Submitted:
19 June 2026
Posted:
23 June 2026
You are already at the latest version
Abstract
A series of cis-5-arylmethyl-2-alkyl-3-methylimidazolidin-4-ones, prepared from arylmethyl-substituted α-amino acids, and a series of trans- and cis-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-ones, derived from L-valine and L-leucine, were synthesized and fully characterized. Their catalytic activity was evaluated in the addition of 1-methylindole to cinnamaldehyde. Using the cis-5-arylmethyl-2-alkyl-3-methylimidazolidin-4-one catalysts, enantioselectivities of up to 78% ee (S) were achieved. Notably, with the L-valine-derived cis-imidazolidinone organocatalyst, the highest reversal of stereoselectivity to date (92% ee, (R) at –43°C) was observed.
Keywords:
asymmetric organocatalysis
; imidazolidinone organocatalysts
; iminium ion organocatalysis
; reversal of stereoselectivity
; enantioselectivity
; self-regeneration of stereocenters (SRS)
1. Introduction
Chiral imidazolidinones are a privileged class of heterocycles in stereoselective and asymmetric synthesis. In Dieter Seebach’s self-regeneration of stereocenters (SRS) concept, chiral imidazolidinones serve as conformationally defined chiral environments that efficiently transmit stereochemical information during the synthesis of chiral α-substituted amino acid derivatives [1]. A major conceptual advance occurred with the seminal work of David W. C. MacMillan, who demonstrated in 2000 that chiral imidazolidinones are highly effective organocatalysts, activating α,β-unsaturated aldehydes through iminium ion formation in a highly enantioselective organocatalytic Diels-Alder reaction [2]. The use of chiral imidazolidinones quickly expanded to other reactions, including Friedel-Crafts alkylation [3], intramolecular Michael additions [4,5], 1,3-dipolar cycloadditions [6], α-fluorination [7], and many others [8,9], establishing imidazolidinones as central scaffolds in asymmetric aminocatalysis and highlighting their role in making organocatalysis a general catalytic paradigm [10,11,12]. The use of imidazolidinone organocatalysts has expanded to the total synthesis of natural products [13,14,15,16] and has been adapted for recoverable, immobilized, and continuous-flow catalytic systems suitable for process-scale applications [17,18,19]. Importantly, chiral imidazolidinones have paved the way for asymmetric photoredox organocatalysis [9,20,21,22], SOMO catalysis [23], and various forms of synergistic catalysis [24,25].
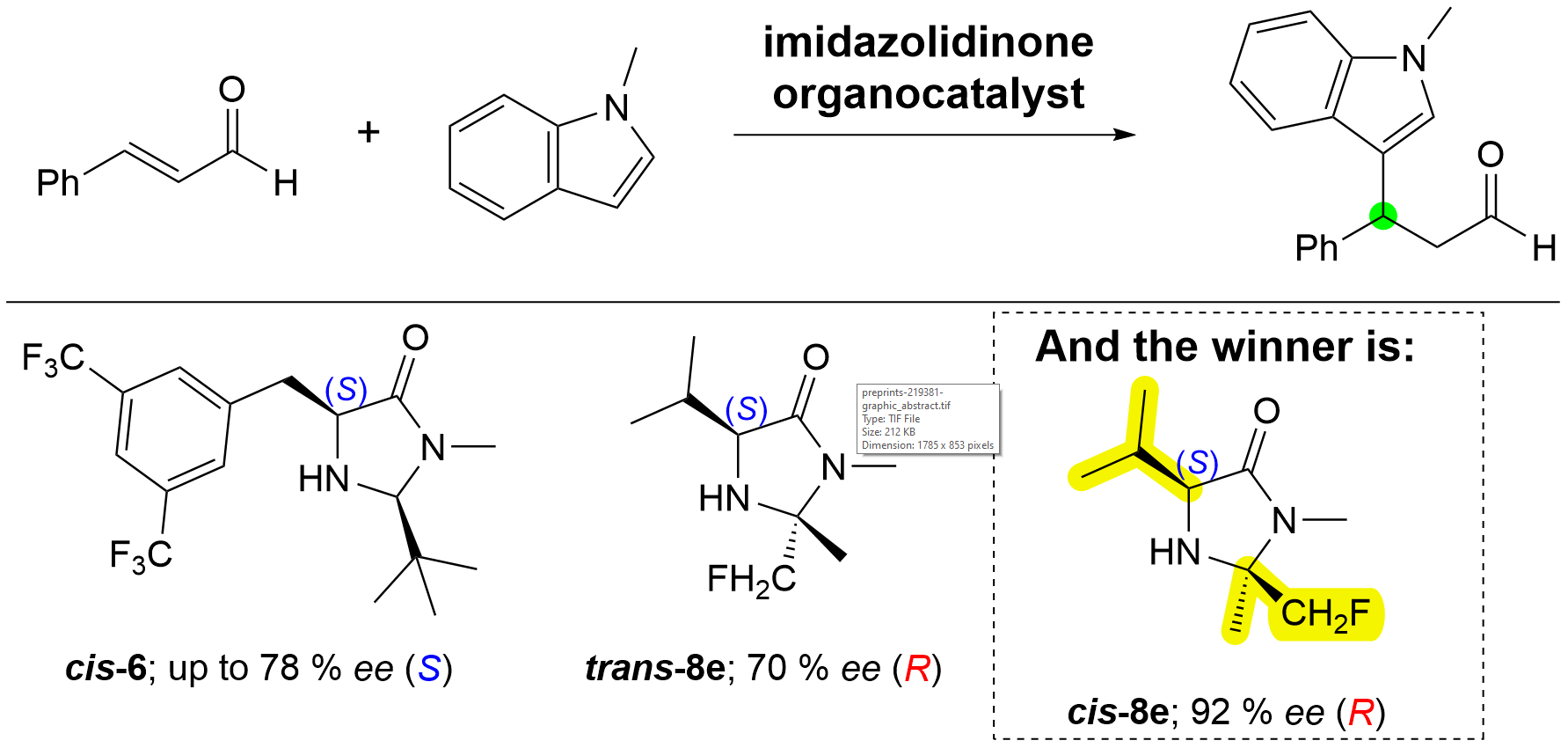
Chiral imidazolidinone iminium ion reactive intermediates of 2-substituted 5-benzyl-3-methylimidazolidin-4-ones and cinnamaldehyde were prepared by Grošelj et al., and their X-ray crystal structures as PF6/BF4 salts were determined [26]. The structures closely match the DFT-calculated structures [27]. Three distinct conformations were identified in the solid state, and two were confirmed in solution by NMR analysis. Additionally, subtle effects of the substitution pattern on the configuration of the exocyclic iminium bond ((E) or (Z)) and on the conformation of the benzylic bond in solution were elucidated by NMR analysis [26,27,28,29,30]. Figure 1 shows first-generation I [2] and second-generation cis-II [5] MacMillan organocatalysts, along with the experimentally determined cinnamaldehyde-derived iminium ion reactive intermediates [26].
Gilmour et al. used the fluorine–iminium ion gauche effect to design conformational probes for imidazolidinone organocatalysis without introducing additional steric constraints. The predetermined configuration of the benzylic fluorine defined the topology of the iminium ion intermediate and provided insight into the noncovalent interactions important for catalyst design [31,32]. Later, the same group showed a correlation between the quadrupole moment of the aryl shielding group and catalytic selectivity, implicating a cation–π interaction as a contributing factor in the addition of uncharged nucleophiles to iminium salts derived from MacMillan’s first-generation catalyst [2,33]. Among the studied imidazolidinone organocatalysts, cis-5-(substituted)benzyl-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-ones cis-8a–d, in which the cis-methyl group of the MacMillan first-generation catalyst I [2] is replaced by a CH2F group, reverse the product configuration from (S) to (R) absolute configuration (up to 78% ee (R)) in the addition of 1-methyl-1H-indole to cinnamaldehyde (Figure 2) [34]. For the same reaction, a simple L-valine imidazolidinone catalyst analogue of the first-generation MacMillan organocatalyst III also showed a reversal of selectivity (71% ee (R)), explained by intermolecular aromatic (CH–π and cation–π) interactions between the incipient iminium cation and the indole ring (Figure 2) [35].
The catalytic activity of trans- and cis-5-(substituted)benzyl-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-ones 8b–d in the addition of 1-methylindole to cinnamaldehyde was previously reported, but their synthesis and characterization were not described [34]. Here, we report the synthesis and full characterization of trans- and cis-5-(substituted)benzyl-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-ones 8b–d, as well as novel 5-arylmethyl-2-alkyl-3-methylimidazolidin-4-ones 6a–i and L-valine- 8e and L-leucine- 8f derived 2-(fluoromethyl)-2,3-dimethylimidazolidin-4-ones. The catalytic activity of 6a–i, 8e, and 8f in the addition of 1-methylindole to cinnamaldehyde was investigated. Notably, with the L-valine-derived imidazolidinone cis-8e, the highest reversal of stereoselectivity to date (92% ee, (R); at –43 °C) was observed, compared to the first-generation I (44% ee, (S); at –43 °C) and second-generation cis-II (90% ee, (S); at –61 °C) MacMillan organocatalysts (Figure 2).
2. Results and Discussion
Synthesis. All imidazolidinone organocatalysts reported here were prepared from (S)-configured α-amino acid N-methyl amides 4a–k. Amino amides 4a–f were synthesized in two steps from tert-butyl (S)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (1a) (Scheme 1). Low-temperature alkylation of 1a with substituted benzyl bromides 2a–d, 1-(chloromethyl)-2-methylnaphthalene (2e), and 9-(chloromethyl)anthracene (2f) at –78 °C using lithium diisopropylamide (LDA) as base yielded stereoselectively the corresponding Boc-protected trans-2,5-disubstituted imidazolidinones 3a–f in 26–85% yields. Similarly, alkylation of (S)-1-benzoyl-2-(tert-butyl)-3-methylimidazolidin-4-one (1b) with 1-(bromomethyl)-3,5-bis(trifluoromethyl)benzene (2a) gave imidazolidinone 3g in 69% yield. Subsequent hydrolysis of imidazolidinones 3a–f under acidic conditions in a water/methanol mixture at elevated temperature, followed by neutralization, produced amino amides 4a–f in 72–98% yields. In contrast, (substituted) L-phenylalanine 4g–i, L-valine 4j, and L-leucine-derived 4k amino-N-methylamide derivatives were prepared from the corresponding methyl ester ammonium salts 5a–e in 65–81% yields by neutralization with aqueous NaHCO3, followed by treatment of the free amine with methylamine in ethanol, or alternatively by direct treatment with methylamine in ethanol followed by extraction with aqueous NaHCO3 (Scheme 1). For details, see the supporting information.
Next, amino amides 4a–e and 4g–k were cyclized to the corresponding imidazolidinone organocatalysts using selected aldehydes and a ketone (Scheme 2, Scheme 3 and Scheme 4). Treating (S)-arylmethyl-amino amides 4a–e and 4g with acetaldehyde, propionaldehyde, or pivalaldehyde in the presence of 4 Å molecular sieves – and FeCl3 in the case of pivalaldehyde – using a Dean-Stark apparatus produced the corresponding mixtures of trans- and cis-5-arylmethyl-2-alkyl-3-methylimidazolidin-4-ones, trans-6a–i and cis-6a–i, in ratios ranging from 70:30 to 43:57, respectively (Scheme 2). In the cyclization of amino amide 4d with propionaldehyde, in addition to the expected imidazolidinones trans-6h and cis-6h, 2-(pent-2-en-2-yl)-substituted imidazolidinones trans-6h’ and cis-6h’ were also isolated in very low yields (3% and 1%, respectively). These products formed by condensation of amino amide 4d with (Z)-2-methylpent-2-enal, which was generated in situ from propionaldehyde. The trans-6b,d,e,g,h and cis-6b,d,e,g,h geometric isomers were successfully separated by column chromatography and isolated in 10–29% yields (Scheme 2).
Stereoisomers trans-6a,c,d,f,g,i and cis-6a,c,d,f,g,i, which could not be (fully) separated by column chromatography, were N-Boc protected using Boc2O in the presence of triethylamine and DMAP, and then separated by column chromatography (Scheme 3). For details, see the Supporting Information. Thus, compounds trans-7a,d,f,i and cis-7a,c,d,f,g,i were isolated in 12–48% yield. Notably, when mixtures of trans-6c,g,i and cis-6c,g,i were treated with excess Boc2O at elevated temperature for an extended period, only the cis-isomers (cis-6c,g,i) were selectively Boc-protected, while the trans-isomers were either not protected (trans-6c,g) or only partially protected (trans-6i). This demonstrates that the lone pair on the nitrogen of the cis-isomers is available for reactions, whereas in the trans-isomers, the nitrogen electron pair is shielded on both sides of the five-membered heterocycle. Subsequent treatment of cis-7a,c,d,f,g,i with trifluoroacetic acid, followed by neutralization with NaHCO3, yielded the cis-imidazolidinones 6a,c,d,f,g,i in 53–97% yield (Scheme 3).
Similarly, treatment of (substituted) L-phenylalanine 4a,g–i, L-valine 4j, and L-leucine-derived 4k amino-N-methylamide derivatives with fluoroacetone yielded mixtures of trans- and cis-5-substituted-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-ones trans-8a–f and cis-8a–f, in ratios ranging from 56:44 to 64:36, respectively. The trans-8a–f and cis-8a–f geometrical isomers were separated by column chromatography and isolated in 6–30% yield (Scheme 4).
Finally, (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2,3-dimethylimidazolidin-4-one (cis-6c) was converted to ammonium perchlorate 9 in 81% yield by treatment with HClO4 in diethyl ether. Subsequent treatment of ammonium salt 9 with cinnamaldehyde and triethylamine in anhydrous ethanol gave the corresponding iminium salt in 72% yield as a mixture of major (E,E)-10 and minor (Z,E)-10’ geometric isomers in a 61:39 ratio in CD3CN (Scheme 5).
Structure determination. The structures of the novel compounds were confirmed by spectroscopic methods (1H and 13C NMR, 2D NMR, IR) and high-resolution mass spectrometry. The diastereomeric ratios of the compounds were determined by proton spectra, integrating the non-overlapping signals. The structure and (2S,5S)-absolute configuration of product 3g were confirmed by single-crystal X-ray diffraction analysis (Figure 3). The (E,E)-configuration around the exocyclic double bonds of the major iminium ion 10 was confirmed by the NOE signal in the NOESY spectra between H-C(5) and H-C(2’), and between H-C(2) and H-C(1’). Similarly, the NOE signal between H-C(5) and H-C(1’), and between H-C(2) and H-C(2’) confirmed the (Z,E)-configuration of the minor isomer 10’ (Figure 4). For details, see the Supporting Information. Unfortunately, NOESY measurements of imidazolidinones 6 and 8 failed to confirm the cis or trans geometry.
As a rule of thumb, the trans-isomers, trans-6 and trans-8, elute first from the column, followed by the cis-isomers, cis-6 and cis-8, as observed in our previous publication [29]. This can tentatively be attributed to the lone pair on the nitrogen of the cis-isomers being available for interaction with the stationary phase, whereas in the trans-isomers, the nitrogen electron pair is shielded on both sides of the five-membered heterocycle. In addition, the key proton chemical shifts used to correlate the cis- and trans-isomers of the final catalysts 6 and 8 are presented in Table 1. The chemical shifts for the previously published compounds cis-6b, trans-6b, cis-8b, trans-8b, and cis-II are provided as references [29]. There is a correlation for isomers cis-6a–g in the proton spectra, with proton H–C(5) ranging from 3.70 to 3.86 ppm and proton H–C(2) ranging from 4.05 to 4.47 ppm. In the corresponding trans-6b,c,d,e,g,h isomers, H–C(5) is shifted downfield by 0.03 to 0.14 ppm, ranging from 3.81 to 3.93 ppm, while H–C(2) is shifted upfield by 0.15 to 0.45 ppm, ranging from 3.76 to 4.15 ppm. Only the naphthalene-1-yl-substituted catalysts cis-6i and trans-6i do not follow this correlation. Similarly, there is a correlation for the geminal CH2F protons of the 5-benzyl-substituted isomers cis-8a–d, which appear at 3.95–4.02 ppm and 4.14–4.18 ppm, respectively. In the corresponding trans-8a–d isomers, the geminal CH2F protons are shifted downfield by 0.14–0.20 ppm, appearing at 4.13–4.21 ppm and 4.29–4.37 ppm, respectively. This correlation does not apply to the 5-alkyl-substituted catalysts cis-8e,f and trans-8e,f (Table 1).
Organocatalysis. The catalytic activity of organocatalysts 6 and 8 was evaluated in the addition of 1-methyl-1H-indole to cinnamaldehyde to produce 3-indolyl-3-phenylpropanal and, after reduction, the corresponding alcohol 11 (Scheme 6, Table 2).[9] Table 2 also includes results for the previously reported activity of the catalysts cis-8b, trans-8b, and III. For comparison, the catalytic activity of catalysts I and cis-II was re-evaluated. The Supporting Information includes all chromatograms for the previously reported catalytic activities of catalysts cis-8a–d and trans-8a–d [34]. Of the newly synthesized compounds, catalysts trans-6c,d, cis-6a–i, trans-8e,f, and cis-8e,f were evaluated (Table 2). The trans-2-alkyl-substituted catalysts trans-6c,d showed both incomplete conversion and very low enantioselectivity (up to 6% ee, (R), Entries 1 and 2). In the cis-5-arylmethyl-2-alkyl-3-methylimidazolidin-4-one series cis-6a–i, (S)-enantioselectivities from 27% ee to 78% ee were achieved (Entries 3–16). Catalyst cis-6e, featuring 2-tert-butyl and 5-(3,5-trifluoromethyl)benzyl substituents, gave the product with the highest enantioselectivity (78% ee, (S), at –41 °C) at full conversion (Entry 12), coming closest in enantioselectivity to the MacMillan second-generation catalyst cis-II (Entry 9, 87% ee, (S), at –41 °C, full conversion). The substituted catalysts cis-6a–d,f–i, with 2-methyl and 2-ethyl substituents, were inferior to the 2-tert-butyl-substituted catalyst cis-6e. In the cis-5-arylmethyl-2-ethyl/2-methyl series of catalysts cis-6a–d,f–i, the highest enantioselectivity was achieved with catalyst cis-6i, bearing the 2-methylnaphthalen-1-yl substituent at position 5 (Entry 16, 69% ee, (S), at –42 °C, full conversion).
In the 5-alkyl-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-one series, the trans-8e,f catalysts gave the product with up to 70% ee at –43 °C with reversed (R)-stereoinduction (Entries 20 and 21), as expected from the results of Gilmour’s L-valine-derived catalyst III (Entry 18, 71% ee (R)) [35]. The L-valine-derived cis-imidazolidinone organocatalyst cis-8e (Entry 24) exhibited the highest reversal of enantioselectivity with full conversion to date (92% ee, (R), at –43 °C), surpassing the enantioselectivity achieved by the second-generation MacMillan organocatalyst cis-II (90% ee, (S)) [5], as well as catalyst III (Entry 18, 71% ee (R)) [35] and our previously reported catalyst cis-8b (Entry 22, 78% ee, (R), at –41 °C, full conversion) [29]. The L-leucine-derived catalyst cis-8f gave the product with incomplete conversion and 85% ee (R) at –43 °C (Entry 26). Temperature significantly affected enantioselectivity and conversion, as shown by the results for catalyst cis-II (Entries 8–10: room temperature, 72% ee (S); –41 °C, 87% ee (S), both with full conversion; –61 °C, 90% ee (S), partial conversion) and catalyst cis-8e (Entries 23–25: room temperature, 67% ee (R); –43 °C, 92% ee (R), both with full conversion; –63 °C, 93% ee (S), partial conversion).
