Submitted:
10 June 2026
Posted:
11 June 2026
You are already at the latest version
Abstract
Post-stroke cognitive impairment (PSCI) affects up to half of survivors and is a leading cause of long-term disability, making it the norm rather than the exception. Despite its prevalence and impact, PSCI remains largely unaddressed in rehabilitation programs, which focus primarily on physical recovery. The subtle nature of cognitive deficits in the acute stages renders them difficult to detect and address, yet they often worsen over time, highlighting the need to understand the biological processes driving PSCI. Growing evidence implicates inflammation not as a singular event, but as a dynamic, phase-specific process that evolves from acute injury to chronic maladaptation, driving cognitive decline. Acute neuroinflammation, involving microglial activation and cytokine release, initiates secondary brain injury and evolves to a chronic state with sustained glial activation and peripheral immune cell involvement, leading to synaptic loss, white matter injury, and network dysfunction. Preclinical models demonstrate that immunomodulatory interventions can mitigate cognitive deficits, highlighting their therapeutic potential. The future management of PSCI will therefore require a dual approach by suppressing maladaptive inflammation through phase-specific immunomodulation, integrated with cognitively focused rehabilitation. Developing inflammatory biomarkers for patient stratification will be essential to personalize these strategies and translate them into successful clinical outcomes.
Keywords:
post-stroke cognitive impairment (PSCI)
; neuroinflammation
; stroke recovery
1. Introduction
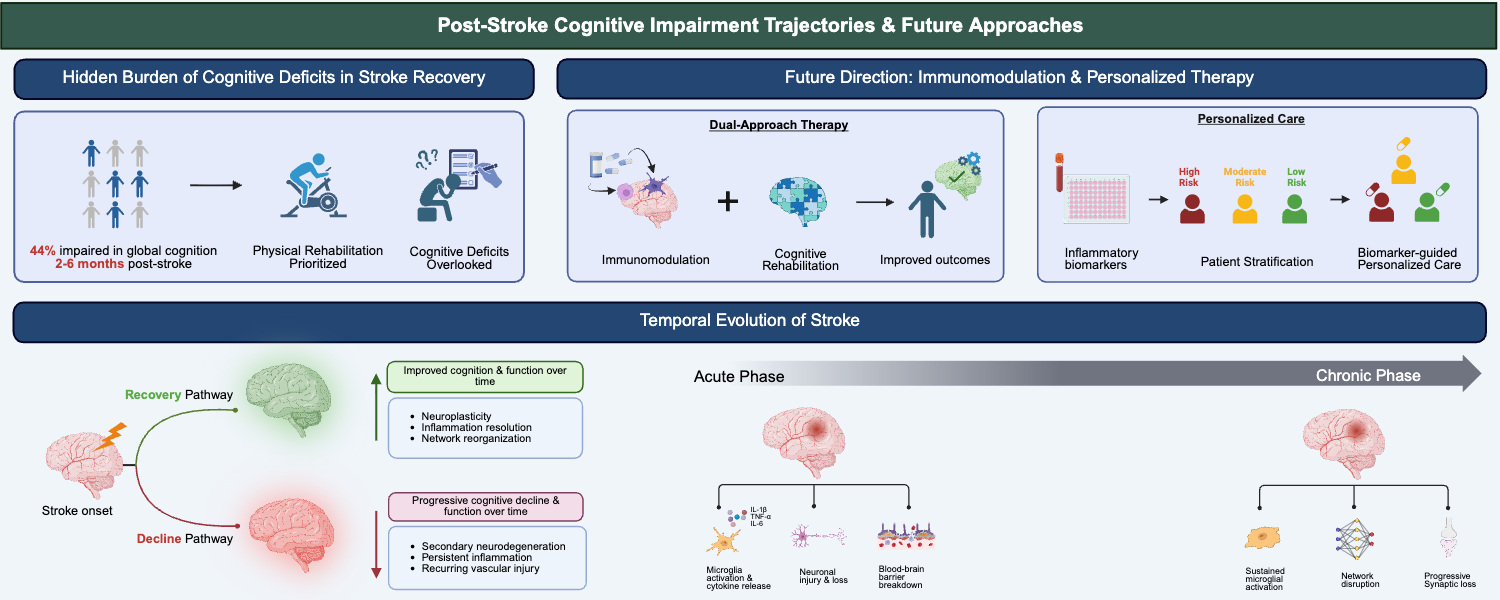
Post-stroke cognitive impairment (PSCI) is a critical yet historically underrecognized consequence of cerebrovascular events, with nearly 44% of stroke survivors exhibiting cognitive deficits within 2–6 months [1]. Stroke management has traditionally focused on motor rehabilitation and the prevention of recurrent vascular events [1]. Yet cognitive deficits, ranging from subtle impairments in attention and executive function to dementia, are increasingly recognized as major contributors to long-term disability [2]. PSCI arises from the interplay between the initial vascular injury and secondary processes that evolve, including neurodegeneration and chronic inflammation, which may not be apparent during the acute phase [1,2,3,4,5]. Preclinical studies indicate that immune-modulating drugs can improve post-stroke cognitive function, supporting further investigation of immunomodulation as a strategy to mitigate cognitive impairment after stroke [6]. This scoping review synthesizes current knowledge from clinical studies, preclinical models, and mechanistic investigations to map the landscape of PSCI research, with particular emphasis on inflammatory pathways and identifying gaps that must be addressed to advance therapeutic development.
1.1. Epidemiology of PSCI
Studies have shown that PSCI affects a substantial proportion of stroke survivors. Estimates vary widely from 20% to 80%, reflecting differences in assessment timing, population characteristics, diagnostic criteria employed, and sensitivity of cognitive instruments used [1,7,8]. Despite this variability, large-scale analyses consistently report that cognitive impairments, including post-stroke dementia, are common following stroke [7,8,9]. A landmark meta-analysis by Pendlebury and Rothwell demonstrated that approximately one-third of stroke survivors develop new dementia, with rates increasing substantially in those with recurrent strokes or pre-existing cognitive vulnerability [7]. Beyond dementia-level impairment, a systematic review of cognitive impairment no dementia (CIND) reported that many stroke survivors develop mild-to-moderate deficits within the first year [10], reinforcing that PSCI spans a spectrum from subtle impairments to overt dementia [8,11]. At the population level, studies from diverse geographic regions have confirmed that PSCI represents a global health burden, affecting both high-income and low-to-middle-income countries, though detection rates may vary based on healthcare infrastructure and screening practices [11]. While methodological differences account for much of the variability in prevalence estimates, the collective evidence makes it clear that cognitive impairment after stroke is the norm rather than the exception. Accordingly, standardized cognitive screening should be an integral component of post-stroke care [12,13].
1.2. Clinical Significance and Impact on Rehabilitation Outcomes
Successful post-stroke rehabilitation depends not only on recovery of motor function but also on cognitive abilities [14,15,16]. PSCI exerts profound effects on functional independence, rehabilitation participation, and long-term disability that often exceed the impact of physical deficits alone [2,17]. Although lesion size and location were once thought to determine the severity of post-stroke cognitive problems, accumulating evidence shows that these factors alone cannot explain the substantial variability in outcomes; in fact, individuals with small infarcts may experience disproportionately severe cognitive impairment [2,7,8,11,18]. Even when controlling for clinical factors such as age and vascular risk, cognitive impairment independently predicts reduced functional recovery and is associated with higher mortality risk [14,19]. When unrecognized or unsupported, cognitive deficits limit patients’ ability to return to work, participate in social and community activities, and successfully resume everyday routines. These impairments also carry significant downstream consequences. PSCI increases caregiver burden, raises the likelihood of institutionalization, and contributes to prolonged hospital stays and higher healthcare utilization [20,21]. Despite this broad and well-documented impact, routine cognitive assessment remains inconsistently implemented across stroke care pathways, and few interventions specifically target cognitive recovery, highlighting a gap between clinical need and current therapeutic options [16].
1.3. Gap Between Clinical Burden and Mechanistic Understanding
Although the clinical importance of PSCI is well established and its mechanisms are increasingly recognized, the underlying processes remain poorly characterized compared to the relatively well-understood pathophysiology of acute ischemic injury [22,23]. While acute stroke research has focused predominantly on neuronal death within the infarct core and penumbra, cognitive outcomes appear to depend on complex interactions between direct tissue injury, remote network dysfunction, white matter damage, and secondary neurodegenerative processes that evolve over weeks to months [24]. The disconnect between lesion size or location and cognitive outcomes has prompted mechanistic studies to investigate secondary processes that may explain interindividual variability in PSCI. Chronic inflammation, blood–brain barrier (BBB) dysfunction, and impaired neuroplasticity are emerging as central drivers of post-stroke cognitive decline, interacting in complex ways to influence recovery and functional outcomes [25]. Translating mechanistic insights into clinical trials has been difficult. Cognitive endpoints are frequently overlooked, and screening tools often fail to detect subtle deficits, leading to underestimation of PSCI prevalence and limiting the incorporation of cognitive endpoints into trial design [26]. Longitudinal, multimodal studies are needed to translate preclinical findings into outcomes that support early detection, personalized treatment, and long-term management [27].
1.4. Justification for Focusing on Inflammation as a Key Mechanism
Inflammation has emerged as a central pathophysiological process linking acute stroke injury to chronic cognitive decline, operating through both direct neurotoxic effects and indirect disruption of neuroplasticity and repair mechanisms [28,29]. Unlike the transient ischemic cascade that resolves within hours to days, inflammatory processes persist for weeks to months after stroke, corresponding temporally with the evolution of cognitive deficits observed in clinical populations [30]. Preclinical studies have demonstrated that microglial activation, peripheral immune cell infiltration, and sustained cytokine production contribute to secondary neurodegeneration in regions remote from the infarct, including hippocampal and cortical areas critical for cognitive function [31,32]. Clinical evidence supports this mechanistic framework, with elevated inflammatory biomarkers in acute stroke patients predicting worse cognitive outcomes at follow-up, independent of infarct volume [33]. The inflammatory response also intersects with other processes implicated in PSCI, including blood-brain barrier (BBB) dysfunction, white matter injury, and impaired adult neurogenesis, positioning it as a potential therapeutic target that could address multiple pathogenic pathways simultaneously [34]. Given that post-stroke inflammation is not static but fluctuates between reparative and neurotoxic states, delineating the temporal point at which these responses become chronically maladaptive may help identify when patients are most likely to benefit from targeted intervention. This prolonged and potentially modifiable trajectory stands in contrast to the narrow window for acute neuroprotection, underscoring the appeal of inflammation-focused strategies for preventing or attenuating PSCI [35].
2. Clinical Literature on PSCI
Over the past two decades, clinical characterization of PSCI has broadened considerably, shifting from an emphasis on vascular dementia to recognition of a spectrum of domain-specific deficits that vary in severity [1,18]. Contemporary studies consistently show prominent impairments in executive function, processing speed, and attention, distinguishing PSCI from the episodic memory predominant patterns typically seen in Alzheimer’s disease (AD) [2,8]. This heterogeneity reflects differences in lesion location, cerebrovascular mechanisms, premorbid cognitive reserve, and coexisting neuropathologies [8,11,18]. Multiple standardized cognitive screening tools and diagnostic criteria have been developed to support early detection and longitudinal monitoring; however, adoption in routine clinical pathways remains inconsistent [1,7]. A clear understanding of PSCI phenotypes, risk factors, and natural history is critical for guiding prognosis, tailoring rehabilitation strategies, and informing the development of targeted therapeutic interventions [9,16].
2.1. Cognitive Domains Affected
PSCI presents a profile distinct from other dementia syndromes, with executive dysfunction and slowed processing speed emerging as the most prominent and consistently affected domains [36,37]. Executive deficits, including planning, organization, cognitive flexibility, and working memory, primarily arise from damage to frontal–subcortical circuits affected by cortical and subcortical infarcts [38]. Slowed processing speed is often associated with white matter damage and disrupted network efficiency. This leads to reduced processing efficiency, reflected in slower information processing and delayed responses across cognitive tasks [39]. Attention is also frequently impaired, particularly for sustained and divided tasks, and may contribute to difficulties in other cognitive domains [40]. Episodic memory is generally less affected than in AD, but difficulties with forming new memories (encoding) and recalling them (retrieval) can occur, particularly after strokes in the left hemisphere or in key areas such as the hippocampus or thalamus [41]. Language deficits beyond classical aphasia, such as word-finding challenges and reduced verbal fluency, are common following left hemisphere strokes [42]. Overall, the specific pattern of cognitive impairment varies with lesion location; cortical strokes generally produce focal deficits, whereas small vessel disease is associated with more diffuse impairments, primarily affecting executive function and processing speed [43].
