Submitted:
14 June 2026
Posted:
22 June 2026
You are already at the latest version
Abstract
Disulfidptosis is a newly identified form of regulated cell death driven by high SLC7A11 expression under glucose starvation. Mechanistically, massive cystine influx via SLC7A11, coupled with NADPH depletion due to glucose deprivation, leads to cystine accumulation, disulfide stress, aberrant crosslinking of actin cytoskeleton (notably at Cys257, Cys284, and Cys373), and eventual membrane rupture. This process is independent of apoptosis, ferroptosis, and necroptosis. This review systematically outlines the molecular regulatory network of disulfidptosis, including upstream triggers, key execution molecules (glutathione and thioredoxin systems, RAC1/WRC/Arp2/3 axis), and downstream effectors. We further evaluate its therapeutic potential across various malignancies (glioblastoma, hepatocellular, gastric, colorectal, pancreatic, breast, lung, and prostate cancers) and other diseases (neurodegenerative, metabolic, orthopedic, cardiovascular). Recent advances in nanodelivery systems for selective disulfidptosis induction are summarized. Key translational challenges are critically analyzed, including targeting specificity, safety, delivery barriers, resistance mechanisms, and biomarker deficiency. By exploiting metabolic vulnerabilities (high SLC7A11 expression and glucose dependence) of pathological cells, disulfidptosis opens a new therapeutic window that can synergize with ferroptosis, cuproptosis, and immunotherapy. This review provides a comprehensive framework for basic research and clinical translation of disulfidptosis-targeted therapies.
Keywords:
disulfidptosis
; SLC7A11
; glucose deprivation
; NADPH depletion
; cytoskeleton
; metabolic reprogramming
; cancer therapy
; nanodelivery
; translational medicine
1. Introduction
1.1. Current Research Status and Metabolic Directions of Regulated Cell Death
Cell death, a fundamental process for maintaining organismal homeostasis, is broadly categorized into non-regulated and regulated forms. Non-regulated cell death occurs accidentally under extreme physical, chemical, or pathological conditions without genetic control. In contrast, regulated cell death (RCD) is actively orchestrated by specific intracellular signaling pathways and molecular mechanisms. For decades, apoptosis—characterized by caspase activation, mitochondrial outer membrane permeabilization, and orderly cellular dismantling—has been considered the primary form of programmed cell death [1]. However, deepening understanding of disease metabolism has revealed the intricate regulatory network between RCD and cellular metabolism as a key research area.
Among novel RCD forms, ferroptosis—an iron-dependent [2] cell death pathway driven by lipid peroxidation [3]—has attracted considerable attention for its critical role in cancer, neurodegenerative diseases, and ischemia-reperfusion injury [4,5]. Elucidation of SLC7A11 as a core regulator of ferroptosis resistance has revealed the critical importance of amino acid metabolism in cell survival [6]. SLC7A11, via system Xc⁻, imports extracellular cystine while exporting glutamate, thereby providing cysteine for glutathione (GSH) synthesis [6]. While protecting cells from oxidative damage, this system creates a metabolic vulnerability exploitable for targeted therapies, suggesting metabolic reprogramming is crucial for understanding novel RCD mechanisms.
Metabolic reprogramming refers to the process by which cells alter bioenergetic patterns by regulating key metabolic enzymes or signaling pathways to adapt to specific physiological or pathological environments [7]. This phenomenon occurs widely in tumors, inflammatory diseases, and metabolic disorders, and associates closely with disease progression, metastasis, and drug resistance [8,9,10].
Notably, many pathological cells (especially tumor and chronic inflammatory cells) upregulate SLC7A11 by activating RAS and NRF2 pathways, thereby enhancing cystine uptake and GSH synthesis to combat oxidative stress and ferroptosis. However, GSH synthesis requires substantial reduced nicotinamide adenine dinucleotide phosphate (NADPH), primarily generated by the pentose phosphate pathway (PPP). Consequently, these cells exhibit significant glucose dependence [11].Under ample glucose conditions, this metabolic reprogramming favors cell survival. Yet, when glucose is restricted (e.g., in solid tumor cores, ischemic tissues, or upon GLUT downregulation), NADPH production rapidly declines, preventing cystine reduction to cysteine and triggering disulfide stress. Concurrently, inhibited GSH synthesis causes thioredoxin system dysfunction, redox buffer collapse, and abnormal disulfide bond formation in cytoskeletal proteins like actin, ultimately causing cell disintegration—a process recently termed disulfidptosis [11,12,13,14].
1.2. Discovery, Characteristics, and Definition of Disulfidptosis
Disulfidptosis, a novel form of regulated cell death first reported by Gan et al. in 2023, specifically occurs in SLC7A11-overexpressing cells under glucose starvation [15]. This study revealed a unique mechanism: glucose deprivation sharply reduces pentose phosphate pathway (PPP) activity, leading to insufficient NADPH production. Meanwhile, SLC7A11-overexpressing cells continuously uptake cystine, rapidly depleting limited intracellular NADPH reserves. Without sufficient NADPH as a reducing equivalent, cystine cannot be reduced to cysteine and abnormally accumulates, triggering disulfide stress [15,16]. This stress induces abnormal intra- and intermolecular disulfide bonds in cytoskeletal proteins such as actin, causing excessive actin network contraction and cross-linking, cytoskeleton disintegration, and ultimately cell membrane integrity loss and lysis [11,12].
Disulfidptosis requires two strict conditions: (1) high SLC7A11 expression, ensuring continuous and substantial cystine uptake; and (2) glucose starvation, blocking the main NADPH source pathway. Both conditions are indispensable, distinguishing it from other metabolic cell death forms [15]. Importantly, this process resists classic cell death inhibitors, including those targeting apoptosis (Z-VAD-FMK), ferroptosis (Ferrostatin-1), necroptosis (Necrostatin-1), and autophagy (3-MA, chloroquine). However, thiol reductants (e.g., dithiothreitol, β-mercaptoethanol) completely block this process [17].These unique pharmacological and molecular characteristics indicate that disulfidptosis does not rely on known cell death pathways but represents an independent mode that directly attacks cytoskeleton structure through redox imbalance, constituting a significant discovery in cell death research.
1.3. Research Significance and Purpose of This Review
The discovery of disulfidptosis represents a paradigm shift in cell death research, offering a new perspective for translating metabolic stress into precise therapeutic strategies. Unlike traditional cell death pathways requiring exogenous stimuli or drug intervention, disulfidptosis exploits the inherent metabolic vulnerability of SLC7A11-overexpressing cells—their high glucose and NADPH dependence. This selective mechanism, based on tumor or pathological cell metabolic characteristics, opens a new therapeutic window for treating diseases characterized by metabolic reprogramming (e.g., malignant tumors and metabolic syndrome-related diseases). Notably, while initially defined by high SLC7A11 expression and glucose starvation, recent research is breaking this strict dependency. Strategies including NADPH metabolism modulation, thioredoxin system targeting, or mitochondrial function interference have expanded pathways for inducing disulfidptosis, enabling broader disease applications.
Importantly, disulfidptosis holds potential for synergistic action with other cell death forms. Within existing therapeutic paradigms, disulfidptosis can form complementary, multi-modal cell death networks with apoptosis, ferroptosis, and cuproptosis. This enhances therapeutic sensitivity by co-targeting different metabolic nodes while circumventing drug resistance from single pathway blockage. Furthermore, disulfidptosis-mediated immunogenic cell death (ICD) provides a theoretical basis for combination with immune checkpoint inhibitors, radiotherapy, and chemotherapy. This promises to overcome clinical challenges including low immunotherapy response rates and chemotherapy resistance.
Given rapid progress in disulfidptosis research and broad translational prospects, this review aims to:
(1) systematically elucidate the molecular regulatory network of disulfidptosis, including upstream signaling pathways, key executioner molecules, and downstream effector mechanisms;
(2) critically assess disulfidptosis therapeutic potential and limitations in cancer, neurodegenerative diseases, and cardiovascular diseases;
(3) summarize emerging nanomedicine delivery strategies for selectively activating disulfidptosis, including glucose oxidase nanosystems, NADPH-depleting nanodrugs, and mitochondria-targeted nanocarriers;
(4) identify key bottlenecks in translating basic research to clinical application, including biomarker development, targeting specificity optimization, and safety evaluation; and
(5) propose priority research directions to accelerate clinical application of disulfidptosis-related therapies.
By integrating current research progress, this review provides a comprehensive knowledge framework and practical guidance for disulfidptosis researchers and clinical translational practitioners.
Figure 1.
Discovery and milestones of disulfidptosis (2023–2026). Short Title: Discovery and milestones of disulfidptosis (2023–2026). Figure Legend: Timeline illustrating the major milestones in disulfidptosis research from its discovery in 2023 to future translational challenges. Key phases include: initial discovery (high SLC7A11 expression + glucose starvation → NADPH depletion → abnormal actin crosslinking → membrane rupture); mechanistic elucidation (RAC1/WRC/Arp2/3 axis, collapse of GSH and thioredoxin systems); expansion to disease spectrum (cancers, neurodegenerative, metabolic, orthopedic, cardiovascular, and inflammatory diseases); nanodelivery and therapeutic strategies (e.g., CYBC NPs, CuSS@876-PEG, sonodynamic nanosystems); and ongoing translational challenges (biomarker deficiency, specificity/safety, resistance mechanisms, lack of clinical trials). Future directions include multi-omics, PDX models, and AI-assisted patient stratification.
Figure 1.
Discovery and milestones of disulfidptosis (2023–2026). Short Title: Discovery and milestones of disulfidptosis (2023–2026). Figure Legend: Timeline illustrating the major milestones in disulfidptosis research from its discovery in 2023 to future translational challenges. Key phases include: initial discovery (high SLC7A11 expression + glucose starvation → NADPH depletion → abnormal actin crosslinking → membrane rupture); mechanistic elucidation (RAC1/WRC/Arp2/3 axis, collapse of GSH and thioredoxin systems); expansion to disease spectrum (cancers, neurodegenerative, metabolic, orthopedic, cardiovascular, and inflammatory diseases); nanodelivery and therapeutic strategies (e.g., CYBC NPs, CuSS@876-PEG, sonodynamic nanosystems); and ongoing translational challenges (biomarker deficiency, specificity/safety, resistance mechanisms, lack of clinical trials). Future directions include multi-omics, PDX models, and AI-assisted patient stratification.

2. Molecular Mechanisms and Regulatory Network of Disulfidptosis
As a newly discovered cell death form, disulfidptosis involves precise synergistic action of multiple metabolic pathways and organelles. Current research indicates that high SLC7A11 expression and glucose starvation constitute core triggers for disulfidptosis: large-scale cystine uptake rapidly depletes NADPH, while glucose starvation severely reduces NADPH generation. These two factors synergistically collapse redox balance, the fundamental driving force of disulfidptosis. Subsequently, cystine accumulation triggers disulfide stress, leading to abnormal disulfide bond cross-linking, contraction, and plasma membrane detachment of cytoskeletal proteins such as actin, ultimately causing cell integrity loss and disintegration. This represents the direct execution pathway of disulfidptosis [12,18]. The following sections systematically elaborate on the molecular mechanisms and regulatory network of disulfidptosis.
2.1. Core Trigger Conditions and Metabolic Dependence
2.1.1. High SLC7A11 Expression and Cystine Overload
High SLC7A11 expression is a critical trigger for disulfidptosis [15]. SLC7A11, the catalytic subunit of system Xc⁻ transporter, mediates 1:1 stoichiometric antiport of extracellular cystine and intracellular glutamate. This ATP-independent process is driven by cell membrane potential and substrate concentration gradients [11]. In pathological states, particularly tumor cells, multiple signaling pathways synergistically regulate SLC7A11 expression. Main regulatory pathways include:
(1) RAS-RAF-MEK-ERK-ETS1 signaling axis: In various cancers, RAS gene mutations act as core oncogenic drivers, continuously activating downstream phosphorylation cascades. Activated ERK directly phosphorylates transcription factor ETS1, enhancing its transcriptional activity and promoting nuclear localization. Phosphorylated ETS1 binds to specific sites in the SLC7A11 promoter region, significantly upregulating its expression [19].
(2) MerTK/ERK/SP1 pathway: In malignancies including hepatocellular carcinoma, oncogenic tyrosine kinase receptor MerTK activates ERK, which phosphorylates transcription factor SP1, promoting SLC7A11 transcription. This inhibits ferroptosis and enhances tumor cell antioxidant capacity [20].
(3) NRF2-ARE pathway: Under oxidative stress, transcription factor NRF2 dissociates from its inhibitor KEAP1 and translocates to the nucleus, where it binds to the antioxidant response element (ARE) in the SLC7A11 promoter, enhancing its transcriptional activity. This represents a core cellular adaptive response to oxidative stress [21].
(4) ATF4-mediated stress response: Under endoplasmic reticulum stress or amino acid starvation, the integrated stress response (ISR) activates transcription factor ATF4. ATF4 directly binds to the SLC7A11 promoter and upregulates its expression, coupling cellular stress signals with amino acid metabolic reprogramming [22].
(5) Epigenetic modifications: Epigenetic modifications including histone acetylation and DNA methylation also participate in SLC7A11 expression regulation. For example, histone deacetylase inhibitors enhance histone acetylation levels in the SLC7A11 promoter region, upregulating its transcription [23,24].
