Submitted:
30 June 2026
Posted:
01 July 2026
You are already at the latest version
Abstract
Physical performance can be understood as a continuum throughout the life course, ranging from peak athletic ability in early life to the preservation of mobility and functional independence in old age. This narrative review explores whether the biological and genetic pathways involved in athletic performance might also modulate the risk of geriatric motor dysfunctions (GMDs), including sarcopenia, frailty and lower-limb weakness. The available evidence suggests a convergence between performance and motor decline in mechanisms such as mitochondrial function and mitophagy, anabolic-catabolic balance, oxidative stress and low-grade chronic inflammation, neuromuscular integrity, satellite cell function, mechanotransduction, myokine-mediated signalling, and the gut-muscle axis. Although classic candidate genes such as ACTN3 or ACE have been useful for formulating mechanistic hypotheses, genome-wide association studies support a highly polygenic architecture for strength, lean mass, muscle weakness and frailty. These effects are strongly modulated by the exposome, particularly by physical activity, nutrition and comorbidities. Overall, the relationship appears consistent with predominantly beneficial pleiotropy, although context-dependent effects cannot be ruled out. Genetics may influence functional reserve and decline trajectories, but exercise, particularly strength and power training, along with adequate nutrition and the management of comorbidities, remain the primary strategies for preventing or delaying sarcopenia, frailty and lower-limb weakness.
Keywords:
sarcopenia
; frailty
; athletic performance
; genetics
; pleiotropy
; functional reserve
; gut microbiota
; ageing
; redox signalling
; inflammageing
1. Introduction: From Elite Performance to Functional Independence: A Lifelong Continuum
Physical performance exists along a continuum ranging from peak physical capabilities in youth to the maintenance of functional independence in old age. This transition reflects not only inevitable age-related changes, but also complex interactions between genetic factors, environmental exposures and lifestyle determinants that shape quality of life and the functionality of the musculoskeletal system throughout a person’s life. Over recent decades, biomedical and sports science research has shown that the biological systems that determine athletic performance (cardiorespiratory capacity, energy metabolism, muscle strength and power, etc.) are essentially the same critical systems that aid in the preservation of mobility, the prevention of falls and the maintenance of independence in older adults [1,2]. Furthermore, the role of physical exercise as one of the most effective ways of both improving a person’s physical condition and maintaining good health as they age has been widely documented [3]. Data from studies on longevity in elite athletes further support the existence of a biological link between athletic performance and healthy ageing. A comprehensive systematic review involving more than 465,000 elite athletes revealed that former athletes, particularly those who participated in endurance and mixed sports, tend to have lower mortality rates and greater longevity compared to the general population [4]. For example, it has been shown that runners capable of completing a mile in under four minutes live approximately five years longer than the general population [5]. These findings suggest that the physiological systems underlying high physical performance may also contribute to functional resilience and long-term survival.
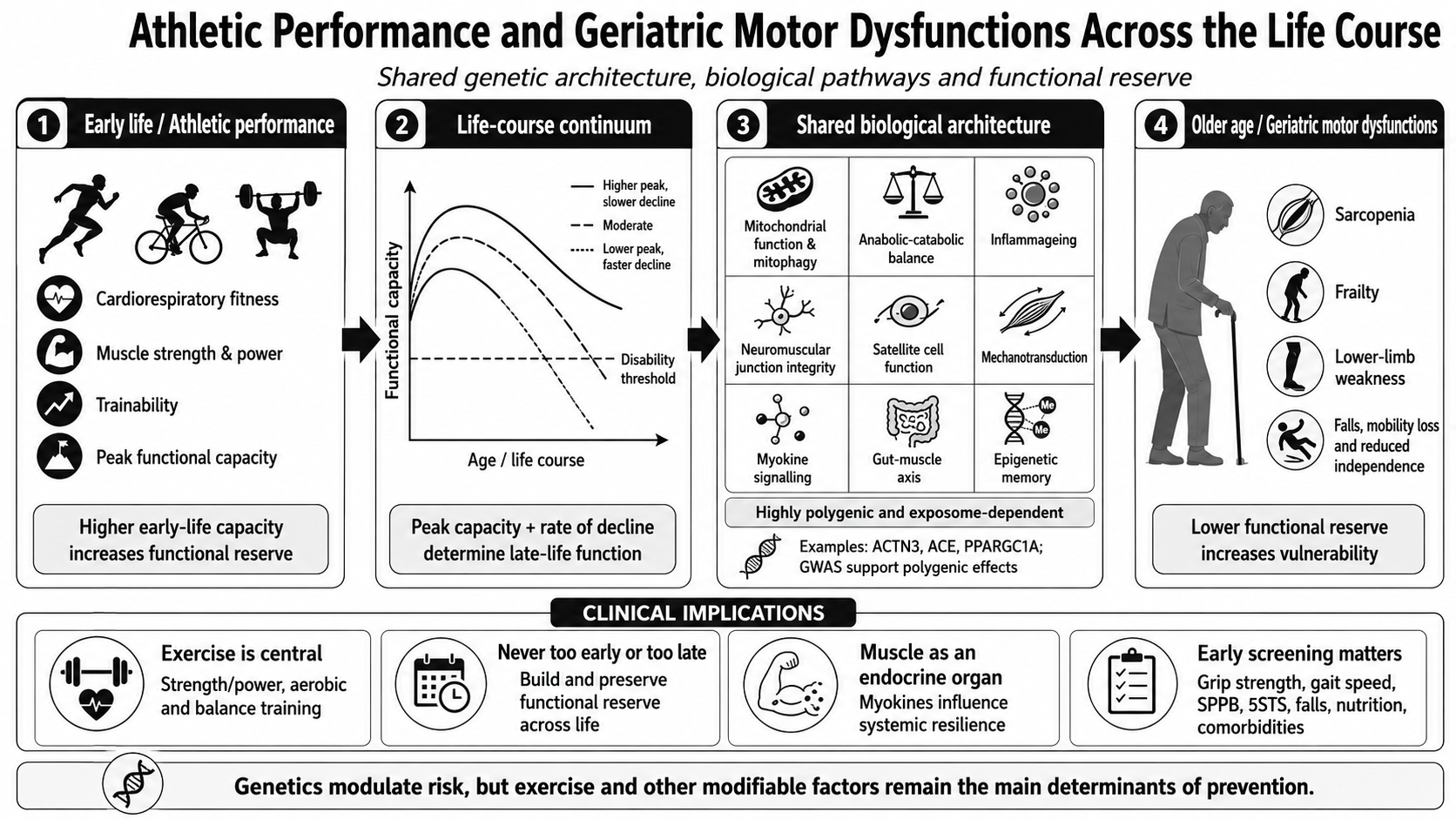
The concept of functional reserve, analogous to cognitive reserve in neuroscience, provides a unifying framework for understanding this continuum. Moreover, functional reserve should be interpreted as the physiological capacity to tolerate stressors while maintaining functional independence. This concept overlaps partially with intrinsic capacity and physical resilience, although it emphasises the dynamic reserve available to preserve mobility and autonomy under conditions of ageing, disease or inactivity [6,7]. Put simply, functional reserve refers to the difference between an individual’s maximum physiological capacity and the minimum threshold required to carry out activities of daily living [8,9]. People who reach higher peaks of aerobic capacity and muscle strength in early adulthood have greater functional reserve, meaning they can lose more absolute function before crossing the threshold into disability [10,11]. This perspective suggests that the peak functional capacity achieved in youth and the subsequent rate of decline are critical determinants of the development of age-related motor dysfunction, such as sarcopenia, frailty or lower-limb weakness, conditions that we group in this review under the term “geriatric motor dysfunctions” (GMDs) (Figure 1).
Furthermore, there is longitudinal evidence to support this framework. In a Swedish cohort of approximately 1.14 million men, higher handgrip strength measured between the ages of 16 and 19 was associated with a substantially lower rate of premature mortality (before the age of 55), even after adjusting for body mass index and blood pressure [12]. Similarly, the CARDIA study showed that higher cardiorespiratory fitness in early adulthood and its maintenance into middle age predicted lower cardiovascular mortality and morbidity decades later [13]. Additional evidence from elite athletic populations further reinforces this perspective, with former elite athletes and individuals capable of exceptional endurance performances exhibiting greater longevity compared with the general population. Collectively, these findings suggest that physical capacity in youth may not only reflect athletic potential, but also broader biological robustness, physiological resilience and the capacity to maintain functional reserve across ageing.
Importantly, the biological systems underlying athletic performance and healthy ageing appear to overlap substantially [14,15]. Pathways regulating mitochondrial function, neuromuscular integrity, anabolic signalling, mechanotransduction, metabolic flexibility, redox homeostasis and chronic low-grade inflammation are involved both in exercise adaptation and in the preservation of mobility and independence during ageing. Within this network, oxidative stress and inflammation should be considered interdependent processes: transient redox-inflammatory signals contribute to tissue repair and training adaptation, whereas chronic activation promotes tissue dysfunction and progressive functional decline [16,17,18,19,20,21,22,23,24]. However, despite growing evidence linking exercise-related phenotypes with ageing trajectories, these domains have traditionally been studied separately, limiting our understanding of the shared mechanisms that may connect peak physical performance with late-life functional outcomes.
Therefore, this narrative review aims to synthesise the current evidence on the biological and genetic overlap between sports performance phenotypes and geriatric motor dysfunctions. Specifically, this review focuses on the role of functional reserve, pleiotropy, polygenic influences and life-course trajectories in shaping musculoskeletal function, physical resilience and functional ageing. In doing so, we propose a conceptual framework in which athletic performance and geriatric motor dysfunctions may represent distinct phenotypic manifestations of common biological systems operating across the lifespan.
2. Literature Review and Selection Strategy
This review has been designed as a narrative and integrative review. The literature was identified through iterative searches in PubMed, Web of Science and Scopus, with the last search conducted on 20 May 2026. To enhance transparency and reproducibility, the search strategy was structured into four conceptual blocks: (i) physical performance phenotypes (e.g., muscle strength, power output, gait speed, VO₂max, athletic performance), (ii) age-related functional decline phenotypes (e.g., sarcopenia, frailty, dynapenia, lower-limb weakness), (iii) biological mechanisms (e.g., mitochondrial function, mitophagy, oxidative stress, redox signalling, antioxidant responses, inflammageing, redox-inflammatory pathways, anabolic signalling, neuromuscular integrity, mechanotransduction and metabolic flexibility), and (iv) genetic and epigenetic determinants (e.g., GWAS, polygenic scores, ACE, ACTN3, PPARGC1A, exercise genomics, epigenetic memory and redox-sensitive regulatory pathways). These blocks were combined using Boolean operators (AND/OR) to maximise sensitivity while preserving conceptual specificity. No time restrictions were applied, although priority was given to publications from the last decade, along with studies relevant to the conceptual development of the field. Preference was given to articles published in English, international consensus documents, genome-wide association studies, meta-analyses of large biobanks, randomised clinical trials, longitudinal studies and high-impact mechanistic reviews.
The selection of references was based on their conceptual relevance, methodological rigour and contribution to the development of the narrative framework of this review. Priority was given to key documents on diagnostic criteria for sarcopenia and frailty, such as EWGSOP2, AWGS2 and SDOC, as well as recent studies on muscle strength, lean body mass, weakness, frailty, mitochondrial biology and the epigenetics of exercise. Given the narrative and integrative nature of this review, no formal risk-of-bias assessment or quantitative synthesis was performed. However, to minimise selection and citation bias, studies were critically appraised based on methodological robustness, sample size, reproducibility of findings, and conceptual contribution to the proposed integrative framework. The synthesis process followed an inductive approach, whereby evidence was organised thematically and iteratively until conceptual saturation was achieved across the domains of physical performance, biological ageing and genetic-environmental interactions.
The evidence identified was therefore organised around three main themes: shared functional phenotypes, converging biological pathways, and genetic-environmental modulators of functional reserve.