2.2. Assessment Tools and Diagnostic Criteria
Accurate diagnosis of PSCI is essential for early identification, targeted intervention, and monitoring of recovery trajectories. This will require sensitive screening tools capable of detecting the executive and attention deficits characteristic of vascular cognitive impairment, along with the standardized diagnostic criteria to ensure consistency across clinical and research settings [44]. The Montreal Cognitive Assessment (MoCA) has become the most widely recommended screening tool for PSCI, demonstrating superior sensitivity compared to the Mini-Mental State Examination (MMSE), particularly for detecting executive dysfunction and mild cognitive impairment [45,46]. The MoCA includes assessments of visuospatial abilities, executive function, attention, and delayed recall, with a recommended cut-off score adjusted for educational level. The Vascular Dementia Assessment Scale – Cognitive subscale (VASD-Cog) was specifically designed to capture the cognitive profile of vascular dementia and shows sensitivity to executive and attention domains [47]. For formal diagnosis, the DSM-5 introduced the category of vascular neurocognitive disorder, with criteria requiring evidence of cognitive decline from a previous level of performance, impairment in one or more cognitive domains, and a temporal relationship to cerebrovascular events [48]. The National Institute of Neurological Disorders and Stroke-Canadian Stroke Network (NINDS-CSN) harmonizes standards to provide additional research criteria emphasizing the importance of comprehensive neuropsychological testing across multiple domains [23]. A comprehensive neuropsychological assessment allows for a detailed evaluation of memory, executive function, attention, language, and visuospatial abilities, which is critical for identifying subtle or domain-specific deficits that brief screening tools may miss.
Despite these advances, significant variability persists in clinical practice regarding the timing of assessment, choice of instruments, and interpretation of results, with many stroke survivors never receiving formal cognitive evaluation [49]. Imaging and biomarker studies may complement cognitive assessments by identifying structural or functional correlates of impairment, although these approaches are not yet part of routine diagnostic criteria. Overall, combining sensitive screening instruments, comprehensive neuropsychological evaluation, and standardized diagnostic frameworks is essential for accurate identification of PSCI, guiding rehabilitation planning, and supporting research into effective interventions.
2.3. Risk Factors and Predictors
Multiple demographic, clinical, and neuroimaging factors predict the development and severity of PSCI, with implications for identifying high-risk patients who may benefit from targeted monitoring and interventions [7]. Advanced age represents one of the most consistent risk factors, with older stroke survivors experiencing higher rates of cognitive decline and dementia [50]. Pre-existing cognitive impairment or low cognitive reserve, often indicated by lower educational attainment, significantly increases vulnerability to PSCI as the brain has diminished capacity to compensate for stroke-related injury [51]. Stroke characteristics, including larger infarct volume, strategic lesion locations such as the thalamus, angular gyrus, and caudate, and recurrent strokes, substantially elevate risk [52]. The burden of white matter hyperintensities on neuroimaging, reflecting chronic small vessel disease, independently predicts cognitive outcomes and may present cumulative cerebrovascular damage that exceeds the impact of the index stroke [53]. Vascular risk factors such as hypertension, diabetes mellitus, atrial fibrillation, and hyperlipidemia contribute to both stroke risk and subsequent cognitive decline through mechanisms involving chronic hypoperfusion and endothelial dysfunction [54]. Genetic factors, including Apolipoprotein E (APOE) ε4 carrier status, modify risk through interactions with vascular pathology and amyloid deposition [55]. Post-stroke depression, occurring in approximately one-third of survivors, represents both a consequence and potential contributor to cognitive impairment through shared pathophysiological mechanisms and reduced engagement in rehabilitation [56]. Biomarkers, including elevated inflammatory markers, brain-derived neurotrophic factor levels, and neuroimaging measures of brain atrophy, are under investigation as predictors of cognitive trajectory [57]. Other clinical comorbidities, such as sleep disorders and cardiovascular disease, may further modulate cognitive recovery by impairing rehabilitation engagement or exacerbating neuroinflammatory processes. Socioeconomic and lifestyle factors, including education, social engagement, physical activity, and adherence to secondary prevention strategies, also influence both the risk and severity of PSCI. Understanding these predictors is critical for early identification of high-risk patients, personalized rehabilitation planning, and the development of preventive and therapeutic strategies targeting the mechanisms underlying PSCI.
2.4. Temporal Patterns
PSCI unfolds along distinct temporal trajectories, reflecting the dynamic interplay between acute injury, recovery processes, and long-term neurodegeneration (Figure 1). In the acute phase, spanning the first days to weeks after stroke, cognitive deficits are highly prevalent, affecting 40% to 70% of patients when systemically assessed [58,59]. At this stage, impairments often reflect the direct effects of neuronal injury, network disruption, and systemic factors, but performance can be influenced by delirium, fatigue, sedation, or difficulty engaging with testing. As patients enter the subacute phase, typically weeks to months post-stroke, recovery patterns become more variable. Some individuals demonstrate remarkable improvement, driven by resolution of diaschisis, neuroplasticity, and functional reorganization, while others continue to experience persistent deficits or even worsening cognition [60,61]. Notably, a subset of patients who appear cognitively intact acutely, approximately 10% to 30%, may develop delayed cognitive decline, suggesting that secondary neurodegenerative processes extend beyond the initial injury [62]. By the chronic phase, beyond six months, cognitive trajectories diverge, where some individuals achieve stable function, while others exhibit progressive decline reminiscent of degenerative dementia [63]. Recurrent strokes or new vascular events during follow-up substantially increase the risk of stepwise deterioration, with each additional event multiplying the likelihood of severe impairment [64]. The fact that cognitive decline can emerge months or years after the infarct highlights the role of chronic inflammation, ongoing white matter injury, and neurodegenerative cascades that persist well beyond the acute insult [65]. Longitudinal studies further show that early cognitive status, particularly within the first three months, provides important prognostic information for long-term outcomes, although accurate prediction at the individual level remains challenging [66]. Understanding these temporal patterns is critical for timing assessments, guiding rehabilitation strategies, and identifying intervention windows that could prevent or slow progressive cognitive decline, emphasizing that PSCI is a dynamic process rather than a static consequence of stroke.
2.5. Distinguishing Cognitive Impairment from Mixed Pathology
Distinguishing PSCI from cognitive decline due to mixed pathology is a complex and clinically important challenge. Many stroke survivors have overlapping pathologies, with cerebrovascular disease and Alzheimer-type changes frequently coexisting in older adults [67,68]. Autopsy studies indicate that more than half of patients with vascular dementia also show Alzheimer’s pathology, such as amyloid plaques and neurofibrillary tangles, while many patients diagnosed with AD exhibit significant cerebrovascular lesions [69]. These pathologies interact synergistically, where vascular injury can lower the threshold for clinical expression of amyloid-related deficits, and amyloid pathology can exacerbate vascular effects [70,71].
Pure vascular cognitive impairment is typically characterized by an abrupt onset of cognitive decline following stroke, stepwise progression with recurrent events, and prominent executive dysfunction relative to memory impairment. Imaging often reveals specific infarcts or extensive white matter disease without marked hippocampal atrophy [72]. However, many stroke patients also harbour preclinical Alzheimer’s pathology, which can influence post-stroke cognitive outcomes. Cerebrospinal fluid analysis and Positron Emission Tomography (PET) imaging of amyloid-β and tau can help detect mixed pathology, though their cost, invasiveness, and limited impact on routine management have prevented widespread adoption [57,73].
Clinically, differentiating pure vascular from mixed pathology is important for prognosis, rehabilitation planning, and potential therapeutic approaches targeting both vascular and amyloid-related mechanisms, although effective treatments remain limited [70,71]. Integrating longitudinal cognitive assessments, neuroimaging, biomarkers, and detailed clinical history offers the best strategy for disentangling PSCI from mixed pathology, enabling more personalized care and realistic prognostic counselling. Recognizing the interplay between vascular and neurodegenerative mechanisms emphasizes that post-stroke cognitive decline is rarely purely vascular and underscores the need for multidimensional assessment strategies.
3. Animal Models
While animal models of stroke have been invaluable for uncovering ischemic mechanisms and testing therapies, their use in studying PSCI has been constrained by methodological challenges and an emphasis on acute neuroprotection over long-term cognitive outcomes [74,75]. Improvements in rodent cognitive testing and stroke induction techniques now allow for systematic investigation of the relationship between cerebrovascular injury and cognitive dysfunction across diverse stroke models [76,77]. These models recapitulate distinct aspects of human disease, from large territorial infarcts caused by proximal arterial occlusion to the diffuse white matter injury characteristic of chronic cerebral hypoperfusion [78,79,80]. The choice of model critically influences the cognitive domains affected, the temporal trajectory, and translational relevance to human populations [75,77,81]. Thus, understanding the strengths and limitations of each model is essential for designing studies that address specific mechanistic questions relevant to PSCI and interpreting preclinical data. This section will focus on the current stroke models used, methods for assessing cognitive function, and the translational challenges of preclinical findings.
3.1. Common Stroke Models and Their Strengths and Limitations
A range of experimental stroke models has been established, including middle cerebral artery occlusion (MCAO), photothrombotic stroke, embolic models, and endothelin-1 (ET-1) models, each with distinct advantages and limitations that make them suited to different aspects of ischemic injury and the cognitive impairments that follow [74,75,76,77,78,79,80,81,82,83,84,85] (Table 1). MCAO is the most widely used model, involving either permanent or transient occlusion of the middle cerebral artery to produce cortical and striatal infarction mimicking large vessel occlusion in humans [74,82]. Transient MCAO, typically using intraluminal filament insertion, allows investigation of reperfusion injury and produces variable infarct sizes depending on occlusion duration. In contrast, permanent MCAO generates more consistent lesions [81]. MCAO produces robust sensorimotor deficits and can impair spatial learning and memory when lesions extend to hippocampal or prefrontal regions, though pure cortical infarcts may require sensitive behavioural testing to detect subtle cognitive effects [76,81,82,86]. Strengths include high face validity for large vessel strokes and relevance to thrombolytic therapy, while limitations include substantial motor impairments that can confound cognitive testing, variability in infarct size, technical complexity, and the need for experienced surgeons [86].
Photothrombotic stroke models use photosensitizing dyes activated by laser or light to induce localized thrombosis, offering precise control over lesion location and size with minimal invasiveness and excellent reproducibility [78]. They produce well-demarcated cortical infarcts with limited penumbra, making them ideal for studying region-specific cognitive functions, though the lack of reperfusion limits relevance to thrombolysis scenarios [78,79,87]. Photothrombotic models generally show more limited inflammatory responses compared with MCAO. Strengths include reproducibility and minimal surgical trauma, while limitations include reduced translational relevance to thromboembolic strokes and the inability to fully model the inflammatory milieu of human stroke. Embolic models, including injection of autologous blood clots or microspheres, closely mimic the thromboembolic etiology of most human strokes and allow investigation of spontaneous or therapeutic recanalization [83]. They generate multifocal lesions affecting cortical and subcortical structures, better capturing the distributed network dysfunction observed in human PSCI, though variability in clot distribution and spontaneous recanalization can complicate interpretation [83,84]. Strengths include pathophysiological relevance and heterogeneity like human strokes, while limitations involve difficulty in standardization and control of lesion distribution.
Endothelin-1 (ET-1) models involve local application or injection of this potent vasoconstrictor peptide to induce dose-dependent, reversible vessel constriction and focal ischemia, with features of both permanent and transient occlusion [75,80]. ET-1–induced striatal strokes produce reliable cognitive deficits with flexible lesion location and severity, making them particularly useful for investigating subcortical contributions to cognitive impairment and executive function [77]. ET-1 models demonstrate prolonged vasoconstriction, which may better replicate microvascular dysfunction, but the mechanism differs from thrombotic or embolic stroke, and ET-1 may have direct inflammatory effects independent of ischemia [75,77,80]. Strengths include flexibility in lesion targeting and consistent cognitive deficits; limitations include mechanistic differences from human thromboembolic stroke. Selecting an appropriate model requires careful consideration of cognitive domains of interest, lesion characteristics, temporal dynamics, and translational relevance [76,84]. A fundamental challenge is dissociating cognitive impairment from motor, sensory, or motivational deficits, as many behavioural tasks rely on intact sensorimotor function. Species differences, such as the smaller proportion of white matter and absence of cortical gyri in rodents, limit modelling of subcortical vascular cognitive impairment and white matter disease [88].
