Submitted:
11 June 2026
Posted:
12 June 2026
You are already at the latest version
Abstract
Personalized oncology seeks to selectively block specific dysregulated pathways to ar-rest cancer development. Increased metabolism of glutamine is a hallmark of cancer, and 6-diazo-5-oxo-L-norleucine (DON), a structural analog of L-glutamine, was the first compound aimed to target the exacerbated nitrogen metabolism observed in can-cer cells. However, it was abandoned due to unacceptable side toxicity. With the same goal of blocking glutamine metabolism, several specific glutaminase inhibitors have been characterized in recent decades, showing promising results. Nevertheless, this strategy frequently induces adaptive metabolic resistance that must be counteracted. In this context, glutaminase has become a key target in combination therapies for sev-eral tumor types aimed at restricting anabolic adaptation when single metabolic ther-apy fails, emerging as a possible synergistic therapeutic intervention. Consequently, combination therapies that include glutaminase inhibition alongside additional drug/s to counteract the metabolic plasticity of cancer have become essential in antitumor personalized pharmacology. Recent findings suggest that novel prodrugs targeting glutamine metabolism can potentiate immune checkpoint inhibitors by reshaping the tumor microenvironment, thereby enhancing cancer immunotherapy while reducing side effects and increasing therapeutic efficacy in certain types of cancer.
Keywords:
DON
; BPTES
; CB‐839
; combination therapy
; DRP‐104
; glutaminase
; synergistic effects
1. Introduction
After glucose, glutamine (Gln) is the second most avidly consumed molecule by cancer cells, with most tumors showing increased Gln metabolism. Thus, approaches aimed at blocking nitrogen metabolism arose as essential antitumor strategies against cancerous growth and proliferation (Altman et al., 2016; Yang et al., 2017). In 1956, the discovery and biological studies of 6-diazo-5-oxo-L-norleucine (DON) were published for the very first time [3], and shortly after its isolation and characterization were also reported [4], all of which proved DON to be a new useful drug in anticancer therapy. In 1957, the first clinical assay using DON as monotherapy for patients with serious malignancies (mainly lung and breast cancers) was published, but very significant side effects were observed, including ulceration of the tongue, mouth, and lips, diarrhea, nausea, and vomiting [5]. DON was later identified as a structural analog of Gln and its chemotherapeutic usefulness was fully established in the 1970s [6], with later experiments proving the synergistic effects of combining Gln depletion strategies and DON (and another Gln analog, acivicin, chemically (2S)-2-amino-2-[(5S)-3-chloro-4,5-dihydro-1,2-oxazol-5-yl]acetic acid) as powerful molecules inhibiting glutaminase (GA) and cancer growth [7]. In the 1980s, several studies reported promising findings in both in vitro and in vivo models, but subsequent clinical trials produced unpromising results due to low specificity (inhibition of other enzymes whose substrate is Gln, amino acid transporters, and transglutaminase), likely leading to the observed generalized toxicity [8]. Nowadays, less toxic agents have been described and characterized as valuable drugs to inhibit GA [9,10,11], including natural products that have shown its interaction by molecular dynamics simulations but still need to be tested in more in-depth studies [12], as well as derivatives of a natural molecule, withangulatin A, that have already shown antitumoral properties in vitro and in mouse xenografts in the highly Gln-dependent triple-negative breast cancer (TNBC) subtype [13]. However, this review will be focused on fully characterized GA inhibitors, including compound 968, BPTES, and CB-839 (telaglenastat), as well as novel prodrugs that release the inhibitor at the tumor site with maximum efficacy while minimizing toxicity. On the other hand, we emphasize the importance of GA in the metabolic network of tumor cells and its crosstalk with other key molecular pathways, as well as its essential role in the regulatory signals that govern cancer metabolism and modulate the energetic and biosynthetic pathways that need to be targeted in anticancer pharmacological therapy [1,9]. In this review, we focus on recent findings and examine how the historical evolution of DON and GA inhibitors can inform current therapeutic development in oncology.
2. Glutaminase Is a Metabolic Target Against Cancer
Cancer metabolism might be described as a large city underground map, where, when one pathway is blocked, another can be activated to meet the energetic and biosynthetic requirements of cancer cells. Metabolic plasticity is a hallmark of tumors and contributes to the emergence of resistance mechanisms that limit therapeutic efficacy. However, although broad, metabolic plasticity is limited, and blocking several related pathways may circumvent resistance, aiming to synergistically arrest cancer development [14,15]. In this context, GA has been fully characterized as a key target in the metabolic therapy of cancer [16,17,18]. GA is the enzyme responsible for catalyzing the conversion of Gln to glutamate, releasing the amide group from Gln as free ammonia, and represents the first step in glutaminolysis [10]. Glutaminolysis is a commonly upregulated pathway in cancer cells, as it can provide both energy (via generation of α-ketoglutarate and incorporation into the tricarboxylic acid cycle) and biosynthetic precursors for the synthesis of proteins, lipids, and nucleic acids, positioning Gln as a crucial metabolite boosting survival, accelerated growth, and proliferation [19]. Given that the reaction catalyzed by GA is the gate to downstream glutaminolytic metabolism, intense focus has been placed on inhibiting GA as an alternative or parallel target for potential cancer treatment regimens, as this strategy might help tackle the heterogeneity among cancer cells [9,20,21,22]. Several human GA proteins have been identified, encoded by two paralogous genes, GLS and GLS2 [9], each of which gives rise to two different isoforms [23]. The transcripts known as KGA (kidney-type glutaminase) and GAC (glutaminase C) arise by alternative splicing of the GLS gene, being collectively named GLS; whereas two GLS2 transcripts have also been identified: the canonical long transcript termed GAB (glutaminase B) and the short transcript LGA (liver-type glutaminase), which was originally identified in rat liver. Both GAB and LGA can also be indistinctly referred to as GLS2 isozymes [19].
GLS is consistently associated with Gln addiction in tumors and has well-established oncogenic properties; however, the role for GLS2 appears to be more complex, as it has been described as a context-dependent tumor suppressor factor [9,16,17,18]. The heterogeneity in the expression of key metabolic genes—like GLS and GLS2—suggests that different tumors might have differential requirements for glutaminolysis [24]. Accordingly, increased Gln catabolism in mouse liver tumors was associated with decreased levels of Gln synthetase (GS) and a shift from GLS2 to GLS. In sharp contrast, MYC-induced lung tumors display increased expression of both GS and GLS and accumulate Gln. These and other findings make it clear that the tumor metabolic profiles depend on both the genotype and tissue of origin, with critical implications for the design of therapies targeting tumor metabolism [25]. Anyhow, specific GLS inhibition has triggered crucial effects in the therapy of different cancers, as will be reviewed in this article. Importantly, GLS not only regulates the availability of Gln to the tumor but also acts as a tumor supportive protein in many types of cancer, as it has an important impact in pro-tumorigenic signaling [26]. The metabolic shift toward glutaminolysis —and the implications that it has for cancer progression— is driven by oncogene c-Myc-mediated GLS overexpression in many cancers [27,28,29]. Notably, c-Myc coordinates key metabolic pathways, including glycolysis and glutaminolysis. Interestingly, in prostate cancer cells, the crosstalk between these two pathways is mediated by GLS. Specifically, c-Myc-induced GLS indirectly repressed thioredoxin-interacting protein (TXNIP), thereby increasing glucose uptake and glycolysis. Inhibition of GLS restored TXNIP expression and reduced glucose uptake in PC3 cells. [30]. Metabolic reprogramming in cancer cells relies on the overexpression of key enzymes in order that upregulate determinant pathways such as glycolysis, glutaminolysis, and fatty acid synthesis [1]. Combined inhibition of three enzymes essential in these pathways —hexokinase-2, GLS and fatty acid synthase— with lonidamine, DON, and orlistat, respectively, results in a significant reduction in cell viability in the human colon cancer SW480 cell line, as well as in vivo in mice. This approach shows good tolerance, overcomes resistance mechanisms, and produces synergistic effects [31]. This example illustrates a widely adopted strategy in cancer research: combination therapy.
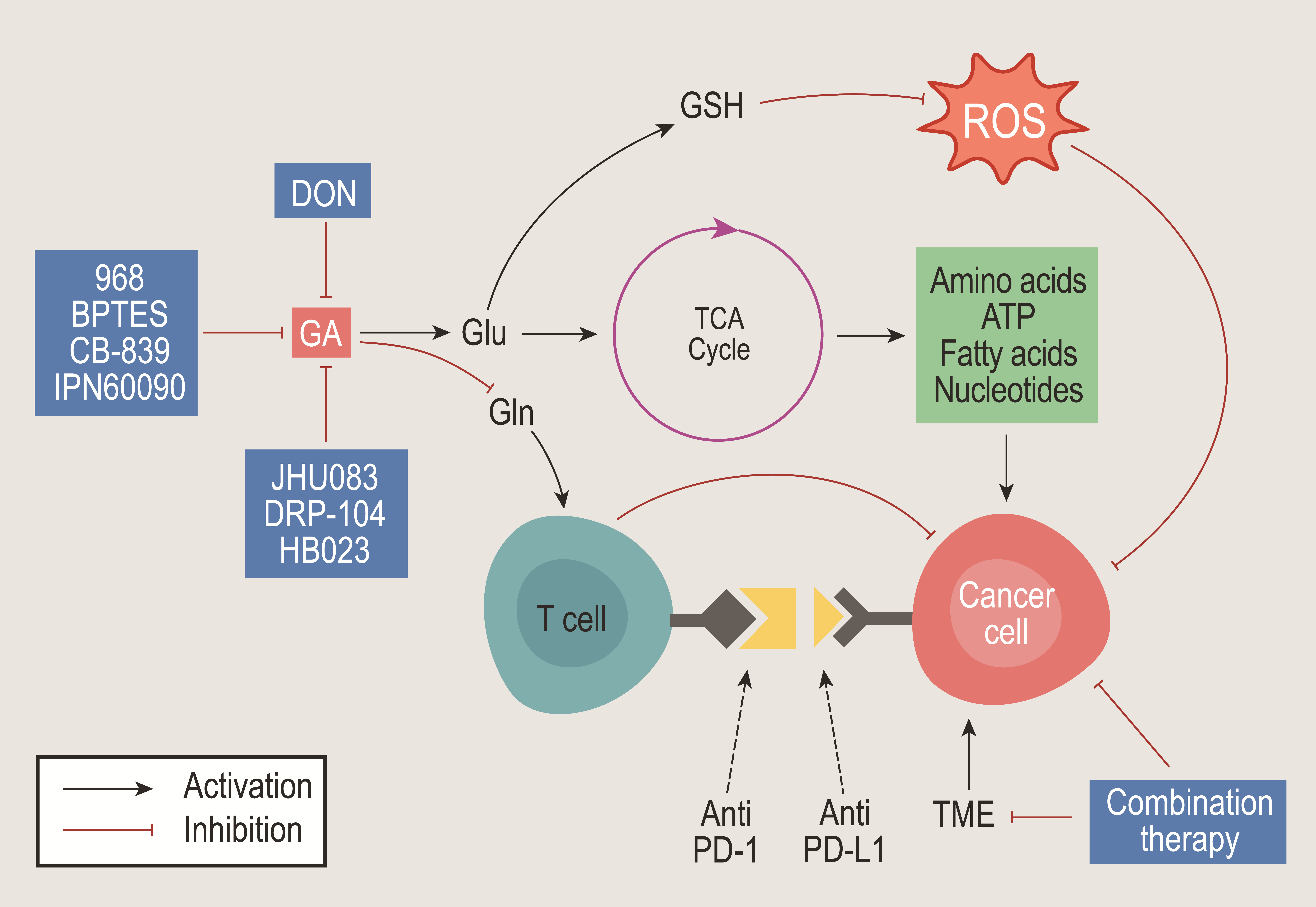
In Figure 1, we depict the main circuits involved in Gln homeostasis in cancer cells, along with major targets susceptible to pharmacological intervention within a multi-target chemotherapeutic approach. On the one hand, and crucially, glutaminolysis is inhibited with a specific inhibitor, among which compound 968, BPTES, and CB-839 have shown the best results (sections 3, 4, and 5). In addition, DON prodrugs and next-generation compounds have emerged as key tools for combination therapies (sections 6, 7, and 8). On the other hand, other key proteins, metabolic pathways, cellular processes, and signaling pathways must also be targeted to effectively constrain tumor growth [10]. In the following sections, we describe GA inhibitors to better understand their effects in monotherapy and, especially, in combination therapies, looking for strategies that not only enhance the antitumor activity of the other drug(s) but also produce synergistic effects, as will be described in detail below.