3. The Link Between Performance and GMDs: Common Phenotypes
In the field of sport, it is common to assess performance in terms of the primary metabolic and physical demands of the activity in question, such as cardiorespiratory endurance, maximum strength, muscular power, training adaptation or mechanical efficiency [15,25]. In people with geriatric motor dysfunctions (GMDs), physical abilities are usually assessed using specific tests, such as the Short Physical Performance Battery (SPPB), the Five Times Sit-to-Stand Test (5STS), measurement of walking speed, or handgrip strength [26]. Although the terminology used may differ, many of these tests or indicators are based on the same biological systems or global physical capability [27,28].
For example, cardiorespiratory endurance depends on the integration of oxygen transport and utilisation, ranging from cardiovascular function to the muscle’s oxidative capacity. In athletes, these capabilities are commonly expressed as maximum oxygen uptake (VO2max), running economy or time to exhaustion [15]. In an older person, the same biology determines the ability to walk, climb stairs or recover from an acute illness [26]. This reflects a systems-level continuity in aerobic capacity, where maximal physiological performance in youth transitions into submaximal functional independence in older age [14]. Similarly, muscle strength and power act as decisive factors in many sports, but also in the ability to stand up from a chair, prevent a fall or maintain a steady gait [29]. The trainability of physical capacities adds yet another layer of information to this continuum. For example, the HERITAGE study showed that the response of VO2max to a standardised aerobic training programme exhibits considerable inter-individual variability, with a genetic component that could account for up to 49% of the variance in VO2max trainability [30]. This inter-individual variability in training responsiveness supports the concept of “functional reserve plasticity”, whereby the adaptive capacity of physiological systems is partially genetically determined and expressed across both athletic and ageing populations [31,32]. In this regard, functional reserve plasticity may also be relevant in older adults: two individuals exposed to the same exercise stimulus, such as a strength training programme, may show different increases in muscle mass, strength or physical function. This variability is likely to reflect the combined influence of previous activity history, baseline functional status, metabolic health, comorbidities, nutrition and genetic predisposition.
When we consider the extreme end of physical capabilities in old age, one of the most defining characteristics is frailty. One possible criterion for identifying it is the Fried Phenotype, which groups together five key criteria: weight loss, exhaustion, weakness, lack of mobility and a low level of physical activity; a person is considered frail if they meet three or more of these criteria [33]. Another widely used measure is the Rockwood Frailty Index, which focuses on the accumulation of various issues affecting an individual’s overall health and enables classification on a frailty scale [34]. These two operational frameworks capture complementary dimensions of frailty, with the Fried phenotype emphasising physical frailty as a biological syndrome, and the Rockwood index conceptualising frailty as an accumulation of multidimensional deficits reflecting systemic decline [33,34]. Both methods reveal two sides of the same coin: on the one hand, measurable physical decline; and on the other, the loss of the ability to perform various everyday tasks or the development of dependency.
Another condition that has gained significant importance in recent years is sarcopenia, initially understood as the loss or lack of muscle mass in older people. According to the consensus established by the European Working Group on Sarcopenia in Older People (EWGSOP2), low muscle strength is the initial criterion, followed by confirmation of low muscle quantity or quality, and finally by an impact on physical performance that increases the severity of the diagnosis [35]. The AWGS2 Asian consensus takes the same approach, adapting the various diagnostic thresholds to Asian anthropometric characteristics using a community-based screening algorithm [36]. Furthermore, the SDOC (Sarcopenia Definition and Outcomes Consortium) proposes that handgrip strength and walking speed should be considered as primary diagnostic criteria for clinical trials, thereby prioritising muscle function over muscle mass [37]. All three consensus statements agree on the importance of strength and speed as key predictors, as these are essential abilities in a wide range of strength- and/or power-based sports. Collectively, these definitions reflect a conceptual shift from a morphology-centred view of sarcopenia towards a function-centred paradigm, aligning clinical diagnosis more closely with performance-based metrics used in sports science.
Finally, weakness in the lower limbs can be identified using tests similar to those mentioned above, such as walking speed, the SPPB or the 5STS; and it is an excellent indicator for predicting disability, institutionalisation and mortality. Taken together, the GMDs reflect a biological reality of the same process: functional and physical decline over the years (Figure 2) [38].
4. The Genetic Architecture Underlying Physical Performance and Decline: From Candidate Genes to Polygenic Traits
Genetics in sports performance began to gain prominence following the identification of the first genetic associations in athletes. Polymorphisms such as the I/D variant in the gene encoding angiotensin-converting enzyme (ACE) or the R577X variant in the gene encoding alpha-actinin-3 (ACTN3) were the first to pave the way for the study of other candidate genes [39,40]. ACE has been studied for its potential role in endurance or strength sports, with results varying over time; whereas ACTN3 is particularly associated with performance in strength and/or power sports [41,42,43].
This same logic can be applied to GMDs: variants that may promote a greater response to hypertrophy, oxidative efficiency or response to training could enhance the ability to maintain strength or mobility in old age. However, candidate gene studies have certain limitations: small sample sizes, small effects, replication issues and a tendency to overinterpret the role of isolated variants. These studies have been useful for identifying potential biological pathways involved, but despite this, they are not sufficient to explain the complexity of performance or its role in GMDs.
The emergence of GWAS studies enabled a shift from a candidate gene approach to a polygenic approach, where specific traits can be studied in an agnostic manner with very broad genomic coverage. Traits related to muscle strength, lean body mass, walking speed, or response to exercise appear to be highly polygenic, depending on many variants with very small individual effects. For example, the GWAS conducted by Willems et al. in over 195,000 participants identified 16 significant loci associated with grip strength, providing the first evidence of Mendelian randomisation of a causal effect of muscle strength on fracture risk [44]. Tikkanen and colleagues, using data from UK Biobank, confirmed a genetic architecture distributed across loci involved in sarcomere organisation, calcium signalling and neurological development [45]. Jones and colleagues recently published one of the first genome-wide association studies (GWAS) specifically focusing on muscle weakness in older adults, involving around 256,000 participants. They identified 15 independent loci with genes implicated in neurological and muscular function and provided evidence, through Mendelian randomisation, of a causal link between weakness and frailty [46]. At the same time, other GWAS studies of appendicular lean body mass in the UK Biobank identified more than 250 associated SNPs, implicating muscular and neurological signalling pathways [47]. Finally, previous studies on body composition have identified the Wnt/β-catenin pathway and bone remodelling as key factors in determining lean body mass [48]. This growing body of evidence highlights the need to move beyond the concept of a ‘performance gene’ or a ‘sarcopenia gene’, and to focus instead on studying the networks underlying musculoskeletal development, neuromuscular signalling, metabolism and cellular regulation.
Importantly, many of the molecular pathways associated with elite physical performance overlap with the biological hallmarks of ageing. Mechanisms involving mitochondrial biogenesis, proteostasis, autophagy, cellular senescence, chronic low-grade inflammation and genomic stability are critically involved both in exercise adaptation and in age-related functional decline [49,50]. Exercise has been proposed as one of the most potent non-pharmacological modulators of these hallmarks, suggesting that some exercise-related genotypes may exert pleiotropic effects across the lifespan by simultaneously influencing athletic capacity and biological ageing trajectories.
Polygenic risk scores (PRS) have emerged as an attempt to synthesise precisely this genetic complexity, integrating the effect of various variants into an estimate of predisposition to a particular trait, phenotype or condition. Theoretically, these scores could help identify individuals with lower muscle reserve or a higher risk of functional decline before any symptoms appear. In practice, their usefulness remains limited, as they can only account for a portion of the variance, have been developed primarily in European populations, and offer only modest improvement over simple functional markers such as grip strength or walking speed. For example, the LASA study found no association between the PRS established for grip strength and frailty in the subjects, whereas direct measures of strength did show a robust association, suggesting that non-genetic factors accounted for much of the association [51]. Therefore, the current value of PRSs related to muscle function is greater as a research tool than as a population-based screening tool.
Recent advances in genome-wide association studies and polygenic risk score methodologies further support the notion that physical performance and ageing phenotypes share a highly polygenic architecture. Rather than depending on single “performance genes”, complex traits such as muscle strength, aerobic fitness, frailty or walking speed are now understood to result from the cumulative interaction of thousands of variants with small individual effects [45,52]. This paradigm shift has substantial implications for exercise prescription and healthy ageing, opening the possibility of precision exercise medicine approaches tailored according to individual biological responsiveness and functional reserve profiles.
Nevertheless, genetic predisposition alone is insufficient to explain variability in performance or ageing outcomes. Increasing evidence indicates that environmental exposures across the lifespan, including nutrition, physical activity, sleep, psychosocial stress and socioeconomic conditions; interact dynamically with the genome through epigenetic and transcriptomic mechanisms [53]. This interaction between genome and exposome may partially explain why individuals with similar genetic backgrounds can exhibit markedly different ageing trajectories or exercise responses. Accordingly, ageing-related motor dysfunctions should not be interpreted solely as consequences of chronological ageing, but rather as emergent phenotypes resulting from cumulative gene–environment interactions over time.
Emerging multi-omics approaches integrating genomics, epigenomics, metabolomics, proteomics and gut microbiome profiling are beginning to provide a more comprehensive understanding of the biological networks underlying both elite performance and functional decline [54,55]. These integrative models may help identify shared molecular signatures associated with adaptability, recovery capacity and neuromuscular resilience, thereby offering novel biomarkers for both sports performance optimisation and early detection of age-related functional deterioration.
5. Key Biological Pathways: The Mechanisms Behind the Overlap
Although the pathways discussed below are presented separately for explanatory purposes, they operate as highly interconnected biological networks rather than independent mechanisms. Recent advances in systems biology suggest that functional reserve emerges from the collective behaviour of multiple interacting systems, including mitochondrial energetics, neuromuscular integrity, immune regulation, mechanotransduction and tissue repair. Consequently, athletic performance and geriatric motor dysfunctions may not represent distinct biological entities but rather different phenotypic expressions of the same underlying network architecture operating under different levels of physiological resilience [14,56]. From this perspective, frailty can be interpreted as a state of network dysregulation characterised by reduced redundancy, impaired adaptability and diminished recovery capacity following stressors, whereas elite athletic performance may represent the opposite extreme of biological robustness and reserve capacity.
Many of the biological pathways studied in the field of sports science are the same as those studied in relation to age-related physical decline [49,57]. Rather than a linear correlation between ‘performance genes’ and ‘sarcopenia genes’, current evidence points to partially overlapping functional networks linking bioenergetics, proteostasis, immunometabolism, neuromuscular integrity, mechanotransduction, muscle endocrine communication and epigenetic regulation. In this context, classic genes such as ACTN3, ACE or PPARGC1A must be interpreted alongside genes identified by GWAS and recent functional studies, such as IRS1, PIK3R1, FTO, STC2, CPNE1, NEB, RIF1, HLA-DRB1/DRB5, IL6R, FOXP1, ZBTB38, PIEZO1, YAP1/WWTR1 and other components of the PINK1/Parkin pathway; these help to explain why strength, power, lean mass, walking speed and frailty share a distributed biological basis [47,58,59].
5.1. Mitochondrial Function, Redox Homeostasis and Mitophagy
In athletes, the oxidative capacity of the muscle determines endurance, recovery and tolerance to repeated exertion. In older adults, mitochondrial dysfunction is associated with reduced ATP production, increased oxidative stress, impaired mitophagy, greater accumulation of mutated mtDNA and a decline in muscle quality [60]. PGC-1α acts as a key regulator of mitochondrial biogenesis and links aerobic exercise, oxidative metabolism and muscle ageing [57]. Exercise activates, in a coordinated manner, mitochondrial biogenesis via PGC-1α and selective mitophagy via PINK1/Parkin, ensuring the renewal of a functional mitochondrial network [61].