An alternative to the ET-1 model is the L-NIO (N5-(1-iminoethyl)-L-ornithine) model. Unlike ET-1, which inconsistently affects white matter, L-NIO produces reproducible lesions localized to white matter regions, such as the corpus callosum (CC) [88,89]. This precise targeting minimizes unintended grey matter damage, which could otherwise influence post-ischemic processes and repair mechanisms in white matter [88,90]. Previous work has demonstrated the utility of this model for studying axonal damage, myelin loss, and glial responses after subcortical white matter stroke in mice [90]. Combining insights across models provides a more comprehensive understanding of PSCI mechanisms and informs the development of targeted therapeutic strategies.
3.2. Cognitive Assessment Methods in Rodents (Morris Water Maze, Novel Object Recognition, Barnes Maze)
A variety of behavioural assays have been developed for rodent stroke models to capture the domain-specific cognitive deficits characteristic of human PSCI. Each test has been designed to probe distinct cognitive processes and differs in its sensitivity to lesion location and severity [91]. Among these assays, the Morris water maze (MWM) remains the gold standard for evaluating spatial learning and memory. In this task, rodents navigate a pool to locate a hidden platform using distal spatial cues, providing insight into hippocampal-dependent learning, working memory, and cognitive flexibility during reversal trials [92,93]. The MWM is particularly sensitive to lesions affecting the hippocampus, medial prefrontal cortex, and their connecting pathways, though its reliance on swimming and motor function can complicate interpretation in animals with significant sensorimotor deficits [94]. Alternatively, the Barnes maze test provides a low-stress, dry-land method for assessing spatial learning and memory, with sensitivity to prefrontal cortex-dependent strategies. In this task, rodents are placed on a circular platform with multiple holes, only one of which leads to an escape hole. They must use spatial cues to navigate the platform, locate the correct hole, and escape the platform [95].
Novel Object Recognition (NOR) is a simple and low-stress test used to measure recognition memory in rodent stroke models. It takes advantage of the natural tendency of rodents to spend more time exploring something new than something they have seen before. In the test, animals first explore two identical objects, and later are shown one familiar object and one new one. If memory is intact, they spend more time exploring the new object. NOR does not require training, rewards, food restriction, or stressful setups, which makes it good for detecting subtle memory problems after stroke [96,97]. The test mainly reflects the function of the perirhinal cortex and partly the hippocampus, both of which are regions commonly affected in PSCI [96,97]. The main limitation is that results can be influenced by post-stroke motor problems, anxiety, or reduced exploration, so these factors need to be controlled when interpreting the data.
Beyond these classic tasks, more specialized paradigms have been developed to target executive function, attention, and working memory. Attentional set-shifting tasks (ASTs) are rodent analogs of the Wisconsin Card Sorting Test in humans and are used to assess cognitive flexibility and executive function [91,98,99]. Importantly, executive dysfunction is one of the most common and functionally disabling cognitive deficits observed in PSCI, making ASTs a particularly translational and clinically relevant measure in stroke models. In these tasks, animals first learn to associate a reward with a specific cue, such as a particular odour or digging medium. They are then challenged to adapt when the rule changes: intra-dimensional (ID) shifts require applying the learned rule to new stimuli of the same type, while extra-dimensional (ED) shifts require switching attention to a different type of stimulus [91,98,99]. Some paradigms also include reversal learning, where the reward contingency is reversed within the same dimension. Performance depends on striatal and prefrontal circuits, and studies show that striatal stroke selectively impairs ED shifts while sparing ID learning, highlighting the distinct contributions of these brain regions to cognitive flexibility. ASTs, therefore, provide a sensitive method to dissect domain-specific executive deficits following stroke [98,99,100].
Five-choice serial reaction time (5-CSRTT) tasks provide a window into sustained attention and impulse control, requiring rodents to detect brief, randomly presented light stimuli across multiple apertures and respond promptly to receive a reward, with incorrect or premature responses revealing deficits in attentional vigilance and inhibitory control [101]. Operant reversal learning paradigms evaluate behavioural flexibility by requiring animals to adapt their responses to changing reward contingencies, a process dependent on orbitofrontal and medial prefrontal cortices [102]. Temporal order memory tasks assess working memory and sequence memory, measuring rodents’ ability to distinguish the temporal order of previously encountered objects [103]. Spontaneous alternation in Y-mazes or radial arm mazes provides an additional measure of working and spatial memory with minimal training, exploiting rodents’ natural tendency to explore novel arms [103]. Selecting an appropriate battery of cognitive tests requires careful consideration of lesion location, expected deficits, timing of assessment, and potential confounds such as motor impairment, fatigue, or anxiety, which may influence performance independently of cognitive function [104].
3.3. Translational Challenges
The translation of preclinical stroke research to effective clinical therapies for PSCI remains challenging. Although numerous neuroprotective agents have shown promising effects in experimental models, most fail to produce meaningful cognitive improvement in clinical trials [103,104]. The widespread use of young, healthy animal models in preclinical studies limits translation, as their vascular, inflammatory, and regenerative responses differ markedly from older adults, the population most at risk for stroke and PSCI [107,108]. Experimental designs further exacerbate these issues; treatments are often administered immediately post-stroke under tightly controlled conditions, in contrast to clinical settings where treatment delays of hours are common, and patients present with variable symptom onset [109]. Additionally, sample sizes are small, randomization or blinding is inconsistent, and outcomes traditionally emphasize infarct size or short-term motor recovery rather than long-term cognitive and functional trajectories, creating a disconnect between preclinical efficacy and clinical meaningfulness [110]. These factors, together with variability in protocols across laboratories, contribute to poor reproducibility, inflated effect sizes, and failure to capture the chronic evolution of cognitive deficits [111,112]. Sex differences have been largely neglected, with most studies conducted exclusively in male animals despite evidence of sexual dimorphism in inflammatory responses and neuroplasticity [113,114]. To address these gaps, the Stroke Therapy Academic Industry Roundtable (STAIR) recommends rigorous approaches, including testing in aged animals with comorbidities, inclusion of both sexes, standardized methodologies across laboratories, and longitudinal assessment of functional and cognitive outcomes. Aligning preclinical models with clinically relevant populations, outcomes, and timeframes is essential to translate experimental findings into therapies that meaningfully improve patient outcomes.
4. Inflammatory Mechanisms
Inflammation is a central mechanism linking acute ischemic injury to chronic cognitive decline in PSCI [1,4,8,32]. Following stroke, activation of resident brain immune cells, disruption of the BBB, and infiltration of peripheral immune cells initiate a cascade of cytokines, chemokines, and reactive oxygen species that extend beyond the infarct core to regions critical for cognition, including the hippocampus and prefrontal cortex [22,31,115,116,117]. Microglia, initially protective, can adopt a chronic pro-inflammatory phenotype that impairs synaptic plasticity, neurogenesis, and network connectivity. In contrast, peripheral immune cells, including neutrophils, monocytes, and lymphocytes, further amplify local and remote neurodegeneration. Elevated cytokines such as interleukin-1 beta (IL-1β), tumour necrosis factor-alpha (TNF-α), and interleukin-6 (IL-6) are associated with worse cognitive outcomes, underscoring their mechanistic and prognostic significance [5,33,118]. Inflammation also interacts with white matter injury, vascular dysfunction, and amyloid accumulation, compounding deficits in memory, executive function, and attention [25,57,119]. The dual nature of post-stroke inflammation, simultaneously mediating tissue repair and neurodegeneration, presents both challenges and opportunities for therapy [32,120,121]. Experimental studies show that targeting specific inflammatory pathways or cellular mediators can mitigate cognitive deficits in rodent models, highlighting the translational potential of mechanism-based interventions [6,29,122,123]. Understanding the temporal dynamics, cellular actors, and molecular mediators of post-stroke inflammation is therefore critical for developing therapies that selectively modulate immune responses and improve cognitive recovery.
4.1. Acute Neuroinflammation
The acute inflammatory response to cerebral ischemia begins within minutes of vessel occlusion, representing a critical early determinant of PSCI [28,30]. Resident microglia rapidly activate, undergo morphological transformation from ramified to amoeboid phenotypes, and proliferate extensively in peri-infarct regions, releasing pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-6 that amplify inflammatory signalling and promote neuronal death [124,125]. This cytokine cascade extends beyond direct neurotoxicity, activating endothelial cells and astrocytes to create a feed-forward loop that exacerbates tissue injury [118]. Compromise of BBB integrity occurs in a biphasic fashion, where an early opening within hours is mediated by oxidative stress and matrix metalloproteinase activation. This is then followed by a delayed phase several days later, associated with infiltration of peripheral immune cells [115,126]. BBB breakdown allows neutrophils, monocytes, and lymphocytes to enter the brain, contributing additional cytokines, cytotoxic factors, and serum proteins that exacerbate vasogenic edema and secondary injury [85]. Necrotic neurons release damage-associated molecular patterns, including high-mobility group box 1 protein (HMGB1) and heat shock proteins, which activate pattern recognition receptors such as toll-like receptors on immune cells, perpetuating inflammatory signalling [127,128]. The spatial pattern of acute neuroinflammation is heterogeneous. The infarct core exhibits intense necrosis and inflammatory activation, whereas peri-infarct and remote regions display subtler, yet potentially persistent, inflammatory responses that may contribute to delayed cognitive deficits [129]. While acute neuroinflammation is essential for debris clearance and initiation of repair, excessive or dysregulated activation impairs synaptic plasticity, neurogenesis, and network connectivity, establishing the foundation for long-term cognitive impairment. Experimental studies show that early modulation of these inflammatory pathways through cytokine inhibition or microglial phenotype shifts can reduce acute tissue injury and improve later cognitive outcomes. This highlights the translational potential of targeting this early temporal window. Understanding the cellular, molecular, and regional dynamics of acute neuroinflammation is therefore critical for designing interventions that mitigate secondary injury while preserving beneficial repair processes.
4.2. Peripheral vs. Central Inflammation
PSCI arises from complex interactions between central nervous system (CNS) immune responses and systemic inflammatory processes, with peripheral immune activation both responding to and contributing to brain injury through multiple pathways [30,130]. Central inflammation is initiated by resident microglia, astrocytes, and endothelial cells, which respond to ischemic injury with rapid cytokine release, reactive oxygen species production, and morphological changes that disrupt synaptic function and plasticity. Microglia transform from a surveillant ramified state to an activated amoeboid phenotype, releasing pro-inflammatory mediators such as IL-1β, TNF-α, and IL-6 that propagate local tissue injury. Astrocytes undergo reactive gliosis, producing both neuroprotective and neurotoxic factors that influence neuronal survival and network remodelling. BBB disruption during the acute and subacute phases permits infiltration of peripheral immune cells, linking central and systemic inflammation. Peripheral inflammation involves neutrophils, monocytes, lymphocytes, and platelets mobilized through systemic cytokine production, hypothalamic-pituitary-adrenal axis activation, and sympathetic signalling, including splenic contraction and lymphocyte apoptosis [131,132,133,134]. Circulating immune cells exhibit altered adhesion, activation, and trafficking, enabling their entry into the brain through the compromised BBB, where they exacerbate neuronal injury via cytokine release, oxidative stress, and proteolytic enzymes [85].
The gut-brain axis further modulates peripheral inflammation, as stroke-induced dysbiosis, increased intestinal permeability, and bacterial translocation amplify systemic inflammatory burden and potentially influence central neuroinflammation [135,136]. Peripheral markers such as C-reactive protein, IL-6, and TNF-α correlate with infarct size, functional outcomes, and cognitive performance, though whether they reflect CNS inflammation or independent systemic effects remains debated [33,137]. Paradoxically, stroke-induced immunosuppression syndrome develops in many patients, with reduced lymphocyte counts and impaired cell-mediated immunity coexisting alongside ongoing inflammatory activation, highlighting the complex balancing act of systemic immune responses [138,139]. Temporal dynamics are critical; acute peripheral inflammation can exacerbate early injury, while chronic low-grade systemic inflammation sustains neuroinflammation, contributing to secondary neurodegeneration, white matter injury, and cognitive decline [140]. The bidirectional crosstalk between peripheral and central compartments amplifies maladaptive processes, including synaptic dysfunction, impaired neurogenesis, and disrupted network connectivity, underscoring why some patients develop progressive cognitive deficits despite relatively small infarcts. Experimental studies show that targeting peripheral immune activation through modulation of neutrophils, monocytes, or cytokine signalling can attenuate central neuroinflammation and improve cognitive outcomes, though timing and specificity are critical to preserve beneficial immune functions such as tissue repair and debris clearance [141].