Under glucose-sufficient conditions, SLC7A11 upregulation enhances cellular antioxidant capacity and ferroptosis resistance. However, when glucose supply is restricted, NADPH generation drastically decreases. The large cystine influx mediated by highly expressed SLC7A11 becomes a fatal metabolic burden—impaired cystine reduction leads to abnormal accumulation, triggering disulfide stress and ultimately disulfidptosis [11]. This reveals the “double-edged sword” effect of SLC7A11 in different metabolic environments.
2.1.2. Glucose Deprivation and NADPH Depletion
After glucose is transported into cells by glucose transporters (GLUTs), it is metabolized via the pentose phosphate pathway (PPP) to produce NADPH. The PPP consists of oxidative and non-oxidative branches, with the oxidative branch being the primary intracellular NADPH source. This branch is initiated by the rate-limiting enzyme glucose-6-phosphate dehydrogenase (G6PD), generating two NADPH molecules per glucose-6-phosphate molecule [25]. When glucose supply is insufficient, PPP activity declines, severely impairing NADPH production.
NADPH is an essential reducing equivalent donor for cystine reduction to cysteine, a process primarily dependent on glutathione reductase (GR) and thioredoxin reductase (TXNRD) systems [14]. In SLC7A11-overexpressing cells, large amounts of internalized cystine require rapid reduction to prevent toxic accumulation, rapidly depleting limited NADPH reserves [26]. When glucose starvation reduces NADPH production, cells enter “NADPH starvation,” where cystine cannot be effectively reduced and abnormally accumulates, triggering disulfide stress.
Critically, NADPH deficiency impairs both thioredoxin and glutathione systems, the two major redox buffering systems: thioredoxin (TRX) cannot be reduced and loses activity, while inhibited glutathione (GSH) synthesis decreases the GSH/GSSG ratio, collapsing cellular redox homeostasis [27]. This vicious cycle ultimately causes abnormal oxidative modification and disulfide bond cross-linking of cytoskeletal proteins, triggering disulfidptosis execution [28,29].
Figure 2.
Metabolic fate of SLC7A11-overexpressing cells under glucose-replete versus glucose-deprived conditions. Figure Legend: Comparative illustration of cell fate decisions based on glucose availability in cells with high SLC7A11 expression. Left panel – Glucose-replete state: High NADPH production via the pentose phosphate pathway (PPP) supports reduction of imported cystine to cysteine, driving GSH synthesis and maintaining GPX4 activity. This results in ferroptosis resistance, low disulfide stress, and cell survival. Right panel – Glucose-deprived state: NADPH levels fall sharply, decreasing the NADPH/cystine ratio below a critical threshold. Cystine cannot be effectively reduced and accumulates intracellularly, triggering disulfide stress and aberrant disulfide bond formation in the actin cytoskeleton. Although GPX4 becomes inactive, lipid peroxidation is not the primary execution mechanism; instead, cells undergo disulfidptosis. The “critical threshold” of the NADPH/cystine ratio determines whether cells survive or undergo disulfidptosis.
Figure 2.
Metabolic fate of SLC7A11-overexpressing cells under glucose-replete versus glucose-deprived conditions. Figure Legend: Comparative illustration of cell fate decisions based on glucose availability in cells with high SLC7A11 expression. Left panel – Glucose-replete state: High NADPH production via the pentose phosphate pathway (PPP) supports reduction of imported cystine to cysteine, driving GSH synthesis and maintaining GPX4 activity. This results in ferroptosis resistance, low disulfide stress, and cell survival. Right panel – Glucose-deprived state: NADPH levels fall sharply, decreasing the NADPH/cystine ratio below a critical threshold. Cystine cannot be effectively reduced and accumulates intracellularly, triggering disulfide stress and aberrant disulfide bond formation in the actin cytoskeleton. Although GPX4 becomes inactive, lipid peroxidation is not the primary execution mechanism; instead, cells undergo disulfidptosis. The “critical threshold” of the NADPH/cystine ratio determines whether cells survive or undergo disulfidptosis.

2.1.3. Disulfide Stress and ROS Positive Feedback Amplification
Under severe NADPH depletion, large cystine influx can neither be converted to cysteine via the thioredoxin (Trx)/glutaredoxin (Grx)-dependent cytosolic reduction pathway nor maintain intracellular thiol homeostasis through the GSH system, leading to continuous free cystine accumulation. Excessively accumulated cystine directly undergoes mixed disulfide exchange reactions with free protein thiols (-SH), including key thiol targets such as the Cys-173 site of redox-sensing protein PRDX1, causing abnormal covalent modifications and triggering disulfide stress [30].
Concurrently, blocked cystine-to-cysteine conversion synergistically dismantles cellular antioxidant defense at two levels: First, cysteine, the rate-limiting substrate for de novo GSH synthesis, is directly deprived, interrupting GSH synthesis substrate supply. Second, severe NADPH depletion causes both glutathione reductase (GR) and thioredoxin reductase (TrxR) to simultaneously lose their electron donors, collapsing both major antioxidant systems (the GSH/GSSG cycle and Trx1/TrxR1 axis) and sharply increasing intracellular reactive oxygen species (ROS) levels.
Elevated ROS directly oxidizes highly conserved free thiols on cytoskeletal proteins (especially β/γ-actin), inducing abnormal intermolecular disulfide bond formation between adjacent molecules and accelerating pathological covalent cross-linking and aggregation of F-actin filaments. This irreversible cross-linking not only directly impairs cytoskeletal network dynamic remodeling capability but also triggers secondary ROS bursts through mitochondrial and endoplasmic reticulum functional damage, forming an “oxidative damage → disulfide bond cross-linking → organelle damage → ROS regeneration” redox positive feedback loop [31]. Continuous amplification of this positive feedback loop ultimately drives irreversible F-actin network contraction and depolymerization, leading to cellular morphology disintegration and constituting the core disulfidptosis execution mechanism.
2.2. Key Molecular Regulatory Nodes (Molecular Event Sequences)
2.2.1. The Glutathione System
The glutathione system comprises three core components—glutathione (GSH), glutathione reductase (GR), and glutaredoxin (Grx)—that collectively maintain intracellular thiol redox homeostasis. GSH is a γ-glutamyl-cysteinyl-glycine tripeptide, and the free thiol group (-SH) on its second cysteine residue confers strong reducing potential, making it the most abundant low-molecular-weight intracellular redox buffer [32]. During reduction, GSH cysteine thiol directly attacks disulfide bonds in proteins or small molecules, reducing them to free thiols while being oxidized to glutathione disulfide (GSSG) [33]. To maintain GSH/GSSG dynamic balance, GR utilizes NADPH reducing equivalents to reduce GSSG disulfide bonds, regenerating and releasing two GSH molecules, completing the redox cycle [34].
At the protein redox regulation level, Grx1 and Grx2 catalyze protein deglutathionylation via thiol-disulfide exchange reactions, reversing S-glutathionylation modifications on protein cysteine residues. S-glutathionylation, a reversible oxidative post-translational modification, participates in protein functional regulation as a stress signal while protecting critical cysteine thiols from irreversible terminal oxidative modifications such as sulfonation (-SO₃H) [35]. Thus, Grx-mediated deglutathionylation constitutes a crucial step by which the GSH system maintains protein thiolome homeostasis.
All components of this system directly or indirectly depend on NADPH: GR-catalyzed GSSG reduction uses NADPH as the sole electron donor, and the Grx catalytic cycle requires continuous GSH regeneration. Consequently, under glucose deprivation leading to inhibited PPP flux and severe NADPH depletion, GR activity is limited, GSH regeneration stalls, and GSSG significantly accumulates. Simultaneously, Grx system activity declines due to GSH scarcity, impeding protein S-glutathionylation modification removal. These combined effects lead to complete collapse of intracellular reducing buffering capacity and uncontrolled accumulation of inter- and intramolecular protein disulfide bonds, providing critical biochemical basis for disulfidptosis.
2.2.2. The Thioredoxin System
The thioredoxin system is another core cellular system maintaining thiol redox homeostasis, composed of thioredoxin (Trx), thioredoxin reductase (TXNRD), and NADPH [36]. Its mechanism relies on classic thiol-disulfide exchange: the two cysteine residues in the Cys-Gly-Pro-Cys sequence within Trx’s active site, in their reduced state (-SH), attack abnormal disulfide bonds in target proteins, reducing them to free thiols, while Trx itself converts to its oxidized state (Trx-S₂). Oxidized Trx is then re-reduced by TXNRD using NADPH as the final electron donor, completing the catalytic cycle and continuously clearing excessively accumulated intracellular protein disulfide bonds.
Within the disulfidptosis regulatory framework, TXNRD1 function is particularly critical. Under normal physiological conditions, TXNRD1 dynamically clears intracellular protein disulfide modifications by maintaining continuous Trx1 reducing activity, acting as a core “safety valve” that negatively regulates disulfide stress and inhibits disulfidptosis initiation [37]. However, normal operation of this system highly depends on NADPH as the ultimate electron source. When large-scale cystine uptake driven by SLC7A11 overexpression leads to inhibited PPP flux and severe NADPH depletion, TXNRD1 becomes functionally paralyzed due to electron donor depletion. Trx1 then remains stalled in its oxidized state, unable to regenerate, and system disulfide clearance capacity consequently drops to zero. In this context, simultaneous collapse of the GSH/GSSG system completely eradicates intracellular reducing power, leading to massive protein disulfide modification accumulation and irreversible disulfide stress amplification [38]. Therefore, TXNRD1 functional state largely determines cellular sensitivity threshold to disulfidptosis, making it a potential tumor metabolic intervention target.
2.2.3. Actin Cytoskeleton: The Direct Execution Substrate of Disulfidptosis
Actin is a core cytoskeleton network structural protein, and its function relies on multiple cysteine residues remaining in reduced state. Under homeostatic conditions, the GSH and thioredoxin systems work synergistically to continuously protect free thiols on actin molecules, ensuring normal folded conformation, polymerization dynamics, and interactions with membrane-anchoring complexes, thereby maintaining cytoskeleton structural integrity and dynamic remodeling capability [39].
When these two major antioxidant systems synergistically collapse due to NADPH depletion, disulfide stress ensues. Rapid intracellular oxidative environment deterioration exposes several highly conserved actin cysteine residues—especially Cys257, Cys284, and Cys373—to oxidative attack by excessive cystine and ROS. This leads to spontaneous formation of numerous abnormal intramolecular and intermolecular disulfide covalent cross-links between adjacent residues or molecules [15,40]. Notably, Cys257 and Cys284 are located near the hydrophobic interface where actin participates in fiber lateral contacts, and their abnormal cross-linking directly interferes with G-actin polymerization into F-actin. Cys373, however, is situated in a functional region where actin interacts with various actin-binding proteins (ABPs) and integrin adhesion complexes, and its oxidative modification disrupts mechanical connections between actin and cell membrane anchoring sites.
This multi-site covalent cross-linking disrupts F-actin filament polymerization stability, causes widespread cytoskeleton detachment from membrane adhesion sites, and triggers overall actin network contraction and depolymerization [41]. Cellular morphology rapidly disintegrates, plasma membrane integrity is lost, and intracellular contents leak. This abnormal actin cross-linking-driven cytoskeleton disintegration process constitutes the morphological and molecular characteristics distinguishing disulfidptosis from other programmed cell death forms including apoptosis and ferroptosis, and represents its ultimate direct execution mechanism.
2.3. Signaling Pathways and Regulatory Networks
2.3.1. The RAC1/WRC/Arp2/3 Complex Axis
Recent research reveals that the RAC1/WRC/Arp2/3 signaling axis plays a critical role in regulating disulfidptosis susceptibility. This pathway directly influences cellular vulnerability to disulfide stress by modulating actin cytoskeleton dynamic remodeling [12,41].
RAC1 Activation and Signal Transduction:
RAC1 is a core Rho family small GTPase member, functioning as a molecular switch cycling between GTP-bound (active) and GDP-bound (inactive) states. Upon GTP binding, RAC1 undergoes conformational change, exposing an interface for downstream effector protein interaction. Activated RAC1-GTP specifically recognizes and binds to the WAVE regulatory complex (WRC), a critical molecular event for initiating the actin polymerization cascade [20,42].
Conformational Activation Mechanism of WRC:
WRC is a heteropentameric complex composed of five subunits: WAVE family members (WAVE1/2/3), Abi, Nap1, Sra1, and HSPC300. In its resting state, WRC adopts an autoinhibited conformation where its crucial WCA domain (WH2-connector-acidic domain) is masked by Sra1 and Abi subunits, preventing Arp2/3 complex interaction. When RAC1-GTP binds to the WRC Sra1 subunit, it induces large-scale WRC conformational rearrangement, releasing the autoinhibited state and allowing WCA domain liberation from the complex core and full exposure [42]. This conformational change is an irreversible activation process, and the liberated WCA domain subsequently directly binds to and activates the Arp2/3 complex.
Arp2/3 Complex-Mediated Branched Actin Nucleation:
The Arp2/3 complex is the core actin network assembly machinery, consisting of seven subunits: two actin-related proteins (Arp2 and Arp3) and five auxiliary subunits. Upon activated WCA domain binding to the Arp2/3 complex, Arp2 and Arp3 are induced to form an actin-dimer-like conformation, providing nucleation sites for nascent actin filaments. This process initiates on pre-existing mother actin filament sides, generating a branched actin network at 70-degree angles, forming a dense dendritic structure supporting lamellipodia formation and cell edge protrusion [12].