At the genetic level, this pathway can be organised into three modules. The first relates to mitochondrial biogenesis, regulated by PPARGC1A/PGC-1α, NRF1, NRF2/GABPA, TFAM, ESRRA and SIRT1/AMPK, which control the expansion of the mitochondrial network, mitochondrial genome transcription and oxidative capacity. Importantly, NRF2/GABPA, involved in mitochondrial biogenesis, is mechanistically distinct from NFE2L2/Nrf2, the KEAP1-regulated transcription factor that coordinates antioxidant and cytoprotective responses. Accordingly, NRF2/GABPA is considered here in the context of mitochondrial biogenesis, whereas NFE2L2/Nrf2 is discussed in relation to redox homeostasis and antioxidant defence. The second corresponds to mitochondrial dynamics, dependent on fusion and fission genes such as MFN1, MFN2, OPA1, DNM1L/DRP1 and FIS1, whose balance maintains a mitochondrial network that is adaptable to training and resistant to metabolic stress. The third module relates to mitophagy and mitochondrial quality control, where PINK1, PRKN/Parkin, BNIP3, BNIP3L/NIX, FUNDC1, SQSTM1/p62, OPTN and LC3/MAP1LC3B enable the recognition and elimination of damaged mitochondria [59,62]. Mitochondrial biogenesis promotes endurance, recovery and energy efficiency in early life, whilst the loss of mitophagy, mitochondrial fragmentation and the accumulation of damaged mtDNA contribute to fatigue, reduced anabolic capacity, sterile inflammation and a decline in muscle quality in old age. Furthermore, the PPARGC1A–AMPK–SIRT1–PINK1/Parkin pathway directly links exercise, energy restriction, selective autophagy and muscular ageing. Therefore, mitochondrial function should not be viewed solely as a pathway for aerobic performance, but as a cellular maintenance system that determines functional reserve throughout the entire life course.
Other key genes involved in these processes include those of the PPAR (peroxisome proliferator-activated receptor) family, such as PPARA, PPARD and PPARG; and the aforementioned PPARGC1A, which encodes PGC-1α. The peroxisome proliferator-activated receptor alpha is expressed in skeletal muscle and brown adipose tissue, amongst other tissues, playing an important role in fatty acid oxidation and glucose metabolism. Under conditions of energy deprivation, the PPARA gene is activated to promote the uptake and utilisation of fatty acids as an energy source. PPARD is probably the subtype with the greatest direct relevance in skeletal muscle, where it has been linked to the regulation of lipid oxidation, mitochondrial biogenesis, resistance to fatigue and the transition towards a more oxidative phenotype of muscle fibres. PPARG is most highly expressed in adipose tissue and is a key regulator of adipogenesis, lipid storage and insulin sensitivity. Although its direct role in muscle fibre is less dominant than that of PPARA or PPARD, it indirectly influences muscle function by modulating lipid distribution, systemic inflammation, adipokine secretion, and the risk of lipotoxicity and insulin resistance. Finally, PGC-1α acts as an integrator of all these responses. It functions as a co-activator of multiple transcriptional programmes related to mitochondrial biogenesis, oxidative respiration, angiogenesis, defence against oxidative stress, and the conversion to fibres that are more resistant to fatigue. Exercise, particularly aerobic training and repeated contractile stimuli, induces the expression of PGC-1α in skeletal muscle, facilitating the expansion of the mitochondrial network and improving oxidative capacity [63]. Taking all of the above into account, greater efficiency in substrate utilisation, improved mitochondrial plasticity and a more robust adaptive response to training could contribute both to performance and to reduced vulnerability to the onset of age-related motor dysfunctions.
Beyond their role in energy production, mitochondria act as signalling organelles capable of communicating metabolic stress to distant tissues through the release of mitochondria-derived peptides (MDPs), including humanin, mitochondrial open reading frame of the 12S rRNA-c (MOTS-c) and small humanin-like peptides (SHLPs). These molecules have emerged as regulators of insulin sensitivity, inflammation, exercise adaptation and healthy ageing. In particular, circulating MOTS-c concentrations have been associated with exercise responsiveness and metabolic resilience, suggesting that mitochondrial communication may constitute an additional link between athletic performance and functional ageing trajectories [64,65,66].
A further dimension of this mitochondrial pathway is redox signalling and its relationship with the endogenous antioxidant response. Mitochondrial reactive oxygen species (ROS) are not merely toxic by-products; at low or moderate levels, they act as second messengers that activate adaptive programmes, including mitochondrial biogenesis and stress resistance [67,68,69]. However, the age-related imbalance between ROS production and antioxidant neutralisation leads to a state of chronic oxidative stress that progressively compromises mitochondrial and cellular function. The Nrf2/KEAP1 pathway is fundamental to the endogenous antioxidant response: under conditions of oxidative stress, Nrf2 translocates to the nucleus and induces the transcription of cytoprotective genes encoding superoxide dismutase, catalase, glutathione peroxidase and heme oxygenase-1 [70,71]. With ageing, both the basal activity of Nrf2 and its inducibility decline, reducing the capacity to buffer mitochondrial ROS and contributing to the accumulation of oxidative damage in skeletal muscle [72,73]. It is crucial to note that mitochondrial ROS also act as upstream activators of the NLRP3 inflammasome, providing a direct mechanistic link between mitochondrial dysfunction, oxidative stress and the chronic inflammatory state characteristic of inflammageing [74,75]. Exercise, by transiently elevating ROS and activating Nrf2 in a hormetic manner, represents a major non-pharmacological strategy for supporting redox homeostasis and mitigating this mitochondria-to-inflammation cascade across the life course [76,77,78].
5.2. Anabolic-Catabolic Balance
Muscle mass and strength depend on the balance between protein synthesis and breakdown, which is regulated by the IGF-1/PI3K/Akt/mTOR, myostatin/activin, ubiquitin-proteasome (FoxO-regulated atrogenes) and autophagy pathways [79]. In early life, efficient anabolic signalling facilitates hypertrophy and adaptation to training. In old age, anabolic resistance due to reduced activation of mTORC1 and myofibrillar protein synthesis in response to equivalent stimuli, reduced mechanical stimulation and the activation of catabolic pathways (atrogin-1, MuRF-1) contribute to the loss of muscle strength and mass.
At the genetic level, this pathway includes a central anabolic axis comprising IGF1, IGF1R, INSR, IRS1, PIK3R1, AKT1, mTOR, RPTOR, RHEB, EIF4EBP1 and RPS6KB1, responsible for activating protein translation and myofibrillar hypertrophy. Conversely, the catabolic axis includes FOXO1, FOXO3, FBXO32/atrogin-1, TRIM63/MuRF1, MSTN, ACVR2A/ACVR2B, SMAD2/3, GDF15 and autophagy genes such as BECN1, ATG5, ATG7 and ULK1. Recent genetic evidence identifies IRS1 and PIK3R1 as particularly relevant genes, as their variants are associated with lean mass, fat mass and body composition, supporting a muscle-adipose pleiotropy linking sarcopenia, sarcopenic obesity and insulin resistance [58,80,81]. This metabolic link is reinforced by Mendelian randomisation studies, which suggest a bidirectional relationship between traits associated with sarcopenia and type 2 diabetes, supporting the view that insulin resistance is not merely a comorbidity, but a potential causal modulator of muscle function decline [82].
Therefore, strength performance and the prevention of age-related muscle loss do not depend solely on ‘having more muscle mass’, but on maintaining the muscle’s sensitivity to mechanical, nutritional and hormonal anabolic signals. Anabolic resistance in old age can be understood as a loss of efficiency in the insulin/IGF-1–PI3K–Akt–mTOR axis, exacerbated by inflammation, inactivity, gut dysbiosis and reduced availability of essential amino acids. Conversely, strength and power training acts as an intervention capable of reactivating this axis, partially inhibiting FOXO/atrogenic factors and restoring myofibrillar protein synthesis, even in older age.
Recent geroscience frameworks propose that alterations in proteostasis represent a central hallmark linking anabolic resistance and age-related functional decline. Beyond reduced protein synthesis, ageing muscle exhibits impairments in protein folding, chaperone activity, autophagic flux and proteasomal degradation efficiency, leading to the accumulation of dysfunctional proteins [57]. Exercise appears capable of partially restoring proteostatic mechanisms through coordinated activation of autophagy, heat-shock proteins and lysosomal pathways, suggesting that muscle quality may depend as much on protein turnover efficiency as on net muscle mass accretion [83].
5.3. Inflammageing and Immunometabolism
Intense exercise triggers transient inflammatory responses that form part of the adaptation process; ageing, on the other hand, is accompanied by a persistent state of low-grade inflammatory activation known as inflammageing [24,84]. When pro-inflammatory cytokines such as IL-6 or TNF-α remain chronically elevated, they interfere with anabolic signalling, promote catabolism and contribute to functional decline. This link between ageing, cardiovascular disease, frailty and inflammation positions GMDs as a multisystemic syndrome, not just a muscular one [24].
GWAS studies on frailty and muscle weakness have identified the involvement of regions within the major histocompatibility complex, such as HLA-DRB1, HLA-DRB5, HLA-DQB1, BTNL2 and TNXB; as well as inflammatory signalling genes such as IL6, IL6R, IL10, TNF, TNFSF9, NFKB1, RELA, TLR4, NLRP3, JAK2 and STAT3. The IL6R rs2228145 variant, for example, has been used in Mendelian randomisation studies to explore the causal contribution of IL-6 signalling to inflammatory diseases and age-related traits. At the level of muscle function, these pathways link antigen presentation, immunosenescence, systemic inflammation, anabolic resistance and neuromuscular deterioration [46,85,86].
A very illustrative example is that of interleukin IL-6: it is important to distinguish between IL-6 as an acute exercise-induced myokine, with context-dependent metabolic and anti-inflammatory effects; and chronically elevated IL-6 associated with inflammageing, which is linked to catabolism, frailty and poorer physical performance. This duality is important in our context: the same molecule can contribute to beneficial adaptation or functional decline depending on the temporal pattern, tissue source and metabolic context. Thus, the overlap between performance and GMDs is not explained solely by muscle genes, but also by immunometabolic genes that regulate the body’s ability to resolve inflammation following mechanical stress and prevent repair from becoming chronic damage.
The relationship between inflammation and functional ageing extends beyond cytokine concentrations. Ageing is accompanied by profound remodeling of the immune system, including thymic involution, T-cell exhaustion, altered macrophage polarization and expansion of senescent immune cell populations [87]. This phenomenon, termed immunosenescence, contributes to impaired tissue repair, chronic inflammation and reduced responsiveness to exercise interventions. Interestingly, lifelong physical activity appears to attenuate several features of immunosenescence, suggesting that exercise may preserve immune adaptability in a manner analogous to its effects on skeletal muscle and cardiovascular function [88].
This immune remodelling is closely coupled to redox imbalance. ROS generated by dysfunctional mitochondria and activated immune cells can oxidise DNA, lipids and proteins, generating damage-associated molecular patterns (DAMPs) that activate pattern-recognition receptors and downstream inflammatory cascades, including NF-κB and the NLRP3 inflammasome [75,89,90]. Activated NF-κB promotes the transcription of pro-inflammatory cytokines (IL-6, TNF-α, IL-1β) whilst suppressing antioxidant gene programmes, perpetuating a redox-inflammatory loop that self-amplifies and intensifies with age. This cycle, conceptualised as ‘oxy-inflamm-ageing’, is further sustained by a decline in endogenous antioxidant capacity, including reduced activity of superoxide dismutase, catalase and glutathione peroxidase, and by the age-related attenuation of the Nrf2/KEAP1 pathway, which normally coordinates the transcriptional induction of cytoprotective and antioxidant genes [70,72]. In the context of muscle ageing, oxidative stress may impair insulin/IGF-1–PI3K–Akt signalling through oxidative modifications of proteins and lipids, including protein carbonylation and lipid peroxidation, thereby contributing to anabolic resistance in parallel with cytokine-driven catabolic signalling [91].