3. Compound 968 Alone or in Combination Therapy
Compound 968 is a dibenzophenanthridine, chemically 5-[3-bromo-4-(dimethylamino)phenyl]-2,3,5,6-tetrahydro-2,2-dimethyl-benzo[a]phenanthridin-4(1H)-one (Figure 2), which binds GLS and, unlike other inhibitors, also targets the GLS2 isoenzymes [23]. It is an allosteric modulator of GA, which induces apoptosis in cancer cells [26]. The pro-apoptotic effects of compound 968 have been demonstrated in both monotherapy and combination therapy settings [32]. For instance, the presence of compound 968 alone activates the release of the pro-apoptotic serine protease granzyme B, which is primarily secreted by activated T lymphocytes (CD3+, CD4+, and CD8+), boosting tumor infiltration and immune stimulation. This effect is further potentiated in vivo when combined with anti-PD-L1 antibodies. In another study, the competitive mTORC allosteric inhibitor PP242 induced an increase in Gln metabolism. A combination of PP242 and GA inhibition by compound 968 in glioblastoma (GBM) patient–derived xenograft (PDX) models effectively blocked tumor growth. Notably, no significant induction of cell death was observed in normal tissues, including the brain (cortex and hippocampus), liver, and kidney in mice treated with PP242, compound 968 alone, or the combination. These results demonstrate that GA inhibition can reverse mTORC-targeted therapy resistance in vivo by impairing tumor bioenergetics along with PP242 (Table 1), while maintaining low toxicity in normal tissues [33]. These findings may have important implications for combining mTORC kinase inhibitors with GA inhibition in patients with GBM and potentially other mTORC-activated cancers.
Another drug demonstrating greater efficacy in tumor cells than in normal ones, dihydroartemisinin—a semisynthetic derivative of artemisinin—has been shown to exert potent anticancer effects by increasing reactive oxygen species (ROS) levels (Table 1). Importantly, glutaminolysis and, therefore, GA activity, play a critical role in maintaining redox homeostasis, as GA-generated Glu serves for glutathione (GSH) synthesis both through direct incorporation and also by favoring cysteine (cystine) uptake via the xCT transporter (SLC7A11) as depicted in Figure 1. Cancer cells typically rely on glutaminolysis to boost antioxidant systems and support survival. Hence, the combination of dihydroartemisinin with compound 968 showed good efficacy, decreasing antioxidant capacity by disrupting redox homeostasis in cancer cells, while sparing normal cells [34]. Similarly, inhibition of proliferation was selectively enhanced in cancer cells by compound 968, which inhibited tumor growth through down-regulation of the epidermal growth factor receptor (EGFR)/extracellular signal-regulated kinase (ERK) pathway and induction of G1/G0-phase cell cycle arrest [35]. Interestingly, in the case of overcoming adriamycin chemoresistance, the effect appears not to be exclusively linked to GLS inhibition, since the same outcome is not achieved when compound 968 is replaced with another allosteric inhibitor, such as the drug CB-839, which specifically targets GLS isoforms [36].
4. BPTES as an Effective GLS Inhibitor Against Cancer
BPTES is an allosteric inhibitor of GLS isoforms, chemically identified as bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl (Figure 2). The efficacy of BPTES as a single antitumor agent has been demonstrated both in vitro and in vivo across several cancer types [37], although its highest potential is typically observed when used in combination with other drugs in multiple malignancies (Table 2). Tumor heterogeneity is a key factor in treatment design and optimization [25]. Therefore, understanding the gene expression profile of each specific tumor type is essential to decide which therapeutic strategy —or combination thereof— can achieve the greatest efficacy [21]. A study conducted at Columbia University provides a compelling example. Mutated NOTCH1, a common alteration in T-cell acute lymphoblastic leukemia (T-ALL), drives cell proliferation by regulating multiple pathways, including glutaminolysis. NOTCH1 signaling channels Gln metabolism into the tricarboxylic acid (TCA) cycle, promoting its use as a carbon source in NOTCH1-induced T-ALL. Dibenzozepine (DBZ), a gamma-secretase inhibitor (GSI), effectively blocks NOTCH1 activity. However, DBZ-treated leukemia cells that overexpress GLS show increased Gln utilization, suggesting that GLS overexpression may represent a potential mechanism of resistance to NOTCH1 inhibition. Combined treatment with DBZ and BPTES exhibited pronounced synergistic effects, impairing cell proliferation and sensitizing cells to NOTCH1 inhibition, with glutaminolysis assuming a dominant metabolic role over glycolysis. In xenograft mouse models, administration of BPTES plus DBZ resulted in strong tumor growth suppression. Notably, the Pten phenotype also appears to be a critical predictive factor for the efficacy of combined DBZ-BPTES therapy, as Pten-deleted T-ALL cells failed to response to GSI alone or in combination with GLS inhibition. Consequently, both GLS expression and Pten status could serve as predictive elements to guide treatment strategies in T-ALL [38].
5. CB-839 Is the Most Successful GLS Inhibitor in Cancer Therapy
CB-839 (telaglenastat) is a derivative of BPTES, chemically 2-(pyridin-2-yl)-N-(5-(4-(6-(2-(3-(trifluoromethoxy)phenyl)acetamido)pyridazin-3-yl)butyl)-1,3,4-thiadiazol-2-yl)acetamide (Figure 2). CB-839 monotherapy has demonstrated significant antitumor activity both in vitro and in vivo [46,47]. However, it is important to note that dual or triple combination therapy has shown a synergistic increase in antiproliferative effects without causing greater damage to healthy cells, while also allowing dose reduction, for example with doxorubicin, which is associated with severe side effects in patients [48]. Although combination therapy is the preferred strategy in most antitumor approaches (Table 3), some studies report that a single nitrogen metabolism inhibitor, such as CB-839, can be even more effective than combining different inhibitors. For instance, in patient-derived xenografts (PDX) of thyroid squamous cell carcinoma (SCC), CB-839 reduced the levels of the antioxidant enzyme NAD(P)H quinone oxidoreductase 1 (NQO1), thereby targeting and diminishing the master regulator nuclear factor erythroid 2-related factor 2 (NRF2) activity, which maintains redox homeostasis and protects cells from oxidative stress and damage. As an anticancer monotherapy, CB-839 was even more effective than the combination of the multireceptor tyrosine kinase inhibitor cabozantinib and the PI3K inhibitor GDC-0326 [49].
Interestingly, a recent study found that colorectal cancer (CRC) models developed resistance to CDK4/6 inhibitor palbociclib by upregulating glutaminolysis, and that combination with CB-839 overcame this resistance, producing synergistic effects [55]. Similar results were reported in lung adenocarcinoma models [56]. These studies highlight the relevance of Gln metabolism as a resistant and survival mechanism in cancer cells, due to its capacity to sustain multiple essential pathways. Therefore, GA inhibition appears to be a relevant adjuvant strategy, even in previously unsuspected contexts such as the CDK4/6 axis. In this case, although mechanisms are not yet fully elucidated, they likely involve downregulation of the E2F/Myc axis following CDK4/6 inhibition, thereby creating a synthetic lethal relationship with glutaminolysis and, consequently, with GA.
6. DON Returns Like a Trojan Horse
Although some recent studies have successfully used DON in combination therapy, for example, with MDiVi-1, a mitochondrial fission inhibitor that demonstrated antitumor efficacy in vitro and in vivo in ovarian cancer models [78], toxicity concerns have prevented DON from becoming the anti-Gln drug of choice. In 2016, a breakthrough opened a new avenue for DON use in the form of a prodrug, ethyl 2-(2-amino-4-methylpentanamido)-DON, named JHU083, with the critical advantage of inactive systemic administration of the compound and preferential accumulation and activation in the tumor microenvironment, allowing adequate concentrations at the site of action while minimizing side effects [79]. In 2019, another DON prodrug, (S)-isopropyl 2-((S)-2-acetamido-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate, known as DRP-104 (sirpiglenastat), was characterized, retaining the benefits of DON but avoiding side toxicity [80]. Notably, DRP-104 is bioactivated by serine proteases in the tumor, where it releases the antitumor drug DON, while it is bioinactivated by carboxylesterases in healthy gastrointestinal tissue to a non-toxic molecule [81]. Since then, several studies have confirmed the utility of both compounds in different tumor models (Table 4 and Table 5).
And, as if completing a circle, we return to the beginning with the chemical structure of DON (Figure 3). After decades of neglect, the inhibitory potential of DON has reemerged in the form of chimeric prodrugs that release the active compound—the Gln analog itself—directly at the tumor site to perform its precise function, while avoiding the drawbacks of its intrinsic toxicity. DON forms an irreversible covalent complex with GA by binding to the active-site serine residue and releasing the diazo group [96]. Thus, masked as precursors of the active agent, these prodrugs have once again highlighted the strong antitumor activity of DON. Interestingly, monotherapy with JHU083 for 14 days successfully reprogrammed metabolism in mouse models of melanoma, lymphoma, and colon cancer [82]. Notably, clinical trials are already underway for DRP-104 (NCT04471415, NCT06027086) and trials for JHU083 are also expected.
7. Targeting GA in Clinical Trails
As described above, the antitumor activity of CB-839 has been extensively characterized in multiple cell-based cancer models and in vivo (section 5). Highly significant effects have been observed with both monotherapy [68,69] and combination therapy (Table 1). One of the most frequently reported antitumor effects is increased oxidative stress, often combined with radiotherapy for enhanced efficacy [68,69,97]. However, the options are very diverse, as demonstrated across various models confirming its value as an antitumor drug (Table 1). CB-839 has been the most studied GA inhibitor to date. Although its solubility and pharmacokinetic properties are suboptimal, its high specificity and potency have driven its inclusion in numerous clinical trials. These include: as monotherapy in solid tumors, specifically metastatic or unresectable malignant peripheral nerve sheath tumors (NCT03872427) [98]; in combination therapy with the monoclonal antibody panitumumab (anti-EGFR) in CRC (NCT03263429), with the antimetabolite azacytidine in myelodysplastic syndromes (NCT03047993), using a 5-fluorouracil (5-FU) prodrug as capecitabine in CRC (NCT02861300) [98], with a taxane like paclitaxel in triple negative breast cancer (TNBC) (NCT03057600), with the tyrosine kinase inhibitor (TKI) cabozantinib in renal cell carcinoma (RCC) (NCT03428217) [99], together with the TKI palbociclib in metastatic solid tumors (NCT03965845), with the TKI osimertinib in non-small cell lung cancer (NSCLC) (NCT03831932), with the mTOR inhibitor everolimus in RCC (NCT03163667), together with the mTORC inhibitor sapanisertib in NSCLC (NCT04250545) [100], with the proteasome inhibitor carfilzomib in multiple myeloma (NCT03798678), and with radiation therapy and temozolomide in treating patients with isocitrate dehydrogenase (IDH)-mutated diffuse astrocytoma or anaplastic astrocytoma (NCT03528642) [101]. Additionally, it was used in a phase II clinical trial with radiotherapy and cisplatin in cervical cancer (NCT05521997), a phase I/II trial using the antibody nivolumab for targeting programmed cell death protein 1 (PD-1) in advanced cancers, i.e., melanoma, NSCLC and RCC [97], and in a phase II clinical trial with pembrolizumab to equally target PD-1 in NSCLC (NCT04265534) [73].
Many articles highlight the need for a combined therapeutic approach that includes GA targeting, as Gln metabolism affects multiple cellular processes and metabolic pathways, including oxidative stress [102], DNA damage [103], response to radiotherapy [97], alterations in the harsh tumor microenvironment (TME) [104], and changes in the immune response [90,105]. A significant step toward clinical translation occurred in 2020 with the development of IPN60090 (or IACS-6274), a BPTES-derived molecule with the chemical structure 1-[(2R)-4-[6-[[2-[4-(3,3-difluorocyclobutyl)oxy-6-methylpyridin-2-yl]acetyl]amino]pyridazin-3-yl]-2-fluorobutyl]-N-methyltriazole-4-carboxamide. IPN60090 showed greater potency and selectivity than CB-839, with optimal pharmacokinetic properties characterized in mouse, rat, and dog [106]. This drug is currently being evaluated in a phase I clinical trial (NCT03894540). In epithelial ovarian cancer, IPN60090 has been shown to inhibit Zinc finger SWIM-type containing 4 (ZSWIM4), a transcription factor that induces drug resistance. Its effects have been characterized in vitro and in mouse xenografts, where it sensitized tumors to a CBP inhibitor (targeting CREB-binding protein and its paralog p300), and reduced tumor volume through an apoptosis/glutathione-dependent mechanism. These antitumor effects were synergistically augmented when combined with paclitaxel in patient-derived organoids (PDO) models [107]. In acute myeloid leukemia (AML) cells, IPN60090 induced apoptosis, inhibited tumor growth, and suppressed NADPH and ATP synthesis. Accordingly, it exhibited synergistic antiproliferative effects when combined with with venetoclax, a BCL-2 inhibitor [67]. In a novel nanostrategy, IPN60090 was encapsulated in nanospheres targeting mitochondria, together with copper and IR780. While IRP60090 blocks GSH-mediated antioxidant defense, copper accentuates oxidative stress through cuproptosis, and IRP780 enhances the immune response and immunogenic cell death, achieving substantial synergistic antitumor effects in breast cancer cells both in vitro and in vivo [108].