Special Role in Disulfidptosis:
Notably, the RAC1/WRC/Arp2/3 axis paradoxically increases cellular disulfidptosis susceptibility by promoting actin network polymerization. This seemingly contradictory phenomenon may stem from: (1) highly polymerized F-actin networks providing more disulfide cross-linking sites, facilitating abnormal intermolecular disulfide bond formation under disulfide stress; (2) dense actin networks, after abnormal cross-linking, being more prone to overall contraction and plasma membrane detachment, accelerating cytoskeletal collapse; and (3) active actin dynamic remodeling consuming ATP and NADPH, further exacerbating energy and redox crises under glucose starvation.
Regulatory Role and Specificity of NCKAP1:
NCKAP1 (NCK-associated protein 1, also known as NAP1 or Hem2) encodes a crucial WRC complex scaffold subunit, essential for maintaining WRC structural integrity and regulating its activation threshold. Loss-of-function experiments show that NCKAP1 knockout does not affect basal SLC7A11 expression levels, demonstrating that NCKAP1 is not involved in upstream cystine transport regulation [41]. However, NCKAP1 deficiency significantly attenuates abnormal disulfide bond formation induced under glucose starvation and inhibits pathological F-actin network contraction and plasma membrane detachment. These results indicate that the NCKAP1/WRC/Arp2/3 axis specifically regulates disulfidptosis execution phase (i.e., disulfide bond-mediated actin cytoskeleton disintegration) rather than trigger phase (cystine overload and NADPH depletion). This finding provides potential targets for selectively intervening in disulfidptosis execution while preserving normal SLC7A11-mediated antioxidant function.
Potential Therapeutic Implications:
The RAC1/WRC/Arp2/3 signaling axis offers multiple intervention points for therapeutic disulfidptosis regulation: (1) RAC1 inhibitors (e.g., NSC23766) can block signal initiation; (2) directly targeting WRC or Arp2/3 complex can disrupt actin network polymerization; (3) enhancing NCKAP1 function might protect normal cells from disulfidptosis in specific contexts. These strategies’ clinical translational potential warrants further validation.
2.3.2. GSH Synthesis Pathway
Glutathione (GSH) biosynthesis occurs through two consecutive enzymatic reactions. The first step is rate-limiting: glutamate-cysteine ligase (GCL) catalyzes glutamate and cysteine condensation to produce γ-glutamylcysteine; the second step, catalyzed by glutathione synthetase (GS), links glycine to the γ-glutamylcysteine C-terminus, forming the GSH tripeptide structure [43,44]. Due to the distinctly rate-limiting nature of the GCL-catalyzed first reaction, intracellular cysteine supply level directly determines the upper flux limit for the entire synthesis pathway.
SLC7A11, the core functional subunit of system Xc⁻ transporter, transports extracellular cystine into cells at 1:1 stoichiometric ratio while simultaneously releasing glutamate extracellularly. Internalized cystine is rapidly reduced to two cysteine molecules, driven by the thioredoxin system and intracellular reducing power; this cysteine then enters the GCL-catalyzed step as direct substrate for GSH synthesis. Therefore, SLC7A11-mediated cystine uptake rate is the primary rate-limiting node determining intracellular cysteine supply and GSH synthesis rate [11,45].
However, cystine reduction to cysteine relies on NADPH-provided reducing equivalents. Under glucose deprivation, inhibited PPP flux leads to severe NADPH depletion, sharply decreasing cystine reduction efficiency to cysteine, interrupting cysteine supply, and halting de novo GSH synthesis due to substrate scarcity [46]. This process forms a self-reinforcing vicious feedback loop: declining GSH levels cause the GRx system to lose its electron carrier, further weakening cellular ability to reduce abnormal protein disulfide bonds; meanwhile, continuous deepening of protein thiol oxidation accelerates further residual GSH depletion, with both factors mutually promoting and continuously amplifying disulfide stress intensity, ultimately driving irreversible disulfidptosis onset. This positive feedback mechanism reveals that NADPH depletion is not only a triggering event for disulfidptosis but also continuously amplifies death signals by disrupting the GSH synthesis pathway, endowing disulfidptosis with unique irreversibility and autocatalytic characteristics.
2.3.3. Cross-Regulation and Distinctions Between Disulfidptosis, Ferroptosis, and Cuproptosis
Ferroptosis is an iron-dependent, lipid peroxidation-driven programmed cell death form characterized by massive phospholipid hydroperoxide (PLOOH) accumulation and plasma membrane lipid damage due to GPX4 inactivation. Disulfidptosis, in contrast, is characterized by disulfide stress-driven covalent cross-linking and mechanical disintegration of actin cytoskeleton. Both exhibit distinct morphological and biochemical fingerprints [47,48]. Although both are regulated by SLC7A11, the core system Xc⁻ transporter subunit, they differ fundamentally in regulatory direction, metabolic dependencies, and downstream effects [14,26].
In ferroptosis, high SLC7A11 expression plays a protective role: large cystine influx is reduced to cysteine, which, as the rate-limiting substrate for de novo GSH synthesis, drives substantial GSH production. GSH, an essential GPX4 cofactor, reduces toxic phospholipid hydroperoxides (PLOOH) to harmless phospholipid alcohols, effectively blocking lipid peroxidation chain reactions and antagonizing ferroptosis initiation [49]. Thus, high SLC7A11 expression exerts anti-death protective effects in ferroptosis.
However, in disulfidptosis, high SLC7A11 expression itself does not directly induce cell death but constitutes potential “metabolic vulnerability.” Under glucose-replete conditions, the pentose phosphate pathway (PPP) continuously supplies NADPH, maintaining normal cystine reduction and allowing cells to tolerate high cystine influx levels without damage. However, once glucose deprivation leads to inhibited PPP flux and severe NADPH depletion, excessively internalized cystine accumulates intracellularly due to inability to be reduced, triggering widespread abnormal protein disulfide cross-linking, especially covalent modifications at critical sites including actin Cys257, Cys284, and Cys373, ultimately driving cytoskeleton disintegration and cell death. [50] Therefore, high SLC7A11 expression plays a conditional “pro-death” role in disulfidptosis—transforming its transport advantage into lethal burden under specific metabolic stress conditions.
In summary, high SLC7A11 expression exhibits diametrically opposite functional logic in these two death modalities: it is a protective factor in ferroptosis but becomes a death driver under glucose restriction-induced disulfidptosis. This “dual identity” suggests that when assessing tumor cell death sensitivity solely based on SLC7A11 expression levels, metabolic context influence, such as glucose availability, must be fully considered.
At the effector mechanism level, ferroptosis relies on GPX4 inactivation and lipid peroxidation accumulation, and can be blocked by iron chelators or lipid peroxidation inhibitors (e.g., ferrostatin-1). Disulfidptosis, conversely, does not rely on iron or lipid peroxidation but can be reversed by exogenous thiol reductants (e.g., DTT, N-acetylcysteine), possessing distinct pharmacological characteristics. Despite distinct pathways, both share several key metabolic intersection points: NADPH depletion synergistically promotes ferroptosis by weakening the TrxR/GPX4 axis and directly triggers disulfidptosis by blocking cystine reduction. Furthermore, GSH inhibits both cell death forms by maintaining GPX4 activity and sustaining Trx/GRx system reducing power, respectively, making GSH metabolism a common regulatory hub for both pathways.
Notably, because ferroptosis and disulfidptosis are mechanistically independent, cells developing resistance to radiotherapy-induced ferroptosis—often by upregulating ferroptosis-protective pathways including GPX4, SLC7A11, or NRF2/HO-1—may retain or even enhance their disulfidptosis sensitivity due to sustained high SLC7A11 expression. This logic suggests that within radiotherapy combined with metabolic intervention strategies, inducing disulfidptosis through glucose restriction or PPP inhibitors (e.g., 6-AN) holds promise as a novel sensitization strategy to overcome radiotherapy-induced ferroptosis resistance [51].
Potential Interactions with Cuproptosis
Cuproptosis is a copper ion-dependent cell death form whose core mechanism involves ferredoxin 1 (FDX1)-mediated abnormal aggregation of lipoylated proteins in the tricarboxylic acid cycle and subsequent mitochondrial dysfunction [52]. Although FDX1 has not been directly confirmed to be involved in disulfidptosis, both pathways exhibit significant molecular-level commonalities: both culminate in abnormal protein modification events (Cu⁺-bound aggregation of lipoylated proteins in cuproptosis vs. abnormal actin disulfide cross-linking in disulfidptosis), and both are closely related to redox imbalance.
In cuproptosis, FDX1-mediated Cu²⁺→Cu⁺ reduction disrupts mitochondrial redox balance, while in disulfidptosis, NADPH depletion leads to overall cellular redox homeostasis collapse. Both pathways utilize redox imbalance as a death signal amplifier. Furthermore, SLC7A11-mediated high cystine uptake may enhance mitochondrial stress by consuming NADPH, potentially increasing cuproptosis susceptibility. Conversely, reactive oxygen species generated by copper ion accumulation may oxidize cystine residues and promote abnormal disulfide bond formation, leading to “copper-disulfide” synergistic toxicity.
Future research needs to systematically analyze these two pathway interaction networks, including whether FDX1 indirectly affects disulfidptosis sensitivity by regulating redox status, and whether spatiotemporal synergy exists between lipoylated protein aggregation and actin disulfide cross-linking. Understanding these cross-regulatory mechanisms will help design rational combination therapy strategies, such as sequentially inducing cuproptosis to disrupt mitochondria followed by triggering disulfidptosis, or synergistically targeting FDX1 and glutathione reductase to achieve super-additive killing effects at the redox hub.

Table 1.
Comparison of Ferroptosis, Disulfidptosis, and Cuproptosis.
| Characteristics | Ferroptosis | Disulfidptosis | Cuproptosis |
|---|---|---|---|
| Core triggering conditions | Iron overload / GPX4 inactivation | Glucose starvation + SLC7A11 overexpression | Copper ion overload |
| Key regulatory molecules | GPX4, ACSL4, SLC7A11, FTH1, Fe²⁺ | SLC7A11, NADPH, actin (ACTB, etc.) | FDX1, LIAS, DLAT, lipoylated proteins, Cu²⁺ |
| Metabolic dependencies | Iron, polyunsaturated fatty acids (PUFAs) | Glucose (PPP pathway), cystine | Copper, mitochondrial respiration (TCA cycle) |
| Terminal effector events | Lipid peroxidation → membrane rupture | Aberrant disulfide bonds in F-actin → cytoskeleton collapse | Lipoylated protein aggregation → loss of mitochondrial function |
| Inhibitors | Iron chelators (DFO), Lip-1, Fer-1 | Thiol-reducing agents (DTT, NAC, β-mercaptoethanol)* | Copper chelators (TTM, ETC) |
| Relationship with SLC7A11 | High expression inhibits cell death (via GSH synthesis) | High expression promotes cell death (under glucose deprivation) | Not yet clarified |
| References | [1,2] |
[41] | [52] |
Note: *These thiol-reducing agents can effectively block disulfidptosis but are not specific inhibitors unique to disulfidptosis; they also affect other redox pathways.
Figure 3.
Molecular crosstalk among ferroptosis, disulfidptosis, and cuproptosis. Figure Legend: Schematic diagram depicting the central molecular network linking three metabolic cell death pathways: ferroptosis, disulfidptosis, and cuproptosis. The system Xc⁻ transporter (SLC7A11-SLC3A2) imports extracellular cystine in exchange for glutamate. Under glucose-sufficient conditions, cystine is reduced to cysteine (fueling GSH synthesis) and NADPH is abundant, which supports GPX4 activity to reduce lipid hydroperoxides (PL-OOH) and inhibit ferroptosis. Under glucose deprivation, NADPH depletion prevents cystine reduction, leading to cystine accumulation, disulfide stress, and aberrant disulfide crosslinking of the actin cytoskeleton (disulfidptosis). Concurrently, GPX4 becomes inactive, but lipid peroxidation is not the dominant death mechanism. RAC1-GTP activates the WRC/Arp2/3 complex to promote cytoskeletal rearrangement. The pathway to cuproptosis (involving other substances) is also indicated, highlighting shared metabolic nodes (NADPH, GSH) but distinct execution mechanisms.
Figure 3.
Molecular crosstalk among ferroptosis, disulfidptosis, and cuproptosis. Figure Legend: Schematic diagram depicting the central molecular network linking three metabolic cell death pathways: ferroptosis, disulfidptosis, and cuproptosis. The system Xc⁻ transporter (SLC7A11-SLC3A2) imports extracellular cystine in exchange for glutamate. Under glucose-sufficient conditions, cystine is reduced to cysteine (fueling GSH synthesis) and NADPH is abundant, which supports GPX4 activity to reduce lipid hydroperoxides (PL-OOH) and inhibit ferroptosis. Under glucose deprivation, NADPH depletion prevents cystine reduction, leading to cystine accumulation, disulfide stress, and aberrant disulfide crosslinking of the actin cytoskeleton (disulfidptosis). Concurrently, GPX4 becomes inactive, but lipid peroxidation is not the dominant death mechanism. RAC1-GTP activates the WRC/Arp2/3 complex to promote cytoskeletal rearrangement. The pathway to cuproptosis (involving other substances) is also indicated, highlighting shared metabolic nodes (NADPH, GSH) but distinct execution mechanisms.