Furthermore, oxidative stress is a primary trigger of cellular senescence: accumulated oxidative damage to DNA activates the p53/p21 and p16INK4a tumour suppressor pathways, leading cells into irreversible senescence and triggering the secretion of the senescence-associated secretory phenotype (SASP), which amplifies systemic inflammation and compromises the regenerative microenvironment available to satellite cells [92]. At the biomarker level, circulating markers of oxidative damage, including F2-isoprostanes, malondialdehyde (MDA) and 8-hydroxy-2′-deoxyguanosine (8-OHdG), have been reported alongside elevated levels of IL-6 and C-reactive protein in older adults with frailty or sarcopenia, reinforcing the notion that redox dysregulation and inflammatory activation are concurrent, mutually reinforcing processes, rather than independent phenomena [93,94].
5.4. Neuromuscular Integrity and Satellite Cell Reserve
Strength does not depend solely on muscle size: it requires motor neurons, a functional neuromuscular junction (NMJ), efficient motor unit recruitment and motor control. The neuromuscular junction is one of the most vulnerable aspects of musculoskeletal ageing: partial denervation, motor end-plate fragmentation and the loss of motor units explain why strength declines more rapidly than muscle mass [95]. At the same time, satellite cells lose their regenerative capacity with age. Sousa-Victor and colleagues demonstrated that satellite cells in older adults transition from reversible quiescence to irreversible senescence mediated by p16INK4a, compromising muscle regeneration and providing a causal link between cellular senescence and functional decline [96]. In a sporting context, small differences in neuromuscular coordination and satellite cell reserve can distinguish between different levels of performance; in old age, those same differences can, for example, increase or decrease the risk of falling.
The stability of the motor endplate depends on the AGRN–LRP4–MUSK–DOK7 axis, together with RAPSN and subunits of the nicotinic acetylcholine receptor such as CHRNA1, CHRNB1, CHRND and CHRNE. These molecules regulate acetylcholine receptor clustering, postsynaptic maturation and the stability of the neuromuscular synapse. Ageing, by altering this architecture, reduces the reliability of neuromuscular transmission and contributes to denervation, the loss of type II fibres and a decline in muscle strength [97]. At the same time, the muscle’s regenerative reserve depends on genes such as PAX7, MYOD1, MYF5, MYOG, MEF2C, NOTCH1, DLL1, WNT7A, TGFB1 and CDKN2A/p16INK4a. In early stages, efficient activation of satellite cells enables hypertrophy, repair following injury and adaptation to training; in old age, the senescence of these cells, reduced Notch signalling, increased TGF-β and the inflammatory environment reduce the capacity for regeneration. All this suggests that geriatric motor dysfunctions (GMDs) do not result solely from the loss of muscle tissue, but from the progressive degradation of the entire neuromuscular axis.
Emerging evidence suggests that neuromuscular ageing should not be viewed exclusively as a peripheral phenomenon. Structural and functional alterations within cortical motor networks, corticospinal pathways and sensorimotor integration circuits also contribute to age-related declines in strength, coordination and mobility [98]. Consequently, preservation of motor function across the lifespan may depend not only on muscle regenerative capacity but also on lifelong maintenance of neuroplasticity and central motor control [99,100].
5.5. Mechanotransduction
Muscle is a tissue that is sensitive to mechanical stress: it requires tension, stretching and contraction to maintain its structure. Mechanosensitive channels such as PIEZO1 are involved in translating physical forces into cellular signals; during myogenesis, PIEZO1 regulates the fusion and elongation of myotubes via Ca²⁺ flux and the RhoA/ROCK pathway [101]. Disuse of muscle tissue leads to atrophy, whereas strength training remains effective even in old age: mechanical loading is not merely an external intervention, but an essential molecular signal for maintaining function.
The mechanotransduction pathway can be divided into three genetic levels. The first corresponds to direct mechanical sensors, such as PIEZO1, PIEZO2, ITGA7/ITGB1 integrins, DMD dystrophin, costamers and sarcomeric proteins such as ACTN3, TTN and NEB. The second corresponds to intracellular transducers such as PTK2/FAK, RHOA, ROCK1/2, MAPK, mTORC1 and the Hippo effectors YAP1 and WWTR1/TAZ. The third corresponds to final adaptive responses: protein synthesis, extracellular matrix remodelling, myogenic differentiation and local angiogenesis [102].
Mechanical sensitivity determines the extent to which a muscle ‘translates’ a given external load into growth, repair or maintenance. A young muscle subjected to training activates mTOR, YAP/TAZ and hypertrophy programmes; whereas an aged, inflamed or anabolism-resistant muscle may receive the same stimulus but with a diminished response. On the other hand, NEB encodes nebulin, a structural protein that regulates the length of thin filaments and sarcomeric stability; RIF1 is involved in repair and genomic stability. Its association with muscle mass suggests that the mechanical quality of the muscle depends both on sarcomeric integrity and on the ability to maintain the cellular genome under repeated stress. Thus, mechanotransduction integrates not only the response to load, but also the structural capacity of the muscle to tolerate it without deteriorating.
Recent studies indicate that the extracellular matrix (ECM) should be considered an active participant in mechanotransduction rather than a passive structural scaffold [103]. Ageing-associated fibrosis, altered collagen cross-linking and changes in ECM stiffness modify force transmission and mechanosensory signalling, potentially reducing the effectiveness of anabolic stimuli [104]. Therefore, age-related impairments in muscle adaptation may arise not only from intracellular resistance to mechanical loading but also from alterations in the extracellular mechanical environment itself.
5.6. Myokines, Exerkines and Muscle-Organ Communication
Skeletal muscle is not only an effector organ but also an endocrine organ. The review by Severinsen and Pedersen lists more than 650 factors released by muscle during exercise, including IL-6 (which paradoxically has acute anti-inflammatory effects on myocytes), irisin, BDNF, myonectin, decorin and SPARC, with endocrine and paracrine actions on the liver, bone, adipose tissue and the brain [105]. This broadens the link between physical activity and health to a systemic level, integrating muscle into communication networks between organs, the disruption of which with age contributes to multisystemic frailty.
The myokine network can be viewed as a muscle-organ communication system that translates muscle contraction into systemic signals. Among the most relevant genes/factors are IL6, FNDC5/irisin, BDNF, APLN/apelin, LIF, METRNL, SPARC, DCN/decorin, FST/folistatin, MSTN/myostatin, GDF15, ANGPTL4 and CTRP15/mionectin. In the context of performance, these signals promote metabolic adaptation, angiogenesis, substrate oxidation, neural plasticity and bone remodelling. In ageing, the loss of contractile mass, inactivity and inflammageing reduce the quality of the muscle secretome and disrupt communication with the liver, bone, adipose tissue, immune system and brain [106,107,108].
Sarcopenia, therefore, is not merely the loss of muscle as a mechanical tissue, but the loss of an endocrine organ. The decline in beneficial myokines and the increase in catabolic signals such as myostatin or GDF15 may contribute to multisystemic frailty, age-related anorexia, metabolic decline, reduced neuroplasticity and a poorer response to exercise. Conversely, regular training not only improves strength or VO₂max, but also partially restores a more favourable secretory profile, transforming active muscle into a systemic modulator of health.
The concept of myokines has recently evolved into the broader framework of exerkines, encompassing bioactive molecules released during physical activity from multiple tissues including skeletal muscle, adipose tissue, liver, bone and the vascular endothelium [109]. This integrated signalling network mediates many of the systemic benefits of exercise and may explain why physical activity influences cognitive function, immune competence, metabolic health and longevity beyond its effects on muscle tissue alone [110,111] .
Beyond their role as intercellular messengers, myokines and other exerkines may also influence the systemic redox environment [105,110]. Exercise-induced muscle contractions generate a transient and controlled increase in reactive oxygen species (ROS), which, through hormetic mechanisms, can activate the Nrf2/KEAP1 antioxidant pathway and PGC-1α-mediated adaptive programmes, thereby reinforcing endogenous antioxidant defences in skeletal muscle and, potentially, in distal tissues [67,112]. This exercise-driven antioxidant response contrasts with the less favourable secretory profile of inactive or ageing muscle, which may be characterised by increased catabolic or stress-associated factors such as myostatin and GDF15, together with altered expression of exercise-responsive myokines such as irisin and apelin [113,114]. Thus, the decline in muscle mass and contractile activity with ageing may progressively attenuate exercise-induced antioxidant signalling, contributing to systemic redox dysregulation and inflammageing. Regular strength and endurance training should therefore not be viewed solely as a strategy for preserving muscle function and the anabolic secretome, but also as a means of supporting systemic redox homeostasis and mitigating components of oxi-inflamm-ageing across the life course [77].
5.7. Epigenetic Memory of Exercise
Exercise leaves stable epigenetic marks in skeletal muscle. Lindholm and colleagues described a coordinated reprogramming of the muscle methylome and transcriptome following aerobic exercise in humans, with changes at loci regulating oxidative metabolism [115]. Seaborne and colleagues demonstrated that muscle possesses an epigenetic memory of hypertrophy: previous episodes of training lead to persistent hypomethylation of anabolic genes, which facilitates muscle ‘recovery’ following periods of inactivity [116]. This capability has direct implications for veteran athletes, for rehabilitation following periods of immobilisation, and, conceptually, for the functional reserve model: early physical activity may leave molecular imprints that facilitate an adaptive response decades later.
In the study by Seaborne et al., genes such as UBR5, RPL35A, SETD3, HEG1 and PLA2G16 showed hypomethylation and increased expression following loading, with amplified responses during reloading; other genes such as AXIN1, GRIK2, CAMK4 and TRAF1 retained hypomethylated signatures during the unloading period, suggesting a molecular memory of the previous stimulus. Therefore, it is not merely a matter of ‘having trained before’, but of maintaining a muscle that is molecularly primed to respond better to new loads [116,117].
In addition to DNA methylation, epigenetic regulation of muscle involves histone remodelling, miRNAs and epitranscriptomic regulation via m⁶A. In this latter context, FTO emerges as a gene of particular interest for this manuscript, as it integrates adiposity, energy metabolism, muscle differentiation, mTOR–PGC-1α signalling and the cellular stress response. FTO, therefore, should not be confined to its classical role in obesity, but rather interpreted as an epitranscriptomic modulator of muscle homeostasis and a potential bridge between metabolism, hypoxia, myocellular senescence and GMDs.
From a conceptual standpoint, epigenetic memory provides molecular support for the life-course model: exercise undertaken in youth or adulthood may increase peak functional reserve, but may also leave adaptive marks that facilitate recovery following periods of inactivity in later life. This hypothesis does not imply determinism nor does it guarantee protection against the onset of any GMD, but it does reinforce the idea that the history of physical activity is inscribed in partially persistent molecular layers.
The biological pathways mentioned above are summarized in Figure 3.
Recent developments in biological age estimation have further strengthened the link between exercise and epigenetic regulation. DNA methylation-based epigenetic clocks consistently demonstrate associations between higher physical activity levels, superior cardiorespiratory fitness and slower biological ageing [118,119]. These findings suggest that exercise-induced epigenetic remodelling may not only influence muscle adaptation but also contribute to systemic modulation of ageing trajectories, thereby providing a potential mechanistic bridge between athletic performance and long-term functional preservation [120,121].
6. Gut Microbiota: The Modulatory Layer of the Gut-Muscle Axis
The gut microbiota has a direct impact on metabolic, inflammatory and neurological processes, as well as on muscle function. The gut-muscle axis has gained prominence following observations that microbial composition affects both muscle mass and function in older adults, as well as their response to training [122,123]. Although this field is still under development, it highlights a key point: not all the biological factors that influence muscle function are directly encoded in the human genome.