8. Future Perspectives
Targeting Gln metabolism has been extensively validated as a powerful anticancer strategy [9,10,11,101,102,103,104,105]. In this context, targeting GA appears particularly promising, as it represents the main rate-limiting step of glutaminolysis, a pathway that provides bioenergetics, biosynthetic intermediates, and antioxidant protection. In fact, many cancers overexpress GA, particularly GLS, making its inhibition a prime opportunity to reverse cancer cell metabolic reprogramming, limit proliferation and render cells vulnerable to death induction, thereby enhancing sensitivity to chemoradiotherapy and improving therapeutic responses [10]. For immunotherapy-nonresponsive cancers, the prodrug JHU083 emerges as a promising novel treatment, even in the presence of immune-suppressing macrophages and scarce T-cells, positioning it as a potent option for tumors unresponsive to checkpoint inhibitors. Besides, it has demonstrated substantial therapeutic benefits in urological cancers, particularly by reducing angiogenesis in the TME [90].
Combinations aimed at boosting immune response are of particular interest here, since Gln depletion in the TME, caused by unrestrained incorporation and metabolism by Gln-addicted tumors, critically limits lymphocyte activation and growth, which critically rely on Gln for clonal expansion, thereby constituting an immune escape mechanism for cancer cells. Hence, limiting glutaminolysis by tumors increases Gln availability to immune cells in the TME, boosting immune response. Very recently, a new DON chimera has been designed by fusing DON with JQ1 to leverage JQ1’s well-established downregulation of PD-L1; chemically (6S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno [2-f]triazolo [3-a]diazepine-6-acetic acid 1,1-dimethylethyl ester, resulting in the new compound HB023 [109]. This new drug has shown great chemotherapeutic capacity in a murine model of colon cancer, overcoming tumor immune evasion and increasing anticancer efficacy by cooperatively activating both adaptive and innate antitumor immunity. Similar results have been observed in a murine model of clear cell renal cell carcinoma (ccRCC) with DRP-104, and with the GLS inhibitor CB-839 combined with PD-L1, achieving synergistic increases in antitumor immunity [110]. Indeed, anti-PD-1 and anti-PD-L1 therapies are revolutionizing cancer treatment, as drug resistance remains a major challenge; immune checkpoint inhibitors thus emerge as valuable tools to overcome multidrug resistance and collaborate with Gln metabolic reprogramming to achieve durable anticancer responses [32,33,72,73,82,85,86,90,93,105,110]. Collectively, the investigations described above emphasize the importance of combination therapy and the optimization of synergistic approaches, in which targeting Gln metabolism serves as a valuable tool.
Notably, Gln-based positron emission tomography (Gln-PET) imaging using (2S, 4R)-4-[18F]Gln has proven effective for tumor diagnosis [111], disease tracking [112], therapeutic monitoring [113], and pharmacodynamic of drugs such as CB-839 [78]. This technique exhibits the highest reproducibility and repeatability in preclinical cancer models [114], and has been validated in a phase I/II clinical trial of CB-839 (NCT03263429) in patients with metastatic CRC [75]. In personalized oncology, Gln-PET enables organ- and patient-specific monitoring based on the organ being studied and the patient [77], including analysis of the TME profiling, as demonstrated in a mouse model and a patient with brain metastases from invasive ductal breast carcinoma [115]. In addition to addressing some of the main challenges in antitumor therapy, such as cancer heterogeneity, drug resistance, and immune evasion, nano-delivery strategies will be essential for modulating the TME through combination therapy and immunometabolic crosstalk [116]. Of particular interest are chimeric compounds that exhibit immunometabolic bifunctionality. For instance, Zhang et al. combined the prodrug JHU083 (which blocks Gln metabolism) with MSA-2, an agonist of the interferon gene activation pathway that regulates innate immunity in the TME and elicits a robust immunogenic response [117]. This bifunctional construct has demonstrated very promising results in vitro and in vivo in colon cancer models., with synergistic enhancement when combined with 5-FU or anti-PD-L1 therapy [117]. Nevertheless, future research will must study how, for every single cancer, Gln blockage can support or impair antitumor immunity, depending on tumor type, immune context, dosage, compartment and treatment combination.
9. Conclusions
Many tumors exhibit significant intrinsic plasticity, triggering metabolic reprogramming to develop drug resistance and enhance biosynthetic and signaling pathways that sustain their proliferation program [118,119]. Therefore, combination therapy represents one of the few viable strategies for successful neoplasm treatment (Table 1, Table 2, Table 3, Table 4 and Table 5). Several reviews have described the activity of multiple GA inhibitors [9,10,11,101], as Gln metabolism constitutes a central network for tumor growth and survival, providing energy and essential substrates [23,105,120]. In this work we emphasize the need to design targeted therapeutic interventions based on blocking and/or activating different metabolic and/or cell signaling pathways looking to counteract the great adaptability of tumors to oxidative and/or metabolic stress conditions. Inhibition of GA, either through allosteric inhibitors (i.e.,: compound 968, BPTES, CB-839, IPN60090) or Gln analogs (i.e.,: DON, JHU083, DRP-104, HB023) represents a useful intervention targeting glutaminolysis, which as discussed constitutes an essential pathway for cancer survival and progression. This approach offers dual advantages: synergistic efficacy with a broad range of drugs, and reduced dosing requirements for certain agents, thereby minimizing toxicity to healthy cells. Combination therapy can integrate Gln metabolism inhibitors with other chemotherapeutic drugs, radiotherapy, or immunotherapy, optimizing overall efficacy by targeting multiple pathways while mitigating resistance development [121]. Recent research and ongoing clinical trials position CB-839 and DRP-104 as the most promising drugs targeting nitrogen metabolism, either by blocking GA (CB-839) or inhibiting Gln across multiple enzymes (DRP-104) [23]. Both demonstrate efficacy as monotherapy but excel in combination regimens, with CB-839 notably restricting TCA cycle flux, and DRP-104 broadly suppressing proliferative nodes like purine biosynthesis [122]. Moreover, DRP-104 uniquely modulates antitumor immune activity [81] by reshaping the TME [90,93]. A dysregulated, tumor-supportive TME represents a hallmark of cancer [123], and targeting Gln metabolism shows great potential for abrogating the tumor-supportive environment in both preclinical models [78] and patients [115]. Efforts to optimize inhibitors—including prodrug formulations—continue, exemplified by a tert-butyl ester derivative of DRP-104 that offers improved solubility and in vivo stability [124].
The above findings underscore the value of identifying possible targeted therapy-induced resistance mechanisms to design novel combination approaches that achieves the previously discussed advantages. Recent strategies have proposed integrating Gln metabolism into cancer diagnosis, classification, treatment, and monitoring [14]. In animal models, many tumors are especially dependent on Gln metabolism [21]. This observation suggests that imaging, quantifying, or blocking Gln metabolism in human cancers could be incorporated into the diagnosis and management of the disease [125], including PET-based technologies [77,110,126]. In most cases, it would be beneficial to use in vivo perioperative administration of isotope-labeled biomarkers (glucose and Gln) to cancer patients to differentiate metabolic pathways between tumors and benign tissue [127]. Additionally, in vivo Gln metabolic studies may help predict which tumors will respond to metabolism-targeted therapies [2].
In contrast, it should not be overlooked that several studies have identified important limitations and challenges associated with glutaminase inhibitors, including: (i) the need for isoenzyme-specific GLS inhibition when GLS2 acts as a tumor suppressor [17]; (ii) intestinal side effects caused by mucosal damage and cell death following GA inhibition [128]; (iii) metabolic reprogramming in some tumors through increased lipid oxidation to compensate for reduced glutaminolysis, thereby limiting the efficacy of GA inhibition [23]; (iv) compensatory increases in glucose metabolism through pyruvate carboxylase [14]; (v) the ability of some KRAS-mutant cancers to acquire Gln through alternative mechanisms, reducing the effectiveness of GA as a therapeutic target [75,121]; (vi) the incompatibility of GA inhibition with KEAP1 and STK11/Lkb1 co-mutations in KRAS-mutant lung adenocarcinoma because glutamate is required for CD8 T-cell activation [129]; and (vii) the high dependence of immune-system T cells on Gln metabolism for proliferation, requiring careful evaluation of GA blockade to achieve a balanced therapeutic response [130]. Hence, further research is needed to elucidate the metabolic consequences of GLS inhibition in combination with immunotherapy, as well as potential resistance mechanisms and side effects. However, there are also highly promising therapeutic findings, for example in ovarian cancer [32] as well as in lung and colon cancer [45]. For possible personalized use in the future a strong and in deep study must be done, including tumor type, selected biomarkers, immune context, glutamine dependency, GLS versus GLS2 biology, metabolic plasticity, resistance mechanisms, and adequate patient selection. In addition, understanding the genetic and epigenetic circuits regulating GLS will facilitate the development of improved combination therapies that exploits Gln metabolism reprogramming for immune evasion, ultimately leading to novel anticancer drugs and precision oncology approaches [82,105,118,131,132]. In this field, the most promising research strategies appear to integrate anti-PD-1/PD-L1 therapies with targeting of metabolic interactions within the TME that promote tumorigenesis (Figure 4) [133]. Indeed, several very recent analyses have identified compelling combination therapy approaches showing clinical efficacy [134].
Author Contributions
Conceptualization, J.A.C.-S., J.D.L.S.-J., and J.M.M.; methodology, J.A.C.-S., J.D.L.S.-J., and J.M.M.; resources, J.M. and J.M.M.; writing—original draft preparation, J.M.M.; writing—review and editing, J.A.C.-S., J.D.L.S.-J., J.M., and J.M.M.; project administration, J.M. and J.M.M.; funding acquisition, J.M. and J.M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by Ministerio de Ciencia e Innovación of Spain, PID20221403880B-I00 (to JMM).
Data Availability Statement
All data generated in this study are available from the corresponding authors upon reasonable request.