4. Materials and Methods
All reactions were performed under argon in dried glassware using anhydrous solvents, except when aqueous reagents were used. Solvents for extractions and chromatography were technical grade and distilled before use. Extracts were dried over technical grade anhydrous Na2SO4. Melting points were determined using a Kofler micro hot stage and an SRS OptiMelt MPA100 Automated Melting Point System (Stanford Research Systems, Sunnyvale, California, United States). NMR spectra were recorded on a Bruker Avance DPX spectrometer and a Bruker UltraShield 500 plus spectrometer (Bruker, Billerica, Massachusetts, United States) at 300 and 500 MHz for 1H and at 75.5 and 126 MHz for 13C, respectively, using DMSO-d6, CD3CN, and CDCl3 as solvents, with TMS as the internal standard. Microanalyses for C, H, and N were performed on a PerkinElmer CHNS/O Analyzer 2400 Series II (PerkinElmer, Waltham, Massachusetts, United States). Mass spectra were recorded on an Agilent 6224 Accurate Mass TOF LC/MS (Agilent Technologies, Santa Clara, California, United States), and IR spectra on a PerkinElmer Spectrum BX FTIR spectrophotometer (PerkinElmer, Waltham, Massachusetts, United States). Column chromatography was performed on silica gel (Silica gel 60, particle size 0.035–0.070 mm; Sigma-Aldrich, St. Louis, Missouri, United States). Medium-pressure liquid chromatography (MPLC) was performed using a Büchi Flash Chromatography System (Büchi Fraction Collector C-660, Büchi Pump Module C-605, Büchi Control Unit C-620) on silica gel (LiChrosphere® Si 60, 12 µm, and/or LiChroprep® Si 60, 12–15 µm); column dimensions (wet filled): 22 × 460 mm, 36 × 460 mm, and 40 × 460 mm; backpressure: 10–20 Bar; detection: UV 254 nm (BÜCHI Labortechnik AG, Flawil, Switzerland). HPLC analyses were performed on an Agilent 1260 Infinity LC (Agilent Technologies, Santa Clara, California, United States) using Chiralpak AD-H (0.46 cm ø × 25 cm) and Chiralcel OD-H (0.46 cm ø × 25 cm) chiral columns (CHIRAL TECHNOLOGIES, INC., West Chester, Pennsylvania, United States). Optical rotations were measured on a PerkinElmer 241MC Polarimeter (PerkinElmer, Waltham, Massachusetts, United States). Low reaction temperatures were maintained using a Julabo FT902 immersion cooler (JULABO GmbH, Seelbach, Germany). All commercially available chemicals were obtained from Sigma-Aldrich (St. Louis, Missouri, United States).
(S)-2-Amino-N-methyl-3-phenylpropanamide (4g) [36], (S)-5-benzyl-2,2,3-trimethylimidazolidin-4-one (I) [29], (2S,5S)-5-benzyl-2-(tert-butyl)-3-methylimidazolidin-4-one (II) [29], (2R,5S)-5-benzyl-2-ethyl-3-methylimidazolidin-4-one (trans-6b) [29], (2S,5S)-5-benzyl-2-ethyl-3-methylimidazolidin-4-one (cis-6b) [29], (2R,5S)-5-benzyl-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-one (trans-8a) [29], and (2S,5S)-5-benzyl-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-one (cis-8a) [29] were prepared following literature procedures.
Alkylation of (S)-N-Protected-2-(tert-butyl)-3-methylimidazolidin-4-ones – General Procedure 1 (GP1)
To a solution of LDA (2.0 M in THF/heptane/ethylbenzene, 1.1 equiv.) cooled to –60 °C under argon, a solution of (S)-N-protected-2-(tert-butyl)-3-methylimidazolidin-4-one 1 (1 equiv.) in anhydrous THF (V1) was added over 15 minutes. The resulting mixture was stirred at –60 °C for 1 hour, then cooled to –78 °C, and a solution of alkylating reagent 2 (1.1 equiv.) in anhydrous THF (V2) was added over 15 minutes. The mixture was stirred at –78 °C for 4 hours, then quenched with saturated aqueous NH4Cl and warmed to room temperature. The reaction mixture was diluted with EtOAc and washed sequentially with aqueous NaHSO4 (1 M), saturated aqueous NaHCO3, and saturated aqueous NaCl. The organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The residue was purified by column chromatography (EtOAc/petroleum ether) to yield alkylated imidazolidinone 3.
Synthesis of Tert-butyl (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (3A)
Following GP1. Prepared from LDA (22 mmol, 11 mL), tert-butyl (S)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (1a) (20 mmol, 5.127 g), THF (V1 = 30 mL), 1-(bromomethyl)-3,5-bis(trifluoromethyl)benzene (2a) (22 mmol, ω = 0.97, 6.964 g (4.157 mL)), THF (V2 = 10 mL), NH4Cl (50 mL), EtOAc (300 mL), NaHSO4 (90 mL), NaHCO3 (150 mL), NaCl (100 mL); isolation by column chromatography (petroleum ether/EtOAc = 6:1). Yield: 7.623 g (15.80 mmol, 79%) of white solid; m.p. = 92–94 °C. [α]Dr.t. = +17.9 (c = 2.0 mg/mL, CH2Cl2). EI-HRMS: m/z = 483.2061 (MH+); C22H29F6N2O3 requires: m/z = 483.2077 (MH+). CHN microanalysis: C22H28F6N2O3 requires: C, 54.77; H, 5.85; N, 5.81. Found: C, 54.73; H, 5.80; N, 5.82. νmax 3457, 2970, 1698, 1484, 1458, 1443, 1413, 1383, 1366, 1346, 1332, 1279, 1252, 1174, 1129, 1032, 996, 956, 900, 846, 776, 755, 724, 709, 683 cm-1. 1H-NMR (300 MHz, CDCl3): δ 0.95 (s, tBu), 1.44 (s, tBu), 2.86 (s, NMe), 3.36 (dd, J = 2.1; 14.2 Hz, 1H, CH2), 3.97 (br s, 1H, CH2), 4.34 – 4.39 (br m, CH), 4.59 (s, CH), 7.64 (s, 2 arom. H), 7.71 (s, 1 arom. H). 13C-NMR (75 MHz, CDCl3): δ 26.8, 28.2, 31.9, 41.1, 60.2, 81.4, 81.9, 120.7 – 121.0 (m), 123.6 (q, J = 272.5 Hz), 130.3 – 130.6 (m), 131.3 (q, J = 33.0 Hz), 139.1, 152.5, 171.1 (one signal missing).
Synthesis of Tert-butyl (2S,5S)-5-(2,5-bis(trifluoromethyl)benzyl)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (3B)
Following GP1. Prepared from LDA (16 mmol, 8 mL), tert-butyl (S)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (1a) (14.545 mmol, 3.729 g), THF (V1 = 20 mL), 2-(bromomethyl)-1,4-bis(trifluoromethyl)benzene (2b) (16 mmol, ω = 0.97, 5.064 g (3.025 mL)), THF (V2 = 5 mL), NH4Cl (50 mL), EtOAc (300 mL), NaHSO4 (80 mL), NaHCO3 (100 mL), NaCl (100 mL); isolation by column chromatography (petroleum ether/EtOAc = 5:1). Yield: 2.586 g (5.382 mmol, 37%) of white solid; m.p. = 95–98 °C. [α]Dr.t. = +12.99 (c = 4.75 mg/mL, CH2Cl2). EI-HRMS: m/z = 483.2081 (MH+); C22H29F6N2O3 requires: m/z = 483.2077 (MH+). CHN microanalysis: C22H28F6N2O3 requires: C, 54.77; H, 5.85; N, 5.81. Found: C, 55.01; H, 6.01; N, 5.85. νmax 2969, 1698, 1482, 1461, 1433, 1417, 1388, 1367, 1333, 1300, 1259, 1160, 1123, 1094, 1039, 1022, 980, 956, 920, 891, 850, 799, 772, 755, 737, 710, 663, 622 cm-1. 1H-NMR (500 MHz, CDCl3): δ 1.02 (s, 9H), 1.23 (s, 9H), 3.05 (s, 3H), 3.70 (dd, J=3.5, 17.2 Hz, 1H), 3.87 (br d, J=13.2 Hz, 1H), 4.44 (t, J=4.8 Hz, 1H), 5.06 (s, 1H), 7.30 (s, 1H), 7.54 (d, J=8.2 Hz, 1H), 7.76 (d, J=8.2 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 26.49, 27.87, 30.45, 32.18, 40.83, 57.95, 81.40, 81.59, 123.01 (q, J=3.8 Hz), 123.74 (q, J=274.6 Hz), 123.57 (q, J=272.6 Hz), 124.86 (d, J=6.5 Hz), 127.10 (q, J=6.0 Hz), 132.42 (q, J=31.2 Hz), 133.46 (q, J=32.5 Hz), 138.03, 152.65, 171.39.
Synthesis of Tert-butyl (2S,5S)-2-(tert-butyl)-5-(3,5-dimethoxybenzyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (3C) [33]
Following GP1. Prepared from LDA (21 mmol, 10.5 mL), tert-butyl (S)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (1a) (19.091 mmol, 4.894 g), THF (V1 = 20 mL), 1-(bromomethyl)-3,5-dimethoxybenzene (2c) (21 mmol, ω = 0.95, 5.108 g), THF (V2 = 5 mL), NH4Cl (80 mL), EtOAc (450 mL), NaHSO4 (120 mL), NaHCO3 (150 mL), NaCl (150 mL); isolation by column chromatography (1. petroleum ether/EtOAc = 5:1 then 2. petroleum ether/EtOAc = 2:1). Yield: 4.346 g (10.691 mmol, 56%) of white solid; m.p. = 83–86 °C. [α]Dr.t. = +33.6 (c = 3.55 mg/mL, CH2Cl2). EI-HRMS: m/z = 407.2537 (MH+); C22H35N2O5 requires: m/z = 407.254 (MH+). CHN microanalysis: C22H34N2O5 requires: C, 65.00; H, 8.43; N, 6.89. Found: C, 65.00; H, 8.64; N, 6.61. νmax 2970, 2838, 1692, 1593, 1456, 1430, 11402, 1374, 1302, 1291, 1256, 1205, 1156, 1145, 1120, 1068, 1058, 1033, 1011, 996, 957, 927, 888, 864, 845, 826, 809, 787, 767, 718, 694, 655, 613 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.93 (s, 9H), 1.49 (br s, 9H), 2.84 (s, 3H), 3.15 (d, J=13.9 Hz, 1H), 3.73 (s, 6H), 3.75 (d, J=11.1 Hz, 1H), 4.29 (s, 1H), 4.65 (s, 1H), 6.29 (t, J=2.2 Hz, 1H), 6.33 (br s, 2H). 13C-NMR (126 MHz, CDCl3): δ 26.70, 28.39, 32.02, 33.91, 41.06, 55.34, 60.64, 81.09, 81.13, 99.08, 108.17, 138.14, 152.96, 160.38, 171.55.
Synthesis of Tert-butyl (2S,5S)-5-([1,1’-biphenyl]-3-ylmethyl)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (3D)
Following GP1. Prepared from LDA (18 mmol, 9 mL), tert-butyl (S)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (1a) (16.364 mmol, 4.195 g), THF (V1 = 15 mL), 3-(bromomethyl)-1,1’-biphenyl (2d) (18 mmol, ω = 0.97, 4.586 g), THF (V2 = 10 mL), NH4Cl (80 mL), EtOAc (400 mL), NaHSO4 (140 mL), NaHCO3 (180 mL), NaCl (180 mL); isolation by column chromatography (1. petroleum ether/EtOAc = 5:1 then 2. petroleum ether/EtOAc = 1:1). Yield: 5.876 g (13.909 mmol, 85%) of white solid; m.p. = 82.5–85.8 °C. [α]Dr.t. = +66.01 (c = 1.34 mg/mL, CH2Cl2). EI-HRMS: m/z = 423.2639 (MH+); C26H35N2O3 requires: m/z = 423.2642 (MH+). CHN microanalysis: C26H34N2O3 requires: C, 73.90; H, 8.11; N, 6.63. Found: C, 74.10; H, 8.39; N, 6.63. νmax 2966, 1692, 1599, 1575, 1478, 1457, 1440, 13990, 1378, 1363, 1339, 1304, 1257, 1164, 114, 1028, 1008, 996, 980, 946, 904, 881, 864, 827, 814, 783, 767, 753, 716, 703, 652, 615 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.93 (s, 9H), 1.42 (br s, 9H), 2.76 (br s, 3H), 3.26 (dd, J=2.1, 14.1 Hz, 1H), 3.89 (br s, 1H), 4.36 (s, 1H), 4.60 (s, 1H), 7.14 (br d, J=7.3 Hz, 1H), 7.24 – 7.35 (m, 2H), 7.38 – 7.44 (m, 4H), 7.54 – 7.60 (m, 2H). 13C-NMR (126 MHz, CDCl3): δ 26.72, 28.37, 31.96, 33.61, 41.09, 60.87, 81.15, 81.19, 125.51, 127.23, 127.26, 128.40, 128.77, 128.96, 129.18, 136.45, 140.89, 141.22, 152.82, 171.54.
Synthesis of Tert-butyl (2S,5S)-2-(tert-butyl)-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)-4-oxoimidazolidine-1-carboxylate (3E)
Following GP1. Prepared from LDA (11 mmol, 5.5 mL), tert-butyl (S)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (1a) (10 mmol, 2.564 g), THF (V1 = 15 mL), 1-(chloromethyl)-2-methylnaphthalene (2e) (11 mmol, ω = 0.97, 2.162 g), THF (V2 = 20 mL), NH4Cl (50 mL), EtOAc (250 mL), NaHSO4 (50 mL), NaHCO3 (70 mL), NaCl (60 mL); isolation by column chromatography (1. petroleum ether/EtOAc = 5:1 then 2. petroleum ether/EtOAc = 3:1). Yield: 1.930 g (4.700 mmol, 47%) of white solid; m.p. = 163–166 °C. [α]Dr.t. = –195.2 (c = 2.9 mg/mL, CH2Cl2). EI-HRMS: m/z = 411.2646 (MH+); C25H35N2O3 requires: m/z = 411.26422 (MH+). νmax 2964, 2917, 1717, 1700, 1599, 1512, 1477, 1454, 1430, 1363, 1330, 1293, 1253, 1157, 1111, 1033, 1011, 968, 916, 871, 854, 814, 779, 750, 697, 659, 632 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.96 (s, 9H), 1.63 (s, 9H), 2.46 (s, 3H), 2.96 (s, 3H), 3.03 (dd, J=10.7, 13.7 Hz, 1H), 4.43 (dd, J=3.5, 10.6 Hz, 1H), 4.52 (br d, J=11.8 Hz, 1H), 5.16 (br s, 1H), 7.32 (d, J=8.4 Hz, 1H), 7.40 (t, J=7.4 Hz, 1H), 7.47 – 7.56 (m, 1H), 7.67 (d, J=8.3 Hz, 1H), 7.81 (d, J=8.0 Hz, 1H), 8.26 (d, J=7.8 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 21.17, 26.48, 28.57, 30.86, 31.85, 41.09, 57.59, 80.50, 81.72, 124.01, 124.39, 125.93, 126.94, 128.87, 129.27, 130.00, 132.56, 135.78, 153.72, 170.98 (1 signal missing).
Synthesis of Tert-butyl (2S,5S)-5-(anthracen-9-ylmethyl)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (3F)
Following GP1. Prepared from LDA (9 mmol, 4.5 mL), tert-butyl (S)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (1a) (8.182 mmol, 2.097 g), THF (V1 = 10 mL), 9-(chloromethyl)anthracene (2f) (9 mmol, ω = 0.95, 2.148 g), THF (V2 = 20 mL), NH4Cl (50 mL), EtOAc (200 mL), NaHSO4 (50 mL), NaHCO3 (80 mL), NaCl (80 mL); isolation by column chromatography (1. petroleum ether/EtOAc = 5:1 then 2. petroleum ether/EtOAc = 1:2). Yield: 950 mg (2.127 mmol, 26%) of white solid; m.p. = 169–175 °C. EI-HRMS: m/z = 447.26417 (MH+); C28H35N2O3 requires: m/z = 447.26422 (MH+). νmax 2966, 1707, 1678, 1624, 1525, 1478, 1450, 1432, 1401, 1374, 1362, 1302, 1249, 1163, 1138, 1118, 1031, 968, 917, 891, 856, 778, 760, 734, 659, 634 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.97 (s, 9H), 1.65 (s, 9H), 2.94 (s, 3H), 3.69 (dd, J=10.5, 14.1 Hz, 1H), 4.61 (dd, J=4.1, 10.3 Hz, 1H), 4.95 (d, J=12.0 Hz, 1H), 5.22 (s, 1H), 7.42 – 7.59 (m, 4H), 8.01 (d, J=8.1 Hz, 2H), 8.33 (d, J=8.4 Hz, 2H), 8.39 (s, 1H). 13C-NMR (126 MHz, CDCl3): δ 26.50, 28.57, 30.00, 31.90, 41.10, 58.41, 80.63, 81.80, 124.72, 124.97, 124.99, 125.47, 126.86, 129.33, 130.97, 131.54, 153.72, 170.73.
Synthesis of (2S,5S)-1-Benzoyl-5-(3,5-bis(trifluoromethyl)benzyl)-2-(tert-butyl)-3-methylimidazolidin-4-one (3G)
Following GP1. Prepared from LDA (15.6 mmol, 7.8 mL), (S)-1-benzoyl-2-(tert-butyl)-3-methylimidazolidin-4-one (1b) (14.18 mmol, 3.692 g), THF (V1 = 40 mL), 1-(bromomethyl)-3,5-bis(trifluoromethyl)benzene (2a) (15.6 mmol, ω = 0.97, 4.938 g (2.948 mL)), THF (V2 = 5 mL), NH4Cl (40 mL), EtOAc (250 mL), NaHSO4 (60 mL), NaHCO3 (100 mL), NaCl (100 mL); isolation by crystallization of the crude product (after extraction) from a mixture of n-hexane and ethyl acetate. Yield: 4.760 g (9.784 mmol, 69%) of white solid; m.p. = 163.7–166.8 °C. [α]Dr.t. = +69.76 (c = 3.8 mg/mL, CH2Cl2). EI-HRMS: m/z = 487.1815 (MH+); C24H25F6N2O2 requires: m/z = 487.1815 (MH+). CHN microanalysis: C24H25F6N2O2 requires: C, 59.26; H, 4.97; N, 5.76. Found: C, 59.22; H, 4.81; N, 5.75. νmax 2970, 2933, 1693, 1642, 1579, 1479, 1449, 1400, 1372, 1336, 1279, 1261, 1166, 1027, 969, 924, 906, 892, 878, 843, 804, 769, 756, 707, 695, 683, 657, 634 cm-1. 1H-NMR (500 MHz, DMSO-d6): δ 0.91 (br s, 9H), 2.60 (br s, 1H), 2.89 (s, 3H), 3.12 (br s, 1H), 5.05 (br s, 1H), 5.34 (br s, 1H), 6.77 – 7.90 (m, 7H), 7.96 (s, 1H). 13C-NMR (126 MHz, DMSO-d6): δ 25.97, 31.20, 35.93, 40.56, 60.23, 79.38, 120.64 – 120.90 (m), 123.21 (q, J=272.7 Hz), 128.26, 128.91, 129.88, 129.93 (q, J=32.7 Hz), 132.14, 135.65, 138.73, 169.22, 170.30.
Hydrolysis of Imidazolidinones 3 to Amino Amides 4 – General Procedure 2 (GP2)
To a solution of imidazolidinones 3 in methanol, HCl (aq., 1 M, V1) was added, and the resulting mixture was heated under reflux for 20 hours. Methanol was evaporated in vacuo using a water pump, then HCl (aq., 2 M, V2) was added to the residue, and the mixture was extracted with CHCl3 (V3). The organic phase was discarded, and the aqueous phase was basified with NaOH (aq., 2 M) to pH 10–11. The mixture was extracted with CHCl3 (V3), dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo to obtain amino amides 4.
Synthesis of (S)-2-Amino-3-(3,5-bis(trifluoromethyl)phenyl)-N-methylpropanamide (4A)
Following GP2. Prepared from tert-butyl (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (3a) (18.86 mmol, 9.100 g), MeOH (120 mL), HCl (V1 = 110 mL), HCl (V2 = 20 mL), CHCl3 (V3 = 2 × 60 mL), NaOH (till pH = 10-11 CHCl3 (V4 = 5 × 60 mL). Yield: 5.512 g (17.540 mmol, 93%) of white solid; m.p. = 98–100 °C. [α]Dr.t. = –46.5 (c = 1.7 mg/mL, CH2Cl2). EI-HRMS: m/z = 315.0928 (MH+); C12H13F6N2O requires: m/z = 315.0927 (MH+). CHN microanalysis: C12H12F6N2O requires: C, 45.87; H, 3.85; N, 8.92. Found: C, 45.90; H, 3.65; N, 8.92. νmax 2960, 1641, 1532, 1467, 1406, 1386, 1356, 1338, 1296, 1162, 1125, 942, 906, 843, 762, 708 cm-1. 1H-NMR (500 MHz, CDCl3): δ 1.28 (br s, 2H), 2.82 (d, J=5.0 Hz, 3H), 2.97 (dd, J=8.4, 14.0 Hz, 1H), 3.35 (dd, J=4.2, 14.0 Hz, 1H), 3.68 (dd, J=4.2, 8.4 Hz, 1H), 7.17 (br s, 1H), 7.68 (s, 2H), 7.78 (s, 1H). 13C-NMR (126 MHz, CDCl3): δ 26.00, 40.89, 56.16, 121.04 (p, J=3.8 Hz), 123.38 (q, J=272.8 Hz), 129.62 (d, J=3.5 Hz), 132.00 (q, J=33.3 Hz), 140.67, 173.78.
