & Development 2019, 33(15–16), 1008–1026. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.; Piazza, A.; Bermejo, R.; Kriegsman, B.; Colosio, A.; Teulade-Fichou, M.-P.; Foiani, M.; Nicolas, A. G-quadruplex-induced instability during leading-strand replication. The EMBO Journal 2011, 30(19), 4033–4046. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S. R.; Loeb, L. A.; Herr, A. J. Somatic mutations in aging, cancer and neurodegeneration. Mechanisms of Ageing and Development 2012, 133(4), 118–126. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T. B. L. DNA, mutations and aging. Mutation Research/DNAging 1989, 219(1), 1–7. [Google Scholar] [CrossRef] [PubMed]
- Turan, Z. G.; Parvizi, P.; Dönertaş, H. M.; Tung, J.; Khaitovich, P.; Somel, M. Molecular footprint of Medawar’s mutation accumulation process in mammalian aging. Aging Cell 2019, 18(4). [Google Scholar] [CrossRef] [PubMed]
- Vijg, J. Somatic mutations and aging: a re-evaluation. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 2000, 447(1), 117–135. [Google Scholar] [CrossRef] [PubMed]
- Rahbari, R.; Wuster, A.; Lindsay, S. J.; Hardwick, R. J.; Alexandrov, L. B.; Al Turki, S.; Dominiczak, A.; Morris, A.; Porteous, D.; Smith, B.; Stratton, M. R.; Hurles, M. E. Timing, rates and spectra of human germline mutation. Nature Genetics 2016, 48(2), 126–133. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J. B.; Freyer, C.; Elson, J. L.; Wredenberg, A.; Cansu, Z.; Trifunovic, A.; Larsson, N.-G. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biology 2008, 6(1), e10. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K. F.; Pezic, D.; Stuwe, E.; Webster, A. The piRNA Pathway Guards the Germline Genome Against Transposable Elements; 2016; pp. 51–77. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V. V.; Hochwagen, A. The Meiotic Checkpoint Network: Step-by-Step through Meiotic Prophase. Cold Spring Harbor Perspectives in Biology 2014, 6(10), a016675–a016675. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Frigge, M. L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S. A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; Wong, W. S. W.; Sigurdsson, G.; Walters, G. B.; Steinberg, S.; Helgason, H.; Thorleifsson, G.; Gudbjartsson, D. F.; Helgason, A.; Magnusson, O. Th.; Stefansson, K. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488(7412), 471–475. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.; Ackerman, M. S.; Miller, S. F.; Doak, T. G.; Lynch, M. Drift-barrier hypothesis and mutation-rate evolution. Proceedings of the National Academy of Sciences 2012, 109(45), 18488–18492. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Ackerman, M. S.; Gout, J.-F.; Long, H.; Sung, W.; Thomas, W. K.; Foster, P. L. Genetic drift, selection and the evolution of the mutation rate. Nature Reviews Genetics 2016, 17(11), 704–714. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P. Aging, DNA methylation and cancer. Critical Reviews in Oncology/Hematology 1999, 32(1), 31–43. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. DNA methylation and epigenetic inheritance. Philosophical Transactions of the Royal Society of London. B, Biological Sciences 1990, 326(1235), 329–338. [Google Scholar] [CrossRef] [PubMed]
- Booth, L. N.; Brunet, A. The Aging Epigenome. Molecular Cell 2016, 62(5), 728–744. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P. P.; Nativio, R.; Berger, S. L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166(4), 822–839. [Google Scholar] [CrossRef] [PubMed]
- Koch, Z.; Li, A.; Evans, D. S.; Cummings, S.; Ideker, T. Somatic mutation as an explanation for epigenetic aging. Nature Aging 2025, 5(4), 709–719. [Google Scholar] [CrossRef] [PubMed]
- Challen, G. A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J. S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; Liang, S.; Lu, Y.; Darlington, G. J.; Meissner, A.; Issa, J.-P. J.; Godley, L. A.; Li, W.; Goodell, M. A. Dnmt3a is essential for hematopoietic stem cell differentiation. Nature Genetics 2012, 44(1), 23–31. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P. V.; Mar, B. G.; Lindsley, R. C.; Mermel, C. H.; Burtt, N.; Chavez, A.; Higgins, J. M.; Moltchanov, V.; Kuo, F. C.; Kluk, M. J.; Henderson, B.; Kinnunen, L.; Koistinen, H. A.; Ladenvall, C.; Getz, G.; Ebert, B. L. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. New England Journal of Medicine 2014, 371(26), 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Yang, J. H.; Hayano, M.; Griffin, P. T.; Amorim, J. A.; Bonkowski, M. S.; Apostolides, J. K.; Salfati, E. L.; Blanchette, M.; Munding, E. M.; Bhakta, M.; Chew, Y. C.; Guo, W.; Yang, X.; Maybury-Lewis, S.; Tian, X.; Ross, J. M.; Coppotelli, G.; Meer, M. V.; Rogers-Hammond, R.; Sinclair, D. A. Loss of epigenetic information as a cause of mammalian aging. Cell 2023, 186(2), 305–326.e27. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N. J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408(6809), 239–247. [Google Scholar] [CrossRef] [PubMed]
- Gerschman, R.; Gilbert, D. L.; Nye, S. W.; Dwyer, P.; Fenn, W. O. Oxygen Poisoning and X-irradiation: A Mechanism in Common. Science 1954, 119(3097), 623–626. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. Journal of Gerontology 1956, 11(3), 298–300. [Google Scholar] [CrossRef] [PubMed]
- HARMAN, D. The Biologic Clock: The Mitochondria? Journal of the American Geriatrics Society 1972, 20(4), 145–147. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Rusling, J. F. Oxidation Chemistry of DNA and p53 Tumor Suppressor Gene. ChemistryOpen 2019, 8(3), 252–265. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, L.; Wild, S.; Brunoldi, G.; Piceni, A.; Ceppi, I.; Kummer, S.; Lutz, R. E.; Cejka, P.; Gari, K. The iron–sulphur cluster in human DNA2 is required for all biochemical activities of DNA2. Communications Biology 2020, 3(1), 322. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A. G.; Siebler, H. M.; Pavlov, Y. I.; Tahirov, T. H. Iron–Sulfur Clusters in DNA Polymerases and Primases of Eukaryotes; 2018; pp. 1–20. [Google Scholar] [CrossRef] [PubMed]
- Jozwiakowski, S. K.; Kummer, S.; Gari, K. Human DNA polymerase delta requires an iron–sulfur cluster for high-fidelity DNA synthesis. Life Science Alliance 2019, 2(4), e201900321. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Holt, M. E.; Thompson, M. K.; Salay, L. E.; Ehlinger, A. C.; Chazin, W. J.; Barton, J. K. The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science 2017, 355(6327). [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Bertini, I.; Rosato, A. Metalloproteomes: A Bioinformatic Approach. Accounts of Chemical Research 2009, 42(10), 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C. C. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicology Letters 1995, 82–83, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Keyer, K.; Imlay, J. A. Superoxide accelerates DNA damage by elevating free-iron levels. Proceedings of the National Academy of Sciences 1996, 93(24), 13635–13640. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochemical Journal 1973, 134(3), 707–716. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews 1979, 59(3), 527–605. [Google Scholar] [CrossRef] [PubMed]
- Davies, K. J. Protein damage and degradation by oxygen radicals. I. general aspects. Journal of Biological Chemistry 1987, 262(20), 9895–9901. [Google Scholar] [CrossRef]
- MITCHELL, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism. Nature 1961, 191(4784), 144–148. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S. E.; Linford, N. J.; Martin, G. M.; Treuting, P.; Ogburn, C. E.; Emond, M.; Coskun, P. E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; Wallace, D. C.; Rabinovitch, P. S. Medecine: Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308(5730), 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Goehring, L.; Huang, T. T.; Smith, D. J. Transcription–Replication Conflicts as a Source of Genome Instability. Annual Review of Genetics 2023, 57(1), 157–179. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Bocek, M. J.; Saldivar, J. C.; Swigut, T.; Cimprich, K. A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170(4), 774–786.e19. [Google Scholar] [CrossRef] [PubMed]
- Sabatinos, S.; Forsburg, S. Managing Single-Stranded DNA during Replication Stress in Fission Yeast. Biomolecules 2015, 5(3), 2123–2139. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J. C.; Cortez, D.; Cimprich, K. A. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nature Reviews Molecular Cell Biology 2017, 18(10), 622–636. [Google Scholar] [CrossRef] [PubMed]
- Gyenis, A.; Chang, J.; Demmers, J. J. P. G.; Bruens, S. T.; Barnhoorn, S.; Brandt, R. M. C.; Baar, M. P.; Raseta, M.; Derks, K. W. J.; Hoeijmakers, J. H. J.; Pothof, J. Genome-wide RNA polymerase stalling shapes the transcriptome during aging. Nature Genetics 2023, 55(2), 268–279. [Google Scholar] [CrossRef] [PubMed]
- Stoeger, T.; Grant, R. A.; McQuattie-Pimentel, A. C.; Anekalla, K. R.; Liu, S. S.; Tejedor-Navarro, H.; Singer, B. D.; Abdala-Valencia, H.; Schwake, M.; Tetreault, M.-P.; Perlman, H.; Balch, W. E.; Chandel, N. S.; Ridge, K. M.; Sznajder, J. I.; Morimoto, R. I.; Misharin, A. V.; Budinger, G. R. S.; Nunes Amaral, L. A. Aging is associated with a systemic length-associated transcriptome imbalance. Nature Aging 2022, 2(12), 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Saxowsky, T. T.; Meadows, K. L.; Klungland, A.; Doetsch, P. W. 8-Oxoguanine-mediated transcriptional mutagenesis causes Ras activation in mammalian cells. Proceedings of the National Academy of Sciences 2008, 105(48), 18877–18882. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, C.; Gout, J.-F.; Haroon, S.; Towheed, A.; Chung, C.; LaGosh, J.; McGann, E.; Zhang, X.; Song, Y.; Simpson, S.; Danthi, P. S.; Benayoun, B. A.; Wallace, D.; Thomas, K.; Lynch, M.; Vermulst, M. Genome-wide surveillance of transcription errors in response to genotoxic stress. Proceedings of the National Academy of Sciences 2021, 118(1). [Google Scholar] [CrossRef] [PubMed]
- Vermulst, M.; Denney, A. S.; Lang, M. J.; Hung, C.-W.; Moore, S.; Moseley, M. A.; Thompson, J. W.; Madden, V.; Gauer, J.; Wolfe, K. J.; Summers, D. W.; Schleit, J.; Sutphin, G. L.; Haroon, S.; Holczbauer, A.; Caine, J.; Jorgenson, J.; Cyr, D.; Kaeberlein, M.; Erie, D. A. Transcription errors induce proteotoxic stress and shorten cellular lifespan. Nature Communications 2015, 6(1), 8065. [Google Scholar] [CrossRef] [PubMed]
- Debès, C.; Papadakis, A.; Grönke, S.; Karalay, Ö.; Tain, L. S.; Mizi, A.; Nakamura, S.; Hahn, O.; Weigelt, C.; Josipovic, N.; Zirkel, A.; Brusius, I.; Sofiadis, K.; Lamprousi, M.; Lu, Y.-X.; Huang, W.; Esmaillie, R.; Kubacki, T.; Späth, M. R.; Beyer, A. Ageing-associated changes in transcriptional elongation influence longevity. Nature 2023, 616(7958), 814–821. [Google Scholar] [CrossRef] [PubMed]
- Soheili-Nezhad, S.; Ibáñez-Solé, O.; Izeta, A.; Hoeijmakers, J. H. J.; Stoeger, T. Time is ticking faster for long genes in aging. Trends in Genetics 2024, 40(4), 299–312. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, M.; Howell, P.; Dutta, S.; Heintz, C.; Mair, W. B. Alternative splicing in aging and longevity. Human Genetics 2020, 139(3), 357–369. [Google Scholar] [CrossRef] [PubMed]
- Sale, J. E. Translesion DNA Synthesis and Mutagenesis in Eukaryotes. Cold Spring Harbor Perspectives in Biology 2013, 5(3), a012708–a012708. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L. B.; Nik-Zainal, S.; Wedge, D. C.; Aparicio, S. A. J. R.; Behjati, S.; Biankin, A. V.; Bignell, G. R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; Boyault, S.; Burkhardt, B.; Butler, A. P.; Caldas, C.; Davies, H. R.; Desmedt, C.; Eils, R.; Eyfjörd, J. E.; Foekens, J. A.; Stratton, M. R. Signatures of mutational processes in human cancer. Nature 2013, 500(7463), 415–421. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347(6217), 78–81. [Google Scholar] [CrossRef] [PubMed]
- White, R. R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Molecular Cell 2016, 63(5), 729–738. [Google Scholar] [CrossRef] [PubMed]
- Swain, U.; Rao, K. S. Age-dependent decline of DNA base excision repair activity in rat cortical neurons. Mechanisms of Ageing and Development 2012, 133(4), 186–194. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Matanoski, G. M.; Farmer, E. R.; Hedayati, M. A.; Grossman, L. DNA repair and aging in basal cell carcinoma: a molecular epidemiology study. Proceedings of the National Academy of Sciences 1993, 90(4), 1614–1618. [Google Scholar] [CrossRef] [PubMed]
- Langie, S.; Cameron, K.; Ficz, G.; Oxley, D.; Tomaszewski, B.; Gorniak, J.; Maas, L.; Godschalk, R.; Van Schooten, F.; Reik, W.; Von Zglinicki, T.; Mathers, J. The Ageing Brain: Effects on DNA Repair and DNA Methylation in Mice. Genes 2017, 8(2), 75. [Google Scholar] [CrossRef] [PubMed]
- Sliwinska, A.; Sitarek, P.; Toma, M.; Czarny, P.; Synowiec, E.; Krupa, R.; Wigner, P.; Bialek, K.; Kwiatkowski, D.; Korycinska, A.; Majsterek, I.; Szemraj, J.; Galecki, P.; Sliwinski, T. Decreased expression level of BER genes in Alzheimer’s disease patients is not derivative of their DNA methylation status. Progress in Neuro-Psychopharmacology and Biological Psychiatry 2017, 79, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Helbock, H. J.; Beckman, K. B.; Shigenaga, M. K.; Walter, P. B.; Woodall, A. A.; Yeo, H. C.; Ames, B. N. DNA oxidation matters: The HPLC–electrochemical detection assay of 8-oxo-deoxyguanosine and 8-oxo-guanine. Proceedings of the National Academy of Sciences 1998, 95(1), 288–293. [Google Scholar] [CrossRef] [PubMed]
- Sastre, J.; Pallardó, F. V.; García de la Asunción, J.; Viña, J. Mitochondria, oxidative stress and aging. Free Radical Research 2000, 32(3), 189–198. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Park, J. W.; Ames, B. N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proceedings of the National Academy of Sciences 1988, 85(17), 6465–6467. [Google Scholar] [CrossRef] [PubMed]
- AMES, B. N. Delaying the Mitochondrial Decay of Aging. Annals of the New York Academy of Sciences 2004, 1019(1), 406–411. [Google Scholar] [CrossRef] [PubMed]
- Medawar, P. B. An unsolved problem of biology; 1952. [Google Scholar]
- Williams, G. C. Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution 1957, 11(4), 398. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T. B. L. Evolution of ageing. Nature 1977, 270(5635), 301–304. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J.; Economos, A. C.; Fleming, J.; Johnson, J. E. Mitochondrial role in cell aging. Experimental Gerontology 1980, 15(6), 575–591. [Google Scholar] [CrossRef] [PubMed]
- Hämäläinen, R. H.; Landoni, J. C.; Ahlqvist, K. J.; Goffart, S.; Ryytty, S.; Rahman, M. O.; Brilhante, V.; Icay, K.; Hautaniemi, S.; Wang, L.; Laiho, M.; Suomalainen, A. Defects in mtDNA replication challenge nuclear genome stability through nucleotide depletion and provide a unifying mechanism for mouse progerias. Nature Metabolism 2019, 1(10), 958–965. [Google Scholar] [CrossRef] [PubMed]
- Pearl, R. The Rate of Living: Being an Account of Some Experimental Studies on the Biology of Life Duration; A.A. Knopf, 1928; Available online: https://books.google.com/books?id=P3jwAAAAMAAJ.
- Szilard, L. ON THE NATURE OF THE AGING PROCESS. Proceedings of the National Academy of Sciences 1959, 45(1), 30–45. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J.; Suh, Y. Genome Instability and Aging. Annual Review of Physiology 2013, 75(1), 645–668. [Google Scholar] [CrossRef] [PubMed]
- Orgel, L. E. The Maintenance of the Accuracy of Protein Synthesis and its Relevance to Aging. Proceedings of the National Academy of Sciences 1963, 49(4), 517–521. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. The Inheritance of Epigenetic Defects. Science 1987, 238(4824), 163–170. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y. R.; Tian, X.; Sinclair, D. A. The Information Theory of Aging. Nature Aging 2023, 3(12), 1486–1499. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R. I.; Cuervo, A. M. Proteostasis and the Aging Proteome in Health and Disease. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 2014, 69 (Suppl 1), S33–S38. [Google Scholar] [CrossRef] [PubMed]
- Greider, C. W.; Blackburn, E. H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 1985, 43(2), 405–413. [Google Scholar] [CrossRef] [PubMed]
- Van Meter, M.; Kashyap, M.; Rezazadeh, S.; Geneva, A. J.; Morello, T. D.; Seluanov, A.; Gorbunova, V. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nature Communications 2014, 5(1), 5011. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D. C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 146(5), 682–695. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P. S. The serial cultivation of human diploid cell strains. Experimental Cell Research 1961, 25(3), 585–621. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell 2005, 120(4), 513–522. [Google Scholar] [CrossRef] [PubMed]
- van Deursen, J. M. The role of senescent cells in ageing. Nature 2014, 509(7501), 439–446. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D. J.; Jamieson, C. H. M.; Weissman, I. L. Stems Cells and the Pathways to Aging and Cancer. Cell 2008, 132(4), 681–696. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D. W.; Fasano, A.; Miller, G. W.; Miller, A. H.; Mantovani, A.; Weyand, C. M.; Barzilai, N.; Goronzy, J. J.; Rando, T. A.; Effros, R. B.; Lucia, A.; Kleinstreuer, N.; Slavich, G. M. Chronic inflammation in the etiology of disease across the life span. Nature Medicine 2019, 25(12), 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- FRANCESCHI, C.; BONAFÈ, M.; VALENSIN, S.; OLIVIERI, F.; DE LUCA, M.; OTTAVIANI, E.; DE BENEDICTIS, G. Inflamm-aging: An Evolutionary Perspective on Immunosenescence. Annals of the New York Academy of Sciences 2000, 908(1), 244–254. [Google Scholar] [CrossRef] [PubMed]
- Walford, R. L. The Immunologic Theory of Aging. Immunological Reviews 1969, 2(1), 171–171. [Google Scholar] [CrossRef]
- Goronzy, J. J.; Weyand, C. M. Understanding immunosenescence to improve responses to vaccines. Nature Immunology 2013, 14(5), 428–436. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. T cells and aging january 2002 update. Frontiers in Bioscience 2002, 7(4), A831. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V. M.; Cerami, A. Nonenzymatic Browning in Vivo: Possible Process for Aging of Long-Lived Proteins. Science 1981, 211(4481), 491–493. [Google Scholar] [CrossRef] [PubMed]
- Cerami, A. Hypothesis: glucose as a mediator of aging. Journal of the American Geriatrics Society 1985, 33(9), 626–634. [Google Scholar] [CrossRef] [PubMed]
- Dilman, V. M. Age-associated elevation of hypothalamic, threshold to feedback control, and its role in development, ageing, and disease. The Lancet 1971, 297(7711), 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, S. W. J.; van den Beld, A. W.; van der Lely, A.-J. The Endocrinology of Aging. Science 1997, 278(5337), 419–424. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366(6454), 461–464. [Google Scholar] [CrossRef] [PubMed]
- Kapahi, P.; Zid, B. M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of Lifespan in Drosophila by Modulation of Genes in the TOR Signaling Pathway. Current Biology 2004, 14(10), 885–890. [Google Scholar] [CrossRef] [PubMed]
- Rando, T. A.; Chang, H. Y. Aging, Rejuvenation, and Epigenetic Reprogramming: Resetting the Aging Clock. Cell 2012, 148(1–2), 46–57. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P. W.; Jeffery, I. B. Gut microbiota and aging. Science 2015, 350(6265), 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T. S.; Shanahan, F.; O’Toole, P. W. The gut microbiome as a modulator of healthy ageing. Nature Reviews Gastroenterology & Hepatology 2022, 19(9), 565–584. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Kirkwood, T. B. L. A network theory of ageing: the interactions of defective mitochondria, aberrant proteins, free radicals and scavengers in the ageing process. Mutation Research/DNAging 1996, 316(5–6), 209–236. [Google Scholar] [CrossRef] [PubMed]
- Kriete, A.; Bosl, W. J.; Booker, G. Rule-Based Cell Systems Model of Aging using Feedback Loop Motifs Mediated by Stress Responses. PLoS Computational Biology 2010, 6(6), e1000820. [Google Scholar] [CrossRef] [PubMed]
- GAVRILOV, L. A.; GAVRILOVA, N. S. The Reliability Theory of Aging and Longevity. Journal of Theoretical Biology 2001, 213(4), 527–545. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M. V. Aging: ROS or TOR. Cell Cycle 2008, 7(21), 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M. V. Aging and Immortality: Quasi-Programmed Senescence and Its Pharmacologic Inhibition. Cell Cycle 2006, 5(18), 2087–2102. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V. P. Aging is a specific biological function rather than the result of a disorder in complex living systems: biochemical evidence in support of Weismann’s hypothesis. Biochemistry. Biokhimiia 1997, 62(11), 1191–1195. [Google Scholar] [PubMed]
- Goldsmith, T. C. Aging as an evolved characteristic – Weismann’s theory reconsidered. Medical Hypotheses 2004, 62(2), 304–308. [Google Scholar] [CrossRef] [PubMed]
- Longo, V. D.; Mitteldorf, J.; Skulachev, V. P. Programmed and altruistic ageing. Nature Reviews Genetics 2005, 6(11), 866–872. [Google Scholar] [CrossRef] [PubMed]
- Hopfield, J. J. Kinetic Proofreading: A New Mechanism for Reducing Errors in Biosynthetic Processes Requiring High Specificity. Proceedings of the National Academy of Sciences 1974, 71(10), 4135–4139. [Google Scholar] [CrossRef] [PubMed]
- De Bont, R. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 2004, 19(3), 169–185. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, J. A.; Adzhubei, I.; Thurman, R. E.; Kryukov, G. V.; Mirkin, S. M.; Sunyaev, S. R. Human mutation rate associated with DNA replication timing. Nature Genetics 2009, 41(4), 393–395. [Google Scholar] [CrossRef] [PubMed]
- Tomkova, M.; Tomek, J.; Kriaucionis, S.; Schuster-Böckler, B. Mutational signature distribution varies with DNA replication timing and strand asymmetry. Genome Biology 2018, 19(1), 129. [Google Scholar] [CrossRef] [PubMed]
- Frederico, L. A.; Kunkel, T. A.; Shaw, B. R. A sensitive genetic assay for the detection of cytosine deamination: determination of rate constants and the activation energy. Biochemistry 1990, 29(10), 2532–2537. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L. B.; Kim, J.; Haradhvala, N. J.; Huang, M. N.; Tian Ng, A. W.; Wu, Y.; Boot, A.; Covington, K. R.; Gordenin, D. A.; Bergstrom, E. N.; Islam, S. M. A.; Lopez-Bigas, N.; Klimczak, L. J.; McPherson, J. R.; Morganella, S.; Sabarinathan, R.; Wheeler, D. A.; Mustonen, V.; Alexandrov, L. B.; von Mering, C. The repertoire of mutational signatures in human cancer. Nature 2020, 578(7793), 94–101. [Google Scholar] [CrossRef] [PubMed]
- Cagan, A.; Baez-Ortega, A.; Brzozowska, N.; Abascal, F.; Coorens, T. H. H.; Sanders, M. A.; Lawson, A. R. J.; Harvey, L. M. R.; Bhosle, S.; Jones, D.; Alcantara, R. E.; Butler, T. M.; Hooks, Y.; Roberts, K.; Anderson, E.; Lunn, S.; Flach, E.; Spiro, S.; Januszczak, I.; Martincorena, I. Somatic mutation rates scale with lifespan across mammals. Nature 2022, 604(7906), 517–524. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; Nijman, I. J.; Martincorena, I.; Mokry, M.; Wiegerinck, C. L.; Middendorp, S.; Sato, T.; Schwank, G.; Nieuwenhuis, E. E. S.; Verstegen, M. M. A.; van Boxtel, R. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538(7624), 260–264. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M. P. How mitochondria produce reactive oxygen species. Biochemical Journal 2009, 417(1), 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brand, M. D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radical Biology and Medicine 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Pontel, L. B.; Rosado, I. V.; Burgos-Barragan, G.; Garaycoechea, J. I.; Yu, R.; Arends, M. J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; Swenberg, J. A.; Crossan, G. P.; Patel, K. J. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Molecular Cell 2015, 60(1), 177–188. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiological Reviews 2007, 87(1), 245–313. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Fahimi, H. D. Peroxisomes and oxidative stress. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2006, 1763(12), 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, B.; Lindahl, T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. The EMBO Journal 1982, 1(2), 211–216. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L. J. Lipid peroxidation—DNA damage by malondialdehyde. Mutation Research - Fundamental and Molecular Mechanisms of Mutagenesis 1999, 424(1–2), 83–95. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J. A. Pathways of Oxidative Damage. Annual Review of Microbiology 2003, 57(1), 395–418. [Google Scholar] [CrossRef] [PubMed]