4.3. Specific Pathways: Complement System, Inflammasomes, Astrogliosis, and Microglial Phenotypes
While acute inflammation after stroke typically resolves within days in most tissues, ischemic injury in the brain triggers a prolonged and maladaptive inflammatory response that can persist for weeks to months. This chronic neuroinflammation not only affects the infarcted region but also extends to connected structures, including the peri-infarct cortex, hippocampus, thalamus, and white matter tracts [31,32,142,143]. This sustained immune activation drives secondary neurodegeneration, network-level dysfunction, and contributes to cognitive decline. The mechanism underlying this chronic inflammation is temporally dynamic, region-specific, and involves a complex interplay of cellular and molecular pathways.
Following a stroke, inflammation is orchestrated by multiple cellular and molecular pathways, each representing a potential target for interventions aimed at reducing cognitive deficits without the risks associated with broad immunosuppression [30,32]. Microglia in these regions adopt a sustained activated state, characterized by amoeboid morphology and persistent expression of pro-inflammatory markers, contributing to ongoing production of cytokines, reactive oxygen species, and proteolytic enzymes that disrupt neuronal function, synaptic plasticity, and adult neurogenesis [144]. Microglia, the brain’s primary immune cells, exhibit highly dynamic phenotypic changes in response to ischemic injury, shaping both tissue outcomes and recovery trajectories [120]. Traditionally, activated microglia were classified as pro-inflammatory M1, producing mediators such as nitric oxide, TNF-α, IL-1β or anti-inflammatory M2, which release neurotrophic factors and support repair. However, transcriptomic analyses reveal a spectrum of intermediate and context-dependent states, reflecting greater heterogeneity than the M1/M2 dichotomy [125,145]. Microglial responses vary across brain regions and over time, with cells in peri-infarct areas displaying different profiles from those in remote regions like the hippocampus and thalamus [142,146]. These cells contribute to debris clearance, synaptic remodelling, and modulation of plasticity, yet prolonged activation can perpetuate neuroinflammation and drive secondary neuronal damage [121,144].
Secondary neurodegeneration propagates along anatomically connected pathways through mechanisms such as Wallerian degeneration, transneuronal atrophy, and diaschisis, processes that are amplified by chronic inflammatory signalling [147,148]. The hippocampus is particularly sensitive, exhibiting reduced dentate gyrus neurogenesis, diminished synaptic density, and impaired long-term potentiation, which collectively correlate with deficits in spatial learning and memory [149,150]. White matter is also vulnerable, with activated microglia and reactive astrocytes producing factors that hinder oligodendrocyte survival and myelin repair, driving progressive demyelination and axonal injury [151,152]. Chronic inflammation further compromises the neurovascular unit, producing sustained BBB leakiness, impaired autoregulation of cerebral blood flow, and reduced angiogenesis, collectively limiting metabolic support and perfusion [153].
The transition from acute to chronic inflammation involves complex shifts in immune cell phenotypes, where insufficient resolution and inadequate anti-inflammatory signalling reinforce maladaptive activation [125,145]. Importantly, both the intensity and duration of chronic neuroinflammation vary considerably between individuals, offering a potential explanation for heterogeneity in cognitive outcomes and highlighting a targetable mechanism for interventions aimed at improving long-term recovery [154].
The complement cascade, typically associated with pathogen defence, is rapidly activated post-stroke through classical and alternative pathways. Deposition of components such as C3 and the C5b-9 membrane attack complex occurs on neurons and synapses in affected regions, marking synapses for microglial pruning and potentially promoting cognitive impairment through excessive synaptic loss [155,156,157]. Experimental models demonstrate that blocking complement activity reduces infarct volume and improves functional outcomes [158]. Inflammasomes, particularly NLR family pyrin domain containing 3 (NLRP3), sense ischemia-related damage signals and activate caspase-1, facilitating maturation of IL-1β and interleukin-18 (IL-18). Activation occurs in microglia, astrocytes, and infiltrating immune cells, driving inflammation and pyroptotic cell death, while inhibition mitigates neuronal injury and cognitive decline in preclinical studies [123,159,160,161,162].
Astrocytes also respond to ischemic injury through reactive transformation, a process marked by hypertrophy, proliferation, and altered gene expression. Astrogliosis can limit the spread of inflammation via glial scar formation but may simultaneously impede synaptic remodelling, and astrocytes secrete factors that variably support or harm neuronal survival depending on context [163,164]. Collectively, these interacting pathways illustrate the complexity and spatial-temporal dynamics of post-stroke neuroinflammation, highlighting opportunities for targeted interventions that preserve cognitive function while minimizing collateral tissue damage.
4.4. Network Dysfunction and Diaschisis Related to Inflammation
Network dysfunction and diaschisis are central contributors to PSCI, highlighting that those deficits extend far beyond the primary site of injury. Stroke-induced cognitive impairment cannot be fully explained by direct tissue loss within the infarct, as dysfunction propagates to anatomically connected brain regions through mechanisms collectively termed diaschisis, which are increasingly recognized to involve inflammatory mediation [22,165]. Diaschisis, originally described as reduced metabolism and blood flow in regions remote from but connected to the infarct, reflects disruption of neural networks and loss of excitatory input to structurally intact tissue [166]. Inflammatory mechanisms contribute to diaschisis through multiple pathways, including microglial activation in distant brain regions that spreads along white matter tracts and synaptic connections, even in the absence of direct ischemic injury [116,148]. The hippocampus and thalamus are particularly vulnerable, exhibiting robust microglial activation, cytokine production, and synaptic alterations despite being outside the primary lesion territory [167,168]. Inflammation-mediated degeneration spreading through synaptically connected neuronal networks (transneuronal degeneration) drives progressive neuronal loss in these connected structures, contributing to delayed cognitive decline over weeks to months post-stroke [169]. Similarly, Wallerian degeneration of axons whose cell bodies were destroyed in the infarct triggers inflammatory responses along the entire axonal pathway, producing secondary white matter damage that further disrupts connectivity between cognitive networks [88,170]. Functional imaging studies using resting-state functional magnetic resonance imaging (fMRI) in stroke patients demonstrate widespread alterations in network coherence that closely correlate with cognitive performance, suggesting that inflammatory disruption of network function may be as impactful as structural damage in determining outcomes [117,171]. Large-scale networks, including the default mode, salience, and executive control networks, show vulnerability, with reduced connectivity linked to impairments in attention, executive function, and memory [172]. At the molecular level, inflammatory mediators such as cytokines and complement proteins alter synaptic transmission, modulate neuronal excitability, and impair long-term potentiation in regions remote from the infarct, providing a mechanistic link between inflammation and network dysfunction [156,173,174]. Chronic inflammation may perpetuate this network disruption by limiting compensatory plasticity and reorganization, creating a self-reinforcing cycle of impaired connectivity and persistent cognitive deficits [174]. Together, these findings emphasize that post-stroke inflammation acts not only locally but across distributed networks, driving diaschisis and shaping long-term cognitive outcomes.
4.5. White Matter Injury and Inflammation
White matter injury is a key yet underappreciated contributor to post-stroke pathology, with inflammatory processes driving both acute damage and chronic demyelination that disproportionately affect cognitive function relative to motor outcomes [88,152]. The unique vascular architecture of white matter, characterized by long penetrating arterioles with limited collateral flow, combined with the high metabolic demands of oligodendrocytes and axons and the susceptibility of myelin lipids to oxidative stress, renders these regions especially vulnerable to ischemic insult [151,175]. Acute injury triggers oligodendrocyte death and myelin disruption, accompanied by axonal swelling and activation of resident microglia, while inflammatory mediators, including TNF-α, glutamate excitotoxicity, and reactive oxygen species, amplify cell loss and damage [151,175,176,177,178]. Microglia and infiltrating macrophages clear myelin debris, a process necessary for repair, yet this phagocytic activity can propagate local inflammation to neighbouring intact fibres [179]. Over time, Wallerian degeneration of axons originating from infarcted cortical or subcortical neurons contributes to progressive white matter degeneration, with serial inflammatory activation along these tracts sustaining injury [180,181]. Periventricular white matter is particularly susceptible, as chronic hypoperfusion after stroke accelerates small vessel disease and promotes the development of white matter hyperintensities visible on neuroimaging [25]. Disruption of the BBB in these regions allows plasma proteins and peripheral immune cells to enter the parenchyma, creating a hostile environment that impedes oligodendrocyte precursor maturation and remyelination [119,182]. Persistent inflammatory signals, including leucine-rich repeat and Ig domain-containing Nogo receptor-interacting protein 1(LINGO-1), hyaluronan accumulation and inhibitory cytokines maintain progenitor cells in a quiescent state, limiting repair [183,184]. Functionally, white matter integrity measured by diffusion tensor imaging correlates strongly with cognitive outcomes, particularly executive function and processing speed, reflecting the dependence of these domains on long-range association fibers connecting frontal, parietal, and temporal cortices [2,185]. Emerging therapeutic strategies that modulate microglial activation, complement pathways, or inflammasome signalling show promise in preserving white matter, maintaining connectivity, and mitigating long-term cognitive decline. Collectively, these observations frame white matter injury as a dynamic, inflammation-driven process that bridges cellular pathology and network-level dysfunction in PSCI.
5. Future Direction and Therapeutic Implications
Although our understanding of the mechanisms driving PSCI has grown substantially, translating these insights into effective therapies remains a major challenge. Current stroke care remains heavily focused on acute interventions, such as recanalization and neuroprotection, while largely overlooking cognitive outcomes and strategies to prevent secondary neurodegenerative changes. Recognizing that inflammation is a prolonged, modifiable process that extends beyond the acute phase opens new therapeutic opportunities across multiple time windows. Developing effective anti-inflammatory interventions, however, demands careful navigation of the immune system’s dual roles in injury and repair, aiming to suppress harmful responses while promoting beneficial ones. Beyond drug-based approaches, progress in biomarker development, precision medicine, and routine cognitive assessment will be crucial for identifying patients at greatest risk and tracking treatment effects. This section explores promising therapeutic strategies, highlights key research priorities, and outlines frameworks to advance PSCI care toward meaningful clinical impact.
5.1. Biomarker Development
Accurate and reliable biomarkers are critical for addressing the clinical and mechanistic complexity of PSCI [11,186]. Early inflammatory signals in the blood, such as high-sensitivity C-reactive protein (hs-CRP), IL-6, and TNF-α detected during the acute phase of stroke, suggest that systemic immune responses influence subsequent cognitive trajectories, yet their predictive power for individual patients remains limited [33,137]. This limitation has motivated investigation of complementary biomarkers that capture distinct aspects of brain pathology, including BBB disruption (matrix metalloproteinase-9, S100B), neuronal injury (neurofilament light chain, tau), and central inflammatory cascades such as NLRP3 inflammasome components [187,188,189]. Cerebrospinal fluid measures provide a direct window into central nervous system pathology but are constrained by invasiveness and the feasibility of repeated sampling, highlighting the value of non-invasive imaging biomarkers [190]. Structural and functional neuroimaging captures the cumulative impact of vascular and neurodegenerative injury, linking white matter hyperintensity burden, medial temporal lobe atrophy, and network connectivity alterations to cognitive outcomes [13,53]. Advanced magnetic resonance imaging (MRI) modalities, including diffusion tensor imaging to assess white matter integrity, arterial spin labelling to measure cerebral perfusion, and susceptibility-weighted imaging to detect microbleeds, offer mechanistic insight into tissue integrity and perfusion [185,191]. PET targeting the 18 kDa translocator protein on activated microglia enables in vivo visualization of neuroinflammation, though cost and tracer availability limit widespread use [142,192]. Genetic susceptibility, including apolipoprotein E (APOE) genotype, inflammatory gene polymorphisms, and polygenic risk scores for dementia, further modulates these processes, refining risk stratification and informing preventive strategies [55].
Integrating molecular, imaging, and genetic markers offers the potential to construct predictive models that reflect the multifactorial nature of PSCI. By combining these complementary approaches, researchers can advance precision medicine frameworks that identify high-risk patients, tailor interventions to underlying pathophysiology, and dynamically monitor treatment effects, ultimately bridging the gap between mechanistic understanding and clinical impact [193].
5.2. Immunomodulatory Interventions
Immunomodulatory therapies targeting specific inflammatory pathways offer promise for reducing PSCI while avoiding the complications associated with broad immunosuppression that could impair beneficial repair processes and increase infection risk [30,32]. Pharmacological approaches include anti-cytokine therapies such as interleukin-1 receptor antagonist (IL-1Ra; anakinra) and monoclonal antibodies against TNF-α or IL-6, which have shown efficacy in preclinical stroke models by reducing infarct size and improving functional outcomes, with early-phase clinical trials demonstrating safety and potential efficacy signals [137,194,195]. Complement inhibition represents another attractive target given its role in synapse loss and neuroinflammation, with C3 and C5 inhibitors demonstrating neuroprotective effects in animal models, although translation to clinical trials has been limited by concerns about infection risk and timing of intervention [158,196].