Acknowledgments
Generative AI and AI-assisted technologies were NOT used in the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
| 5-FU | 5-fluorouracilo |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| ATP | Adenosine triphosphate |
| ccRCC | Clear cell renal cell carcinoma |
| CRC | Colorectal cancer |
| DBZ | Dibenzozepine |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular signal-regulated kinase |
| GA | Glutaminase |
| GBM | Glioblastoma |
| Gln | Glutamine |
| Glu | Glutamate |
| GLS | Glutaminase isoenzyme 1 |
| GLS2 | Glutaminase isoenzyme 2 |
| GS | Glutamine synthetase |
| GSI | Gamma-secretase inhibitor |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NSCLC | Non-small cell lung cancer |
| PD-1 | Programmed cell death protein 1 |
| PET | Positron emission tomography |
| PDO | Patient-derived organoids |
| PDX | Patient-derived xenografts |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death ligand 1 |
| ROS | Reactive oxygen species |
| SCC | Squamous cell carcinoma |
| TCA | Tricarboxylic acid |
| TKI | Tyrosine kinase inhibitor |
| TME | Tumor microenvironment |
| TNBC | Triple negative breast cancer |
References
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16(10), 619–634. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu Rev. BioMed Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Coffey, G.L.; Ehrlich, J.; Fisher, M.W.; Hillegas, A.B.; Kohberger, D.L.; Machamer, H.E.; Rightsel, W.A.; Roerner, F.R. 6-Diazo-5-oxo-L-norleucine, a new tumor-inhibitory substance. I. Biologic studies. Antibiot. Chemother. 1956, 6(8), 487–97. [Google Scholar]
- Dion, H.W.; Fusari, S.A.; Jakubowski, Z.L.; Zora, J.G.; Bartz, Q.R. 6-Diazo-5-oxo-L-norleucine, A New Tumor-inhibitory Substance. II. Isolation and Characterization. J. Am. Chem. Soc. 1956, 78(13), 3075–3077. [Google Scholar] [CrossRef]
- Magill, G.B.; Myers, W.P.; Reilly, H.C.; Putnam, R.C.; Magill, J.W.; Sykes, M.P.; et al. Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease. Cancer 1957, 10(6), 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Le Page, G.A.; Loo, T.L. Purine antagonists. In Cancer Medicine; Holland, J. F., Frei, E., III, Eds.; Lea and Febiger: Philadelphia, 1973; pp. 754–756. [Google Scholar]
- Rosenfeld, H.; Roberts, J. Enhancement of antitumor activity of glutamine antagonists 6-diazo-5-oxo-L-norleucine and acivicin in cell culture by glutaminase-asparaginase. Cancer Res. 1981, 41, 1324–1328. [Google Scholar] [PubMed]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018, 17(9), 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Campos-Sandoval, J.A.; Márquez, J. Glutaminase isoenzymes in the metabolic therapy of cancer. Biochim Biophys. Acta Rev. Cancer 2018, 1870, 158–164. [Google Scholar] [CrossRef]
- Matés, J.M.; Campos-Sandoval, J.A.; de Los Santos-Jiménez, J.; Segura, J.A.; Alonso, F.J.; Márquez, J. Metabolic Reprogramming of Cancer by Chemicals that Target Glutaminase Isoenzymes. Curr. Med. Chem. 2020, 27(32), 5317–5339. [Google Scholar] [CrossRef]
- Anthony, J.; Varalakshmi, S.; Sekar, A.K.; Devarajan, N.; Janakiraman, B.; Peramaiyan, R. Glutaminase—A potential target for cancer treatment. Biomedicine 2024, 14(2), 29–37. [Google Scholar] [CrossRef] [PubMed]
- Tabrez, S.; Zughaibi, T.A.; Hoque, M.; Suhail, M.; Khan, M.I.; Khan, A.U. Targeting Glutaminase by Natural Compounds: Structure-Based Virtual Screening and Molecular Dynamics Simulation Approach to Suppress Cancer Progression. Molecules 2022, 27(15), 5042. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.X.; Chen, C.; Liu, X.Q.; Li, Y.; Lin, Y.L.; Wu, X.T.; Kong, L.Y.; Luo, J.G. Discovery and optimization of withangulatin A derivatives as novel glutaminase 1 inhibitors for the treatment of triple-negative breast cancer. Eur. J. Med. Chem. 2021, 210, 112980. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368(6487), eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- El-Tanani, M.; Rabbani, S.A.; El-Tanani, Y.; Matalka, I.I. Metabolic vulnerabilities in cancer: A new therapeutic strategy. Crit. Rev. Oncol. Hematol. 2024, 201, 104438. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Peña, E.; Arnold, J.; Shivakumar, V.; Joseph, R.; Vidhya Vijay, G.; den Hollander, P.; Bhangre, N.; Allegakoen, P.; Prasad, R.; Conley, Z.; Matés, J.M.; Márquez, J.; Chang, J.T.; Vasaikar, S.; Soundararajan, R.; Sreekumar, A.; Mani, S.A. The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression. Cancers 2019, 11(10), 1610. [Google Scholar] [CrossRef] [PubMed]
- De Los Santos-Jiménez, J.; Campos-Sandoval, J.A.; Márquez-Torres, C.; Urbano-Polo, N.; Brøndegaard, D.; Martín-Rufián, M.; Lobo, C.; Peñalver, A.; Gómez-García, M.C.; Martín-Campos, J.; Cardona, C.; Castilla, L.; da Costa Souza, F.; Cheng, T.; Segura, J.A.; Alonso, F.J.; Curi, R.; Colquhoun, A.; DeBerardinis, R.J.; Márquez, J.; Matés, J.M. Glutaminase isoforms expression switches microRNA levels and oxidative status in glioblastoma cells. J. BioMed Sci. 2021, 28(1), 14. [Google Scholar] [CrossRef] [PubMed]
- De Los Santos-Jiménez, J.; Rosales, T.; Ko, B.; Campos-Sandoval, J.A.; Alonso, F.J.; Márquez, J.; DeBerardinis, R.J.; Matés, J.M. Metabolic Adjustments following Glutaminase Inhibition by CB-839 in Glioblastoma Cell Lines. Cancers 2023, 15(2), 531. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Segura, J.A.; Martín-Rufián, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Márquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534. [Google Scholar] [CrossRef] [PubMed]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3(3), 169–180. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30(3), 434–446. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Wu, S.; Shen, H.; Luo, K.; Huang, Z.; Lu, N.; Li, Y.; Lan, Q.; Xian, Y. Glutamine Metabolism Heterogeneity in Glioblastoma Unveils an Innovative Combination Therapy Strategy. J. Mol. Neurosci. 2024, 74(2), 52. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Cao, D.; Zhang, Y.; Yu, X.; Wu, Y.; Jia, Z.; Jiang, J.; Cao, X. Integrative pan-cancer analysis and experiment validation identified GLS as a biomarker in tumor progression, prognosis, immune microenvironment, and immunotherapy. Sci. Rep. 2025, 15(1), 525. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; Chen, X.; Bishop, J.M. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M.; Di Paola, F.J.; Campos-Sandoval, J.A.; Mazurek, S.; Márquez, J. Therapeutic targeting of glutaminolysis as an essential strategy to combat cancer. Semin Cell Dev. Biol. 2020, 98, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; Dang, C.V. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458(7239), 762–5. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 2014, 24(19), 2274–80. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Braas, D.; Go, J.; Graeber, T.G.; Parlati, F.; Demo, S.; Li, R.; Walser, T.C.; Gricowski, M.; Shuman, R.; Ibarra, J.; Fridman, D.; Phelps, M.E.; Badran, K.; St John, M.; Bernthal, N.M.; Federman, N.; Yanagawa, J.; Dubinett, S.M.; Sadeghi, S.; Christofk, H.R.; Shackelford, D.B. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 2018, 33(5), 905–921.e5. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Sun, J.; Zhang, Y.; Li, J.; Hu, J.; Li, K.; Gao, L.; Shen, L. c-Myc-driven glycolysis via TXNIP suppression is dependent on glutaminase-MondoA axis in prostate cancer. Biochem Biophys. Res. Commun. 2018, 504, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Madrid, D.; Dominguez-Gomez, G.; Gonzalez-Fierro, A.; Perez-Cardenas, E.; Taja-Chayeb, L.; Trejo-Becerril, C.; Duenas-Gonzalez, A. Feasibility and antitumor efficacy in vivo, of simultaneously targeting glycolysis, glutaminolysis and fatty acid synthesis using lonidamine, 6-diazo-5-oxo-L-norleucine and orlistat in colon cancer. Oncol. Lett. 2017, 13, 1905–1910. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Siu, M.K.; Jiang, Y.X.; Leung, T.H.; Chan, D.W.; Wang, H.G.; Ngan, H.Y.; Chan, K.K. A Combination of Glutaminase Inhibitor 968 and PD-L1 Blockade Boosts the Immune Response against Ovarian Cancer. Biomolecules 2021, 11(12), 1749. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; Masui, K.; Gini, B.; Yang, H.; Hosoda, K.; Sasaki, R.; Mischel, P.S.; Kohmura, E. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Invest. 2015, 125, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Meng, G.; Zheng, M.; Zhang, Y.; Chen, A.; Wu, J.; Wei, J. The Glutaminase-1 Inhibitor 968 Enhances Dihydroartemisinin-Mediated Antitumor Efficacy in Hepatocellular Carcinoma Cells. PLoS ONE 2016, 11, e0166423. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Guo, M.; Zhang, T.; Gan, M.; Xie, C.; Wang, J.B. A novel Glutaminase inhibitor-968 inhibits the migration and proliferation of non-small cell lung cancer cells by targeting EGFR/ERK signaling pathway. Oncotarget 2016, 8, 28063–28073. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Guo, Z.; Zhao, Y.; Ma, L.; Li, B.; Yang, C. Compound 968 reverses adriamycin resistance in breast cancer MCF-7(ADR) cells via inhibiting P-glycoprotein function independently of glutaminase. Cell Death Discov. 2021, 7(1), 204. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, H.J.; Figueira, M.I.; Vaz, C.V.; Carvalho, T.M.A.; Brás, L.A.; Madureira, P.A.; Oliveira, P.J.; Sardão, V.A.; Socorro, S. Glutaminolysis is a metabolic route essential for survival and growth of prostate cancer cells and a target of 5α-dihydrotestosterone regulation. Cell Oncol. (Dordr) 2021, 44(2), 385–403. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Ambesi-Impiombato, A.; Sudderth, J.; Sánchez-Martín, M.; Belver, L.; Tosello, V.; Xu, L.; Wendorff, A.A.; Castillo, M.; Haydu, J.E.; Márquez, J.; Matés, J.M.; Kung, A.L.; Rayport, S.; Cordon-Cardo, C.; DeBerardinis, R.J.; Ferrando, A.A. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in acute lymphoblastic leukemia. Nat. Med. 2015, 21, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kang, J.H.; Lee, S.H.; Lee, C.H.; Son, J.; Kim, S.Y. Glutaminase 1 inhibition reduces thymidine synthesis in NSCLC. Biochem Biophys. Res. Commun. 2016, 477, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cui, H.; Fang, J.; Deng, H.; Kuang, P.; Guo, H.; Wang, X.; Zhao, L. Glutamine deprivation plus BPTES alters etoposide- and cisplatin-induced apoptosis in triple negative breast cancer cells. Oncotarget 2016, 7, 54691–54701. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, C.A.; Joudiou, N.; Brusa, D.; Corbet, C.; Feron, O.; Gallez, B. Acidosis-induced metabolic reprogramming in tumor cells enhances the anti-proliferative activity of the PDK inhibitor dichloroacetate. Cancer Lett. 2020, 470, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Deng, H.; Wang, W.; Xiao, S.; Zheng, R.; Lv, L.; Wang, H.; Chen, J.; Zhang, B. LRPPRC promotes glycolysis by stabilising LDHA mRNA and its knockdown plus glutamine inhibitor induces synthetic lethality via m(6) A modification in triple-negative breast cancer. Clin. Transl. Med. 2024, 14(2), e1583. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Hayashi, S.; Otsuka, T.; Kamiya, T.; Ishikawa, K.; Hara, H. Inhibition of glutamine metabolism increases sensitivity to plasma-activated medium-induced cytotoxicity. Free Radic. Res. 2024, 58(3), 170–179. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Li, J.; Wang, T.; Zhang, X.; Du, P.; Dong, Y.; Jiao, Z. Self-assembled metal-polyphenolic based multifunctional nanomedicine to improve therapy treatment of pancreatic cancer by inhibition of glutamine metabolism. Colloids Surf. B Biointerfaces 2024, 244, 114162. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.K.; Park, M.; Lee, S.; Yun, J.W.; Lee, J.; Kim, J.S.; Cho, S.J.; Jeon, H.J.; Lee, I.K.; Choi, Y.K.; Park, K.G. Inhibition of glutamine utilization synergizes with immune checkpoint inhibitor to promote antitumor immunity. Mol. Cell. 2020, 80, 592–606.e8. [Google Scholar] [CrossRef] [PubMed]