- Gedik, C. M.; Collins, A. Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. The FASEB Journal 2005, 19(1), 82–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Clauson, C. L.; Robbins, P. D.; Niedernhofer, L. J.; Wang, Y. The oxidative DNA lesions 8,5′-cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell 2012, 11(4), 714–716. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P. J. The 8,5′-cyclopurine-2′-deoxynucleosides: Candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA Repair 2008, 7(7), 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Dollé, M. E. T.; Busuttil, R. A.; Garcia, A. M.; Wijnhoven, S.; van Drunen, E.; Niedernhofer, L. J.; van der Horst, G.; Hoeijmakers, J. H. J.; van Steeg, H.; Vijg, J. Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice. Mutation Research - Fundamental and Molecular Mechanisms of Mutagenesis 2006, 596(1–2), 22–35. [Google Scholar] [CrossRef] [PubMed]
- Brégeon, D.; Doetsch, P. W. Transcriptional mutagenesis: causes and involvement in tumour development. Nature Reviews Cancer 2011, 11(3), 218–227. [Google Scholar] [CrossRef] [PubMed]
- Crochemore, C.; Chica, C.; Garagnani, P.; Lattanzi, G.; Horvath, S.; Sarasin, A.; Franceschi, C.; Bacalini, M. G.; Ricchetti, M. Epigenomic signature of accelerated ageing in progeroid Cockayne syndrome. Aging Cell 2023, 22(10). [Google Scholar] [CrossRef] [PubMed]
- Gonzalo-Hansen, C.; Steurer, B.; Janssens, R. C.; Zhou, D.; van Sluis, M.; Lans, H.; Marteijn, J. A. Differential processing of RNA polymerase II at DNA damage correlates with transcription-coupled repair syndrome severity. Nucleic Acids Research 2024, 52(16), 9596–9612. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.; Sitko, L. K.; Khoirunnisa, R.; Navarro-Aguad, F.; Samuel, D. M.; Park, H.; Cheon, B.; Mutsnaini, L.; Lee, J.; Otlu, B.; Takeda, S.; Lee, S.; Ivanov, D.; Gartner, A. Comprehensive whole-genome sequencing reveals origins of mutational signatures associated with aging, mismatch repair deficiency and temozolomide chemotherapy. Nucleic Acids Research 2025, 53(1). [Google Scholar] [CrossRef] [PubMed]
- Spisak, N.; de Manuel, M.; Przeworski, M. Collateral mutagenesis funnels multiple sources of DNA damage into a ubiquitous mutational signature. 2025. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; Guo, Z. OGG1 is essential in oxidative stress induced DNA demethylation. Cellular Signalling 2016, 28(9), 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A. M.; Ding, Y.; Burrows, C. J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proceedings of the National Academy of Sciences 2017, 114(10), 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A. M.; Burrows, C. J. 8-Oxo-7,8-dihydroguanine, friend and foe: Epigenetic-like regulator versus initiator of mutagenesis. DNA Repair 2017, 56, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Hahm, J. Y.; Park, J.; Jang, E.-S.; Chi, S. W. 8-Oxoguanine: from oxidative damage to epigenetic and epitranscriptional modification. Experimental & Molecular Medicine 2022, 54(10), 1626–1642. [Google Scholar] [CrossRef] [PubMed]
- Frey, Y.; Haj, M.; Ziv, Y.; Elkon, R.; Shiloh, Y. Broad repression of DNA repair genes in senescent cells identified by integration of transcriptomic data. Nucleic Acids Research 2025, 53(1). [Google Scholar] [CrossRef] [PubMed]
- Robinson, A. R.; Yousefzadeh, M. J.; Rozgaja, T. A.; Wang, J.; Li, X.; Tilstra, J. S.; Feldman, C. H.; Gregg, S. Q.; Johnson, C. H.; Skoda, E. M.; Frantz, M.-C.; Bell-Temin, H.; Pope-Varsalona, H.; Gurkar, A. U.; Nasto, L. A.; Robinson, R. A. S.; Fuhrmann-Stroissnigg, H.; Czerwinska, J.; McGowan, S. J.; Niedernhofer, L. J. Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biology 2018, 17, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L. J.; Garinis, G. A.; Raams, A.; Lalai, A. S.; Robinson, A. R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; van Leeuwen, W.; Theil, A. F.; Vermeulen, W.; van der Horst, G. T. J.; Meinecke, P.; Kleijer, W. J.; Vijg, J.; Jaspers, N. G. J.; Hoeijmakers, J. H. J. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444(7122), 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Hazra, T. K.; Papaconstantinou, J.; Mitra, S.; Boldogh, I. Age-dependent deficiency in import of mitochondrial DNA glycosylases required for repair of oxidatively damaged bases. Proceedings of the National Academy of Sciences 2003, 100(19), 10670–10675. [Google Scholar] [CrossRef] [PubMed]
- Cabelof, D. C.; Raffoul, J. J.; Yanamadala, S.; Ganir, C.; Guo, Z.; Heydari, A. R. Attenuation of DNA polymerase β-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutation Research - Fundamental and Molecular Mechanisms of Mutagenesis 2002, 500(1–2), 135–145. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; MacGarvey, U.; Kaufman, A. E.; Koontz, D.; Shoffner, J. M.; Wallace, D. C.; Beal, M. F. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Annals of Neurology 1993, 34(4), 609–616. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M. L.; Van Remmen, H.; Drake, J. A.; Yang, H.; Guo, Z. M.; Kewitt, K.; Walter, C. A.; Richardson, A. Does oxidative damage to DNA increase with age? Proceedings of the National Academy of Sciences 2001, 98(18), 10469–10474. [Google Scholar] [CrossRef] [PubMed]
- Maltseva, D. V.; Baykov, A. A.; Jeltsch, A.; Gromova, E. S. Impact of 7,8-Dihydro-8-oxoguanine on Methylation of the CpG Site by Dnmt3a. Biochemistry 2009, 48(6), 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Cedar, H.; Razin, A. Substrate and sequence specificity of a eukaryotic DNA methylase. Nature 1982, 295(5850), 620–622. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R. Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Research 2006, 34(4), 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Challen, G.; Goodell, M. A. Clonal Hematopoiesis: Mechanisms Driving Dominance of Stem Cell Clones; Blood, 2020. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Marteijn, J. A.; Schumacher, B.; Hoeijmakers, J. H. J.; Jansen, G.; Vermeulen, W. Involvement of Global Genome Repair, Transcription Coupled Repair, and Chromatin Remodeling in UV DNA Damage Response Changes during Development. PLoS Genetics 2010, 6(5), e1000941. [Google Scholar] [CrossRef] [PubMed]
- Sabatella, M.; Thijssen, K. L.; Davó-Martínez, C.; Vermeulen, W.; Lans, H. Tissue-Specific DNA Repair Activity of ERCC-1/XPF-1. Cell Reports 2021, 34(2), 108608. [Google Scholar] [CrossRef] [PubMed]
- Rieckher, M.; Gallrein, C.; Alquezar-Artieda, N.; Bourached-Silva, N.; Vaddavalli, P. L.; Mares, D.; Backhaus, M.; Blindauer, T.; Greger, K.; Wiesner, E.; Pontel, L. B.; Schumacher, B. Distinct DNA repair mechanisms prevent formaldehyde toxicity during development, reproduction and aging. Nucleic Acids Research 2024, 52(14), 8271–8285. [Google Scholar] [CrossRef] [PubMed]
- Ghaddar, A.; Mony, V. K.; Mishra, S.; Berhanu, S.; Johnson, J. C.; Enriquez-Hesles, E.; Harrison, E.; Patel, A.; Horak, M. K.; Smith, J. S.; O’Rourke, E. J. Increased alcohol dehydrogenase 1 activity promotes longevity. Current Biology 2023, 33(6), 1036–1046.e6. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V. E.; Zhou, S.; Diaz, L. A.; Kinzler, K. W. Cancer Genome Landscapes. Science 2013, 339(6127), 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Nouspikel, T.; Hanawalt, P. C. Terminally Differentiated Human Neurons Repair Transcribed Genes but Display Attenuated Global DNA Repair and Modulation of Repair Gene Expression. Molecular and Cellular Biology 2000, 20(5), 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P. J.; Cheng, T.-F.; Cooper, L. Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage? DNA Repair 2008, 7(6), 834–848. [Google Scholar] [CrossRef] [PubMed]
- Barlow, R. E.; Proschan, F. Mathematical Theory of Reliability; Society for Industrial and Applied Mathematics, 1996. [Google Scholar] [CrossRef]
- Gompertz, B. On the nature of the function expressive of the law of human mortality, and on a new mode of determining the value of life contingencies. In a letter to Francis Baily, Esq. F. R. S. &c. By Benjamin Gompertz, Esq. F. R. S. Abstracts of the Papers Printed in the Philosophical Transactions of the Royal Society of London; 1833; Volume (2), pp. 252–253. [Google Scholar] [CrossRef]
- Sekhar, R. V.; Patel, S. G.; Guthikonda, A. P.; Reid, M.; Balasubramanyam, A.; Taffet, G. E.; Jahoor, F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. The American Journal of Clinical Nutrition 2011, 94(3), 847–853. [Google Scholar] [CrossRef] [PubMed]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Agerelated Diseases. Current Genomics 2014, 15(1), 38–51. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A. M. Autophagy and aging: keeping that old broom working. Trends in Genetics 2008, 24(12), 604–612. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I. I.; Holmström, K. M.; Fergusson, M. M.; Yoo, Y. H.; Combs, C. A.; Finkel, T. Measuring In Vivo Mitophagy. Molecular Cell 2015, 60(4), 685–696. [Google Scholar] [CrossRef] [PubMed]
- Killilea, D. W.; Atamna, H.; Liao, C.; Ames, B. N. Iron Accumulation During Cellular Senescence in Human Fibroblasts In Vitro. Antioxidants & Redox Signaling 2003, 5(5), 507–516. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K. H.; Lu, X. Live or let die: the cell’s response to p53. Nature Reviews Cancer 2002, 2(8), 594–604. [Google Scholar] [CrossRef] [PubMed]
- Wilmut, I.; Beaujean, N.; de Sousa, P. A.; Dinnyes, A.; King, T. J.; Paterson, L. A.; Wells, D. N.; Young, L. E. Somatic cell nuclear transfer. Nature 2002, 419(6907), 583–587. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, T.; Perry, A. C. F.; Zuccotti, M.; Johnson, K. R.; Yanagimachi, R. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 1998, 394(6691), 369–374. [Google Scholar] [CrossRef] [PubMed]
- Tomaskovic, I.; Prieto-Garcia, C.; Boskovic, M.; Glumac, M.; Tsai, T.-L.; Mosler, T.; Kazi, R.; Rathore, R.; Andrade, J.; Hoffmann, M.; Giuliani, G.; Jacomin, A.-C.; Pereira, R. S.; Knop, E.; Wachsmuth, L.; Beli, P.; Husnjak, K.; Pasparakis, M.; Ablasser, A.; Dikic, I. DNA-protein cross-links promote cGAS-STING–driven premature aging and embryonic lethality. Science 2026, 391(6784). [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J. I.; Crossan, G. P.; Langevin, F.; Daly, M.; Arends, M. J.; Patel, K. J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489(7417), 571–575. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, J. W.; Carey, J. R.; Christensen, K.; Johnson, T. E.; Yashin, A. I.; Holm, N. V.; Iachine, I. A.; Kannisto, V.; Khazaeli, A. A.; Liedo, P.; Longo, V. D.; Zeng, Y.; Manton, K. G.; Curtsinger, J. W. Biodemographic Trajectories of Longevity. Science 1998, 280(5365), 855–860. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, W. D. The moulding of senescence by natural selection. Journal of Theoretical Biology 1966, 12(1), 12–45. [Google Scholar] [CrossRef] [PubMed]
- Milholland, B.; Dong, X.; Zhang, L.; Hao, X.; Suh, Y.; Vijg, J. Differences between germline and somatic mutation rates in humans and mice. Nature Communications 2017, 8(1), 15183. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, L. A.; Besenbacher, S.; Zheng, J.; Li, P.; Bertelsen, M. F.; Quintard, B.; Hoffman, J. I.; Li, Z.; St. Leger, J.; Shao, C.; Stiller, J.; Gilbert, M. T. P.; Schierup, M. H.; Zhang, G. Evolution of the germline mutation rate across vertebrates. Nature 2023, 615(7951), 285–291. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P. S.; Coorens, T. H. H.; Palles, C.; Mitchell, E.; Abascal, F.; Olafsson, S.; Lee, B. C. H.; Lawson, A. R. J.; Lee-Six, H.; Moore, L.; Sanders, M. A.; Hewinson, J.; Martin, L.; Pinna, C. M. A.; Galavotti, S.; Rahbari, R.; Campbell, P. J.; Martincorena, I.; Tomlinson, I.; Stratton, M. R. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nature Genetics 2021, 53(10), 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, L.; Fritzell, J. A.; Baker, S. M.; Liskay, R. M.; Glazer, P. M. Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2. Proceedings of the National Academy of Sciences 1997, 94(7), 3122–3127. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, K.; Fernandez-Tajes, J.; Gül, G.; Thorn, S.; Ward, J. C.; Wilson, J. F.; Palles, C.; Bishop, D. T.; Houlston, R. S.; Dunlop, M. G.; Tomlinson, I. P. M. Germline haploinsufficiency for the base excision repair gene MUTYH. causes mutational signature SBS18 in multiple tumour types, specifically leading to an increased risk of colorectal cancer 2025. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V. D. Extending Healthy Life Span—From Yeast to Humans. Science 2010, 328(5976), 321–326. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, E.; Spencer Chapman, M.; Williams, N.; Dawson, K. J.; Mende, N.; Calderbank, E. F.; Jung, H.; Mitchell, T.; Coorens, T. H. H.; Spencer, D. H.; Machado, H.; Lee-Six, H.; Davies, M.; Hayler, D.; Fabre, M. A.; Mahbubani, K.; Abascal, F.; Cagan, A.; Vassiliou, G. S.; Campbell, P. J. Clonal dynamics of haematopoiesis across the human lifespan. Nature 2022, 606(7913), 343–350. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Han, C.; Luo, W.; Wang, X.; Li, H.; Luo, H.; Zhou, J.; Qi, J.; He, R. Accumulated hippocampal formaldehyde induces age-dependent memory decline. AGE 2013, 35(3), 583–596. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, Y.; Sasaki, K.; Mitsutake, N.; Matsuse, M.; Shimada, M.; Nardo, T.; Takahashi, Y.; Ohyama, K.; Ito, K.; Mishima, H.; Nomura, M.; Kinoshita, A.; Ono, S.; Takenaka, K.; Masuyama, R.; Kudo, T.; Slor, H.; Utani, A.; Tateishi, S.; Ogi, T. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nature Genetics 2012, 44(5), 586–592. [Google Scholar] [CrossRef] [PubMed]
- Demin, A. A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; Sasanuma, H.; Hanzlikova, H.; Takeda, S.; Caldecott, K. W. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Molecular Cell 2021, 81(14), 3018–3030.e5. [Google Scholar] [CrossRef] [PubMed]
- Lodato, M. A.; Rodin, R. E.; Bohrson, C. L.; Coulter, M. E.; Barton, A. R.; Kwon, M.; Sherman, M. A.; Vitzthum, C. M.; Luquette, L. J.; Yandava, C. N.; Yang, P.; Chittenden, T. W.; Hatem, N. E.; Ryu, S. C.; Woodworth, M. B.; Park, P. J.; Walsh, C. A. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 2018, 359(6375), 555–559. [Google Scholar] [CrossRef] [PubMed]
- Wijnhoven, S. W. P.; Beems, R. B.; Roodbergen, M.; van den Berg, J.; Lohman, P. H. M.; Diderich, K.; van der Horst, G. T. J.; Vijg, J.; Hoeijmakers, J. H. J.; van Steeg, H. Accelerated aging pathology in ad libitum fed XpdTTD mice is accompanied by features suggestive of caloric restriction. DNA Repair 2005, 4(11), 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, W. P.; Dollé, M. E. T.; Reiling, E.; Jaarsma, D.; Payan-Gomez, C.; Bombardieri, C. R.; Wu, H.; Roks, A. J. M.; Botter, S. M.; van der Eerden, B. C.; Youssef, S. A.; Kuiper, R. V.; Nagarajah, B.; van Oostrom, C. T.; Brandt, R. M. C.; Barnhoorn, S.; Imholz, S.; Pennings, J. L. A.; de Bruin, A.; Hoeijmakers, J. H. J. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 2016, 537(7620), 427–431. [Google Scholar] [CrossRef] [PubMed]
- Das, M. M.; Godoy, M.; Chen, S.; Moser, V. A.; Avalos, P.; Roxas, K. M.; Dang, I.; Yáñez, A.; Zhang, W.; Bresee, C.; Arditi, M.; Liu, G. Y.; Svendsen, C. N.; Goodridge, H. S. Young bone marrow transplantation preserves learning and memory in old mice. Communications Biology 2019, 2(1), 73. [Google Scholar] [CrossRef] [PubMed]
- Jazbec, K.; Jež, M.; Švajger, U.; Smrekar, B.; Miceska, S.; Rajčevič, U.; Justin, M.; Završnik, J.; Malovrh, T.; Švara, T.; Gombač, M.; Ramšak, Ž.; Rožman, P. The Influence of Heterochronic Non-Myeloablative Bone Marrow Transplantation on the Immune System, Frailty, General Health, and Longevity of Aged Murine Recipients. Biomolecules 2022, 12(4), 595. [Google Scholar] [CrossRef] [PubMed]
- Lountzi, D.; Henzel, K.; Jazbec, K.; Bano, D.; Krauss, S.; Rožman, P.; Ehninger, D. Effects of heterochronic, non-myeloablative bone marrow transplantation on age-related behavioural changes in mice. Mechanisms of Ageing and Development 2020, 191, 111327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lee, D. E.; Trapp, A.; Tyshkovskiy, A.; Lu, A. T.; Bareja, A.; Kerepesi, C.; McKay, L. K.; Shindyapina, A. V.; Dmitriev, S. E.; Baht, G. S.; Horvath, S.; Gladyshev, V. N.; White, J. P. Multi-omic rejuvenation and lifespan extension on exposure to youthful circulation. Nature Aging 2023, 3(8), 948–964. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; Araoka, T.; Vazquez-Ferrer, E.; Donoso, D.; Roman, J. L.; Xu, J.; Rodriguez Esteban, C.; Nuñez, G.; Nuñez Delicado, E.; Campistol, J. M.; Izpisua Belmonte, J. C. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167(7), 1719–1733.e12. [Google Scholar] [CrossRef] [PubMed]
- Paine, P. T.; Rechsteiner, C.; Morandini, F.; Desdín-Micó, G.; Mrabti, C.; Parras, A.; Haghani, A.; Brooke, R.; Horvath, S.; Seluanov, A.; Gorbunova, V.; Ocampo, A. Initiation phase cellular reprogramming ameliorates DNA damage in the ERCC1 mouse model of premature aging. Frontiers in Aging 2024, 4. [Google Scholar] [CrossRef] [PubMed]
- Macip, C. C.; Hasan, R.; Hoznek, V.; Kim, J.; Lu, Y. R.; Metzger, L. E.; Sethna, S.; Davidsohn, N. Gene Therapy-Mediated Partial Reprogramming Extends Lifespan and Reverses Age-Related Changes in Aged Mice. Cellular Reprogramming 2024, 26(1), 24–32. [Google Scholar] [CrossRef] [PubMed]
- Browder, K. C.; Reddy, P.; Yamamoto, M.; Haghani, A.; Guillen, I. G.; Sahu, S.; Wang, C.; Luque, Y.; Prieto, J.; Shi, L.; Shojima, K.; Hishida, T.; Lai, Z.; Li, Q.; Choudhury, F. K.; Wong, W. R.; Liang, Y.; Sangaraju, D.; Sandoval, W.; Izpisua Belmonte, J. C. In vivo partial reprogramming alters age-associated molecular changes during physiological aging in mice. Nature Aging 2022, 2(3), 243–253. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, A.; Ghanegolmohammadi, F.; Wang, Y.; Leng, J.; Kreymerman, A.; Gamboa Varela, J.; Garbern, J.; Elwell, H.; Cao, F.; Ricci-Blair, E. M.; Liang, C.; Balamkundu, S.; Vidoudez, C.; DeMott, M. S.; Bedi, K.; Margulies, K. B.; Bennett, D. A.; Palmer, A. A.; Barkley-Levenson, A.; Dedon, P. C. Discovery adductomics provides a comprehensive portrait of tissue-, age- and sex-specific DNA modifications in rodents and humans. Nucleic Acids Research 2023, 51(20), 10829–10845. [Google Scholar] [CrossRef] [PubMed]
- Zhavoronkov, A.; Bhullar, B. Classifying aging as a disease in the context of ICD-11. Frontiers in Genetics 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Khaltourina, D.; Matveyev, Y.; Alekseev, A.; Cortese, F.; Ioviţă, A. Aging Fits the Disease Criteria of the International Classification of Diseases. Mechanisms of Ageing and Development 2020, 189, 111230. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Witkowski, J. M.; McElhaney, J.; Loeb, M.; Mitnitski, A.; Pawelec, G. Aging, frailty and age-related diseases. Biogerontology 2010, 11(5), 547–563. [Google Scholar] [CrossRef] [PubMed]
- Rattan, S. Aging is not a disease: implications for intervention; Aging and Disease, 2014. [Google Scholar] [CrossRef] [PubMed]
- Rabheru, K.; Byles, J. E.; Kalache, A. How “old age” was withdrawn as a diagnosis from ICD-11. The Lancet Healthy Longevity 2022, 3(7), e457–e459. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T. A. Evolving Views of DNA Replication (In)Fidelity. Cold Spring Harbor Symposia on Quantitative Biology 2009, 74(0), 91–101. [Google Scholar] [CrossRef] [PubMed]
- Muller, H. J. The relation of recombination to mutational advance. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 1964, 1(1), 2–9. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E. J.; Madden, R.; Paul, G.; Taddei, F. Aging and death in an organism that reproduces by morphologically symmetric division. PLoS Biology 2005, 3(2), e45. [Google Scholar] [CrossRef] [PubMed]
- Piraino, S.; Boero, F.; Aeschbach, B.; Schmid, V. Reversing the Life Cycle: Medusae Transforming into Polyps and Cell Transdifferentiation in Turritopsis nutricula (Cnidaria, Hydrozoa). The Biological Bulletin 1996, 190(3), 302–312. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Torner, M.; Carrero, D.; Pérez-Silva, J. G.; Álvarez-Puente, D.; Roiz-Valle, D.; Bretones, G.; Rodríguez, D.; Maeso, D.; Mateo-González, E.; Español, Y.; Mariño, G.; Acuña, J. L.; Quesada, V.; López-Otín, C. Comparative genomics of mortal and immortal cnidarians unveils novel keys behind rejuvenation. Proceedings of the National Academy of Sciences 2022, 119(36). [Google Scholar] [CrossRef] [PubMed]
- Póti, Á.; Szüts, D.; Vermezovic, J. Mutational profile of the regenerative process and de novo genome assembly of the planarian Schmidtea polychroa. Nucleic Acids Research 2024, 52(4), 1779–1792. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T. B. L.; Holliday, R. The evolution of ageing and longevity. Proceedings of the Royal Society of London. Series B. Biological Sciences 1979, 205(1161), 531–546. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, N.; Kaneko, K.; Koonin, E. V. Horizontal Gene Transfer Can Rescue Prokaryotes from Muller’s Ratchet: Benefit of DNA from Dead Cells and Population Subdivision. G3 Genes|Genomes|Genetics 2014, 4(2), 325–339. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, O.; Barrick, J. E.; Ribeck, N.; Deatherage, D. E.; Blanchard, J. L.; Dasgupta, A.; Wu, G. C.; Wielgoss, S.; Cruveiller, S.; Médigue, C.; Schneider, D.; Lenski, R. E. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature 2016, 536(7615), 165–170. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T. B. L. Deciphering death: a commentary on Gompertz (1825) ‘On the nature of the function expressive of the law of human mortality, and on a new mode of determining the value of life contingencies’. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 370, (1666). [Google Scholar] [CrossRef] [PubMed]
- Jones, O. R.; Scheuerlein, A.; Salguero-Gómez, R.; Camarda, C. G.; Schaible, R.; Casper, B. B.; Dahlgren, J. P.; Ehrlén, J.; García, M. B.; Menges, E. S.; Quintana-Ascencio, P. F.; Caswell, H.; Baudisch, A.; Vaupel, J. W. Diversity of ageing across the tree of life. Nature 2014, 505(7482), 169–173. [Google Scholar] [CrossRef] [PubMed]
- W. Vaupel, J., Baudisch, A., Dölling, M., A. Roach, D., & Gampe, J. (2004). The case for negative senescence. Theoretical Population Biology, 65(4), 339–351. [CrossRef] [PubMed]
- Roark, E. B.; Guilderson, T. P.; Dunbar, R. B.; Fallon, S. J.; Mucciarone, D. A. Extreme longevity in proteinaceous deep-sea corals. Proceedings of the National Academy of Sciences 2009, 106(13), 5204–5208. [Google Scholar] [CrossRef] [PubMed]
- Martı́nez, D. E. Mortality Patterns Suggest Lack of Senescence in Hydra. Experimental Gerontology 1998, 33(3), 217–225. [Google Scholar] [CrossRef] [PubMed]
- Ruby, J. G.; Smith, M.; Buffenstein, R. Naked mole-rat mortality rates defy Gompertzian laws by not increasing with age. ELife 2018, 7, e31157. [Google Scholar] [CrossRef] [PubMed]
- Bosch, T. C. G. Hydra and the evolution of stem cells. BioEssays 2009, 31(4), 478–486. [Google Scholar] [CrossRef] [PubMed]
- Lu, A. T.; Fei, Z.; Haghani, A.; Robeck, T. R.; Zoller, J. A.; Li, C. Z.; Lowe, R.; Yan, Q.; Zhang, J.; Vu, H.; Ablaeva, J.; Acosta-Rodriguez, V. A.; Adams, D. M.; Almunia, J.; Aloysius, A.; Ardehali, R.; Arneson, A.; Baker, C. S.; Banks, G.; Horvath, S. Universal DNA methylation age across mammalian tissues. Nature Aging 2023, 3(9), 1144–1166. [Google Scholar] [CrossRef] [PubMed]
- Kerepesi, C.; Meer, M. V.; Ablaeva, J.; Amoroso, V. G.; Lee, S.-G.; Zhang, B.; Gerashchenko, M. V.; Trapp, A.; Yim, S. H.; Lu, A. T.; Levine, M. E.; Seluanov, A.; Horvath, S.; Park, T. J.; Gorbunova, V.; Gladyshev, V. N. Epigenetic aging of the demographically non-aging naked mole-rat. Nature Communications 2022, 13(1), 355. [Google Scholar] [CrossRef] [PubMed]
- The evolution of ageing and longevity. Proceedings of the Royal Society of London. Series B. Biological Sciences 1979, 205(1161), 531–546. [CrossRef] [PubMed]
- Reznick, D. N.; Bryant, M. J.; Roff, D.; Ghalambor, C. K.; Ghalambor, D. E. Effect of extrinsic mortality on the evolution of senescence in guppies. Nature 2004, 431(7012), 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Seifert, U. Stochastic thermodynamics, fluctuation theorems and molecular machines. Reports on Progress in Physics 2012, 75(12), 126001. [Google Scholar] [CrossRef] [PubMed]
- Goldbeter, A. Dissipative structures in biological systems: bistability, oscillations, spatial patterns and waves. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences 2018, 376(2124), 20170376. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, E.; Penrose, R. What is Life? Cambridge University Press, 1992. [Google Scholar] [CrossRef]
- Sartori, P.; Pigolotti, S. Kinetic versus Energetic Discrimination in Biological Copying. Physical Review Letters 2013, 110(18), 188101. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T. B. L.; Austad, S. N. Why do we age? Nature 2000, 408(6809), 233–238. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M. P.; Joyce, G. F. The Origins of the RNA World. Cold Spring Harbor Perspectives in Biology 2012, 4(5), a003608–a003608. [Google Scholar] [CrossRef] [PubMed]
- Joyce, G. F. The antiquity of RNA-based evolution. Nature 2002, 418(6894), 214–221. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R. B. [URE3] as an Altered URE2 Protein: Evidence for a Prion Analog in Saccharomyces cerevisiae. Science 1994, 264(5158), 566–569. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S. B. Novel Proteinaceous Infectious Particles Cause Scrapie. Science 1982, 216(4542), 136–144. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447(7143), 396–398. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. The Origins of Genome Architecture; Sinauer, 2007; Available online: https://books.google.com/books?id=7NAPAQAAMAAJ.