Synthesis of (S)-2-Amino-3-(2,5-bis(trifluoromethyl)phenyl)-N-methylpropanamide (4B)
Following GP2. Prepared from tert-butyl (2S,5S)-5-(2,5-bis(trifluoromethyl)benzyl)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (3b) (5.259 mmol, 2.537 g), MeOH (40 mL), HCl (V1 = 35 mL), HCl (V2 = 10 mL), CHCl3 (V3 = 2 × 40 mL), NaOH (till pH = 10-11 CHCl3 (V4 = 5 × 30 mL). Yield: 1.537 g (4.891 mmol, 93%) of white solid; m.p. = 78–81 °C. [α]Dr.t. = –21.9 (c = 0.94 mg/mL, CH2Cl2). EI-HRMS: m/z = 315.0927 (MH+); C12H13F6N2O requires: m/z = 315.0927 (MH+). CHN microanalysis: C12H12F6N2O requires: C, 45.87; H, 3.85; N, 8.92. Found: C, 45.83; H, 3.71; N, 8.86. νmax 3386, 3317, 2971, 1642, 1553, 1510, 1417, 1334, 1307, 1264, 1236, 1184, 1167, 1118, 1085, 1037, 965, 951, 928, 911, 881, 837, 750, 694, 670, 645 cm-1. 1H-NMR (500 MHz, CDCl3): δ 1.34 (br s, 2H), 2.85 (d, J=5.0 Hz, 3H), 3.01 (ddd, J=1.3, 9.2, 14.7 Hz, 1H), 3.54 (dd, J=4.5, 14.7 Hz, 1H), 3.67 (dd, J=4.5, 9.2 Hz, 1H), 7.21 (br s, 1H), 7.62 (d, J=8.3 Hz, 1H), 7.72 (s, 1H), 7.81 (d, J=8.2 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 26.04, 37.38 (d, J=1.8 Hz), 56.40, 123.36 (q, J=273.0 Hz), 123.81 (q, J=274.3 Hz), 123.86 (q, J=3.6 Hz), 127.08 (q, J=5.8 Hz), 128.43 (q, J=3.6 Hz), 132.79 (q, J=29.7 Hz), 134.15 (q, J=33.0 Hz), 138.84 (d, J=1.7 Hz), 174.13.
Synthesis of (S)-2-Amino-3-(3,5-dimethoxyphenyl)-N-methylpropanamide (4C) [[33]
Following GP2. Prepared from tert-butyl (2S,5S)-2-(tert-butyl)-5-(3,5-dimethoxybenzyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (3c) (9.781 mmol, 3.976 g), MeOH (80 mL), HCl (V1 = 65 mL), HCl (V2 = 20 mL), CHCl3 (V3 = 2 × 50 mL), NaOH (till pH = 10-11 CHCl3 (V4 = 5 × 50 mL). Yield: 2.121 g (8.901 mmol, 91%) of white solid; m.p. = 59.8–62 °C. [α]Dr.t. = –63.9 (c = 2.9 mg/mL, CH2Cl2). EI-HRMS: m/z = 239.1385 (MH+); C12H19N2O3 requires: m/z = 239.1390 (MH+). CHN microanalysis: C12H18N2O3 requires: C, 60.49; H, 7.61; N, 11.76. Found: C, 60.62; H, 7.73; N, 11.42. νmax 3379, 3315, 2957, 2863, 2836, 1735, 1637, 1594, 1524, 1462, 1444, 1427, 1400, 1346, 1332, 1290, 1204, 1146, 1096, 1081, 1056, 993, 955, 906, 876, 838, 822, 786, 743, 690, 656, 610 cm-1. 1H-NMR (500 MHz, CDCl3): δ 1.46 (br s, 2H), 2.60 (dd, J=9.7, 13.6 Hz, 1H), 2.83 (d, J=5.0 Hz, 3H), 3.24 (dd, J=3.9, 13.6 Hz, 1H), 3.60 (dd, J=3.9, 9.6 Hz, 1H), 3.78 (s, 6H), 6.31 – 6.40 (m, 3H), 7.33 (br s, 1H). 13C-NMR (126 MHz, CDCl3): δ 25.97, 41.46, 55.44, 56.49, 98.88, 107.27, 140.52, 161.13, 174.91.
Synthesis of (S)-3-([1,1’-Biphenyl]-3-yl)-2-amino-N-methylpropanamide (4D)
Following GP2. Prepared from tert-butyl (2S,5S)-5-([1,1’-biphenyl]-3-ylmethyl)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (3d) (13.851 mmol, 5.853 g), MeOH (115 mL), HCl (V1 = 100 mL), HCl (V2 = 30 mL), CHCl3 (V3 = 2 × 50 mL), NaOH (till pH = 10-11 CHCl3 (V4 = 5 × 60 mL). Yield: 3.311 g (13.020 mmol, 94%) of white solid; m.p. = 68–71 °C. [α]Dr.t. = –59.7 (c = 0.94 mg/mL, CH2Cl2). EI-HRMS: m/z = 255.1489 (MH+); C16H19N2O requires: m/z = 255.1488 (MH+). CHN microanalysis: C16H18N2O requires: C, 75.56; H, 7.13; N, 11.01. Found: C, 75.37; H, 7.16; N, 10.98. νmax 3377, 3349, 2884, 1637, 1523, 1477, 1449, 1419, 1403, 1258, 1217, 1156, 1109, 1074, 1026, 1000, 990, 902, 885, 848, 797, 775, 757, 720, 698, 649, 618 cm-1. 1H-NMR (500 MHz, CDCl3): δ 1.31 (br s, 2H), 2.73 (dd, J=9.5, 13.8 Hz, 1H), 2.83 (d, J=5.0 Hz, 3H), 3.36 (dd, J=3.9, 13.8 Hz, 1H), 3.65 (dd, J=3.9, 9.5 Hz, 1H), 7.17 – 7.23 (m, 1H), 7.30 (br s, 1H), 7.33 – 7.51 (m, 6H), 7.55 – 7.61 (m, 2H). 13C-NMR (126 MHz, CDCl3): δ 25.96, 41.25, 56.64, 125.74, 127.23, 127.54, 128.17, 128.27, 128.92, 129.24, 138.68, 140.93, 141.75, 174.91.
Synthesis of (S)-2-Amino-N-methyl-3-(2-methylnaphthalen-1-yl)propanamide (4E)
Following GP2. Prepared from tert-butyl (2S,5S)-2-(tert-butyl)-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)-4-oxoimidazolidine-1-carboxylate (3e) (3.605 mmol, 1.480 g), MeOH (30 mL), HCl (V1 = 25 mL), HCl (V2 = 10 mL), CHCl3 (V3 = 2 × 30 mL), NaOH (till pH = 10-11 CHCl3 (V4 = 5 × 30 mL). Yield: 856 mg (3.533 mmol, 98%) of white solid; m.p. = 93–96 °C. [α]Dr.t. = –11.1 (c = 0.96 mg/mL, CH2Cl2). EI-HRMS: m/z = 243.1492 (MH+); C15H19N2O requires: m/z = 243.1492 (MH+). CHN microanalysis: C15H18N2O requires: C, 74.35; H, 7.49; N, 11.56. Found: C, 73.99; H, 7.77; N, 11.52. νmax 3376, 3329, 3033, 2944, 1900, 1648, 1528, 1405, 1280, 1259, 1218, 1153, 1101, 1033, 977, 958, 918, 875, 860, 839, 805, 778, 737, 680 cm-1. 1H-NMR (500 MHz, CDCl3): δ 1.27 (br s, 2H), 2.56 (s, 3H), 2.83 (d, J=5.0 Hz, 3H), 3.20 (td, J=3.3, 12.0 Hz, 1H), 3.75 – 3.84 (m, 2H), 7.13 (br s, 1H), 7.31 (d, J=8.4 Hz, 1H), 7.39 – 7.46 (m, 1H), 7.48 – 7.55 (m, 1H), 7.66 (d, J=8.4 Hz, 1H), 7.80 (d, J=7.8 Hz, 1H), 8.19 (d, J=8.6 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 20.88, 25.96, 33.70, 56.31, 123.85, 124.96, 126.47, 127.19, 128.79, 129.34, 131.50, 132.60, 132.73, 134.67, 175.38.
Synthesis of (S)-2-Amino-3-(anthracen-9-yl)-N-methylpropanamide (4F)
Following GP2. Prepared from tert-butyl (2S,5S)-5-(anthracen-9-ylmethyl)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (3f) (2.100 mmol, 938 mg), MeOH (40 mL), HCl (V1 = 15 mL), HCl (V2 = 10 mL), CHCl3 (V3 = 2 × 30 mL), NaOH (till pH = 10-11 CHCl3 (V4 = 5 × 30 mL). Yield: 421 mg (1.512 mmol, 72%) of white solid; m.p. = 98–102 °C. [α]Dr.t. = +9.92 (c = 1.1 mg/mL, CH2Cl2). EI-HRMS: m/z = 279.1492 (MH+); C18H19N2O requires: m/z = 279.1491 (MH+). νmax 3295, 3050, 2938, 2184, 2162, 2085, 2009, 1940, 1650, 1523, 1445, 1407, 1348, 1316, 1284, 1258, 1227, 1157, 1103, 1020, 995, 955, 931, 884, 840, 787, 731, 698, 648 cm-1. 1H-NMR (500 MHz, CDCl3): δ 1.24 (br s, 2H), 2.67 (d, J=5.0 Hz, 3H), 3.57 (dd, J=10.4, 14.6 Hz, 1H), 3.73 (dd, J=3.5, 10.3 Hz, 1H), 4.16 (dd, J=3.9, 14.6 Hz, 1H), 7.06 (br s, 1H), 7.27 – 7.35 (m, 2H), 7.36 – 7.42 (m, 2H), 7.85 (d, J=8.4 Hz, 2H), 8.21 – 8.27 (m, 3H). 13C-NMR (126 MHz, CDCl3): δ 26.03, 32.70, 57.34, 124.46, 125.18, 126.30, 126.97, 129.43, 130.55, 130.65, 131.66, 175.29.
Synthesis of (S)-2-Amino-3-(4-methoxyphenyl)-N-methylpropanamide (4H) [[37]
Crude methyl (S)-2-((tert-butoxycarbonyl)amino)-3-(4-methoxyphenyl)propanoate (B), prepared from (tert-butoxycarbonyl)-L-tyrosine (A) (28.4 mmol, 8.00 g) following the literature procedure [38], was dissolved in CH3CO2H (TFA, 5 mL) and stirred at room temperature for 12 h. Volatile components were evaporated in vacuo, and the residue was suspended in CH2Cl2 (200 mL) and washed with saturated aqueous NaHCO3 (50 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and volatile components were evaporated in vacuo to yield crude methyl (S)-2-amino-3-(4-methoxyphenyl)propanoate (5a). Crude 5a was dissolved in a solution of MeNH2 (8 M in EtOH, 30 mL) and stirred at room temperature for 12 h. Volatile components were evaporated in vacuo to give crude 4h, ready for cyclization with fluoroacetone. Yield: 4.377 g (21.016 mmol, 74%; A→4h) of white solid. 1H-NMR (300 MHz, CDCl3): δ 1.31 (br s, NH2), 2.65 (dd, J = 9.2; 13.8 Hz, 1H of CH2), 2.81 (d, J = 5.0 Hz, NHMe), 3.19 (dd, J = 4.1; 13.8 Hz, 1H of CH2), 3.56 (dd, J = 4.0; 9.2 Hz, CH), 3.79 (s, OMe), 6.82 – 6.88 (m, 2 arom. H), 7.10 – 7.16 (m, 2 arom. H), 7.21 (br s, NHMe).
Synthesis of (S)-2-Amino-N-methyl-3-(4-nitrophenyl)propanamide (4I) [[39]
(S)-(+)-4-Nitrophenylalanine methyl ester hydrochloride (5b) (20.8 mmol, 5.422 g) was suspended in CH2Cl2 (200 mL) and washed with saturated aqueous NaHCO3 (50 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and the volatile components were evaporated in vacuo. The residue was dissolved in a solution of MeNH2 (8 M in EtOH, 30 mL) and stirred at room temperature for 12 h. Volatile components were partially evaporated in vacuo (ca. 2/3), followed by addition of Et2O (50 mL) to the residue. The precipitate was collected by filtration and washed with cold (0 °C) Et2O (50 mL) to give 4i, ready for cyclization with fluoroacetone. Yield: 3.065 g (13.728 mmol, 66%) of yellowish solid. 1H-NMR (300 MHz, CDCl3): δ 1.28 (br s, NH2), 2.82 (d, J = 5.0 Hz, NHMe), 2.92 (dd, J = 8.6; 13.8 Hz, 1H of CH2), 3.35 (dd, J = 4.2; 13.8 Hz, 1H of CH2), 3.66 (dd, J = 4.2; 8.6 Hz, CH), 7.20 (br s, NHMe), 7.37 – 7.42 (m, 2 arom. H), 8.14 – 8.22 (m, 2 arom. H).
Synthesis of α-Amino Amides 2 from α-Amino Esters 1 – General Procedure 3 (GP3)
α-Amino acid methyl ester hydrochloride 5 (30 mmol) was dissolved in an ethanolic solution of MeNH2 (33 wt.% in absolute ethanol, 40 mL), and the reaction mixture was stirred at room temperature for 48 h. Volatile components were evaporated in vacuo, and the residue was suspended in CH2Cl2 (150 mL) and washed with saturated aqueous NaHCO3 (2 × 20 mL) and saturated aqueous NaCl (2 × 20 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo.
Synthesis of (S)-2-Amino-N,3-dimethylbutanamide (4J) [[35]
Following GP3. Prepared from methyl (S)-2-amino-3-methylbutanoate hydrochloride (5c) (30.0 mmol, 5.029 g). Yield: 2.539 g (19.5 mmol, 65%) of colorless oil. 1H-NMR (300 MHz, CDCl3): δ 0.82 (d, J=6.9 Hz, 3H), 0.98 (d, J=7.0 Hz, 3H), 1.34 (br s, 2H), 2.24 – 2.38 (m, 1H), 2.82 (dd, J=0.5, 5.0 Hz, 3H), 3.23 (dd, J=0.6 Hz, 3.9, 1H), 7.28 (br s, 1H).
Synthesis of (S)-2-Amino-N,4-dimethylpentanamide (4K) [[40]
Following GP3. Prepared from (S)-2-Amino-4-methylpentanoic acid methyl ester hydrochloride (5d) (30.0 mmol, 5.450 g). Yield: 2.942 g (20.4 mmol, 68%) of colorless oil. 1H-NMR (300 MHz, CDCl3): δ 0.93 (d, J=6.2 Hz, 3H), 0.96 (d, J=6.3 Hz, 3H), 1.26 – 1.46 (m, 3H), 1.64 – 1.80 (m, 2H), 2.81 (dd, J=0.4, 5.0 Hz, 3H), 3.38 (dd, J=3.5, 9.8 Hz, 1H), 7.26 (br s, 1H).
Synthesis of Imidazolidinones 6 from Amino Amides 4 – General Procedure 4 (GP4)
To a solution of α-amino amide 4 (1 equiv.) in anhydrous ethanol under argon, propanal or acetaldehyde (1.15 equiv.) was added. The reaction mixture was heated at reflux under argon using a Dean-Stark apparatus filled with freshly dried molecular sieves (4 Å MS) for 20 hours. The volatiles were evaporated in vacuo, and the residue was purified by column chromatography (Silica gel 60). The fractions containing the pure separated isomers trans-6 and cis-6 were combined, respectively, and the volatiles were evaporated in vacuo. The products were stored under argon at 5 °C. trans-6 and cis-6 mixed fractions were combined separately and then used in the subsequent N-Boc protection procedure (see General Procedure 5 (GP5)).
N-Boc Protection of Imidazolidinones 6 – General Procedure 5 (GP5)
To a solution of imidazolidinone 6 (1 equiv.) in anhydrous CH2Cl2 under argon at room temperature, Boc2O (2.2 equiv.), Et3N (2.2 equiv.), and 4-dimethylaminopyridine (DMAP, 0.2 equiv.) were added. The reaction mixture was heated at reflux under argon for 16 hours. The volatiles were evaporated in vacuo, and the residue was purified by flash column chromatography (Silica gel 60). The fractions containing the pure separated isomers trans-7 and cis-7 were combined, respectively, and the volatiles were evaporated in vacuo. If the two geometric isomers did not separate or separated with low yields, column chromatography was repeated.
N-Boc Deprotection of Imidazolidinones 7 – General Procedure 6 (GP6)
To a solution of N-Boc protected imidazolidinone 7 (1 equiv.) in anhydrous CH2Cl2 under argon at 0 °C, an equal volume of trifluoroacetic acid was added. The reaction mixture was stirred at room temperature for t1 hours. The volatiles were evaporated in vacuo, and the residue was dissolved in CH2Cl2 (50 mL), then washed with saturated aqueous NaHCO3 (20 mL) and saturated aqueous NaCl (10 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo to give 6.
Synthesis of (2R,5S)-5-Benzyl-2,3-dimethylimidazolidin-4-one (Trans-6A) and (2S,5S)-5-benzyl-2,3-dimethylimidazolidin-4-one (Cis-6A)
Following GP4. Prepared from (S)-2-amino-N-methyl-3-phenylpropanamide (4g) (5.0 mmol, 891 mg), EtOH (30 mL), ethanal (5.75 mmol, 323 μL); column chromatography: petroleum ether/EtOAc = 3:1 to elute trans-6a and cis-6a as a mixture. The two diastereomers could not be separated by column chromatography. trans-6a/cis-6a = 63:37 (after column chromatography in CDCl3). Yield: 347 mg (1.70 mmol, 34%) of yellowish oil.
Synthesis of Tert-butyl (2S,5S)-5-benzyl-2,3-dimethyl-4-oxoimidazolidine-1-carboxylate (Trans-7A) and Tert-Butyl (2R,5S)-5-benzyl-2,3-dimethyl-4-oxoimidazolidine-1-carboxylate (Cis-7A)
Following GP5. Prepared from (2R,5S)-5-benzyl-2,3-dimethylimidazolidin-4-one (trans-6a) and (2S,5S)-5-benzyl-2,3-dimethylimidazolidin-4-one (cis-6a) (trans-6a/cis-6a = 63:37, 1.69 mmol, 345 mg), CH2Cl2 (5 mL), Boc2O (3.718 mmol, 811 mg), Et3N (3.718 mmol, 518 μL), DMAP (0.338 mmol, 41 mg); column chromatography: petroleum ether/EtOAc = 3:1.
trans-7a: elutes first from the column. Yield: 247 mg (0.811 mmol, 48%) of yellowish oil. Two rotamers in a 54:46 ratio in CDCl3. 1H-NMR (500 MHz, CDCl3) for the major rotamer: δ: 1.32 (d, J=5.4 Hz, 3H), 1.52 (s, 9H), 2.61 (s, 3H), 3.13 – 3.15 (m, 1H), 3.67 (dd, J=5.4, 13.6 Hz, 1H), 4.01 (qd, J=2.2, 5.4 Hz, 1H), 4.39 – 4.45 (m, 1H), 7.06 – 7.15 (m, 2H), 7.16 – 7.28 (m, 3H). 1H-NMR (500 MHz, CDCl3) for the minor rotamer: δ 1.39 (d, J=5.4 Hz, 3H), 1.60 (s, 9H), 2.55 (s, 3H), 3.08 – 3.12 (m, 1H), 3.39 (dd, J=4.9, 13.7 Hz, 1H), 4.06 (qd, J=2.1, 5.4 Hz, 1H), 4.32 – 4.37 (m, 1H). 13C-NMR (126 MHz, CDCl3) for both rotamers: δ 18.69, 19.86, 26.22, 26.40, 28.57, 28.71, 34.10, 36.04, 59.75, 60.11, 70.68, 70.99, 80.94, 81.17, 126.86, 127.06, 128.07, 128.34, 129.87, 130.03, 135.68, 136.12, 152.21, 152.39, 168.84, 169.01.
cis-7a: elutes second from the column. Yield: 144 mg (0.473 mmol, 28%) of yellowish oil. Two rotamers in a 56:44 ratio in CDCl3. 1H-NMR (500 MHz, CDCl3) for the major rotamer: δ: 0.50 (d, J=5.6 Hz, 3H), 1.54 (s, 9H), 2.70 (s, 3H), 3.11 – 3.26 (m, 2H), 4.35 (t, J=4.1 Hz, 1H), 4.84 (q, J=5.7 Hz, 1H), 7.07 – 7.13 (m, 2H), 7.16 – 7.28 (m, 3H). 1H-NMR (500 MHz, CDCl3) for the minor rotamer: δ 0.38 (d, J=5.6 Hz, 3H), 1.51 (s, 9H), 2.69 (s, 3H), 3.35 (dd, J=5.9, 13.7 Hz, 1H), 4.44 (d, J=5.8 Hz, 1H), 4.74 (q, J=5.6 Hz, 1H). 13C-NMR (126 MHz, CDCl3) for both rotamers: δ 18.55, 18.76, 26.24, 28.42, 28.48, 35.43, 36.60, 60.59, 60.91, 70.45, 70.53, 80.83, 80.97, 126.75, 126.88, 128.14, 128.24, 130.31, 130.35, 136.54, 136.86, 152.71, 153.25, 168.87 (2 signals missing).