- Basu, H.S.; Wilganowski, N.; Robertson, S.; Reuben, J.M.; Cohen, E.N.; Zurita, A.; Ramachandran, S.; Xiao, L.C.; Titus, M.; Wilding, G. Prostate cancer cells survive anti-androgen and mitochondrial metabolic inhibitors by modulating glycolysis and mitochondrial metabolic activities. Prostate 2021, 81(12), 799–811. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Li, X.; Diao, Y.; Du, B.; Li, Y. Targeted inhibition of glutamine metabolism enhances the antitumor effect of seluºmetinib in KRAS-mutant NSCLC. Transl. Oncol. 2021, 14(1), 100920. [Google Scholar] [CrossRef] [PubMed]
- Draguet, A.; Tagliatti, V.; Colet, J.M. Targeting Metabolic Reprogramming to Improve Breast Cancer Treatment: An In Vitro Evaluation of Selected Metabolic Inhibitors Using a Metabolomic Approach. Metabolites 2021, 11(8), 556. [Google Scholar] [CrossRef] [PubMed]
- Schoultz, E.; Dahlberg, J.; Nilsson, L.M.; Dzanan, J.J.; Carlsson, T.; Dahr, N.; Andersson, E.; Muhammad, G.; Muth, A.; Elias, E.; Fagman, H.; Sayin, V.I.; Nilsson, J.A.; Nilsson, M. Involvement of KEAP1/NRF2 pathway in non-BRAF mutated squamous cell carcinoma of the thyroid. J. Pathol. 2025, 266(4-5), 481–494. [Google Scholar] [CrossRef] [PubMed]
- Lampa, M.; Arlt, H.; He, T.; Ospina, B.; Reeves, J.; Zhang, B.; Murtie, J.; Deng, G.; Barberis, C.; Hoffmann, D.; Cheng, H.; Pollard, J.; Winter, C.; Richon, V.; Garcia-Escheverria, C.; Adrian, F.; Wiederschain, D.; Srinivasan, L. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS ONE 2017, 12, e0185092. [Google Scholar] [CrossRef] [PubMed]
- Emberley, E.; Pan, A.; Chen, J.; Dang, R.; Gross, M.; Huang, T.; Li, W.; MacKinnon, A.; Singh, D.; Sotirovska, N.; Steggerda, S.M.; Wang, T.; Parlati, F. The glutaminase inhibitor telaglenastat enhances the antitumor activity of signal transduction inhibitors everolimus and cabozantinib in models of renal cell carcinoma. PLoS ONE 2021, 16(11), e0259241. [Google Scholar] [CrossRef] [PubMed]
- Brady, M.R.; Matulionis, N.; Christofk, H.R.; Garon, E.B.; Lisberg, A.; Shackelford, D.B.; Momcilovic, M. Macropinocytosis and Vascularization Determine Response to mTOR Inhibitors in Lung Squamous Cell Carcinoma. Cancer Res. 2025. [Google Scholar] [CrossRef] [PubMed]
- Lang, L.; Wang, F.; Ding, Z.; Zhao, X.; Loveless, R.; Xie, J.; Shay, C.; Qiu, P.; Ke, Y.; Saba, N.F.; Teng, Y. Blockade of glutamine-dependent cell survival augments antitumor efficacy of CPI-613 in head and neck cancer. J. Exp. Clin. Cancer Res. 2021, 40(1), 393. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.J.; Miao, D.L.; Su, Q.Y.; Tang, X.L.; Wang, X.L.; Deng, L.B.; Shi, H.D.; Xin, H.B. THZ1 suppresses human non-small-cell lung cancer cells in vitro through interference with cancer metabolism. Acta Pharmacol. Sin. 2019, 40(6), 814–822. [Google Scholar] [CrossRef] [PubMed]
- Tarrado-Castellarnau, M.; Foguet, C.; Tarragó-Celada, J.; Palobart, M.; Hernández-Carro, C.; Perarnau, J.; Zodda, E.; Polat, I.H.; Marin, S.; Suarez-Bonnet, A.; Lozano, J.J.; Yuneva, M.; Thomson, T.M.; Cascante, M. Glutaminase as a metabolic target of choice to counter acquired resistance to Palbociclib by colorectal cancer cells. Oncogene 2025, 44(36), 3386–3406. [Google Scholar] [CrossRef] [PubMed]
- Conroy, L.R.; Lorkiewicz, P.; He, L.; Yin, X.; Zhang, X.; Rai, S.N.; Clem, B.F. Palbociclib treatment alters nucleotide biosynthesis and glutamine dependency in A549 cells. Cancer Cell Int. 2020, 20, 280. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, S.; Zhao, Y.; Dinh, T.; Jiang, D.; Selfridge, J.E.; Myers, G.; Wang, Y.; Zhao, X.; Tomchuck, S.; Dubyak, G.; Lee, R.T.; Estfan, B.; Shapiro, M.; Kamath, S.; Mohamed, A.; Huang, S.C.; Huang, A.Y.; Conlon, R.; Krishnamurthi, S.; Eads, J.; Willis, J.E.; Khorana, A.A.; Bajor, D.; Wang, Z. Neutrophil extracellular traps induced by chemotherapy inhibit tumor growth in murine models of colorectal cancer. J. Clin. Invest. 2024, 134(5), e175031. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, X.Y.; Ji, J.B.; Zheng, Z.X.; Shang, P.F.; Guo, X.L. GLS1 inhibitor CB-839 inhibits the malignant progression of 5-FU resistant hepatoma cells by regulating glutamine metabolism. Chem. Biol. Interact. 2026, 423, 111812. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Niu, X.; Chang, J.; Shao, M.; Peng, L.; Xi, Y.; Lin, A.; Wang, C.; Cui, Q.; Luo, Y.; Fan, W.; Chen, Y.; Sun, Y.; Guo, W.; Tan, W.; Lin, D.; Wu, C. Metabolic remodeling by TIGAR overexpression is a therapeutic target in esophageal squamous-cell carcinoma. Theranostics 2020, 10(8), 3488–3502. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Cui, J.; Wang, W.; Hu, Q.; Xue, Y.; Liu, X.; Gong, T.; Lu, Y.; Ma, H.; Yang, X.; Feng, B.; Wang, Q.; Zhang, N.; Xu, Y.; Liu, M.; Nussinov, R.; Cheng, F.; Ji, H.; Huang, J. PPIA dictates NRF2 stability to promote lung cancer progression. Nat. Commun. 2024, 15(1), 4703. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, K.; Matsumoto, T.; Yamamoto, N.; Yamasaki, T.; Takami, T. Metabolic Analysis of DFO-Resistant Huh7 Cells and Identification of Targets for Combination Therapy. Metabolites 2023, 13(10), 1073. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, D.J.; Park, S.J.; Jang, W.J.; Jeong, C.H. Inhibition of GLS1 and ASCT2 Synergistically Enhances the Anticancer Effects in Pancreatic Cancer Cells. J. Microbiol. Biotechnol. 2025, 35, e2412032. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.M.; Kim, J.; Lee, J.Y.; Lee, J.S.; Lee, J.M. Regulation of glucose and glutamine metabolism to overcome cisplatin resistance in intrahepatic cholangiocarcinoma. BMB Rep. 2023, 56(11), 600–605. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Niu, Y.; Chen, X.; Kong, X.; Yan, G.; Zhuang, A.; Li, X.; Lian, L.; Qin, D.; Zhang, Q.; Zhang, R.; Yang, K.; Xia, X.; Chen, K.; Xiao, M.; Yang, C.; Wu, T.; Shen, Y.; Yu, C.; Luo, C.; Lin, S.H.; Li, W. Ursodeoxycholic acid inhibits the uptake of cystine through SLC7A11 and impairs de novo synthesis of glutathione. J. Pharm. Anal. 2025, 15(1), 101068. [Google Scholar] [CrossRef] [PubMed]
- Holt, A.K.; Najumudeen, A.K.; Collard, T.J.; Li, H.; Millett, L.M.; Hoskin, A.J.; Legge, D.N.; Mortensson, E.M.H.; Flanagan, D.J.; Jones, N.; Kollareddy, M.; Timms, P.; Hitchings, M.D.; Cronin, J.; Sansom, O.J.; Williams, A.C.; Vincent, E.E. Aspirin reprogrammes colorectal cancer cell metabolism and sensitises to glutaminase inhibition. Cancer Metab. 2023, 11(1), 18. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Feng, Y.; Wang, W.; Xu, L.; Zhang, M.; Yao, Y.; Wu, X.; Zhang, Q.; Huang, W.; Wang, X.; Li, X.; Ying, P.; Shang, L. Targeting Glutaminolysis to Treat Multiple Myeloma: An In Vitro Evaluation of Glutaminase Inhibitors Telaglenastat and Epigallocatechin-3-gallate. Anticancer Agents Med. Chem. 2023, 23(7), 779–785. [Google Scholar] [CrossRef] [PubMed]
- Okabe, S.; Moriyama, M.; Arai, Y.; Gotoh, A. Glutaminase 1 plays critical roles in myelodysplastic syndrome and acute myeloid leukemia cells. Cancer Biomark. 2024, 41(1), 55–68. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, R.; Jayachandran, K.; Zhang, J.; Menon, V.; Muhammad, N.; Zahner, M.; et al. Glutaminase inhibitors induce thiol-mediated oxidative stress and radiosensitization in treatment-resistant cervical cancers. Mol. Cancer Ther. 2020, 19(12), 2465–75. [Google Scholar] [CrossRef] [PubMed]
- Wicker, C.A.; Hunt, B.G.; Krishnan, S.; Aziz, K.; Parajuli, S.; Palackdharry, S.; et al. Glutaminase inhibition with telaglenastat (Cb-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett. 2021, 502, 180–8. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wang, S.; Zaal, E.A.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; Beijersbergen, R.; Berkers, C.R.; Qin, W.; Bernards, R. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. Elife 2020, 9, e56749. [Google Scholar] [CrossRef] [PubMed]
- Ochi, N.; Miyake, N.; Takeyama, M.; Yamane, H.; Fukazawa, T.; Nagasaki, Y.; Kawahara, T.; Ichiyama, N.; Kosaka, Y.; Mimura, A.; Nakanishi, H.; Hiraki, A.; Kiura, K.; Takigawa, N. The combined inhibition of SLC1A3 and glutaminase in osimertinib-resistant EGFR mutant cells. Biochim Biophys. Acta Gen. Subj. 2024, 1868(10), 130675. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.; Chen, J.; Englert, J.; Janes, J.; Leone, R.; MacKinnon, A.; et al. Abstract 2329: Glutaminase inhibition with cb-839 enhances anti-tumor activity of pd-1 and pd-L1 antibodies by overcoming a metabolic checkpoint blocking T cell activation. Cancer Res. (2016) 76((14_) Supplement. [CrossRef]
- Varghese, S.; Pramanik, S.; Williams, L.J.; Hodges, H.R.; Hudgens, C.W.; Fischer, G.M.; et al. The glutaminase inhibitor cb-839 (Telaglenastat) enhances the antimelanoma activity of T-Cell-Mediated immunotherapies. Mol. Cancer Ther. 2021, 20(3), 500–11. [Google Scholar] [CrossRef] [PubMed]
- Glassman, D.; Kim, M.S.; Spradlin, M.; Badal, S.; Taki, M.; Bhattacharya, P.; Dutta, P.; Kingsley, C.V.; Foster, K.I.; Animasahun, O.; Jeon, J.H.; Achreja, A.; Jayaraman, A.; Kumar, P.; Nenwani, M.; Wuchu, F.; Bayraktar, E.; Wu, Y.; Stur, E.; Mangala, L.; Lee, S.; Yap, T.A.; Westin, S.N.; Eberlin, L.S.; Nagrath, D.; Sood, A.K. Exploiting metabolic vulnerabilities after anti-VEGF antibody therapy in ovarian cancer. iScience 2023, 26(2), 106020. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Bae, S.W.; Whisenant, J.G.; Ayers, G.D.; Sheng, Q.; Peterson, T.E.; Smith, G.T.; Lin, K.; Chowdhury, S.; Kanikarla Marie, P.; Sorokin, A.; Cohen, A.S.; Goff, L.W.; Cardin, D.B.; Shen, J.P.; Kopetz, S.; Eng, C.; Shyr, Y.; Berlin, J.; Manning, H.C. Results of the Phase I/II Study and Preliminary B-cell Gene Signature of Combined Inhibition of Glutamine Metabolism and EGFR in Colorectal Cancer. Clin. Cancer Res. 2025, 31(8), 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.S.; Geng, L.; Zhao, P.; Fu, A.; Schulte, M.L.; Graves-Deal, R.; Washington, M.K.; Berlin, J.; Coffey, R.J.; Manning, H.C. Combined blockade of EGFR and glutamine metabolism in preclinical models of colorectal cancer. Transl. Oncol. 2020, 13(10), 100828. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ren, L.; Beck, J.A.; Phelps, T.E.; Olkowski, C.; Ton, A.; Roy, J.; White, M.E.; Adler, S.; Wong, K.; Cherukuri, A.; Zhang, X.; Basuli, F.; Choyke, P.L.; Jagoda, E.M.; LeBlanc, A.K. Exploration of Imaging Biomarkers for Metabolically-Targeted Osteosarcoma Therapy in a Murine Xenograft Model. Cancer Biother. Radiopharm. 2023, 38(7), 475–485. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Ghosh, S.; Roy, S.S. Glutamine deficiency promotes stemness and chemoresistance in tumor cells through DRP1-induced mitochondrial fragmentation. Cell Mol. Life Sci. 2021, 78(10), 4821–4845. [Google Scholar] [CrossRef] [PubMed]