- Hoeijmakers, J. H. J. DNA Damage, Aging, and Cancer. New England Journal of Medicine 2009, 361(15), 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, S. D.; Kunkel, T. A. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Research 2008, 18(1), 148–161. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Popodi, E.; Tang, H.; Foster, P. L. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proceedings of the National Academy of Sciences 2012, 109(41). [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T. A.; Erie, D. A. DNA MISMATCH REPAIR. Annual Review of Biochemistry 2005, 74(1), 681–710. [Google Scholar] [CrossRef] [PubMed]
- Lee-Six, H.; Øbro, N. F.; Shepherd, M. S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Osborne, R. J.; Huntly, B. J. P.; Martincorena, I.; Anderson, E.; O’Neill, L.; Stratton, M. R.; Laurenti, E.; Green, A. R.; Kent, D. G.; Campbell, P. J. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561(7724), 473–478. [Google Scholar] [CrossRef] [PubMed]
- Polyanskiy, Y.; Poor, H. V.; Verdu, S. Channel Coding Rate in the Finite Blocklength Regime. IEEE Transactions on Information Theory 2010, 56(5), 2307–2359. [Google Scholar] [CrossRef]
- Murugan, A.; Huse, D. A.; Leibler, S. Speed, dissipation, and error in kinetic proofreading. Proceedings of the National Academy of Sciences 2012, 109(30), 12034–12039. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C. O. Quasispecies theory in the context of population genetics. BMC Evolutionary Biology 2005, 5(1), 44. [Google Scholar] [CrossRef] [PubMed]
- Bull, J. J.; Sanjuán, R.; Wilke, C. O. Theory of Lethal Mutagenesis for Viruses. Journal of Virology 2007, 81(6), 2930–2939. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B. K.; Berger, S. L.; Brunet, A.; Campisi, J.; Cuervo, A. M.; Epel, E. S.; Franceschi, C.; Lithgow, G. J.; Morimoto, R. I.; Pessin, J. E.; Rando, T. A.; Richardson, A.; Schadt, E. E.; Wyss-Coray, T.; Sierra, F. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159(4), 709–713. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annual Review of Pathology: Mechanisms of Disease 2010, 5(1), 99–118. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nature Reviews Endocrinology 2018, 14(10), 576–590. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J. N.; Rovio, A. T.; Bruder, C. E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; Törnell, J.; Jacobs, H. T.; Larsson, N.-G. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429(6990), 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G. C.; Hiona, A.; Pugh, T. D.; Someya, S.; Panzer, K.; Wohlgemuth, S. E.; Hofer, T.; Seo, A. Y.; Sullivan, R.; Jobling, W. A.; Morrow, J. D.; Van Remmen, H.; Sedivy, J. M.; Yamasoba, T.; Tanokura, M.; Weindruch, R.; Leeuwenburgh, C.; Prolla, T. A. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309(5733), 481–484. [Google Scholar] [CrossRef] [PubMed]
- Craver, C. F. Explaining the Brain: Mechanisms and the Mosaic Unity of Neuroscience; Clarendon Press, 2007; Available online: https://books.google.com/books?id=NhNREAAAQBAJ.
- Machamer, P.; Darden, L.; Craver, C. F. Thinking about Mechanisms. Philosophy of Science 2000, 67(1), 1–25. Available online: http://www.jstor.org/stable/188611. [CrossRef]
- Woese, C. R.; Kandler, O.; Wheelis, M. L. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proceedings of the National Academy of Sciences 1990, 87(12), 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Watson, J. D. Molecular Biology of the Gene; Pearson/Benjamin Cummings, 2004; Available online: https://books.google.com/books?id=S3sEcX7Fbt0C.
- Griffith, F. The Significance of Pneumococcal Types. Journal of Hygiene 1928, 27(2), 113–159. [Google Scholar] [CrossRef] [PubMed]
- Avery, O. T.; MacLeod, C. M.; McCarty, M. STUDIES ON THE CHEMICAL NATURE OF THE SUBSTANCE INDUCING TRANSFORMATION OF PNEUMOCOCCAL TYPES. Journal of Experimental Medicine 1944, 79(2), 137–158. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. Journal of Molecular Biology 1961, 3(3), 318–356. [Google Scholar] [CrossRef] [PubMed]
- Meselson, M.; Stahl, F. W. The replication of DNA in Escherichia coli. Proceedings of the National Academy of Sciences 1958, 44(7), 671–682. [Google Scholar] [CrossRef] [PubMed]
- Nirenberg, M. W.; Matthaei, J. H. The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proceedings of the National Academy of Sciences 1961, 47(10), 1588–1602. [Google Scholar] [CrossRef] [PubMed]
- Arber, W. Host-Controlled Modification of Bacteriophage. Annual Review of Microbiology 1965, 19(1), 365–378. [Google Scholar] [CrossRef] [PubMed]
- Smith, H. O.; Welcox, K. W. A Restriction enzyme from Hemophilus influenzae. Journal of Molecular Biology 1970, 51(2), 379–391. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D. A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315(5819), 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K. A.; Tomita, M.; Wanner, B. L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular Systems Biology 2006, 2(1). [Google Scholar] [CrossRef] [PubMed]
- Wagers, A. J. The Stem Cell Niche in Regenerative Medicine. Cell Stem Cell 2012, 10(4), 362–369. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Goodell, M. A.; Rando, T. A. Ageing and rejuvenation of tissue stem cells and their niches. Nature Reviews Molecular Cell Biology 2023, 24(1), 45–62. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. SATELLITE CELL OF SKELETAL MUSCLE FIBERS. The Journal of Cell Biology 1961, 9(2), 493–495. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Price, F.; Rudnicki, M. A. Satellite Cells and the Muscle Stem Cell Niche. Physiological Reviews 2013, 93(1), 23–67. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126(4), 663–676. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T. B. L. Understanding the Odd Science of Aging. Cell 2005, 120(4), 437–447. [Google Scholar] [CrossRef] [PubMed]
- Bjorksten, J. A COMMON MOLECULAR BASIS FOR THE AGING SYNDROME. Journal of the American Geriatrics Society 1958, 6(10), 740–748. [Google Scholar] [CrossRef] [PubMed]
- Zieman, S. J.; Melenovsky, V.; Kass, D. A. Mechanisms, Pathophysiology, and Therapy of Arterial Stiffness. Arteriosclerosis, Thrombosis, and Vascular Biology 2005, 25(5), 932–943. [Google Scholar] [CrossRef] [PubMed]
- Verzijl, N.; DeGroot, J.; Thorpe, S. R.; Bank, R. A.; Shaw, J. N.; Lyons, T. J.; Bijlsma, J. W. J.; Lafeber, F. P. J. G.; Baynes, J. W.; TeKoppele, J. M. Effect of Collagen Turnover on the Accumulation of Advanced Glycation End Products. Journal of Biological Chemistry 2000, 275(50), 39027–39031. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. D. M. ADVANCED PROTEIN GLYCOSYLATION IN DIABETES AND AGING. Annual Review of Medicine 1995, 46(1), 223–234. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M.; Vlassara, H.; Kooney, A.; Ulrich, P.; Cerami, A. Aminoguanidine Prevents Diabetes-Induced Arterial Wall Protein Cross-Linking. Science 1986, 232(4758), 1629–1632. [Google Scholar] [CrossRef] [PubMed]
- Semba, R. D.; Nicklett, E. J.; Ferrucci, L. Does Accumulation of Advanced Glycation End Products Contribute to the Aging Phenotype? The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 2010, 65A(9), 963–975. [Google Scholar] [CrossRef] [PubMed]
- Kahn, A. Regaining Lost Youth: The Controversial and Colorful Beginnings of Hormone Replacement Therapy in Aging. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 2005, 60(2), 142–147. [Google Scholar] [CrossRef] [PubMed]
- Biagetti, B.; Puig-Domingo, M. Age-Related Hormones Changes and Its Impact on Health Status and Lifespan. Aging and Disease 2023, 14(3), 605. [Google Scholar] [CrossRef] [PubMed]
- Stott, D. J.; Rodondi, N.; Kearney, P. M.; Ford, I.; Westendorp, R. G. J.; Mooijaart, S. P.; Sattar, N.; Aubert, C. E.; Aujesky, D.; Bauer, D. C.; Baumgartner, C.; Blum, M. R.; Browne, J. P.; Byrne, S.; Collet, T.-H.; Dekkers, O. M.; den Elzen, W. P. J.; Du Puy, R. S.; Ellis, G.; Gussekloo, J. Thyroid Hormone Therapy for Older Adults with Subclinical Hypothyroidism. New England Journal of Medicine 2017, 376(26), 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bravata, D. M.; Olkin, I.; Nayak, S.; Roberts, B.; Garber, A. M.; Hoffman, A. R. Systematic Review: The Safety and Efficacy of Growth Hormone in the Healthy Elderly. Annals of Internal Medicine 2007, 146(2), 104–115. [Google Scholar] [CrossRef] [PubMed]
- Writing Group for the Women’s Health Initiative Investigators. Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal Women: Principal Results From the Women’s Health Initiative Randomized Controlled Trial. JAMA: The Journal of the American Medical Association 2002, 288(3), 321–333. [Google Scholar] [CrossRef]
- Cushman, M. Estrogen Plus Progestin and Risk of Venous Thrombosis. JAMA 2004, 292(13), 1573. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C. J. The genetics of ageing. Nature 2010, 464(7288), 504–512. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A. Growth Hormone and Aging: Updated Review. The World Journal of Men’s Health 2019, 37(1), 19. [Google Scholar] [CrossRef]
- Samaras, N.; Papadopoulou, M.-A.; Samaras, D.; Ongaro, F. Off-label use of hormones as an antiaging strategy: a review. Clinical Interventions in Aging 2014, 1175. [Google Scholar] [CrossRef]
- Pataky, M. W.; Young, W. F.; Nair, K. S. Hormonal and Metabolic Changes of Aging and the Influence of Lifestyle Modifications. Mayo Clinic Proceedings 2021, 96(3), 788–814. [Google Scholar] [CrossRef] [PubMed]
- Walford, R. L. The immunologic theory of aging. Immunological Reviews 1969, 2(1), 171. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. The twilight of immunity: emerging concepts in aging of the immune system. Nature Immunology 2018, 19(1), 10–19. [Google Scholar] [CrossRef] [PubMed]
- Williams, G. C. Pleiotropy, Natural Selection, and the Evolution of Senescence. Science of Aging Knowledge Environment 2001, 2001(1). [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: when bad things happen to good cells. Nature Reviews Molecular Cell Biology 2007, 8(9), 729–740. [Google Scholar] [CrossRef] [PubMed]
- Baker, D. J.; Childs, B. G.; Durik, M.; Wijers, M. E.; Sieben, C. J.; Zhong, J.; A. Saltness, R.; Jeganathan, K. B.; Verzosa, G. C.; Pezeshki, A.; Khazaie, K.; Miller, J. D.; van Deursen, J. M. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530(7589), 184–189. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A. C.; Ding, H.; Giorgadze, N.; Palmer, A. K.; Ikeno, Y.; Hubbard, G. B.; Lenburg, M.; O’Hara, S. P.; LaRusso, N. F.; Miller, J. D.; Roos, C. M.; Verzosa, G. C.; LeBrasseur, N. K.; Wren, J. D.; Farr, J. N.; Khosla, S.; Kirkland, J. L. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 2015, 14(4), 644–658. [Google Scholar] [CrossRef] [PubMed]
- Rose, M. R. Evolutionary Biology of Aging; Oxford University Press, 1994; Available online: https://books.google.com/books?id=LZ1mY1G-myoC.
- Smith, J. M.; Szathmary, E. The Major Transitions in Evolution; OUP Oxford, 1997; Available online: https://books.google.com/books?id=dUzLngEACAAJ.
- Abegglen, L. M.; Caulin, A. F.; Chan, A.; Lee, K.; Robinson, R.; Campbell, M. S.; Kiso, W. K.; Schmitt, D. L.; Waddell, P. J.; Bhaskara, S.; Jensen, S. T.; Maley, C. C.; Schiffman, J. D. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA 2015, 314(17), 1850. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, J. M.; Lynch, V. J. Pervasive duplication of tumor suppressors in Afrotherians during the evolution of large bodies and reduced cancer risk. ELife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.; Semeiks, J.; Webb, A. E.; Li, Y. I.; Quesada, V.; Craig, T.; Madsen, L. B.; van Dam, S.; Brawand, D.; Marques, P. I.; Michalak, P.; Kang, L.; Bhak, J.; Yim, H.-S.; Grishin, N. V.; Nielsen, N. H.; Heide-Jørgensen, M. P.; Oziolor, E. M.; Matson, C. W.; de Magalhães, J. P. Insights into the Evolution of Longevity from the Bowhead Whale Genome. Cell Reports 2015, 10(1), 112–122. [Google Scholar] [CrossRef] [PubMed]
- Firsanov, D.; Zacher, M.; Tian, X.; Sformo, T. L.; Zhao, Y.; Tombline, G.; Lu, J. Y.; Zheng, Z.; Perelli, L.; Gurreri, E.; Zhang, L.; Guo, J.; Korotkov, A.; Volobaev, V.; Biashad, S. A.; Zhang, Z.; Heid, J.; Maslov, A. Y.; Sun, S.; Gorbunova, V. Evidence for improved DNA repair in the long-lived bowhead whale. Nature 2025, 648(8094), 717–725. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Pavard, S.; Promislow, D. E. L. The Size–Life Span Trade-Off Decomposed: Why Large Dogs Die Young. The American Naturalist 2013, 181(4), 492–505. [Google Scholar] [CrossRef] [PubMed]
- Sutter, N. B.; Bustamante, C. D.; Chase, K.; Gray, M. M.; Zhao, K.; Zhu, L.; Padhukasahasram, B.; Karlins, E.; Davis, S.; Jones, P. G.; Quignon, P.; Johnson, G. S.; Parker, H. G.; Fretwell, N.; Mosher, D. S.; Lawler, D. F.; Satyaraj, E.; Nordborg, M.; Lark, K. G.; Ostrander, E. A. A Single IGF1 Allele Is a Major Determinant of Small Size in Dogs. Science 2007, 316(5821), 112–115. [Google Scholar] [CrossRef] [PubMed]
- Manov, I.; Hirsh, M.; Iancu, T. C.; Malik, A.; Sotnichenko, N.; Band, M.; Avivi, A.; Shams, I. Pronounced cancer resistance in a subterranean rodent, the blind mole-rat, Spalax: in vivo and in vitroevidence. BMC Biology 2013, 11(1), 91. [Google Scholar] [CrossRef] [PubMed]
- Buffenstein, R. Negligible senescence in the longest living rodent, the naked mole-rat: insights from a successfully aging species. Journal of Comparative Physiology B 2008, 178(4), 439–445. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T. W.; Buffenstein, R.; Hulbert, A. J. Membrane phospholipid composition may contribute to exceptional longevity of the naked mole-rat (Heterocephalus glaber): A comparative study using shotgun lipidomics. Experimental Gerontology 2007, 42(11), 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, A. J.; Faulks, S. C.; Buffenstein, R. Oxidation-Resistant Membrane Phospholipids Can Explain Longevity Differences Among the Longest-Living Rodents and Similarly-Sized Mice. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 2006, 61(10), 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Andziak, B.; O’Connor, T. P.; Buffenstein, R. Antioxidants do not explain the disparate longevity between mice and the longest-living rodent, the naked mole-rat. Mechanisms of Ageing and Development 2005, 126(11), 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. N.; Andziak, B.; Yang, T.; Buffenstein, R. The Naked Mole-Rat Response to Oxidative Stress: Just Deal with It. Antioxidants & Redox Signaling 2013, 19(12), 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E. H.; Greider, C. W.; Szostak, J. W. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nature Medicine 2006, 12(10), 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Karlseder, J.; Broccoli, D.; Dai, Y.; Hardy, S.; de Lange, T. p53- and ATM-Dependent Apoptosis Induced by Telomeres Lacking TRF2. Science 1999, 283(5406), 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, A. G.; Ouellette, M.; Frolkis, M.; Holt, S. E.; Chiu, C.-P.; Morin, G. B.; Harley, C. B.; Shay, J. W.; Lichtsteiner, S.; Wright, W. E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279(5349), 349–352. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M. A. Telomeres and human disease: ageing, cancer and beyond. Nature Reviews Genetics 2005, 6(8), 611–622. [Google Scholar] [CrossRef]
- Gomes, N. M. V.; Ryder, O. A.; Houck, M. L.; Charter, S. J.; Walker, W.; Forsyth, N. R.; Austad, S. N.; Venditti, C.; Pagel, M.; Shay, J. W.; Wright, W. E. Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell 2011, 10(5), 761–768. [Google Scholar] [CrossRef] [PubMed]
- Demanelis, K.; Jasmine, F.; Chen, L. S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; Ramirez, E.; Oliva, M.; Kim-Hellmuth, S.; Stranger, B. E.; Lai, T.-P.; Aviv, A.; Ardlie, K. G.; Aguet, F.; Ahsan, H.; Volpi, S. Determinants of telomere length across human tissues. Science 2020, 369(6509). [Google Scholar] [CrossRef] [PubMed]
- Hipp, M. S.; Kasturi, P.; Hartl, F. U. The proteostasis network and its decline in ageing. Nature Reviews Molecular Cell Biology 2019, 20(7), 421–435. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R. C.; Dillin, A. Aging as an Event of Proteostasis Collapse. Cold Spring Harbor Perspectives in Biology 2011, 3(5), a004440–a004440. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R. I. Repression of the Heat Shock Response Is a Programmed Event at the Onset of Reproduction. Molecular Cell 2015, 59(4), 639–650. [Google Scholar] [CrossRef] [PubMed]
- Toyama, B. H.; Savas, J. N.; Park, S. K.; Harris, M. S.; Ingolia, N. T.; Yates, J. R.; Hetzer, M. W. Identification of Long-Lived Proteins Reveals Exceptional Stability of Essential Cellular Structures. Cell 2013, 154(5), 971–982. [Google Scholar] [CrossRef] [PubMed]
- Truscott, R. J. W.; Schey, K. L.; Friedrich, M. G. Old Proteins in Man: A Field in its Infancy. Trends in Biochemical Sciences 2016, 41(8), 654–664. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Van Meter, M.; Ablaeva, J.; Ke, Z.; Gonzalez, R. S.; Taguchi, T.; De Cecco, M.; Leonova, K. I.; Kogan, V.; Helfand, S. L.; Neretti, N.; Roichman, A.; Cohen, H. Y.; Meer, M. V.; Gladyshev, V. N.; Antoch, M. P.; Gudkov, A. V.; Sedivy, J. M.; Seluanov, A.; Gorbunova, V. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metabolism 2019, 29(4), 871–885.e5. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Woodbine, L.; Marchetti, C.; Dawson, S.; Lambe, T.; Liu, C.; Rodrigues, N. P.; Crockford, T. L.; Cabuy, E.; Vindigni, A.; Enver, T.; Bell, J. I.; Slijepcevic, P.; Goodnow, C. C.; Jeggo, P. A.; Cornall, R. J. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 2007, 447(7145), 686–690. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D. J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I. L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447(7145), 725–729. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K. J.; Hämäläinen, R. H.; Yatsuga, S.; Uutela, M.; Terzioglu, M.; Götz, A.; Forsström, S.; Salven, P.; Angers-Loustau, A.; Kopra, O. H.; Tyynismaa, H.; Larsson, N.-G.; Wartiovaara, K.; Prolla, T.; Trifunovic, A.; Suomalainen, A. Somatic Progenitor Cell Vulnerability to Mitochondrial DNA Mutagenesis Underlies Progeroid Phenotypes in Polg Mutator Mice. Cell Metabolism 2012, 15(1), 100–109. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G. C.; Hiona, A.; Pugh, T. D.; Someya, S.; Panzer, K.; Wohlgemuth, S. E.; Hofer, T.; Seo, A. Y.; Sullivan, R.; Jobling, W. A.; Morrow, J. D.; Van Remmen, H.; Sedivy, J. M.; Yamasoba, T.; Tanokura, M.; Weindruch, R.; Leeuwenburgh, C.; Prolla, T. A. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309(5733), 481–484. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A. T.; Dufour, E.; Khvorostov, I.; Spelbrink, J. N.; Wibom, R.; Jacobs, H. T.; Larsson, N.-G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proceedings of the National Academy of Sciences 2005, 102(50), 17993–17998. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.; Shabalina, I. G.; Prime, T. A.; Rogatti, S.; Kalinovich, A. V.; Hartley, R. C.; Budd, R. C.; Cannon, B.; Murphy, M. P. In vivo levels of mitochondrial hydrogen peroxide increase with age in mt DNA mutator mice. Aging Cell 2014, 13(4), 765–768. [Google Scholar] [CrossRef] [PubMed]
- Kovina, M. V.; Zuev, V. A.; Kagarlitskiy, G. O.; Khodarovich, Y. M. Effect on lifespan of high yield non-myeloablating transplantation of bone marrow from young to old mice. Frontiers in Genetics 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.; Zhang, C.; Van, H. Q. T.; Seino, T.; Zhang, Y. Reducing functionally defective old HSCs alleviates aging-related phenotypes in old recipient mice. Cell Research 2025, 35(1), 45–58. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M. C.; Sinclair, D. A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annual Review of Pathology: Mechanisms of Disease 2010, 5(1), 253–295. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D. G.; Schaffer, B. E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends in Cell Biology 2016, 26(3), 190–201. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; Fussi, H.; Deszcz, L.; Hartl, R.; Schraml, E.; Criollo, A.; Megalou, E.; Weiskopf, D.; Laun, P.; Heeren, G.; Madeo, F. Induction of autophagy by spermidine promotes longevity. Nature Cell Biology 2009, 11(11), 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K. F.; Yoon, M. J.; Imai, S. Nicotinamide Mononucleotide, a Key NAD+ Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metabolism 2011, 14(4), 528–536. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD + in aging, metabolism, and neurodegeneration. Science 2015, 350(6265), 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; van der Pluijm, I.; Moorhouse, M. J.; Kosteas, T.; Robinson, A. R.; Suh, Y.; Breit, T. M.; van Steeg, H.; Niedernhofer, L. J.; van IJcken, W.; Bartke, A.; Spindler, S. R.; Hoeijmakers, J. H. J.; van der Horst, G. T. J.; Garinis, G. A. Delayed and Accelerated Aging Share Common Longevity Assurance Mechanisms. PLoS Genetics 2008, 4(8), e1000161. [Google Scholar] [CrossRef] [PubMed]
- Villeda, S. A.; Plambeck, K. E.; Middeldorp, J.; Castellano, J. M.; Mosher, K. I.; Luo, J.; Smith, L. K.; Bieri, G.; Lin, K.; Berdnik, D.; Wabl, R.; Udeochu, J.; Wheatley, E. G.; Zou, B.; Simmons, D. A.; Xie, X. S.; Longo, F. M.; Wyss-Coray, T. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nature Medicine 2014, 20(6), 659–663. [Google Scholar] [CrossRef] [PubMed]
- Rebo, J.; Mehdipour, M.; Gathwala, R.; Causey, K.; Liu, Y.; Conboy, M. J.; Conboy, I. M. A single heterochronic blood exchange reveals rapid inhibition of multiple tissues by old blood. Nature Communications 2016, 7(1), 13363. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.; Romano, S.; Ansorge, R.; Aboelnour, A.; Le Gall, G.; Savva, G. M.; Pontifex, M. G.; Telatin, A.; Baker, D.; Jones, E.; Vauzour, D.; Rudder, S.; Blackshaw, L. A.; Jeffery, G.; Carding, S. R. Fecal microbiota transfer between young and aged mice reverses hallmarks of the aging gut, eye, and brain. Microbiome 2022, 10(1), 68. [Google Scholar] [CrossRef] [PubMed]
- Curtis, H. J. A Composite Theory of Aging. The Gerontologist 1966, 6(3 Part 1), 143–149. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biology 2013, 14(10), 3156. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J.; Suh, Y. Genome Instability and Aging. Annual Review of Physiology 2013, 75(1), 645–668. [Google Scholar] [CrossRef] [PubMed]
- Pérez, V. I.; Van Remmen, H.; Bokov, A.; Epstein, C. J.; Vijg, J.; Richardson, A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 2009, 8(1), 73–75. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L. L.; Simonetti, R. G.; Gluud, C. Mortality in Randomized Trials of Antioxidant Supplements for Primary and Secondary Prevention. JAMA 2007, 297(8), 842. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J. K. Epigenetics and aging. Science Advances 2016, 2(7). [Google Scholar] [CrossRef] [PubMed]
- Kane, A. E.; Sinclair, D. A. Epigenetic changes during aging and their reprogramming potential. Critical Reviews in Biochemistry and Molecular Biology 2019, 54(1), 61–83. [Google Scholar] [CrossRef] [PubMed]
- Garinis, G. A.; van der Horst, G. T. J.; Vijg, J.; & H.J. Hoeijmakers, J. DNA damage and ageing: new-age ideas for an age-old problem. Nature Cell Biology 2008, 10(11), 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W. Origin of life: The RNA world. Nature 1986, 319(6055), 618–618. [Google Scholar] [CrossRef]
- Dawkins, R. The Selfish Gene; Oxford University Press, 2016; Available online: https://books.google.com/books?id=ekonDAAAQBAJ.