2.4. Bidirectional Regulation of Disulfidptosis by the Tissue Microenvironment
Disulfidptosis occurrence depends not only on cellular SLC7A11 expression level and NADPH reserve status but is also multi-dimensionally regulated by three tissue microenvironment layers: metabolic, cellular, and physicochemical. These microenvironmental factors exhibit bidirectional regulatory nature for disulfidptosis—acting as pro-death signals driving its initiation under specific conditions while exerting protective effects to antagonize its onset in other contexts.
Regulation by the Metabolic Microenvironment
Tissue regions with microcirculatory disorders leading to local glucose deprivation (e.g., solid tumor necrotic edges, avascular central regions of degenerated intervertebral discs) naturally constitute glucose-starved triggering background, significantly increasing local cell risk of undergoing disulfidptosis. Furthermore, abnormal metabolite accumulation plays a regulatory role: both lactate accumulation and glutamine depletion exacerbate disulfidptosis susceptibility by disrupting NADPH homeostasis. In the typical tumor immune microenvironment context, tumor-infiltrating CD8⁺ T cells, despite compensatory lactate dehydrogenase B (LDHB) upregulation after prolonged exposure to high-lactate, low-glucose metabolic competitive environments, experience inhibited G6PD activity, decreased PPP flux, NADPH depletion, and halted cystine reduction due to continuous metabolic stress, ultimately driving CD8⁺ T cells themselves to undergo disulfidptosis. This mechanism constitutes a molecular pathway by which tumors exploit metabolic competition to weaken immune surveillance and achieve immune evasion [15,16].
Regulation by the Cellular Microenvironment
At the intercellular signaling level, pro-inflammatory factors including TNF-α and IL-17A secreted by M1 macrophages and Th17 cells upregulate SLC7A11 transcriptional expression in keratinocytes and tumor cells by activating the NF-κB signaling pathway, thereby enhancing cystine influx into target cells [48,49]. Cancer-associated fibroblasts (CAFs) enhance adjacent tumor cell cystine uptake capacity through two mechanisms: directly secreting cystine into the extracellular environment to expand substrate supply, or releasing extracellular vesicles (EVs) carrying functional system Xc⁻ components, thereby transferring transport capacity to target cells. These pathway superpositions drive abnormal intracellular cystine accumulation, laying substrate foundation for disulfidptosis initiation under glucose restriction.
Moreover, extracellular matrix (ECM) mechanical stiffness influences disulfidptosis threshold. Stiff ECM commonly found in highly fibrotic tissues places actin cytoskeleton in high-tension pre-stressed state through integrin-mediated mechanotransduction (including sustained Rho/ROCK pathway activation). When disulfide stress is superimposed, high-tension cytoskeletal network sensitivity to abnormal disulfide bond-induced covalent cross-linking and structural disintegration is significantly enhanced, lowering disulfidptosis initiation threshold [15].
Regulation by Physicochemical Properties
Hypoxia and acidic low pH are two typical tumor microenvironment physicochemical perturbing factors that synergistically promote disulfidptosis by regulating SLC7A11 expression and inhibiting NADPH generation. Under hypoxia, activated HIF-1α transcriptionally upregulates SLC7A11 expression, driving increased cystine uptake; concurrently, HIF-1α impairs mitochondrial-dependent NADPH regeneration pathways by reprogramming cellular metabolism and inhibiting mitochondrial oxidative phosphorylation. Acidic microenvironment (low pH) enhances SLC7A11 transport activity by affecting its transport kinetics, while its inhibitory effect on mitochondrial function further reduces NADPH supply. These two physicochemical perturbations synergistically create “double metabolic pressure” of increased cystine influx and insufficient NADPH reserves, establishing sufficient microenvironmental conditions for disulfidptosis initiation [16,26].
In intervertebral disc degeneration (IDD) pathological context, the nucleus pulposus (NP) region is chronically nutrient-deprived due to its naturally avascular nature, and continuous abnormal mechanical load application further exacerbates local glucose and oxygen supply. Concurrently, mechanical stimulation may upregulate SLC7A11 expression levels in nucleus pulposus cells (NPCs) via TGF-β signaling or oxidative stress pathways, forming a metabolically vulnerable state of “high cystine uptake, low NADPH regeneration” against severe glucose deficiency background, ultimately driving NPCs to undergo disulfidptosis, thereby participating in and accelerating intervertebral disc degeneration pathological progression [16,46].
3. Therapeutic Potential of Disulfidptosis in Multiple Diseases
Disulfidptosis therapeutic targets primarily focus on the core pathogenic axis: “redox imbalance-disulfide stress-actin cytoskeleton collapse.” This cell death modality offers unique bidirectional disease regulation: in proliferative diseases (e.g., malignant tumors), induced disulfidptosis selectively eliminates pathological cells by leveraging cancer cell vulnerabilities—high SLC7A11 expression and metabolic reprogramming. Conversely, in cell-damaging diseases (e.g., ischemia-reperfusion injury, neurodegenerative diseases), inhibiting disulfidptosis protects functional cells from redox stress and cytoskeletal collapse damage.
This bidirectional capacity stems from unique disulfidptosis molecular characteristics: triggering depends on specific metabolic contexts (glucose deprivation with high cystine uptake), execution involves intervenable signaling pathways (RAC1/WRC/Arp2/3 axis), and clear biomarkers (SLC7A11 expression) enable patient stratification. Therefore, research into dual disulfidptosis regulatory mechanisms will enable tumor-targeted therapies based on metabolic vulnerabilities and provide theoretical frameworks and intervention targets for neuroprotection and organ protection, with significant implications for major disease treatment.
3.1. Malignant Tumors
Metabolic cell death pathways represent unique cancer vulnerabilities, and disulfidptosis induction particularly targets tumors with high SLC7A11 expression, glucose dependence, aberrant glutathione and thioredoxin reductase regulation, and immunotherapy resistance [53].High SLC7A11 expression typically helps cancer cells resist ferroptosis, shortening patient overall survival and contributing to chemoresistance and targeted therapy resistance [53,54]. Therefore, disulfidptosis serves as a signaling target in malignant tumor treatment and can combine with other cell death mechanisms (e.g., ferroptosis, cuproptosis) and therapeutic modalities (e.g., immunotherapy, radio-chemotherapy) to enhance efficacy and patient benefit.
3.1.1. Neurological Tumors
Glioblastoma (GBM):
TrxR1 inhibition induces glioblastoma disulfidptosis. This cell death is reversed by disulfide-reducing agents but not ROS scavengers or apoptosis, ferroptosis, or necroptosis inhibitors; in orthotopic xenograft GBM models, TrxR1 deficiency inhibited tumor growth, suggesting TrxR1 as a druggable glioblastoma disulfidptosis node [38].
3.1.2. Digestive System Tumors
Hepatocellular Carcinoma (HCC):
Mendelian randomization studies on HBV-related HCC found that disulfidptosis-related genes (GYS1, RPN1, SLC7A11, LRPPRC, CAPZB) effectively predict prognosis, with functional experiments confirming GYS1 silencing inhibits tumor proliferation and metastasis [55]. Pan-cancer mRNA expression data show MYH9 inhibition alleviates sorafenib resistance in HCC through disulfidptosis-like changes [56]. Thus, GYS1 and MYH9 are two functionally validated HCC therapeutic targets associated with disulfidptosis. In HCC, elevated disulfidptosis-related gene (DRG) expression correlates with increased induced regulatory T cells (iTregs), macrophages, natural killer (NK) cells, and T cells, possibly due to enhanced TME immune activation, leading to better immune checkpoint inhibitor (ICI) response in high-risk patients [57]. This suggests disulfidptosis can modulate TME and enhance tumor cell killing.
Gastric Cancer (GC):
NCKAP1 and SLC7A11 are overexpressed in GC and associate with actin activity, GTPase energy metabolism, immune infiltration, and immunotherapy [58,59]. The disulfidptosis-related lncRNA AL359182.1, a novel prognostic biomarker, may interact with the MYH10-driven actin cytoskeleton signaling axis pathway, with its inhibition potentially suppressing gastric cancer metastasis [60].
Colorectal Cancer (CRC):
FLNA mediates colorectal cancer disulfidptosis. FLNA knockdown significantly increases mouse CD4⁺ and CD8⁺ T cells and inhibits tumor cell migration and invasion in vitro. Combined glucose transporter 1 (GLUT1) and anti-programmed cell death protein 1 (PD-1) therapy enhances CD8⁺ T cell recruitment and inhibits EMT [61], suggesting disulfidptosis modulation with immunotherapy as a promising colorectal cancer therapeutic target. GPX4 is a key cellular redox homeostasis regulator. Dipeptidyl peptidase 7 (DPP7) stabilizes GPX4 protein, helping cells resist disulfidptosis and NK cell killing through GPX4-dependent mechanisms, offering new colorectal cancer treatment possibilities [62].
Pancreatic Cancer (PAAD):
TMEM105, an lncRNA associated with pancreatic cancer disulfidptosis, alleviates disulfidptosis by inducing GLUT1 transcriptional activity, suggesting TMEM105 as a druggable PAAD disulfidptosis-inducing node. Notably, pancreatic cancer’s dense desmoplastic stroma creates glucose-deprived microenvironments, potentially sensitizing tumors to disulfidptosis inducers [63]. G6PD is overexpressed in PAAD and associates with clinical stage, histological grade, and prognosis. In vitro experiments show G6PD inhibitor RRx-001 induces disulfidptosis-like features in PAAD cells, preliminarily indicating G6PD as a PAAD disulfidptosis activation target [64].
Pancreatic Ductal Adenocarcinoma (PDAC):
Increased CASC8 expression in PDAC interacts with c-Myc to activate the pentose phosphate pathway, reducing NADP⁺/NADPH ratio and inhibiting disulfidptosis under glucose starvation, driving PDAC progression. This suggests CASC8 as a potential PDAC therapeutic target [65].
3.1.3. Gynecological Tumors
Ovarian Cancer:
SLC7A11 expression is significantly elevated in ovarian cancer, enabling disulfidptosis induction under glucose starvation. This vulnerability enables cancer cell killing by controlling intracellular glucose levels using GLUT inhibitors [66,67]. The p53-GYS1 feedback loop promotes NADPH production, counteracting disulfidptosis and promoting platinum resistance. Targeting GYS1 pathway restores disulfidptosis and enhances drug sensitivity, improving ovarian cancer treatment [68].
Breast Cancer:
Breast cancer types exhibit varying expression profiles, with higher SLC7A11 mRNA in TNBC and Luminal B subtypes [69,70]. High SLC7A11-expressing breast cancer cells maintain GSH by enhancing cystine uptake, inhibiting disulfidptosis. GLUT1 inhibition blocking NADPH supply induces breast cancer disulfidptosis [71]. For instance, disulfidptosis nano-inducers (CYBC NPs) loaded with cystine and GLUT1 inhibitor BAY-876 selectively induce triple-negative breast cancer disulfidptosis, reverse immunosuppressive tumor microenvironment (ITME), and inhibit tumor growth [72]. Additionally, targeting GYS1 may trigger disulfidptosis by inducing F-actin contraction in TNBC [73]. PTTG1IP is highly expressed in reast cancer cell lines MDA-MB-231, MCF-7, and breast cancer tissues. PTTG1IP knockdown induces disulfidptosis and reduces cancer cell proliferation [74]. These studies provide references for utilizing disulfidptosis in breast cancer treatment.
3.1.4. Respiratory System Tumors
Lung Cancer:
KEAP1-mutated lung cancer cells, through NRF2 transcription factor activation, exhibit high SLC7A11 expression and increased SLC7A11-mediated cystine uptake, causing abnormal disulfide bond accumulation. This sensitizes these tumors to glucose transporter inhibitors (GLUT inhibitors), suggesting this target for KEAP1-mutated lung cancer precision treatment [75,76]. FGA is a tumor suppressor. In vitro experiments show FGA inhibition attenuates disulfidptosis by downregulating SLC7A11/xCT while enhancing malignant phenotype in lung adenocarcinoma (LUAD) models, providing new LUAD treatment strategies by regulating disulfidptosis [77]. G6PD associates with poor prognosis and pro-cancer effects in LUAD. G6PD inhibitors induce disulfidptosis by reducing NADPH production, significantly inhibiting proliferative capacity [78,79]. OGFRP1 is a disulfidptosis-related lncRNA. OGFRP1 inhibition significantly suppresses lung cancer cell invasion and migration [16]. ZIC5 is highly expressed in LUAD and associates with poor patient prognosis. ZIC5 silencing increases NADP⁺/NADPH ratio, decreases GSH levels and GSSG/GSH ratio, exhibiting typical disulfidptosis features [80]. These findings provide basis for lung cancer targeted therapy using disulfidptosis.
3.1.5. Urological Tumors
Prostate Cancer:
CCNB2 is a novel disulfidptosis regulator. CCNB2 knockdown triggers disulfide peptide formation and inhibits prostate cancer proliferation, migration, and invasion, indicating CCNB2-mediated disulfidptosis regulation can limit prostate cancer progression [81]. FOXA1 is a key SLC7A11 transcriptional regulator. Reducing FOXA1 and SLC7A11 expression protects cells from disulfidptosis, highlighting the SE/FOXA1/SLC7A11 axis critical role in regulating disulfidptosis and tumor progression. Targeting this pathway, especially in glucose-deprived tumor environments, may offer promising prostate cancer therapeutic strategies [82].