- Rais, R.; Jančařík, A.; Tenora, L.; Nedelcovych, M.; Alt, J.; Englert, J.; Rojas, C.; Le, A.; Elgogary, A.; Tan, J.; Monincová, L.; Pate, K.; Adams, R.; Ferraris, D.; Powell, J.; Majer, P.; Slusher, B.S. Discovery of 6-Diazo-5-oxo-l-norleucine (DON) Prodrugs with Enhanced CSF Delivery in Monkeys: A Potential Treatment for Glioblastoma. J. Med. Chem. 2016, 59(18), 8621–33. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Nedelcovych, M.; Wild, R. drp-104, a novel broad acting glutamine antagonist, induces distinctive immune modulation mechanisms and synergistic efficacy in combination with immune checkpoint blockade. 34th annual meeting & pre-conference programs of the society for immunotherapy of cancer (Sitc 2019): Part 1. J. Immunother. Cancer (2019) 7 (suppl 1), 282. [CrossRef]
- Rais, R.; Lemberg, K.M.; Tenora, L.; Arwood, M.L.; Pal, A.; Alt, J.; Wu, Y.; Lam, J.; Aguilar, J.M.H.; Zhao, L.; Peters, D.E.; Tallon, C.; Pandey, R.; Thomas, A.G.; Dash, R.P.; Seiwert, T.; Majer, P.; Leone, R.D.; Powell, J.D.; Slusher, B.S. Discovery of DRP-104, a tumor-targeted metabolic inhibitor prodrug. Sci. Adv. 2022, 8(46), eabq5925. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.-H.; Oh, M.-H.; Sun, I.-H.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366(6468), 1013–21. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.S.; da Costa Rosa, M.; Stumpo, V.; Rais, R.; Slusher, B.S.; Riggins, G.J. The glutamine antagonist prodrug JHU-083 slows malignant glioma growth and disrupts mTOR signaling. Neurooncol Adv. 2020, 3(1), vdaa149. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Xiong, D.; Pan, J.; Zhang, Q.; Sei, S.; Shoemaker, R.H.; Lubet, R.A.; Montuenga, L.M.; Wang, Y.; Slusher, B.S.; You, M. Targeting Glutamine Metabolism to Enhance Immunoprevention of EGFR-Driven Lung Cancer. Adv. Sci. (Weinh) 2022, 9(26), e2105885. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.-H.; Sun, I.-H.; Zhao, L.; Leone, R.D.; Sun, I.-M.; Xu, W.; et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Invest. 2020, 130(7), 3865–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Q.; Xi, C.; Ju, N.T.; Shen, C.T.; Qiu, Z.L.; Song, H.J.; Luo, Q.Y. Targeting glutamine metabolism exhibits anti-tumor effects in thyroid cancer. J. Endocrinol. Invest. 2024, 47(8), 1953–1969. [Google Scholar] [CrossRef] [PubMed]
- Udutha, S.; Taglang, C.; Batsios, G.; Gillespie, A.M.; Tran, M.; Hoeve, J.T.; Graeber, T.G.; Viswanath, P. Combined inhibition of de novo glutathione and nucleotide biosynthesis is synthetically lethal in glioblastoma. Cell Rep. 2025, 44(5), 115596. [Google Scholar] [CrossRef] [PubMed]
- Chortis, V.; Borges, K.S.; Yao, C.H.; Ribeiro, C.; Nagano, L.F.; Berber, M.; Prete, A.; Najdekr, L.; Klontzas, M.E.; Jankevics, A.; Vendramini, P.; Kremer, J.L.; Kelley, L.; Raveenthiraraj, S.; Tsagarakis, S.; Macech, M.; Pupovac, I.D.; Papathomas, T.G.; Haykir, B.; Winder, C.; Quinkler, M.; Dennedy, M.C.; Ueland, G.Å.; Beuschlein, F.; Tabarin, A.; Fassnacht, M.; Taylor, A.E.; Kastelan, D.; Ambroziak, U.; Vassiliadi, D.A.; Kiseljak-Vassiliades, K.; Bancos, I.; Carlone, D.L.; Dunn, W.B.; Arlt, W.; Haigis, M.C.; Breault, D.T. Glutamine antagonism suppresses tumor growth in adrenocortical carcinoma through inhibition of de novo nucleotide biosynthesis. bioRxiv [Preprint] 2025, 2025.09.28.674326. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, M.; Shen, F.; Lee, A.J.; Zhao, L.; Nirschl, T.R.; Theodros, D.; Singh, A.K.; Wang, X.; Adusei, K.M.; Lombardo, K.A.; Williams, R.A.; Sena, L.A.; Thompson, E.A.; Tam, A.; Yegnasubramanian, S.; Pearce, E.J.; Leone, R.D.; Alt, J.; Rais, R.; Slusher, B.S.; Pardoll, D.M.; Powell, J.D.; Zarif, J.C. Metabolic Reprogramming of Tumor-Associated Macrophages Using Glutamine Antagonist JHU083 Drives Tumor Immunity in Myeloid-Rich Prostate and Bladder Cancers. Cancer Immunol. Res. 2024, 12(7), 854–875. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Estok, T.M.; Wild, R. Sirpiglenastat (DRP-104) Induces Antitumor Efficacy through Direct, Broad Antagonism of Glutamine Metabolism and Stimulation of the Innate and Adaptive Immune Systems. Mol. Cancer Ther. 2022, 21(10), 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Encarnación-Rosado, J.; Sohn, A.S.W.; Biancur, D.E.; Lin, E.Y.; Osorio-Vasquez, V.; Rodrick, T.; González-Baerga, D.; Zhao, E.; Yokoyama, Y.; Simeone, D.M.; Jones, D.R.; Parker, S.J.; Wild, R.; Kimmelman, A.C. Targeting pancreatic cancer metabolic dependencies through glutamine antagonism. Nat. Cancer 2024, 5(1), 85–99. [Google Scholar] [CrossRef] [PubMed]
- Allevato, M.M.; Trinh, S.; Koshizuka, K.; Nachmanson, D.; Nguyen, T.C.; Yokoyama, Y.; Wu, X.; Andres, A.; Wang, Z.; Watrous, J.; Molinolo, A.A.; Mali, P.; Harismendy, O.; Jain, M.; Wild, R.; Gutkind, J.S. A genome-wide CRISPR screen reveals that antagonism of glutamine metabolism sensitizes head and neck squamous cell carcinoma to ferroptotic cell death. Cancer Lett. 2024, 598, 217089. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.; LeBoeuf, S.E.; Hao, Y.; New, C.; Blum, J.L.E.; Rashidfarrokhi, A.; Huang, S.M.; Bahamon, C.; Wu, W.L.; Karadal-Ferrena, B.; Herrera, A.; Ivanova, E.; Cross, M.; Bossowski, J.P.; Ding, H.; Hayashi, M.; Rajalingam, S.; Karakousi, T.; Sayin, V.I.; Khanna, K.M.; Wong, K.K.; Wild, R.; Tsirigos, A.; Poirier, J.T.; Rudin, C.M.; Davidson, S.M.; Koralov, S.B.; Papagiannakopoulos, T. Glutamine antagonist DRP-104 suppresses tumor growth and enhances response to checkpoint blockade in KEAP1 mutant lung cancer. Sci. Adv. 2024, 10(13), eadm9859. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.; Hauck, J.S.; Jiang, X.; Quang, H.; Xu, L.; Zhang, F.; Gao, X.; Wild, R.; Everitt, J.I.; Macias, E.; He, Y.; Huang, J. Targeting glutamine dependence with DRP-104 inhibits proliferation and tumor growth of castration-resistant prostate cancer. Prostate 2024, 84(4), 349–357. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Jin, C.; Su, C.; Moon, D.; Sun, M.A.; Zhang, H.; Jiang, X.; Zhang, F.; Tserentsoodol, N.; Bowie, M.L.; Pirozzi, C.J.; George, D.J.; Wild, R.; Gao, X.; Ashley, D.M.; He, Y.; Huang, J. Resilience and Vulnerabilities of Tumor Cells under Purine Shortage Stress. Clin. Cancer Res. 2025, 31(20), 4345–4360. [Google Scholar] [CrossRef] [PubMed]
- Thangavelu, K.; Qing Yun Chong, Q.Y.; Low, B.C.; Sivaraman, J. Structural basis for the active site inhibition mechanism of human kidney-type glutaminase (KGA). Sci. Rep. 2024, 4, 3827. [Google Scholar] [CrossRef]
- Alden, R.S.; Kamran, M.Z.; Bashjawish, B.A.; Simone, B.A. Glutamine metabolism and radiosensitivity: Beyond the Warburg effect. Front Oncol. 2022, 12, 1070514. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Feng, X.; Chen, Y.; Selfridge, J.E.; Gorityala, S.; Du, Z.; Wang, J.M.; Hao, Y.; Cioffi, G.; Conlon, R.A.; Barnholtz-Sloan, J.S.; Saltzman, J.; Krishnamurthi, S.S.; Vinayak, S.; Veigl, M.; Xu, Y.; Bajor, D.L.; Markowitz, S.D.; Meropol, N.J.; Eads, J.R.; Wang, Z. 5-fluorouracil enhances the anti-tumor activity of the glutaminase inhibitor CB-839 against PIK3CA-mutant colorectal cancers. Cancer Res. 2020, 80(21), 4815–4827. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Lee, R.J.; Carthon, B.C.; Iliopoulos, O.; Mier, J.W.; Patel, M.R.; et al. CB-839, a glutaminase inhibitor, in combination with cabozantinib in patients with clear cell and papillary metastatic renal cell cancer (mRCC): Results of a phase I study. JCO 2019, 37, 549–9. [Google Scholar] [CrossRef]
- Riess, J.W.; Frankel, P.; Shackelford, D.; Dunphy, M.; Badawi, R.D.; Nardo, L.; Cherry, S.R.; Lanza, I.; Reid, J.; Gonsalves, W.I.; Kunos, C.; Gandara, D.R.; Lara, P.N.; Newman, E.; Paik, P.K. Phase 1 Trial of MLN0128 (Sapanisertib) and CB-839 HCl (Telaglenastat) in Patients With Advanced NSCLC (NCI 10327): Rationale and Study Design. Clin. Lung Cancer 2021, 22(1), 67–70. [Google Scholar] [CrossRef] [PubMed]
- Vagaggini, C.; D’Ursi, P.; Poggialini, F.; Fossa, P.; Francesconi, V.; Trombetti, G.; Orro, A.; Dreassi, E.; Schenone, S.; Tonelli, M.; Carbone, A. Deciphering the landscape of allosteric glutaminase 1 inhibitors as anticancer agents. Bioorg Chem. 2025, 161, 108523. [Google Scholar] [CrossRef] [PubMed]
- De Los Santos-Jiménez, J.; Campos-Sandoval, J.A.; Alonso, F.J.; Márquez, J.; Matés, J.M. GLS and GLS2 Glutaminase Isoenzymes in the Antioxidant System of Cancer Cells. Antioxidants 2024, 13(6), 745. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Gwak, J.; Lee, E.K.; Jeong, S.M. Mitochondrial Glutamine Metabolism Determines Senescence Induction After Chemotherapy. Anticancer Res. 2020, 40(12), 6891–6897. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Yan, X.; Song, J. Glutamine metabolism remodels tumor-associated macrophage: mechanistic explorations and new strategies in translational medicine. Front Immunol. 2026, 16, 1715170. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Zhao, Y.; Han, Y.; Li, M.; Wang, G. Targeting glutamine metabolism crosstalk with tumor immune response. Biochim Biophys. Acta Rev. Cancer 2025, 1880(1), 189257. [Google Scholar] [CrossRef] [PubMed]
- Soth, M.J.; Le, K.; Di Francesco, M.E.; Hamilton, M.M.; Liu, G.; Burke, J.P.; Carroll, C.L.; Kovacs, J.J.; Bardenhagen, J.P.; Bristow, C.A.; Cardozo, M.; Czako, B.; de Stanchina, E.; Feng, N.; Garvey, J.R.; Gay, J.P.; Do, M.K.G.; Greer, J.; Han, M.; Harris, A.; Herrera, Z.; Huang, S.; Giuliani, V.; Jiang, Y.; Johnson, S.B.; Johnson, T.A.; Kang, Z.; Leonard, P.G.; Liu, Z.; McAfoos, T.; Miller, M.; Morlacchi, P.; Mullinax, R.A.; Palmer, W.S.; Pang, J.; Rogers, N.; Rudin, C.M.; Shepard, H.E.; Spencer, N.D.; Theroff, J.; Wu, Q.; Xu, A.; Yau, J.A.; Draetta, G.; Toniatti, C.; Heffernan, T.P.; Jones, P. Discovery of IPN60090, a Clinical Stage Selective Glutaminase-1 (GLS-1) Inhibitor with Excellent Pharmacokinetic and Physicochemical Properties. J. Med. Chem. 2020, 63(21), 12957–12977. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Huang, Y.; Zheng, Y.; Hao, W.; Shi, K. ZSWIM4 inhibition improves chemosensitivity in epithelial ovarian cancer cells by suppressing intracellular glycine biosynthesis. J. Transl. Med. 2024, 22(1), 192. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Zhang, Y.; Ren, Y.; Luo, F.; Wu, Y.; Ran, H.; Cao, Y. Unleashing the Potent Antitumor Force: A Reactive Oxygen Species (ROS) Storm Formation by Ultrasound-Activatable Metal Nanosonosensitizers. Small 2026, 22(11), e13649. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Chen, Y.; Zhang, Y.; Hai, Y.; Fan, R.; Dou, J.; Lu, X.; Wang, W.; Zhang, B.; Hou, Z.; Liang, L.; Liu, Y.; Wei, G. HB023: A glutamine antagonist prodrug boosting antitumor lmmunity via PD-L1 suppression and mitochondrial membrane remodeling. J. Adv. Res. 2026, S2090-1232(26)00101-3. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Jia, H.; Tian, X.; Wang, G.; Zhang, X.; Mi, Z.; Jia, Y.; Wang, L.; Liang, Z.; Li, D.; Mao, X.; Liang, Y.; Niu, H. Inhibition of glutamine metabolism blocks tumor growth and sensitizes ccRCC to immune checkpoint blockade. J. Transl. Med. 2026. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ploessl, K.; Zhou, R.; Mankoff, D.; Kung, H.F. Metabolic Imaging of Glutamine in Cancer. J. Nucl. Med. 2017, 58(4), 533–537. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, F.; Hou, X.; Zhang, Q.; Ren, Y.; Zhu, H.; Yang, Z.; Xu, X. KRAS Mutation Detection with (2S,4R)-4-[18F]FGln for Noninvasive PDAC Diagnosis. Mol. Pharm. 2024, 21(4), 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.W.; Wang, J.; Georgiou, D.K.; Wen, X.; Cohen, A.S.; Geng, L.; Tantawy, M.N.; Manning, H.C. Feasibility of [(18)F]FSPG PET for Early Response Assessment to Combined Blockade of EGFR and Glutamine Metabolism in Wild-Type KRAS Colorectal Cancer. Tomography 2023, 9(2), 497–508. [Google Scholar] [CrossRef] [PubMed]