- Smith, J. M. The Concept of Information in Biology. Philosophy of Science 2000, 67(2), 177–194. [Google Scholar] [CrossRef]
- Orgel, L. E.; Crick, F. H. C. Selfish DNA: the ultimate parasite. Nature 1980, 284(5757), 604–607. [Google Scholar] [CrossRef] [PubMed]
- Ninio, J. Kinetic amplification of enzyme discrimination. Biochimie 1975, 57(5), 587–595. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C. H. The thermodynamics of computation—a review. International Journal of Theoretical Physics 1982, 21(12), 905–940. [Google Scholar] [CrossRef]
- Fisher, R. A. The genetical theory of natural selection; Clarendon Press, 1930. [Google Scholar] [CrossRef]
- Tenaillon, O. The Utility of Fisher’s Geometric Model in Evolutionary Genetics. Annual Review of Ecology, Evolution, and Systematics 2014, 45(1), 179–201. [Google Scholar] [CrossRef] [PubMed]
- Good, B. H.; McDonald, M. J.; Barrick, J. E.; Lenski, R. E.; Desai, M. M. The dynamics of molecular evolution over 60,000 generations. Nature 2017, 551(7678), 45–50. [Google Scholar] [CrossRef] [PubMed]
- Lenski, R. E.; Wiser, M. J.; Ribeck, N.; Blount, Z. D.; Nahum, J. R.; Morris, J. J.; Zaman, L.; Turner, C. B.; Wade, B. D.; Maddamsetti, R.; Burmeister, A. R.; Baird, E. J.; Bundy, J.; Grant, N. A.; Card, K. J.; Rowles, M.; Weatherspoon, K.; Papoulis, S. E.; Sullivan, R.; Hajela, N. Sustained fitness gains and variability in fitness trajectories in the long-term evolution experiment with Escherichia coli. Proceedings of the Royal Society B: Biological Sciences 2015, 282(1821), 20152292. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Raine, K. M.; Gerstung, M.; Dawson, K. J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M. R.; Campbell, P. J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171(5), 1029–1041.e21. [Google Scholar] [CrossRef] [PubMed]
- Lindner, A. B.; Madden, R.; Demarez, A.; Stewart, E. J.; Taddei, F. Asymmetric segregation of protein aggregates is associated with cellular aging and rejuvenation. Proceedings of the National Academy of Sciences 2008, 105(8), 3076–3081. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, J.; Uphoff, S.; Sherratt, D. J. Nonrandom segregation of sister chromosomes by Escherichia coli MukBEF. Proceedings of the National Academy of Sciences 2021, 118(33). [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Santos, A. L.; Xu, L.; Lotton, C.; Taddei, F.; Lindner, A. B. Temporal scaling of aging as an adaptive strategy of Escherichia coli. Science Advances 2019, 5(5). [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karin, O.; Mayo, A.; Song, X.; Chen, P.; Santos, A. L.; Lindner, A. B.; Alon, U. Damage dynamics and the role of chance in the timing of E. coli cell death. Nature Communications 2023, 14(1), 2209. [Google Scholar] [CrossRef] [PubMed]
- Drake, J. W. A constant rate of spontaneous mutation in DNA-based microbes. Proceedings of the National Academy of Sciences 1991, 88(16), 7160–7164. [Google Scholar] [CrossRef] [PubMed]
- Horibata, K.; Iwamoto, Y.; Kuraoka, I.; Jaspers, N. G. J.; Kurimasa, A.; Oshimura, M.; Ichihashi, M.; Tanaka, K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proceedings of the National Academy of Sciences 2004, 101(43), 15410–15415. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, G. T. J.; van Steeg, H.; Berg, R. J. W.; van Gool, A. J.; de Wit, J.; Weeda, G.; Morreau, H.; Beems, R. B.; van Kreijl, C. F.; de Gruijl, F. R.; Bootsma, D.; Hoeijmakers, J. H. J. Defective Transcription-Coupled Repair in Cockayne Syndrome B Mice Is Associated with Skin Cancer Predisposition. Cell 1997, 89(3), 425–435. [Google Scholar] [CrossRef] [PubMed]
- Laugel, V.; Dalloz, C.; Stary, A.; Cormier-Daire, V.; Desguerre, I.; Renouil, M.; Fourmaintraux, A.; Velez-Cruz, R.; Egly, J.-M.; Sarasin, A.; Dollfus, H. Deletion of 5′ sequences of the CSB gene provides insight into the pathophysiology of Cockayne syndrome. European Journal of Human Genetics 2008, 16(3), 320–327. [Google Scholar] [CrossRef] [PubMed]
- Chatre, L.; Biard, D. S. F.; Sarasin, A.; Ricchetti, M. Reversal of mitochondrial defects with CSB-dependent serine protease inhibitors in patient cells of the progeroid Cockayne syndrome. Proceedings of the National Academy of Sciences 2015, 112(22). [Google Scholar] [CrossRef] [PubMed]
- Crochemore, C.; Fernández-Molina, C.; Montagne, B.; Salles, A.; Ricchetti, M. CSB promoter downregulation via histone H3 hypoacetylation is an early determinant of replicative senescence. Nature Communications 2019, 10(1), 5576. [Google Scholar] [CrossRef] [PubMed]
- van der Pluijm, I.; Garinis, G. A.; Brandt, R. M. C.; Gorgels, T. G. M. F.; Wijnhoven, S. W.; Diderich, K. E. M.; de Wit, J.; Mitchell, J. R.; van Oostrom, C.; Beems, R.; Niedernhofer, L. J.; Velasco, S.; Friedberg, E. C.; Tanaka, K.; van Steeg, H.; Hoeijmakers, J. H. J.; van der Horst, G. T. J. Impaired Genome Maintenance Suppresses the Growth Hormone–Insulin-Like Growth Factor 1 Axis in Mice with Cockayne Syndrome. PLoS Biology 2006, 5(1), e2. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Enokido, Y.; Inamura, N.; Yoshino, M.; Nakatsu, Y.; van der Horst, G. T. J.; Hoeijmakers, J. H. J.; Tanaka, K.; Hatanaka, H. Early postnatal ataxia and abnormal cerebellar development in mice lacking Xeroderma pigmentosum Group A and Cockayne Syndrome Group B DNA repair genes. Proceedings of the National Academy of Sciences 2001, 98(23), 13379–13384. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J. I.; Crossan, G. P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; Stratton, M. R.; Patel, K. J. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553(7687), 171–177. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H. T.; de la Chapelle, A. Hereditary Colorectal Cancer. New England Journal of Medicine 2003, 348(10), 919–932. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.-B.; Howarth, K. M.; Domingo, E.; Jones, A. M.; Broderick, P.; Kemp, Z.; Spain, S. L.; Guarino, E.; Salguero, I.; Sherborne, A.; Chubb, D.; Carvajal-Carmona, L. G.; Ma, Y.; Kaur, K.; Dobbins, S.; Barclay, E.; Gorman, M.; Martin, L.; Tomlinson, I. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature Genetics 2013, 45(2), 136–144. [Google Scholar] [CrossRef] [PubMed]
- Cheadle, J. P.; Sampson, J. R. MUTYH-associated polyposis—From defect in base excision repair to clinical genetic testing. DNA Repair 2007, 6(3), 274–279. [Google Scholar] [CrossRef] [PubMed]
- David, S. S.; O’Shea, V. L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447(7147), 941–950. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G. P.; Bruce, A. T.; Ikeda, H.; Old, L. J.; Schreiber, R. D. Cancer immunoediting: from immunosurveillance to tumor escape. Nature Immunology 2002, 3(11), 991–998. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H. Senescence, ageing and death of the whole plant. New Phytologist 2013, 197(3), 696–711. [Google Scholar] [CrossRef] [PubMed]
- Stahle, D. W.; Edmondson, J. R.; Howard, I. M.; Robbins, C. R.; Griffin, R. D.; Carl, A.; Hall, C. B.; Stahle, D. K.; Torbenson, M. C. A. Longevity, climate sensitivity, and conservation status of wetland trees at Black River, North Carolina. Environmental Research Communications 2019, 1(4), 041002. [Google Scholar] [CrossRef]
- DeWoody, J.; Rowe, C. A.; Hipkins, V. D.; Mock, K. E. “Pando” Lives: Molecular Genetic Evidence of a Giant Aspen Clone in Central Utah. Western North American Naturalist 2008, 68(4), 493–497. [Google Scholar] [CrossRef]
- Schmid-Siegert, E.; Sarkar, N.; Iseli, C.; Calderon, S.; Gouhier-Darimont, C.; Chrast, J.; Cattaneo, P.; Schütz, F.; Farinelli, L.; Pagni, M.; Schneider, M.; Voumard, J.; Jaboyedoff, M.; Fankhauser, C.; Hardtke, C. S.; Keller, L.; Pannell, J. R.; Reymond, A.; Robinson-Rechavi, M.; Reymond, P. Low number of fixed somatic mutations in a long-lived oak tree. Nature Plants 2017, 3(12), 926–929. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R. Do plants have a segregated germline? PLOS Biology 2018, 16(5), e2005439. [Google Scholar] [CrossRef] [PubMed]
- Schaible, R.; Scheuerlein, A.; Dańko, M. J.; Gampe, J.; Martínez, D. E.; Vaupel, J. W. Constant mortality and fertility over age in Hydra. Proceedings of the National Academy of Sciences 2015, 112(51), 15701–15706. [Google Scholar] [CrossRef] [PubMed]
- Vlamakis, H.; Aguilar, C.; Losick, R.; Kolter, R. Control of cell fate by the formation of an architecturally complex bacterial community. Genes & Development 2008, 22(7), 945–953. [Google Scholar] [CrossRef] [PubMed]
- Shaulsky, G.; Kessin, R. H. The Cold War of the Social Amoebae. Current Biology 2007, 17(16), R684–R692. [Google Scholar] [CrossRef] [PubMed]
- Kirk, D. L. Germ–Soma Differentiation in Volvox. Developmental Biology 2001, 238(2), 213–223. [Google Scholar] [CrossRef] [PubMed]
- Crespi, B. J.; Yanega, D. The definition of eusociality. Behavioral Ecology 1995, 6(1), 109–115. [Google Scholar] [CrossRef]
- Jarvis, J. U. M. Eusociality in a Mammal: Cooperative Breeding in Naked Mole-Rat Colonies. Science 1981, 212(4494), 571–573. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dai, H.; Martos, S. N.; Xu, B.; Gao, Y.; Li, T.; Zhu, G.; Schones, D. E.; Wang, Z. Distinct roles of DNMT1-dependent and DNMT1-independent methylation patterns in the genome of mouse embryonic stem cells. Genome Biology 2015, 16(1), 115. [Google Scholar] [CrossRef] [PubMed]
- Probst, A. V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nature Reviews Molecular Cell Biology 2009, 10(3), 192–206. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z. D.; Meissner, A. DNA methylation: roles in mammalian development. Nature Reviews Genetics 2013, 14(3), 204–220. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R. A. Transgenerational Epigenetic Inheritance: Myths and Mechanisms. Cell 2014, 157(1), 95–109. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, Y.; Hara, Y.; Oka, Y.; Komine, O.; van den Heuvel, D.; Guo, C.; Daigaku, Y.; Isono, M.; He, Y.; Shimada, M.; Kato, K.; Jia, N.; Hashimoto, S.; Kotani, Y.; Miyoshi, Y.; Tanaka, M.; Sobue, A.; Mitsutake, N.; Suganami, T.; Ogi, T. Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 2020, 180(6), 1228–1244.e24. [Google Scholar] [CrossRef] [PubMed]
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P. B.; Steitz, T. A. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science 2000, 289(5481), 920–930. [Google Scholar] [CrossRef] [PubMed]
- Fica, S. M.; Tuttle, N.; Novak, T.; Li, N.-S.; Lu, J.; Koodathingal, P.; Dai, Q.; Staley, J. P.; Piccirilli, J. A. RNA catalyses nuclear pre-mRNA splicing. Nature 2013, 503(7475), 229–234. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, D. Expression of animal virus genomes. Bacteriological Reviews 1971, 35(3), 235–241. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press, 1983. [Google Scholar] [CrossRef]
- Freeland, S. J.; Hurst, L. D. The Genetic Code Is One in a Million. Journal of Molecular Evolution 1998, 47(3), 238–248. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D. A.; Wilke, C. O. Mistranslation-Induced Protein Misfolding as a Dominant Constraint on Coding-Sequence Evolution. Cell 2008, 134(2), 341–352. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Mason, S. P.; Barabási, A.-L.; Oltvai, Z. N. Lethality and centrality in protein networks. Nature 2001, 411(6833), 41–42. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B. Molecular Biology of the Cell; W.W. Norton, 2017; Available online: https://books.google.com/books?id=2xIwDwAAQBAJ.
- Jung, Y.; Brack, A. S. Cellular Mechanisms of Somatic Stem Cell Aging; 2014; pp. 405–438. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Dong, X.; Vijg, J. Age-related somatic mutation burden in human tissues. Frontiers in Aging 2022, 3. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, Y.; Maslov, A. Y.; Dong, X.; Vijg, J. SomaMutDB: a database of somatic mutations in normal human tissues. Nucleic Acids Research 2022, 50(D1), D1100–D1108. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I. Somatic mutation and clonal expansions in human tissues. Genome Medicine 2019, 11(1), 35. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D. C.; Fullam, A.; Alexandrov, L. B.; Tubio, J. M.; Stebbings, L.; Menzies, A.; Widaa, S.; Stratton, M. R.; Jones, P. H.; Campbell, P. J. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348(6237), 880–886. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Fowler, J. C.; Wabik, A.; Lawson, A. R. J.; Abascal, F.; Hall, M. W. J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M. R.; Fitzgerald, R. C.; Handford, P. A.; Campbell, P. J.; Saeb-Parsy, K.; Jones, P. H. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362(6417), 911–917. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Campbell, P. J. Somatic mutation in cancer and normal cells. Science 2015, 349(6255), 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Gaboriaud, J.; Wu, P.-Y. J. Insights into the Link between the Organization of DNA Replication and the Mutational Landscape. Genes 2019, 10(4), 252. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Polak, P.; Nemesh, J.; Michaelson, J. J.; Sebat, J.; Sunyaev, S. R.; McCarroll, S. A. Differential Relationship of DNA Replication Timing to Different Forms of Human Mutation and Variation. The American Journal of Human Genetics 2012, 91(6), 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G. C. Mechanisms of DNA damage, repair, and mutagenesis. Environmental and Molecular Mutagenesis 2017, 58(5), 235–263. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J. R. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harbor Perspectives in Biology 2013, 5(2), a012559–a012559. [Google Scholar] [CrossRef] [PubMed]
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mechanisms of Ageing and Development 2013, 134(5–6), 161–170. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, W.; Scott, R. J.; Rodgers, S.; Müller, H. J.; Cole, J.; Arlett, C. F.; Kleijer, W. J.; Bootsma, D.; Hoeijmakers, J. H.; Weeda, G. Clinical heterogeneity within xeroderma pigmentosum associated with mutations in the DNA repair and transcription gene ERCC3. American Journal of Human Genetics 1994, 54(2), 191–200. [Google Scholar] [PubMed]
- Tebbs, R. S.; Flannery, M. L.; Meneses, J. J.; Hartmann, A.; Tucker, J. D.; Thompson, L. H.; Cleaver, J. E.; Pedersen, R. A. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Developmental Biology 1999, 208(2), 513–529. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.-L.; D’Andrea, A. D. How the Fanconi Anemia Pathway Guards the Genome. Annual Review of Genetics 2009, 43(1), 223–249. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanna, J. J.; Kraemer, K. H. Shining a Light on Xeroderma Pigmentosum. Journal of Investigative Dermatology 2012, 132(3), 785–796. [Google Scholar] [CrossRef] [PubMed]
- Klungland, A.; Rosewell, I.; Hollenbach, S.; Larsen, E.; Daly, G.; Epe, B.; Seeberg, E.; Lindahl, T.; Barnes, D. E. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proceedings of the National Academy of Sciences 1999, 96(23), 13300–13305. [Google Scholar] [CrossRef] [PubMed]
- de Boer, J.; Andressoo, J. O.; de Wit, J.; Huijmans, J.; Beems, R. B.; van Steeg, H.; Weeda, G.; van der Horst, G. T. J.; van Leeuwen, W.; Themmen, A. P. N.; Meradji, M.; Hoeijmakers, J. H. J. Premature Aging in Mice Deficient in DNA Repair and Transcription. Science 2002, 296(5571), 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Al-Tassan, N.; Chmiel, N. H.; Maynard, J.; Fleming, N.; Livingston, A. L.; Williams, G. T.; Hodges, A. K.; Davies, D. R.; David, S. S.; Sampson, J. R.; Cheadle, J. P. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nature Genetics 2002, 30(2), 227–232. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, W.; Yang, K.; Umar, A.; Heyer, J.; Lau, K.; Fan, K.; Liedtke, W.; Cohen, P. E.; Kane, M. F.; Lipford, J. R.; Yu, N.; Crouse, G. F.; Pollard, J. W.; Kunkel, T.; Lipkin, M.; Kolodner, R.; Kucherlapati, R. Mutation in the Mismatch Repair Gene Msh6 Causes Cancer Susceptibility. Cell 1997, 91(4), 467–477. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Cerosaletti, K. M.; Girard, P.-M.; Dai, Y.; Stumm, M.; Kysela, B.; Hirsch, B.; Gennery, A.; Palmer, S. E.; Seidel, J.; Gatti, R. A.; Varon, R.; Oettinger, M. A.; Neitzel, H.; Jeggo, P. A.; Concannon, P. DNA Ligase IV Mutations Identified in Patients Exhibiting Developmental Delay and Immunodeficiency. Molecular Cell 2001, 8(6), 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Vogel, H.; Lim, D.-S.; Karsenty, G.; Finegold, M.; Hasty, P. Deletion of Ku86 causes early onset of senescence in mice. Proceedings of the National Academy of Sciences 1999, 96(19), 10770–10775. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D. A.; Smith, S.; Uziel, T.; Sfez, S.; Ashkenazi, M.; Pecker, I.; Frydman, M.; Harnik, R.; Patanjali, S. R.; Simmons, A.; Clines, G. A.; Sartiel, A.; Gatti, R. A.; Shiloh, Y. A Single Ataxia Telangiectasia Gene with a Product Similar to PI-3 Kinase. Science 1995, 268(5218), 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Weedon, M. N.; Ellard, S.; Prindle, M. J.; Caswell, R.; Allen, H. L.; Oram, R.; Godbole, K.; Yajnik, C. S.; Sbraccia, P.; Novelli, G.; Turnpenny, P.; McCann, E.; Goh, K. J.; Wang, Y.; Fulford, J.; McCulloch, L. J.; Savage, D. B.; O’Rahilly, S.; Kos, K.; Hattersley, A. T. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nature Genetics 2013, 45(8), 947–950. [Google Scholar] [CrossRef] [PubMed]
- Murdocca, M.; Spitalieri, P.; De Masi, C.; Udroiu, I.; Marinaccio, J.; Sanchez, M.; Talarico, R. V.; Fiorillo, C.; D’Adamo, M.; Sbraccia, P.; D’Apice, M. R.; Novelli, G.; Sgura, A.; Sangiuolo, F. Functional analysis of POLD1 p.ser605del variant: the aging phenotype of MDPL syndrome is associated with an impaired DNA repair capacity. Aging 2021, 13(4), 4926–4945. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 2003, 85(11), 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Nardo, T.; Oneda, R.; Spivak, G.; Vaz, B.; Mortier, L.; Thomas, P.; Orioli, D.; Laugel, V.; Stary, A.; Hanawalt, P. C.; Sarasin, A.; Stefanini, M. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proceedings of the National Academy of Sciences 2009, 106(15), 6209–6214. [Google Scholar] [CrossRef] [PubMed]
- Donnio, L.; Giglia-Mari, G. Keep calm and reboot – how cells restart transcription after DNA damage and DNA repair. FEBS Letters 2025, 599(2), 275–294. [Google Scholar] [CrossRef] [PubMed]
- Jauregui-Lozano, J.; Escobedo, S.; Easton, A.; Lanman, N. A.; Weake, V. M.; Hall, H. Proper control of R-loop homeostasis is required for maintenance of gene expression and neuronal function during aging. Aging Cell 2022, 21(2). [Google Scholar] [CrossRef] [PubMed]
- van Sluis, M.; Yu, Q.; van der Woude, M.; Gonzalo-Hansen, C.; Dealy, S. C.; Janssens, R. C.; Somsen, H. B.; Ramadhin, A. R.; Dekkers, D. H. W.; Wienecke, H. L.; Demmers, J. J. P. G.; Raams, A.; Davó-Martínez, C.; Llerena Schiffmacher, D. A.; van Toorn, M.; Häckes, D.; Thijssen, K. L.; Zhou, D.; Lammers, J. G.; Marteijn, J. A. Transcription-coupled DNA–protein crosslink repair by CSB and CRL4CSA-mediated degradation. Nature Cell Biology 2024, 26(5), 770–783. [Google Scholar] [CrossRef] [PubMed]
- Carnie, C. J.; Acampora, A. C.; Bader, A. S.; Erdenebat, C.; Zhao, S.; Bitensky, E.; van den Heuvel, D.; Parnas, A.; Gupta, V.; D’Alessandro, G.; Sczaniecka-Clift, M.; Weickert, P.; Aygenli, F.; Götz, M. J.; Cordes, J.; Esain-Garcia, I.; Melidis, L.; Wondergem, A. P.; Lam, S.; Stingele, J. Transcription-coupled repair of DNA–protein cross-links depends on CSA and CSB. Nature Cell Biology 2024, 26(5), 797–810. [Google Scholar] [CrossRef] [PubMed]
- Oka, Y.; Nakazawa, Y.; Shimada, M.; Ogi, T. Endogenous aldehyde-induced DNA–protein crosslinks are resolved by transcription-coupled repair. Nature Cell Biology 2024, 26(5), 784–796. [Google Scholar] [CrossRef] [PubMed]
- Mulderrig, L.; Garaycoechea, J. I.; Tuong, Z. K.; Millington, C. L.; Dingler, F. A.; Ferdinand, J. R.; Gaul, L.; Tadross, J. A.; Arends, M. J.; O’Rahilly, S.; Crossan, G. P.; Clatworthy, M. R.; Patel, K. J. Aldehyde-driven transcriptional stress triggers an anorexic DNA damage response. Nature 2021, 600(7887), 158–163. [Google Scholar] [CrossRef] [PubMed]
- Swenberg, J. A.; Lu, K.; Moeller, B. C.; Gao, L.; Upton, P. B.; Nakamura, J.; Starr, T. B. Endogenous versus Exogenous DNA Adducts: Their Role in Carcinogenesis, Epidemiology, and Risk Assessment. Toxicological Sciences 2011, 120 (Supplement 1), S130–S145. [Google Scholar] [CrossRef] [PubMed]
- van de Grint, J.; Raseta, M.; Brandt, R.; van Loon, Y.; Demmers, J.; Dealy, S.; Chang, J.; Hoeijmakers, J.; Pothof, J. Transcriptional stress in aging: integrating experimental data and modeling to quantify DNA damage accumulation. Frontiers in Molecular Biosciences 2025, 12. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, A.; Amarante, T. D.; Czarnecki, J.; Shooter, S.; Zou, X.; Glodzik, D.; Morganella, S.; Nanda, A. S.; Badja, C.; Koh, G.; Momen, S. E.; Georgakopoulos-Soares, I.; Dias, J. M. L.; Young, J.; Memari, Y.; Davies, H.; Nik-Zainal, S. A practical framework and online tool for mutational signature analyses show intertissue variation and driver dependencies. Nature Cancer 2020, 1(2), 249–263. [Google Scholar] [CrossRef] [PubMed]
- Kucab, J. E.; Zou, X.; Morganella, S.; Joel, M.; Nanda, A. S.; Nagy, E.; Gomez, C.; Degasperi, A.; Harris, R.; Jackson, S. P.; Arlt, V. M.; Phillips, D. H.; Nik-Zainal, S. A Compendium of Mutational Signatures of Environmental Agents. Cell 2019, 177(4), 821–836.e16. [Google Scholar] [CrossRef] [PubMed]
- Boysen, G.; Alexandrov, L. B.; Rahbari, R.; Nookaew, I.; Ussery, D.; Chao, M.-R.; Hu, C.-W.; Cooke, M. S. Investigating the origins of the mutational signatures in cancer. Nucleic Acids Research 2025, 53(1). [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nature Reviews Genetics 2014, 15(9), 585–598. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L. A.; Harris, C. C. Advances in Chemical Carcinogenesis: A Historical Review and Prospective. Cancer Research 2008, 68(17), 6863–6872. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, C. D.; Williams, N.; Dawson, K. J.; Watson, C.; Yousefzadeh, M. J.; Le, D.; Nyamondo, K.; Kodavali, S.; Cagan, A.; Waldvogel, S.; Zhang, X.; De La Fuente, J.; Leongamornlert, D.; Mitchell, E.; Florez, M. A.; Sosnowski, K.; Aguilar, R.; Martell, A.; Guzman, A.; Nangalia, J. Clonal dynamics and somatic evolution of haematopoiesis in mouse. Nature 2025, 641(8063), 681–689. [Google Scholar] [CrossRef] [PubMed]
- Jones, P. A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nature Reviews Genetics 2009, 10(11), 805–811. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R. Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Research 2006, 34(4), 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Slatkin, M. Epigenetic Inheritance and the Missing Heritability Problem. Genetics 2009, 182(3), 845–850. [Google Scholar] [CrossRef] [PubMed]
- Sale, J. E.; Lehmann, A. R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nature Reviews Molecular Cell Biology 2012, 13(3), 141–152. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M. T.; Humbert, R.; Rynes, E.; Thurman, R. E.; Haugen, E.; Wang, H.; Reynolds, A. P.; Sandstrom, R.; Qu, H.; Brody, J.; Shafer, A.; Neri, F.; Lee, K.; Kutyavin, T.; Stehling-Sun, S.; Johnson, A. K.; Canfield, T. K.; Giste, E.; Diegel, M.; Stamatoyannopoulos, J. A. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337(6099), 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Tewhey, R.; Kotliar, D.; Park, D. S.; Liu, B.; Winnicki, S.; Reilly, S. K.; Andersen, K. G.; Mikkelsen, T. S.; Lander, E. S.; Schaffner, S. F.; Sabeti, P. C. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 2016, 165(6), 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Polo, S. E.; Almouzni, G. Chromatin dynamics after DNA damage: The legacy of the access–repair–restore model. DNA Repair 2015, 36, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Polo, S. E.; Almouzni, G. Prime, Repair, Restore: The Active Role of Chromatin in the DNA Damage Response. Molecular Cell 2012, 46(6), 722–734. [Google Scholar] [CrossRef] [PubMed]
- Huang, J. C.; Svoboda, D. L.; Reardon, J. T.; Sancar, A. Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5’ and the 6th phosphodiester bond 3’ to the photodimer. Proceedings of the National Academy of Sciences 1992, 89(8), 3664–3668. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.; Park, H. J.; Celik, H.; Ostrander, E. L.; Reyes, J. M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; Guryanova, O. A.; Li, W.; Goodell, M. A.; Challen, G. A. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Reports 2018, 23(1), 1–10. [Google Scholar] [CrossRef] [PubMed]
- Capuano, F.; Mülleder, M.; Kok, R.; Blom, H. J.; Ralser, M. Cytosine DNA Methylation Is Found in Drosophila melanogaster but Absent in Saccharomyces cerevisiae, Schizosaccharomyces pombe, and Other Yeast Species. Analytical Chemistry 2014, 86(8), 3697–3702. [Google Scholar] [CrossRef] [PubMed]
- Simpson, V. J.; Johnson, T. E.; Hammen, R. F. Caenorhabditis elegans DNA does not contain 5-methylcytosine at any time during development or aging. Nucleic Acids Research 1986, 14(16), 6711–6719. [Google Scholar] [CrossRef] [PubMed]
- Seisenberger, S.; Andrews, S.; Krueger, F.; Arand, J.; Walter, J.; Santos, F.; Popp, C.; Thienpont, B.; Dean, W.; Reik, W. The Dynamics of Genome-wide DNA Methylation Reprogramming in Mouse Primordial Germ Cells. Molecular Cell 2012, 48(6), 849–862. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Huang, B.; Zhang, B.; Xiang, Y.; Du, Z.; Xu, Q.; Li, Y.; Wang, Q.; Ma, J.; Peng, X.; Xu, F.; Xie, W. Resetting Epigenetic Memory by Reprogramming of Histone Modifications in Mammals. Molecular Cell 2016, 63(6), 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Oberdoerffer, P.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, S.-K.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; Wright, S. M.; Mills, K. D.; Bonni, A.; Yankner, B. A.; Scully, R.; Prolla, T. A.; Alt, F. W.; Sinclair, D. A. SIRT1 Redistribution on Chromatin Promotes Genomic Stability but Alters Gene Expression during Aging. Cell 2008, 135(5), 907–918. [Google Scholar] [CrossRef] [PubMed]
- Mills, K. D.; Sinclair, D. A.; Guarente, L. MEC1-Dependent Redistribution of the Sir3 Silencing Protein from Telomeres to DNA Double-Strand Breaks. Cell 1999, 97(5), 609–620. [Google Scholar] [CrossRef] [PubMed]
- Grzelak, A.; Macierzyńska, E.; Bartosz, G. Accumulation of oxidative damage during replicative aging of the yeast Saccharomyces cerevisiae. Experimental Gerontology 2006, 41(9), 813–818. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Brown, A. J.; Malc, E. P.; Mieczkowski, P. A.; Smerdon, M. J.; Roberts, S. A.; Wyrick, J. J. Genome-wide maps of alkylation damage, repair, and mutagenesis in yeast reveal mechanisms of mutational heterogeneity. Genome Research 2017, 27(10), 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Robert, C.; Jang, Y.-Y.; Liu, H.; Sharkis, S.; Baylin, S. B.; Rassool, F. V. Human induced pluripotent cells resemble embryonic stem cells demonstrating enhanced levels of DNA repair and efficacy of nonhomologous end-joining. Mutation Research - Fundamental and Molecular Mechanisms of Mutagenesis 2011, 713(1–2), 8–17. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Swistowska, A. M.; Lee, J. W.; Liu, Y.; Liu, S.-T.; Da Cruz, A. B.; Rao, M.; de Souza-Pinto, N. C.; Zeng, X.; Bohr, V. A. Human Embryonic Stem Cells Have Enhanced Repair of Multiple Forms of DNA Damage. Stem Cells 2008, 26(9), 2266–2274. [Google Scholar] [CrossRef] [PubMed]
- Paine, P. T.; Nguyen, A.; Ocampo, A. Partial cellular reprogramming: A deep dive into an emerging rejuvenation technology. Aging Cell 2024, 23(2). [Google Scholar] [CrossRef] [PubMed]
- Timmons, J. A.; Brenner, C. The information theory of aging has not been tested. Cell 2024, 187(5), 1101–1102. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-H.; Hayano, M.; Rajman, L. A.; Sinclair, D. A. Response to: The information theory of aging has not been tested. Cell 2024, 187(5), 1103–1105. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; García-Muse, T. R Loops: From Transcription Byproducts to Threats to Genome Stability. Molecular Cell 2012, 46(2), 115–124. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K. A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, P. C.; Spivak, G. Transcription-coupled DNA repair: two decades of progress and surprises. Nature Reviews Molecular Cell Biology 2008, 9(12), 958–970. [Google Scholar] [CrossRef] [PubMed]
- Lindsey-Boltz, L. A.; Sancar, A. The Transcription-Repair Coupling Factor Mfd Prevents and Promotes Mutagenesis in a Context-Dependent Manner. Frontiers in Molecular Biosciences 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P. M. J.; Kunkel, T. A. Eukaryotic DNA Replication Fork. Annual Review of Biochemistry 2017, 86(1), 417–438. [Google Scholar] [CrossRef] [PubMed]
- Makridakis, N. M.; Reichardt, J. K. V. Translesion DNA Polymerases and Cancer. Frontiers in Genetics 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Flusberg, B. A.; Webster, D. R.; Lee, J. H.; Travers, K. J.; Olivares, E. C.; Clark, T. A.; Korlach, J.; Turner, S. W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nature Methods 2010, 7(6), 461–465. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C. J.; Talmane, L.; Luft, J.; Connelly, J.; Nicholson, M. D.; Verburg, J. C.; Pich, O.; Campbell, S.; Giaisi, M.; Wei, P.-C.; Sundaram, V.; Connor, F.; Ginno, P. A.; Sasaki, T.; Gilbert, D. M.; Aitken, S.; Arnedo-Pac, C.; Daunesse, M.; Drews, R. M.; Taylor, M. S. Strand-resolved mutagenicity of DNA damage and repair. Nature 2024, 630(8017), 744–751. [Google Scholar] [CrossRef] [PubMed]
- Charlet-Berguerand, N.; Feuerhahn, S.; Kong, S. E.; Ziserman, H.; Conaway, J. W.; Conaway, R.; Egly, J. M. RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. The EMBO Journal 2006, 25(23), 5481–5491. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Lima, L. C.; Veloso, A.; Paulsen, M. T.; Menck, C. F. M.; Ljungman, M. DNA repair and recovery of RNA synthesis following exposure to ultraviolet light are delayed in long genes. Nucleic Acids Research 2015, 43(5), 2744–2756. [Google Scholar] [CrossRef] [PubMed]
- Kisby, G. E.; Kohama, S. G.; Olivas, A.; Churchwell, M.; Doerge, D.; Spangler, E.; de Cabo, R.; Ingram, D. K.; Imhof, B.; Bao, G. Effect of caloric restriction on base-excision repair (BER) in the aging rat brain. Experimental Gerontology 2010, 45(3), 208–216. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, L.; Zhang, W.; Meng, D.; Zhang, H.; Jiang, Y.; Xu, X.; Van Meter, M.; Seluanov, A.; Gorbunova, V.; Mao, Z. SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle 2015, 14(2), 269–276. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, S.-I.; Ray, S.; Tarone, R. E.; Kraemer, K. H.; Grossman, L. The effect of donor age on the processing of UV-damaged DNA by cultured human cells: Reduced DNA repair capacity and increased DNA mutability. Mutation Research/DNA Repair 1996, 364(2), 117–123. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.-D.; Gao, Q.; Zhang, W.; Zhang, B.; Ma, Y.; Zhang, L.-X.; Muer, C.; Xie, Y.; Liu, Y. The age-related expression decline of ERCC1 and XPF for forensic age estimation: A preliminary study. Journal of Forensic and Legal Medicine 2017, 49, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Zhang, L.; Bai, J.; Ge, H.; Liu, P. Expression changes in DNA repair enzymes and mitochondrial DNA damage in aging rat lens. Molecular Vision 2010, 16, 1754–1763. [Google Scholar] [PubMed]
- Lautrup, S.; Myrup Holst, C.; Yde, A.; Asmussen, S.; Thinggaard, V.; Larsen, K.; Laursen, L. S.; Richner, M.; Vægter, C. B.; Prieto, G. A.; Berchtold, N.; Cotman, C. W.; Stevnsner, T. The role of aging and brain-derived neurotrophic factor signaling in expression of base excision repair genes in the human brain. Aging Cell 2023, 22(9). [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R. J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biology and Medicine 1991, 11(1), 81–128. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G. P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P. R. J.; Barrera, G. DNA damage by lipid peroxidation products: implications in cancer, inflammation and autoimmunity. AIMS Genetics 2017, 04(02), 103–137. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, H.; Nair, J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbeck’s Archives of Surgery 2006, 391(5), 499–510. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.-F.; Santana, L. F.; Vermulst, M.; Tomazela, D. M.; Emond, M. J.; MacCoss, M. J.; Gollahon, K.; Martin, G. M.; Loeb, L. A.; Ladiges, W. C.; Rabinovitch, P. S. Overexpression of Catalase Targeted to Mitochondria Attenuates Murine Cardiac Aging. Circulation 2009, 119(21), 2789–2797. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, A. J.; Pamplona, R.; Buffenstein, R.; Buttemer, W. A. Life and Death: Metabolic Rate, Membrane Composition, and Life Span of Animals. Physiological Reviews 2007, 87(4), 1175–1213. [Google Scholar] [CrossRef] [PubMed]
- Oka, Y.; Hamada, M.; Nakazawa, Y.; Muramatsu, H.; Okuno, Y.; Higasa, K.; Shimada, M.; Takeshima, H.; Hanada, K.; Hirano, T.; Kawakita, T.; Sakaguchi, H.; Ichimura, T.; Ozono, S.; Yuge, K.; Watanabe, Y.; Kotani, Y.; Yamane, M.; Kasugai, Y.; Ogi, T. Digenic mutations in ALDH2 and ADH5 impair formaldehyde clearance and cause a multisystem disorder, AMeD syndrome. Science Advances 2020, 6(51). [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R. J. The Role of Mitochondria in Apoptosis. Annual Review of Genetics 2009, 43(1), 95–118. [Google Scholar] [CrossRef] [PubMed]
- Pang, W. W.; Price, E. A.; Sahoo, D.; Beerman, I.; Maloney, W. J.; Rossi, D. J.; Schrier, S. L.; Weissman, I. L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proceedings of the National Academy of Sciences 2011, 108(50), 20012–20017. [Google Scholar] [CrossRef] [PubMed]
- Geiger, H.; de Haan, G.; Florian, M. C. The ageing haematopoietic stem cell compartment. Nature Reviews Immunology 2013, 13(5), 376–389. [Google Scholar] [CrossRef] [PubMed]
- Netz, D. J. A.; Stith, C. M.; Stümpfig, M.; Köpf, G.; Vogel, D.; Genau, H. M.; Stodola, J. L.; Lill, R.; Burgers, P. M. J.; Pierik, A. J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nature Chemical Biology 2012, 8(1), 125–132. [Google Scholar] [CrossRef] [PubMed]
- Lisova, A. E.; Baranovskiy, A. G.; Morstadt, L. M.; Babayeva, N. D.; Stepchenkova, E. I.; Tahirov, T. H. The iron-sulfur cluster is essential for DNA binding by human DNA polymerase ε. Scientific Reports 2022, 12(1), 17436. [Google Scholar] [CrossRef] [PubMed]
- ter Beek, J.; Parkash, V.; Bylund, G. O.; Osterman, P.; Sauer-Eriksson, A. E.; Johansson, E. Structural evidence for an essential Fe–S cluster in the catalytic core domain of DNA polymerase ϵ. Nucleic Acids Research 2019, 47(11), 5712–5722. [Google Scholar] [CrossRef] [PubMed]
- Linn, S. DNA Damage by Iron and Hydrogen Peroxide. In Advances in DNA Damage and Repair; Springer US, 1999; pp. 259–266. [Google Scholar] [CrossRef]
- Imlay, J. A.; Linn, S. DNA Damage and Oxygen Radical Toxicity. Science 1988, 240(4857), 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G. P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochimica et Biophysica Acta (BBA) - General Subjects 2014, 1840(5), 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Schoebel, S.; Schmitz, F.; Dong, H.; Hedfalk, K. Characterization of aquaporin-driven hydrogen peroxide transport. Biochimica et Biophysica Acta (BBA) - Biomembranes 2020, 1862(2), 183065. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D. G. Bioenergetics; Academic Press, 2013; Available online: https://books.google.com/books?id=b3fTWHBTHAAC.