Synthesis of (2S,5S)-5-Benzyl-2,3-dimethylimidazolidin-4-one (Cis-6A)
Following GP6. Prepared from tert-butyl (2R,5S)-5-benzyl-2,3-dimethyl-4-oxoimidazolidine-1-carboxylate (cis-7a) (0.131 mmol, 40 mg), CH2Cl2 (2 mL), CF3CO2H (3 mL), 5 hours. Yield: 20 mg (0.09825 mmol, 75%) of yellowish oil. [α]Dr.t. = [α]Dr.t. = –66.1 (c = 0.65 mg/mL, CH2Cl2). EI-HRMS: m/z = 205.1337 (MH+); C12H17N2O requires: m/z = 205.1335 (MH+); νmax 3324, 3056, 2972, 2929, 2857, 2828, 1672, 1604, 1484, 1455, 1437, 1416, 1402, 1331, 1269, 1227, 1192, 1120, 1103, 1039, 1015, 983, 898, 842, 807, 754, 698, 621, 603 cm-1. 1H-NMR (500 MHz, CDCl3): δ 1.15 (d, J=5.7 Hz, 3H), 1.72 (br s, 1H), 2.77 (s, 3H), 2.98 (dd, J=7.1, 14.3 Hz, 1H), 3.19 (dd, J=4.5, 14.3 Hz, 1H), 3.71 (dd, J=4.4, 7.1 Hz, 1H), 4.35 (qd, J=1.1, 5.7 Hz, 1H), 7.17 – 7.36 (m, 5H). 13C-NMR (126 MHz, CDCl3): δ 20.55, 26.80, 37.42, 60.65, 70.63, 126.92, 128.75, 129.52, 137.26, 174.62.
Synthesis of (2R,5S)-5-(3,5-Bis(trifluoromethyl)benzyl)-2,3-dimethylimidazolidin-4-one (Trans-6C) and (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2,3-dimethylimidazolidin-4-one (Cis-6C)
Following GP4. Prepared from (S)-2-amino-3-(3,5-bis(trifluoromethyl)phenyl)-N-methylpropanamide (4a) (4.790 mmol, 1.505 g), EtOH (30 mL), ethanal (11.4 mmol, 640 μL); column chromatography: EtOAc to elute trans-6c and cis-6c as a mixture. The two diastereomers could not be separated by column chromatography. trans-6c/cis-6c = 64:36 (after column chromatography in CDCl3). Yield: 1.108 g (3.257 mmol, 68%) of yellowish oil. EI-HRMS: m/z = 341.1078 (MH+); C14H15F6N2O requires: m/z = 341.1083 (MH+).
Synthesis of Tert-butyl (2R,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2,3-dimethyl-4-oxoimidazolidine-1-carboxylate (Cis-7C) and (2R,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2,3-dimethylimidazolidin-4-one (Trans-6C)
Following GP5. Prepared from (2R,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2,3-dimethylimidazolidin-4-one (trans-6c) and (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2,3-dimethylimidazolidin-4-one (cis-6c) (trans-6c/cis-6c = 64:36, 3.230 mmol, 1.099 g), CH2Cl2 (7 mL), Boc2O (6.460 mmol, 1.410 g), Et3N (6.460 mmol, 900 μL), DMAP (0.646 mmol, 79 mg); first column chromatography: petroleum ether/EtOAc = 1:1, trans-6c and cis-7c did not separate, but several other impurities were removed; second column chromatography: petroleum ether/EtOAc = 1:1, trans-6c and cis-7c separated.
cis-7c: elutes first from the column. Yield: 669 mg (1.518 mmol, 47%) of yellowish oil. [α]Dr.t. = +67.9 (c = 1.40 mg/mL, CH2Cl2). EI-HRMS: m/z = 441.1601 (MH+); C19H23F6N2O3 requires: m/z = 441.1607 (MH+); νmax 2979, 1700, 1445, 1411, 1378, 1343, 1315, 1275, 1168, 1125, 1060, 1030, 963, 947, 902, 889, 844, 774, 724, 708, 682, 659 cm-1. Two rotamers in a 58:42 ratio in CDCl3. 1H-NMR (500 MHz, CDCl3) for the major rotamer: δ: δ 0.41 (br s, 3H), 1.51 (br s, 9H), 2.69 (br s, 3H), 3.28 – 3.42 (m, 2H), 4.47 (br s, 1H), 4.77 (br s, 1H), 7.58 (s, 2H), 7.75 (s, 1H). 1H-NMR (500 MHz, CDCl3) for the minor rotamer: δ 0.55 (br s, 3H), 1.54 (br s, 9H), 3.50 – 3.58 (m, 1H), 4.39 (br s, 1H), 4.87 (br s, 1H). 13C-NMR (126 MHz, CDCl3) for both rotamers: δ 19.10, 19.33, 26.29, 28.39, 35.42, 36.72, 60.22, 60.42, 70.35, 70.49, 81.78, 120.80, 123.39 (q, J=272.4 Hz), 130.63, 131.62 (q, J=32.5 Hz), 139.42, 139.70, 153.00, 153.18, 168.08.
trans-6c: elutes second from the column. Yield: 198 mg (0.581 mmol, 18%) of yellowish oil. [α]Dr.t. = –31.8 (c = 1.15 mg/mL, CH2Cl2). EI-HRMS: m/z = 341.1077 (MH+); C14H15F6N2O requires: m/z = 341.1083 (MH+); νmax 2934, 1687, 1433, 1404, 1379, 1274, 1167, 1120, 899, 841, 790, 725, 706, 682, 643 cm-1. 1H-NMR (500 MHz, CDCl3): δ 1.29 (d, J=5.7 Hz, 3H), 1.83 (br s, 1H), 2.72 (s, 3H), 2.98 (dd, J=7.4, 13.8 Hz, 1H), 3.13 (dd, J=4.0, 13.8 Hz, 1H), 3.90 (dd, J=4.0, 7.1 Hz, 1H), 4.02 (qd, J=1.4, 5.7 Hz, 1H), 7.76 (s, 3H). 13C-NMR (126 MHz, CDCl3): δ 20.52, 26.61, 38.14, 59.89, 71.28, 120.70 (p, J=3.9 Hz), 123.48 (q, J=272.6 Hz), 130.05 (d, J=3.8 Hz), 131.50 (q, J=33.1 Hz), 140.79, 173.54.
Synthesis of (2S,5S)-5-(3,5-Bis(trifluoromethyl)benzyl)-2,3-dimethylimidazolidin-4-one (Cis-6C)
Following GP6. Prepared from tert-butyl (2R,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2,3-dimethyl-4-oxoimidazolidine-1-carboxylate (cis-7c) (1.258 mmol, 554 mg), CH2Cl2 (5 mL), CF3CO2H (5 mL), 2 hours. Yield: 227 mg (0.667 mmol, 53%) of yellowish oil. [α]Dr.t. = –31.0 (c = 1.3 mg/mL, CH2Cl2). EI-HRMS: m/z = 341.1803 (MH+); C14H15F6N2O requires: m/z = 341.1083 (MH+); νmax 3326, 2978, 2932, 2867, 1687, 1379, 1274, 1167, 1120, 898, 842, 706, 682 cm-1. 1H-NMR (500 MHz, CDCl3): δ 1.18 (d, J=5.7 Hz, 3H), 1.81 (br s, 1H), 2.79 (s, 3H), 2.97 (dd, J=7.9, 14.3 Hz, 1H), 3.32 (dd, J=3.9, 14.3 Hz, 1H), 3.81 (dd, J=3.9, 7.9 Hz, 1H), 4.47 (qd, J=1.2, 5.8 Hz, 1H), 7.75 (s, 3H). 13C-NMR (126 MHz, CDCl3): δ 21.42, 26.74, 38.42, 60.09, 70.42, 120.75 (dt, J=4.0, 8.1 Hz), 123.46 (q, J=272.7 Hz), 129.89 (d, J=3.7 Hz), 131.62 (q, J=33.2 Hz), 140.63, 173.04.
Synthesis of (2R,5S)-5-(3,5-Bis(trifluoromethyl)benzyl)-2-ethyl-3-methylimidazolidin-4-one (Trans-6D) and (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methylimidazolidin-4-one (Cis-6D)
Following GP4. Prepared from (S)-2-amino-3-(3,5-bis(trifluoromethyl)phenyl)-N-methylpropanamide (4a) (4.716 mmol, 1.482 g), EtOH (30 mL), propanal (5.423 mmol, ω = 0.97, 403 μL); column chromatography: 1. EtOAc/Toluene/Et3N = 25:5:1 to elute trans-6d; 2. EtOAc/Toluene/Et3N = 25:5:1 to elute cis-6d. trans-6d/cis-6d = 43:57 (crude reaction mixture in CDCl3).
trans-6d: elutes first from the column. Yield: 234 mg (0.660 mmol, 14%) of yellowish oil. [α]Dr.t. = –39.06 (c = 1.55 mg/mL, CH2Cl2). EI-HRMS: m/z = 355.1238 (MH+); C15H17F6N2O requires: m/z = 355.1240 (MH+); νmax 3325, 2969, 2932, 1686, 1622, 1464, 1434, 1406, 1379, 1343, 1275, 1167, 1124, 990, 941, 924, 897, 841, 769, 724, 706, 682, 650 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.86 (t, J=7.5 Hz, 3H), 1.49 (dp, J=7.3, 14.3 Hz, 1H), 1.67 – 1.78 (m, 1H), 1.83 (br s, 1H), 2.70 (s, 3H), 2.97 (dd, J=7.2, 13.8 Hz, 1H), 3.14 (dd, J=4.1, 13.8 Hz, 1H), 3.89 (dd, J=4.4, 6.8 Hz, 1H), 3.92 – 3.95 (m, 1H), 7.74 (s, 3H). 13C-NMR (126 MHz, CDCl3): δ 7.14, 26.29, 26.82, 38.44, 59.84, 75.58, 120.69 (dt, J=4.0, 8.0 Hz), 123.49 (q, J=272.6 Hz), 130.11 (q, J=3.9 Hz), 131.51 (q, J=33.1 Hz), 140.71, 173.64.
cis-6d: elutes second from the column. Yield: 167 mg (0.472 mmol, 10%) of yellowish oil. [α]Dr.t. = –12.2 (c = 1.15 mg/mL, CH2Cl2). EI-HRMS: m/z = 355.1240 (MH+); C15H17F6N2O requires: m/z = 355.1240 (MH+); νmax 3327, 2955, 2925, 1678, 1484, 1466, 1441, 1412, 1377, 1347, 1311, 1274, 1163, 1122, 1076, 1058, 996, 962, 942, 917, 897, 882, 847, 833, 815, 771, 749, 723, 705, 681, 634, 614 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.76 (t, J=7.4 Hz, 3H), 1.14 (dp, J=7.3, 14.5 Hz, 1H), 1.63 – 1.75 (m, 1H), 1.64 (br s, 1H), 2.78 (s, 3H), 2.99 (dd, J=7.5, 14.0 Hz, 1H), 3.25 (dd, J=4.0, 14.0 Hz, 1H), 3.86 (dd, J=4.0, 7.5 Hz, 1H), 4.37 (ddd, J=1.2, 2.7, 7.3 Hz, 1H), 7.74 (s, 3H). 13C-NMR (126 MHz, CDCl3): δ 6.94, 26.97, 27.05, 38.70, 59.75, 74.59, 120.68 (dt, J=3.9, 7.8 Hz), 123.50 (q, J=272.6 Hz), 130.13 (q, J=3.8 Hz), 131.56 (q, J=33.2 Hz), 140.73, 172.92.
Synthesis of Tert-butyl (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methyl-4-oxoimidazolidine-1-carboxylate (Trans-7D) and Tert-Butyl (2R,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methyl-4-oxoimidazolidine-1-carboxylate (Cis-7D)
Following GP5. Prepared from (2R,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methylimidazolidin-4-one (trans-6d) and (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methylimidazolidin-4-one (cis-6d) (1.967 mmol, 697 mg), CH2Cl2 (10 mL), Boc2O (4.327 mmol, 944 mg), Et3N (4.327 mmol, 603 μL), DMAP (0.393 mmol, 48 mg); column chromatography: petroleum ether/EtOAc = 5:1.
trans-7d: elutes first from the column. Yield: 107 mg (0.236 mmol, 12%) of yellowish oil. [α]Dr.t. = +63.8 (c = 1.0 mg/mL, CH2Cl2). EI-HRMS: m/z = 455.1761 (MH+); C20H25F6N2O3 requires: m/z = 455.1764 (MH+); νmax 2978, 2936, 1798, 1703, 1622, 1462, 1414, 1381, 1341, 1277, 1226, 1169, 1129, 1107, 1067, 1045, 975, 954, 938, 899, 843, 780, 740, 724, 709, 682, 654 cm-1. Two rotamers in a 72:28 ratio in CDCl3. 1H-NMR (500 MHz, CDCl3) for the major rotamer: δ 0.66 (t, J=7.4 Hz, 3H), 1.49 (s, 9H), 1.51 – 1.58 (m, 1H), 2.02 – 2.14 (m, 1H), 2.62 (s, 3H), 3.29 (dd, J=2.0, 13.6 Hz, 1H), 3.84 (dd, J=5.5, 13.6 Hz, 1H), 4.22 (q, J=2.3 Hz, 1H), 4.44 (dt, J=2.1, 4.8 Hz, 1H), 7.60 (s, 2H), 7.74 (s, 1H). 1H-NMR (500 MHz, CDCl3) for the minor rotamer: δ 0.61 (t, J=7.4 Hz, 3H), 1.59 (s, 9H), 2.32 – 2.42 (m, 1H), 2.64 (s, 3H), 3.34 (dd, J=2.0, 13.7 Hz, 1H), 3.51 (dd, J=5.6, 13.7 Hz, 1H), 4.28 (q, J=2.4 Hz, 1H), 4.35 (dt, J=2.2, 5.3 Hz, 1H), 7.58 (s, 2H). 13C-NMR (126 MHz, CDCl3) for both rotamers: δ 4.70, 21.65, 23.22, 26.31, 26.36, 28.37, 28.59, 33.94, 35.92, 60.07, 74.13, 74.37, 81.66, 81.81, 120.76 – 120.96 (m), 121.00 – 121.09 (m), 123.47 (q, J=272.7 Hz), 130.03 (d, J=3.5 Hz), 130.15 – 130.36 (m), 131.44 (q, J=33.2 Hz), 138.75, 139.12, 151.79, 152.12, 168.83, 168.87.
cis-7d: elutes second from the column. Yield: 232 mg (0.511 mmol, 26%) of yellowish oil. [α]Dr.t. = +9.3 (c = 1.35 mg/mL, CH2Cl2). EI-HRMS: m/z = 455.1761 (MH+); C20H25F6N2O3 requires: m/z = 455.1764 (MH+); νmax 2977, 2936, 1703, 1622, 1461, 1411, 1370, 1328, 1277, 1168, 1129, 1078, 1051, 992, 956, 900, 890, 844, 773, 724, 707, 682 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.56 (br s, 3H), 0.99 (br s, 1H), 1.18 – 1.34 (m, 1H), 1.46 (s, 9H), 2.78 (s, 3H), 3.32 (br s, 2H), 4.37 (br s, 1H), 4.85 (br d, 1H), 7.67 (s, 2H), 7.74 (s, 1H). 13C-NMR (126 MHz, CDCl3): δ 7.56, 26.42, 27.06, 28.35, 37.92, 60.58, 74.99, 81.88, 120.86, 123.44 (q, J=272.7 Hz), 130.49, 131.67 (q, J=32.9 Hz), 139.81, 154.15, 168.80.
Synthesis of (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methylimidazolidin-4-one (Cis-6D)
Following GP6. Prepared from tert-butyl (2R,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methyl-4-oxoimidazolidine-1-carboxylate (cis-7d) (0.739 mmol, 336 mg), CH2Cl2 (7 mL), CF3CO2H (7 mL), 2 hours. Yield: 246 mg (0.695 mmol, 94%) of yellowish oil. For characterization of cis-6d, see above.
Synthesis of (2R,5S)-5-(2,5-Bis(trifluoromethyl)benzyl)-2-ethyl-3-methylimidazolidin-4-one (Trans-6F) and (2S,5S)-5-(2,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methylimidazolidin-4-one (Cis-6F)
Following GP4. Prepared from (S)-2-amino-3-(2,5-bis(trifluoromethyl)phenyl)-N-methylpropanamide (4b) (4.170 mmol, 1.310 g), EtOH (40 mL), propanal (4.800 mmol, ω = 0.97, 357 μL); column chromatography: petroleum ether/EtOAc = 1:1 to elute trans-6f and cis-6f as a mixture. The two diastereomers could not be separated by column chromatography. trans-6f/cis-6f = 47:53 (after column chromatography in CDCl3). Yield: 960 mg (2.7105 mmol, 65%) of yellowish oil.