Bladder Cancer (BCa):
Chen et al. identified disulfidptosis-related genes (DRG) POU5F1 and CTSE as promising BCa clinical treatment intervention targets [83]. Furthermore, combining nanotechnology with sonodynamic therapy, utilizing GLUT1 inhibitors and cystine-containing polymers to induce disulfidptosis, improves bladder cancer immunotherapy efficacy [83].
3.1.6. Other Tumors
Head and Neck Cancer (HNC):
HNC cells exhibit higher SLC7A11 expression versus normal cells [84,85,86]. HNC cells show resistance to ferroptosis inducers, cuproptosis inducers, and combined immunotherapy but sensitivity to disulfidptosis. Although in vitro and in vivo validation remains limited, this provides new directions [26,87,88]. Peroxisome proliferator-activated receptor gamma (PPARγ) is a ligand-dependent nuclear transcription factor with significant tumor development implications [89,90]. PPARγ inhibition upregulates solute carrier family 7 member 11 (SLC7A11), promoting disulfidptosis in oral squamous cell carcinoma (OSCC), a common head and neck squamous cell carcinoma subtype [91].
Uveal Melanoma (UM):
FTO inhibitor meclofenamic acid (MA) restores N6-methyladenosine (m6A) RNA methylation levels in uveal melanoma cells, upregulates SLC7A11, and induces disulfidptosis. This supports disulfidptosis mechanisms as potential UM therapeutic strategies [92].
Table 2.
Disulfidptosis in Cancer: Key Molecules and Mechanisms.
| Cancer Type | Key Molecules | Mechanism of Action | Main Evidence | References |
|---|---|---|---|---|
| Nervous System Tumors | ||||
| Glioblastoma (GBM) | TrxR1 | Inhibition of TrxR1 induces disulfidptosis, which can be reversed by disulfide bond reducing agents but not rescued by ROS scavengers or apoptosis/ferroptosis/necroptosis inhibitors | TrxR1 deletion inhibits tumor growth in orthotopic xenograft GBM mouse models | [38] |
| Digestive System Tumors | ||||
| Hepatocellular carcinoma (HCC) - HBV-related | GYS1, RPN1, SLC7A11, LRPPRC, CAPZB | Combination of disulfidptosis-related genes predicts prognosis | Mendelian randomization study; silencing GYS1 inhibits tumor proliferation and metastasis | [55] |
| Hepatocellular carcinoma (HCC) - Sorafenib-resistant | MYH9 | Inhibition of MYH9 alleviates sorafenib resistance through disulfidptosis-like changes | Study based on pan-cancer mRNA expression data | [56] |
| Hepatocellular carcinoma (HCC) - Immunotherapy | DRG gene set | Elevated DRG expression correlates with increased levels of iTregs, macrophages, NK cells, and T cells | High-risk patients respond better to ICI | [57] |
| Gastric cancer (GC) | NCKAP1, SLC7A11 | Overexpression associated with actin activity, GTPase energy metabolism, immune infiltration, and immunotherapy | Clinical correlation studies | [58,59] |
| Gastric cancer (GC) | lncRNA AL359182.1 | Prognostic marker that interacts with MYH10-driven actin cytoskeleton signaling axis | Its inhibition may suppress gastric cancer metastasis | [60] |
| Colorectal cancer (CRC) | FLNA | Key mediator of disulfidptosis; FLNA knockdown increases CD4⁺ and CD8⁺ T cells | Combined inhibition of GLUT1 and anti-PD-1 enhances CD8⁺ T cell recruitment and inhibits EMT | [61] |
| Colorectal cancer (CRC) | DPP7-GPX4 axis | DPP7 stabilizes GPX4 protein, helping cells resist disulfidptosis and NK cell killing | GPX4-dependent mechanism | [62] |
| Pancreatic cancer (PAAD) | TMEM105 | lncRNA alleviates disulfidptosis by inducing GLUT1 transcriptional activity | Dense stroma creates glucose-deprived microenvironment, increasing sensitivity to disulfidptosis | [63] |
| Pancreatic cancer (PAAD) | G6PD | Overexpression correlates with clinical stage, histological grade, and prognosis | G6PD inhibitor RRx-001 induces disulfidptosis-like features | [64] |
| Pancreatic ductal adenocarcinoma (PDAC) | CASC8 | Interacts with c-Myc to activate pentose phosphate pathway, decreasing NADP⁺/NADPH ratio and inhibiting disulfidptosis | Leads to PDAC progression | [65] |
| Gynecological Tumors | ||||
| Ovarian cancer | SLC7A11 | Significantly upregulated; glucose starvation induces disulfidptosis | GLUT inhibitors control glucose levels to kill cancer cells | [66,67] |
| Ovarian cancer | p53-GYS1 axis | Promotes NADPH production, counteracts disulfidptosis, and promotes platinum resistance | Targeting GYS1 restores disulfidptosis and enhances drug sensitivity | [68] |
| Breast cancer | SLC7A11 | Highly expressed in TNBC and Luminal B; maintains GSH to inhibit disulfidptosis | Inhibiting GLUT1 blocks NADPH supply and induces disulfidptosis | [26,69] |
| Breast cancer - TNBC | CYBC NPs | Nanoinducer loaded with cystine and GLUT1 inhibitor BAY-876 | Selectively induces disulfidptosis, reverses ITME, and inhibits tumor growth | [72] |
| Breast cancer - TNBC | GYS1 | Targeting GYS1 induces F-actin contraction | Triggers disulfidptosis | [73] |
| Breast cancer | PTTG1IP | Highly expressed in cancer cell lines and tissues | Knockdown leads to disulfidptosis and reduced cancer cell proliferation | [74] |
| Respiratory System Tumors | ||||
| Lung cancer - KEAP1 mutant | KEAP1-NRF2-SLC7A11 axis | NRF2 activation leads to SLC7A11 overexpression and increased cystine uptake | More sensitive to GLUT inhibitors, enabling precision therapy | [21,75] |
| Lung adenocarcinoma (LUAD) | FGA | Tumor suppressor; FGA inhibition downregulates SLC7A11/xCT and attenuates disulfidptosis | Enhances malignant phenotype | [77] |
| Lung adenocarcinoma (LUAD) | G6PD | Associated with poor prognosis and pro-oncogenic effects | G6PD inhibitors reduce NADPH production and induce disulfidptosis | [101] |
| Lung cancer | lncRNA OGFRP1 | Disulfidptosis-related lncRNA | OGFRP1 inhibition significantly suppresses invasion and migration capacity | [16,79] |
| Lung adenocarcinoma (LUAD) | ZIC5 | Highly expressed and associated with poor prognosis | ZIC5 silencing exhibits typical disulfidptosis features (NADP⁺/NADPH↑, GSH↓, GSSG/GSH↓) | [80] |
| Urinary System Tumors | ||||
| Prostate cancer | CCNB2 | Novel regulator of disulfidptosis | Knockdown triggers disulfide peptide formation and inhibits proliferation, migration, and invasion | [81] |
| Prostate cancer | SE/FOXA1/SLC7A11 axis | FOXA1 is a key transcriptional regulator of SLC7A11 | Reduced expression protects cells from disulfidptosis; targeting this axis shows promise in glucose-depleted environments | [82] |
| Bladder cancer (BCa) | POU5F1, CTSE | Disulfidptosis-related genes (DRG) | Serve as potential intervention targets for clinical treatment | [83] |
| Bladder cancer (BCa) | Nanotechnology + Sonodynamic therapy | GLUT1 inhibitor and cystine-containing polymer induce disulfidptosis | Improves immunotherapy outcomes | [102] |
| Other Tumors | ||||
| Head and neck cancer (HNC) | SLC7A11 | Highly expressed; resistant to ferroptosis and cuproptosis inducers | Shows sensitivity to disulfidptosis | [26,87,88] |
| Oral squamous cell carcinoma (OSCC) | PPARγ | Ligand-dependent nuclear transcription factor | PPARγ inhibition leads to SLC7A11 upregulation and promotes disulfidptosis | [91] |
| Uveal melanoma (UM) | FTO | RNA demethylase | FTO inhibitor MA restores m6A levels, upregulates SLC7A11, and triggers disulfidptosis | [92] |
3.2. Other Diseases
Beyond malignant tumor applications, disulfidptosis finds relevance in neurodegenerative diseases (Alzheimer’s disease, Parkinson’s disease); metabolic diseases (type 2 diabetes, metabolic dysfunction-associated steatotic liver disease); orthopedic diseases (intervertebral disc degeneration, osteoporosis, osteoarthritis, osteosarcoma); cardiovascular diseases (heart failure, dilated cardiomyopathy); and other conditions (psoriasis, ulcerative colitis, ischemic stroke). The author compiles key molecular pathways and primary effects for these diseases in a table for reader reference.
Table 3.
Role of Disulfidptosis in Non-Cancer Diseases.
| Disease Category | Specific Disease | Key Molecules/Pathways | Main Effects | References |
|---|---|---|---|---|
| Neurodegenerative Diseases | Alzheimer’s disease | SLC7A11, NADPH, actin dynamics | Regulates Aβ accumulation, tau phosphorylation, and oxidative stress | [12,103] |
| Parkinson’s disease | ACTB, ACTN4, INF2, MYL6 | Closely associated with PD; NAC can inhibit disulfidptosis | [12,104,105] | |
| Metabolic Diseases | Type 2 diabetes mellitus | CXCL6, CD48, C1QB, and COL6A3 | Disulfidptosis signaling pathways may regulate the expression of these genes | [106] |
| MASLD | SLC7A11 | Compensatory SLC7A11 overexpression + NADPH depletion → disulfidptosis exacerbates liver injury | [107,108] | |
| DRGs | Disrupts metabolic homeostasis and reshapes hepatic immune microenvironment | [107] | ||
| Orthopedic Diseases | Intervertebral disc degeneration | SLC7A11, GLUT1-4, NADPH | Glucose deprivation + SLC7A11 overexpression → disulfidptosis in nucleus pulposus cells | [109,110] |
| Osteoporosis | SLC7A11, TXNRD1, NFATc1 | NFATc1 activates SLC7A11 → enhances osteoclast sensitivity to TXNRD1 inhibitors → induces disulfidptosis | [111,112] | |
| PGRMC2 | Regulates monocyte-to-macrophage differentiation and modulates BM-MSC activity | [113] | ||
| Osteoarthritis | PDLIM1, ACTN4 | PDLIM1 overexpression → competitively binds to ACTN4 → F-actin collapse → disulfidptosis | [15,114] | |
| Osteosarcoma | ACTB | Reduces osteosarcoma cell viability | [115] | |
| MYH9 | Downregulation attenuates migration and invasion capacity of osteosarcoma cells | [116] | ||
| LRPPRC | High expression may exert immunosuppressive effects | [116] | ||
| Cardiovascular Diseases | Heart failure (HF) | DRGs (e.g., RPN) | Regulates IME in HF | [117] |
| Dilated cardiomyopathy | TLN1, TLN2 | Loss may lead to β-1 integrin reduction and cardiomyocyte membrane damage | [118] | |
| DSTN | Regulates interaction between cytoskeleton and nuclear structure | [119] | ||
| Inflammatory/Immune-Related Diseases | Psoriasis | SLC7A11, PPP pathway | Basal cells: glucose deprivation + abnormal SLC7A11 → NADPH depletion → disulfidptosis | [11,120] |
| Ulcerative colitis | DRGs, SLC7A11, SLC26A2 | DRGs affect immune infiltration; SLC7A11 and SLC26A2 synergistically regulate disulfidptosis | [11,121,122,123] | |
| Cerebrovascular Diseases | Ischemic stroke | SLC7A11, NADPH | Glucose deprivation + abnormal SLC7A11 → NADPH depletion → disulfidptosis | [124,125] |
| PRDX1 | Regulates disulfidptosis and ischemic postconditioning (IPostC) to provide neuroprotection against stroke | [126] |
Figure 4.
Integrated molecular network of disulfidptosis incorporating upstream signaling, core metabolic pathways, downstream execution machinery and pan-cancer specific targets.abbreviations. Green lines indicate activating regulation; red blocking lines represent inhibitory effects; red solid lines denote core disulfidptosis effector cascades. Nodes are categorized into upstream transcription factors, core metabolic hubs, execution-related effectors and cancer-specific target genes with indicated tumor origin abbreviations.
Figure 4.
Integrated molecular network of disulfidptosis incorporating upstream signaling, core metabolic pathways, downstream execution machinery and pan-cancer specific targets.abbreviations. Green lines indicate activating regulation; red blocking lines represent inhibitory effects; red solid lines denote core disulfidptosis effector cascades. Nodes are categorized into upstream transcription factors, core metabolic hubs, execution-related effectors and cancer-specific target genes with indicated tumor origin abbreviations.