- Lenaz, G.; Genova, M. L. Structural and functional organization of the mitochondrial respiratory chain: A dynamic super-assembly. The International Journal of Biochemistry & Cell Biology 2009, 41(10), 1750–1772. [Google Scholar] [CrossRef] [PubMed]
- Brégeon, D.; Peignon, P.-A.; Sarasin, A. Transcriptional Mutagenesis Induced by 8-Oxoguanine in Mammalian Cells. PLoS Genetics 2009, 5(7), e1000577. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Verheijen, B. M.; Zhang, X.; Huang, B.; Coakley, A.; McGann, E.; Wade, E.; Dinep-Schneider, O.; LaGosh, J.; Anagnostou, M.-E.; Simpson, S.; Thomas, K.; Ernst, M.; Rattray, A.; Lynch, M.; Kashlev, M.; Benayoun, B. A.; Li, Z.; Strathern, J.; Vermulst, M. The fidelity of transcription in human cells. Proceedings of the National Academy of Sciences 2023, 120(5). [Google Scholar] [CrossRef] [PubMed]
- Bell, A. C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405(6785), 482–485. [Google Scholar] [CrossRef] [PubMed]
- Spisak, N.; de Manuel, M.; Milligan, W.; Sella, G.; Przeworski, M. The clock-like accumulation of germline and somatic mutations can arise from the interplay of DNA damage and repair. PLOS Biology 2024, 22(6), e3002678. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Gao, F.; Pfenning, A. R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T. X.; Yamakawa, H.; Pao, P.-C.; Stott, R. T.; Gjoneska, E.; Nott, A.; Cho, S.; Kellis, M.; Tsai, L.-H. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161(7), 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R. M.; Weindruch, R. Metabolic reprogramming, caloric restriction and aging. Trends in Endocrinology & Metabolism 2010, 21(3), 134–141. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J. A.; Colman, R. J.; Beasley, T. M.; Allison, D. B.; Kemnitz, J. W.; Roth, G. S.; Ingram, D. K.; Weindruch, R.; de Cabo, R.; Anderson, R. M. Caloric restriction improves health and survival of rhesus monkeys. Nature Communications 2017, 8(1), 14063. [Google Scholar] [CrossRef] [PubMed]
- Brown-Borg, H. M. Hormonal control of aging in rodents: The somatotropic axis. Molecular and Cellular Endocrinology 2009, 299(1), 64–71. [Google Scholar] [CrossRef] [PubMed]
- Spencer Chapman, M.; Mitchell, E.; Yoshida, K.; Williams, N.; Fabre, M. A.; Ranzoni, A. M.; Robinson, P. S.; Kregar, L. D.; Wilk, M.; Boettcher, S.; Mahbubani, K.; Saeb Parsy, K.; Gowers, K. H. C.; Janes, S. M.; Ng, S. W. K.; Hoare, M.; Green, A. R.; Vassiliou, G. S.; Cvejic, A.; Campbell, P. J. Prolonged persistence of mutagenic DNA lesions in somatic cells. Nature 2025, 638(8051), 729–738. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E. C.; Microbiology.; for, A. S. DNA repair and mutagenesis, 2nd ed.; ASM Press, 2006. [Google Scholar]
- Extavour, C. G.; Akam, M. Mechanisms of germ cell specification across the metazoans: epigenesis and preformation. Development 2003, 130(24), 5869–5884. [Google Scholar] [CrossRef] [PubMed]
- MacQueen, A. J.; Hochwagen, A. Checkpoint mechanisms: the puppet masters of meiotic prophase. Trends in Cell Biology 2011, 21(7), 393–400. [Google Scholar] [CrossRef] [PubMed]
- Seisenberger, S.; Peat, J. R.; Hore, T. A.; Santos, F.; Dean, W.; Reik, W. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philosophical Transactions of the Royal Society B: Biological Sciences 2013, 368(1609), 20110330. [Google Scholar] [CrossRef] [PubMed]
- Siomi, M. C.; Sato, K.; Pezic, D.; Aravin, A. A. PIWI-interacting small RNAs: the vanguard of genome defence. Nature Reviews Molecular Cell Biology 2011, 12(4), 246–258. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, V. C. T.; Otto, S. P.; Aitken, S. N. Somatic mutations substantially increase the per-generation mutation rate in the conifer Picea sitchensis. Evolution Letters 2019, 3(4), 348–358. [Google Scholar] [CrossRef] [PubMed]
- Ganz, J.; Luquette, L. J.; Bizzotto, S.; Miller, M. B.; Zhou, Z.; Bohrson, C. L.; Jin, H.; Tran, A. V.; Viswanadham, V. V.; McDonough, G.; Brown, K.; Chahine, Y.; Chhouk, B.; Galor, A.; Park, P. J.; Walsh, C. A. Contrasting somatic mutation patterns in aging human neurons and oligodendrocytes. Cell 2024, 187(8), 1955–1970.e23. [Google Scholar] [CrossRef] [PubMed]
- Gompertz, B. XXIV. On the nature of the function expressive of the law of human mortality, and on a new mode of determining the value of life contingencies. In a letter to Francis Baily, Esq. F. R. S. &c. Philosophical Transactions of the Royal Society of London 1825, (115), 513–583. [Google Scholar] [CrossRef]
- Prigogine, I.; Nicolis, G. Self-Organisation in Nonequilibrium Systems: Towards A Dynamics of Complexity. In Bifurcation Analysis; Springer Netherlands, 1985; pp. 3–12. [Google Scholar] [CrossRef]
- Morowitz, H. J. Energy flow in biology: biological organization as a problem in thermal physics; Academic Press, 1968. [Google Scholar]
- O’Connor, P. D. T.; Kleyner, A. Practical Reliability Engineering; Wiley, 2011. [Google Scholar] [CrossRef]
- Kapur, K. C.; Lamberson, L. R. Reliability in Engineering Design; Wiley, 1977; Available online: https://books.google.com/books?id=N4RRAAAAMAAJ.
- Strehler, B. L.; Mildvan, A. S. General Theory of Mortality and Aging. Science 1960, 132(3418), 14–21. [Google Scholar] [CrossRef] [PubMed]
- Sacher, G. A. On the Statistical Nature of Mortality, with Especial Reference to Chronic Radiation Mortality. Radiology 1956, 67(2), 250–258. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, J. W.; Manton, K. G.; Stallard, E. The impact of heterogeneity in individual frailty on the dynamics of mortality. Demography 1979, 16(3), 439–454. [Google Scholar] [CrossRef] [PubMed]
- Yashin, A. I.; Manton, K. G.; Vaupel, J. W. Mortality and aging in a heterogeneous population: A stochastic process model with observed and unobserved variables. Theoretical Population Biology 1985, 27(2), 154–175. [Google Scholar] [CrossRef] [PubMed]
- Gnedenko, B. Sur La Distribution Limite Du Terme Maximum D’Une Serie Aleatoire. The Annals of Mathematics 1943, 44(3), 423. [Google Scholar] [CrossRef]
- de Haan, L.; Ferreira, A. Extreme Value Theory; Springer New York, 2006. [Google Scholar] [CrossRef]
- Gavrilov, L. A.; Gavrilova, N. S. The Quest for a General Theory of Aging and Longevity. Science of Aging Knowledge Environment 2003, 2003(28). [Google Scholar] [CrossRef] [PubMed]
- Finch, C. E.; Crimmins, E. M. Inflammatory Exposure and Historical Changes in Human Life-Spans. Science 2004, 305(5691), 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Tai, T. H.; Noymer, A. Models for estimating empirical Gompertz mortality: With an application to evolution of the Gompertzian slope. Population Ecology 2018, 60(1–2), 171–184. [Google Scholar] [CrossRef]
- Ricklefs, R. E.; Scheuerlein, A. Biological Implications of the Weibull and Gompertz Models of Aging. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 2002, 57(2), B69–B76. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, S.; Finch, C. E.; Mesle, F.; Vallin, J. Differential Patterns of Age-Related Mortality Increase in Middle Age and Old Age. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 2003, 58(6), B495–B507. [Google Scholar] [CrossRef] [PubMed]
- Burger, O.; Missov, T. I. Evolutionary theory of ageing and the problem of correlated Gompertz parameters. Journal of Theoretical Biology 2016, 408, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Simons, M. J. P.; Koch, W.; Verhulst, S. Dietary restriction of rodents decreases aging rate without affecting initial mortality rate – a meta-analysis. Aging Cell 2013, 12(3), 410–414. [Google Scholar] [CrossRef] [PubMed]
- de Magalhães, J. P.; Cabral, J. A. S.; Magalhães, D. The Influence of Genes on the Aging Process of Mice. Genetics 2005, 169(1), 265–274. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.; Austad, S. N.; Ikeno, Y.; Unnikrishnan, A.; McCarter, R. J. Significant life extension by ten percent dietary restriction. Annals of the New York Academy of Sciences 2016, 1363(1), 11–17. [Google Scholar] [CrossRef] [PubMed]
- GAVRILOV, L. A.; GAVRILOVA, N. S. Early-Life Programming of Aging and Longevity: The Idea of High Initial Damage Load (the HIDL Hypothesis). Annals of the New York Academy of Sciences 2004, 1019(1), 496–501. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L. L.; Simonetti, R. G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database of Systematic Reviews 2012, 2012(3). [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, C. Meta-Regression Analyses, Meta-Analyses, and Trial Sequential Analyses of the Effects of Supplementation with Beta-Carotene, Vitamin A, and Vitamin E Singly or in Different Combinations on All-Cause Mortality: Do We Have Evidence for Lack of Harm? PLoS ONE 2013, 8(9), e74558. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose-Response 2014, 12(2). [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C. R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proceedings of the National Academy of Sciences 2009, 106(21), 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Rossman, M. J.; Santos-Parker, J. R.; Steward, C. A. C.; Bispham, N. Z.; Cuevas, L. M.; Rosenberg, H. L.; Woodward, K. A.; Chonchol, M.; Gioscia-Ryan, R. A.; Murphy, M. P.; Seals, D. R. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71(6), 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, A. J.; Nagulan, R.; Somerville, V. The Effect of MitoQ on Aging-Related Biomarkers: A Systematic Review and Meta-Analysis. Oxidative Medicine and Cellular Longevity 2018, 2018(1). [Google Scholar] [CrossRef] [PubMed]
- Cragg, L.; Hebbel, R. P.; Miller, W.; Solovey, A.; Selby, S.; Enright, H. The iron chelator L1 potentiates oxidative DNA damage in iron-loaded liver cells. Blood 1998, 92(2), 632–638. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, S.; Dhénaut, A.; Eckert, I.; Radicella, J. P.; Epe, B. Overexpression of Ogg1 in mammalian cells: effects on induced and spontaneous oxidative DNA damage and mutagenesis. Carcinogenesis 1999, 20(9), 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, C.; Han, J.; Xue, Y.; Zheng, X.; Wang, R.; Radak, Z.; Nakabeppu, Y.; Boldogh, I.; Ba, X. Reassessing the roles of oxidative DNA base lesion 8-oxoGua and repair enzyme OGG1 in tumorigenesis. Journal of Biomedical Science 2025, 32(1), 1. [Google Scholar] [CrossRef] [PubMed]
- Rinne, M. L. N-methylpurine DNA glycosylase overexpression increases alkylation sensitivity by rapidly removing non-toxic 7-methylguanine adducts. Nucleic Acids Research 2005, 33(9), 2859–2867. [Google Scholar] [CrossRef] [PubMed]
- Pao, P.-C.; Patnaik, D.; Watson, L. A.; Gao, F.; Pan, L.; Wang, J.; Adaikkan, C.; Penney, J.; Cam, H. P.; Huang, W.-C.; Pantano, L.; Lee, A.; Nott, A.; Phan, T. X.; Gjoneska, E.; Elmsaouri, S.; Haggarty, S. J.; Tsai, L.-H. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nature Communications 2020, 11(1), 2484. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, H. M.; Wang, W.; Sen, S.; DeStefano Shields, C.; Lee, S. S.; Zhang, Y. W.; Clements, E. G.; Cai, Y.; Van Neste, L.; Easwaran, H.; Casero, R. A.; Sears, C. L.; Baylin, S. B. Oxidative Damage Targets Complexes Containing DNA Methyltransferases, SIRT1, and Polycomb Members to Promoter CpG Islands. Cancer Cell 2011, 20(5), 606–619. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Bonham, E. M.; Hannon, B. E.; Amick, T. R.; Baylin, S. B.; O’Hagan, H. M. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. Journal of Molecular Cell Biology 2016, 8(3), 244–254. [Google Scholar] [CrossRef] [PubMed]
- Damsma, G. E.; Cramer, P. Molecular Basis of Transcriptional Mutagenesis at 8-Oxoguanine. Journal of Biological Chemistry 2009, 284(46), 31658–31663. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.-H.; Wang, R.; Wang, Y.; Kung, C.-P.; Weber, J. D.; Patti, G. J. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. ELife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Havens, C. G.; Ho, A.; Yoshioka, N.; Dowdy, S. F. Regulation of Late G 1 /S Phase Transition and APC Cdh1 by Reactive Oxygen Species. Molecular and Cellular Biology 2006, 26(12), 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Di Palo, G.; Scala, G.; Castrignanò, T.; Gorini, F.; Cocozza, S.; Moresano, A.; Pucci, P.; Ma, B.; Stepanov, I.; Lania, L.; Pelicci, P. G.; Dellino, G. I.; Majello, B. Genome-wide mapping of 8-oxo-7,8-dihydro-2′-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Research 2019, 47(1), 221–236. [Google Scholar] [CrossRef] [PubMed]
- Gorini, F.; Scala, G.; Di Palo, G.; Dellino, G. I.; Cocozza, S.; Pelicci, P. G.; Lania, L.; Majello, B.; Amente, S. The genomic landscape of 8-oxodG reveals enrichment at specific inherently fragile promoters. Nucleic Acids Research 2020, 48(8), 4309–4324. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K. F.; Chen, R.; Gu, H.; Bock, C.; Meissner, A.; Göttgens, B.; Darlington, G. J.; Li, W.; Goodell, M. A. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 2014, 14(5), 673–688. [Google Scholar] [CrossRef] [PubMed]
- Radyuk, S. N.; Michalak, K.; Klichko, V. I.; Benes, J.; Rebrin, I.; Sohal, R. S.; Orr, W. C. Peroxiredoxin 5 confers protection against oxidative stress and apoptosis and also promotes longevity in Drosophila. Biochemical Journal 2009, 419(2), 437–445. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, P.; Miccio, A.; Brusson, M. Base and Prime Editing Technologies for Blood Disorders. Frontiers in Genome Editing 2021, 3. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M. P.; Krishnakumar, R.; Timlin, J. A.; Carney, J. P.; Butler, K. S. Gene editing and CRISPR in the clinic: current and future perspectives. Bioscience Reports 2020, 40(4). [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nature Reviews Materials 2021, 6(12), 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P. S.; Rudra, A.; Miao, L.; Anderson, D. G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Molecular Therapy 2019, 27(4), 710–728. [Google Scholar] [CrossRef] [PubMed]
- Garrett-Bakelman, F. E.; Darshi, M.; Green, S. J.; Gur, R. C.; Lin, L.; Macias, B. R.; McKenna, M. J.; Meydan, C.; Mishra, T.; Nasrini, J.; Piening, B. D.; Rizzardi, L. F.; Sharma, K.; Siamwala, J. H.; Taylor, L.; Vitaterna, M. H.; Afkarian, M.; Afshinnekoo, E.; Ahadi, S.; Turek, F. W. The NASA Twins Study: A multidimensional analysis of a year-long human spaceflight. Science 2019, 364(6436). [Google Scholar] [CrossRef] [PubMed]
- Durante, M.; Cucinotta, F. A. Heavy ion carcinogenesis and human space exploration. Nature Reviews Cancer 2008, 8(6), 465–472. [Google Scholar] [CrossRef] [PubMed]
- Stahl-Rommel, S.; Li, D.; Sung, M.; Li, R.; Vijayakumar, A.; Atabay, K. D.; Bushkin, G. G.; Castro, C. L.; Foley, K. D.; Copeland, D. S.; Castro-Wallace, S. L.; Alvarez Saavedra, E.; Gleason, E. J.; Kraves, S. A CRISPR-based assay for the study of eukaryotic DNA repair onboard the International Space Station. PLOS ONE 2021, 16(6), e0253403. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P. H.; King, E. M. On the genesis of the Earth’s magnetism. Reports on Progress in Physics 2013, 76(9), 096801. [Google Scholar] [CrossRef] [PubMed]
- Borovsky, J. E.; Valdivia, J. A. The Earth’s Magnetosphere: A Systems Science Overview and Assessment. Surveys in Geophysics 2018, 39(5), 817–859. [Google Scholar] [CrossRef] [PubMed]
- Neale, R. E.; Lucas, R. M.; Byrne, S. N.; Hollestein, L.; Rhodes, L. E.; Yazar, S.; Young, A. R.; Berwick, M.; Ireland, R. A.; Olsen, C. M. The effects of exposure to solar radiation on human health. Photochemical & Photobiological Sciences 2023, 22(5), 1011–1047. [Google Scholar] [CrossRef] [PubMed]
- Sources and effects of ionizing radiation: United Nations Scientific Committee on the Effects of Atomic Radiation: UNSCEAR 2008 report to the General Assembly, with scientific annexes; United Nations, 2008.
- Pinheiro, V. B.; Holliger, P. The XNA world: progress towards replication and evolution of synthetic genetic polymers. Current Opinion in Chemical Biology 2012, 16(3–4), 245–252. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, V. B.; Taylor, A. I.; Cozens, C.; Abramov, M.; Renders, M.; Zhang, S.; Chaput, J. C.; Wengel, J.; Peak-Chew, S.-Y.; McLaughlin, S. H.; Herdewijn, P.; Holliger, P. Synthetic Genetic Polymers Capable of Heredity and Evolution. Science 2012, 336(6079), 341–344. [Google Scholar] [CrossRef] [PubMed]
- Hoshika, S.; Leal, N. A.; Kim, M.-J.; Kim, M.-S.; Karalkar, N. B.; Kim, H.-J.; Bates, A. M.; Watkins, N. E.; SantaLucia, H. A.; Meyer, A. J.; DasGupta, S.; Piccirilli, J. A.; Ellington, A. D.; SantaLucia, J.; Georgiadis, M. M.; Benner, S. A. Hachimoji DNA and RNA: A genetic system with eight building blocks. Science 2019, 363(6429), 884–887. [Google Scholar] [CrossRef] [PubMed]
- Fredens, J.; Wang, K.; de la Torre, D.; Funke, L. F. H.; Robertson, W. E.; Christova, Y.; Chia, T.; Schmied, W. H.; Dunkelmann, D. L.; Beránek, V.; Uttamapinant, C.; Llamazares, A. G.; Elliott, T. S.; Chin, J. W. Total synthesis of Escherichia coli with a recoded genome. Nature 2019, 569(7757), 514–518. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C. A.; Chuang, R.-Y.; Noskov, V. N.; Assad-Garcia, N.; Deerinck, T. J.; Ellisman, M. H.; Gill, J.; Kannan, K.; Karas, B. J.; Ma, L.; Pelletier, J. F.; Qi, Z.-Q.; Richter, R. A.; Strychalski, E. A.; Sun, L.; Suzuki, Y.; Tsvetanova, B.; Wise, K. S.; Smith, H. O.; Venter, J. C. Design and synthesis of a minimal bacterial genome. Science 2016, 351(6280). [Google Scholar] [CrossRef] [PubMed]
- Richardson, S. M.; Mitchell, L. A.; Stracquadanio, G.; Yang, K.; Dymond, J. S.; DiCarlo, J. E.; Lee, D.; Huang, C. L. V.; Chandrasegaran, S.; Cai, Y.; Boeke, J. D.; Bader, J. S. Design of a synthetic yeast genome. Science 2017, 355(6329), 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Organick, L.; Ang, S. D.; Chen, Y.-J.; Lopez, R.; Yekhanin, S.; Makarychev, K.; Racz, M. Z.; Kamath, G.; Gopalan, P.; Nguyen, B.; Takahashi, C. N.; Newman, S.; Parker, H.-Y.; Rashtchian, C.; Stewart, K.; Gupta, G.; Carlson, R.; Mulligan, J.; Carmean, D.; Strauss, K. Random access in large-scale DNA data storage. Nature Biotechnology 2018, 36(3), 242–248. [Google Scholar] [CrossRef] [PubMed]
- Erlich, Y.; Zielinski, D. DNA Fountain enables a robust and efficient storage architecture. Science 2017, 355(6328), 950–954. [Google Scholar] [CrossRef] [PubMed]
- Church, G. M.; Gao, Y.; Kosuri, S. Next-Generation Digital Information Storage in DNA. Science 2012, 337(6102), 1628–1628. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The Inevitability of Information Corruption. Information cannot be indefinitely read or reproduced without error. The accumulation of errors corrupts the information. Most corrupted information undermines the interpretation of that information, rendering the message misinterpreted with partial information loss but uninterpretable or equivalent to background noise as corruption increases. By analogy, life’s nucleic acid houses the information to order, which can be loosely defined as existence (life) with “purpose or intent.” To maintain its permanence through time, the nucleic acid must be read, a process that converts the chemical information into biological function, that function largely designed to protect the information and ensure its transmission through time (the prime directive). However, slight introductions of chaos in the original information either during nucleic acid maintenance, reading, or copying (replication) by “corruptors” changes the information content of the original molecule. Over time, the original parental instructions are lost, the subsequent vessel is unable to remember how to efficiently order, and the resulting increase in disfunction escalates up and through biological hierarchies of organization, physically manifesting as aging in the most organized of organisms and, after the most crucial of information to support that hierarchy is lost, death. .