Inflammasome inhibitors targeting NLRP3 or caspase-1 reduce brain injury and preserve cognitive function in preclinical studies, with several small-molecule NLRP3 inhibitors now advancing toward clinical testing for inflammatory conditions [161,197]. Microglial modulation strategies aim to shift microglial phenotypes toward reparative states rather than globally suppressing activation, with approaches including colony-stimulating factor 1 receptor modulation, peroxisome proliferator-activated receptor gamma agonists, and enhancement of the triggering receptor expressed on myeloid cells 2 (TREM2) pathway, showing preclinical promise [120,198]. Minocycline, a tetracycline antibiotic with anti-inflammatory properties that crosses the BBB and reduces microglial activation, has shown mixed results in clinical trials, with some studies suggesting benefits for stroke outcomes, though cognitive endpoints have rarely been assessed [199,200]. Statins exhibit pleiotropic immunomodulatory effects beyond lipid lowering, including reduced microglial activation and enhanced endothelial function, with observational data suggesting associations with reduced cognitive decline, though definitive trial evidence is lacking [201,202]. Fingolimod, an approved therapy for multiple sclerosis, prevents lymphocyte egress from lymph nodes and modulates sphingosine-1-phosphate signalling; early stroke trials showed promise, but larger studies failed to meet primary endpoints, possibly due to timing and patient selection issues [122,203]. Therapeutic plasma exchange and intravenous immunoglobulin represent approaches to remove pathogenic antibodies and modulate systemic inflammation, with potential applications in stroke-related autoimmunity, though evidence remains preliminary [141].
Finally, cell-based therapies, including regulatory T cell infusions and mesenchymal stem cells, exert immunomodulatory effects that could dampen chronic neuroinflammation while promoting tissue repair, representing an active area of investigation [204,205]. Timing, dosing, and patient stratification are critical, as early modulation of inflammation may prevent secondary injury, whereas prolonged or excessive suppression could interfere with endogenous repair mechanisms. Ultimately, integrating immunomodulatory therapies with biomarker-guided monitoring, multimodal assessment of network and white matter integrity, and rehabilitative strategies offers a comprehensive approach to improving cognitive recovery after stroke.
5.3. Optimal Timing Windows for Intervention
Optimal timing of interventions is critical for maximizing the efficacy of therapies targeting post-stroke inflammation, network dysfunction, and white matter injury. The temporal dynamics of post-stroke inflammation present both challenges and opportunities for therapeutic intervention, as the balance between detrimental and beneficial immune responses shifts substantially across acute, subacute, and chronic phases, necessitating careful consideration of when to initiate, modify, or discontinue immunomodulatory treatments [30,32]. Hyperacute interventions within the first hours after stroke face the challenge that inflammatory cascades are only beginning to activate, potentially limiting the impact of anti-inflammatory agents while risking interference with early danger signalling necessary for initiating repair responses [29]. The acute phase, spanning the first few days, exhibits peak pro-inflammatory cytokine production, neutrophil infiltration, and BBB disruption, representing a potential window for interventions targeting these processes, though this period also involves initiation of beneficial clearance mechanisms and neuroprotective responses [28,85]. Preclinical studies demonstrate that timing of anti-inflammatory interventions critically influences outcomes, with early administration sometimes worsening results by impairing debris clearance and tissue remodelling, while delayed treatment may miss the window for preventing secondary injury [125,145]. The subacute phase, from days to weeks post-stroke, shows transition from predominantly pro-inflammatory to mixed inflammatory profiles with emergence of reparative macrophage and microglial phenotypes, suggesting this period may be optimal for interventions that enhance anti-inflammatory responses while dampening persistent harmful inflammation [195,206]. Chronic phase interventions targeting sustained low-grade neuroinflammation that persists for months may offer opportunities to prevent progressive cognitive decline and promote delayed plasticity, though identifying patients with pathological versus physiological chronic inflammation remains challenging [31,142].
The concept of therapeutic time windows becomes more complex when considering cognitive outcomes rather than acute infarct size, as cognitive deficits often emerge or worsen over time through mechanisms potentially amenable to late intervention [65]. Individual variability in inflammatory trajectories based on factors including age, genetics, comorbidities, and stroke characteristics suggests that personalized timing of interventions guided by inflammatory biomarkers may be necessary rather than fixed time windows applied uniformly [207,208]. Serial biomarker monitoring could enable adaptive treatment strategies that initiate, adjust, or discontinue immunomodulatory therapies based on individual inflammatory profiles, though such approaches require validation of biomarkers that reliably reflect central nervous system inflammation and predict treatment response [33,186]. Ultimately, understanding the spatiotemporal evolution of post-stroke inflammation is essential to balance risks and benefits, optimize the timing of interventions, and maximize the potential for cognitive recovery in stroke survivors.
5.4. Personalized Approaches Based on Inflammatory Profiles
Personalized approaches guided by individual inflammatory profiles represent a promising frontier for optimizing post-stroke cognitive recovery. The substantial heterogeneity in inflammatory responses across stroke patients suggests that precision medicine approaches, tailoring interventions to individual inflammatory profiles, could improve efficacy compared to uniform treatment strategies applied to all patients [5,33,137]. Patient stratification based on inflammatory biomarkers could identify subgroups most likely to benefit from specific immunomodulatory interventions, with emerging evidence that baseline cytokine profiles, genetic variants in inflammatory pathways, and neuroimaging markers of inflammation predict both spontaneous recovery and treatment responses [3,5,30,117]. Genetic polymorphisms in genes encoding cytokines, pattern recognition receptors, and immune cell surface markers influence inflammatory responses to stroke, with variants in IL6, TNF, and TLR4 genes associated with stroke outcomes, suggesting that genotype-guided therapy selection could enhance treatment efficacy [28,30,31,35]. Biological variables such as sex critically influence inflammatory responses, with female stroke patients generally exhibiting less robust acute inflammation but potentially more sustained chronic responses compared to males, differences that may necessitate sex-specific therapeutic approaches [113,207,208]. Age profoundly affects inflammatory processes, with older individuals demonstrating exaggerated and prolonged inflammatory responses (“inflammaging”) that correlate with worse outcomes and may require more aggressive or prolonged anti-inflammatory interventions [68,107,207]. Comorbidities, including diabetes, hypertension, and obesity, alter inflammatory responses through metabolic dysregulation and chronic low-grade systemic inflammation, potentially modifying therapeutic windows and optimal drug choices [23,53,167]. The presence of pre-existing cognitive impairment or concurrent neurodegenerative pathology, detectable through cerebrospinal fluid or PET imaging biomarkers, identifies patients with mixed pathology who may require combination approaches targeting both vascular and degenerative mechanisms [11,67,69,71]. Stroke characteristics, including lesion location, size, and etiology, influence inflammatory profiles, with large cortical infarcts producing more robust acute inflammation while small vessel disease generates chronic microvascular inflammation that may respond better to sustained low-dose interventions [88,152].
Biomarker-guided strategies integrating circulating cytokines, complement and inflammasome components, neuroimaging measures of network integrity and white matter health, and electrophysiological markers can provide a multidimensional view of each patient’s inflammatory and network status, informing the timing, type, and dosing of immunomodulatory therapies. Machine learning approaches integrating clinical, genetic, biomarker, and imaging data show promise for developing predictive algorithms that match patients to optimal therapies, though prospective validation in clinical trials remains necessary [106,109,186,209]. Adaptive trial designs, including response-adaptive randomization and enrichment strategies, could accelerate identification of effective personalized approaches by allocating more patients to promising treatment combinations and focusing enrollment on biomarker-defined subgroups [106,109,186]. Ultimately, tailoring interventions to an individual’s inflammatory trajectory offers the potential to enhance neuroplasticity, preserve white matter integrity, prevent network dysfunction, and reduce long-term cognitive decline, moving beyond a “one-size-fits-all” paradigm toward precision neurorehabilitation in stroke survivors.
5.5. Research Gaps and Methodological Recommendations
Despite growing recognition of PSCI as a critical clinical problem, substantial research gaps impede translation of mechanistic insights into effective therapies, necessitating methodological improvements and strategic research priorities to accelerate progress [26,27]. Cognitive outcomes remain underrepresented in clinical stroke trials, with most studies focusing on functional independence scales that emphasize motor recovery and fail to capture domain-specific cognitive deficits, highlighting the need for standardized cognitive assessment batteries validated for stroke populations and sensitive to treatment effects [37,38]. The disconnect between preclinical models and clinical populations represents a fundamental translational barrier, with most animal studies employing young healthy males despite stroke predominantly affecting older individuals with multiple comorbidities, necessitating expanded use of aged animals, females, and models incorporating vascular risk factors [109,207]. Longitudinal studies with extended follow-up periods are essential for understanding delayed cognitive decline and identifying late intervention opportunities, yet most clinical trials assess outcomes within 90 days, and preclinical studies rarely extend beyond weeks [7,65].
Mechanistic studies linking specific inflammatory pathways to cognitive domains in humans remain sparse, requiring integration of detailed cognitive phenotyping with inflammatory biomarker profiling, advanced neuroimaging, and, when feasible, cerebrospinal fluid or autopsy tissue analysis [33]. The lack of validated outcome measures for anti-inflammatory interventions targeting cognitive preservation complicates trial design, with uncertainty about appropriate primary endpoints, timing of assessments, and minimal clinically important differences [16,209]. Reproducibility concerns plague preclinical stroke research due to inadequate reporting, small sample sizes, lack of randomization and blinding, and publication bias favouring positive results, necessitating adoption of rigorous standards including preregistration, transparent reporting following Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines, and multicenter replication [112,210,211]. Sex as a biological variable requires systematic investigation, with trials adequately powered for sex-stratified analyses and preclinical studies routinely including both sexes to identify sex-specific mechanisms and treatment responses [211]. Patient and caregiver engagement in research priority setting could ensure that studies address outcomes most valued by those affected, potentially accelerating adoption of cognitive screening and interventions in clinical practice [212]. Funding mechanisms that support high-risk innovative approaches, interdisciplinary collaborations bridging neurology, immunology, and cognitive neuroscience, and long-term cohort studies are essential for addressing these gaps [106]. Collectively, addressing these methodological challenges will enhance reproducibility, enable mechanistic insights to inform targeted therapies, and accelerate translation of precision medicine approaches for preventing and mitigating PSCI.
Funding and Acknowledgements: SNW received funding from the Heart and Stroke Foundation of Canada, and the Canadian Institutes for Health Research. S.Z received training funding from the Vascular Training (VAST) Platform and is an Ontario Graduate Scholar.
Appendix A
Box A1: Key Translational Challenges in PSCI Research
PRECLINICAL-CLINICAL DISCONNECT
Model Limitations
- Preclinical studies predominantly use young, healthy rodents, which do not reflect the older adult population most affected by PSCI
- Single focal infarct models fail to capture the mixed vascular and neurodegenerative pathology frequently observed in patients
- Controlled laboratory conditions cannot reproduce the heterogeneity of human stroke presentations, treatment delays, and environmental factors
Outcome Misalignment
- Infarct size or location alone poorly predicts cognitive outcomes, underscoring the need to examine secondary processes such as network disruption and white matter injury
- Short-term motor recovery metrics in rodents do not translate directly to long-term cognitive function in patients
- Acute neuroprotection does not necessarily correlate with improvements in chronic cognitive trajectories
- Rodent behavioural tasks often target simple learning/memory endpoints and may not capture domain-specific deficits seen in humans (executive function, processing speed, attention).
METHODOLOGICAL GAPS
Study design issues
- Many studies include only male animals, overlooking sex differences in neuroinflammation and cognitive recovery.
- Small sample sizes and publication bias may inflate the effect size.