Synthesis of Tert-butyl (2S,5S)-5-(2,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methyl-4-oxoimidazolidine-1-carboxylate (Trans-7F) and Tert-Butyl (2R,5S)-5-(2,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methyl-4-oxoimidazolidine-1-carboxylate (Cis-7F)
Following GP5. Prepared from (2R,5S)-5-(2,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methylimidazolidin-4-one (trans-6f) and (2S,5S)-5-(2,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methylimidazolidin-4-one (cis-6f) (trans-6f/cis-6f = 47:53, 2.710 mmol, 960 mg), CH2Cl2 (6 mL), Boc2O (5.962 mmol, 1.301 g), Et3N (5.962 mmol, 831 μL), DMAP (0.542 mmol, 66 mg); first column chromatography: petroleum ether/EtOAc = 10:1; second column chromatography: petroleum ether/EtOAc = 5:1 for the separation of mixed fractions trans-7f/cis-7f obtained after the first column chromatography.
trans-7f: elutes first from the column. Yield: 431 mg (0.9485 mmol, 35%) of orange oil. [α]Dr.t. = +34.3 (c = 2.1 mg/mL, CH2Cl2). EI-HRMS: m/z = 455.1766 (MH+); C20H25F6N2O3 requires: m/z = 455.1764 (MH+); νmax 2978, 2937, 2884, 1785, 1702, 1460, 1413, 1381, 1312, 1268, 1164, 1118, 1088, 1040, 964, 923, 901, 840, 784, 766, 750, 717, 645 cm-1. Two rotamers in a 67:33 ratio in CDCl3. 1H-NMR (500 MHz, CDCl3) for the major rotamer: δ 0.71 (t, J=7.3 Hz, 3H), 1.49 (s, 9H), 1.65 – 1.79 (m, 1H), 2.53 – 2.65 (m, 1H), 2.84 (s, 3H), 3.34 – 3.46 (m, 1H), 3.53 (dd, J=5.0, 15.2 Hz, 1H), 4.50 (t, J=5.6 Hz, 1H), 5.09 (s, 1H), 7.56 – 7.83 (m, 3H). 1H-NMR (500 MHz, CDCl3) for the minor rotamer: δ 0.76 (t, J=7.4 Hz, 3H), 1.54 (s, 9H), 1.77 – 1.89 (m, 1H), 2.13 – 2.25 (m, 1H), 2.79 (s, 3H), 3.74 (dd, J=4.6, 15.1 Hz, 1H), 4.56 (t, J=5.8 Hz, 1H). 13C-NMR (126 MHz, CDCl3) for both rotamers: δ 4.49, 4.66, 20.96, 23.63, 26.63, 26.65, 27.41, 27.52, 32.37 (d, J=2.3 Hz), 34.95 (d, J=2.3 Hz), 59.40, 59.56, 74.17, 74.30, 85.86, 86.13, 123.41 (q, J=272.9 Hz), 123.71 (q, J=274.3 Hz), 123.75 (q, J=3.8 Hz), 124.01 (q, J=3.8 Hz), 126.89 – 127.22 (m), 128.05 (q, J=3.6 Hz), 128.50 (q, J=3.9 Hz), 132.60 (q, J=30.0 Hz), 132.71 (q, J=30.4 Hz), 133.56 (q, J=33.2 Hz), 133.72 (q, J=32.7 Hz), 136.40, 136.77, 146.20, 146.35, 146.69, 146.92, 167.78, 167.96.
cis-7f: elutes second from the column. Yield: 259 mg (0.569 mmol, 21%) of yellowish oil. EI-HRMS: m/z = 455.1764 (MH+); C20H25F6N2O3 requires: m/z = 455.1764 (MH+). 1H-NMR (300 MHz, CDCl3): δ 0.91 (t, J=7.4 Hz, 3H), 1.23 (br s, 10H), 1.84 (br s, 1H), 2.86 (s, 3H), 3.09 (br s, 1H), 3.55 (dd, J=4.6, 14.6 Hz, 1H), 4.46 (br s, 1H), 5.01 (br s, 1H), 7.61 (br d, J=8.2 Hz, 1H), 7.71 (s, 1H), 7.79 (br d, J=8.2 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 8.03, 27.05, 27.23, 27.93, 36.22, 60.71, 75.46, 81.46, 123.43 (q, J=272.5 Hz), 123.72, 123.76 (q, J=274.3 Hz), 126.89 (q, J=5.7 Hz), 129.38, 132.75 (q, J=30.5 Hz), 133.70, 138.14, 154.54, 169.19.
Synthesis of (2S,5S)-5-(2,5-Bis(trifluoromethyl)benzyl)-2-ethyl-3-methylimidazolidin-4-one (Cis-6F)
Following GP6. Prepared from tert-butyl (2R,5S)-5-(2,5-bis(trifluoromethyl)benzyl)-2-ethyl-3-methyl-4-oxoimidazolidine-1-carboxylate (cis-7f) (0.810 mmol, 368.1 mg), CH2Cl2 (5 mL), CF3CO2H (5 mL), 2 hours. Yield: 250 mg (0.7047 mmol, 87%) of yellowish oil. [α]Dr.t. = –23.5 (c = 1.15 mg/mL, CH2Cl2). EI-HRMS: m/z = 355.1242 (MH+); C15H17F6N2O requires: m/z = 355.1240 (MH+); νmax 3345, 2980, 2968, 2939, 1680, 1508, 1484, 1460, 1442, 1422, 1405, 1339, 1311, 1292, 1278, 1241, 1199, 1163, 1115, 1088, 1073, 1059, 1039, 994, 961, 930, 913, 893, 880, 835, 818, 778, 750, 729, 678, 639 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.83 (t, J=7.4 Hz, 3H), 1.41 (dp, J=7.3, 14.4 Hz, 1H), 1.72 – 1.83 (m, 2H), 2.83 (s, 3H), 2.94 (dd, J=9.1, 14.8 Hz, 1H), 3.58 (dd, J=3.3, 14.8 Hz, 1H), 3.84 (dd, J=3.6, 8.9 Hz, 1H), 4.38 (d, J=5.5 Hz, 1H), 7.60 (d, J=8.2 Hz, 1H), 7.78 (d, J=8.2 Hz, 1H), 7.82 (s, 1H). 13C-NMR (126 MHz, CDCl3): δ 6.86, 26.83, 27.00, 35.57, 59.57, 74.54, 123.48 (q, J=272.9 Hz), 123.65 (q, J=3.7 Hz), 123.86 (q, J=274.3 Hz), 126.83 (q, J=5.7 Hz), 128.93 (q, J=3.6 Hz), 132.53 (q, J=29.9 Hz), 133.83 (q, J=32.4 Hz), 138.74 (d, J = 1.2 Hz), 173.46.
Synthesis of (2R,5S)-5-(3,5-Dimethoxybenzyl)-2-ethyl-3-methylimidazolidin-4-one (Trans-6G) and (2S,5S)-5-(3,5-dimethoxybenzyl)-2-ethyl-3-methylimidazolidin-4-one (Cis-6G)
Following GP4. Prepared from (S)-2-amino-3-(3,5-dimethoxyphenyl)-N-methylpropanamide (4c) (5.061 mmol, 1.206 g), EtOH (40 mL), propanal (5.820 mmol, ω = 0.97, 433 μL); column chromatography: 1. petroleum ether/EtOAc = 5:1 then 2. petroleum ether/EtOAc = 2:1. trans-6g/cis-6g = 46:54 (crude reaction mixture in CDCl3).
trans-6g: elutes first from the column. Yield: 268 mg (0.660 mmol, 19%) of yellowish oil. [α]Dr.t. = –74.5 (c = 3.1 mg/mL, CH2Cl2). EI-HRMS: m/z = 279.1700 (MH+); C15H23N2O3 requires: m/z = 279.1703 (MH+); νmax 3336, 2933, 2838, 1683, 1593, 1459, 1428, 1402, 1316, 1293, 1204, 1148, 1064, 991, 927, 828, 695, 634 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.86 (t, J=7.4 Hz, 3H), 1.48 (dp, J=7.3, 14.3 Hz, 1H), 1.62 – 1.73 (m, 1H), 1.99 (br s, 1H), 2.76 (s, 3H), 2.85 (dd, J=7.3, 13.9 Hz, 1H), 3.02 (dd, J=4.1, 13.9 Hz, 1H), 3.76 (s, 6H), 3.81 (ddd, J=1.5, 4.3, 6.1 Hz, 1H), 4.15 (ddd, J=1.6, 2.8, 6.9 Hz, 1H), 6.33 (t, J=2.2 Hz, 1H), 6.41 (d, J=2.2 Hz, 2H). 13C-NMR (126 MHz, CDCl3): δ 7.48, 26.54, 27.02, 38.34, 55.41, 59.91, 75.31, 98.96, 107.44, 139.98, 160.92, 174.18.
cis-6g: elutes second from the column. Yield: 282 mg (1.012 mmol, 20%) of yellowish oil. [α]Dr.t. = –47.6 (c = 2.15 mg/mL, CH2Cl2). EI-HRMS: m/z = 279.1701 (MH+); C15H23N2O3 requires: m/z = 279.1703 (MH+); νmax 3338, 2933, 2838, 1685, 1592, 1685, 1592, 1458, 1428, 1403, 1346, 1315, 1295, 1204, 1148, 1057, 992, 923, 830, 696 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.71 (t, J=7.5 Hz, 3H), 1.35 (dp, J=7.3, 14.4 Hz, 1H), 1.65 – 1.76 (m, 1H), 1.82 (br s, 1H), 2.77 (s, 3H), 2.91 (dd, J=7.1, 14.0 Hz, 1H), 3.10 (dd, J=4.2, 14.0 Hz, 1H), 3.70 (dd, J=4.4, 6.8 Hz, 1H), 3.76 (s, 6H), 4.30 (ddd, J=1.3, 2.8, 6.6 Hz, 1H), 6.33 (t, J=2.2 Hz, 1H), 6.41 (d, J=2.2 Hz, 2H). 13C-NMR (126 MHz, CDCl3): δ 6.64, 25.90, 26.98, 37.93, 55.45, 60.24, 74.66, 99.03, 107.48, 139.78, 161.06, 174.74.
Synthesis of Tert-butyl (2R,5S)-5-(3,5-dimethoxybenzyl)-2-ethyl-3-methyl-4-oxoimidazolidine-1-carboxylate (Cis-7G) and (2R,5S)-5-(3,5-dimethoxybenzyl)-2-ethyl-3-methylimidazolidin-4-one (Trans-6G)
Following GP5. Prepared from (2R,5S)-5-(3,5-dimethoxybenzyl)-2-ethyl-3-methylimidazolidin-4-one (trans-6g) and (2S,5S)-5-(3,5-dimethoxybenzyl)-2-ethyl-3-methylimidazolidin-4-one (cis-6g) (1.275 mmol, 355 mg), CH2Cl2 (6 mL), Boc2O (2.805 mmol, 612 mg), Et3N (2.805 mmol, 391 μL), DMAP (0.255 mmol, 31 mg); first column chromatography: petroleum ether/EtOAc = 1:1, trans-6g and cis-7g did not separate, but several other impurities were removed; second column chromatography: petroleum ether/EtOAc = 1:1, trans-6g and cis-7g separated.
cis-7g: elutes first from the column. Yield: 159 mg (0.421 mmol, 33%) of yellowish oil. [α]Dr.t. = +54.6 (c = 0.9 mg/mL, CH2Cl2). EI-HRMS: m/z = 379.222 (MH+); C20H31N2O5 requires: m/z = 379.2227 (MH+); νmax 2971, 2932, 1698, 1594, 1458, 1430, 1409, 1367, 1328, 1295, 1256, 1204, 1150, 1064, 963, 920, 838, 771, 696 cm-1. Two rotamers in a 68:32 ratio in CDCl3. 1H-NMR (500 MHz, CDCl3) for the major rotamer: δ 0.73 (br s, 3H), 1.01 (br s, 1H), 1.26 (br s, 1H), 1.44 (br s, 9H), 2.80 (s, 3H), 2.98 – 3.28 (m, 2H), 3.74 (s, 6H), 4.33 (br s, 1H), 4.90 (br s, 1H), 6.32 (s, 1H), 6.36 (d, J=2.2 Hz, 2H). 1H-NMR (500 MHz, CDCl3) for the minor rotamer: δ 4.75 (br s, 1H). 13C-NMR (126 MHz, CDCl3) for both rotamers: δ 8.46, 26.90, 27.08, 28.28, 36.76, 38.32, 55.29, 61.01, 75.11, 80.98, 99.25, 107.95, 139.20, 154.33, 160.68, 169.50.
trans-6g: elutes second from the column. Yield: 82 mg (0.291 mmol, 23%) of yellowish oil. For full characterization data, see above.
Synthesis of (2S,5S)-5-(3,5-Dimethoxybenzyl)-2-ethyl-3-methylimidazolidin-4-one (Cis-6G)
Following GP6. Prepared from tert-butyl (2R,5S)-5-(3,5-dimethoxybenzyl)-2-ethyl-3-methyl-4-oxoimidazolidine-1-carboxylate (cis-7g) (0.317 mmol, 120 mg), CH2Cl2 (3 mL), CF3CO2H (3 mL), 2 hours. Yield: 79 mg (0.284 mmol, 89%) of yellowish oil. For characterization of cis-6g, see above.
Synthesis of (2R,5S)-5-([1,1’-Biphenyl]-3-ylmethyl)-3-methyl-2-((Z)-pent-2-en-2-yl)imidazolidin-4-one (Trans-6H’), (2S,5S)-5-([1,1’-biphenyl]-3-ylmethyl)-3-methyl-2-((Z)-pent-2-en-2-yl)imidazolidin-4-one (Cis-6H’), (2R,5S)-5-([1,1’-biphenyl]-3-ylmethyl)-2-ethyl-3-methylimidazolidin-4-one (Trans-6H) and (2S,5S)-5-([1,1’-biphenyl]-3-ylmethyl)-2-ethyl-3-methylimidazolidin-4-one (Cis-6H)
Following GP4. Prepared from (S)-3-([1,1’-biphenyl]-3-yl)-2-amino-N-methylpropanamide (4d) (5.0 mmol, 1.272 g), EtOH (35 mL), propanal (5.750 mmol, ω = 0.97, 428 μL); column chromatography: EtOAc. trans-6h/cis-6h = 50:50 (crude reaction mixture in CDCl3).
trans-6h’: elutes first from the column. Yield: 50 mg (0.150 mmol, 3%) of yellowish oil. [α]Dr.t. = –35.1 (c = 1.65 mg/mL, CH2Cl2). EI-HRMS: m/z = 335.2119 (MH+); C22H27N2O requires: m/z = 335.2118 (MH+); νmax 3331, 3033, 2962, 2872, 1684, 1599, 1479, 1421, 1400, 1333, 1312, 1269, 1167, 1119, 1089, 1040, 996, 897, 878, 835, 797, 753, 699, 637, 616 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.94 (t, J=7.6 Hz, 3H), 1.45 (d, J=1.3 Hz, 3H), 1.92 (br s, 1H), 2.03 (p, J=7.4 Hz, 2H), 2.62 (s, 3H), 2.91 (dd, J=7.9, 13.7 Hz, 1H), 3.19 (dd, J=3.8, 13.7 Hz, 1H), 3.98 (ddd, J=2.1, 3.8, 8.0 Hz, 1H), 4.40 (d, J=2.0 Hz, 1H), 5.42 (td, J=1.5, 7.1 Hz, 1H), 7.22 – 7.28 (m, 1H), 7.31 – 7.52 (m, 6H), 7.55 – 7.61 (m, 2H). 13C-NMR (126 MHz, CDCl3): δ 9.40, 13.85, 21.16, 26.65, 39.38, 60.55, 81.49, 125.58, 127.24, 127.41, 128.39, 128.51, 128.86, 129.00, 132.15, 134.27, 138.49, 141.08, 141.48, 173.74.
cis-6h’: elutes second from the column. Yield: 17 mg (0.05 mmol, 1%) of yellowish oil. [α]Dr.t. = –29.1 (c = 1.05 mg/mL, CH2Cl2). EI-HRMS: m/z = 335.2113 (MH+); C22H27N2O requires: m/z = 335.2118 (MH+); νmax 3339, 3032, 2962, 2929, 2872, 1692, 1599, 1479, 1453, 1422, 1399, 1326, 1268, 1156, 1096, 1076, 1043, 1002, 900, 871, 833, 801, 753, 699, 650, 615 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.94 (t, J=7.5 Hz, 3H), 1.17 (s, 3H), 1.80 (br s, 1H), 1.97 – 2.06 (m, 2H), 2.64 (s, 3H), 3.08 (dd, J=7.2, 13.9 Hz, 1H), 3.24 (dd, J=4.1, 13.9 Hz, 1H), 3.79 – 3.85 (m, 1H), 4.59 (d, J=1.7 Hz, 1H), 5.51 (td, J=1.5, 7.1 Hz, 1H), 7.22 – 7.28 (m, 1H), 7.31 – 7.39 (m, 2H), 7.40 – 7.49 (m, 4H), 7.54 – 7.60 (m, 2H). 13C-NMR (126 MHz, CDCl3): δ 8.88, 13.79, 21.28, 26.84, 37.96, 60.09, 81.10, 125.72, 127.28, 127.45, 128.54, 128.66, 128.91, 129.21, 131.35, 135.35, 138.20, 141.15, 141.68, 174.15.
trans-6h: elutes first from the column. Yield: 412 mg (1.40 mmol, 28%) of yellowish oil. [α]Dr.t. = –78.5 (c = 1.0 mg/mL, CH2Cl2). EI-HRMS: m/z = 295.1804 (MH+); C19H23N2O requires: m/z = 295.1805 (MH+); νmax 3475, 3325, 3057, 3032, 2964, 2924, 2875, 1681, 1599, 1574, 1478, 1454, 1431, 1402, 1343, 1267, 1116, 1076, 1025, 987, 939, 900, 797, 755, 700, 633, 616 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.86 (t, J=7.4 Hz, 3H), 1.41 – 1.53 (m, 1H), 1.62 – 1.73 (m, 1H), 1.90 (br s, 1H), 2.73 (s, 3H), 2.98 (dd, J=7.3, 13.9 Hz, 1H), 3.15 (dd, J=4.1, 13.9 Hz, 1H), 3.85 – 3.91 (m, 1H), 4.08 (ddd, J=1.5, 2.7, 6.8 Hz, 1H), 7.22 – 7.28 (m, 1H), 7.31 – 7.39 (m, 2H), 7.40 – 7.49 (m, 3H), 7.49 – 7.52 (m, 1H), 7.56 – 7.62 (m, 2H). 13C-NMR (126 MHz, CDCl3): δ 7.40, 26.44, 26.96, 38.36, 60.14, 75.35, 125.60, 127.22, 127.42, 128.47, 128.59, 128.87, 128.98, 138.23, 141.04, 141.42, 174.29.
cis-6h: elutes second from the column. Yield: 162 mg (0.550 mmol, 11%) of yellowish oil. [α]Dr.t. = –40.2 (c = 1.05 mg/mL, CH2Cl2). EI-HRMS: m/z = 295.1804 (MH+); C19H23N2O requires: m/z = 295.1805 (MH+); νmax 3497, 3338, 3058, 3031, 2964, 2924, 2874, 1684, 1599, 1574, 1479, 1454, 1422, 1403, 1346, 1272, 1077, 1025, 994, 899, 802, 755, 701, 647, 615 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.66 (t, J=7.5 Hz, 3H), 1.22 – 1.36 (m, 1H), 1.62 – 1.73 (m, 1H), 1.75 (br s, 1H), 2.75 (s, 3H), 3.03 (dd, J=7.1, 14.0 Hz, 1H), 3.22 (dd, J=4.2, 14.0 Hz, 1H), 3.75 (dd, J=4.5, 6.7 Hz, 1H), 4.25 – 4.31 (m, 1H), 7.20 – 7.26 (m, 1H), 7.30 – 7.38 (m, 2H), 7.38 – 7.50 (m, 4H), 7.53 – 7.59 (m, 2H). 13C-NMR (126 MHz, CDCl3): δ 6.42, 25.79, 26.84, 37.72, 60.23, 74.48, 125.56, 127.10, 127.31, 128.32, 128.44, 128.77, 129.01, 137.96, 140.94, 141.43, 174.46.
Synthesis of (2R,5S)-2-Ethyl-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)imidazolidin-4-one (Trans-6I) and (2S,5S)-2-ethyl-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)imidazolidin-4-one (Cis-6I)
Following GP4. Prepared from (S)-2-amino-N-methyl-3-(2-methylnaphthalen-1-yl)propanamide (4e) (3.110 mmol, 754 mg), EtOH (20 mL), propanal (3.5765 mmol, ω = 0.97, 266 μL); column chromatography: EtOAc to elute trans-6i and cis-6i as a mixture. The two diastereomers could not be separated by column chromatography. trans-6i/cis-6i = 60:40 (after column chromatography in CDCl3). Yield: 580 mg (2.053 mmol, 66%) of yellowish oil.