Figure 1.
The Inevitability of Information Corruption. Information cannot be indefinitely read or reproduced without error. The accumulation of errors corrupts the information. Most corrupted information undermines the interpretation of that information, rendering the message misinterpreted with partial information loss but uninterpretable or equivalent to background noise as corruption increases. By analogy, life’s nucleic acid houses the information to order, which can be loosely defined as existence (life) with “purpose or intent.” To maintain its permanence through time, the nucleic acid must be read, a process that converts the chemical information into biological function, that function largely designed to protect the information and ensure its transmission through time (the prime directive). However, slight introductions of chaos in the original information either during nucleic acid maintenance, reading, or copying (replication) by “corruptors” changes the information content of the original molecule. Over time, the original parental instructions are lost, the subsequent vessel is unable to remember how to efficiently order, and the resulting increase in disfunction escalates up and through biological hierarchies of organization, physically manifesting as aging in the most organized of organisms and, after the most crucial of information to support that hierarchy is lost, death. .

Figure 2.
The Replicator. a) Any molecule that embeds information in its chemistry and structure that guides or protects its copying process can be considered a replicator. On Earth, this is primarily achieved via a linear stretch of nucleic acid bases whose sequence encodes information, what we all know as DNA or RNA. The hierarchy of replicators spans simple short nucleic acids (transposons) all the way up to replicators that use technology to advance the copying process, such as humans. There are even replicators within replicators, such as non-coding or parasitic nucleic acid that likely has a benefit for the creature but is mostly along for the replicative ride. b) The five tenants of any replicator, which are: (i) Replication is an intrinsic directive; (ii) Chemical memory (“information that drives copying and/or conversion of the information to biology”); (iii) Information corruption; (iv) Selection of useful changes in information; (v) Continuous will to replicate. .
Figure 2.
The Replicator. a) Any molecule that embeds information in its chemistry and structure that guides or protects its copying process can be considered a replicator. On Earth, this is primarily achieved via a linear stretch of nucleic acid bases whose sequence encodes information, what we all know as DNA or RNA. The hierarchy of replicators spans simple short nucleic acids (transposons) all the way up to replicators that use technology to advance the copying process, such as humans. There are even replicators within replicators, such as non-coding or parasitic nucleic acid that likely has a benefit for the creature but is mostly along for the replicative ride. b) The five tenants of any replicator, which are: (i) Replication is an intrinsic directive; (ii) Chemical memory (“information that drives copying and/or conversion of the information to biology”); (iii) Information corruption; (iv) Selection of useful changes in information; (v) Continuous will to replicate. .

Figure 3.
Corruptible Information Types Hypothesized to be Essential for Intropy Loss. All biomolecules harbor intrinsic chemical information that facilitates their function. Whereas the corruption (chemical modification) of ubiquitous cellular molecules such as proteins, lipids, and metabolites (everything except coding nucleic information) is common and possible, this corruption is not permanent or heritable if a new version of that molecule can be made by untarnished and unchanged information in nucleic acid. However, the modification of the nucleic acid core structure, nucleobase sequence, or nucleobase epigenome may impact the generation of every biological entity it encodes by altering the readability and conversion of that information to biological order and function (the information divide). Thus, while modification of non-encoding molecules may contribute to aging, the effect may be acute and is likely the byproduct of a deeper level of permanent corruption of the information systems from either which they arise or interact with or both.
Figure 3.
Corruptible Information Types Hypothesized to be Essential for Intropy Loss. All biomolecules harbor intrinsic chemical information that facilitates their function. Whereas the corruption (chemical modification) of ubiquitous cellular molecules such as proteins, lipids, and metabolites (everything except coding nucleic information) is common and possible, this corruption is not permanent or heritable if a new version of that molecule can be made by untarnished and unchanged information in nucleic acid. However, the modification of the nucleic acid core structure, nucleobase sequence, or nucleobase epigenome may impact the generation of every biological entity it encodes by altering the readability and conversion of that information to biological order and function (the information divide). Thus, while modification of non-encoding molecules may contribute to aging, the effect may be acute and is likely the byproduct of a deeper level of permanent corruption of the information systems from either which they arise or interact with or both.

Figure 4.
The Protector. The replicator utilizes an informational, structural, cellular, social and technological hierarchy of systems to protect its information content and safeguard the prime directive. These systems increase in complexity as information content grows, fueled by limitless challenges wrought by a changing environment and a never-ending competition among all replicators for limited resources. At the base level is protection of information via back-up copies or molecular repair/structural stabilization. The next level includes all cellular protective mechanisms, including control of physical shields like membranes, structure or organization of cellular scaffolds, the cytoplasmic environment, energy generation, acquisition or storage, and enzymatic functions. The next level includes the grouping of cells to tissues with further specialization and function, tissues into organisms with unique adaptations to the environment, and organisms into complex social units that cooperate, specialize, and form integrated ecosystems. As a result of this increasing selection of protective mechanisms, some higher organisms evolved a sophisticated hierarchy of levels of protection, each dependent on the lower levels. At the base of this hierarchy is the unit of information, the replicator, which if corrupted translates the error up the organizational chain of function and order. .
Figure 4.
The Protector. The replicator utilizes an informational, structural, cellular, social and technological hierarchy of systems to protect its information content and safeguard the prime directive. These systems increase in complexity as information content grows, fueled by limitless challenges wrought by a changing environment and a never-ending competition among all replicators for limited resources. At the base level is protection of information via back-up copies or molecular repair/structural stabilization. The next level includes all cellular protective mechanisms, including control of physical shields like membranes, structure or organization of cellular scaffolds, the cytoplasmic environment, energy generation, acquisition or storage, and enzymatic functions. The next level includes the grouping of cells to tissues with further specialization and function, tissues into organisms with unique adaptations to the environment, and organisms into complex social units that cooperate, specialize, and form integrated ecosystems. As a result of this increasing selection of protective mechanisms, some higher organisms evolved a sophisticated hierarchy of levels of protection, each dependent on the lower levels. At the base of this hierarchy is the unit of information, the replicator, which if corrupted translates the error up the organizational chain of function and order. .

Figure 5.
The Corruptor. a) Corruptors modify nucleic acid, thereby altering the original information content. Some of the modified information changes in ways that are not productive or efficient (corrupted information), thereby compromising life’s reading and interpretation of that information and driving a loss of the originally intended biological order. The source of the information corruptor can be varied and diverse, but we favor those associated with handling the energy systems of the cell. b) Shown is a partial list of possible corruptors driving intropy loss. Anything that alters life’s information systems from its designed or adapted intent is technically a corruptor. However, due to the perceived notion that corruptors are likely to have certain features to consistently induce informational changes, we favor small, highly reactive particles, atoms, or molecules with unpaired or imbalanced electrons (or may form readily such a species) whose production is constant or common, may be ubiquitous in the cell or environment (likely in close contact with nucleic acid) and are small and diffusible. Thus, the agents at the top right hand corner of the table, which we term “corruptagens”, are the most probable candidates satisfying most if not all the requirements set forth for information modifiers. c) Types of information modification, including sequence changes (mutation), outright damage (breaks/lesions), and intentional and non-intentional covalent or electronic modification of the DNA/RNA.
Figure 5.
The Corruptor. a) Corruptors modify nucleic acid, thereby altering the original information content. Some of the modified information changes in ways that are not productive or efficient (corrupted information), thereby compromising life’s reading and interpretation of that information and driving a loss of the originally intended biological order. The source of the information corruptor can be varied and diverse, but we favor those associated with handling the energy systems of the cell. b) Shown is a partial list of possible corruptors driving intropy loss. Anything that alters life’s information systems from its designed or adapted intent is technically a corruptor. However, due to the perceived notion that corruptors are likely to have certain features to consistently induce informational changes, we favor small, highly reactive particles, atoms, or molecules with unpaired or imbalanced electrons (or may form readily such a species) whose production is constant or common, may be ubiquitous in the cell or environment (likely in close contact with nucleic acid) and are small and diffusible. Thus, the agents at the top right hand corner of the table, which we term “corruptagens”, are the most probable candidates satisfying most if not all the requirements set forth for information modifiers. c) Types of information modification, including sequence changes (mutation), outright damage (breaks/lesions), and intentional and non-intentional covalent or electronic modification of the DNA/RNA.

Figure 6.
Eden’s Apple and the Paradox of Life. Life is simply a natural process to conserve order via the encoding of self-replicating information – the replicator. During the process of maintaining, reading, and duplicating the information, a stochastic and chaotic microenvironment induces errors in the information content, some of which become fixed and permanent via mutation. Thus ensues a continuous struggle against entropic increase, the errors occasionally being used to build a better protector for the replicator but more often driving disorder and breaking biologic efficiency, especially in the individual unit carrying the replicator. As the protections grew larger and more complex, more energetic capability was needed. This comes at a cost for as increased energy is harnessed to build better protectors, increasingly dangerous chemistry is used, thereby driving the production of more corruptors. The corruptors being ever present (EAI), the best chance at indefinite existence for the replicator is to advance a relatively unaltered copy via germline transmission, a process that both rejuvenates the information against EAI but also increases the possibility of beneficial information changes that drive new protections. The parent vessel, unable to indefinitely repair the information, continuously absorbs detrimental information changes, which likely accumulate and synergize. The extent and location of the corrupted information will determine the strength of the phenotypic “hit” against the parental vessel, the worst of this manifesting in visible disease. Less detrimental but still important corruption of other information drives a reduction in functional efficiency and increase in biological disorder, much of which might be identified as conventional aging. Once the corruption reaches a readable threshold, the information degrades close to the noise or background boundary, a point at which the only outcome is complete collapse of biological order, a small sacrifice to entropy, resulting in the onset of death. But life continues, its immortality manifested in the informational diversity of the replicator, that diversity the best local solution to repel a nearly infinite combination of environmental assaults and challenges.
Figure 6.
Eden’s Apple and the Paradox of Life. Life is simply a natural process to conserve order via the encoding of self-replicating information – the replicator. During the process of maintaining, reading, and duplicating the information, a stochastic and chaotic microenvironment induces errors in the information content, some of which become fixed and permanent via mutation. Thus ensues a continuous struggle against entropic increase, the errors occasionally being used to build a better protector for the replicator but more often driving disorder and breaking biologic efficiency, especially in the individual unit carrying the replicator. As the protections grew larger and more complex, more energetic capability was needed. This comes at a cost for as increased energy is harnessed to build better protectors, increasingly dangerous chemistry is used, thereby driving the production of more corruptors. The corruptors being ever present (EAI), the best chance at indefinite existence for the replicator is to advance a relatively unaltered copy via germline transmission, a process that both rejuvenates the information against EAI but also increases the possibility of beneficial information changes that drive new protections. The parent vessel, unable to indefinitely repair the information, continuously absorbs detrimental information changes, which likely accumulate and synergize. The extent and location of the corrupted information will determine the strength of the phenotypic “hit” against the parental vessel, the worst of this manifesting in visible disease. Less detrimental but still important corruption of other information drives a reduction in functional efficiency and increase in biological disorder, much of which might be identified as conventional aging. Once the corruption reaches a readable threshold, the information degrades close to the noise or background boundary, a point at which the only outcome is complete collapse of biological order, a small sacrifice to entropy, resulting in the onset of death. But life continues, its immortality manifested in the informational diversity of the replicator, that diversity the best local solution to repel a nearly infinite combination of environmental assaults and challenges.

Figure 7.
Aging – information loss and functional inefficiency up the organizational hierarchy of life. Here, aging is presented as being driven by the slow corruption of nucleic acid information by corruptors that induce low-fidelity replication, a situation that amplifies functional inefficiency up the organizational chain of life. Altered information alters all post-replicational activity including epigenomic, transcriptional, and translational efficiency. This scrambled message, manifested permanently in protein function and all activities they execute, as well as regulatory signals controlling gene expression and cellular timing, permeates up a hierarchical ladder to every level of life (molecular, cellular, tissue, and organ). As the protectosphere falters, the rate of information corruption likely increases, thereby accelerating the process. Different areas of functional loss also synergize with each other in countless ways, amplifying the phenotypic effects. Once the functional inefficiency declines enough for stochastic challenges to penetrate protections and cross a threshold, the system undergoes catastrophic collapse, and the aged creature dies.
Figure 7.
Aging – information loss and functional inefficiency up the organizational hierarchy of life. Here, aging is presented as being driven by the slow corruption of nucleic acid information by corruptors that induce low-fidelity replication, a situation that amplifies functional inefficiency up the organizational chain of life. Altered information alters all post-replicational activity including epigenomic, transcriptional, and translational efficiency. This scrambled message, manifested permanently in protein function and all activities they execute, as well as regulatory signals controlling gene expression and cellular timing, permeates up a hierarchical ladder to every level of life (molecular, cellular, tissue, and organ). As the protectosphere falters, the rate of information corruption likely increases, thereby accelerating the process. Different areas of functional loss also synergize with each other in countless ways, amplifying the phenotypic effects. Once the functional inefficiency declines enough for stochastic challenges to penetrate protections and cross a threshold, the system undergoes catastrophic collapse, and the aged creature dies.

Figure 8.
Information corruption amplifies through biological hierarchies to produce aging phenotypes. Two states of the same cellular network are shown. (A) Young cell. Endogenous corruptors (NICs, orange) generated by mitochondrial ROS, inflammation, and metabolic activity reach nuclear DNA at a low steady-state rate. Modifications that arise are efficiently cleared by DNA repair and maintenance (red cross), so the flux through downstream channels (transcription, with Pol II stalling and transcriptional mutagenesis as distinct routes; splicing; regulation; epigenetic maintenance; replicative mutation; and double-strand break generation) remains low (thin red arrows). Transcription proceeds without substantial Pol II stalling or transcriptional mutagenesis, producing an accurate transcriptome and a functional proteome. Post-mitotic cells retain their specialized function, stem cell pools remain intact, and the outputs that suppress malignant transformation and inappropriate apoptosis operate normally. Secondary amplifiers (SASP, inflammation, protein aggregates) are minimal. (B) Old cell. NIC flux increases while repair capacity declines (darkened cross), shifting the balance toward a rising standing modification burden. The resulting signal propagates through every downstream channel simultaneously (thick red arrows): transcription is corrupted by Pol II stalling at bulky modifications and by miscoding at permissive modifications, generating truncated and aberrant transcripts; splicing fidelity falls; regulatory logic becomes unreliable (question mark); epigenetic maintenance drifts; and modifications encountered at replication forks convert into heritable mutations or fork-collapse double-strand breaks. These molecular changes corrupt the proteome, drive protein aggregation, trigger SASP and chronic inflammation, produce DDR-mediated stem cell attrition, and either eliminate cells through apoptosis or allow clonal expansion of mutation-bearing cells toward cancer. The molecular event at the replicator (center) remains the same class of chemistry in both panels; what changes with age is the rate of corruption, the capacity for repair, and the extent to which downstream channels amplify rather than buffer the signal.
Figure 8.
Information corruption amplifies through biological hierarchies to produce aging phenotypes. Two states of the same cellular network are shown. (A) Young cell. Endogenous corruptors (NICs, orange) generated by mitochondrial ROS, inflammation, and metabolic activity reach nuclear DNA at a low steady-state rate. Modifications that arise are efficiently cleared by DNA repair and maintenance (red cross), so the flux through downstream channels (transcription, with Pol II stalling and transcriptional mutagenesis as distinct routes; splicing; regulation; epigenetic maintenance; replicative mutation; and double-strand break generation) remains low (thin red arrows). Transcription proceeds without substantial Pol II stalling or transcriptional mutagenesis, producing an accurate transcriptome and a functional proteome. Post-mitotic cells retain their specialized function, stem cell pools remain intact, and the outputs that suppress malignant transformation and inappropriate apoptosis operate normally. Secondary amplifiers (SASP, inflammation, protein aggregates) are minimal. (B) Old cell. NIC flux increases while repair capacity declines (darkened cross), shifting the balance toward a rising standing modification burden. The resulting signal propagates through every downstream channel simultaneously (thick red arrows): transcription is corrupted by Pol II stalling at bulky modifications and by miscoding at permissive modifications, generating truncated and aberrant transcripts; splicing fidelity falls; regulatory logic becomes unreliable (question mark); epigenetic maintenance drifts; and modifications encountered at replication forks convert into heritable mutations or fork-collapse double-strand breaks. These molecular changes corrupt the proteome, drive protein aggregation, trigger SASP and chronic inflammation, produce DDR-mediated stem cell attrition, and either eliminate cells through apoptosis or allow clonal expansion of mutation-bearing cells toward cancer. The molecular event at the replicator (center) remains the same class of chemistry in both panels; what changes with age is the rate of corruption, the capacity for repair, and the extent to which downstream channels amplify rather than buffer the signal.


Table 1.
Theories of Aging.
|
GROUP A: Evolutionary Constraints Why Aging Persists | ||
| These theories explain why natural selection tolerates aging rather than eliminating it. In our framework, they describe the evolutionary logic governing how the protectosphere is calibrated: evolution optimizes protection to sustain information integrity through the reproductive window, not indefinitely. | ||
|
Theory (canonical ref/name) |
Original Scope | Role in our unified framework |
| Mutation Accumulation (Medawar 1952)[163] | Selection pressure weakens after reproduction, allowing late-acting deleterious alleles to accumulate in the population. | This theory explains why the protectosphere is optimized for the reproductive window. Late-life intropy loss goes unchecked not because protection fails catastrophically but because selection does not invest in maintaining it beyond the period needed to execute the prime directive. |
| Antagonistic Pleiotropy (Williams 1957)[164,165] | Genes beneficial early in life become harmful later; selection favors early-life fitness over late-life maintenance. | Antagonistic Pleiotropy maps to the energy-protection tradeoff at the heart of our model. Pathways such as mTOR that drive growth and reproduction early in life become sources of damage amplification later, not because they are programmed to cause aging but because selection favored their early benefits despite late costs. |
| Disposable Soma (Kirkwood 1977)[165] | Organisms allocate finite resources between reproduction and somatic maintenance; perfect repair is energetically prohibitive. | Kirkwood’s Disposable Soma theory formalizes the thermodynamic constraint underlying our framework. The protectosphere cannot achieve perfect information fidelity because doing so would require infinite energy. Evolution allocates just enough repair capacity to sustain order through the reproductive window, leaving the soma disposable once the prime directive is executed. |
|
GROUP B: The Initiating Cause Corruptor Chemistry | ||
| These Theories identify the molecular species and chemical processes that initiate information corruption. In our framework, they describe the first step of the cascade: reactive molecules modify DNA nucleobases, altering the information content of the replicator. | ||
|
Theory (canonical ref/name) |
Original Scope | Role in our unified framework |
| Free Radical Theory (Harman 1956; Gerschman et al. 1954)[123,124] | Reactive oxygen species generated during metabolism causes progressive molecular damage to proteins, lipids, and DNA. | This theory identifies one major class of corruptors (ROS/RNS) but overclaims their scope. In our framework, radicals are one subset of nucleobase information corruptors (NICs). The theory’s key contribution is the mechanism: aerobic metabolism unavoidably generates reactive species that modify nucleic acid. Its key limitation is that only modifications to nucleic acid information drive aging; damage to proteins and lipids is transient unless it feeds back to alter the replicator. Antioxidant supplementation fails because it does not address the full spectrum of NICs (alkylation, deamination, aldehyde adducts) or the spatial relationship between NICs and replicating DNA. |
| Mitochondrial Theories of Aging (Harman 1972; Miquel et al. 1980)[125,166] | Mitochondria are both the primary energy source and the primary source of ROS; mitochondrial DNA damage creates a vicious cycle of declining function and increasing oxidant production. | Mitochondria are the major endogenous NIC production site: electron leakage at Complexes I and III generates superoxide, which dismutates to H2O2 and drives Fenton chemistry near DNA. This is the energetic tradeoff at the core of Eden’s Apple: endogenous metabolism expanded biological possibility but made NIC production unavoidable. mtDNA is itself a replicator subject to the corruption cascade, and its proximity to the electron transport chain makes it especially vulnerable. Mitochondrial ROS also reach nuclear DNA indirectly through lipid peroxidation, generating diffusible aldehydes (4-HNE, malondialdehyde, acrolein) that form etheno and propano adducts beyond the short diffusion range of hydroxyl radicals. A second ROS-independent route also operates: compensatory mtDNA replication in response to mutation load depletes cellular nucleotide pools, causing replicative stress and nuclear double-strand breaks in dividing cells,[167] which trigger DDR-mediated stem cell elimination in the POLG mutator mouse (see EN36). Both routes converge on corruption or depletion of the nuclear genome. |
| Rate-of-Living Hypothesis (Pearl 1928)[168] | Lifespan inversely correlates with metabolic rate; organisms that burn energy faster die sooner. | This is a coarse empirical observation that our framework mechanistically explains. Higher metabolic rate means greater electron transport chain throughput, more ROS production, more NIC generation, and faster information corruption. The correlation breaks when organisms invest disproportionately in the protectosphere (e.g., birds have high metabolic rates but long lifespans, potentially due to enhanced repair and more peroxidation-resistant membrane composition). Metabolic rate sets the NIC production rate; the protectosphere determines how much of that production translates into transient and permanent information loss. |
|
GROUP C: Intermediate Steps From Modifications to Permanent Information Loss | ||
| These theories describe how initial chemical modifications become irreversible changes in the replicator’s information content. | ||
|
Theory (canonical ref/name) |
Original Scope | Role in our unified framework |
| DNA Damage Accumulation Theory (Szilard 1959; Vijg & Suh 2013)[169,170] | Somatic DNA damage accumulates with age, impairing gene function and cellular fitness. | This theory describes the central intermediate step of the cascade: somatic damage to nucleic acid accumulates with age and degrades gene function and cellular fitness in proportion to its burden, with Vijg, Suh, and colleagues establishing the modern accumulation evidence across tissues and cell types. Our framework retains the accumulation logic but distinguishes the initiating event, nucleobase modification by NICs, from its heritable consequence, permanent mutation produced when a modification is encountered at the replication fork or processed by error-prone repair. The most comprehensive contemporary synthesis of this lineage is Schumacher et al. 2021, which we engage in detail in Group F. |
| Eigen Error Threshold/Quasispecies Theory (Eigen 1971)[14] | For any genome, there is a maximum tolerable mutation rate; exceeding it causes information collapse into randomness. | Sets the theoretical upper bound on tolerable information corruption in asexual quasispecies replicators. Our framework generalizes this into an Eigen-like informational collapse ceiling that bounds the corridor of viable error rates for any replicator system, multicellular organisms included. Aging occurs well below this ceiling as a gradual rate of efficiency loss; intropic collapse, the threshold-crossing event, is what the framework identifies with death. The ceiling constrains protectosphere design: evolution must hold germline corruption well below it to preserve lineage viability, while the soma is allowed to drift higher because selection acts weakly past the reproductive window, except where intra-organism selection re-engages, as in cancer and clonal expansion. |