- The timing of cognitive assessments is inconsistent across studies, limiting cross-study comparability
- Standardized cognitive batteries are rarely implemented in preclinical PSCI research
Assessment Challenges
- Motor deficits can confound cognitive tests in rodents, obscuring true cognitive impairment
- Brief clinical screening tools (e.g.,
Box A2: Inflammation-Specific Challenges
Temporal Complexity
- Identifying when inflammatory responses are protective versus harmful remains a major challenge
- The mechanisms marking the shift from acute to chronic inflammation are poorly defined
- Optimal timing for therapeutic intervention to maximize benefit is not yet established
Mechanistic Heterogeneity
- Patients show widely different inflammatory trajectories after stroke
- Multiple overlapping pathways, including complement activation, inflammasomes, and glial signalling, contribute to cognitive outcomes
- The relative roles of central vs peripheral inflammation are unclear
- Network-level interactions are difficult to reproduce in animal models
Clinical Implementation Barriers
- There are no validated biomarkers to stratify patients for targeted interventions
- Concerns about infection risk limit the adoption of anti-inflammatory strategies
- Cognitive rehabilitation remains largely focused on motor recovery rather than cognition
- Current healthcare studies often do not incentivize routine cognitive screening
Box A3: Recommendations for Advancing PSCI Research
Preclinical Research
- Use aged animals with relevant comorbidities (e.g., hypertension, diabetes) to better model the human population
- Include both sexes with adequate power for stratified analyses
- Extend follow-up to capture delayed cognitive decline
- Implement standardized, domain-specific cognitive batteries across laboratories
- Report negative findings to reduce publication bias and improve reproducibility
Clinical Research
- Incorporate domain-specific cognitive endpoints in all stroke trials
- Develop and validate inflammatory biomarker panels for prognosis and target engagement
- Design adaptive trials with biomarker-guided patient selection
- Extend outcome assessment to > 1 year post stroke to capture long-term trajectories
- Integrate cognitive rehabilitation strategies with immunomodulatory interventions
Translational Integration
- Establish bidirectional feedback between bench and bedside
- Use human biomarker and imaging data to refine preclinical targets
- Validate preclinical findings in human tissue and clinical studies
- Foster interdisciplinary collaboration (neurology, immunology, rehabilitation)
- Engage patients and caregivers in setting research priorities
Outstanding Questions
- Can biomarkers identify patients with “treatable” inflammatory profiles?
- Which cognitive domains are most amenable to anti-inflammatory interventions?
- How do we balance suppressing harmful inflammation while preserving repair?
- What combination of pharmacological and rehabilitative strategies is optimal?
Can late intervention reverse chronic neuroinflammation and restore function?
References
- El Husseini, N.; et al. Cognitive Impairment After Ischemic and Hemorrhagic Stroke: A Scientific Statement From the American Heart Association/American Stroke Association. Stroke 2023, 54. [Google Scholar] [CrossRef]
- Jokinen, H.; et al. Post-stroke cognitive impairment is common even after successful clinical recovery. Eur. J. Neurol. 2015, 22, 1288–1294. [Google Scholar] [CrossRef]
- Tack, R. W. P.; et al. Inflammation, Anti-inflammatory Interventions, and Post-stroke Cognitive Impairment: a Systematic Review and Meta-analysis of Human and Animal Studies. Transl. Stroke Res. 2025, 16, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Devlin, P.; Urayama, A.; Ritzel, R. M. Models and mechanisms of post-stroke dementia and cognitive impairment. Front. Stroke 2025, 4. [Google Scholar] [CrossRef]
- Sandvig, H. V.; et al. Plasma Inflammatory Biomarkers Are Associated With Poststroke Cognitive Impairment: The Nor-COAST Study. Stroke 2023, 54, 1303–1311. [Google Scholar] [CrossRef]
- Myers, S. J.; et al. Acute minocycline treatment inhibits microglia activation, reduces infarct volume, and has domain-specific effects on post-ischemic stroke cognition in rats. Behav. Brain Res. 2023, 455, 114680. [Google Scholar] [CrossRef]
- Pendlebury, S. T.; Rothwell, P. M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 2009, 8, 1006–1018. [Google Scholar] [CrossRef]
- Sun, J.H.; Tan, L.; Yu, J.T. Post-stroke cognitive impairment: epidemiology, mechanisms and management. Ann. Transl. Med. 2014, 2, 80. [Google Scholar] [PubMed]
- Dowling, N. M.; Johnson, S.; Nadareishvili, Z. Poststroke Cognitive Impairment and the Risk of Recurrent Stroke and Mortality: Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2024, 13. [Google Scholar] [CrossRef] [PubMed]
- Sexton, E.; et al. Systematic review and meta-analysis of the prevalence of cognitive impairment no dementia in the first year post-stroke. Eur. Stroke J. 2019, 4, 160–171. [Google Scholar] [CrossRef]
- Kalaria, R. N.; Akinyemi, R.; Ihara, M. Stroke injury, cognitive impairment and vascular dementia. Biochim. Et. Biophys. Acta (BBA) -Mol. Basis Dis. 2016, 1862, 915–925. [Google Scholar] [CrossRef]
- Barbay, M.; Diouf, M.; Roussel, M.; Godefroy, O. Systematic Review and Meta-Analysis of Prevalence in Post-Stroke Neurocognitive Disorders in Hospital-Based Studies. Dement Geriatr. Cogn. Disord. 2018, 46, 322–334. [Google Scholar] [CrossRef]
- Dichgans, M.; Leys, D. Vascular Cognitive Impairment. Circ. Res. 2017, 120, 573–591. [Google Scholar] [CrossRef]
- Zinn, S.; et al. The effect of poststroke cognitive impairment on rehabilitation process and functional outcome. Arch. Phys. Med. Rehabil. 2004, 85, 1084–1090. [Google Scholar] [CrossRef]
- Barker-Collo, S.; Feigin, V. L.; Parag, V.; Lawes, C. M. M.; Senior, H. Auckland Stroke Outcomes Study. Neurology 2010, 75, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Cicerone, K. D.; et al. Evidence-Based Cognitive Rehabilitation: Systematic Review of the Literature From 2009 Through 2014. Arch. Phys. Med. Rehabil. 2019, 100, 1515–1533. [Google Scholar] [CrossRef]
- Patel, M.; Coshall, C.; Rudd, A. G.; DA Wolfe, C. Natural history of cognitive impairment after stroke and factors associated with its recovery. Clin. Rehabil. 2003, 17, 158–166. [Google Scholar] [CrossRef]
- Mijajlović, M. D.; et al. Post-stroke dementia – a comprehensive review. BMC Med. 2017, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Obaid, M.; Flach, C.; Marshall, I.D.A.; Wolfe, C.; Douiri, A. Long-Term Outcomes in Stroke Patients with Cognitive Impairment: A Population-Based Study. Geriatrics 2020, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Patel, M. D.; et al. Relationships between long-term stroke disability, handicap and health-related quality of life. Age Ageing 2006, 35, 273–279. [Google Scholar] [CrossRef]
- Kwok, T.; et al. Quality of Life of Stroke Survivors: A 1-Year Follow-Up Study. Arch. Phys. Med. Rehabil. 2006, 87, 1177–1182. [Google Scholar] [CrossRef]
- Lim, J.-S.; Kang, D.-W. Stroke Connectome and Its Implications for Cognitive and Behavioral Sequela of Stroke. J. Stroke 2015, 17, 256–267. [Google Scholar] [CrossRef]
- Gorelick, P. B.; et al. Vascular Contributions to Cognitive Impairment and Dementia. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef]
- Corbett, A.; Bennett, H.; Kos, S. Cognitive Dysfunction Following Subcortical Infarction. Arch. Neurol. 1994, 51, 999–1007. [Google Scholar] [CrossRef]
- Ihara, M.; Yamamoto, Y. Emerging Evidence for Pathogenesis of Sporadic Cerebral Small Vessel Disease. Stroke 2016, 47, 554–560. [Google Scholar] [CrossRef]
- Quinn, T. J.; et al. European Stroke Organisation and European Academy of Neurology joint guidelines on post-stroke cognitive impairment. Eur. J. Neurol. 2021, 28, 3883–3920. [Google Scholar] [CrossRef]
- Brainin, M.; et al. Post-stroke cognitive decline: an update and perspectives for clinical research. Eur. J. Neurol. 2015, 22, 229. [Google Scholar] [CrossRef]
- Anrather, J.; Iadecola, C. Inflammation and Stroke: An Overview. Neurotherapeutics 2016, 13, 661–670. [Google Scholar] [CrossRef]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A. M. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: from mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Doyle, K. P.; Simon, R. P.; Stenzel-Poore, M. P. Mechanisms of ischemic brain damage. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef]
- Lambertsen, K. L.; Finsen, B.; Clausen, B. H. Post-stroke inflammation—target or tool for therapy? Acta Neuropathol. 2019, 137, 693–714. [Google Scholar] [CrossRef]
- Rothenburg, L. S.; et al. The Relationship Between Inflammatory Markers and Post Stroke Cognitive Impairment. J. Geriatr. Psychiatry Neurol. 2010, 23, 199–205. [Google Scholar] [CrossRef]
- Vahidy, F. S.; et al. Acute splenic responses in patients with ischemic stroke and intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2016, 36, 1012–1021. [Google Scholar]
- Becker, K. J. Modulation of the Postischemic Immune Response to Improve Stroke Outcome. Stroke 2010, 41. [Google Scholar] [CrossRef]
- Sachdev, P. S.; et al. The neuropsychological profile of vascular cognitive impairment in stroke and TIA patients. Neurology 2004, 62, 912–919. [Google Scholar] [CrossRef]
- Nys, G. M. S.; et al. The prognostic value of domain-specific cognitive abilities in acute first-ever stroke. Neurology 2005, 64, 821–827. [Google Scholar] [CrossRef]
- Cumming, T. B.; Marshall, R. S.; Lazar, R. M. Stroke, Cognitive Deficits, and Rehabilitation: Still an Incomplete Picture. Int. J. Stroke 2013, 8, 38–45. [Google Scholar] [CrossRef]
- Pohjasvaara, T.; et al. Post-stroke depression, executive dysfunction and functional outcome. Eur. J. Neurol. 2002, 9, 269–275. [Google Scholar] [CrossRef]
- Planton, M.; et al. Neuropsychological outcome after a first symptomatic ischaemic stroke with ‘good recovery. Eur. J. Neurol. 2012, 19, 212–219. [Google Scholar] [CrossRef]
- Snaphaan, L.; de Leeuw, F.-E. Poststroke Memory Function in Nondemented Patients. Stroke 2007, 38, 198–203. [Google Scholar] [CrossRef]
- Leśniak, M.; Bak, T.; Czepiel, W.; Seniów, J.; Członkowska, A. Frequency and Prognostic Value of Cognitive Disorders in Stroke Patients. Dement Geriatr. Cogn. Disord. 2008, 26, 356–363. [Google Scholar] [CrossRef]
- Jokinen, H. Cognitive profile of subcortical ischaemic vascular disease. J. Neurol. Neurosurg. Psychiatry 2006, 77, 28–33. [Google Scholar] [CrossRef]
- Hachinski, V.; et al. National Institute of Neurological Disorders and Stroke–Canadian Stroke Network Vascular Cognitive Impairment Harmonization Standards. Stroke 2006, 37, 2220–2241. [Google Scholar] [CrossRef]
- Nasreddine, Z. S.; et al. The Montreal Cognitive Assessment, MoCA: A Brief Screening Tool For Mild Cognitive Impairment. J. Am. Geriatr. Soc. 2005, 53, 695–699. [Google Scholar] [CrossRef]
- Pendlebury, S. T.; Cuthbertson, F. C.; Welch, S. J. V.; Mehta, Z.; Rothwell, P. M. Underestimation of Cognitive Impairment by Mini-Mental State Examination Versus the Montreal Cognitive Assessment in Patients With Transient Ischemic Attack and Stroke. Stroke 2010, 41, 1290–1293. [Google Scholar] [CrossRef]
- Ferris, S. H.; et al. ADCS Prevention Instrument Project: Overview and Initial Results. Alzheimer Dis. Assoc. Disord. 2006, 20, S109–S123. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association, 2013. [Google Scholar] [CrossRef]
- Pendlebury, S. T.; et al. Underfunding of Stroke Research. Stroke 2004, 35, 2368–2371. [Google Scholar] [CrossRef]
- Tatemichi, T. K.; et al. Cognitive impairment after stroke: frequency, patterns, and relationship to functional abilities. J. Neurol. Neurosurg. Psychiatry 1994, 57, 202–207. [Google Scholar] [CrossRef]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef]