Microbial metabolites are the main focus of interest. Short-chain fatty acids (SCFAs), particularly butyrate, modulate inflammation, insulin sensitivity, the integrity of the intestinal barrier and gene expression, amongst other factors. In old age, a less diverse microbiota, with a loss of butyrate-producing bacteria and an increase in pro-inflammatory signals, could contribute to the onset of inflammageing and anabolic resistance. Furthermore, in the context of sport, a more diverse microbiota that is functionally oriented towards energy metabolism could promote recovery and the availability of substrates.
Urolithin A is a prime example of the interaction between diet, the gut microbiota and mitochondrial function. This metabolite, produced by the gut microbiota from dietary ellagitannins, has been linked to the activation of mitophagy and improvements in mitochondrial biomarkers in humans [124]. The randomised clinical trial by Liu et al. in adults aged 65 and over showed that supplementation with 1,000 mg/day for 4 months significantly improved muscle strength (assessed by the number of knee extension repetitions performed) and biomarkers of mitochondrial health [125]. Beyond the significance of this specific finding, urolithin A demonstrates how a biological pathway can be modulated by mechanisms that depend on the host, diet and gut ecology.
The comparison between athletes and sedentary individuals reinforces the impact of the microbiome on the body. Studies in athletes have described greater microbial diversity and an abundance of short-chain fatty acid producers, alongside distinct functional profiles in terms of energy metabolism [126,127]. Using metabolomics, Scheiman and colleagues identified a higher abundance of Veillonella atypica, a bacterium capable of converting lactate into propionate, in Olympic marathon runners, providing the first experimental evidence of a microbial metabolite with an ergogenic effect [128]. Exercise interventions in sedentary individuals have also shown that physical activity can reversibly alter the composition and function of the gut microbiota [129].
Genetic predisposition does not, therefore, operate in a vacuum: the microbiota modulates pathways that are also relevant to performance or metabolic health, such as inflammation, energy metabolism and mitochondrial function. The gut-muscle axis broadens the genetic framework and makes it more dependent on the life course and lifestyle.
7. Pleiotropy, Trade-Offs and the Exposome: How to Interpret the Relationship Between Performance and GMDs
Pleiotropy explains why a single variant or biological pathway can influence multiple phenotypes. Favourable pleiotropy would be the most intuitive scenario: variants that promote greater mitochondrial efficiency, a better response to training, adequate anabolic signalling or reduced chronic inflammation could be associated with both improved physical performance and a lower risk of developing GMDs. Under this interpretation, athletic performance would not be at odds with functional health, but rather an early expression of robust biological systems. The Mendelian randomisation of the GWAS by Jones et al. supports a causal link between muscle weakness and frailty [46].
ACTN3 partly illustrates this logic, although it also highlights its limitations. The genotype associated with power may favour certain sporting demands in young people [40], but its isolated effect is unlikely to predict a person’s muscle ageing. Something similar applies to ACE or PPARGC1A: these are biologically plausible genes that are useful for understanding mechanisms, but they do not, on their own, determine a person’s life course. Pleiotropy must be interpreted at the level of networks and pathways, not as a simple relationship between a variant and a clinical outcome.
Antagonistic pleiotropy could introduce a further, more complex possibility. Certain signals that are beneficial for growth, reproduction or performance may entail costs at later stages if they remain chronically activated or if the environmental context changes. mTOR is a conceptual example: its activation is necessary for protein synthesis and hypertrophy, but the regulation of cell growth, autophagy and ageing depends on a delicate balance. Similarly, an effective inflammatory response may be necessary to repair tissue following exercise, whilst chronic inflammatory activation may contribute to an increased likelihood of developing frailty [24,84]. In humans, however, there is virtually no direct evidence that variants associated with improved athletic performance significantly increase the risk of GMD.
8. Empirical Evidence: Longitudinal Trajectories and Common Markers
Studies have been conducted linking objective physical capacity to mortality, showing that strength, speed and functional performance are not merely sporting or geriatric measures, but indicators of systemic health [28]. Grip strength follows typical patterns throughout the life course and is associated with adverse outcomes when it falls below the expected values for a given age and sex [130]. The LASA study mentioned above confirms precisely that grip strength predicts frailty and functional limitation, illustrating the significance of the exposome [51].
A person’s sporting history and sustained physical activity over time are also significant. Former athletes and people who have maintained high levels of activity tend to have better physical function in later life, although these studies should be interpreted with caution due to potential selection bias: those who went on to become elite athletes may already have had genetic, social or health-related advantages. Master athletes provide a particularly useful analytical framework by distinguishing intrinsic ageing from factors attributable to inactivity or chronic disease [131]; however, regardless of this, the practical message remains the same: training for strength, power, balance and aerobic capacity improves function in older adults and can partially reverse factors attributable to sarcopenia or frailty [1,2].
Furthermore, functional markers provide a common language. Grip strength summarises overall strength levels and is associated with prognosis; walking speed incorporates lower-limb power, balance, neuromotor control and cardiorespiratory fitness; the SPPB combines balance, walking and the ability to stand up from a chair, and therefore predicts mortality and disability [51,131]. These tools are valuable because they enable complex molecular mechanisms to be translated into observable clinical outcomes.
At the genetic level, GWAS and PRS are beginning to enable cross-analyses between performance traits, body composition and physical function [44,45,46,47,48]. There is still a lack of studies directly linking genotypes associated with athletic performance in youth to diagnoses of sarcopenia or frailty decades later. This absence does not invalidate the hypothesis, but it does mean that it must be framed as a research programme rather than a definitive conclusion.
The microbiota provides a second line of complementary evidence. Differences between athletes and sedentary individuals [126,127], findings on microorganisms involved in lactate metabolism [128] and the ability of exercise to alter the microbiota [129] suggest that physical activity reshapes not only muscle but also associated systems. In older adults, the relationship between the microbiota, muscle mass and frailty [122] points in the same direction: functional decline may depend on biological networks that go beyond the muscle as an isolated organ.
9. Practical Implications: Preserving Functional Reserve Through Exercise, Early Screening and Risk Communication
The proposed framework has practical implications, but it should also be interpreted with caution. In research, integrating genetics, physical function, inflammatory biomarkers, body composition, the microbiota and longitudinal follow-up can help to identify risk subgroups and dominant mechanisms. In current clinical practice, however, the most useful tools remain the simplest: grip strength, walking speed, SPPB, history of falls, level of physical activity, nutritional status and presence of comorbidities.
From a clinical perspective, sport and exercise should not be viewed solely as tools for improving performance, but also as preventive measures against GMDs. Sports activities and programmes tailored to an individual’s specific circumstances can incorporate aerobic training, strength, power, balance, coordination and social engagement, all of which are important for maintaining functional reserve. International physical activity guidelines are clear on this point, recognising the benefits for adults and older adults in terms of mortality, cardiovascular disease, type 2 diabetes, mental health, cognitive function, falls and body composition. Furthermore, clinical trials such as the LIFE Study have shown that a structured physical activity programme can reduce the onset of major mobility disability in older adults at functional risk [132]. Therefore, sport adapted to age, clinical condition and functional level can be considered a relatively low-cost clinical strategy with high potential impact for delaying the transition from preserved function to sarcopenia, frailty or lower-limb weakness.
Furthermore, it is important to emphasise that it is never too early or too late to start or maintain a physically active lifestyle. In early life, physical activity and training help to maximise peak functional capacity, increasing the reserve capacity against subsequent decline. In older age, even when there is already some loss of strength, mobility or balance, exercise is still capable of inducing significant adaptations. The World Health Organization’s guidelines highlight that any amount of physical activity is better than none, that all activity counts, and that muscle strengthening brings benefits across all age groups; this is particularly important given that inactivity increases from the age of 60 onwards, precisely at the stage when maintaining strength, power and balance becomes most crucial for preventing falls, dependency or loss of independence [133].
The preventive benefits of exercise must also be understood in light of the role of skeletal muscle as an endocrine organ, as mentioned above. Muscle contraction triggers the release of myokines and other secreted factors capable of modulating systemic metabolism, inflammation, vascular function, bone homeostasis, muscle-brain communication and adaptation to physiological stress [105,106]. The loss of muscle mass and function represents not only a mechanical limitation but also a reduction in the body’s endocrine and metabolic capacity. Therefore, interventions such as strength, power and endurance training can act simultaneously on the muscle as both contractile tissue and a secretory organ, promoting a systemic profile more compatible with functional resilience and reinforcing the need to consider sarcopenia and frailty as multisystemic syndromes, not merely as localised problems at the level of muscle mass [107,108].
Furthermore, in a context where life expectancy continues to rise, the functional tests used in clinical practice should be adapted to a more ambitious goal: not only to identify established disability, but also to detect early losses of intrinsic capacity and functional reserve. Classic tests such as handgrip strength, walking speed, the SPPB, the 5STS or the Timed Up and Go (TUG) remain useful due to their low cost, reproducibility and prognostic value. However, in the context of an ageing population, these measures should be complemented by a more dynamic and longitudinal interpretation, based on individual trajectories, intra-individual changes and the early detection of subtle declines. The WHO has already proposed a model of integrated care designed to guide healthcare systems towards optimising intrinsic capacity and functional ability, rather than limiting themselves to the late diagnosis of a disease or disability [134].
Genetic screening could prove useful for risk stratification before symptoms appear, particularly when combined with functional and environmental data. However, the available PRSs have significant limitations: moderate predictive power, insufficient validation in non-European populations, and unestablished incremental utility compared to inexpensive and reproducible functional tests [51]. For this reason, their use should, for the time being, be confined to research or very specific contexts, and not serve as a substitute for clinical assessment.
Preventive interventions should prioritise components that are broadly beneficial. Strength training is key to maintaining muscle mass, strength and function [2]; strength, power and balance training help to reduce the risk of falls; aerobic exercise preserves cardiorespiratory capacity and oxidative metabolism; and nutrition should ensure optimal protein synthesis, while supporting metabolic health and limiting chronic low-grade inflammatory burden. In this regard, dietary patterns rich in anti-inflammatory and redox-active bioactive compounds, including polyphenols found in berries, green tea and extra virgin olive oil, carotenoids and vitamins C and E, may contribute to a more favourable inflammatory and oxidative profile, although they should be interpreted as part of an integrated lifestyle strategy rather than as isolated antioxidant interventions [19,135,136,137,138,139]. Some emerging interventions based on microbial metabolites, such as urolithin A, are expanding the therapeutic repertoire by targeting mitochondrial quality control and potentially modulating redox-inflammatory pathways. These compounds provide a useful example of how diet–microbiota interactions may influence muscle ageing; however, their long-term clinical relevance for the preservation of muscle function remains to be established [125]. Overall, although genetics may influence the magnitude of the response, mechanical, metabolic and nutritional stimuli remain necessary.
Finally, it is worth noting that communicating genetic risk requires particular care: presenting a variant or a score as an inevitable fate can lead to a sense of fatalism, anxiety or even stigmatisation. The correct framing is that of a modifiable predisposition: genetics may shift the starting point or sensitivity to certain stimuli, but it does not negate the effect of exercise, nutrition or disease management. This distinction is ethically and clinically essential, because the aim should not be to classify individuals as predestined to develop a GMD, but rather to identify early opportunities for prevention.
10. Limitations and Research Agenda
The main limitation in this field is phenotypic heterogeneity: Firstly, athletic performance encompasses very different disciplines, such as endurance, power, technical sports, team sports, combined events, etc.; whilst sarcopenia and frailty vary according to the definitions provided by the various consensus groups (EWGSOP2, AWGS2, SDOC or FNIH), cut-off points and populations [35,36,37]. This heterogeneity makes it difficult to compare studies and may explain some of the inconsistencies observed in the genetics of performance.
A second limitation is the lack of life-course cohorts with in-depth functional phenotyping: many biobanks have genotypes and large sample sizes, but do not always include detailed measures of strength, muscle composition, sporting history, training or neuromuscular function, amongst other factors. Conversely, studies with more comprehensive functional testing tend to have fewer participants or shorter follow-up periods. To robustly test the shared architecture hypothesis, cohorts are needed that span youth, middle age and old age, with repeated measurements of performance, strength, gait, sarcopenia, frailty, physical activity, nutrition, microbiota or comorbidities.