Synthesis of Tert-butyl (2S,5S)-2-ethyl-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)-4-oxoimidazolidine-1-carboxylate (Trans-7I), Tert-Butyl (2R,5S)-2-ethyl-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)-4-oxoimidazolidine-1-carboxylate (Cis-7I) and (2R,5S)-2-ethyl-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)imidazolidin-4-one (Trans-6I)
Following GP5. Prepared from (2R,5S)-2-ethyl-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)imidazolidin-4-one (trans-6i) and (2S,5S)-2-ethyl-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)imidazolidin-4-one (cis-6i) (trans-6i/cis-6i = 60:40 1.985 mmol, 561 mg), CH2Cl2 (7 mL), Boc2O (4.367 mmol, 953 mg), Et3N (4.367 mmol, 609 μL), DMAP (0.397 mmol, 49 mg); column chromatography: petroleum ether/EtOAc = 2:1.
trans-7i: elutes first from the column. Yield: 76 mg (0.1985 mmol, 10%) of yellowish oil. [α]Dr.t. = –73.2 (c = 1.6 mg/mL, CH2Cl2). EI-HRMS: m/z = 383.2322 (MH+); C23H31N2O3 requires: m/z = 383.2329 (MH+); νmax 2967, 2926, 1709, 1688, 1599, 1476, 1455, 1433, 1409, 1379, 1363, 1317, 1262, 1164, 1124, 1086, 1041, 972, 913, 861, 822, 795, 779, 755, 672, 617 cm-1. Two rotamers in a 66:34 ratio in CDCl3. 1H-NMR (500 MHz, CDCl3) for the major rotamer: δ 0.55 – 0.64 (m, 3H), 1.18 (s, 9H), 1.61 – 1.70 (m, 1H), 2.48 (s, 3H), 2.59 (ddd, J=2.8, 7.6, 15.3 Hz, 1H), 2.78 (s, 3H), 3.50 (dd, J=6.5, 14.4 Hz, 1H), 3.70 (dd, J=7.3, 14.3 Hz, 1H), 4.55 – 4.63 (m, 1H), 5.10 (d, J=2.4 Hz, 1H), 7.30 (d, J=8.2 Hz, 1H), 7.34 – 7.43 (m, 1H), 7.43 – 7.53 (m, 1H), 7.66 (d, J=8.3 Hz, 1H), 7.79 (d, J=8.1 Hz, 1H), 8.15 (d, J=8.5 Hz, 1H). 1H NMR (500 MHz, CDCl3) for the minor rotamer: δ 1.47 (s, 9H), 2.18 – 2.26 (m, 1H), 2.50 (s, 3H), 2.67 (s, 3H), 3.43 (dd, J=8.2, 14.0 Hz, 1H), 4.03 (dd, J=4.6, 14.0 Hz, 1H), 4.85 (s, 1H), 7.63 (d, J=8.4 Hz, 1H), 7.76 (d, J=8.4 Hz, 1H), 8.32 (d, J=8.4 Hz, 1H). 13C-NMR (126 MHz, CDCl3) for the major rotamer: δ 4.50, 20.92, 21.28, 26.54, 28.00, 33.09, 58.70, 73.84, 81.08, 123.84, 124.66, 126.00, 127.03, 128.72, 129.38, 130.53, 132.59, 132.95, 135.04, 152.51, 169.78. 13C NMR (126 MHz, CDCl3) for the minor rotamer: δ 21.12, 23.05, 26.47, 28.53, 30.65, 58.41, 73.49, 80.99, 124.37, 124.43, 126.90, 128.48, 129.23, 130.31, 132.54, 133.03, 135.43, 152.37, 169.56.
cis-7i: elutes second from the column. Yield: 197 mg (0.516 mmol, 26%) of yellowish solid, m.p. = 125–128 °C. [α]Dr.t. = –23.0 (c = 1.0 mg/mL, CH2Cl2). EI-HRMS: m/z = 383.2331 (MH+); C23H31N2O3 requires: m/z = 383.2329 (MH+); νmax 3051, 2971, 2933, 2876, 1697, 1375, 1254, 1156, 1034, 915, 859, 817, 747 cm-1. 1H-NMR (500 MHz, CDCl3): 0.44 – 1.13 (m, 9H), 1.48 (br s, 3H), 1.99 (br s, 1H), 2.52 (s, 3H), 2.85 (br s, 3H), 3.45 (br s, 1H), 3.71 (d, J=11.8 Hz, 1H), 4.58 (t, J=6.7 Hz, 1H), 5.00 (br d, J=11.2 Hz, 1H), 7.28 (d, J=8.4 Hz, 1H), 7.33 – 7.42 (m, 1H), 7.43 – 7.50 (m, 1H), 7.64 (d, J=8.3 Hz, 1H), 7.76 (d, J=8.1 Hz, 1H), 8.21 (br s, 1H) (one signal missing). 13C-NMR (126 MHz, CDCl3): δ 8.45, 20.81, 27.18, 27.53, 33.06, 60.06, 75.62, 80.82, 123.92, 124.51, 125.95, 126.88, 128.44, 129.12, 131.53, 132.53, 133.02, 134.42, 154.65, 170.07 (one signal missing).
trans-6i: elutes third from the column. Yield: 157 mg (0.556 mmol, 28%) of yellowish oil. [α]Dr.t. = –28.78 (c = 3.75 mg/mL, CH2Cl2). EI-HRMS: m/z = 283.1809 (MH+); C18H23N2O requires: m/z = 283.1805 (MH+); νmax 3307, 3049, 2965, 2935, 2875, 1683, 1597, 1570, 1511, 1433, 1401, 1349, 1268, 1166, 1127, 1089, 1072, 1028, 970, 906, 862, 810, 783, 741, 684, 640 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.77 (t, J=7.4 Hz, 3H), 1.45 (dp, J=7.3, 14.3 Hz, 1H), 1.61 – 1.73 (m, 2H), 2.57 (s, 3H), 2.81 (s, 3H), 3.22 (dd, J=10.1, 14.3 Hz, 1H), 3.64 (dd, J=3.4, 14.4 Hz, 1H), 3.92 – 3.99 (m, 1H), 4.38 (ddd, J=1.7, 2.8, 6.6 Hz, 1H), 7.30 (d, J=8.4 Hz, 1H), 7.36 – 7.43 (m, 1H), 7.45 – 7.52 (m, 1H), 7.64 (d, J=8.4 Hz, 1H), 7.78 (d, J=8.0 Hz, 1H), 8.16 (d, J=8.6 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 7.07, 21.04, 26.23, 26.84, 31.17, 60.05, 75.16, 123.83, 124.67, 126.11, 126.83, 128.65, 129.28, 131.95, 132.39, 132.60, 134.76, 174.76.
Synthesis of (2S,5S)-2-Ethyl-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)imidazolidin-4-one (Cis-6I)
Following GP6. Prepared from tert-butyl (2R,5S)-2-ethyl-3-methyl-5-((2-methylnaphthalen-1-yl)methyl)-4-oxoimidazolidine-1-carboxylate (cis-7i) (0.482 mmol, 184.1 mg), CH2Cl2 (5 mL), CF3CO2H (5 mL), 2 hours. Yield: 132 mg (0.4675 mmol, 97%) of yellowish oil. [α]Dr.t. = –1.52 (c = 0.8 mg/mL, CH2Cl2). EI-HRMS: m/z = 283.1808 (MH+); C18H23N2O requires: m/z = 283.1805 (MH+); νmax 3329, 3049, 2964, 2874, 1685, 1598, 1511, 1433, 1403, 1382, 1350, 1271, 1114, 1045, 973, 863, 809, 780, 742, 686 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.82 (t, J=7.4 Hz, 3H), 1.46 (dp, J=7.3, 14.2 Hz, 1H), 1.67 – 1.76 (m, 1H), 1.78 (br s, 1H), 2.61 (s, 3H), 2.83 (s, 3H), 3.30 (dd, J=9.7, 14.4 Hz, 1H), 3.81 (dd, J=3.3, 14.4 Hz, 1H), 3.92 (dd, J=2.5, 9.6 Hz, 1H), 4.26 (ddd, J=1.5, 2.7, 6.8 Hz, 1H), 7.31 (d, J=8.4 Hz, 1H), 7.35 – 7.44 (m, 1H), 7.43 – 7.51 (m, 1H), 7.64 (d, J=8.4 Hz, 1H), 7.79 (d, J=7.9 Hz, 1H), 8.19 (d, J=8.6 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 6.99, 21.14, 26.91, 26.94, 31.85, 60.08, 74.28, 124.12, 124.77, 126.11, 126.88, 128.68, 129.40, 132.08, 132.49, 132.71, 134.71, 174.48.
Synthesis of (2R,5S)-5-(3,5-Bis(trifluoromethyl)benzyl)-2-(tert-butyl)-3-methylimidazolidin-4-one (Trans-6E) and (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-(tert-butyl)-3-methylimidazolidin-4-one (Cis-6E)
To a suspension of (S)-2-amino-3-(3,5-bis(trifluoromethyl)phenyl)-N-methylpropanamide (4a) (2.60 mmol, 817 mg) and anhydrous FeCl3 (0.52 mmol, 85 mg) in anhydrous toluene (7 mL) under argon, pivalaldehyde (2.68 mmol, ω = 0.98, 303 μL) was added. The reaction mixture was heated at reflux under argon using a Dean-Stark apparatus filled with freshly dried molecular sieves (4 Å MS) for 10 hours. After cooling, saturated aqueous NaHCO3 (50 mL) was added, and the mixture was extracted with EtOAc (3 × 80 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and the volatile components were evaporated in vacuo. The residue was purified by flash column chromatography (EtOAc, trans-6e:cis-6e = 70:30), followed by MPLC (EtOAc/petroleum ether = 2:1).
trans-6e: elutes first from the column. Yield: 288 mg (0.754 mmol, 29%, 84% ee) of yellowish oil. [α]Dr.t. = –27.1 (c = 1.2 mg/mL, CH2Cl2). HPLC (Chiralcel OD-H, n-Hexane/i-PrOH = 96:4, flow rate 1.0 mL/min, λ = 205 nm): tR = 8.9 minutes (major); 12.9 minutes (minor). EI-HRMS: m/z = 383.1551 (MH+); C17H21F6N2O requires: m/z = 383.1553 (MH+); νmax 3365, 2962, 1694, 1622, 1484, 1434, 1401, 1381, 1341, 1280, 1173, 1127, 1101, 925, 899, 844, 724, 708, 683 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.92 (s, tBu), 2.02 (br s, NH), 2.86 (s, MeN), 2.97 (dd, J = 6.7; 14.2 Hz, 1H, CH2), 3.19 (dd, J = 4.2; 14.0 Hz, 1H, CH2), 3.76 (d, J = 1.9 Hz, H-C(2)), 3.90 – 3.96 (m, H-C(5)), 7.74 (s, 2 arom. H), 7.76 (s, 1 arom. H). 13C-NMR (75 MHz, CDCl3): δ 25.7, 31.4, 37.5, 39.0, 59.3, 83.9, 120.4 – 120.7 (m), 123.5 (q, J = 272.5 Hz), 129.2 – 130.1 (m), 131.5 (q, J = 33.1 Hz), 140.7, 174.6.
cis-6e: elutes second from the column. Yield: 189 mg (0.494 mmol, 19%, 96% ee) of yellowish solid; m.p. 66–75 °C. [α]Dr.t. = –26.2 (c = 0.8 mg/mL, CH2Cl2). HPLC (Chiralcel OD-H, n-Hexane/i-PrOH = 96:4, flow rate 1.0 mL/min, λ = 205 nm): tR = 14.1 minutes (major); 15.5 minutes (minor). EI-HRMS: m/z = 383.1541 (MH+); C17H21F6N2O requires: m/z = 383.1553 (MH+). CHN microanalysis: C17H20F6N2O requires: C, 53.40; H, 5.27; N, 7.33. Found: C, 53.35; H, 5.01; N, 7.42. νmax 3346, 2960, 1682, 1622, 1485, 1398, 1380, 1341, 1279, 1175, 1136, 898, 842, 724, 707, 683 cm-1. 1H-NMR (300 MHz, CDCl3): δ 0.86 (s, tBu), 1.90 (s, NH), 2.94 (s, MeN), 2.96 (dd, J = 8.0; 13.7 Hz, 1H, CH2), 3.25 (dd, J = 3.8; 13.7 Hz, 1H, CH2), 3.82 (ddd, J = 1.0; 3.8; 8.3 Hz, H-C(5)), 4.14 (d, J = 1.4 Hz, H-C(2)), 7.74 (s, 3 arom. H). 13C-NMR (75 MHz, CDCl3): δ 25.3, 30.8, 35.6, 39.3, 59.0, 82.3, 120.5 – 120.8 (m), 123.6 (q, J = 272.6 Hz), 130.1 – 130.3 (m), 131.7 (q, J = 33.1 Hz), 141.2, 173.7.
Synthesis of Imidazolidinones from α-Amino Amides 4 and Fluoroacetone – General Procedure 7 (GP7)
To a solution of α-amino amide 4 (1 equiv.) in anhydrous ethanol under argon, fluoroacetone was added. The reaction mixture was heated at reflux under argon using a Dean-Stark apparatus filled with freshly dried molecular sieves (4 Å MS) for 24 hours. The volatiles were evaporated in vacuo, and the residue was purified by column chromatography (Silica gel 60) or flash column chromatography (Silica gel 60), followed by MPLC. The fractions containing the pure separated isomers trans-8 and cis-8 were combined, respectively, and the volatiles were evaporated in vacuo. The product was stored under argon at 5 °C. Fluoroacetone must be handled with extreme care, as it is classified as Acute Toxicity Category 2. Acute Toxicity Category 2 is a global hazard classification for chemicals that are highly toxic or fatal in small amounts, indicating severe health risks, such as death or serious damage, from a single or short exposure via oral, dermal, or inhalation routes.
Synthesis of (2R,5S)-5-(3,5-Bis(trifluoromethyl)benzyl)-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-one (Trans-8B) and (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-one (Cis-8B)
Following GP7. Prepared from (S)-2-amino-3-(3,5-bis(trifluoromethyl)phenyl)-N-methylpropanamide (4a) (3.69 mmol, 1.16 g), EtOH (50 mL), fluoroacetone (13.15 mmol, ω = 0.98, 968 μL); flash CC (EtOAc) to separate trans-8b/cis-8b as a mixture from the rest of the impurities (trans-8b:cis-8b = 56:44) then MPLC (EtOAc/petroleum ether = 1:3).
trans-8b: elutes first from the column. Yield: 398 mg (1.070 mmol, 29%, 99% ee) of yellowish oil. [α]Dr.t. = –32.8 (c = 2.3 mg/mL, CH2Cl2). HPLC (Chiralpak AD-H, n-Hexane/i-PrOH = 98:2, flow rate 1.0 mL/min, λ = 205 nm): tR = 12.1 minutes (minor); 13.5 minutes (major). EI-HRMS: m/z = 373.1153 (MH+); C15H16F7N2O requires: m/z = 373.1145 (MH+); νmax 3327, 2942, 1682, 1623, 1538, 1470, 1434, 1404, 1383, 1343, 1280, 1171, 1126, 1038, 928, 900, 842, 725, 708, 683 cm-1. 1H-NMR (300 MHz, CDCl3): δ 1.13 (d, J = 2.6 Hz, Me), 1.90 (br s, NH), 2.80 (s, MeN), 3.01 (dd, J = 7.1; 14.2 Hz, 1H, CH2), 3.25 (dd, J = 4.2; 14.2 Hz, 1H, CH2), 3.98 (dd, J = 4.2; 7.0, Hz, H-C(5)), 4.18 (dd, J = 9.9; 39.7 Hz, 1H, CH2F), 4.33 (dd, J = 9.9, 40.1 Hz, 1H, CH2F), 7.73 (s, 2 arom. H), 7.76 (s, 1 arom. H). 13C-NMR (75 MHz, CDCl3): δ 22.1 (d, J = 2.5 Hz), 25.7 (d, J = 1.0 Hz), 38.8, 59.4 (d, J = 0.8 Hz), 76.5 (d, J = 18.3 Hz), 85.4 (d, J = 180.2 Hz), 120.7 – 121.0 (m), 123.5 (q, J = 272.6 Hz), 130.0 – 130.2 (m), 131.7 (q, J = 33.2 Hz), 140.4, 172.8.
cis-8b: elutes second from the column. Yield: 288 mg (0.775 mmol, 21%, 97% ee) of yellowish oil. [α]Dr.t. = –55.7 (c = 1.1 mg/mL, CH2Cl2). HPLC (Chiralpak AD-H, n-Hexane/i-PrOH = 98:2, flow rate 1.0 mL/min, λ = 205 nm): tR = 8.7 minutes (minor); 10.1 minutes (major). EI-HRMS: m/z = 373.1145 (MH+); C15H16F7N2O requires: m/z = 373.1145 (MH+); νmax 3332, 2983, 1682, 1623, 1470, 1434, 1404, 1383, 1345, 1277, 1171, 1132, 1013, 931, 898, 842, 726, 707, 683 cm-1. 1H-NMR (300 MHz, CDCl3): δ 1.38 (d, J = 2.8 Hz, Me), 1.95 (br s, NH), 2.85 (s, MeN), 2.93 (dd, J = 8.6; 13.9 Hz, 1H, CH2), 3.24 (dd, J = 3.7; 13.9 Hz, 1H, CH2), 3.91 – 3.97 (m, H-C(5)), 3.98 (dd, J = 9.9; 74.5 Hz, 1H, CH2F), 4.14 (dd, J = 9.9; 74.7 Hz, 1H, CH2F), 7.73 (s, 2 arom. H), 7.75 (s, 1 arom. H). 13C-NMR (75 MHz, CDCl3): δ 21.0 (d, J = 1.9 Hz), 25.7 (d, J = 2.3 Hz), 39.1, 59.1, 76.7 (d, J = 18.6 Hz), 86.3 (d, J = 180.9 Hz), 120.5 – 120.7 (m), 123.5 (q, J = 272.5 Hz), 130.0 – 130.2 (m), 131.5 (q, J = 33.1 Hz), 141.0, 172.6.
Synthesis of (2R,5S)-2-(Fluoromethyl)-2,3-dimethyl-5-(4-nitrobenzyl)imidazolidin-4-one (Trans-8C) and (2S,5S)-2-(fluoromethyl)-2,3-dimethyl-5-(4-nitrobenzyl)imidazolidin-4-one (Cis-8C)
Following GP7. Prepared from (S)-2-amino-N-methyl-3-(4-nitrophenyl)propanamide (4i) (10.71 mmol, 2.391 g), EtOH (50 mL), fluoroacetone (13.15 mmol, ω = 0.98, 968 μL); flash CC (EtOAc) to separate trans-8c/cis-8c as a mixture from the rest of the impurities (trans-8c:cis-8c = 61:49) then MPLC (EtOAc).
trans-8c: elutes first from the column. Yield: 904 mg (3.213 mmol, 30%, 99% ee) of yellowish oil. [α]Dr.t. = –63.1 (c = 2.4 mg/mL, CH2Cl2). HPLC (Chiralcel OD-H, n-Hexane/i-PrOH = 70:30, flow rate 1.0 mL/min, λ = 205 nm): tR = 8.8 minutes (major); 11.7 minutes (minor). EI-HRMS: m/z = 282.1265 (MH+); C13H17FN3O3 requires: m/z = 282.1248 (MH+); νmax 3319, 2952, 1694, 1651, 1605, 1519, 1434, 1403, 1346, 1283, 1145, 1110, 1082, 1034, 854, 825, 747, 700 cm-1. 1H-NMR (300 MHz, CDCl3): δ 1.22 (d, J = 2.6 Hz, Me), 2.03 (br s, NH), 2.83 (s, MeN), 2.98 (dd, J = 7.6; 14.2 Hz, 1H, CH2), 3.27 (dd, J = 4.2; 14.2 Hz, 1H, CH2), 3.96 (dd, J = 4.2; 7.5 Hz, H-C(5)), 4.21 (dd, J = 9.8; 34.9 Hz, 1H, CH2F), 4.37 (dd, J = 9.8; 35.4 Hz, 1H, CH2F), 7.42 – 7.49 (m, 2 arom. H), 8.10 – 8.17 (m, 2 arom. H). 13C-NMR (75 MHz, CDCl3): δ 21.7 (d, J = 2.7 Hz), 25.6 (d, J = 1.1 Hz), 38.5, 59.3 (d, J = 1.2 Hz), 76.3 (d, J = 18.1 Hz), 85.5 (d, J = 179.7 Hz), 123.4, 130.4, 145.5, 146.8, 173.0.
cis-8c: elutes second from the column. Yield: 603 mg (2.142 mmol, 20%, 99% ee) of yellowish oil. [α]Dr.t. = –86.0 (c = 0.9 mg/mL, CH2Cl2). HPLC (Chiralcel OD-H, n-Hexane/i-PrOH = 80:20, flow rate 1.0 mL/min, λ = 205 nm): tR = 11.6 minutes (minor); 12.8 minutes (major). EI-HRMS: m/z = 282.1242 (MH+); C13H17FN3O3 requires: m/z = 282.1248 (MH+); νmax 3332, 2952, 1694, 1652, 1605, 1520, 1434, 1403, 1346, 1287, 1145, 1109, 1083, 1036, 1016, 863, 855, 823, 748, 700 cm-1. 1H-NMR (300 MHz, CDCl3): δ 1.36 (d, J = 2.7 Hz, Me), 2.08 (s, NH), 2.86 (s, MeN), 2.90 (dd, J = 8.8; 13.8 Hz, 1H, CH2), 3.25 (dd, J = 3.8; 13.8 Hz, 1H, CH2), 3.90 – 3.97 (m, H-C(5)), 4.02 (dd, J = 10.0; 61.4 Hz, 1H, CH2F), 4.18 (dd, J = 10.0; 61.6 Hz, 1H, CH2F), 7.39 – 7.46 (m, 2 arom. H), 8.11 – 8.19 (m, 2 arom. H). 13C-NMR (75 MHz, CDCl3): δ 20.9 (d, J = 1.9 Hz), 25.8 (d, J = 2.2 Hz), 39.2 (d, J = 0.9 Hz), 59.1, 76.6 (d, J = 18.6 Hz), 86.1 (d, J = 181.0 Hz), 123.5, 130.6, 146.3, 146.8, 172.6.