- Nys, G. M. S.; et al. Cognitive Disorders in Acute Stroke: Prevalence and Clinical Determinants. Cerebrovasc. Dis. 2007, 23, 408–416. [Google Scholar] [CrossRef]
- Debette, S.; Markus, H. S. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ 2010, 341, c3666–c3666. [Google Scholar] [CrossRef]
- Gorelick, P. B. Risk Factors for Vascular Dementia and Alzheimer Disease. Stroke 2004, 35, 2620–2622. [Google Scholar] [CrossRef]
- Ballard, C.; Rowan, E.; Stephens, S.; Kalaria, R.; Kenny, R. A. Prospective Follow-Up Study Between 3 and 15 Months After Stroke. Stroke 2003, 34, 2440–2444. [Google Scholar] [CrossRef] [PubMed]
- Kauhanen, M.-L.; et al. Poststroke Depression Correlates With Cognitive Impairment and Neurological Deficits. Stroke 1999, 30, 1875–1880. [Google Scholar] [CrossRef]
- Kalaria, R. N. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol. 2016, 131, 659–685. [Google Scholar] [CrossRef]
- Sachdev, P. S.; et al. Risk Profiles of Subtypes of Mild Cognitive Impairment: The S ydney Memory and Ageing Study. J. Am. Geriatr. Soc. 2012, 60, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Mellon, L.; et al. Cognitive impairment six months after ischaemic stroke: a profile from the ASPIRE-S study. BMC Neurol. 2015, 15, 31. [Google Scholar] [CrossRef]
- Hochstenbach, J.; Mulder, T.; van Limbeek, J.; Donders, R.; Schoonderwaldt, H. Cognitive Decline Following Stroke: A Comprehensive Study of Cognitive Decline Following Stroke*. J. Clin. Exp. Neuropsychol. 1998, 20, 503–517. [Google Scholar] [CrossRef]
- SACHDEV, P. S.; et al. The determinants and longitudinal course of post-stroke mild cognitive impairment. J. Int. Neuropsychol. Soc. 2009, 15, 915–923. [Google Scholar] [CrossRef]
- Pendlebury, S. T.; Wadling, S.; Silver, L. E.; Mehta, Z.; Rothwell, P. M. Transient Cognitive Impairment in TIA and Minor Stroke. Stroke 2011, 42, 3116–3121. [Google Scholar] [CrossRef]
- Douiri, A.; Rudd, A. G.; Wolfe, C. D. A. Prevalence of Poststroke Cognitive Impairment. Stroke 2013, 44, 138–145. [Google Scholar] [CrossRef]
- Kokmen, E.; Whisnant, J. P.; O’Fallon, W. M.; Chu, C.-P.; Beard, C. M. Dementia after ischemic stroke. Neurology 1996, 46, 154–159. [Google Scholar] [CrossRef]
- Levine, D. A.; et al. Trajectory of Cognitive Decline After Incident Stroke. JAMA 2015, 314, 41. [Google Scholar] [CrossRef]
- Rasquin, S. M. C. Demographic and CT scan features related to cognitive impairment in the first year after stroke. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1562–1567. [Google Scholar] [CrossRef]
- Schneider, J. A.; Arvanitakis, Z.; Bang, W.; Bennett, D. A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef]
- Jellinger, K. A. Pathology and pathogenesis of vascular cognitive impairment—a critical update. Front Aging Neurosci. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M. M.; Nagy, Z.; Smith, M. Z.; Barnetson, L.; Smith, A. D. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. The Lancet 1999, 354, 919–920. [Google Scholar] [CrossRef] [PubMed]
- Snowdon, D. A. Brain Infarction and the Clinical Expression of Alzheimer Disease<subtitle>The Nun Study</subtitle>. JAMA J. Am. Med. Assoc. 1997, 277, 813. [Google Scholar]
- Zlokovic, B. V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J. T.; Thomas, A. Vascular dementia. The Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef]
- Villeneuve, S.; et al. Vascular risk and Aβ interact to reduce cortical thickness in AD vulnerable brain regions. Neurology 2014, 83, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Fluri, F.; Schuhmann, M. Animal models of ischemic stroke and their application in clinical research. Drug Des. Devel Ther. 2015, 3445. [Google Scholar] [CrossRef] [PubMed]
- Macrae, I. Preclinical stroke research – advantages and disadvantages of the most common rodent models of focal ischaemia. Br. J. Pharmacol. 2011, 164, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Schaar, K. L.; Brenneman, M. M.; Savitz, S. I. Functional assessments in the rodent stroke model. Exp. Transl. Stroke Med. 2010, 2, 13. [Google Scholar] [CrossRef]
- Whitehead, S.; Cheng, G.; Hachinski, V.; Cechetto, D. F. Interaction Between a Rat Model of Cerebral Ischemia and β-Amyloid Toxicity. Stroke 2005, 36, 1782–1789. [Google Scholar] [CrossRef]
- Watson, B. D.; Dietrich, W. D.; Busto, R.; Wachtel, M. S.; Ginsberg, M. D. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann. Neurol. 1985, 17, 497–504. [Google Scholar] [CrossRef]
- Labat-gest, V.; Tomasi, S. Photothrombotic Ischemia: A Minimally Invasive and Reproducible Photochemical Cortical Lesion Model for Mouse Stroke Studies. J. Vis. Exp. [CrossRef]
- Biernaskie, J.; Corbett, D.; Peeling, J.; Wells, J.; Lei, H. A serial MR study of cerebral blood flow changes and lesion development following endothelin-1-induced ischemia in rats. Magn. Reson Med. 2001, 46, 827–830. [Google Scholar] [CrossRef]
- Carmichael, S. T. Rodent models of focal stroke: Size, mechanism, and purpose. NeuroRX 2005, 2, 396–409. [Google Scholar] [CrossRef]
- Longa, E. Z.; Weinstein, P. R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef]
- Zhang, R. L.; Chopp, M.; Zhang, Z. G.; Jiang, Q.; Ewing, J. R. A rat model of focal embolic cerebral ischemia. Brain Res. 1997, 766, 83–92. [Google Scholar] [CrossRef]
- Hossmann, K.-A. The Two Pathophysiologies of Focal Brain Ischemia: Implications for Translational Stroke Research. J. Cereb. Blood Flow Metab. 2012, 32, 1310–1316. [Google Scholar] [CrossRef]
- Gelderblom, M.; et al. Temporal and Spatial Dynamics of Cerebral Immune Cell Accumulation in Stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef]
- Liu, F.; McCullough, L. D. Middle Cerebral Artery Occlusion Model in Rodents: Methods and Potential Pitfalls. In Biomed Res Int; 2011. [Google Scholar]
- Uzdensky, A. B. Photothrombotic Stroke as a Model of Ischemic Stroke. Transl. Stroke Res. 2018, 9, 437–451. [Google Scholar] [CrossRef]
- Sozmen, E. G.; Hinman, J. D.; Carmichael, S. T. Models That Matter: White Matter Stroke Models. Neurotherapeutics 2012, 9, 349–358. [Google Scholar] [CrossRef]
- Huuskonen, M. T.; et al. Protection of ischemic white matter and oligodendrocytes in mice by 3K3A-activated protein C. J. Exp. Med. 2022, 219. [Google Scholar] [CrossRef]
- Sozmen, E. G.; Kolekar, A.; Havton, L. A.; Carmichael, S. T. A white matter stroke model in the mouse: Axonal damage, progenitor responses and MRI correlates. J. Neurosci. Methods 2009, 180, 261–272. [Google Scholar] [CrossRef]
- Langdon, K. D.; Corbett, D. Improved Working Memory Following Novel Combinations of Physical and Cognitive Activity. Neurorehabil Neural Repair 2012, 26, 523–532. [Google Scholar] [CrossRef]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Vorhees, C. V.; Williams, M. T. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef]
- D’Hooge, R.; De Deyn, P. P. Applications of the Morris water maze in the study of learning and memory. Brain Res. Rev. 2001, 36, 60–90. [Google Scholar] [CrossRef]
- Barnes, C. A. Memory deficits associated with senescence: A neurophysiological and behavioral study in the rat. J. Comp. Physiol. Psychol. 1979, 93, 74–104. [Google Scholar] [CrossRef]
- Ennaceur, A.; Delacour, J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 1988, 31, 47–59. [Google Scholar] [CrossRef]
- Antunes, M.; Biala, G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn. Process 2012, 13, 93–110. [Google Scholar] [CrossRef]
- Birrell, J. M.; Brown, V. J. Medial Frontal Cortex Mediates Perceptual Attentional Set Shifting in the Rat. J. Neurosci. 2000, 20, 4320–4324. [Google Scholar] [CrossRef]
- Tennant, K. A.; Jones, T. A. Sensorimotor behavioral effects of endothelin-1 induced small cortical infarcts in C57BL/6 mice. J. Neurosci. Methods 2009, 181, 18–26. [Google Scholar] [CrossRef]
- Langdon, K. D.; Granter-Button, S.; Corbett, D. Persistent behavioral impairments and neuroinflammation following global ischemia in the rat. Eur. J. Neurosci. 2008, 28, 2310–2318. [Google Scholar] [CrossRef]
- Bari, A.; Dalley, J. W.; Robbins, T. W. The application of the 5-choice serial reaction time task for the assessment of visual attentional processes and impulse control in rats. Nat. Protoc. 2008, 3, 759–767. [Google Scholar] [CrossRef]
- Izquierdo, A.; et al. Reversal-Specific Learning Impairments After a Binge Regimen of Methamphetamine in Rats: Possible Involvement of Striatal Dopamine. Neuropsychopharmacology 2010, 35, 505–514. [Google Scholar] [CrossRef]
- Dere, E.; Huston, J. P.; De Souza Silva, M. A. The pharmacology, neuroanatomy and neurogenetics of one-trial object recognition in rodents. Neurosci. Biobehav Rev. 2007, 31, 673–704. [Google Scholar] [CrossRef]
- Modo, M.; Stroemer, R. P.; Tang, E.; Patel, S.; Hodges, H. Effects of Implantation Site of Stem Cell Grafts on Behavioral Recovery From Stroke Damage. Stroke 2002, 33, 2270–2278. [Google Scholar] [CrossRef] [PubMed]
- O’Collins, V. E.; et al. 1,026 Experimental treatments in acute stroke. Ann. Neurol. 2006, 59, 467–477. [Google Scholar] [CrossRef]
- Dirnagl, U. Bench to Bedside: The Quest for Quality in Experimental Stroke Research. J. Cereb. Blood Flow Metab. 2006, 26, 1465–1478. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; et al. Accelerated infarct development, cytogenesis and apoptosis following transient cerebral ischemia in aged rats. Acta Neuropathol. 2007, 113, 277–293. [Google Scholar] [CrossRef]
- Ay, I.; Sorensen, A. G.; Ay, H. Vagus nerve stimulation reduces infarct size in rat focal cerebral ischemia: An unlikely role for cerebral blood flow. Brain Res. 2011, 1392, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; et al. Update of the Stroke Therapy Academic Industry Roundtable Preclinical Recommendations. Stroke 2009, 40, 2244–2250. [Google Scholar] [CrossRef]
- Corbett, D.; Jeffers, M.; Nguemeni, C.; Gomez-Smith, M.; Livingston-Thomas, J. Lost in translation; 2015; pp. 413–434. [Google Scholar] [CrossRef]
- Macleod, M. R.; et al. Good Laboratory Practice. Stroke 2009, 40. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W. J.; Cuthill, I. C.; Emerson, M.; Altman, D. G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Manwani, B.; McCullough, L. D. Sexual Dimorphism in Ischemic Stroke: Lessons from the Laboratory. Women’s Health 2011, 7, 319–339. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; et al. Sex Differences in the Response to Poly(ADP-ribose) Polymerase-1 Deletion and Caspase Inhibition After Stroke. Stroke 2011, 42, 1090–1096. [Google Scholar] [CrossRef]
- Rosenberg, G. A.; Yang, Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg. Focus 2007, 22, 1–9. [Google Scholar] [CrossRef]
- Block, F.; Dihné, M.; Loos, M. Inflammation in areas of remote changes following focal brain lesion. Prog. Neurobiol. 2005, 75, 342–365. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J. S.; et al. Disruptions of network connectivity predict impairment in multiple behavioral domains after stroke. Proc. Natl. Acad. Sci. 2016, 113. [Google Scholar] [CrossRef] [PubMed]
- Allan, S. M.; Rothwell, N. J. Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2001, 2, 734–744. [Google Scholar] [CrossRef]
- Rosenberg, G. A. Inflammation and White Matter Damage in Vascular Cognitive Impairment. Stroke 2009, 40. [Google Scholar] [CrossRef]
- Hu, X.; et al. Microglial and macrophage polarization—new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M. R.; Franklin, R. J. M. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 2008, 132, 288–295. [Google Scholar] [CrossRef]
- Zhu, Z.; et al. Combination of the Immune Modulator Fingolimod With Alteplase in Acute Ischemic Stroke. Circulation 2015, 132, 1104–1112. [Google Scholar] [CrossRef]