| Orgel’s Error Catastrophe (Orgel 1963)[171] | Translation errors produce defective proteins, including defective polymerases and repair enzymes, creating a positive feedback loop that accelerates further errors. | This theory describes a potential positive feedback mechanism within the cascade. In our framework, corruption of repair genes (themselves part of the protectosphere) reduces repair capacity, which accelerates further modification accumulation, leading to a repair-decline feedback loop. However, Orgel’s specific prediction of exponential translational error accumulation has not been observed empirically; the feedback is real but dampened by redundancy and proteostasis. The loop becomes significant only when the protectosphere capacity falls below a critical threshold. |
|
GROUP D: Downstream Amplifiers How Corruption Propagates Through the Hierarchical Model | ||
| These theories describe processes that are consequences of upstream information corruption but that amplify functional decline once initiated. | ||
|
Theory (canonical ref/name) |
Original Scope | Role in our unified framework |
| Epigenetic Drift / Information Theory of Aging (Holliday 1987; Lu, Tian, & Sinclair 2023)[172,173] | Age-related changes in DNA methylation, histone modifications, and chromatin structure dysregulate gene expression and drive functional decline. Sinclair proposed that epigenetic information loss alone is a reversible cause of aging. | These theories identify a component of intropy (epigenetics) as both locus and reversibility point; the intropy framework treats that component as one input among many into a scalar capacity, and its loss as a downstream signature of upstream template corruption, not the root. In other words, they describe a downstream amplifier, not the initiating cause. In our causal ordering, NIC-driven modifications alter the epigenome through two routes. Directly, modifications such as 8-oxoguanine at CpG sites recruit repair machinery that demethylates adjacent 5-methylcytosine, meaning a single modification event rewrites the local epigenetic state without requiring a mutation. Indirectly, when modifications seed mutations at CpG sites, they permanently remodel the surrounding methylome (Koch et al. 2025, who showed mutations may account for more than half of epigenetic age variation). Epigenetic drift is thus functionally consequential and accelerates decline by dysregulating gene expression, but it follows from upstream modification and sequence corruption rather than arising independently. The partial and temporary reversal achieved by reprogramming factors (OSK/OSKM) is consistent with this ordering: reprogramming activates TET1/2 and base excision repair, potentially clearing some modifications and allowing the underlying sequence to re-specify appropriate methylation patterns, but it cannot restore sequence integrity, placing a floor on reversal. In our view, the rapid rebound in epigenetic age after reprogramming cessation is better explained by the reassertion of standing modification and/or mutational pressure than by autonomous epigenetic drift or retrieval from a backup archive. |
| Proteostasis Collapse (Morimoto & Cuervo 2014)[174] | Decline in chaperone function, ubiquitin-proteasome system, and autophagy leads to accumulation of damaged or misfolded proteins, contributing to age-related pathologies. | Our framework views this theory as a downstream consequence of information corruption at the nucleic acid level. Corrupted genes produce corrupted proteins; as the informational template degrades, the proteome increasingly diverges from functional specifications. Proteostasis machinery is itself encoded in the genome and subject to the same corruption cascade; its decline reflects information loss in the genes encoding chaperones, proteasome subunits, and autophagy regulators. Proteostasis collapse amplifies aging by allowing damaged proteins to persist and interfere with cellular function, but it cannot be the initiating cause because proteins turn over while DNA information loss is permanent. |
| Telomere Attrition / Replicative Senescence (Olovnikov 1973; Blackburn, Greider, & Szostak 1985)[43,175] | Progressive shortening of chromosome ends during DNA replication triggers cell cycle arrest (senescence) or apoptosis, limiting the replicative capacity of somatic cells. | We view this as a replicator-intrinsic information boundary. Telomere shortening reflects the inherent imperfection of the copying process (the end-replication problem) and serves as a protectosphere checkpoint that limits the proliferation of cells whose information content may be corrupted. In our framework, telomere attrition is a proxy for accumulated replicative history rather than a direct cause of aging, consistent with the observation that telomere length varies widely across species without corresponding lifespan differences. Exposed telomeric DNA is also especially vulnerable to oxidative modification (G-rich repeats are targets for 8-oxoG), linking telomere erosion to NIC-driven corruption. |
| Transposable Element Activation (Van Meter et al. 2014 ; De Cecco et al. 2019)[46,176] | Age-related derepression of transposable elements (LINE-1, Alu, etc.) triggers genomic instability, inflammation via cytoplasmic DNA sensing, and interferon responses. | An endogenous corruptor source that amplifies the cascade. Transposable elements are silenced by the epigenome (DNA methylation, heterochromatin); as upstream corruption erodes epigenetic control, these elements mobilize and generate additional insertional mutations, structural rearrangements, and innate immune activation. In our framework, transposable element activation is predicted as a downstream consequence of epigenetic drift, which is itself downstream of modification-driven sequence corruption, and therefore a tertiary amplifier in the cascade. |
| Disabled Macroautophagy (Rubinsztein, Mariño, & Kroemer 2011)[177] | Macroautophagy, the lysosomal degradation pathway that removes damaged organelles, protein aggregates, and non-proteinaceous macromolecules, declines with age. This decline contributes to accumulation of dysfunctional mitochondria, misfolded proteins, and cellular debris, accelerating functional decline. Genetic or pharmacological enhancements of autophagy extends lifespan in multiple models organisms. | Autophagy is a protectosphere mechanism that operates at the organelle and macromolecular level, clearing damaged components before they can amplify dysfunction. Its age-related decline follows from upstream information corruption: the genes encoding autophagy regulators (ATGs, TFEB, Beclin-1) are themselves subject to modification and mutation, and their expression is further compromised by epigenetic drift and mTOR-driven suppression. The consequence is a positive feedback loop: as autophagy declines, damaged mitochondria accumulate and produce more ROS (more NICs), which accelerate further information corruption, which further impairs autophagy gene expression. This is one concrete instance of the repair-decline feedback loop our model predicts. Disabled autophagy is separated from proteostasis collapse because autophagy degrades entire organelles and non-protein macromolecules, making it a broader quality-control system than chaperone-mediated protein maintenance alone. |
| GROUP E: Emergent Systems-Level Consequences | ||
| These theories describe aging phenotypes that emerge from the hierarchical propagation of information corruption across biological levels of organization. | ||
|
Theory (canonical ref/name) |
Original Scope | Role in our unified framework |
| Cellular Senescence (Hayflick 1961; Campisi 2005; van Deursen 2014)[178,179,180] | Cells enter irreversible growth arrest in response to damage or stress, accumulating with age and secreting inflammatory factors (SASP) that damage surrounding tissue. | In our framework, this is a protectosphere response to information corruptors that become pathological at scale. Senescence evolved as a tumor-suppressive checkpoint, removing cells whose information content is too corrupted to safely replicate. However, accumulation of senescent cells and their SASP creates a pro-inflammatory microenvironment that accelerates NIC production in neighboring cells, exemplifying the positive feedback between damage and inflammation our model predicts. Senescence is thus both a protector (preventing corrupted replication) and an amplifier (accelerating corruption in bystander cells). |
| Stem Cell Exhaustion / Mosaicism (Rossi et al. 2008; Goodell 2024)[48,181] | Decline in the number or functional capacity of tissue-resident stem cells impairs regeneration and tissue homeostasis with age. | Stem cell exhaustion is a critical node in the hierarchical cascade. Stem cells are the information custodians for their tissue; their corruption has outsized consequences because every daughter cell inherits the corrupted template. Clonal hematopoiesis demonstrates this empirically: mutations in DNMT3A, TET2, or ASXL1 create stem cell clones with altered behavior that progressively dominates the tissue, producing the mosaicism increasingly recognized as a hallmark of aging (Goodell 2024). Our framework predicts that stem cell corruption is the rate-limiting step for tissue-level decline, and that replacing corrupted stem cells with less corrupted ones (heterochronic transplantation) should partially rescue tissue function downstream of the replaced cells. This is consistent with existing evidence that young bone marrow transplantation preserves cognitive function and bone integrity in old mice. However, the replacement cells enter an old NIC environment and will themselves accumulate modifications, predicting that the rescue is temporary and proportional to the completeness of stem cell replacement. |
| Inflammaging (Franceschi et al. 2000; Furman et al. 2019)[182,183] | Chronic, low-grade sterile inflammation increases with age and contributes to virtually all age-related diseases. | We view this as an emergent property of the corruption cascade operating across hierarchical levels. In our framework, inflammaging likely arises from multiple converging sources: SASP from senescent cells, innate immune activation by cytoplasmic DNA from transposable element mobilization, altered cytokine output from corrupted stem cell clones (the DNMT3A-inflammation feedback loop), and declining immune surveillance (immunosenescence). Inflammation itself generates ROS/RNS, additional NICs that accelerate information corruption, creating a self-reinforcing positive feedback loop. Inflammaging is a systems-level amplifier, not a root cause. |
| Immunosenescence (Walford 1969; Pawelec et al. 2002; Goronzy & Weyand 2013)[184,185,186] | Age-related decline in immune function reduces defenses against pathogens, impairing tumor surveillance, and contributes to chronic inflammation. | Immunosenescence, in our view, is a tissue-specific manifestation of the hierarchical cascade in the hematopoietic/immune system. HSC corruption (driven by modification-mutation accumulation) produces skewed differentiation, reduced lymphoid output, clonal dominance, and impaired immune function. This reduces the organism’s capacity to clear senescent cells, damaged tissue, and pathogens, effectively degrading a critical layer of the protectosphere. The resulting immune dysfunction then feeds back to accelerate systemic decline. |
| Wear and Tear Hypothesis (Weismann 1882)[20] | Aging results from cumulative physical damage to tissues and organs, analogous to mechanical wear on a machine. | The wear and tear hypothesis describes the crude macroscopic phenotype of aging but misidentifies the causal level. In our framework, tissue “wear” is the visible consequence of information corruption propagating up the hierarchy: molecular-level damage produces cellular dysfunction, which produces tissue level pathology, which manifests as the gross anatomical changes Weismann described. Unlike a machine, biological tissues can repair and regenerate, but only as long as the informational template directing that repair remains intact and uncorrupted. Wear and tear is an effect, not a cause. |
| Glycation / Cross-linking Hypothesis (Monnier & Cerami 1981; Cerami 1985)[187,188] | Non-enzymatic glycation of proteins produces advanced glycation end-products (AGEs) that cross-link macromolecules, contributing to tissue stiffness, inflammation, and age-related pathology. | We view glycation and cross-linking as a downstream consequence of corrupted metabolic and proteostatic regulation. AGE accumulation reflects both the chemistry of long-lived proteins (collagen, crystallins) and the declining capacity of systems that prevent or clear cross-linked products. In our framework, glycation is one manifestation of the broader principle that corrupted information produces corrupted function across all cellular processes, including metabolic control. AGEs are an effect of aging, not a cause, because the systems controlling protein quality and glucose metabolism are themselves directed by information that is being progressively corrupted. |
| Hormonal/Endocrine Theory (Dilman 1971; Lamberts et al. 1997)[189,190] | Age-related changes in hormone levels (growth hormone, IGF-1, sex steroids, thyroid) drive aging phenotypes. | This theory describes regulatory changes that follow from upstream information corruption. Hormonal output depends on the integrity of the genes encoding hormones, receptors, and signaling cascades, all subject to modification-driven corruption. Furthermore, hormonal systems are not universal across kingdoms of life; bacteria, fungi, and plants lack endocrine systems yet still age, disqualifying hormonal decline as a universal cause. In our framework, endocrine changes are species-specific manifestations of the general principle that corrupted information produces corrupted regulatory output. |
| Deregulated Nutrient Sensing (Kenyon et al. 1993; Kapahi et al. 2004)[191,192] | Four interconnected nutrient-sensing pathways (insulin/IGF-1 signaling (IIS), mTOR, AMPK, and sirtuins) become dysregulated with age, shifting the balance between anabolic growth and catabolic maintenance. Attenuation of the pro-growth pathways (IIS, mTOR) or activation of the energy-scarcity sensors (AMPK, sirtuins) extend lifespan across model organisms from yeast to mice. | Nutrient-sensing pathways are encoded regulatory circuits that translate metabolic state into cellular behavior. Their dysregulation with age is a downstream consequence of information corruption in the genes encoding these pathways and their regulators. In our framework, the reason caloric restriction extends lifespan is mechanistically straightforward: reduced metabolic throughput means fewer electrons through the electron transport chain, less ROS generation, slower NIC production, and consequently slower information corruption. The nutrient-sensing pathways are the regulatory interface through which this reduction is transduced into cellular responses: reduced protein synthesis (less energetic cost, less proteotoxic stress), enhanced autophagy (better clearance of damaged organelles), and shifted metabolic flux. However, caloric restriction cannot halt information corruption entirely because endogenous NIC production from sources other than mitochondrial respiration (spontaneous depurination, deamination, endogenous alkylation) continues regardless of metabolic rate. This explains why caloric restriction extends lifespan but does not abolish aging. |
| Altered Intercellular Communication (Conboy et al. 2005; Rando & Chang 2012)[51,193] | Aging involves progressive deterioration of the signaling environment between cells, including endocrine, neuroendocrine, and paracrine communication. The senescence-associated secretory phenotype (SASP) converts local damage signals into tissue-wide and systemic inflammatory cascades. Extracellular vesicles, gap junction signaling, and circulating factors all shift towards pro-inflammatory, pro-senescent profiles with age. | Intercellular communication is the mechanism through which information corruption at the cellular level propagates to the tissue and organismal levels, the connective tissue of the hierarchical cascade. When senescent cells broadcast SASP factors, or when damaged neurons release aberrant signals, the information corruption that began in individual genomes becomes a systemic force affecting cells whose own genomes may still be relatively intact. This is how the hierarchy amplifies: a single corrupted cell can alter the behavior of neighbors through paracrine signaling. The heterochronic parabiosis experiments, which show that young blood can partially rejuvenate old tissues and old blood can accelerate aging in young tissues, demonstrating that systemic signaling environment is a powerful modulator of the corruption cascade, even though it is not the initiating cause. |
| Dysbiosis (O’Toole & Jeffery 2015; Ghosh, Shanahan & O’Toole 2022)[194,195] | The gut microbiome undergoes age-related shifts in composition and function: decreased diversity, loss of beneficial commensals (Bifidobacterium, butyrate-producing Firmicutes), and expansion of pro-inflammatory taxa (Proteobacteria). These changes increase intestinal permeability, activate systemic immune responses, and alter microbial metabolite profiles, contributing to inflammaging, metabolic dysfunction, and even neurodegeneration via the gut-brain axis. Recognized as one of the three new hallmarks of aging in the 2023 update. | The gut microbiome represents a large, semi-autonomous population of replicators (bacterial genomes) whose information integrity and compositional balance are maintained by the host’s protectosphere (immune surveillance, epithelial barrier function, antimicrobial peptides). As the host’s information corruption degrades immune function and epithelial integrity, the microbiome shifts towards dysbiosis, another instance of corruption propagating across hierarchical levels. In turn, dysbiotic microbiota produce metabolites and signals that increase systemic inflammation, generate additional ROS, and may directly influence host epigenetic state through short-chain fatty acid signaling. This creates a host-microbiome feedback loop consistent with our model’s prediction that corruption at one hierarchical level accelerates corruption at others. Notably, dysbiosis is not universal across all life (it requires a gut microbiome), making it an amplifier specific to organisms with complex digestive systems rather than a universal cause of aging. |
| GROUP F: Integrative Frameworks and Mathematical Models | ||
|
Theory (canonical ref/name) |
Original Scope | Role in our unified framework |
| Network Theory of Aging (Kowald & Kirkwood 1996; Kriete, Bosl & Booker 2010)[196,197] | Aging arises from interconnected feedback loops between multiple damage and response processes rather than from any single cause. Kowald and Kirkwood computationally linked the free radical theory to the protein error theory, showing that ROS-driven mitochondrial damage and aberrant protein accumulation form a self-amplifying vicious cycle. Kriete et al. extended this with a fuzzy-logic cell model incorporating positive feedback loops (damage amplification) and negative feedback loops (adaptive stress responses via NF-κB and mTOR), demonstrating that the aging phenotype emerges from the interplay between damage accumulation and protective countermeasures. | The Network Theory correctly identified that aging theories should not be treated as competing alternatives and that feedback loops between damage processes produce emergent aging dynamics. Our framework improves on this foundation in three ways. First, we assign causal and temporal order to the network, identifying nucleobase modification as the initiating event, mutation as its heritable consequence, and epigenetic drift as its regulatory amplifier, rather than treating all damage types as co-equal nodes. Second, we distinguish the molecular substrate that matters: only nucleic acid corruption drives aging permanently because proteins and lipids turn over, while sequence changes propagate through all descendant cells. Third, we provide the evolutionary and thermodynamic grounding (the prime directive, the disposable soma logic, Shannon/Eigen/Landauer/drift barrier constraints on copying fidelity) that explains why the network is structured as it is and why evolution permits its eventual failure. The Network Theory models what happens during aging; this framework proposes why, in what order, and at what molecular level each node contributes to the loss of intropy. |
| Hallmarks of Aging (López-Otín et al. 2013, 2023)[18,19] | Taxonomy of nine (later twelve) hallmarks: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, disabled macroautophagy, chronic inflammation, dysbiosis. | López-Otín’s work is a comprehensive catalog of aging phenomena that our framework orders causally. Each hallmark maps to a specific position in the corruption cascade: genomic instability and telomere attrition are intermediate steps; epigenetic alterations are downstream amplifiers; loss of proteostasis, mitochondrial dysfunction, and deregulated nutrients sensing reflect information-directed functional decline; cellular senescence and stem cell exhaustion are hierarchical consequences; inflammation and dysbiosis are emergent system-level amplifiers. The hallmarks framework catalogs what happens during aging; this framework explains why, places each hallmark in causal order, and identifies each as a facet of the progressive loss of intropy. |
| Reliability Theory of Aging (Gavrilov & Gavrilova 2001)[198] | Applies engineering reliability theory to biological systems: aging results from progressive failure of redundant components, producing increasing hazard rates consistent with Gompertz dynamics. | The Reliability Theory of Aging is mathematically compatible with our hierarchical model. Their theory predicts that systems with redundant components and imperfect elements show increasing failure rates, which is precisely what our hierarchical cascade produces. In our framework, the “components” are cells, tissues, and organs whose functional capacity degrades as information corruption in the underlying template accumulates; redundancy (diploidy, stem cells, tissue reserves) delays but cannot prevent eventual crossing of failure thresholds. Reliability theory provides the mathematical shape (Gompertz); our framework provides the molecular mechanisms driving component failure (see EN86 for how the reliability framework’s “initial flaws” and component degradation rate map onto the standing intropy deficit at I₀ and the rate of intropy loss, dI/dt). |
| Hyperfunction / Quasi-programmed Aging (Blagosklonny 2006, 2008)[199,200] | Aging is driving by the continued activity of developmental growth programs (especially mTOR) beyond their useful period, causing cellular hypertrophy, senescence, and organ pathology. | This theory describes an amplification mechanism consistent with our framework. mTORC1 activation promotes mitochondrial biogenesis and respiration, increasing ROS production, while simultaneously suppressing autophagy, reducing clearance of damaged mitochondria and protein aggregates. The combined effect is increased NIC production alongside decreased protectosphere function. In our framework, this is an instance of antagonistic pleiotropy at the molecular level: growth programs selected for early-life fitness become damage amplifiers later. Rapamycin’s lifespan-extending effects are consistent with this interpretation, as mTOR inhibition reduces oxidative DNA damage, enhances autophagic clearance, and suppresses the senescence-associated secretory phenotype, each of which maps to either reducing NIC production or restoring protectosphere capacity. However, rapamycin does not address the underlying modification burden, consistent with the observation that its effects, while significant, are partial. |
| Programmed Aging Theories (Skulachev 1997, Goldsmith 2004, Longo, Mitteldorf & Skulachev 2005)[201,202,203] | Aging is an evolved program, actively triggered by genetic mechanisms to benefit the population by removing older individuals. | Our framework does not support programmed aging. No dedicated pathway has been identified whose sole function is to initiate organismal decline. Pathways invoked as “aging programs” (mTOR, insulin/IGF-1 signaling) are growth and nutrient-sensing programs whose late-life effects are better explained by antagonistic pleiotropy. In our framework, aging is not programmed but is instead the inevitable consequence of thermodynamic constraints on information fidelity and the evolutionary logic of disposable soma. |
| DNA Damage Theory of Aging (Schumacher, Pothof, Vijg & Hoeijmakers 2021)[45] | Positions DNA damage as the initiating and unifying driver of aging, arguing that inherited repair defects (progeroid syndromes), the age-associated rise in lesions and strand breaks, gene-length-biased transcriptional decline, and lifespan extension produced by enhanced repair together identify nuclear DNA damage as causally upstream of every recognized hallmark. | Schumacher, Pothof, Vijg, Hoeijmakers, and colleagues’ synthesis is the closest empirical neighbor to the intropy framework and, like our framework, places nuclear DNA integrity at the root of the hallmarks of aging. We extend the framework at the level of mechanism and formalization. First, we distinguish the initiating event (covalent nucleobase modification) from its heritable consequence (mutation) and from its structural extremes (e.g., strand breaks), three categories that DNA damage theory tends to treat as a single class; the separation resolves why replication-fidelity defects accelerate cancer without accelerating aging while transcription-blocking modifications produce progeroid pathology. Second, we ground the inevitability of corruption in Shannon and Landauer as theoretical-level constraints and in the drift barrier and an Eigen-like informational collapse threshold as practical limits on selectable fidelity, supplying a thermodynamic rationale for why corruption cannot be eliminated and a rate-setting argument for why its accumulation is species-specific. Third, we connect molecular-level corruption to Gompertz-Makeham mortality through the stochastrophe argument, deriving exponential age-specific mortality from the stochastic accumulation of linear intropy loss. Fourth, we extend the integrative reach beyond the López-Otín hallmarks to roughly thirty theories of aging, positioning each as a node in a causal cascade (see Table 1). Fifth, we decompose the path from modification to functional decline into seven readout channels (see EN81), providing mechanistic granularity that a “damage affects most hallmarks” formulation does not. Sixth, we distinguish two routes to stem cell decline, quality loss through information corruption and quantity loss through DDR-mediated exhaustion, and identify empirical tests (DNMT3A clonal hematopoiesis versus POLG mutator mice) that separate them (see EN36). Seventh, we develop a protectosphere calibration argument extending Kirkwood’s disposable soma (see EN29b): actively maintained defenses are tuned to just past the reproductive window, producing coordinated late-life deterioration that presents as many independent mechanisms but reflects a single scaling variable, the rate of intropy loss. Eighth, we reframe the underlying biology as an interaction among three characters, the replicator, the protector, and the corruptor, which separates intervention points at three levels rather than collapsing them into damage and repair. DNA damage theory describes the empirical consequences of corruption inside the cell; the intropy framework proposes the variable whose loss those consequences reflect (realized intropy, I(t)), the evolutionary calibration of its maintenance, the mortality mathematics that couples its linear loss to exponential death, the readout taxonomy through which its loss is effected, and the conceptual architecture from which all those consequences follow. |
Table 2.
Steps and evidence for the intropy framework. The framework presented herein lends itself to several suppositions. Listed here are these suppositions and the most compelling evidence to support them. Where evidence is lacking, the status of the step is referenced. Whereas most of the individual steps are falsifiable with one or a series of experiments, the framework as a whole may not be from any single experiment. “Premise” means foundational claims we assert. “Mechanism” means how the system works that follows from the premises. “Corollary” means the logical consequences of the mechanisms. “Prediction” means empirically testable claims that follow from the framework.
Table 2.
Steps and evidence for the intropy framework. The framework presented herein lends itself to several suppositions. Listed here are these suppositions and the most compelling evidence to support them. Where evidence is lacking, the status of the step is referenced. Whereas most of the individual steps are falsifiable with one or a series of experiments, the framework as a whole may not be from any single experiment. “Premise” means foundational claims we assert. “Mechanism” means how the system works that follows from the premises. “Corollary” means the logical consequences of the mechanisms. “Prediction” means empirically testable claims that follow from the framework.