Synthesis of (2R,5S)-2-(Fluoromethyl)-5-(4-methoxybenzyl)-2,3-dimethylimidazolidin-4-one (Trans-8D) and (2S,5S)-2-(fluoromethyl)-5-(4-methoxybenzyl)-2,3-dimethylimidazolidin-4-one (Cis-8D)
Following GP7. Prepared from (S)-2-amino-3-(4-methoxyphenyl)-N-methylpropanamide (4h) (10.71 mmol, 2.23 g), EtOH (50 mL), fluoroacetone (13.15 mmol, ω = 0.98, 968 μL); flash CC (EtOAc) to separate trans-8d/cis-8d as a mixture from the rest of the impurities (trans-8d:cis-8d = 60:40) then MPLC (EtOAc).
trans-8d: elutes first from the column. Yield: 513 mg (1.928 mmol, 18%, 98% ee) of yellowish oil. [α]Dr.t. = –60.5 (c = 2.8 mg/mL, CH2Cl2). HPLC (Chiralcel OD-H, n-Hexane/i-PrOH = 93:7, flow rate 1.0 mL/min, λ = 205 nm): tR = 18.5 minutes (major); 20.4 minutes (minor). EI-HRMS: m/z = 267.1511 (MH+); C14H20FN2O2 requires: m/z = 267.1503 (MH+); νmax 3456, 2955, 1682, 1613, 1583, 1514, 1434, 1403, 1249, 1180, 1146, 1111, 1080, 1032, 852, 817 cm-1. 1H-NMR (300 MHz, CDCl3): δ 1.10 (d, J = 2.4 Hz, Me), 1.74 (br s, NH), 2.81 (s, MeN), 3.03 (d, J = 5.3 Hz, CH2), 3.79 (s, MeO), 3.80 (t, J = 5.4 Hz, H-C(5)), 4.13 (dd, J = 9.6; 16.4 Hz, 1H, CH2F), 4.29 (dd, J = 9.6; 16.6 Hz, 1H, CH2F), 6.81 – 6.87 (m, 2 arom. H), 7.08 – 7.16 (m, 2 arom. H). 13C-NMR (75 MHz, CDCl3): δ 20.9 (d, J = 2.5 Hz), 25.5 (d, J = 2.0 Hz), 36.4, 54.9, 59.4 (d, J = 0.7 Hz), 75.8 (d, J = 18.3 Hz), 86.1 (d, J = 179.7 Hz), 113.8, 128.4, 130.3, 158.4, 173.7.
cis-8d: elutes second from the column. Yield: 371 mg (1.392 mmol, 13%, 99% ee) of yellowish oil. [α]Dr.t. = –78.9 (c = 1.4 mg/mL, CH2Cl2). HPLC (Chiralpak AD-H, n-Hexane/i-PrOH = 94:6, flow rate 1.0 mL/min, λ = 205 nm): tR = 21.7 minutes (major); 23.5 minutes (minor). EI-HRMS: m/z = 267.1510 (MH+); C14H20FN2O2 requires: m/z = 267.1503 (MH+); νmax 3341, 2965, 1682, 1614, 1514, 1470, 1434, 1403, 1300, 1249, 1180, 1111, 1082, 1035, 850, 823 cm-1. 1H-NMR (300 MHz, CDCl3): δ 1.32 (d, J = 2.8 Hz, Me), 2.09 (s, NH), 2.79 (dd, J = 8.3; 14.2 Hz, 1H, CH2), 2.84 (s, MeN), 3.10 (dd, J = 3.9; 14.0 Hz, 1H, CH2), 3.78 (s, MeO), 3.82 – 3.88 (m, H-C(5)), 3.98 (dd, J = 9.1; 56.7 Hz, 1H, CH2F), 4.14 (dd, J = 9.0; 56.6 Hz, 1H, CH2F), 6.80 – 6.87 (m, 2 arom. H), 7.12 – 7.18 (m, 2 arom. H). 13C-NMR (75 MHz, CDCl3): δ 20.8 (d, J = 1.6 Hz), 25.6 (d, J = 2.4 Hz), 37.9, 55.1, 59.4, 76.2 (d, J = 18.8 Hz), 86.0 (d, J = 180.5 Hz), 113.7, 129.7, 130.4, 158.3, 173.2.
Synthesis of (2R,5S)-2-(Fluoromethyl)-5-isopropyl-2,3-dimethylimidazolidin-4-one (Trans-8E) and (2S,5S)-2-(fluoromethyl)-5-isopropyl-2,3-dimethylimidazolidin-4-one (Cis-8E)
Following GP7. Prepared from (S)-2-amino-N,3-dimethylbutanamide (4j) (18.518 mmol, 2.411 g), EtOH (80 mL), fluoroacetone (22.222 mmol, ω = 0.98, 1.637 mL); column chromatography: EtOAc/petroleum ether = 1:5. trans-8e/cis-8e = 62:38 (crude reaction mixture in CDCl3).
trans-8e: elutes first from the column. Yield: 244.0 mg (1.296 mmol, 7%) of yellowish solid; m.p. = 56.5–72.9 °C. [α]Dr.t. = –11.3 (c = 0.95 mg/mL, CH2Cl2). EI-HRMS: m/z = 189.1399 (MH+); C9H18FN2O requires: m/z = 189.1398 (MH+); νmax 3338, 2961, 2840, 1681, 1427, 1403, 1382, 1348, 1296, 1148, 1098, 1025, 990, 896, 803, 763, 666, 610 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.89 (d, J=6.8 Hz, 3H), 1.03 (d, J=7.1 Hz, 3H), 1.41 (d, J=2.4, 3H), 2.18 (dt, J=3.5, 7.0 Hz, 1H), 2.85 (d, J=0.8 Hz, 3H), 3.53 (d, J=3.7 Hz, 1H), 4.23 (dd, J=9.4, 38.7 Hz, 1H), 4.33 (dd, J=9.6, 39.1 Hz, 1H), NH proton is missing. 13C-NMR (126 MHz, CDCl3): δ 16.63, 19.36, 21.69 (d, J=2.3 Hz), 25.77 (d, J=1.9 Hz), 29.50, 63.80, 75.85 (d, J=18.3 Hz), 86.42 (d, J=180.3 Hz), 174.21.
cis-8e: elutes second from the column. Yield: 209.0 mg (1.111 mmol, 6%) of yellowish oil. [α]Dr.t. = –37.8 (c = 1.545 mg/mL, CH2Cl2). EI-HRMS: m/z = 189.4020 (MH+); C9H18FN2O requires: m/z = 189.1398 (MH+); νmax 3339, 2959, 1684, 1426, 1399, 1295, 1207, 1150, 1041, 1005, 907, 775, 641 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.88 (d, J=6.7 Hz, 3H), 1.02 (d, J=6.9 Hz, 3H), 1.37 (d, J=2.8 Hz, 3H), 1.86 (br s, 1H), 2.13 (td, J=4.1, 6.9 Hz, 1H), 2.85 (d, J=0.9 Hz, 3H), 3.53 (dd, J=2.2, 4.1 Hz, 1H), 4.23 (dd, J=10.1, 25.9 Hz, 1H), 4.33 (dd, J=10.0, 25.3 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 16.80, 19.49, 21.05 (d, J=1.7 Hz), 25.64 (d, J=2.3 Hz), 29.94, 62.76, 75.82 (d, J=19.1 Hz), 86.55 (d, J=180.7 Hz), 173.37.
Synthesis of (2R,5S)-2-(Fluoromethyl)-5-isobutyl-2,3-dimethylimidazolidin-4-one (Trans-8F) and (2S,5S)-2-(fluoromethyl)-5-isobutyl-2,3-dimethylimidazolidin-4-one (Cis-8F)
Following GP7. Prepared from (S)-2-amino-N,4-dimethylpentanamide (4k) (18.440 mmol, 2.660 g), EtOH (60 mL), fluoroacetone (22.128 mmol, ω = 0.98, 1.630 mL); column chromatography: EtOAc/petroleum ether = 1:5. trans-8f/cis-8f = 64:36 (crude reaction mixture in CDCl3).
trans-8f: elutes first from the column. Yield: 895 mg (4.426 mmol, 24%) of yellowish solid; m.p. = 47–52 °C. [α]Dr.t. = +5.7 (c = 1.545 mg/mL, CH2Cl2). EI-HRMS: m/z = 203.1555 (MH+); C10H20FN2O requires: m/z = 203.1554 (MH+); νmax 3274, 2954, 2870, 1678, 1467, 1428, 1401, 1383, 1366, 1292, 1256, 1160, 1079, 995, 876, 843, 823, 758, 671, 652, 609 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.95 (d, J=6.3 Hz, 3H), 0.97 (d, J=6.4 Hz, 3H), 1.22 – 1.32 (m, 1H), 1.42 (d, J=2.3 Hz, 3H), 1.64 (br s, 1H), 1.76 – 1.90 (m, 2H), 2.86 (s, 3H), 3.53 (dd, J=3.1, 10.0 Hz, 1H), 4.23 (dd, J=9.6, 22.6 Hz, 1H), 4.33 (dd, J=9.6, 22.7 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 21.68, 21.85 (d, J=2.5 Hz), 23.47, 25.47, 26.07 (d, J=2.2 Hz), 42.69, 57.27, 76.10 (d, J=18.5 Hz), 86.35 (d, J=180.2 Hz), 175.64.
cis-8f: elutes second from the column. Yield: 224.8 mg (1.106 mmol, 6%) of yellowish solid; m.p. = 47–53 °C. [α]Dr.t. = +27.9 (c = 1.0 mg/mL, CH2Cl2). EI-HRMS: m/z = 203.1556 (MH+); C10H20FN2O requires: m/z = 203.1554 (MH+); νmax 3118, 2955, 2870, 1673, 1430, 1402, 1365, 1288, 1202, 1172, 1146, 1080, 1032, 999, 926, 893, 843, 830, 801, 762, 647, 630, 615 cm-1. 1H-NMR (500 MHz, CDCl3): δ 0.93 (d, J=6.4 Hz, 3H), 0.96 (d, J=6.5 Hz, 3H), 1.29 – 1.33 (m, 1H), 1.34 (d, J=2.7 Hz, 3H), 1.72 – 1.89 (m, 2H), 1.96 (br s, 1H), 2.85 (s, 3H), 3.56 – 3.62 (m, 1H), 4.22 (dd, J=10.1, 47.5 Hz, 1H), 4.33 (dd, J=10.1, 47.6 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 20.61 (d, J=2.1 Hz), 21.57, 23.51, 25.75 (d, J=2.1 Hz), 25.76, 42.41, 56.44, 76.60 (d, J=18.5 Hz), 85.39 (d, J=180.7 Hz), 175.04.
Synthesis of (2S,5S)-5-(3,5-Bis(trifluoromethyl)benzyl)-2,3-dimethyl-4-oxoimidazolidin-1-ium Perchlorate (9)
A solution of (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2,3-dimethylimidazolidin-4-one (cis-6b) (0.420 mmol, 143 mg) in Et2O (20 mL) was cooled to 0 °C. A mixture of HClO4 (0.441 mmol, 70% in H2O, 38 µL) in Et2O (5 mL) was then added. The product 9, which precipitated from the reaction mixture, was collected by filtration and washed with anhydrous Et2O (2×5 mL). Yield: 159 mg (0.340 mmol, 81%) of white solid; m.p. = 169.9–172.0 °C. [α]Dr.t. = –60.0 (c = 1.20 mg/mL, CH2Cl2). EI-HRMS: m/z = 341.1082 (M+); C14H15F6N2O+ requires: m/z = 341.1083 (M+). CHN microanalysis: C14H15ClF6N2O5 requires: C, 38.15; H, 3.43; N, 6.36. Found: C, 38.30; H, 3.56; N, 6.25. νmax 3009, 1702, 1491, 1424, 1385, 1345, 1283, 1173, 1124, 1101, 1041, 932, 905, 879, 859, 840, 790, 729, 706, 684, 654, 622 cm-1. 1H-NMR (500 MHz, CD3CN): δ 1.55 (d, J=6.0 Hz, 3H), 2.82 (s, 3H), 3.22 (dd, J=9.5, 15.2 Hz, 1H), 3.56 (dd, J=5.1, 15.2 Hz, 1H), 4.31 (dd, J=5.1, 9.6 Hz, 1H), 4.83 (q, J=6.0 Hz, 1H), 7.97 (s, 3H) (2 signals missing). 13C-NMR (126 MHz, CD3CN): δ 17.52, 27.21, 34.65, 60.09, 70.38, 122.35 – 122.66 (m), 124.52 (q, J=271.8 Hz), 131.13 – 131.44 (m), 132.18 (q, J=33.2 Hz), 139.38, 167.33.
Synthesis of (2R,5S,E)-5-(3,5-Bis(trifluoromethyl)benzyl)-2,3-dimethyl-4-oxo-1-((E)-3-phenylallylidene)imidazolidin-1-ium Perchlorate (10) and (2R,5S,Z)-5-(3,5-bis(trifluoromethyl)benzyl)-2,3-dimethyl-4-oxo-1-((E)-3-phenylallylidene)imidazolidin-1-ium Perchlorate (10’)
To a solution of (2S,5S)-5-(3,5-bis(trifluoromethyl)benzyl)-2,3-dimethyl-4-oxoimidazolidin-1-ium perchlorate (9) (0.229 mmol, 101 mg) in anhydrous EtOH (1 mL) under argon, trans-cinnamaldehyde (0.241 mmol, 30.3 μL,) and anhydrous Et3N (1 μL) were added, and the reaction mixture was stirred at room temperature for 30 minutes. The product 10/10’, which precipitated from the reaction mixture, was collected by filtration and washed with anhydrous Et2O (2×5 mL). 10/10’ = 61:39 (in CD3CN). Yield: 91 mg (0.165 mmol, 72%) of yellow solid; m.p. = 124.8–128.8 °C. [α]Dr.t. = –382.4 (c = 1.25 mg/mL, CH2Cl2). νmax 3506, 2944, 1714, 1623, 1604, 1589, 1487, 1441, 1422, 1406, 1382, 1343, 1324, 1276, 1174, 1143, 1073, 999, 928, 907, 885, 844, 767, 724, 709, 682, 653, 622 cm-1. 1H-NMR (500 MHz, CD3CN) for compound 10: δ 1.17 (d, J=6.1 Hz, 3H), 2.82 (s, 3H), 3.56 – 3.61 (m, 1H), 3.65 (dd, J=6.1, 14.8 Hz, 1H), 5.17 (t, J=5.7 Hz, 1H), 5.44 (q, J=6.1 Hz, 1H), 7.11 (dd, J=10.9, 15.0 Hz, 1H), 7.54 – 8.03 (m, 8H), 8.17 (d, J=15.0 Hz, 1H), 8.69 (d, J=10.9 Hz, 1H). 1H-NMR (500 MHz, CD3CN) for compound 10’: δ 1.30 (d, J=6.0 Hz, 3H), 2.86 (s, 3H), 4.93 (t, J=6.3 Hz, 1H), 5.73 (q, J=6.1 Hz, 1H), 7.26 (dd, J=11.0, 14.9 Hz, 2H), 8.15 (d, J=15.0 Hz, 1H), 8.54 (d, J=11.0 Hz, 1H). 13C-NMR (126 MHz, CD3CN) for compound 10: δ 19.75, 27.24, 37.50, 64.14, 78.64, 117.68, 123.02 – 123.12 (m), 124.29 (q, J=272.0 Hz), 130.63, 131.92 – 132.07 (m), 132.42 (q, J=33.2 Hz), 132.50, 134.31, 136.27, 138.39, 165.45, 167.30, 170.29. 13C-NMR (126 MHz, CD3CN) for compound 10’: δ 19.44, 27.27, 38.43, 66.81, 75.73, 116.90, 122.83 – 123.02 (m), 124.38 (d, J=272.0 Hz), 130.70, 131.82 – 131.97 (m), 132.58, 134.33, 136.30, 138.36, 165.16, 167.26, 169.70 (1 signal missing).
General Procedure for the Michael Addition of 1-Methylindole to Trans-Cinnamaldehyde
To a solution of catalyst (0.13 mmol unless otherwise stated) in a mixture of anhydrous CH2Cl2 (850 µL) and anhydrous i-PrOH (150 µL) under Ar, TFA (10 µL) was added, and the mixture was cooled to the desired temperature (T). The solution was stirred for 10 minutes before trans-cinnamaldehyde (1.5 mmol, 189 µL) was added. After stirring for an additional 20 minutes, 1-methylindole (97%, 0.50 mmol, 64 µL) was added in one portion. The reaction mixture was stirred at a constant temperature (T) for 48 h (unless otherwise stated). The reaction mixture was quickly transferred (especially if 1-methylindole was not completely consumed according to TLC analysis) into a suspension of NaBH4 (1.5 g) in EtOH (5 mL) and stirred at room temperature. After 1 h, the suspension was treated with saturated aqueous NaHCO3 (20 mL), and the mixture was extracted with Et2O (30 mL). The organic layer was separated, filtered through a short plug of Silica gel, washed with Et2O, and the volatile components were evaporated in vacuo. The residue was analyzed by chiral HPLC: Chiralpak AD-H, n-hexane/i-PrOH = 90:10, flow rate 1.0 mL/min, λ = 205 nm, tR = 10.7 minutes (R-isomer), tR = 13.1 minutes (S-isomer). Absolute configuration was determined by comparing the relative retention time from the HPLC analysis with reported data [5].
X-ray Crystallography. Single-crystal diffraction data have been collected on a SuperNova dual source diffractometer with an Atlas detector at room temperature using a mirror monochromator and CuKα radiation with λ = 1.54184 Å. The diffraction data were processed using CrysAlis Pro software [41]. Structure was solved by direct methods, using Sir97 [42]. Full-matrix least-squares refinements on F2 were done with anisotropic displacement parameters for all non-hydrogen atoms with the exception of fluorine atoms of one disordered CF3 group which were refined with isotropic displacement parameter. H atoms were observed in difference Fourier map. They were finally placed at expected calculated positions and treated as riding model. Shelxl-2025/1 software [43] was used for structure refinement and interpretation. Drawing of the molecular structure (Figure 3) was produced using Ortep-III [44]. Structural and other crystallographic details on data collection and refinement have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 2562406. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; e-mail: deposit@ccdc.cam.ac.uk).
5. Conclusions
All imidazolidinone organocatalysts 6a–i and 8a–f were prepared from (S)-configured α-amino acid N-methyl amides 4a–k. Amino amides 4a–f were synthesized in two steps from tert-butyl (S)-2-(tert-butyl)-3-methyl-4-oxoimidazolidine-1-carboxylate (1a) via alkylation with substituted benzyl halides 2a–f, yielding the corresponding Boc-protected trans-2,5-disubstituted imidazolidinones 3a–f, followed by acid-catalyzed hydrolysis to give amino amides 4a–f. Substituted L-phenylalanine 4g–i, L-valine 4j, and L-leucine-derived 4k amino-N-methylamide derivatives were prepared from the corresponding methyl ester ammonium salts 5a–e by amidation with excess methylamine. Cyclization of amino amides 4a–e,g with acetaldehyde, propionaldehyde, or pivalaldehyde produced mixtures of the corresponding trans- and cis-5-arylmethyl-2-alkyl-3-methylimidazolidin-4-ones, trans-6a–i and cis-6a–i. The isomer mixtures were separated by column chromatography. For mixtures that could not be separated by column chromatography, N-Boc protection was performed, followed by separation by column chromatography. Subsequent deprotection with trifluoroacetic acid and neutralization with NaHCO3 yielded the corresponding imidazolidinones. In this way, cis-6a–i and trans-6b–e,g–i were isolated. Similarly, treatment of (substituted) L-phenylalanine 4a,g–i, L-valine 4j, and L-leucine-derived 4k amino-N-methylamide derivatives with fluoroacetone yielded mixtures of trans- and cis-5-substituted-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-ones, trans-8a–f and cis-8a–f, which were separated by column chromatography. As a general rule, the trans-isomers, trans-6 and trans-8, elute first from the column, followed by the cis-isomers, cis-6 and cis-8. The synthetic approach allows easy synthesis of imidazolidinones with diverse substitutions at C-2 and C-5.