4. Translational Challenges of Disulfidptosis-Targeted Therapy
4.1. Specificity and Safety Issues
A primary disulfidptosis-targeted therapy challenge is achieving selective pathological cell toxicity while protecting normal tissues. Many normal tissues (neurons, cardiomyocytes, renal tubular epithelial cells) exhibit high metabolic activity and off-target susceptibility [26,93]. For instance, glucose is widely required by almost all tissues, so effective therapeutic strategies must target high SLC7A11-expressing tumor cells. Global GLUT1 inhibition could affect normal tissues; therefore, targeted GLUT1-targeting drug delivery to cancer cells is crucial to avoid systemic metabolic toxicity. Nanodelivery systems, antibody-drug conjugates (ADCs), and conditionally activated drugs may address this [94,95,96]. These technologies achieve precise drug delivery by identifying tumor-specific biomarkers (e.g., CD24, FAP-α), minimizing normal tissue damage.
Furthermore, narrow therapeutic windows necessitate precise dosing and delivery strategies. Low-dose induction agents may cause incomplete cell death, with sublethal stress promoting persistent inflammation, cellular senescence, or oncogenic transformation [97]. Conversely, high-dose induction may cause metabolically active tissue acute toxicity, leading to severe adverse reactions. Therefore, identifying patient populations most likely to benefit and optimal effective doses based on biomarkers (SLC7A11 expression, glucose dependence, redox state) is crucial. Future GLP-compliant toxicological, pharmacokinetic, and safety pharmacology studies in relevant animal models will establish comprehensive preclinical safety evaluation systems. Subsequently, First-in-Human studies can progressively initiate in biomarker-screened patient populations. This precision medicine strategy enhances therapeutic efficacy and minimizes treatment-related toxic side effects, laying solid foundations for disulfidptosis-targeted therapy clinical translation.
4.2. Technical Bottlenecks in Delivery Systems
Many disulfidptosis modulators (e.g., SLC7A11 modulators) suffer from poor water solubility, short half-lives, and low bioavailability, limiting systemic administration effectiveness [12,98]. Nanodelivery systems have partially addressed these limitations by precisely delivering effector molecules to pathological sites, maintaining drug stability, and overcoming physiological barriers, effectively inducing disulfidptosis.
4.2.1. Progress in Nanoplatform Applications
Researchers have developed various innovative nanodelivery systems for inducing disulfidptosis. For instance, copper-based metal-organic framework nanoparticle CuSS@876-PEG triggers both cuproptosis and disulfidptosis through glutathione depletion and copper ion and glucose transporter inhibitor BAY-876 release, achieving synergistic dual death pathway activation [99]. This innovative design highlights nanoscale therapy immense potential in overcoming single-agent treatment limitations. Furthermore, combining nanotechnology with sonodynamic therapy to deliver GLUT1 inhibitors directly creates disulfidptosis trigger conditions in bladder cancer. This method effectively integrates with immunotherapy (PD-1 antibodies), establishing clear therapeutic paradigms [1]. The table below summarize reported nanoplatform applications in inducing disulfidptosis:
Table 4.
Disulfidptosis-Targeting Nanoplatforms for Cancer Therapy.
| Nanoplatform | Loaded Cargo | Targeting/Responsive Features | Disease Model | Main Effects | References |
|---|---|---|---|---|---|
| CYBC NPs | Cystine + GLUT1 inhibitor BAY-876 | --- | Triple-negative breast cancer | Selectively induces disulfidptosis and reverses immunosuppressive tumor microenvironment | [72] |
| CuSS@876-PEG | BAY-876 + Cu²⁺ | --- | Cancer (pan-cancer) | Synergistically activates disulfidptosis and cuproptosis, dual-pathway anticancer effect | [99] |
| Nanotechnology combined with sonodynamic therapy | GLUT1 inhibitor + cystine-containing polymer | Sonodynamic (ultrasound)-responsive release | Bladder cancer | Induces disulfidptosis and improves immunotherapy | [127] |
| FTO-targeting nanodrug | Meclofenamic acid | GSH-responsive release | Uveal melanoma | Upregulates SLC7A11 expression and induces disulfidptosis | [92] |
| Cys-hMnO₂@GOx@EM-CD24 | Cystine + glucose oxidase (GOx) | Active targeting via CD24 antibody | Neuroblastoma | Decreased NADPH levels + cystine overload, synergistically induces disulfidptosis | [94] |
4.2.2. Challenges and Future Directions
While nanodelivery systems demonstrate effectiveness in various disease models, they introduce new challenges including scaled-up production, stability, and regulatory approval issues. Moreover, specialized tissue barriers complicate delivery: blood-brain barrier restricts CNS penetration in neurodegenerative disease applications; dense tumor stroma and elevated interstitial pressure impede solid tumor nanoparticle penetration; skin and mucosal barriers hinder pathological tissue drug penetration. These factors significantly impact drug efficacy.
Intelligent responsive nanomedicine platform design should be further developed. These platforms should integrate multiple functions: (1) active targeting ligands for tissue selectivity and specialized barrier crossing; (2) stimulus-responsive release mechanisms (pH, ROS, enzymes, external stimuli like near-infrared light); (3) multi-drug co-delivery systems for combined effects; (4) real-time drug release and therapeutic response monitoring. Such multifunctionally integrated nanoplatforms will provide more precise and efficient disulfidptosis-targeted therapy delivery solutions, accelerating clinical translation.
4.3. Resistance Mechanisms and Combination Therapy Strategies
When one pathway is targeted for inhibition, cells often upregulate alternative survival mechanisms evading death [100]. From this perspective, disulfidptosis complex cross-regulatory and potential resistance mechanisms require further investigation. Cells may develop disulfidptosis resistance through: upregulating alternative redox buffering systems (e.g., enhancing Trx system activity when GSH system is inhibited); increasing NADPH production to maintain redox balance; altering metabolic patterns to reduce glucose and cystine dependence; or activating stress response pathways enhancing cell survival.
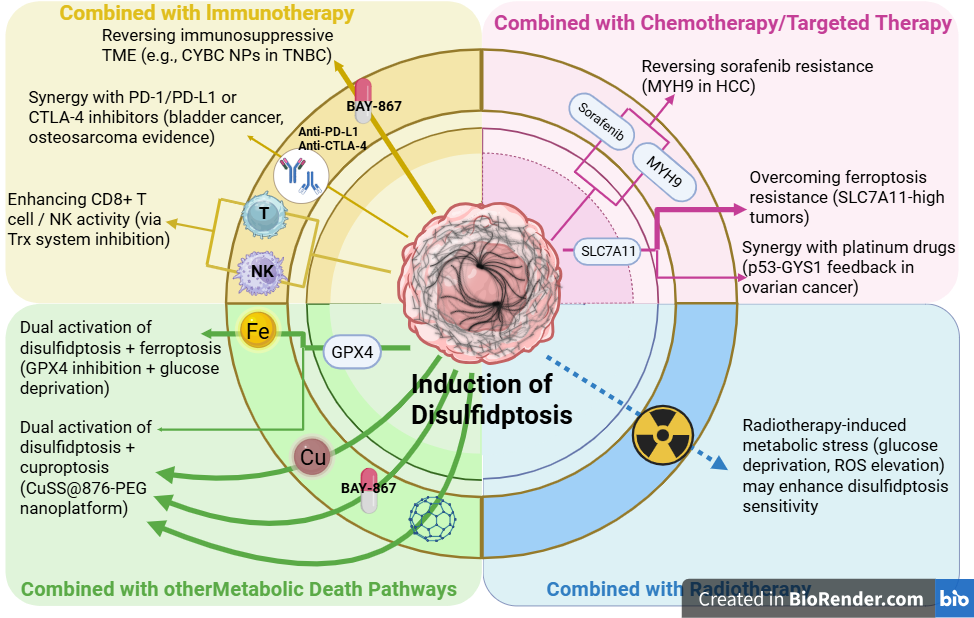
Studies show combining disulfidptosis with other cell death forms (ferroptosis, cuproptosis), radiochemotherapy strategies, or immunotherapy may improve therapeutic outcomes. This multiple death pathway combination strategy effectively overcomes single-targeted therapy limitations by simultaneously blocking multiple survival pathways, reducing resistance development. For instance, combined SLC7A11 and GPX4 inhibition simultaneously induces disulfidptosis and ferroptosis; combining disulfidptosis inducers with copper chelators triggers synergistic cuproptosis and disulfidptosis effects. Furthermore, disulfidptosis induction enhances immune checkpoint inhibitor efficacy, as disulfidptosis-triggered immunogenic cell death activates anti-tumor immune responses.
Refining these cross-regulatory mechanisms and designing rational multi-pathway combination therapy strategies will significantly impact disulfidptosis disease treatment applications. Comprehensive disulfidptosis regulatory network analysis using systems biology approaches will identify key compensatory mechanisms and resistance nodes, enabling targeted combination therapy regimen design. Concurrently, establishing predictive biomarkers will screen patient populations most likely to benefit from combination therapy, achieving precise individualized treatment.
4.4. Lack of Clinical Translation
To date, no drugs directly targeting disulfidptosis have entered clinical trials, contrasting sharply with rapid fundamental research progress. This gap has multifaceted reasons. Firstly, at biomarker level, despite SLC7A11 and GLUT1 confirmation as closely related to disulfidptosis occurrence, systematic, accurate, dynamically traceable, clinically validated biomarker systems are lacking. This hinders effective patient stratification, efficacy monitoring, and resistance early warning. Single biomarker sensitivity and specificity are insufficient for precise clinical diagnosis demands, and disulfidptosis dynamic and heterogeneous nature further complicates accurate clinical sample identification. Secondly, at efficacy evaluation level, existing research lacks unified disulfidptosis extent quantification methods and recognized functional readout indicators, making reliable individual patient-level drug intervention effect assessment difficult.
To address these bottlenecks, future work should integrate multi-omics technologies (single-cell transcriptomics, spatial metabolomics, proteomics). This will systematically identify unique disulfidptosis molecular characteristics in different disease contexts and construct clinically practical patient stratification predictive models. Concurrently, at disease model level, most preclinical studies rely on standard cell lines or simple animal models struggling to replicate inherent human disease genetic heterogeneity and microenvironmental complexity. Future work should systematically introduce engineered mouse models with specific genetic modifications and patient-derived xenograft (PDX) models to more realistically simulate clinical scenarios, providing experimental platforms with greater drug development translational value. Only by establishing complete chains from mechanistic discovery to clinical validation can disulfidptosis therapeutic potential truly translate into patient benefit.
4.5. Expansion into Emerging Disease Fields
Beyond cancer and aforementioned diseases, disulfidptosis holds noteworthy potential relevance in several emerging fields. In infectious diseases, pathogen infection profoundly reshapes host cell metabolic states. Certain viral or bacterial infections may induce high SLC7A11 expression and NADPH depletion, rendering infected cells metabolically vulnerable, providing theoretical basis for selectively clearing infected cells using disulfidptosis. In rare genetic diseases, cystinosis is characterized by pathological cystine accumulation within lysosomes. This condition shares natural mechanistic intersections with disulfidptosis at substrate accumulation level, and deeper understanding of their connection could broaden intervention strategies for such diseases. In aging, age-accumulated oxidative stress progressively weakens cellular NADPH production capacity and GSH reduction reserves, increasing aging cell susceptibility to disulfidptosis. This may partly explain age-related cellular functional decline and suggests modulating disulfidptosis threshold could become new anti-aging intervention dimensions.
5. Conclusion
Disulfidptosis, a novel cell death form driven by SLC7A11 overactivation, NADPH depletion, and abnormal protein disulfide bond accumulation, has demonstrated significant pathophysiological importance and therapeutic intervention value across cancer, neurodegenerative diseases, metabolic diseases, and orthopedic diseases since its conceptualization. Its core mechanistic advantage lies in transforming cellular metabolic vulnerability—especially high SLC7A11 tumor cell absolute glucose dependence—into precisely exploitable therapeutic windows, theoretically enabling selective specific cell subpopulation killing without affecting normal metabolic phenotype healthy cells.
However, translating this mechanistic potential into clinical reality requires overcoming multiple systemic challenges. At mechanistic level, cross-talk between disulfidptosis and other cell death programs (ferroptosis, apoptosis, autophagy) has not been fully elucidated, and precise boundaries of specific triggering conditions remain undefined. At technical level, targeted delivery system development, intervention strategies for “undruggable” targets like cytoskeletal proteins, and dynamic biomarker system establishment suitable for in vivo detection constitute urgent technical bottlenecks. At clinical translation level, pathways from in vitro discovery to patient benefit still require rigorous model validation and safety assessment.
Looking ahead, with in-depth single-cell multi-omics technology application, gradual AI-assisted target discovery and patient stratification model maturation, and continuous interdisciplinary collaboration deepening, disulfidptosis-centric targeted therapeutic strategies are expected to move from concept to practice. Particularly, combined applications with immune checkpoint blockade, nanodrug delivery, and metabolic reprogramming interventions may generate new-generation multimodal therapeutic solutions that are specific, effective, and safe, offering viable pathways to address unmet clinical needs current therapies struggle to satisfy.
Author Contributions
Ting Wangprepared the figures and the manuscript, including searching the literature, wring original draft and editing. Sihan Liu revised the details of this review. Dehuan Zhang contributed to study supervision, conceptual framework design, and data visualization, edited the manuscript. All authors have read and approved the final manuscript.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Conflicts of interest
The authors have no conflicts of interest to report.
Acknowledgments
The figures were created with BioRender.com,Python3.14 and Adobe Illustrator 2020. AI was used for language polishing of this manuscript, while all content was originally created and reviewed by the authors.
Ethics approval
Not applicable.