Population transferability is another challenge: a large proportion of genetic studies have been conducted in populations of European descent, which limits the application of PRS to other groups. Furthermore, muscle mass, strength or the rate of decline vary according to sex, age, hormonal status or sociocultural context; therefore, the development of precision medicine for muscle function will require more diverse studies and stratified analyses that do not obscure certain relevant interactions.
Finally, causality remains difficult to establish: the fact that two phenotypes share genetic pathways or correlations does not imply that a variant that improves performance directly protects against any of the GMDs mentioned above. Longitudinal analyses and Mendelian randomisation are required where possible [44,46], intervention studies and mechanistic models that enable the separation of genetic predisposition, athletic selection, behaviour, cumulative training and environmental exposures. The growing availability of multi-omic data (genomic, transcriptomic, methylomic, metabolomic or microbiomic) in longitudinal cohorts presents an opportunity to integrate these layers of information into predictive models of functional trajectories.
11. Conclusions: From the Genetics of Performance to Functional Medicine
The relationship between athletic performance and age-related changes can be understood as a continuum of muscle function throughout the life course. The variants and pathways that influence strength, power, endurance or trainability do not disappear with age: they are expressed in a different physiological context, characterised by reduced reserve capacity, comorbidity, inflammageing, neuromuscular changes, deterioration of satellite cells and cumulative exposure to physical activity or inactivity.
The available evidence supports the existence of shared biological pathways linking sporting performance to age-related motor decline: mitochondrial function and mitophagy, the anabolic-catabolic balance, oxidative stress and chronic low-grade inflammation (‘oxi-inflamm-ageing’), neuromuscular integrity and satellite cell reserve, mechanotransduction, endocrine communication via myokines, epigenetic memory and, increasingly, the gut microbiota. Within this context, mitochondrial reactive oxygen species (ROS) may contribute to the activation of NF-κB and the NLRP3 inflammasome, thereby promoting cytokine secretion, impairing antioxidant defences and favouring cellular senescence and the senescence-associated secretory phenotype (SASP). These processes may create a self-amplifying redox-inflammatory loop that accelerates tissue dysfunction and functional decline with ageing. Much of this relationship appears consistent with favourable pleiotropy: systems that promote greater functional capacity in the early stages of life may also help to preserve mobility in old age. Antagonistic pleiotropy and trade-offs remain plausible hypotheses, particularly in pathways related to cell growth, inflammation and redox signalling, but require more direct evidence from human studies.
The practical message is clear: genetics predispose, but do not determine. Functional reserve is built by maximising peak capacity at a young age and slowing the rate of decline throughout adulthood and old age. Exercise—particularly strength and power training—along with proper nutrition and the management of comorbidities, remain key strategies, regardless of genetic profile. The promise for the future lies in functional medicine that integrates genetics, functional phenotypes, biomarkers and the exposome to anticipate risk and personalise interventions, whilst never losing sight of the fact that preserving autonomy, mobility and quality of life is the ultimate clinical goal.
Author Contributions
Conceptualization, S.F.L., J.G.B. and J.E.C.; methodology, C.M.P. and R.I.L.; investigation, S.F.L., L.P.R., R.I.L. and C.M.P.; resources, J.G.B., F.J.M.N. and J.E.C.; writing—original draft preparation, S.F.L., L.P.R. and F.J.M.N.; writing—review and editing, C.M.P., J.G.B., F.J.M.N. and J.E.C.; visualization, S.F.L., L.P.R. and R.I.L.; supervision, J.G.B., F.J.M.N. and J.E.C.; project administration, F.J.M.N. and J.E.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported within the framework of an Industrial Doctorate project between the High Performance Sports Research Centre at Universidad Católica San Antonio de Murcia and Sabartech S.L. ; In addition, this work has received partial support from the AVI-Generalitat Valenciana, through grant INNEST/2021/292; from the MICIU/AEI/10.13039/501100011033; and from the EU’s NextGenerationEU/PRTR program, through grant PLEC2022-009352; the CDTI/ISCIII-CDTI Joint Action and the EU’s NextGenerationEU/PRTR program, through grant IDI-20230068 / EIS-20220014; and the IVACE/FEDER, through grant IMIDTA/2023/43; The funders had no role in the design of the review, literature interpretation, manuscript preparation, decision to submit the work for publication or preparation of the manuscript for publication.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were generated or analysed in this study.
Acknowledgments
The authors acknowledge the institutional support of the High Performance Sports Research Centre, Universidad Católica San Antonio de Murcia, and Sabartech S.L. during the development of this work.
Conflicts of Interest
Samuel Fernández Lorenzo is conducting an Industrial Doctorate project at Sabartech S.L. in collaboration with Universidad Católica San Antonio de Murcia and the High Performance Sports Research Centre. Sabartech S.L. has participated in government-funded research projects related to sarcopenia and frailty. The remaining authors declare no conflicts of interest. The authors declare that these relationships did not influence the interpretation, content or conclusions of the manuscript.; Declaration of Generative AI and AI-Assisted Technologies; While preparing this manuscript, the authors used artificial intelligence tools to assist with linguistic review and the creation of figures. The authors have reviewed and edited all results generated with the help of AI and assume full responsibility for the content of this publication.
Abbreviations
| 1-RM | One-repetition maximum |
| 5STS | Five Times Sit-to-Stand Test |
| ACE | Angiotensin-converting enzyme |
| ACTN3 | Alpha-actinin-3 |
| ADLs | Activities of daily living |
| AMPK | AMP-activated protein kinase |
| ATP | Adenosine triphosphate |
| AWGS2 | Asian Working Group for Sarcopenia 2 |
| CARDIA | Coronary Artery Risk Development in Young Adults |
| DNA | Deoxyribonucleic acid |
| DXA | Dual-energy X-ray absorptiometry |
| ECM | Extracellular matrix |
| EWGSOP2 | European Working Group on Sarcopenia in Older People 2 |
| FNIH | Foundation for the National Institutes of Health |
| FTO | Fat mass and obesity-associated gene |
| GMD | Geriatric motor dysfunction |
| GMDs | Geriatric motor dysfunctions |
| GWAS | Genome-wide association studies |
| HERITAGE | Health, Risk Factors, Exercise Training and Genetics Family Study |
| I/D | Insertion/deletion |
| ICOPE | Integrated Care for Older People |
| IGF-1 | Insulin-like growth factor 1 |
| IL-6 | Interleukin-6 |
| LASA | Longitudinal Aging Study Amsterdam |
| LIFE | Lifestyle Interventions and Independence for Elders |
| m⁶A | N6-methyladenosine |
| MDPs | Mitochondria-derived peptides |
| miRNAs | MicroRNAs |
| MOTS-c | Mitochondrial open reading frame of the 12S rRNA-c |
| MR | Mendelian randomization |
| mTOR | Mechanistic target of rapamycin |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| mtDNA | Mitochondrial DNA |
| NMJ | Neuromuscular junction |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K | Phosphoinositide 3-kinase |
| PINK1 | PTEN-induced kinase 1 |
| PPAR | Peroxisome proliferator-activated receptor |
| PPARGC1A | Gene encoding PGC-1α |
| PRS | Polygenic risk score |
| SCFAs | Short-chain fatty acids |
| SDOC | Sarcopenia Definition and Outcomes Consortium |
| SHLPs | Small humanin-like peptides |
| SNP | Single-nucleotide polymorphism |
| SPPB | Short Physical Performance Battery |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| TUG | Timed Up and Go |
| VO₂max | Maximal oxygen uptake |
| WHO | World Health Organization |
References
- Cartee GD, Hepple RT, Bamman MM, Zierath JR. Exercise Promotes Healthy Aging of Skeletal Muscle. Cell Metab. 2016;23(6):1034-1047. [CrossRef]
- Fragala MS, Cadore EL, Dorgo S, et al. Resistance Training for Older Adults: Position Statement From the National Strength and Conditioning Association. J Strength Cond Res. 2019;33(8):2019-2052. [CrossRef]
- McPhee JS, French DP, Jackson D, Nazroo J, Pendleton N, Degens H. Physical activity in older age: perspectives for healthy ageing and frailty. Biogerontology. 2016;17(3):567-580. [CrossRef]
- Lemez S, Baker J. Do Elite Athletes Live Longer? A Systematic Review of Mortality and Longevity in Elite Athletes. Sports Med - Open. 2015;1(1):16. [CrossRef]
- Foulkes S, Hewitt D, Skow R, et al. Outrunning the grim reaper: longevity of the first 200 sub-4 min mile male runners. Br J Sports Med. 2024;58(13):717-721. [CrossRef]
- Whitson HE, Cohen HJ, Schmader KE, Morey MC, Kuchel G, Colon-Emeric CS. Physical Resilience: Not Simply the Opposite of Frailty. J Am Geriatr Soc. 2018;66(8):1459-1461. [CrossRef]
- Hadley EC, Kuchel GA, Newman AB, Workshop Speakers and Participants. Report: NIA Workshop on Measures of Physiologic Resiliencies in Human Aging. J Gerontol A Biol Sci Med Sci. 2017;72(7):980-990. [CrossRef]
- Cesari M, Calvani R, Marzetti E. Frailty in Older Persons. Clin Geriatr Med. 2017;33(3):293-303. [CrossRef]
- Kraal AZ, Massimo L, Fletcher E, et al. Functional reserve: The residual variance in instrumental activities of daily living not explained by brain structure, cognition, and demographics. Neuropsychology. 2021;35(1):19-32. [CrossRef]
- Mandsager K, Harb S, Cremer P, Phelan D, Nissen SE, Jaber W. Association of Cardiorespiratory Fitness With Long-term Mortality Among Adults Undergoing Exercise Treadmill Testing. JAMA Netw Open. 2018;1(6):e183605. [CrossRef]
- Leong DP, Teo KK, Rangarajan S, et al. Prognostic value of grip strength: findings from the Prospective Urban Rural Epidemiology (PURE) study. Lancet. 2015;386(9990):266-273. [CrossRef]
- Ortega FB, Silventoinen K, Tynelius P, Rasmussen F. Muscular strength in male adolescents and premature death: cohort study of one million participants. BMJ. 2012;345:e7279. [CrossRef]
- Carnethon MR, Gidding SS, Nehgme R, Sidney S, Jacobs DR, Liu K. Cardiorespiratory fitness in young adulthood and the development of cardiovascular disease risk factors. JAMA. 2003;290(23):3092-3100. [CrossRef]
- Seals DR, Justice JN, LaRocca TJ. Physiological geroscience: targeting function to increase healthspan and achieve optimal longevity. J Physiol. 2016;594(8):2001-2024. [CrossRef]
- Joyner MJ, Coyle EF. Endurance exercise performance: the physiology of champions. J Physiol. 2008;586(1):35-44. [CrossRef]
- Hood DA, Memme JM, Oliveira AN, Triolo M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu Rev Physiol. 2019;81:19-41. [CrossRef]
- Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci. 1995;50 Spec No:11-16. [CrossRef]
- Power GA, Dalton BH, Rice CL. Human neuromuscular structure and function in old age: A brief review. J Sport Health Sci. 2013;2(4):215-226. [CrossRef]