| # | Supposition | Evidence/Status |
| 1 | All life stores heritable information in nucleic acid. The linear sequence of nucleobases constitutes a form of chemical memory that directs the ordering of biological processes. (Premise) | Universal across all known cellular life.[65,92] No known exceptions. |
| 2 | The fundamental imperative of all nucleic acid replicators is to copy (the prime directive). All biological structures and functions, from enzymes to organisms to ecosystems, can be viewed as protections enabling this copying (Premise). | Built from Dawkins’ selfish gene framework and Hamilton’s inclusive fitness; the prime directive is a selection-filter shorthand for the observation that biological structures persist only when they support replication of the heritable substrate (FN7). Replication is thermodynamically favored once self-catalyzing chemistry arises. The achievability premise is supported by ribozyme-mediated templated polymerization, autocatalytic networks, and the early appearance of life in the geological record. |
| 3 | Perfect biological fidelity is unattainable. Shannon’s noisy-channel theorem allows arbitrarily low error through sufficient redundancy below channel capacity, but Landauer’s principle and kinetic proofreading impose physical costs that finite biological systems cannot fully afford. Life exploits redundancy extensively (duplex structure, diploidy, cascaded repair) but residual error remains irreducible. (Premise) | Shannon (1948);[12] Landauer (1961);[8] Hopfield (1974).[204] Polymerase error rates 10⁻⁷ to 10⁻¹⁰ per base per replication even with proofreading and mismatch repair.[75] |
| 4 | Two constraints set the practical floor on error rate. The drift barrier prevents selection from improving fidelity once the fitness benefit of further reduction falls below ~1/Ne (effective population size), and some minimal error rate is required to generate the variation on which adaptive selection depends. Complex life therefore operates within a bounded window between an Eigen-like informational error ceiling and the drift barrier’s floor, with errors accumulating at non-zero rates throughout life. (Premise) | Across the tree of life, total per-site mutation rates per generation scale inversely with Ne, with bacteria around 10⁻¹⁰ and multicellular eukaryotes around 10⁻⁹ to 10⁻⁸,[14,15,112] consistent with the drift barrier acting on the integrated output of fidelity and repair systems rather than on polymerase fidelity in isolation. Eigen (1971) sets the upper bound, with empirical support from viral quasispecies experiments demonstrating error catastrophe at high mutagen doses.[14] |
| 5 | Nucleobases are chemically reactive. They undergo covalent modification by electronically imbalanced atoms and molecules (corruptors), producing adducts that alter information content. (Premise) | Tens of thousands of endogenous modifications per cell per day: ~10,000 AP sites, ~500 8-oxoG, plus alkylation, deamination, aldehyde adducts, lipid peroxidation products.[63,205] The molecular evidence for this is very strong. |
| 6 | Nucleobase reactivity is highest when bases are physically exposed: during replication, transcription, and chromatin remodeling. The model does not preclude modified incoming nucleotides being incorporated into the growing strand, nor other points of DNA exposure. (Premise) | Mutational signatures show replication-timing and strand biases; late-replicating regions accumulate more mutations.[206,207] Cytosine deamination is ~100-fold faster in single-stranded DNA.[208] Methylated CpG sites are particular hotspots because 5-methylcytosine deamination produces thymine, evading uracil-glycosylase repair (source of SBS1). |
| 7 | Adduct formation is sequence- and nucleobase-dependent. Some atoms and positions on the nucleobase ring are far more vulnerable to modification than others. (Premise) | Guanine’s low redox potential makes it the primary oxidation target; CpG sites are deamination hotspots; trinucleotide context determines mutational signatures.[209] Strong molecular evidence and follows logically from core principles of reactive chemistry. |
| 8 | The rate of nucleic acid modification depends on the local chemical environment, including corruptor concentration, detoxification capacity, and metal availability, and varies by cell type, tissue, and species. (Premise) | Tissue-specific mutation rates vary several-fold within a single organism.[210,211] Cross-species somatic mutation rates inversely correlate with lifespan.[210] Strong evidence and also follows from basic principles of chemistry/buffering and local environmental conditions. |
| 9 | Energy metabolism is a primary source of corruptors, an energetic tradeoff we term Eden’s Apple: extracting chemical energy from substrates necessarily generates reactive byproducts. Reactive oxygen, nitrogen, and sulfur species, endogenous aldehydes, and alkylating agents are produced through oxidative phosphorylation, NADPH oxidase activity, and peroxisomal fatty acid oxidation. Other endogenous processes (histone demethylation, SAM-driven methylation, spontaneous hydrolysis) contribute additional corruptor flux. (Mechanism) | Mitochondrial ROS production.[212,213] Endogenous formaldehyde from histone demethylation comparable to exogenous exposure.[214] NADPH oxidase-derived ROS.[215] Peroxisomal fatty acid oxidation.[216] SAM-derived non-enzymatic methylation.[217] Lipid peroxidation products.[218] Strong evidence that energy metabolism is a primary source of endogenous corruptors; relative contributions of specific subsystems remain an active area of investigation. |
| 10 | The framework predicts that the most probable corruptors are small, stable, highly diffusible, and ubiquitous molecules whose concentration cannot be locally suppressed without disrupting metabolism. (Mechanism) | Reactive oxygen species, endogenous aldehydes, and alkylating agents satisfy these criteria and are the dominant identified endogenous corruptors. Diffusion-limited damage chemistry produces effects within nanometers of generation,[219] so corruptor proximity to DNA matters as much as bulk flux. Specific corruptor identities and their relative contributions are an active area of investigation. |
| 11 | Most adducts are repaired by dedicated pathways (BER, NER, direct reversal). Only a small fraction persists at any given time, constituting the standing modification burden. (Mechanism) | BER processes tens of thousands of lesions per cell per day. Steady-state 8-oxoG is ~1-2 per 10⁶ guanines.[220] NER-exclusive endogenous substrates, particularly 8,5’-C from hydroxyl radical attack, are less frequent but transcription-blocking and accumulate with age in a tissue-specific manner.[221,222] Total endogenous DNA damage estimated at 10⁴-10⁵ events per cell per day.[63,205] Very strong evidence exists. |
| 12 | The standing modification burden is functionally consequential even before any mutation occurs. Aging-relevant impact is set by the steady-state burden of readout-disruptive modifications, weighted by tissue context and persistence. Transcription-blocking modifications (bulky adducts, DPCs, ICLs, AP sites, SSBs from incomplete BER) cause the strongest acute effects through Pol II stalling and gene silencing. Transcription-permissive modifications (such as 8-oxoG read-through) contribute through transcriptional mutagenesis, epigenetic remodeling at CpG sites, and conversion to mutations during replication. Severity depends on abundance, persistence, and gene context. (Mechanism) | Csb−/− and XpdTTD mice develop premature aging without elevated mutation frequencies,[223] inconsistent with mutation accumulation as the sole driver (FN51, FN76). Modified bases cause RNA Pol II pausing and misincorporation.[224] Cockayne syndrome and UV-sensitive syndrome (UVSS) share TC-NER defects, but only CS shows progeria, with the difference tracking Pol II stall duration rather than mutation rate.[225,226] Multiple repair-deficient progeroid syndromes (CS, XPA, ERCC1-XPF, FA, SPRTN) all involve impaired processing of nucleic acid modifications, with phenotype severity apparently tracking persistence of unrepaired lesions rather than mutation rate. |
| 13 | A fraction of unrepaired modifications cause replicative polymerases to misincorporate a nucleotide, converting a lesion into a permanent mutation. This conversion is mediated primarily by translesion synthesis polymerases and generates the clock-like mutational signature SBS5/SBSB-like. (Mechanism) | REV7 knockout eliminates SBS40 in TK6 cells, closest in-vitro relative of SBS5.[227] SBS1 arises from spontaneous deamination of 5-methylcytosine, a purely chemical modification process. SBS5/SBSB captures multiple damage sources funneled through shared TLS.[227,228] |
| 14 | Once a mutation is fixed on both DNA strands, it is irreversible by normal cellular mechanisms and inherited by all descendant cells. Mutations are the most stable form of nucleic acid information corruption, arising from unrepaired modifications or from replication-fidelity errors. (Mechanism) | Foundational molecular biology. Confirmed by single-cell sequencing showing clonal expansion of somatic mutations.[210,211] Thus, mutation is one way in which information irreversibly changes/corrupts. |
| 15 | Modifications alter the epigenome directly: for example, OGG1 at oxidized CpG sites can recruit TET1 to drive demethylation or recruit DNMT to induce methylation, depending on context. Mutations alter the epigenome indirectly: permanent sequence changes at or near CpG sites remodel local methylation patterns. The epigenome is a downstream reporter of both modification burden and mutational load. (Mechanism) | Mutations at CpG sites coincide with extensive methylome remodeling; mutation-based age predictions parallel epigenetic clock estimates.[118] 8-oxoG/OGG1 drives demethylation via TET1[229,230,231] or methylation via DNMT [232] depending on context. Cockayne syndrome fibroblasts show approximately 15.5 years of accelerated epigenetic age relative to the pooled non-progeroid group (UVSS plus healthy controls) on the Horvath Skin & Blood clock, despite shared TC-NER defects and no elevated mutation rates.[225] Within this framework, the difference is consistent with the rate of Pol II stalling at unrepaired transcription-blocking lesions as the relevant scaling variable. Strong evidence that epigenetic alterations both disrupt normal gene regulation acutely and produce heritable changes in information integrity that persist across cell divisions. |
| 16 | The combined effect of modifications, mutations, and epigenetic alterations on chromatin structure and gene regulation creates positive feedback: corruption of repair and maintenance genes reduces the cell’s capacity to prevent further corruption, accelerating the cascade. This is the repair-decline feedback loop. (Mechanism) | Direct feedback evidence: DNA repair genes undergo broad transcriptional repression in senescent cells and in fibroblasts from older donors;[233] oxidative damage at repair gene promoters can recruit DNMTs through OGG1, MSH2-MSH6, and EZH2-related machinery (FN89), establishing a route by which corruption suppresses its own repair. Supporting accumulation evidence: ERCC1-deficient mice show accelerated aging with oxidative damage levels comparable to normal aging;[221,234,235] age-related decline in BER enzymes;[236,237] 8-oxodG accumulates with age across tissues;[238,239] cyclopurines accumulate tissue-specifically;[221] somatic mutations accumulate linearly across 16 mammalian species.[210] |
| 17 | Modifications at CpG sites can trigger BER-mediated demethylation or inhibit DNMT activity, creating a hemimethylated intermediate. If the cell divides before re-methylation restores the mark, one daughter inherits a fully unmethylated CpG that DNMT1 will not re-methylate. Transient modifications thus produce irreversible regulatory changes inherited by all descendant cells. (Mechanism) | 8-oxoG within CpG recognition sites decreases Dnmt3a activity up to 25-fold.[240] DNMT1 has strong preference for hemimethylated substrates and very low activity on fully unmethylated CpGs.[241,242] It has also recently been confirmed that mutations at CpG sites drive extensive methylome remodeling.[118] Strong evidence that division-mediated propagation converts transient modification into heritable epigenetic change. |
| 18 | Each successive cell division adds new mutations to the existing burden, while modifications are typically resolved through repair, replication-fork-collapse-induced cell loss, or conversion to mutation during translesion synthesis (FN83). The mutation ratchet is unidirectional: information loss accumulates and cannot be reversed by normal cellular mechanisms. (Corollary) | Muller’s ratchet applied to somatic lineages. Consistent with linear accumulation of clock-like signatures, particularly SBS1, with age across tissues.[210] |
| 19 | Corruption of progenitor cells (stem cells) has outsized consequences in proliferative tissues because every daughter cell in the lineage inherits the corrupted template. Stem cell corruption is a major driver of tissue-level functional decline in tissues that depend on continued cell turnover. (Corollary) | Clonal hematopoiesis: single DNMT3A or TET2 mutations in HSCs produce tissue-wide mosaicism.[48,120,243] Permanent information corruption at progenitor levels propagates up the biological hierarchy because all descendant cells inherit the corrupt template. |
| 20 | Non-dividing (post-mitotic) cells age through standing modification burden causing transcriptional stress and epigenetic drift, bystander effects from corrupted dividing cells, and progressive failure of systemic support, but not through the replication-dependent mutation ratchet. Mutations still accumulate in post-mitotic cells through repair-associated DNA synthesis, but at rates and in patterns distinct from dividing-cell accumulation. This explains why post-mitotic tissues such as brain and heart are long-lived yet eventually fail. (Corollary) | In C. elegans, where all adult somatic cells are post-mitotic, TC-NER is specifically required for somatic maintenance [244] and declines with age in muscle.[245] Formaldehyde tolerance in the post-mitotic adult soma requires TC-NER.[246] ADH-1 overexpression extends lifespan.[247] Post-mitotic tissues demonstrate that modification-driven aging proceeds through readout corruption and standing burden, independently of replication. |
| 21 | The unequal distribution of corruption across the genome, cell types, and tissues means some sites and compartments are disproportionately consequential. There exist driver modifications, driver mutations, and driver tissues (those whose corruption produces outsized organismal consequences, such as brain, heart, and hematopoietic stem cells) whose corruption has outsized effects on organismal function and survival. Driver modifications are most often those that block transcription or replication in critical genes, though transcription-permissive modifications can also drive aging through mutagenesis, epigenetic remodeling, and conversion. (Corollary) | Analogous to the driver/passenger distinction in cancer.[248] GG-NER is attenuated in post-mitotic neurons while TC-NER is maintained,[249] making these cells dependent on transcription-coupled repair for endogenous damage clearance. Neurodegeneration is the hallmark of TC-NER deficiency in CSB and CSA patients.[250] The “driver” terminology is borrowed from cancer biology and used here in an analogical sense, denoting outsized phenotypic consequence rather than clonal selection advantage. |
| 22 | Biological organization is hierarchical: genome → protein → cell → tissue → organ → organism. Functional inefficiency at any level can propagate to higher levels, with amplification when the failed component is poorly redundant or occupies a load-bearing position in the architecture. (Premise) | Standard biological organization. The amplification principle is a foundational result in reliability engineering.[251] This concept is foundational in systems engineering and there is strong evidence it follows for biology. |
| 23 | Approximately linear erosion of protectosphere integrity by NIC-driven corruption produces exponentially increasing mortality (Gompertz dynamics) because progressively thinned protective capacity converts stochastic challenges into threshold failures with increasing probability, a coupling we term stochastrophe. Death occurs when a challenge finds a protectosphere thinned past the threshold required to withstand it. (Mechanism) | Gompertz-Makeham mortality dynamics are observed across nearly all adult animal populations.[252] Gavrilov & Gavrilova (2001) demonstrated that hierarchical redundant systems with constant-rate component failure produce Gompertz kinetics;[198] the present framework reinterprets the biological content of that mathematics, identifying the protectosphere as the shield whose thinning rate (c) determines the Gompertz slope (see FN86). The framework’s specific reinterpretation (linear protectosphere thinning by NIC-driven corruption producing exponential mortality via stochastrophe) is testable through interventions that alter NIC flux, repair capacity, or tolerance, with the prediction that true anti-aging interventions should reduce the Gompertz β parameter (see suppositions 38, 39). |
| 24 | Nucleobase modifications corrupt DNA information through at least seven distinct channels (see FN81 for the full taxonomy), including transcriptional silencing (Pol II stalls at helix-distorting modifications, suppressing gene output), transcriptional mutagenesis (Pol II reads through small modifications with misincorporation, producing miscoded mRNA and aberrant protein), replicative mutagenesis (modifications encountered during replication are converted into permanent heritable mutations), and regulatory disruption (modifications at regulatory sites alter regulation or epigenetic marks). The consequence of any given modification depends on its genomic location, the cellular process engaging that locus, and whether repair clears it before the next readout event. (Mechanism) | Pol II stalling: ~40% of Pol II stalled in aged mouse liver,[143] with corresponding gene-length-dependent transcriptome shifts.[144] Transcriptional mutagenesis: a single 8-oxoG lesion produces continuous miscoded transcripts.[145,146] Replicative mutagenesis: SBS40 eliminated by REV7 knockout in TK6 cells.[227] Regulatory disruption: 8-oxoG at CpG sites triggers BER-mediated demethylation.[229,230] See FN81 for treatment of all seven channels. These channels reflect current evidence on how DNA-level informational corruption propagates. Relative weights, and the existence of additional channels, await further empirical characterization. |
| 25 | Since the information that directs the construction and maintenance of the protectosphere is itself encoded in nucleic acid, corruption of that information reduces the cell’s capacity to protect against further corruption. The ordered integrity of actively maintained protective systems progressively declines. (Corollary) | Age-related decline is documented across multiple protectosphere components: glutathione and antioxidant capacity ,[253] proteasome function,[254] autophagy and mitophagy,[255,256] DNA repair gene expression,[233,236] iron homeostasis,[257] and immune surveillance. ERCC1-deficient mice show dramatically accelerated aging when one critical component (NER) collapses,[235] confirming that protectosphere integrity is required for normal lifespan. |
| 26 | Any catastrophic alteration of the genome leading to severe loss or gain of function is selected against at the cellular level (apoptosis, senescence) or organismal level (lethality). Tissue examined at any time point therefore represents cells that survived prior corruption, not the full history of damage. This generates survivorship bias. (Corollary) | p53-dependent apoptosis and damage-induced senescence eliminate cells carrying excessive DNA damage, so any tissue sample reflects survivors of prior corruption rather than the full damage history.[258] High embryo attrition in mammalian cloning by somatic cell nuclear transfer makes the mismatch directly visible.[259,260] |
| 27 | The corruption channels of Supposition 24 propagate to organismal decline through downstream pathways that vary by tissue, modification type, and repair context. Three are particularly consequential: inflammatory signaling from unresolved modifications activating cGAS-STING; replication failure from fork collapse and DSBs driving stem cell exhaustion; and regulatory dysregulation from corruption of non-coding elements. (Mechanism) | Inflammatory signaling: cGAS knockout in SPRTN-deficient mice rescues kyphosis and lipodystrophy but only partially rescues other features.[261] Replication failure: Fanconi anemia fork collapse drives bone marrow failure;[262] ERCC1-XPF deficiency depletes hematopoietic stem cells through replication stress.[235] Regulatory dysregulation: 8-oxoG within CpG sites inhibits DNMT3a up to 25-fold [240] and triggers BER-mediated demethylation,[229,231] remodeling local methylation patterns at modified regulatory regions. |
| 28 | The effects on cell function are progressive but nonlinear: small increments of information corruption can be buffered, while corruption in driver genes, repair systems, stem cells, or post-mitotic tissues can produce disproportionate functional decline. Aging is the physical manifestation of progressive but tolerable corruption of information. (Corollary) | Consistent with Gompertz-Makeham mortality dynamics. Late-life mortality deceleration in some species may reflect heterogeneity in individual corruption rates.[263] |
| 29 | The corruption is tolerable because intolerable corruption (catastrophic information loss) is lethal and therefore selected against. Any adaptation that promotes tolerance to corruption is favored. Corruption that exceeds the evolved tolerance threshold produces either beneficial adaptation (when heritable and germline) or pathology (when somatic, especially cancer). (Corollary) | Cancer can be viewed as a somatic cell reverting to its own prime directive at the expense of the organismal hierarchy. p53 and DNA damage response pathways represent evolved tolerance mechanisms. The same tolerance-threshold logic extends to other pathologies linked to information change, varying with which information is affected (essential versus regulatory) and which level of selection acts on it (germline versus somatic). |
| 30 | Evolution calibrates the protectosphere to sustain information integrity through the reproductive window, not indefinitely. The soma is disposable once the prime directive is executed. The protectosphere is calibrated, not maximized, because the marginal reproductive benefit of additional repair/protection capacity approaches zero after the reproductive window while the metabolic cost remains constant. (Mechanism) | Kirkwood’s disposable soma (1977).[165] End-of-life mutation burdens converge within ~3-fold across 16 mammalian species despite 30-fold lifespan differences, indicating evolutionary calibration.[210] Hamilton (1966) formalized the declining force of selection with age.[264] This is an evolutionary concept of what would be expected if life was attempting to extend its reproductive window when faced with progressive information corruption. |
| 31 | In species examined, germline lineages accumulate fewer mutations per unit time than somatic lineages. This reflects coordinated adaptations protecting the transmitted copy: reduced replication frequency, piRNA silencing of mobile elements, meiotic checkpoint elimination of damaged germ cells, and two-wave epigenetic reprogramming. Lower replication is not separate from the protection. It is part of it, since copying is itself a major source of corruption. (Mechanism) | Germline mutation rates are 1-2 orders of magnitude lower than somatic rates in mammals.[265,266] Two waves of epigenetic reprogramming (germline and early embryo) reset the methylome. Meiotic checkpoints eliminate germ cells with excessive damage. These coordinated germline protections follow from the corridor of life (FN9, Figure 2). In multicellular organisms, holding the transmission lineage tightly within the corridor while permitting the soma to drift toward and above its upper bound is the efficient allocation when universal corridor-tight fidelity is unaffordable. |
| 32 | A lifespan is the balance between the rate of information corruption and the evolved capacity to tolerate it. Death occurs as cumulative inefficiency thins the protectosphere past the capacity required to withstand stochastic challenges, with the probability of lethal failure rising as capacity declines (see FN86). (Corollary) | Consistent with Gompertz-Makeham dynamics. The capacity-and-stochastic-failure framing is developed in FN86; the framework’s threshold concept is analogous to (rather than a direct application of) Eigen’s quasispecies error catastrophe,[14] since somatic cells do not replicate as quasispecies. Cumulative modification burden may also serve as a clock-like predictor of time-to-failure if tissue-specific tolerance thresholds can be calibrated. |
| 33 | Informational change is also the substrate of adaptation. Changes that enhance the prime directive in the germline are selected; changes that degrade somatic function drive aging. Aging and evolution are thus products of the same underlying process (information change under selection), differing in whether the change is heritable across generations and subject to germline selection, or confined to somatic lineages and subject only to within-organism selection. (Premise) | Standard evolutionary theory applied to this framework. Germline mutations provide adaptation; somatic mutations provide aging. Both arise from the same chemical processes acting on the same substrate. |
| 34 | All life that uses chemical information to construct order is subject to this process. The framework is not restricted to terrestrial biology; any replicator operating under thermodynamic constraints in a chemically reactive environment will experience information corruption and functional decline. (Corollary) | Follows from Suppositions 1, 3, and thermodynamic constraints. The framework’s specific mechanisms apply to nucleic-acid-based life from which intropy arises; the underlying corruption logic extends to any replicator under thermodynamic constraints. |
| 35 | The modification-processing channel and the replication-error channel are mechanistically separable. Defects that increase only replication-fidelity errors should elevate mutation rates without accelerating aging, producing cancer predisposition alone. Defects that impair processing of transcription-blocking or replication-blocking modifications should produce premature aging, sometimes without elevating mutation frequency. (Prediction) | Confirmed in both directions. Mutations without aging: POLE/POLD1 proofreading-domain variants produce 10-100x elevated mutations without premature aging;[267] Pms2-null mice have 100-fold elevated mutations with no accelerated aging.[268] Aging without mutations: Csb−/− and XpdTTD mice show premature aging with normal mutation frequencies (see FN51, FN76).[223] Boundary case: MUTYH heterozygotes show 2.5-fold elevated SBS18 in tumors and modestly elevated CRC risk without premature aging,[269] consistent with elevated lesion-to-mutation conversion in BER without elevated transcription-blocking modification burden. |
| 36 | Mutational signatures (SBS1, SBS5/SBSB, SBS18/SBSC) are archived outputs of lifetime nucleobase modification flux, not direct drivers of aging. For each signature class, observed mutation burden approximates the time integral of lesion production rate multiplied by the probability of lesion-to-mutation conversion, where the conversion probability is itself a function of repair efficiency, replication frequency, and bypass mechanism. Most modifications act transiently through readout failure, repair stress, epigenetic remodeling, or lesion persistence; only a small fraction is archived as fixed mutations. The framework therefore treats SBS signatures as measurable proxies for the otherwise hard-to-measure lifetime modification flux. (Mechanism) | Cagan et al. (2022) found inverse correlation between annual somatic mutation rate and lifespan across 16 mammalian species, with end-of-life burdens converging within ~3-fold despite ~30-fold lifespan variation.[210] The dominant signatures were SBS1 (5mC deamination), SBSB (resembling SBS5), and SBSC (resembling SBS18), with SBS1 and SBSC mechanistically attributable to nucleobase modification chemistry rather than replication-fidelity errors. Hwang et al. (2025) show that REV7 knockout in TK6 cells eliminates the SBS5/SBS40-like background and reveals SBS18 as roughly 32% of remaining mutations, identifying polymerase zeta translesion synthesis as a shared funnel through which distinct lesion classes converge on a common signature footprint.[227] Spisak et al. (2025), in a preprint, extend this funneling logic by modeling multiple endogenous and exogenous damage sources as inputs to the SBS5 signature.[228] |
| 37 | Within architecturally similar groups, somatic mutation rate should inversely correlate with lifespan, extending Cagan et al. (2022) beyond mammals. Across architecturally dissimilar groups, raw correlation should weaken with differences in repair architecture, body temperature, regenerative modularity, genome size, and functional target size; an architecture-corrected corruption index recovering the relationship across classes is a research program rather than a sharp prediction. (Prediction) | Cagan et al. (2022) established the within-mammal inverse correlation across 16 species.[210] Bergeron et al. (2023) found ~40-fold germline mutation-rate variation across 68 vertebrate species, supporting the expectation that mutation-rate architecture varies more between classes than within them.[266] Within-class somatic sequencing in reptiles, birds, and fish would directly test the near-term prediction; the cross-class adjusted index awaits operational specification. |
| 38 | True anti-aging interventions should reduce the slope of SBS5/SBSB and SBS18/SBSC accumulation over time. Interventions that merely reduce external mortality (lifestyle changes that limit exposure without altering endogenous lesion flux, medical interventions that reduce death from specific causes) should extend survival without changing signature accrual rates. (Prediction) | CR reduces oxidative stress, enhances autophagy, and upregulates repair,[270] all of which should reduce SBS5/SBS18 slope; direct measurement under CR is untested. Rapamycin and mitochondrial aldehyde detoxification enhancement are predicted to do the same. Although this uses SBS taxonomy as a readout, the underlying principles should apply to most non-productive information change. |
| 39 | Controlled exposure to agents that produce persistent transcription-blocking or repair-stalling nucleic-acid modifications should cause dose-dependent acceleration of Gompertz-Makeham mortality dynamics, primarily through elevation of β, the age-dependent rate-of-increase parameter. Acute toxicity at high doses should instead appear as increased early/background mortality or a shifted mortality intercept rather than as true aging acceleration. (Prediction) | ERCC1-/Δ mice show accelerated aging from steady-state accumulation of NER substrates. Csb-/- and XpdTTD mice show progeroid phenotypes without elevated mutation frequencies. Chemotherapy survivors show accelerated aging alongside elevated SBS5 in HSPCs.[271] Endogenous formaldehyde rises with age in WT mouse hippocampus and correlates with memory decline.[272] Controlled dose-titration in tractable model organisms with paired Gompertz-Makeham parameter estimation, adductomics, readout-stalling measures, and SBS decomposition would constitute a decisive quantitative test. |
| 40 | The aging severity of any repair deficiency should correlate with the duration of readout blockade rather than with total modification burden or repair pathway identity. Current evidence weights this most heavily on transcriptional readout (Pol II stall duration), with replicative and other readout-blockade mechanisms contributing through parallel but distinct routes. (Prediction) | NER severity gradient: XPC/DDB2 loss (GG-NER only) produces cancer without aging; loss of shared NER factors (XPA, XPD, ERCC1-XPF) produces aging. CS vs UVSS contrast: both share TC-NER defects, but CS traps stalled Pol II for hours while UVSS clears it efficiently;[226] CS patients develop severe progeria, UVSS patients show no aging.[273] Mouse Csb-null phenotypes are mild because alternative Pol II clearance pathways compensate; severe aging emerges only when global-genome NER is co-disrupted, with Csa−/−/Xpa−/− double knockouts dying by ~20 weeks with severe neurodegeneration (FN51). BER paradox: glycosylase knockouts (OGG1, NEIL1/2/3 triple KO) produce no aging; XRCC1 loss creates transcription-blocking intermediates and causes severe aging.[274] ~40% of Pol II stalled in aged mouse liver with long-gene bias.[143] |
| 41 | SBS5/SBSB accumulation reflects modification processing rate, not cell division rate, because replication fork bypass, BER long-patch repair, and NER gap-filling all use error-prone polymerases whose error spectra converge on this signature family. Post-mitotic tissues should therefore accumulate SBS5/SBSB at substantial rates, and TLS-deficient cells should show reduced SBS5/SBSB even under elevated lesion exposure. The family is a biomarker of modification processing, not a driver of aging. (Prediction) | The post-mitotic accumulation prediction is met: SBS5/SBSB accumulates in human cortical neurons at rates comparable to dividing tissues.[275] The TLS-dependence prediction is directly confirmed in TK6 cells, where REV7 knockout eliminates the SBS5/SBS40-like background signature, identifying polymerase zeta translesion synthesis as the polymerase whose error spectrum generates this signature family.[227] Spisak et al. (2025), in a preprint, extend this by reporting that neuronal SBS5 shows little dependence on NER-rate variation along the genome (α ≈ 0, γ > 0), consistent with repair-error-driven generation, whereas more rapidly dividing colonic epithelial cells show stronger repair-rate sensitivity, suggesting a greater replication/TLS-over-lesion contribution in dividing tissue.[228] |
| 42 | Caloric restriction extends lifespan through multiple mechanisms that converge on two framework-compatible effects: reducing NIC production and enhancing the protectosphere. The framework predicts that CR should reduce the rate of SBS5/SBSB accumulation. (Prediction) | CR extends lifespan from yeast to primates.[270] Identified mechanisms (reduced ROS, enhanced autophagy, upregulated repair, reduced IGF-1/insulin signaling) all map to reduced NIC production or enhanced protectosphere. XpdTTD mice spontaneously develop CR-like metabolic features when repair is insufficient,[276] consistent with adaptive downshift of NIC production. Direct test in Ercc1∆/− progeroid mice: 30% dietary restriction tripled both median and maximal remaining lifespan, preserved neurons and motor function, reversed gene-length-biased transcriptional decline, and reduced γH2AX foci, establishing that lowering damage flux rescues a repair-deficient progeroid phenotype.[277] SBS5/SBSB reduction under CR is untested. |
| 43 | Replacing corrupted stem cells with less-corrupted ones (heterochronic transplantation) should partially rescue tissue function, but the rescue should be temporary because transplanted cells enter an environment with elevated NIC burden. The degree of rescue should be proportional to the completeness of replacement. (Prediction) | Young bone marrow transplantation with >90% engraftment preserves cognitive function in old mice.[278] Non-myeloablative transplantation at ~19% chimerism shows limited cognitive and behavioral benefit,[279,280] consistent with proportionality. Heterochronic parabiosis with detachment yields rejuvenation that fades over months (~6-week median lifespan extension),[281] supporting the temporary sub-claim by analogy. Corruption rates in donor cells within old hosts have not been directly measured. An inference from the hierarchy framework: anti-aging solutions are difficult to evaluate because any single intervention is undercut by uncorrected corruption elsewhere, suggesting the most effective interventions may be those acting early in development without disrupting it. |
| 44 | Partial reprogramming (OSK/OSKM) should provide only transient epigenetic benefit because the underlying modification and mutation burden continues to produce new drift. Any sustained benefit likely arises from non-epigenetic effects (repair activation, damaged cell clearance, reduced inflammation) rather than epigenetic rejuvenation alone. (Prediction) | Reprogramming extends lifespan in LAKI mice,[282] an HGPS model where progerin disrupts chromatin through mechanical stress, not modifications; benefit may reflect restoration of structural shielding. In vitro reprogramming of Ercc1-/Δ cells (modification-driven progeria) primarily upregulates DNA repair pathways rather than resetting epigenetic marks, with greater benefit than in WT cells,[283] directly supporting the prediction that repair activation, not epigenetic reset, drives improvement. Reportedly extends lifespan in old WT mice.[284] Epigenetic age reboundsafter factor withdrawal.[285] In vivo lifespan extension in repair-deficient progeroid models has not been tested. |
| 45 | SBS5/SBSB is the most informative single-signature archive of aging-relevant modification flux. Cross-species lifespan and within-tissue accumulation should track this archive more strongly than any single lesion class, repair pathway, or alternative signature. (Prediction) | SBSB scales inversely with lifespan across 16 mammals.[210] SBS5-like accumulates linearly with age in post-mitotic neurons [275] Polζ activity generates the SBS5/SBS40-like background signature;[227] SBS5 is interpreted as collateral mutagenesis funneled through shared TLS/repair pathways.[228] Untargeted adductomics identifies age-dependent endogenous adducts handled by Polζ as candidate upstream lesions.[286] Cross-species adductomics-SBS correlation is the cleanest near-term test of this archive’s primacy (see S48 for the broader paired-measurement program). |
| 46 | No single signature or measure should fully predict biological age. A composite index combining SBS1, SBS5/SBSB, SBS18/SBSC contributions, standing adduct burden (mass spec adductomics), repair gene expression, and tissue tolerance architecture should predict biological age, tissue dysfunction, and mortality more strongly than existing epigenetic clocks or raw mutation burden alone. (Prediction) | Existing biomarkers (Horvath clock, GrimAge, PhenoAge) predict mortality with modest accuracy. The framework predicts that a composite incorporating upstream (lesion flux, adduct burden) and downstream (signature archives, repair status) measures should outperform existing methylation-based clocks. Empirical comparison is untested. |
| 47 | All well-characterized systemic progeroid syndrome involves impaired modification processing, disruption of nuclear architecture supporting DNA maintenance, or telomere dysfunction. The intropy framework predicts that any newly characterized progeroid syndrome will fall into one of these categories, with nucleobase modifications as the upstream causal variable. (Prediction) | Modification-processing: NER deficiency (XPA, XPD, ERCC1-XPF, CSA, CSB), ICL repair (Fanconi anemia, FANCD2), DPC repair (SPRTN). Nuclear architecture: HGPS (LMNA/progerin), Nestor-Guillermo progeria (BANF1). Telomere dysfunction: dyskeratosis congenita (DKC1, TERC, TERT). Replication fidelity defects (POLE, POLD1, MMR) produce cancer without premature aging,[267,268] confirming the modification-processing channel as the aging-relevant axis. Reprogramming temporarily reverses but does not prevent epigenetic drift.[285] Age-related repair gene decline driven by an active transcriptional repressor [233] precedes downstream epigenetic and mutational changes, while CpG-site mutations drive surrounding methylome remodeling,[118] together positioning nucleobase modifications upstream of both mutation accumulation and epigenetic remodeling. |
| 48 | The decisive empirical test of the framework is direct measurement of standing modification burden across tissues, ages, and intervention conditions, paired with mutational signature decomposition. (Prediction) | Tens of thousands of modifications per cell per day [63,205] versus tens to hundreds of somatic mutations per cell per year across mammals [210] (~47 in humans, ~800 in mice) make adductomics the highest-resolution available measure of the framework’s central variable. Existing methods include mass spectrometry-based adductomics for major lesion classes, AP-Seq for abasic sites, OxiDIP-Seq for oxidative damage genome distribution, and emerging single-cell approaches. Paired adductomics and SBS sequencing in matched tissues across age, exposure, and intervention conditions would test whether sustained modification burden, measured longitudinally or in matched same-age subjects with different exposure histories, predicts future SBS5/SBSB and SBS18/SBSC accumulation and functional decline more strongly than chronological age or raw mutation burden alone. |
| 49 | Substantial slowing or prevention of aging will likely require technological control over the base information systems that govern corruption, repair, and readout. (Corollary) | Follows from the framework’s causal ordering. No current intervention addresses the root cause (nucleobase modification) directly. The challenge is distinguishing modifications that disrupt the plan from the normal developmental and regulatory information changes that constitute ordered biology, with the right balance varying by tissue, time, and information class. It is somewhat analogous to servicing and rebuilding a jet engine with the aircraft still in flight. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