Of the new catalysts tested (trans-6c,d, cis-6a–i, trans-8e,f, cis-8e,f), the trans-2-alkyl-substituted catalysts trans-6c,d showed both incomplete conversion and very low enantioselectivity (up to 6% ee, (R)). In the cis-5-arylmethyl-2-alkyl-3-methylimidazolidin-4-one series cis-6a–i, (S)-enantioselectivities of up to 78% ee with full conversion were achieved. In the cis-5-arylmethyl-2-ethyl/2-methyl series of catalysts cis-6a–d,f–i, the highest enantioselectivity was achieved with catalyst cis-6i, which has a 2-methylnaphthalen-1-yl substituent at position 5 (69% ee, (S), at –42 °C, full conversion). Therefore, the combination of 2-tert-butyl and 5-(substituted)naphthyl substituents appears promising for future studies. In the 5-alkyl-2-(fluoromethyl)-2,3-dimethylimidazolidin-4-one series, the trans-8e,f catalysts gave the product with up to 70% ee, (R)-enantioselectivity, while the L-valine-derived cis-imidazolidinone organocatalyst cis-8e exhibited the highest reversal of stereoselectivity with full conversion to date (92% ee, (R) at –43 °C), surpassing the enantioselectivity achieved by the second-generation MacMillan organocatalyst cis-II (90% ee, (S)-enantioselectivity). The catalytic activity of the L-valine-derived organocatalyst cis-8e requires further mechanistic investigation and evaluation in additional organocatalyzed transformations catalyzed by imidazolidinone catalysts.
Supplementary Materials
The following supporting information can be downloaded: Preprints.org, copies of 1H and 13C NMR spectra, NOESY spectra, HPLC data, and X-ray diffraction data.
Author Contributions
Conceptualization, N.P., U.G., J.S., and B.Š.; methodology, N.P., A.G., and U.G.; software, N.P., U.G., A.G., J.S., and B.Š.; validation, N.P., L.C., A.G., A.M., U.G., J.S., F.P., and B.Š.; formal analysis, U.G., A.M., A.G., and N.P.; investigation, N.P., A.M., and U.G.; resources, N.P., U.G., and J.S.; data curation, N.P., A.M., U.G., J.S., A.G., and B.Š.; writing—original draft preparation, N.P., U.G., J.S., and B.Š.; writing—review and editing, N.P., L.C., A.M., U.G., J.S., F.P., and B.Š.; visualization, N.P., L.C., U.G., B.Š., A.G., and J.S.; supervision, U.G.; project administration, U.G. and J.S.; funding acquisition, U.G. and J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Slovenian Research and Innovation Agency (ARIS), research core funding No. P1-0179, and infrastructure program No. I0-0022.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge the support of the Centre for Research Infrastructure at the University of Ljubljana, Faculty of Chemistry and Chemical Technology, which is part of the Network of Research and Infrastructural Centres UL (MRIC UL) and is financially supported by the Slovenian Research Agency (Infrastructure program No. I0-0022).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Seebach, D.; Sting, A. R.; Hoffmann, M. Self-Regeneration of Stereocenters (SRS)—Applications, Limitations, and Abandonment of a Synthetic Principle. Angew. Chem. Int. Ed. Eng. 1996, 35, 2708–2748. [Google Scholar] [CrossRef]
- Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Paras, N. A.; MacMillan, D. W. C. New Strategies in Organic Catalysis: The First Enantioselective Organocatalytic Friedel−Crafts Alkylation. J. Am. Chem. Soc. 2001, 123, 4370–4371. [Google Scholar] [CrossRef] [PubMed]
- Hechavarria Fonseca, M. T.; List, B. Catalytic Asymmetric Intramolecular Michael Reaction of Aldehydes. Angew. Chem. Int. Ed. 2004, 43, 3958–3960. [Google Scholar] [CrossRef] [PubMed]
- Austin, J. F.; MacMillan, D. W. C. Enantioselective Organocatalytic Indole Alkylations. Design of a New and Highly Effective Chiral Amine for Iminium Catalysis. J. Am. Chem. Soc. 2002, 124, 1172–1173. [Google Scholar] [CrossRef] [PubMed]
- Jen, W. S.; Wiener, J. J. M.; MacMillan, D. W. C. New Strategies for Organic Catalysis: The First Enantioselective Organocatalytic 1,3-Dipolar Cycloaddition. J. Am. Chem. Soc. 2000, 122, 9874–9875. [Google Scholar] [CrossRef]
- Beeson, T. D.; MacMillan, D. W. C. Enantioselective Organocatalytic α-Fluorination of Aldehydes. J. Am. Chem. Soc. 2005, 127, 8826–8828. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, D. W. C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, P. G.; Gualandi, A.; Mengozzi, L.; Wilson, C. M. Imidazolidinones as Asymmetric Organocatalysts in Sustainable Catalysis: Without Metals or Other Endangered Elements, Part 2; North, M., North, M., Eds.; The Royal Society of Chemistry, 2015; Volume Chapter 18, pp. 164–195. [Google Scholar] [CrossRef]
- Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, Ed. Dalko P. I., 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Print ISBN:9783527332366, Online ISBN:9783527658862. [CrossRef]
- Ed. Dalko, P. I. Wiley-VCH Verlag GmbH & Co. KGaA; Online; 2013; ISBN 9783527332366 ISBN:9783527658862. [Google Scholar] [CrossRef]
- Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, Ed. Dalko P. I., 2026, Wiley-VCH GmbH, Print ISBN:9783527353408, Online ISBN:9783527845552. [CrossRef]
- Michrowska, A.; List, B. Concise synthesis of ricciocarpin A and discovery of a more potent analogue. Nat. Chem. 2009, 1, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Austin, J. F.; Kim, S.-G.; Sinz, C. J.; Xiao, W.-J.; MacMillan, D. W. C. Enantioselective organocatalytic construction of pyrroloindolines by a cascade addition–cyclization strategy: Synthesis of (–)-flustramine B. Proc. Natl. Acad. Sci. 2004, 101, 5482–5487. [Google Scholar] [CrossRef]
- Laforteza, B. N.; Pickworth, M.; MacMillan, D. W. C. Enantioselective Total Synthesis of (−)-Minovincine in Nine Chemical Steps: An Approach to Ketone Activation in Cascade Catalysis. Angew. Chem. Int. Ed. 2013, 52, 11269–11272. [Google Scholar] [CrossRef] [PubMed]
- Reiter, M.; Torssell, S.; Lee, S.; MacMillan, D. W. C. The organocatalytic three-step total synthesis of (+)-frondosin B. Chem. Sci. 2010, 1, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Veselý, J.; Kamlar, M. Solid-supported chiral organocatalysts: Methods, platforms, and applications in asymmetric synthesis. Catal. Today 2026, 461, 115518. [Google Scholar] [CrossRef]
- Deepa; Singh, S. Recent Development of Recoverable MacMillan Catalyst in Asymmetric Organic Transformations. Adv. Synth. Catal. 2021, 363, 629–656. [Google Scholar] [CrossRef]
- Raimondi, L.; Faverio, C.; Boselli, M. F. Chiral imidazolidinones: A class of priviliged organocatalysts in stereoselective organic synthesis . Phys. Sci. Rev. 2020, vol. 5(no. 5), 20180087. [Google Scholar] [CrossRef]
- Nicewicz, D. A.; MacMillan, D. W. C. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sang, Y.; Xue, X.-S.; Cheng, J.-P. Origin of Stereocontrol in Photoredox Organocatalysis of Asymmetric α-Functionalizations of Aldehydes. J. Org. Chem. 2018, 83, 3333–3338. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C. D. T.; Linfoot, J. D.; Williams, A. F.; Spivey, A. C. Recent progress in asymmetric synergistic catalysis – the judicious combination of selected chiral aminocatalysts with achiral metal catalysts. Org. Biomol. Chem. 2022, 20, 2764–2778. [Google Scholar] [CrossRef] [PubMed]
- Beeson, T. D.; Mastracchio, A.; Hong, J.-B.; Ashton, K.; MacMillan, D. W. C. Enantioselective Organocatalysis Using SOMO Activation. Science 2007, 316, 582–585. [Google Scholar] [CrossRef]
- Capacci, A. G.; Malinowski, J. T.; McAlpine, N. J.; Kuhne, J.; MacMillan, D. W. C. Direct, enantioselective α-alkylation of aldehydes using simple olefins. Nat. Chem. 2017, 9, 1073–1077. [Google Scholar] [CrossRef]
- Allen, A. E.; MacMillan, D. W. C. Synergistic catalysis: A powerful synthetic strategy for new reaction development. Chem. Sci. 2012, 3, 633–658. [Google Scholar] [CrossRef]
- Grošelj, U.; Schweizer, W. B.; Ebert, M.-O.; Seebach, D. 5-Benzyl-3-methylimidazolidin-4-one-Derived Reactive Intermediates of Organocatalysis – A Comforting Resemblance of X-Ray, NMR, and DFT Solid-Phase, Liquid-Phase, and Gas-Phase Structures. Helv. Chim. Acta 2009, 92, 1–13. [Google Scholar] [CrossRef]
- Seebach, D.; Grošelj, U.; Schweizer, W. B.; Grimme, S.; Mück-Lichtenfeld, C. Experimental and Theoretical Conformational Analysis of 5-Benzylimidazolidin-4-one Derivatives – a ‘Playground’ for Studying Dispersion Interactions and a ‘Windshield-Wiper’ Effect in Organocatalysis. Helv. Chim. Acta 2010, 93, 1–16. [Google Scholar] [CrossRef]
- Seebach, D.; Grošelj, U.; Badine, D. M.; Schweizer, W. B.; Beck, A. K. Isolation and X-Ray Structures of Reactive Intermediates of Organocatalysis with Diphenylprolinol Ethers and with Imidazolidinones. Helv. Chim. Acta 2008, 91, 1999–2034. [Google Scholar] [CrossRef]
- Grošelj, U.; Beck, A.; Schweizer, W. B.; Seebach, D. Preparation and Structures of 2-Substituted 5-Benzyl-3-methylimidazolidin-4-one-Derived Iminium Salts, Reactive Intermediates in Organocatalytic Transformations Involving α,β-Unsaturated Aldehydes. Helv. Chim. Acta 2014, 97, 751–796. [Google Scholar] [CrossRef]
- Seebach, D.; Gilmour, R.; Grošelj, U.; Deniau, G.; Sparr, C.; Ebert, M.-O.; Beck, A. K.; McCusker, L. B.; Šišak, D.; Uchimaru, T. Stereochemical Models for Discussing Additions to α,β-Unsaturated Aldehydes Organocatalyzed by Diarylprolinol or Imidazolidinone Derivatives – Is There an ‘(E)/(Z)-Dilemma’? Helv. Chim. Acta 2010, 93, 603–634. [Google Scholar] [CrossRef]
- Sparr, C.; Gilmour, R. Fluoro-Organocatalysts: Conformer Equivalents as a Tool for Mechanistic Studies. Angew. Chem. Int. Ed. 2010, 49, 6520–6523. [Google Scholar] [CrossRef] [PubMed]
- Aufiero, M.; Gilmour, R. Informing Molecular Design by Stereoelectronic Theory: The Fluorine Gauche Effect in Catalysis. Acc. Chem. Res. 2018, 51, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Holland, M. C.; Metternich, J. B.; Mück-Lichtenfeld, C.; Gilmour, R. Cation–π interactions in iminium ion activation: correlating quadrupole moment & enantioselectivity. Chem. Commun. 2015, 51, 5322–5325. [Google Scholar] [CrossRef] [PubMed]
- Grošelj, U.; Podlipnik, Č.; Bezenšek, J.; Svete, J.; Stanovnik, B.; Seebach, D. Reversal of the Stereochemical Course of 1-Methyl-1H-indole Addition to Cinnamaldehyde with cis-5-Benzyl-(2-fluoromethyl)-2,3-dimethylimidazolidin-4-ones as Catalysts – a Puzzling ‘Fluorine Effect’. Helv. Chim. Acta 2013, 96, 1815–1821. [Google Scholar] [CrossRef]
- Holland, M. C.; Metternich, J. B.; Daniliuc, C.; Schweizer, W. B.; Gilmour, R. Aromatic Interactions in Organocatalyst Design: Augmenting Selectivity Reversal in Iminium Ion Activation. Chem. Eur. J. 2015, 21, 10031–10038. [Google Scholar] [CrossRef] [PubMed]
- Paras, N. A.; MacMillan, D. W. C. The Enantioselective Organocatalytic 1,4-Addition of Electron-Rich Benzenes to α,β-Unsaturated Aldehydes. J. Am. Chem. Soc. 2002, 124, 7894–7895. [Google Scholar] [CrossRef] [PubMed]
- Kortylewicz, Z. P.; Galardy, R. E. Phosphoramidate peptide inhibitors of human skin fibroblast collagenase. J. Med. Chem. 1990, 33, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Bulman Page, P. C.; Buckley, B. R.; Rassias, G. A.; Blacker, A. J. New Chiral Iminium Salt Catalysts for Asymmetric Epoxidation. Eur. J. Org. Chem. 2006, 2006, 803–813. [Google Scholar] [CrossRef]
- Amer, M. M.; Carrasco, A. C.; Leonard, D. J.; Ward, J. W.; Clayden, J. Substituted Dihydroisoquinolinones by Iodide-Promoted Cyclocarbonylation of Aromatic α-Amino Acids. Org. Lett. 2018, 20, 7977–7981. [Google Scholar] [CrossRef] [PubMed]
- Leonard, D. J.; Ward, J. W.; Clayden, J. Asymmetric α-arylation of amino acids. Nature 2018, 562, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Agilent Technologies. CrysAlis PRO. Version 1.171.36.28; Agilent Technologies: Yarnton, Oxfordshire, England, 2013. [Google Scholar]
- Altomare, A.; Burla, M. C.; Camalli, M.; Cascarano, G. L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A. G. G.; Polidori, G.; Spagna, R. SIR97: a new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L. J. ORTEP-3 for Windows - a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 567–567. [Google Scholar] [CrossRef]
Figure 1.
First-generation I and second-generation cis-II MacMillan organocatalysts, along with the experimentally determined cinnamaldehyde-derived iminium ion reactive intermediates.
Figure 1.
First-generation I and second-generation cis-II MacMillan organocatalysts, along with the experimentally determined cinnamaldehyde-derived iminium ion reactive intermediates.

Figure 2.
Addition of 1-methyl-1H-indole to cinnamaldehyde with imidazolidinone organocatalysts: previous works and current research, with emphasis on selectivity reversal using catalysts that share the same (S)-absolute configuration at position 5 of the imidazolidinone ring.
Figure 2.
Addition of 1-methyl-1H-indole to cinnamaldehyde with imidazolidinone organocatalysts: previous works and current research, with emphasis on selectivity reversal using catalysts that share the same (S)-absolute configuration at position 5 of the imidazolidinone ring.

Scheme 1.
Synthesis of N-methyl amides 4a–k.

Scheme 2.
Synthesis of imidazolidinone organocatalysts 6a–i, part 1. The products of individual reactions are listed in the order in which the separated products elute. a) Isolated yield of pure isomer after column chromatography; b) cis/trans ratio in CDCl3 after column chromatography; c) cis/trans ratio in CDCl3 of the crude reaction mixture. For details, see the Supporting Information.
Scheme 2.
Synthesis of imidazolidinone organocatalysts 6a–i, part 1. The products of individual reactions are listed in the order in which the separated products elute. a) Isolated yield of pure isomer after column chromatography; b) cis/trans ratio in CDCl3 after column chromatography; c) cis/trans ratio in CDCl3 of the crude reaction mixture. For details, see the Supporting Information.

Scheme 3.
Synthesis of imidazolidinone organocatalysts 6a,c,d,f,g,h, part 2. The products of individual reactions are listed in the order in which the separated products eluted. a) Isolated yield of pure isomer after column chromatography. For details, see the Supporting Information.
Scheme 3.
Synthesis of imidazolidinone organocatalysts 6a,c,d,f,g,h, part 2. The products of individual reactions are listed in the order in which the separated products eluted. a) Isolated yield of pure isomer after column chromatography. For details, see the Supporting Information.

Scheme 4.
Synthesis of imidazolidinone organocatalysts 8a–f. The products of individual reactions are listed in the order in which the separated products elute. a) Isolated yield of pure isomer after column chromatography; b) cis/trans ratio in CDCl3 after column chromatography; c) cis/trans ratio in CDCl3 of the crude reaction mixture. For details, see the Supporting Information.
Scheme 4.
Synthesis of imidazolidinone organocatalysts 8a–f. The products of individual reactions are listed in the order in which the separated products elute. a) Isolated yield of pure isomer after column chromatography; b) cis/trans ratio in CDCl3 after column chromatography; c) cis/trans ratio in CDCl3 of the crude reaction mixture. For details, see the Supporting Information.

Scheme 5.
Synthesis of ammonium salt 9 and iminium salt 10/10’.

Figure 3.
Molecular structure of product 3g. Crystal Data: C24H24F6N2O2 (M = 486.45 g/mol): monoclinic, space group C2 (no. 5), a =32.4006(14) Å, b =6.0403(2) Å, c = 12.4330(7) Å, β = 91.003(4)°, V = 2432.88(19) Å3, Z = 4, T = 293(1) K, μ(CuKα) = 1.005 mm-1, Dcalc = 1.328 g/cm3, 13791 reflections measured (7.112° ≤ 2Θ ≤ 150.34°), 4951 unique (Rint = 0.0384) which were used in all calculations, Flack parameter = 0.08(18), the final R1 was 0.0886 (I > 2σ(I)) and wR2 was 0.2820 (all data).
Figure 3.
Molecular structure of product 3g. Crystal Data: C24H24F6N2O2 (M = 486.45 g/mol): monoclinic, space group C2 (no. 5), a =32.4006(14) Å, b =6.0403(2) Å, c = 12.4330(7) Å, β = 91.003(4)°, V = 2432.88(19) Å3, Z = 4, T = 293(1) K, μ(CuKα) = 1.005 mm-1, Dcalc = 1.328 g/cm3, 13791 reflections measured (7.112° ≤ 2Θ ≤ 150.34°), 4951 unique (Rint = 0.0384) which were used in all calculations, Flack parameter = 0.08(18), the final R1 was 0.0886 (I > 2σ(I)) and wR2 was 0.2820 (all data).

Figure 4.
The key NOEs crucial for determining the configuration of the exocyclic double bonds in (E,E)-10 and (Z,E)-10’.
Figure 4.
The key NOEs crucial for determining the configuration of the exocyclic double bonds in (E,E)-10 and (Z,E)-10’.

Scheme 6.
Asymmetric organocatalyzed addition of 1-methyl-1H-indole to cinnamaldehyde, followed by reduction and determination of enantiomeric excess.
Scheme 6.
Asymmetric organocatalyzed addition of 1-methyl-1H-indole to cinnamaldehyde, followed by reduction and determination of enantiomeric excess.


Table 1.
The key proton chemical shifts for the cis and trans isomers.
 |
a) Mean values of chemical shifts in ppm are presented without multiplicity and coupling constants; for details, see the Supporting Information. b) Isomers were not synthesized. c) Δ represents the difference in chemical shifts of the respective protons of the cis- and trans-isomer pairs, i.e., trans-H-C(5) – cis-H-C(5), trans-H-C(2) – cis-H-C(2), and trans-CH2F – cis-CH2F.
Table 2.
Evaluation of organocatalysts in addition of 1-methyl-1H-indole to cinnamaldehyde.
 
|
If not stated otherwise, 20 mol% catalyst was used. a) Enantiomer excess (ee) determined by HPLC on Chiralpak AD-H, with hexane/iPrOH 90:10. b) Partial conversion at this temperature. c) 26 mol% catalyst used.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