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25(3), 486–541. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012, 149(5), 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Liu, Z.Y.; Wang, M.S.; et al. Mechanisms and regulations of ferroptosis. Front Immunol. 2023, 14, 1269451. [Google Scholar] [CrossRef] [PubMed]
- Long, D.; Mao, C.; Huang, Y.; Xu, Y.; Zhu, Y. Ferroptosis in ulcerative colitis: Potential mechanisms and promising therapeutic targets. BioMed Pharmacother. 2024, 175, 116722. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Chen, X.; Linkermann, A.; et al. A guideline on the molecular ecosystem regulating ferroptosis. Nat. Cell Biol. 2024, 26(9), 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Amino acid metabolism in breast cancer: pathogenic drivers and therapeutic opportunities - PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/39973065/ (accessed on 2 June 2026).
- Very, N.; Staels, B.; Eeckhoute, J. Increased O-GlcNAcylation connects metabolic to transcriptional reprogramming during pathophysiological cell activation. Trends Cell Biol. 2024, 34(12), 988–991. [Google Scholar] [CrossRef] [PubMed]
- Chen Y, Jin GW, He LH, et al. MTFP1 drives pancreatic cancer liver metastatic colonisation by regulating mitochondrial metabolism reprogramming. Gut. Published online February 20, 2026:gutjnl-2025-336323. [CrossRef]
- Chen, Y.; Yang, M.; Huang, W.; et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ. Res. 2019, 125(12), 1087–1102. [Google Scholar] [CrossRef] [PubMed]
- Mercado-Gómez, M.; Goikoetxea-Usandizaga, N.; Giné, A.E.; et al. Role of CNNM4 in the progression of cholangiocarcinoma: implications for ferroptosis and therapeutic potential. Gut 2026, 75(2), 341–352. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhuang, L.; Gan, B. Disulfidptosis: disulfide stress-induced cell death. Trends Cell Biol. 2024, 34(4), 327–337. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Pan, L.; Chen, G.; et al. Disulfidptosis: A novel cell death mechanism with pathological significance and therapeutic potential in diseases. Pharmacol. Rev. 2026, 78(3), 100127. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Fan, K.; et al. Inhibition of Endoplasmic Reticulum Stress Cooperates with SLC7A11 to Promote Disulfidptosis and Suppress Tumor Growth upon Glucose Limitation. Adv. Sci. (Weinh) 2025, 12(7), e2408789. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Wang, M.; Zhuang, L.; Gan, B. Metabolic cell death in cancer: ferroptosis, cuproptosis, disulfidptosis, and beyond. Protein Cell. 2024, 15(9), 642–660. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nie, L.; Zhang, Y.; et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 2023, 25(3), 404–414. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ma, B.; Yang, Y.; Wang, B.; Hao, J.; Zhou, X. Disulfidptosis decoded: a journey through cell death mysteries, regulatory networks, disease paradigms and future directions. Biomark. Res. 2024, 12(1), 45. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; An, Y.; Xu, M.; et al. Disulfidptosis, A Novel Cell Death Pathway: Molecular Landscape and Therapeutic Implications. Aging Dis. 2024, 16(2), 917–945. [Google Scholar] [CrossRef] [PubMed]
- Ru, Q.; Li, Y.; Chen, L.; Wu, Y.; Min, J.; Wang, F. Iron homeostasis and ferroptosis in human diseases: mechanisms and therapeutic prospects. Signal Transduct. Target Ther. 2024, 9(1), 271. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.K.M.; Delaidelli, A.; Minaker, S.W.; et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA 2019, 116(19), 9433–9442. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Wang, G.; Dai, Q.; et al. Targeting novel regulated cell death: disulfidptosis in cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2025, 13(1), 35. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell. 2018, 34(1), 21–43. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Liu, Y.; Wang, L.; Gu, W. Regulation of SLC7A11 as an unconventional checkpoint in tumorigenesis through ferroptosis. Genes Dis. 2025, 12(1), 101254. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Ling, H.; Huang, Y.; et al. AZD1775 synergizes with SLC7A11 inhibition to promote ferroptosis. Sci. China Life Sci. 2025, 68(1), 204–218. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Munoz-Pinto, M.F.; Nunes-Costa, D.; et al. Footprints of a microbial toxin from the gut microbiome to mesencephalic mitochondria. Gut 2023, 72(1), 73–89. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Ralser, M.; Fan, J.; Rabinowitz, J.D. The pentose phosphate pathway in health and disease. Nat. Metab. 2023, 5(8), 1275–1289. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Jiang, D.; Koong, A.C.; Gan, B. Exploiting metabolic cell death for cancer therapy. Nat. Rev. Cancer 2026, 26(1), 27–45. [Google Scholar] [CrossRef] [PubMed]
- Loopmans, S.; Rohlenova, K.; van Brussel, T.; et al. The pentose phosphate pathway controls oxidative protein folding and prevents ferroptosis in chondrocytes. Nat. Metab. 2025, 7(1), 182–195. [Google Scholar] [CrossRef] [PubMed]
- Mi, T.; Kong, X.; Chen, M.; Guo, P.; He, D. Inducing disulfidptosis in tumors:potential pathways and significance. MedComm (2020) 2024, 5(11), e791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, H.; Zhu, X.; et al. Synchronously Evoking Disulfidptosis and Ferroptosis via Systematical Glucose Deprivation Targeting SLC7A11/GSH/GPX4 Antioxidant Axis. ACS Nano 2025, 19(14), 14233–14248. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Teng, H.; Hang, Q.; et al. SLC7A11 expression level dictates differential responses to oxidative stress in cancer cells. Nat. Commun. 2023, 14(1), 3673. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nie, L.; Zhang, Y.; et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 2023, 25(3), 404–414. [Google Scholar] [CrossRef] [PubMed]
- Tossounian, M.A.; Zhao, Y.; Yu, B.Y.K.; et al. Low-molecular-weight thiol transferases in redox regulation and antioxidant defence. Redox Biol. 2024, 71, 103094. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. Oxidative damage in neurodegeneration: roles in the pathogenesis and progression of Alzheimer disease. Physiol. Rev. 2024, 104(1), 103–197. [Google Scholar] [CrossRef] [PubMed]
- Coppo, L.; Mishra, P.; Siefert, N.; Holmgren, A.; Pääbo, S.; Zeberg, H. A substitution in the glutathione reductase lowers electron leakage and inflammation in modern humans. Sci. Adv. 2022, 8(1), eabm1148. [Google Scholar] [CrossRef] [PubMed]
- Corteselli, E.M.; Sharafi, M.; Hondal, R.; et al. Structural and functional fine mapping of cysteines in mammalian glutaredoxin reveal their differential oxidation susceptibility. Nat. Commun. 2023, 14(1), 4550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Zhao, Z.; Cai, W.; Fang, J. Thioredoxin Signaling Pathways in Cancer. Antioxid. Redox Signal. 2023, 38(4-6), 403–424. [Google Scholar] [CrossRef] [PubMed]
- Lühle, J.; Seeberger, P.H.; Moscovitz, O. Cell surface thiol-disulfide regulation in cancer: Mechanisms, implications, and theranostic strategies. Redox Biol. 2026, 92, 104142. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Dirks, K.; Kim, S.Y.; et al. Inhibition of thioredoxin reductase 1 sensitizes glucose-starved glioblastoma cells to disulfidptosis. Cell Death Differ. 2025, 32(4), 598–612. [Google Scholar] [CrossRef] [PubMed]
- Rouyère, C.; Serrano, T.; Frémont, S.; Echard, A. Oxidation and reduction of actin: Origin, impact in vitro and functional consequences in vivo. Eur. J. Cell Biol. 2022, 101(3), 151249. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, S.; Wu, Y.; et al. Molecular signatures of disulfidptosis: interplay with programmed cell death pathways and therapeutic implications in oncology. Cell Mol. Biol. Lett. 2025, 30(1), 66. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nie, L.; Zhang, Y.; et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell Biol. 2023, 25(3), 404–414. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Yang, S.; Schaks, M.; et al. Structures reveal a key mechanism of WAVE regulatory complex activation by Rac1 GTPase. Nat. Commun. 2022, 13(1), 5444. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Asp. Med. 2009, 30(1-2), 86–98. [Google Scholar] [CrossRef] [PubMed]
- Gasmi, A.; Nasreen, A.; Lenchyk, L.; et al. An Update on Glutathione’s Biosynthesis, Metabolism, Functions, and Medicinal Purposes. Curr. Med. Chem. 2024, 31(29), 4579–4601. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, M.N.; Pileggi, C.A.; Karam, C.Y.; et al. Cystine/glutamate antiporter xCT controls skeletal muscle glutathione redox, bioenergetics and differentiation. Redox Biol. 2024, 73, 103213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, Y.; He, W.; et al. Deciphering the rules of disulfidptosis: a genome-wide signature for identifying disulfidptosis-related genes and analyzing hepatocellular carcinoma chemotherapy sensitivities. Free Radic. Biol. Med. 2025, 241, 914–932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, Y.; Guo, D. Application and potential of the tumor microenvironment, ferroptosis, cuproptosis, and disulfidptosis in cancer treatment and monitoring. Crit. Rev. Oncol. Hematol. 2026, 218, 105066. [Google Scholar] [CrossRef] [PubMed]
- Shuai, Y.; Ma, Z.; Yuan, P. Disulfidptosis: disulfide stress-induced novel cell death pathway. MedComm (2020) 2024, 5(7), e579. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Kang, R.; Tang, D.; Liu, J. Ferroptosis: principles and significance in health and disease. J. Hematol. Oncol. 2024, 17(1), 41. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Yang, M.; Liu, S.B. Research progress on morphology and mechanism of programmed cell death. Cell Death Dis. 2024, 15(5), 327. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.; Green, M.D.; Wang, W.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9(12), 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375(6586), 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Srivastava, Y.; Prasad, S.; et al. SLC7A11 as a molecular nexus of prognosis, resistance, and therapeutic roadmap in cancer: A systematic review and meta-analysis. Int. J. Biol. Macromol. 2026, 335 Pt 1, 149257. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Li, Y.; Xu, Y.; et al. SLC7A11 protects luminal A breast cancer cells against ferroptosis induced by CDK4/6 inhibitors. Redox Biol. 2024, 76, 103304. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xiao, K.; Liu, Z.; et al. Unveiling disulfidptosis-related genes in HBV-associated hepatocellular carcinoma: an integrated study incorporating transcriptome and Mendelian randomization analyses. J. Cancer 2024, 15(17). [Google Scholar] [CrossRef]
- Zhang, K.; Zhu, Z.; Zhou, J.; et al. Disulfidptosis-related gene expression reflects the prognosis of drug-resistant cancer patients and inhibition of MYH9 reverses sorafenib resistance. Transl. Oncol. 2024, 49, 102091. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guo, K.; Zhang, D.; et al. Disulfidptosis classification of hepatocellular carcinoma reveals correlation with clinical prognosis and immune profile. Int. Immunopharmacol. 2023, 120, 110368. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Fang, Z.; Shi, M.; et al. Clinical Significance of Disulfidptosis-related Genes and Functional Analysis in Gastric Cancer. J. Cancer 2024, 15(4), 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, T.; Sun, J.; et al. Integrated analysis of disulfidptosis-related immune genes signature to boost the efficacy of prognostic prediction in gastric cancer. Cancer Cell Int. 2024, 24(1), 112. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yin, L.; Li, M.; et al. Disulfidptosis-related LncRNA signatures in gastric cancer: regulation of MYH10-driven cytoskeletal remodeling and therapeutic implications. Discov. Oncol. 2025, 16(1), 1385. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Huang, R.; Lv, L.; et al. FLNA, a disulfidptosis-related gene, modulates tumor immunity and progression in colorectal cancer. Cell Mol. Biol. Lett. 2025, 30(1), 92. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, X.; Liu, J.; et al. DPP7 Promotes Colorectal Cancer Progression Through GPX4-Dependent Suppression of Disulfidptosis and Immune Evasion. J. Cell Mol. Med. 2025, 29(12), e70660. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Sun, Y.; Yao, H.; et al. TMEM105 modulates disulfidptosis and tumor growth in pancreatic cancer via the β-catenin-c-MYC-GLUT1 axis. Int. J. Biol. Sci. 2025, 21(5), 1932–1948. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wu, B.; Han, T.; et al. Identification of the disulfidptosis-related gene G6PD as a potential biomarker and therapeutic target in pancreatic ductal adenocarcinoma by integrative bioinformatics analysis and experimental validation. Discov. Oncol. 2026, 17(1), 556. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.F.; Ge, J.; Chen, J.; et al. CASC8 activates the pentose phosphate pathway to inhibit disulfidptosis in pancreatic ductal adenocarcinoma though the c-Myc-GLUT1 axis. J. Exp. Clin. Cancer Res. 2025, 44(1), 26. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ou, Y.H.; Shi, W. The effect of disulfidptosis induced by glucose starvation in SLC7A11high ovarian cancer cells. Front Cell Dev. Biol. 2025, 13, 1608150. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Yao, Q.; Huang, L.; et al. Glucose Deprivation-Induced Disulfidptosis via the SLC7A11-INF2 Axis: Pan-Cancer Prognostic Exploration and Therapeutic Validation. Adv. Sci. (Weinh) 2025, 12(39), e08556. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.Y.; Luo, R.Z.; Deng, R.; et al. Glycogen stores mediated by the p53-GYS1 feedback circuit engenders platinum resistance in ovarian clear cell carcinoma. Cell Death Differ. 2025, 32(9), 1707–1721. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.; Sharma, P.; Sundaram, S.; Venkatraman, G.; Bera, A.K.; Karunagaran, D. SLC7A11/ xCT is a target of miR-5096 and its restoration partially rescues miR-5096-mediated ferroptosis and anti-tumor effects in human breast cancer cells. Cancer Lett. 2021, 522, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Nath, P.; Alfarsi, L.H.; El-Ansari, R.; et al. The amino acid transporter SLC7A11 expression in breast cancer. Cancer Biol. Ther. 2024, 25(1), 2291855. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Zhou, C.; Ding, Y.; Duan, S. Disulfidptosis: a new target for metabolic cancer therapy. J. Exp. Clin. Cancer Res. 2023, 42(1), 103. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ming, X.; Qi, J.; et al. Disulfidptosis Nanoinducer Interrupts Tumor Metabolic Privilege to Boost Sustained Immunotherapy. ACS Nano 2025, 19(33), 30303–30321. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Deng, X.; Xie, Y.; et al. Multi-omics analysis of disulfidptosis regulators and therapeutic potential reveals glycogen synthase 1 as a disulfidptosis triggering target for triple-negative breast cancer. MedComm (2020) 2024, 5(3), e502. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.; Jiang, S.; Xu, Z.; Luo, M.; Yan, C. Disulfidptosis heterogeneity in breast cancer uncovers PTTG1IP as an actionable therapeutic target. Genes Dis. 2025, 12(1), 101257. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Olszewski, K.; Zhang, Y.; et al. KEAP1 deficiency drives glucose dependency and sensitizes lung cancer cells and tumors to GLUT inhibition. iScience 2021, 24(6), 102649. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell. 2018, 34(1), 21–43. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, Q.; Yang, S.; Guo, D.; Tao, Y.; Jia, Y. FGA modulates immune infiltration and tumor progression via SLC7A11/xCT-mediated disulfidptosis in the tumor microenvironment of lung adenocarcinoma. Front Immunol. 2025, 16, 1595900. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; He, X.; Wang, S.; et al. Integrating single-cell sequencing and machine learning to uncover the role of mitophagy in subtyping and prognosis of esophageal cancer. Apoptosis 2025, 30(3-4), 1021–1041. [Google Scholar] [CrossRef] [PubMed]
- Robelin, P.; Hadoux, J.; Forestier, J.; et al. Characterization, Prognosis, and Treatment of Patients With Metastatic Lung Carcinoid Tumors. J. Thorac. Oncol. 2019, 14(6), 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Huang, D.; Wang, L.; Liang, H.; Ma, X. Silencing ZIC5 suppresses glycolysis and promotes disulfidptosis in lung adenocarcinoma cells. Cancer Biol. Ther. 2025, 26(1), 2501780. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wang, Q.; Zhou, J.; et al. Disulfidptosis-Associated CCNB2: A Prognostic Biomarker and Immune Microenvironment Modulator in Prostate Cancer. J. Cancer 2025, 16(13), 3928–3941. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Lin, B.; Ke, Z.B.; et al. Super-enhancers mediates SLC7A11 via FOXA1 to regulate disulfidptosis in prostate cancer. Cell Death Dis. 2025, 17(1), 63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zheng, D.; Zhu, S.; et al. Leveraging a disulfidptosis-based signature to characterize heterogeneity and optimize treatment in multiple myeloma. Front Immunol. 2025, 16, 1559317. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jin, S.; Zhang, Z.; Ma, H.; Yang, X. Interleukin-6 facilitates tumor progression by inducing ferroptosis resistance in head and neck squamous cell carcinoma. Cancer Lett. 2022, 527, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Savic, D.; Steinbichler, T.B.; Ingruber, J.; et al. Erk1/2-Dependent HNSCC Cell Susceptibility to Erastin-Induced Ferroptosis. Cells 2023, 12(2), 336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhou, J.; Xiong, Y.; et al. Sulfasalazine combined with anti-IL-1β mAb induces ferroptosis and immune modulation in oral squamous cell carcinoma. Cell Mol. Life Sci. 2025, 82(1), 216. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, X.; Huang, X.; et al. Metabolic cell deaths in head and neck cancer: mechanisms, therapeutic potential, and challenges. Ann. Med. 2025, 57(1), 2582906. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Wang, Z.; Zhou, C.; Wen, M.; Zhou, Y.; Huang, S. Based on disulfide accumulation, leveraging a THBS1-signature to improve the outcome of head and neck carcinoma patients. Int. J. Biol. Macromol. 2025, 328 Pt 2, 147679. [Google Scholar] [CrossRef] [PubMed]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. 2021, 184(21), 5309–5337. [Google Scholar] [CrossRef] [PubMed]
- Daniel, Y.; Rauch, C.; Moutaux, L.; et al. PPARγ, a key modulator of metabolic reprogramming, stemness and chemoresistance associated with retrodifferentiation in human hepatocellular carcinomas. Cell Death Dis. 2025, 16(1), 831. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Gu, J.; et al. PPARγ Antagonists Exhibit Antitumor Effects by Regulating Ferroptosis and Disulfidptosis. Biomolecules 2024, 14(5), 596. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Deng, H.; Liu, X.; et al. A novel FTO-targeting nanodrug induces disulfidptosis and ameliorates the suppressive tumor immune environment to treat uveal melanoma. Biomaterials 2025, 319, 123168. [Google Scholar] [CrossRef] [PubMed]
- van der Rijt, S.; Leemans, J.C.; Florquin, S.; Houtkooper, R.H.; Tammaro, A. Immunometabolic rewiring of tubular epithelial cells in kidney disease. Nat. Rev. Nephrol. 2022, 18(9), 588–603. [Google Scholar] [CrossRef] [PubMed]
- Mi, T.; Liu, J.; Luo, J.; et al. CD24-targeted cystine and glucose oxidase cascade catalytic nanosystem triggers disulfidptosis in neuroblastoma. Mater. Today Bio 2025, 35, 102496. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ming, X.; Qi, J.; et al. Disulfidptosis Nanoinducer Interrupts Tumor Metabolic Privilege to Boost Sustained Immunotherapy. ACS Nano 2025, 19(33), 30303–30321. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Huang, Z.; Luo, Y.; et al. A fibroblast activation protein α-activatable nanoagent co-delivering diethyldithiocarbamate and copper for tumor therapy and imaging. Acta Biomater. 2024, 187, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Oberst, A. Cellular memory of sub-lethal stress. Immunity 2026, 59(4), 801–812. [Google Scholar] [CrossRef] [PubMed]
- Pardieu, B.; Pasanisi, J.; Ling, F.; et al. Cystine uptake inhibition potentiates front-line therapies in acute myeloid leukemia. Leukemia 2022, 36(6), 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Gao, M.; Li, Q.; et al. Copper metal-organic framework-based multifaceted strategy for boosting cancer therapy via synergistic cuproptosis and disulfidptosis. Biomaterials 2026, 325, 123592. [Google Scholar] [CrossRef] [PubMed]
- Labrie, M.; Brugge, J.S.; Mills, G.B.; Zervantonakis, I.K. Therapy resistance: opportunities created by adaptive responses to targeted therapies in cancer. Nat. Rev. Cancer 2022, 22(6), 323–339. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.U.; Kim, T.H. Current evidence and the potential role of proton beam therapy for hepatocellular carcinoma. Clin. Mol. Hepatol. 2023, 29(4), 958–968. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hu, F.; Zhang, H.; et al. Ultrasound-Triggered Nanoparticles Induce Cuproptosis for Enhancing Immunogenic Sonodynamic Therapy. Adv. Mater. 2025, 37(29), e2504228. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zheng, N.; Chen, Z.; et al. The emerging role of disulfidptosis in Alzheimer’s disease. Eur. J. Pharmacol. 2025, 1005, 178085. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Kong, X.; Wei, J. Disulfidptosis: A New Target for Parkinson’s Disease and Cancer. Curr. Issues Mol. Biol. 2024, 46(9), 10038–10064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, T.; Wu, H.; Wei, J.; Qu, Q. Target oxidative stress-induced disulfidptosis: novel therapeutic avenues in Parkinson’s disease. Mol. Brain 2025, 18(1), 29. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Zou, Y.; Jin, Y.; et al. Mechanistic Role of Disulfidptosis in Type 2 Diabetes Mellitus. Mol. Nutr. Food Res. 2026, 70(4), e70424. [Google Scholar] [CrossRef] [PubMed]
- Wen, R.; Wang, H.; Wang, S. The roles and mechanisms of sulfur metabolism and disulfidptosis in metabolic dysfunction-associated steatotic liver disease. Biochem Pharmacol. 2026, 249, 117892. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Xie, E.; Shen, S.; et al. Essentiality of SLC7A11-mediated nonessential amino acids in MASLD. Sci. Bull. 2024, 69(23), 3700–3716. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, J.; Wang, M.; et al. Glucose deprivation-induced disulfidptosis in human nucleus pulposus cells: a novel pathological mechanism of intervertebral disc degeneration. Biol. Direct 2024, 19(1), 81. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Han, C.; Mo, S.; Huang, H.; Luo, X. Advancements in programmed cell death research in antitumor therapy: a comprehensive overview. Apoptosis 2025, 30(1-2), 401–421. [Google Scholar] [CrossRef] [PubMed]
- NFATc1-mediated expression of SLC7A11 drives sensitivity to TXNRD1 inhibitors in osteoclast precursors - PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/37148740/ (accessed on 3 June 2026).
- Zhong, Z.; Zhang, C.; Ni, S.; et al. NFATc1-mediated expression of SLC7A11 drives sensitivity to TXNRD1 inhibitors in osteoclast precursors. Redox Biol. 2023, 63, 102711. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, H.; Chen, Y.; et al. PGRMC2 influences the onset of postmenopausal osteoporosis through disulfidptosis in monocytes: Evidence from experimental validation and Mendelian randomization. Heliyon 2024, 10(17), e36570. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhou, J.K.; Wang, K.; et al. PDLIM1 Inhibits Tumor Metastasis Through Activating Hippo Signaling in Hepatocellular Carcinoma. Hepatology 2020, 71(5), 1643–1659. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Tai, J.; et al. Transcriptome and single-cell analysis reveal disulfidptosis-related modification patterns of tumor microenvironment and prognosis in osteosarcoma. Sci. Rep. 2024, 14(1), 9186. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Shen, J. A Disulfidptosis-Related Gene Signature Associated with Prognosis and Immune Cell Infiltration in Osteosarcoma. Bioengineering 2023, 10(10), 1121. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, J.; Song, Q.; Dai, C.; Qin, Y.; Li, A. Comprehensive analysis of disulfidptosis-related genes and the immune microenvironment in heart failure. Front Cell Dev. Biol. 2024, 12, 1516898. [Google Scholar] [CrossRef] [PubMed]
- Manso, A.M.; Okada, H.; Sakamoto, F.M.; et al. Loss of mouse cardiomyocyte talin-1 and talin-2 leads to β-1 integrin reduction, costameric instability, and dilated cardiomyopathy. Proc. Natl. Acad. Sci. U S A 2017, 114(30), E6250–E6259. [Google Scholar] [CrossRef] [PubMed]
- Stanczyk, P.J.; Tatekoshi, Y.; Shapiro, J.S.; et al. DNA Damage and Nuclear Morphological Changes in Cardiac Hypertrophy Are Mediated by SNRK Through Actin Depolymerization. Circulation 2023, 148(20), 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Jiang, J.; Li, Z.; et al. Energy competition remodels the metabolic glucose landscape of psoriatic epidermal cells. Theranostics 2024, 14(8), 3339–3357. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zhang, F.; Bai, X.; Liang, H.; Niu, J.; Miao, Y. Comprehensive analysis of disulfidptosis-related genes reveals the effect of disulfidptosis in ulcerative colitis. Sci. Rep. 2024, 14(1), 15705. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Yue, T.; Jia, Z.; et al. Disulfidptosis links the pathophysiology of ulcerative colitis and immune infiltration in colon adenocarcinoma. Sci. Rep. 2025, 15(1), 5365. [Google Scholar] [CrossRef] [PubMed]
- Mertz, E.L.; Facchini, M.; Pham, A.T.; et al. Matrix disruptions, growth, and degradation of cartilage with impaired sulfation. J. Biol. Chem. 2012, 287(26), 22030–22042. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.P.; Liu, C.; Xu, B.; Zhou, H.; Zhao, H. Disulfidptosis and its Role in Peripheral Blood Immune Cells after a Stroke: A New Frontier in Stroke Pathogenesis. Curr. Neurovasc Res. 2024, 20(5), 608–622. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Singh, T.G.; Mannan, A. Therapeutic implications of glucose transporters (GLUT) in cerebral ischemia. Neurochem Res. 2022, 47(8), 2173–2186. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, Q.; Xu, C.; et al. Ischemic Postconditioning Regulates New Cell Death Mechanisms in Stroke: Disulfidptosis. Biomolecules 2024, 14(11), 1390. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, L.; Liang, G.; Xiao, H.; Zhang, L.; Liu, T. Sonodynamic activated nanoparticles with Glut1 inhibitor and cystine-containing polymer stimulate disulfidptosis for improved immunotherapy in bladder cancer. Biomaterials 2025, 319, 123178. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