- Calder PC, Bosco N, Bourdet-Sicard R, et al. Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing Res Rev. 2017;40:95-119. [CrossRef]
- Wilkinson DJ, Piasecki M, Atherton PJ. The age-related loss of skeletal muscle mass and function: Measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing Res Rev. 2018;47:123-132. [CrossRef]
- Goodpaster BH, Sparks LM. Metabolic Flexibility in Health and Disease. Cell Metab. 2017;25(5):1027-1036. [CrossRef]
- San-Millán I, Brooks GA. Assessment of Metabolic Flexibility by Means of Measuring Blood Lactate, Fat, and Carbohydrate Oxidation Responses to Exercise in Professional Endurance Athletes and Less-Fit Individuals. Sports Med. 2018;48(2):467-479. [CrossRef]
- Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88(4):1243-1276. [CrossRef]
- Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15(9):505-522. [CrossRef]
- Suchomel TJ, Nimphius S, Stone MH. The Importance of Muscular Strength in Athletic Performance. Sports Med. 2016;46(10):1419-1449. [CrossRef]
- Studenski S, Perera S, Patel K, et al. Gait speed and survival in older adults. JAMA. 2011;305(1):50-58. [CrossRef]
- Guralnik JM, Simonsick EM, Ferrucci L, et al. A short physical performance battery assessing lower extremity function: association with self-reported disability and prediction of mortality and nursing home admission. J Gerontol. 1994;49(2):M85-94. [CrossRef]
- Cooper R, Kuh D, Hardy R, Mortality Review Group, FALCon and HALCyon Study Teams. Objectively measured physical capability levels and mortality: systematic review and meta-analysis. BMJ. 2010;341:c4467. [CrossRef]
- Reid KF, Fielding RA. Skeletal muscle power: a critical determinant of physical functioning in older adults. Exerc Sport Sci Rev. 2012;40(1):4-12. [CrossRef]
- Bouchard C, Sarzynski MA, Rice TK, et al. Genomic predictors of the maximal O₂ uptake response to standardized exercise training programs. J Appl Physiol. 2011;110(5):1160-1170. [CrossRef]
- Bouchard C, Antunes-Correa LM, Ashley EA, et al. Personalized preventive medicine: genetics and the response to regular exercise in preventive interventions. Prog Cardiovasc Dis. 2015;57(4):337-346. [CrossRef]
- Coffey VG, Hawley JA. The molecular bases of training adaptation. Sports Med. 2007;37(9):737-763. [CrossRef]
- Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci. 2001;56(3):M146-156. [CrossRef]
- Rockwood K, Song X, MacKnight C, et al. A global clinical measure of fitness and frailty in elderly people. CMAJ Can Med Assoc J J Assoc Medicale Can. 2005;173(5):489-495. [CrossRef]
- Cruz-Jentoft AJ, Bahat G, Bauer J, et al. Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing. 2019;48(1):16-31. [CrossRef]
- Chen LK, Woo J, Assantachai P, et al. Asian Working Group for Sarcopenia: 2019 Consensus Update on Sarcopenia Diagnosis and Treatment. J Am Med Dir Assoc. 2020;21(3):300-307.e2. [CrossRef]
- Bhasin S, Travison TG, Manini TM, et al. Sarcopenia Definition: The Position Statements of the Sarcopenia Definition and Outcomes Consortium. J Am Geriatr Soc. 2020;68(7):1410-1418. [CrossRef]
- Cesari M, Landi F, Vellas B, Bernabei R, Marzetti E. Sarcopenia and physical frailty: two sides of the same coin. Front Aging Neurosci. 2014;6:192. [CrossRef]
- Montgomery HE, Marshall R, Hemingway H, et al. Human gene for physical performance. Nature. 1998;393(6682):221-222. [CrossRef]
- Yang N, MacArthur DG, Gulbin JP, et al. ACTN3 genotype is associated with human elite athletic performance. Am J Hum Genet. 2003;73(3):627-631. [CrossRef]
- Tharabenjasin P, Pabalan N, Jarjanazi H. Association of the ACTN3 R577X (rs1815739) polymorphism with elite power sports: A meta-analysis. PLoS One. 2019;14(5):e0217390. [CrossRef]
- Ma F, Yang Y, Li X, et al. The association of sport performance with ACE and ACTN3 genetic polymorphisms: a systematic review and meta-analysis. PLoS One. 2013;8(1):e54685. [CrossRef]
- Ipekoglu G, Bulbul A, Cakir HI. A meta-analysis on the association of ACE and PPARA gene variants and endurance athletic status. J Sports Med Phys Fitness. 2022;62(6):795-802. [CrossRef]
- Willems SM, Wright DJ, Day FR, et al. Large-scale GWAS identifies multiple loci for hand grip strength providing biological insights into muscular fitness. Nat Commun. 2017;8:16015. [CrossRef]
- Tikkanen E, Gustafsson S, Amar D, et al. Biological Insights Into Muscular Strength: Genetic Findings in the UK Biobank. Sci Rep. 2018;8(1):6451. [CrossRef]
- Jones G, Trajanoska K, Santanasto AJ, et al. Genome-wide meta-analysis of muscle weakness identifies 15 susceptibility loci in older men and women. Nat Commun. 2021;12(1):654. [CrossRef]
- Pei YF, Liu YZ, Yang XL, et al. The genetic architecture of appendicular lean mass characterized by association analysis in the UK Biobank study. Commun Biol. 2020;3(1):608. [CrossRef]
- Zillikens MC, Demissie S, Hsu YH, et al. Large meta-analysis of genome-wide association studies identifies five loci for lean body mass. Nat Commun. 2017;8(1):80. [CrossRef]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194-1217. [CrossRef]
- Booth FW, Roberts CK, Laye MJ. Lack of exercise is a major cause of chronic diseases. Compr Physiol. 2012;2(2):1143-1211. [CrossRef]
- Stringa N, van Schoor NM, Hoogendijk EO, Milaneschi Y, Huisman M. The phenotypic and genotypic association of grip strength with frailty, physical performance and functional limitations over time in older adults. Age Ageing. 2023;52(10):afad189. [CrossRef]
- Bouchard C, Rankinen T, Timmons JA. Genomics and Genetics in the Biology of Adaptation to Exercise. Compr Physiol. 2011;1(3):1603-1648. [CrossRef]
- Vermeulen R, Schymanski EL, Barabási AL, Miller GW. The exposome and health: Where chemistry meets biology. Science. 2020;367(6476):392-396. [CrossRef]
- Hota M, Barber JL, Ruiz-Ramie JJ, et al. Omics-driven investigation of the biology underlying intrinsic submaximal working capacity and its trainability. Physiol Genomics. 2023;55(11):517-543. [CrossRef]
- Muniz-Santos R, Magno-França A, Jurisica I, Cameron LC. From Microcosm to Macrocosm: The -Omics, Multiomics, and Sportomics Approaches in Exercise and Sports. Omics J Integr Biol. 2023;27(11):499-518. [CrossRef]
- Varadhan R, Walston JD, Bandeen-Roche K. Can a Link Be Found Between Physical Resilience and Frailty in Older Adults by Studying Dynamical Systems? J Am Geriatr Soc. 2018;66(8):1455-1458. [CrossRef]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. 2023;186(2):243-278. [CrossRef]
- Semenova EA, Pranckevičienė E, Bondareva EA, Gabdrakhmanova LJ, Ahmetov II. Identification and Characterization of Genomic Predictors of Sarcopenia and Sarcopenic Obesity Using UK Biobank Data. Nutrients. 2023;15(3):758. [CrossRef]
- Lei Y, Gan M, Qiu Y, et al. The role of mitochondrial dynamics and mitophagy in skeletal muscle atrophy: from molecular mechanisms to therapeutic insights. Cell Mol Biol Lett. 2024;29(1):59. [CrossRef]
- Ferri E, Marzetti E, Calvani R, Picca A, Cesari M, Arosio B. Role of Age-Related Mitochondrial Dysfunction in Sarcopenia. Int J Mol Sci. 2020;21(15):5236. [CrossRef]
- Drake JC, Wilson RJ, Yan Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J Off Publ Fed Am Soc Exp Biol. 2016;30(1):13-22. [CrossRef]
- Narendra DP, Youle RJ. The role of PINK1-Parkin in mitochondrial quality control. Nat Cell Biol. 2024;26(10):1639-1651. [CrossRef]
- Jung S, Kim K. Exercise-induced PGC-1α transcriptional factors in skeletal muscle. Integr Med Res. 2014;3(4):155-160. [CrossRef]
- Lee C, Zeng J, Drew BG, et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21(3):443-454. [CrossRef]
- Cobb LJ, Lee C, Xiao J, et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging. 2016;8(4):796-809. [CrossRef]
- Kim KH, Son JM, Benayoun BA, Lee C. The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell Metab. 2018;28(3):516-524.e7. [CrossRef]
- Powers SK, Duarte J, Kavazis AN, Talbert EE. Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Exp Physiol. 2010;95(1):1-9. [CrossRef]
- Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol CB. 2014;24(10):R453-462. [CrossRef]
- Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287(7):4434-4440. [CrossRef]
- Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401-426. [CrossRef]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89-116. [CrossRef]
- Safdar A, deBeer J, Tarnopolsky MA. Dysfunctional Nrf2-Keap1 redox signaling in skeletal muscle of the sedentary old. Free Radic Biol Med. 2010;49(10):1487-1493. [CrossRef]
- Miller CJ, Gounder SS, Kannan S, et al. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. Biochim Biophys Acta. 2012;1822(6):1038-1050. [CrossRef]
- Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci. 2019;20(13):3328. [CrossRef]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221-225. [CrossRef]
- Done AJ, Traustadóttir T. Nrf2 mediates redox adaptations to exercise. Redox Biol. 2016;10:191-199. [CrossRef]
- Ji LL, Kang C, Zhang Y. Exercise-induced hormesis and skeletal muscle health. Free Radic Biol Med. 2016;98:113-122. [CrossRef]
- Radak Z, Chung HY, Koltai E, Taylor AW, Goto S. Exercise, oxidative stress and hormesis. Ageing Res Rev. 2008;7(1):34-42. [CrossRef]
- Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013;280(17):4294-4314. [CrossRef]
- Deane CS, Cox J, Atherton PJ. Critical variables regulating age-related anabolic responses to protein nutrition in skeletal muscle. Front Nutr. 2024;11:1419229. [CrossRef]
- Liu Y, Ran S, Lin Y, et al. Four pleiotropic loci associated with fat mass and lean mass. Int J Obes. 2020;44(10):2113-2123. [CrossRef]
- Chen S, Yan S, Aiheti N, et al. A bi-directional Mendelian randomization study of sarcopenia-related traits and type 2 diabetes mellitus. Front Endocrinol. 2023;14:1109800. [CrossRef]
- Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med. 2015;21(12):1406-1415. [CrossRef]
- Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69 Suppl 1:S4-9. [CrossRef]
- Atkins JL, Jylhävä J, Pedersen NL, et al. A genome-wide association study of the frailty index highlights brain pathways in ageing. Aging Cell. 2021;20(9):e13459. [CrossRef]
- Chen B, Li S, Lin S, Dong H. Causal relationship of interleukin-6 and its receptor on sarcopenia traits using mendelian randomization. Nutr J. 2024;23(1):51. [CrossRef]
- Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E. From discoveries in ageing research to therapeutics for healthy ageing. Nature. 2019;571(7764):183-192. [CrossRef]
- Duggal NA, Niemiro G, Harridge SDR, Simpson RJ, Lord JM. Can physical activity ameliorate immunosenescence and thereby reduce age-related multi-morbidity? Nat Rev Immunol. 2019;19(9):563-572. [CrossRef]
- Zuo L, Prather ER, Stetskiv M, et al. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int J Mol Sci. 2019;20(18):4472. [CrossRef]
- Morgan MJ, Liu Z gang. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21(1):103-115. [CrossRef]
- Meng SJ, Yu LJ. Oxidative stress, molecular inflammation and sarcopenia. Int J Mol Sci. 2010;11(4):1509-1526. [CrossRef]
- Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21(12):1424-1435. [CrossRef]
- Soysal P, Isik AT, Carvalho AF, et al. Oxidative stress and frailty: A systematic review and synthesis of the best evidence. Maturitas. 2017;99:66-72. [CrossRef]
- Soysal P, Stubbs B, Lucato P, et al. Inflammation and frailty in the elderly: A systematic review and meta-analysis. Ageing Res Rev. 2016;31:1-8. [CrossRef]