- Ayers, G.D.; Cohen, A.S.; Bae, S.W.; Wen, X.; Pollard, A.; Sharma, S.; Claus, T.; Payne, A.; Geng, L.; Zhao, P.; Tantawy, M.N.; Gammon, S.T.; Manning, H.C. Reproducibility and repeatability of 18F-(2S, 4R)-4-fluoroglutamine PET imaging in preclinical oncology models. PLoS ONE 2025, 20(1), e0313123. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhao, Y.; Chen, L.; Liu, Y.; Xie, H.; Wu, Z.; Zhang, L.; Fan, D.; Xu, X.; Ai, L.; Liu, Q.; Zhu, H.; Li, D. Distribution of glutamate and glutamine in tumor microenvironments of brain metastasis: Insights from biochemical analysis and molecular imaging. iScience 2025, 28(9), 113371. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Cheng, W.; Tian, H.; Huang, Y.; Huang, C.; Jia, Y.; Xu, L. Nano-purpurin-Cu delivery via TPGS-induced macropinocytosis enables cuproptosis/metabolic synergy to ablate cancer stemness and Boost immunotherapy in colorectal cancer. Biomaterials 2026, 328, 123874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Fan, R.; Hai, Y.; Chen, Y.; Lu, X.; Wang, W.; Dou, J.; Yan, J.; Su, C.; Chen, Y.; Yang, L.; Zhao, M.; Liang, L.; Wei, G. Immunometabolic Rewiring of Dendritic Cells to Overcome Glutamine-Driven Immune Suppression in Colorectal Cancer. Adv. Sci. (Weinh) 2026, 13(5), e13986. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Rodado, V.; Lita, A.; Dowdy, T.; Celiku, O.; Saldana, A.C.; Wang, H.; Yang, C.Z.; Chari, R.; Li, A.; Zhang, W.; Song, H.; Zhang, M.; Ahn, S.; Davis, D.; Chen, X.; Zhuang, Z.; Herold-Mende, C.; Walters, K.J.; Gilbert, M.R.; Larion, M. Metabolic plasticity of IDH1-mutant glioma cell lines is responsible for low sensitivity to glutaminase inhibition. Cancer Metab. 2020, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yang, D.; Chen, X.; Yang, Y.; Zhang, B.; Jiang, X.; Xing, L.; Yang, Y.; Sun, Y.; Li, N. Metabolic reprogramming in cancer: dysregulation of glucose, lipid, and amino acid pathways and therapeutic opportunities. Mol. Biomed. 2026, 7(1), 25. [Google Scholar] [CrossRef] [PubMed]
- Nan, D.; Yao, W.; Huang, L.; Liu, R.; Chen, X.; Xia, W.; Sheng, H.; Zhang, H.; Liang, X.; Lu, Y. Glutamine and cancer: metabolism, immune microenvironment, and therapeutic targets. Cell Commun. Signal 2025, 23(1), 45. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Qin, H.; Shen, J.; An, H.; Cao, Y. Beyond the tumor: Enhancing pancreatic cancer therapy through glutamine metabolism and innovative drug delivery. J. Cell Commun. Signal. 2025, 19(3), e70033. [Google Scholar] [CrossRef] [PubMed]
- Blatt, E.B.; DeBerardinis, R.J. Glutamine antagonists may KEAP lung cancer in check. Sci. Adv. 2024, 10(13), eado7808. [Google Scholar] [CrossRef] [PubMed]
- Djelti, F.; Hani, M.; Chetbani, Y.; Belaadi, A.; Ammarullah, M.I. Surviving the siege: A review on the metabolic hallmarks of cancer dormancy. Cancer Treat. Res. Commun. 2026, 47, 101123. [Google Scholar] [CrossRef] [PubMed]
- Novotná, K.; Tenora, L.; Prchalová, E.; Paule, J.; Alt, J.; Veeravalli, V.; Lam, J.; Wu, Y.; Šnajdr, I.; Gori, S.; Mettu, V.S.; Tsukamoto, T.; Majer, P.; Slusher, B.S.; Rais, R. Discovery of tert-Butyl Ester Based 6-Diazo-5-oxo-l-norleucine Prodrugs for Enhanced Metabolic Stability and Tumor Delivery. J. Med. Chem. 2023, 66(22), 15493–15510. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Pantel, A.R.; Li, S.; Lieberman, B.P.; Ploessl, K.; Choi, H.; Blankemeyer, E.; Lee, H.; Kung, H.F.; Mach, R.H.; Mankoff, D.A. [18F](2S,4R)4-Fluoroglutamine PET Detects Glutamine Pool Size Changes in Triple-Negative Breast Cancer in Response to Glutaminase Inhibition. Cancer Res. 2017, 77, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; DeBerardinis, R.J. In vivo analysis of lung cancer metabolism: nothing like the real thing. J. Clin. Invest. 2015, 125, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Cyriac, R.; Lee, K. Glutaminase inhibition as potential cancer therapeutics: current status and future applications. J. Enzym. Inhib. Med. Chem. 2024, 39, 2290911. [Google Scholar] [CrossRef]
- Best, S.A.; Gubser, P.M.; Sethumadhavan, S.; Kersbergen, A.; Negrón Abril, Y.L.; Goldford, J.; Sellers, K.; Abeysekera, W.; Garnham, A.L.; McDonald, J.A.; Weeden, C.E.; Anderson, D.; Pirman, D.; Roddy, T.P.; Creek, D.J.; Kallies, A.; Kingsbury, G.; Sutherland, K.D. Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metab. 2022, 34(6), 874–887.e6. [Google Scholar] [CrossRef] [PubMed]
- Rukonge, P.A.; Kawuribi, V.; Sheng, Y.; Wei, Q.; Kun, Y.; Niyodukunda, P.; Wang, T.; Lu, Z.; Miao, Y.; Xu, K.; Yu, G. Multi-omics profiling of intercellular immunometabolic heterogeneity highlights in lung cancer: Crosstalk mechanisms and resistance in the tumor-immune interface. Crit. Rev. Oncol. Hematol. 2026, 219, 105094. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hong, S.; Gu, K. A pancancer analysis reveals the oncogenic role of glutaminase 1 (GLS1) in tumor metabolism and immune evasion: a bioinformatics analysis. Discov. Oncol. 2025, 16(1), 2156. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Han, X.; Yang, Y.; Shen, J.; Yin, Y.; Liu, Y. Glutamine: a new strategy for targeted metabolic therapy in the tumor microenvironment. Cell Death Discov. 2025, 11(1), 459. [Google Scholar] [CrossRef] [PubMed]
- Vasan, K.; Chalmers, Z.R.; Kong, H.; Chandel, N.S. Tumor-intrinsic metabolic pathways essential for tumorigenesis and resistance to anti-PD1 in oncogenic Kras-driven lung adenocarcinoma. Cancer Metab. 2026. [Google Scholar] [CrossRef] [PubMed]
- Voor, S.F.; Cooper, A.J.L.; Pinto, J.T. Glutaminase I and glutamine transaminase-omega-amidase pathways in colorectal cancer: Metabolic reprogramming and emerging therapeutic strategies. Anal. Biochem. 2026, 709, 115989. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Main pathways of Gln transport and metabolism in the cytosol and mitochondria of cancer cells. Druggable targets for combination therapy are indicated in red, and corresponding drugs are shown in green. AKC, alpha-ketoglutarate. Asp, aspartate. BSO, L-buthionine sulfoximine. CQ, chloroquine. DHA, dihydroartemisinin. GA, glutaminase. Gln, glutamine. Glu, glutamate. GSH, glutathione. Leu, leucine. mTORC1, mammalian target of rapamycin complex 1. OAA, oxaloacetate. Orn, ornithine. Phe, phenylalanine. ROS, reactive oxygen species. TCA, tricarboxylic acid. Trp, tryptophan. UA, ursodeoxycholic acid.
Figure 1.
Main pathways of Gln transport and metabolism in the cytosol and mitochondria of cancer cells. Druggable targets for combination therapy are indicated in red, and corresponding drugs are shown in green. AKC, alpha-ketoglutarate. Asp, aspartate. BSO, L-buthionine sulfoximine. CQ, chloroquine. DHA, dihydroartemisinin. GA, glutaminase. Gln, glutamine. Glu, glutamate. GSH, glutathione. Leu, leucine. mTORC1, mammalian target of rapamycin complex 1. OAA, oxaloacetate. Orn, ornithine. Phe, phenylalanine. ROS, reactive oxygen species. TCA, tricarboxylic acid. Trp, tryptophan. UA, ursodeoxycholic acid.

Figure 2.
Non-competitive allosteric inhibitors of GA: compound 968 prevents the formation of active tetramers of GLS and GLS2. BPTES and CB-839 are isoform-specific and only bind to the interface between two dimers of the GLS tetramer. The three drugs block the conformational change required for catalytic turnover and GA activity.
Figure 2.
Non-competitive allosteric inhibitors of GA: compound 968 prevents the formation of active tetramers of GLS and GLS2. BPTES and CB-839 are isoform-specific and only bind to the interface between two dimers of the GLS tetramer. The three drugs block the conformational change required for catalytic turnover and GA activity.

Figure 3.
DON, prodrugs and new chemicals to block tumorigenesis: DON, DRP-104, JHU083, HB023, and IPN60090.
Figure 3.
DON, prodrugs and new chemicals to block tumorigenesis: DON, DRP-104, JHU083, HB023, and IPN60090.

Figure 4.
Mechanisms targeting cancer cells in combination therapy, together with ROS enhancement and immunotherapy. Blocking GA constitutes an essential component of the combination therapy, which may also incorporate immunotherapeutic drugs, such as checkpoint inhibitors to prevent PD-1/PD-L1 interaction, thereby enabling the immune system to recognize and attack tumor cells. Additional and synergistic effects can be achieved by targeting metabolic reprogramming that resets the tumor microenvironment for cancer growth. In this context, blocking GA allows for greater availability of Gln in the TME, a needed nutrient for T-cells. ATP, adenosine triphosphate. GA, glutaminase. Gln, glutamine. Glu, glutamate. GSH, glutathione. PD-1, programmed cell death protein 1. PD-L1, programmed cell death ligand 1. ROS, reactive oxygen species. TCA, tricarboxylic acid. TME, tumor microenvironment.
Figure 4.
Mechanisms targeting cancer cells in combination therapy, together with ROS enhancement and immunotherapy. Blocking GA constitutes an essential component of the combination therapy, which may also incorporate immunotherapeutic drugs, such as checkpoint inhibitors to prevent PD-1/PD-L1 interaction, thereby enabling the immune system to recognize and attack tumor cells. Additional and synergistic effects can be achieved by targeting metabolic reprogramming that resets the tumor microenvironment for cancer growth. In this context, blocking GA allows for greater availability of Gln in the TME, a needed nutrient for T-cells. ATP, adenosine triphosphate. GA, glutaminase. Gln, glutamine. Glu, glutamate. GSH, glutathione. PD-1, programmed cell death protein 1. PD-L1, programmed cell death ligand 1. ROS, reactive oxygen species. TCA, tricarboxylic acid. TME, tumor microenvironment.


Table 1.
Relevant drug combination studies involving the GA inhibitor compound 968.
| Drug | Model | Key effect(s)1 | Reference |
| PP242 | GBM2 mice xenografts | Inhibiting mTORC13 | [33] |
| Dihydroartemisinin | HCC4 in vitro | Activating apoptosis by increasing ROS5 | [34] |
| Chloroquine | NSCL56 in vitro | Inhibiting autophagy | [35] |
| Adriamycin | MCF-7 breast cancer cells in vitro | Inhibiting P-gp7 and overcoming drug resistance | [36] |
| Anti-PD-L18 | Ovarian cancer in vitro and in mice xenografts | Increasing apoptosis and immune response | [32] |
1 Key effect(s) in addition to glutaminase inhibition are remarked. 2GBM, glioblastoma multiforme. 3mTORC1, mammalian target of rapamycin complex 1. 4HCC, hepatocellular carcinoma. 5ROS, reactive oxygen species. 6NSCLC, non-small cell lung cancer. 7P-gp, P-glycoprotein. 8PD-L1, programmed cell death ligand 1.