- Gonzalez-Freire M, de Cabo R, Studenski SA, Ferrucci L. The Neuromuscular Junction: Aging at the Crossroad between Nerves and Muscle. Front Aging Neurosci. 2014;6:208. [CrossRef]
- Sousa-Victor P, Gutarra S, García-Prat L, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506(7488):316-321. [CrossRef]
- Fish LA, Fallon JR. Multiple MuSK signaling pathways and the aging neuromuscular junction. Neurosci Lett. 2020;731:135014. [CrossRef]
- Clark BC, Taylor JL. Age-related changes in motor cortical properties and voluntary activation of skeletal muscle. Curr Aging Sci. 2011;4(3):192-199. [CrossRef]
- Hortobágyi T, Rider P, Gruber AH, DeVita P. Age and muscle strength mediate the age-related biomechanical plasticity of gait. Eur J Appl Physiol. 2016;116(4):805-814. [CrossRef]
- Seidler RD, Bernard JA, Burutolu TB, et al. Motor control and aging: links to age-related brain structural, functional, and biochemical effects. Neurosci Biobehav Rev. 2010;34(5):721-733. [CrossRef]
- Tsuchiya M, Hara Y, Okuda M, et al. Cell surface flip-flop of phosphatidylserine is critical for PIEZO1-mediated myotube formation. Nat Commun. 2018;9(1):2049. [CrossRef]
- Xiao B. Mechanisms of mechanotransduction and physiological roles of PIEZO channels. Nat Rev Mol Cell Biol. 2024;25(11):886-903. [CrossRef]
- Gillies AR, Lieber RL. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve. 2011;44(3):318-331. [CrossRef]
- Stearns-Reider KM, D’Amore A, Beezhold K, et al. Aging of the skeletal muscle extracellular matrix drives a stem cell fibrogenic conversion. Aging Cell. 2017;16(3):518-528. [CrossRef]
- Severinsen MCK, Pedersen BK. Muscle-Organ Crosstalk: The Emerging Roles of Myokines. Endocr Rev. 2020;41(4):594-609. [CrossRef]
- Bettariga F, Taaffe DR, Galvão DA, et al. Exercise training mode effects on myokine expression in healthy adults: A systematic review with meta-analysis. J Sport Health Sci. 2024;13(6):764-779. [CrossRef]
- Kostka M, Morys J, Małecki A, Nowacka-Chmielewska M. Muscle-brain crosstalk mediated by exercise-induced myokines - insights from experimental studies. Front Physiol. 2024;15:1488375. [CrossRef]
- Wang B, Liang J, Lu C, Lu A, Wang C. Exercise Regulates Myokines in Aging-Related Diseases through Muscle-Brain Crosstalk. Gerontology. 2024;70(2):193-209. [CrossRef]
- Novelli G, Calcaterra G, Casciani F, Pecorelli S, Mehta JL. “Exerkines”: A Comprehensive Term for the Factors Produced in Response to Exercise. Biomedicines. 2024;12(9):1975. [CrossRef]
- Chow LS, Gerszten RE, Taylor JM, et al. Exerkines in health, resilience and disease. Nat Rev Endocrinol. 2022;18(5):273-289. [CrossRef]
- Safdar A, Saleem A, Tarnopolsky MA. The potential of endurance exercise-derived exosomes to treat metabolic diseases. Nat Rev Endocrinol. 2016;12(9):504-517. [CrossRef]
- St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397-408. [CrossRef]
- Sriram S, Subramanian S, Sathiakumar D, et al. Modulation of reactive oxygen species in skeletal muscle by myostatin is mediated through NF-κB. Aging Cell. 2011;10(6):931-948. [CrossRef]
- Vinel C, Lukjanenko L, Batut A, et al. The exerkine apelin reverses age-associated sarcopenia. Nat Med. 2018;24(9):1360-1371. [CrossRef]
- Lindholm ME, Marabita F, Gomez-Cabrero D, et al. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics. 2014;9(12):1557-1569. [CrossRef]
- Seaborne RA, Strauss J, Cocks M, et al. Human Skeletal Muscle Possesses an Epigenetic Memory of Hypertrophy. Sci Rep. 2018;8(1):1898. [CrossRef]
- Sharples AP, Turner DC. Skeletal muscle memory. Am J Physiol Cell Physiol. 2023;324(6):C1274-C1294. [CrossRef]
- Lu AT, Quach A, Wilson JG, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging. 2019;11(2):303-327. [CrossRef]
- Lu AT, Binder AM, Zhang J, et al. DNA methylation GrimAge version 2. Aging. 2022;14(23):9484-9549. [CrossRef]
- Fitzgerald KN, Hodges R, Hanes D, et al. Potential reversal of epigenetic age using a diet and lifestyle intervention: a pilot randomized clinical trial. Aging. 2021;13(7):9419-9432. [CrossRef]
- Sillanpää E, Heikkinen A, Kankaanpää A, et al. Blood and skeletal muscle ageing determined by epigenetic clocks and their associations with physical activity and functioning. Clin Epigenetics. 2021;13(1):110. [CrossRef]
- Ticinesi A, Nouvenne A, Cerundolo N, et al. Gut Microbiota, Muscle Mass and Function in Aging: A Focus on Physical Frailty and Sarcopenia. Nutrients. 2019;11(7):1633. [CrossRef]
- Przewłócka K, Folwarski M, Kaźmierczak-Siedlecka K, Skonieczna-Żydecka K, Kaczor JJ. Gut-Muscle Axis Exists and May Affect Skeletal Muscle Adaptation to Training. Nutrients. 2020;12(5):1451. [CrossRef]
- Andreux PA, Blanco-Bose W, Ryu D, et al. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019;1(6):595-603. [CrossRef]
- Liu S, D’Amico D, Shankland E, et al. Effect of Urolithin A Supplementation on Muscle Endurance and Mitochondrial Health in Older Adults: A Randomized Clinical Trial. JAMA Netw Open. 2022;5(1):e2144279. [CrossRef]
- Clarke SF, Murphy EF, O’Sullivan O, et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut. 2014;63(12):1913-1920. [CrossRef]
- Barton W, Penney NC, Cronin O, et al. The microbiome of professional athletes differs from that of more sedentary subjects in composition and particularly at the functional metabolic level. Gut. 2018;67(4):625-633. [CrossRef]
- Scheiman J, Luber JM, Chavkin TA, et al. Meta-omics analysis of elite athletes identifies a performance-enhancing microbe that functions via lactate metabolism. Nat Med. 2019;25(7):1104-1109. [CrossRef]
- Allen JM, Mailing LJ, Niemiro GM, et al. Exercise Alters Gut Microbiota Composition and Function in Lean and Obese Humans. Med Sci Sports Exerc. 2018;50(4):747-757. [CrossRef]
- Dodds RM, Syddall HE, Cooper R, et al. Grip strength across the life course: normative data from twelve British studies. PLoS One. 2014;9(12):e113637. [CrossRef]
- Lazarus NR, Harridge SDR. Declining performance of master athletes: silhouettes of the trajectory of healthy human ageing? J Physiol. 2017;595(9):2941-2948. [CrossRef]
- Pahor M, Guralnik JM, Ambrosius WT, et al. Effect of structured physical activity on prevention of major mobility disability in older adults: the LIFE study randomized clinical trial. JAMA. 2014;311(23):2387-2396. [CrossRef]
- World Health Organization. WHO Guidelines on Physical Activity and Sedentary Behaviour. World Health Organization: Geneva, Switzerland, 2020. Available online: https://www.who.int/publications/i/item/9789240015128 (accessed on 04 June 2026).
- World Health Organization. Integrated Care for Older People Approach (ICOPE). Available online: https://www.who.int/teams/maternal-newborn-child-adolescent-health-and-ageing/ageing-and-health/integrated-care-for-older-people-icope (accessed on 04 June 2026).
- Joseph SV, Edirisinghe I, Burton-Freeman BM. Berries: anti-inflammatory effects in humans. J Agric Food Chem. 2014;62(18):3886-3903. [CrossRef]
- Del Rio D, Rodriguez-Mateos A, Spencer JPE, Tognolini M, Borges G, Crozier A. Dietary (poly)phenolics in human health: structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid Redox Signal. 2013;18(14):1818-1892. [CrossRef]
- De la Fuente M, Miquel J. An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr Pharm Des. 2009;15(26):3003-3026. [CrossRef]
- Kaulmann A, Bohn T. Carotenoids, inflammation, and oxidative stress--implications of cellular signaling pathways and relation to chronic disease prevention. Nutr Res. 2014;34(11):907-929. [CrossRef]
- Traber MG, Stevens JF. Vitamins C and E: beneficial effects from a mechanistic perspective. Free Radic Biol Med. 2011;51(5):1000-1013. [CrossRef]
Figure 1.
Life-course model of functional reserve. Functional capacity can be conceptualised as a trajectory across the life course, determined by the maximum physiological capacity reached in youth or early adulthood and the subsequent rate of decline with ageing. Individuals with a higher functional peak and slower decline remain above the disability threshold for longer, thereby delaying or reducing the risk of geriatric motor dysfunctions such as sarcopenia, frailty and lower-limb weakness. Individual genetics, physical activity, nutrition, the microbiota, comorbidities and the exposome in general can influence both the maximum capacity achieved and the rate of functional decline over time.
Figure 1.
Life-course model of functional reserve. Functional capacity can be conceptualised as a trajectory across the life course, determined by the maximum physiological capacity reached in youth or early adulthood and the subsequent rate of decline with ageing. Individuals with a higher functional peak and slower decline remain above the disability threshold for longer, thereby delaying or reducing the risk of geriatric motor dysfunctions such as sarcopenia, frailty and lower-limb weakness. Individual genetics, physical activity, nutrition, the microbiota, comorbidities and the exposome in general can influence both the maximum capacity achieved and the rate of functional decline over time.

Figure 2.
Functional phenotypes common to both athletic performance and geriatric motor dysfunctions. Summary of the main functional domains linking athletic performance to geriatric motor dysfunctions. Although sports science and geriatric assessment often use different terminology, both fields evaluate overlapping physiological systems, including cardiorespiratory fitness, muscle strength, lower-limb power, gait efficiency, balance, performance of functional tasks, trainability, muscle quality, and physiological resilience.
Figure 2.
Functional phenotypes common to both athletic performance and geriatric motor dysfunctions. Summary of the main functional domains linking athletic performance to geriatric motor dysfunctions. Although sports science and geriatric assessment often use different terminology, both fields evaluate overlapping physiological systems, including cardiorespiratory fitness, muscle strength, lower-limb power, gait efficiency, balance, performance of functional tasks, trainability, muscle quality, and physiological resilience.

Figure 3.
Common biological pathways linking athletic performance and geriatric motor dysfunctions. Summary of the main biological pathways proposed to link athletic performance phenotypes with geriatric motor dysfunctions across the life course. Mitochondrial function, redox homeostasis and mitophagy, anabolic–catabolic balance, inflammageing, immunometabolism, oxi-inflamm-ageing, neuromuscular integrity, satellite cell function, mechanotransduction, myokine/exerkine signalling, epigenetic memory and the gut–muscle axis may contribute both to interindividual variability in physical performance and to vulnerability or resilience to sarcopenia, frailty and lower-limb weakness.
Figure 3.
Common biological pathways linking athletic performance and geriatric motor dysfunctions. Summary of the main biological pathways proposed to link athletic performance phenotypes with geriatric motor dysfunctions across the life course. Mitochondrial function, redox homeostasis and mitophagy, anabolic–catabolic balance, inflammageing, immunometabolism, oxi-inflamm-ageing, neuromuscular integrity, satellite cell function, mechanotransduction, myokine/exerkine signalling, epigenetic memory and the gut–muscle axis may contribute both to interindividual variability in physical performance and to vulnerability or resilience to sarcopenia, frailty and lower-limb weakness.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