Table 2.
Chemicals used in combination with the GLS inhibitor BPTES that exhibit promising effects in antitumor therapy.
Table 2.
Chemicals used in combination with the GLS inhibitor BPTES that exhibit promising effects in antitumor therapy.
| Drug(s)/agent(s) | Model(s) | Key effect(s)1 | Reference |
| Dibenzozepine | ALL2 mice xenografts | γ-secretase inhibition, NOTCH13 cleavage, and activation of autophagy | [38] |
| 5-Fluorouracil | NSCLC4 in vitro | Inhibiting thymidylate synthase and CPSII5 | [39] |
| Etoposide and cisplatin | TNBC6 in vitro | Activating apoptosis by a BAX/BCL-2 mechanism | [40] |
| Dichloroacetate | CRC7 and cervical cancer cells in vitro | Decrease of PPP8 and activation of apoptosis | [41] |
| Bicalutamide | Prostate cancer in vitro and in rat xenografts | Blocking AR9 and lipid metabolism | [37] |
| FX-11 | TNBC6 mice xenografts and PDO10 | Inhibiting LDHA11 | [42] |
| PAM12 (ROS13) | TNBC6 in vitro | Activating apoptosis by DNA damage and inhibiting ATP14 production | [43] |
| Doxorubicin, Fe3+, EGCG15 and BPTES nanoparticles | PDAC16 in vitro, mice xenografts, and PDO9 | Activating apoptosis by increasing ROS12 and DNA damage | [44] |
| Anti-PD-L117 | Lung and colon cancer in vitro and in mice xenografts | Increasing Fas expression showing a synergistic antitumor effect | [45] |
1 Key effect(s) in addition to GLS inhibition are indicated. 2ALL, acute lymphoblastic leukemia. 3NOTCH1, neurogenic locus notch homolog protein 1. 4HNSCC, head and neck squamous cell carcinoma. 5CPSII, carbamoyl phosphate synthetase II. 6TNBC, triple-negative breast cancer. 7CRC, colorectal cancer. 8PPP, pentose phosphate pathway. 9AR, androgen receptor. 10PDO, patient-derived organoids. 11LDHA, lactate dehydrogenase A. 12PAM, plasma-activated medium. 13ROS, reactive oxygen species. 14ATP, adenosine triphosphate. 15EGCG, epigallocatechin gallate. 16PDAC, pancreatic ductal adenocarcinoma. 17PD-L1, programmed cell death ligand 1.
Table 3.
Studies demonstrating promising efficacy of GLS inhibitor CB-839 in combination therapy.
| Drug(s) | Model(s) | Key effect(s)1 | Reference |
| AZD8055 | TNBC2 mice xenografts | Inhibiting mTORC13 | [50] |
| Everolimus | RCC4 mice xenografts | Inhibiting mTORC13 | [51] |
| MLN128 | Mice xenografts of lung SCC5, HNSCC6 and osteosarcoma | Inhibiting mTORC13 | [29] |
| TAK228 | Lung SCC5 mice xenografts | Inhibiting mTORC13 | [52] |
| CPI-613 | 2D culture, 3D culture, and mice xenografts of HNSCC6 | Inhibiting TCA7 cycle | [53] |
| THZ1 | NSCLC8 in vitro | Inhibiting CDK79 | [54] |
| Palbociclib | CRC10 mice xenografts | Inhibiting CDK4/611 | [55] |
| Palbociclib | Lung adenocarcinoma cells | Inhibiting CDK4/611 | [56] |
| 5-Fluorouracil | CRC10 mice xenografts | Inhibiting thymidylate synthase and inducing IL-812 to attract neutrophils into the tumor | [57] |
| 5-Fluorouracil | HCC13 mice xenografts | Inhibiting thymidylate synthase, enhancing oxidative stress, and increasing ferroptosis | [58] |
| 5-Fluorouracil and cisplatin | ESCC14 in vitro and mice xenografts | Increasing apoptosis targeting TIGAR15 | [59] |
| Cyclosporin A | NSCLC8 mice xenografts | Inducing NRF216 | [60] |
| Deferoxamine | HCC13 in vitro | Iron deficiency | [61] |
| V-9302 | PDAC17 in vitro | Inhibiting ASCT218 Gln transport | [62] |
| DRB-18 | ICC19 in vitro | Inhibiting glucose transport | [63] |
| Ursodeoxycholic acid | Liposarcoma mice xenografts | Inhibiting SLC7A1120 cystine transport and GSH synthesis | [64] |
| Aspirin | CRC10 mice xenografts | Inhibiting SLC7A1120 and SLC7A521 | [65] |
| EGCG22 | Multiple myeloma in vitro | Activating apoptosis by a BAX/BCL-2 mechanism | [66] |
| Venetoclax | AML23 in vitro | Inhibiting BCL-2 | [67] |
| BSO24, auranofin, RT25 | Cervix cancer in vitro and mice xenografts | Increasing oxidative stress | [68] |
| RT25 | HNSCC6 in vitro and mice xenografts | Increasing oxidative DNA damage and apoptosis | [69] |
| Dihydroartemisinin | GBM26 in vitro | Increasing oxidative stress and apoptosis | [22] |
| Oxamate, D609, doxorubicin | Breast cancer in vitro | Inhibiting LDH27 and PC-PLC28 | [48] |
| ENZA29, IACS30 | Prostate cancer cells in vitro and blood cells from patients with prostate cancer | Increasing ROS31 and decreasing oxidative phosphorylation | [46] |
| V-9202 | HCC13 in vitro and mice xenografts | Lowering Gln transport and GSH32, increasing ROS31, and apoptosis | [70] |
| Selumetinib | NSCLC8 in vitro and mice xenografts | Inhibiting ERK33, increasing ROS31 and autophagy | [47] |
| Osimertinib | Lung adenocarcinoma in vitro | Inhibiting tyrosine kinase | [71] |
| Sunitinib or axitinib | RCC4 mice xenografts | Inhibiting tyrosine kinase | [51] |
| anti-PD-134 or anti-PD-L135 | Mice bearing syngeneic colon carcinoma | Inhibiting immune checkpoint proteins and ligands | [72] |
| anti-CD15236 or anti-PD-134 | Melanoma cells in vitro and mice xenografts | Activating T-cell-mediated immunotherapy | [73] |
| Bevacizumab | Ovarian cancer mice xenografts | Inhibiting VEGF37 | [74] |
| Cabozantinib | RCC3 mice xenografts | Inhibiting VEGFR38 | [51] |
| Panitumumab | Metastatic RCC3 patients | Inhibiting EGFR39 | [75] |
| Cetuximab | CRC10 in vitro and mice xenografts | Inhibiting EGFR39 | [76] |
| Metformin | Osteosarcoma in vitro and in mice xenografts | Disrupting metabolism | [77] |
1 Key effect(s) in addition to glutaminase inhibition are indicated. 2TNBC, triple-negative breast cancer. 3mTORC1, mammalian target of rapamycin complex 1.4 RCC, renal cell carcinoma. 5 SCCC, squamous cell carcinoma. 6HNSCC, head and neck squamous cell carcinoma. 7TCA, tricarboxylic acid. 8NSCLC, non-small cell lung cancer. 9CDK7, cyclin-dependent kinase 7. 10CRC, colorectal cancer. 11CDK4/6, cyclin-dependent kinase 4/6. 12IL-8, interleukin-8. 13HCC, hepatocellular carcinoma. 14ESCC, esophageal squamous cell carcinoma. 15TIGAR, TP53-induced glycolysis and apoptosis regulator. 16NRF2, nuclear factor erythroid 2-related factor 2. 17PDAC, human pancreatic adenocarcinoma. 18ASCT2, Alanine Serine Cysteine Transporter 2. 19ICC, intrahepatic cholangiocarcinoma. 20SLC7A11, Solute Carrier Family 7 Member 11. 21SLC7A5, Solute Carrier Family 7 Member 5. 22EGCG, epigallocatechin gallate. 23AML, acute myeloid leukemia. 24BSO, L-buthionine sulfoximine. 25RT, radiation. 26GBM, glioblastoma multiforme. 27LDH, lactate dehydrogenase, 28PC-PLC, phosphatidylcholine-specific phospholipase C. 29ENZA, anti-androgen enzalutamide. 30IACS, mitochondrial electron transport component complex I inhibitor IACS-010759. 31ROS, reactive oxygen species. 32GSH, glutathione. 33ERK, extracellular signal-regulated kinase. 34PD-1, programmed cell death protein 1. 35PD-L1, programmed cell death ligand 1. 36CD152, cluster of differentiation 152. 37VEGF, vascular endothelial growth factor. 38VEGFR, vascular endothelial growth factor receptor. 39EGFR, epidermal growth factor receptor.
Table 4.
JHU083 as a novel DON prodrug for antitumor therapy.
| Additional Drug | Model | Key effect(s)1 | Reference |
| Anti-PD-12 | Mice models bearing colon cancer, lymphoma, and melanoma | Increasing apoptosis and inhibiting antitumor immune response | [82] |
| None | GBM3 in vitro and in orthotopic mice | Inhibiting mTORC14 | [83] |
| EVax5 | Lung transgenic mice | Inhibiting EGFR6 | [84] |
| Anti-CD1527 or anti-PD-12 | Myeloid cells and mice xenografts | Activating T-cell-mediated immunotherapy | [85] |
| None | Thyroid cancer mice xenografts | Inhibiting CD478 and PD-L19 | [86] |
| BSO10 | GBM3 in vitro and mice intracranial xenografts | Inhibiting GSH11-dependent antioxidant capacity | [87] |
| Elimusertib | ACC12 in vitro and mice xenografts | Inhibiting DNA damage response | [88] |
| None | Prostate and bladder cancer in vitro and syngeneic heterotopic mouse models | Increasing apoptosis and decreasing TCA14 cycle and purine metabolism | [89] |
1Key effect(s) in addition to glutamine metabolism inhibition is remarked. 2PD-1, programmed cell death protein. 3GBM, glioblastoma multiforme. 4mTORC1, mammalian target of rapamycin complex 1. 5EVax, EGFR6 peptide vaccine. 6EGFR, epidermal growth factor receptor. 7CD152, cluster of differentiation 152. 8CD47, cluster of differentiation 47. 9PD-L1, programmed cell death ligand 1. 10BSO, buthionine sulfoximine. 11GSH, glutathione. 13Adrenocortical Carcinoma. 14TCA, tricarboxylic acid.
Table 5.
DRP-104 as a novel DON prodrug for antitumor therapy.
| Additional Drug | Model | Key effect(s)1 | Reference |
| None | GBM2 mice xenografts | Inhibiting mTORC13 | [33] |
| None | HCC4 in vitro | Activating apoptosis by increasing ROS5 | [34] |
| None | NSCL56 in vitro | Inhibiting autophagy | [35] |
| Anti-PD-17 | CADC8 in vitro and mice xenografts | Inhibiting cytokines | [90] |
| Trametinib | PDAC9 in vitro and syngeneic mice model | Inhibiting MAPK10 and ERK11 kinase 1/2 | [91] |
| RSL312 | HNSCC13 in vitro and mice xenografts | Inhibition of GPX414 and activation of ferroptosis | [92] |
| Anti-PD-17 | Orthotopic lung cancer mice | Inhibiting cytokines and increasing antitumor T cell response | [93] |
| None | Prostate cancer in vitro and mice xenografts | Activation of apoptosis, targeting TCA cycle and nucleotide synthesis | [94] |
| MTDIA15 | Prostate cancer in vitro and mice xenografts | Inhibition of MTAP16 | [95] |
1 Key effect(s) in addition to glutamine metabolism inhibition is remarked. 2GBM, glioblastoma multiforme. 3mTORC1, mammalian target of rapamycin complex 1. 4HCC, hepatocellular carcinoma. 5ROS, reactive oxygen species. 6NSCLC, non-small cell lung cancer. 7PD-1, programmed cell death antibody. 8CADC, colon adenocarcinoma. 9PDAC, pancreatic ductal adenocarcinoma. 10MAPK, mitogen-activated protein kinase. 11ERK, extracellular signal-regulated kinase. 12RSL3, RAS-selective letal 3. 13HNSCC, head and neck squamous cell carcinoma. 14GPX4, glutathione peroxidase 4. 15MTDIA, inhibitor of MTAP16. 16MTAP, methylthioadenosine phosphorylase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