- Yang, F.; et al. NLRP3 Deficiency Ameliorates Neurovascular Damage in Experimental Ischemic Stroke. J. Cereb. Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Streit, W. J.; Mrak, R. E.; Griffin, W. S. T. Microglia and neuroinflammation: a pathological perspective. J. Neuroinflammation 2004, 1, 14. [Google Scholar] [CrossRef]
- Hu, X.; et al. Microglia/Macrophage Polarization Dynamics Reveal Novel Mechanism of Injury Expansion After Focal Cerebral Ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef]
- Kuroiwa, T.; Ting, P.; Martinez, H.; Klatzo, I. The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol. 1985, 68, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-B.; et al. HMGB1, a Novel Cytokine-Like Mediator Linking Acute Neuronal Death and Delayed Neuroinflammation in the Postischemic Brain. J. Neurosci. 2006, 26, 6413–6421. [Google Scholar] [CrossRef]
- Tang, S.-C.; et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. 2007, 104, 13798–13803. [Google Scholar] [CrossRef] [PubMed]
- Morioka, T.; Kalehua, A. N.; Streit, W. J. Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J. Comp. Neurol. 1993, 327, 123–132. [Google Scholar] [CrossRef]
- Offner, H.; et al. Experimental Stroke Induces Massive, Rapid Activation of the Peripheral Immune System. J. Cereb. Blood Flow Metab. 2006, 26, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Chopp, M.; Li, Y.; Jiang, N.; Zhang, R. L.; Prostak, J. Antibodies against Adhesion Molecules Reduce Apoptosis after Transient Middle Cerebral Artery Occlusion in Rat Brain. J. Cereb. Blood Flow Metab. 1996, 16, 578–584. [Google Scholar] [CrossRef]
- Prass, K.; et al. Stroke-induced Immunodeficiency Promotes Spontaneous Bacterial Infections and Is Mediated by Sympathetic Activation Reversal by Poststroke T Helper Cell Type 1–like Immunostimulation. J. Exp. Med. 2003, 198, 725–736. [Google Scholar] [CrossRef]
- Ajmo, C. T.; et al. The spleen contributes to stroke-induced neurodegeneration. J. Neurosci. Res. 2008, 86, 2227–2234. [Google Scholar] [CrossRef]
- Seifert, H. A.; et al. A Transient Decrease in Spleen Size Following Stroke Corresponds to Splenocyte Release into Systemic Circulation. J. Neuroimmune Pharmacol. 2012, 7, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; et al. Microbiota Dysbiosis Controls the Neuroinflammatory Response after Stroke. J. Neurosci. 2016, 36, 7428–7440. [Google Scholar] [CrossRef]
- Stanley, D.; et al. Translocation and dissemination of commensal bacteria in post-stroke infection. Nat. Med. 2016, 22, 1277–1284. [Google Scholar] [CrossRef]
- Smith, C. J.; et al. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol. 2004, 4, 2. [Google Scholar] [CrossRef]
- Meisel, C.; Schwab, J. M.; Prass, K.; Meisel, A.; Dirnagl, U. Central nervous system injury-induced immune deficiency syndrome. Nat. Rev. Neurosci. 2005, 6, 775–786. [Google Scholar] [CrossRef]
- Chamorro, A.; Urra, X.; Planas, A. M. Infection After Acute Ischemic Stroke. Stroke 2007, 38, 1097–1103. [Google Scholar] [CrossRef]
- Dénes, Á.; Humphreys, N.; Lane, T. E.; Grencis, R.; Rothwell, N. Chronic Systemic Infection Exacerbates Ischemic Brain Damage via a CCL5 (Regulated on Activation, Normal T-Cell Expressed and Secreted)-Mediated Proinflammatory Response in Mice. J. Neurosci. 2010, 30, 10086–10095. [Google Scholar] [CrossRef]
- Liesz, A.; et al. DAMP Signaling is a Key Pathway Inducing Immune Modulation after Brain Injury. J. Neurosci. 2015, 35, 583–598. [Google Scholar] [CrossRef]
- Thiel, A.; Heiss, W.-D. Imaging of Microglia Activation in Stroke. Stroke 2011, 42, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M. A.; Lo, E. H.; Iadecola, C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Block, M. L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Perego, C.; Fumagalli, S.; De Simoni, M.-G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflammation 2011, 8, 174. [Google Scholar] [CrossRef]
- Morrison, H. W.; Filosa, J. A. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflammation 2013, 10, 782. [Google Scholar] [CrossRef]
- Jones, T.; Schallert, T. Use-dependent growth of pyramidal neurons after neocortical damage. J. Neurosci. 1994, 14, 2140–2152. [Google Scholar] [CrossRef]
- Dihné, M.; Block, F. Focal ischemia induces transient expression of IL-6 in the substantia nigra pars reticulata. Brain Res. 2001, 889, 165–173. [Google Scholar] [CrossRef]
- Kee, N. J.; Preston, E.; Wojtowicz, J. M. Enhanced neurogenesis after transient global ischemia in the dentate gyrus of the rat. Exp. Brain Res. 2001, 136, 313–320. [Google Scholar] [CrossRef]
- Rola, R.; et al. Alterations in hippocampal neurogenesis following traumatic brain injury in mice. Exp. Neurol. 2006, 202, 189–199. [Google Scholar] [CrossRef]
- Dewar, D.; Underhill, S. M.; Goldberg, M. P. Oligodendrocytes and Ischemic Brain Injury. J. Cereb. Blood Flow Metab. 2003, 23, 263–274. [Google Scholar] [CrossRef]
- Pantoni, L.; Garcia, J. H.; Gutierrez, J. A. Cerebral White Matter Is Highly Vulnerable to Ischemia. Stroke 1996, 27, 1641–1647. [Google Scholar] [CrossRef]
- Dalkara, T.; Gursoy-Ozdemir, Y.; Yemisci, M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011, 122, 1–9. [Google Scholar] [CrossRef]
- Chamorro, Á.; et al. The immunology of acute stroke. Nat. Rev. Neurol. 2012, 8, 401–410. [Google Scholar] [CrossRef]
- PEDERSEN, E. D.; WAJE-ANDREASSEN, U.; VEDELER, C. A.; AAMODT, G.; MOLLNES, T. E. Systemic complement activation following human acute ischaemic stroke. Clin. Exp. Immunol. 2004, 137, 117–122. [Google Scholar] [CrossRef]
- Schafer, D. P.; et al. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Hong, S.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (1979) 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Ten, V. S.; et al. C1q-Deficiency Is Neuroprotective Against Hypoxic-Ischemic Brain Injury in Neonatal Mice. Stroke 2005, 36, 2244–2250. [Google Scholar] [CrossRef]
- Yang-Wei Fann, D.; et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013, 4, e790–e790. [Google Scholar] [CrossRef]
- Franke, M.; et al. The NLRP3 inflammasome drives inflammation in ischemia/reperfusion injury after transient middle cerebral artery occlusion in mice. Brain Behav. Immun. 2021, 92, 221–231. [Google Scholar] [CrossRef]
- Ismael, S.; Zhao, L.; Nasoohi, S.; Ishrat, T. Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci. Rep. 2018, 8, 5971. [Google Scholar] [CrossRef]
- Denes, A.; et al. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. 2015, 112, 4050–4055. [Google Scholar] [CrossRef]
- Sofroniew, M. V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Astrocyte Reactivity and Reactive Astrogliosis: Costs and Benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef]
- Carrera, E.; Tononi, G. Diaschisis: past, present, future. Brain 2014, 137, 2408–2422. [Google Scholar] [CrossRef]
- Baron, J. C.; Bousser, M. G.; Comar, D.; Castaigne, P. “Crossed cerebellar diaschisis” in human supratentorial brain infarction. Trans. Am. Neurol. Assoc. 1981, 105, 459–61. [Google Scholar]
- Downer, B.; Raji, M. A.; Markides, K. S. Relationship between metabolic and vascular conditions and cognitive decline among older Mexican Americans. Int. J. Geriatr. Psychiatry 2016, 31, 213–221. [Google Scholar] [CrossRef]
- Doyle, K. P.; Buckwalter, M. S. Does B lymphocyte-mediated autoimmunity contribute to post-stroke dementia? Brain Behav. Immun. 2017, 64, 1–8. [Google Scholar] [CrossRef]
- Tamura, A.; et al. Thalamic atrophy following cerebral infarction in the territory of the middle cerebral artery. Stroke 1991, 22, 615–618. [Google Scholar] [CrossRef]
- Imai, F.; et al. Neuroprotective Effect of Exogenous Microglia in Global Brain Ischemia. J. Cereb. Blood Flow Metab. 2007, 27, 488–500. [Google Scholar] [CrossRef]
- Carter, A. R.; et al. Resting interhemispheric functional magnetic resonance imaging connectivity predicts performance after stroke. Ann. Neurol. 2010, 67, 365–375. [Google Scholar] [CrossRef]
- Dacosta-Aguayo, R.; et al. Impairment of functional integration of the default mode network correlates with cognitive outcome at three months after stroke. Hum. Brain Mapp. 2015, 36, 577–590. [Google Scholar] [CrossRef]
- Stellwagen, D.; Malenka, R. C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef]
- Murphy, T. H.; Corbett, D. Plasticity during stroke recovery: from synapse to behaviour. Nat. Rev. Neurosci. 2009, 10, 861–872. [Google Scholar] [CrossRef]
- McQuillen, P. S.; Ferriero, D. M. Selective vulnerability in the developing central nervous system. Pediatr. Neurol. 2004, 30, 227–235. [Google Scholar] [CrossRef]
- Matute, C.; et al. Excitotoxic damage to white matter. J. Anat. 2007, 210, 693–702. [Google Scholar] [CrossRef]
- Takahashi, J. L.; Giuliani, F.; Power, C.; Imai, Y.; Yong, V. W. Interleukin-1β promotes oligodendrocyte death through glutamate excitotoxicity. Ann. Neurol. 2003, 53, 588–595. [Google Scholar] [CrossRef]
- Ness, J. K.; Valentino, M.; McIver, S. R.; Goldberg, M. P. Identification of oligodendrocytes in experimental disease models. Glia 2005, 50, 321–328. [Google Scholar] [CrossRef]
- Kotter, M. R.; Li, W.-W.; Zhao, C.; Franklin, R. J. M. Myelin Impairs CNS Remyelination by Inhibiting Oligodendrocyte Precursor Cell Differentiation. J. Neurosci. 2006, 26, 328–332. [Google Scholar] [CrossRef]
- Coleman, M. Axon degeneration mechanisms: commonality amid diversity. Nat. Rev. Neurosci. 2005, 6, 889–898. [Google Scholar] [CrossRef]
- Thomalla, G. Time course of wallerian degeneration after ischaemic stroke revealed by diffusion tensor imaging. J. Neurol. Neurosurg. Psychiatry 2005, 76, 266–268. [Google Scholar] [CrossRef]
- Shen, Q.; et al. Endothelial Cells Stimulate Self-Renewal and Expand Neurogenesis of Neural Stem Cells. Science (1979) 2004, 304, 1338–1340. [Google Scholar] [CrossRef]
- Mi, S.; et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat. Neurosci. 2005, 8, 745–751. [Google Scholar] [CrossRef]
- Back, S. A.; et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 2005, 11, 966–972. [Google Scholar] [CrossRef]
- Auriel, E.; et al. Estimating Total Cerebral Microinfarct Burden From Diffusion-Weighted Imaging. Stroke 2015, 46, 2129–2135. [Google Scholar] [CrossRef]
- Jickling, G. C.; Sharp, F. R. Biomarker Panels in Ischemic Stroke. Stroke 2015, 46, 915–920. [Google Scholar] [CrossRef]
- Karuppagounder, S. S.; et al. Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef]
- Khalil, M.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Yang, C.; Hawkins, K. E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol.-Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Hjalmarsson, C.; et al. Neuronal and Glia-Related Biomarkers in Cerebrospinal Fluid of Patients with Acute Ischemic Stroke. J. Cent. Nerv. Syst. Dis. 2014, 6, JCNSD.S13821. [Google Scholar] [CrossRef]
- Chappell, M. A.; et al. Separation of macrovascular signal in multi-inversion time arterial spin labelling MRI. Magn. Reson Med. 2010, 63, 1357–1365. [Google Scholar] [CrossRef]
- Schain, M.; Kreisl, W. C. Neuroinflammation in Neurodegenerative Disorders—a Review. Curr. Neurol. Neurosci. Rep. 2017, 17, 25. [Google Scholar] [CrossRef]
- Rost, N. S.; et al. Stroke Severity Is a Crucial Predictor of Outcome: An International Prospective Validation Study. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Emsley, H. An early and sustained peripheral inflammatory response in acute ischaemic stroke: relationships with infection and atherosclerosis. J. Neuroimmunol. 2003, 139, 93–101. [Google Scholar] [CrossRef]
- Clarkson, A. N.; Huang, B. S.; MacIsaac, S. E.; Mody, I.; Carmichael, S. T. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 2010, 468, 305–309. [Google Scholar] [CrossRef]
- Alawieh, A.; Elvington, A.; Tomlinson, S. Complement in the Homeostatic and Ischemic Brain. Front Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Coll, R. C.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Jay, T. R.; von Saucken, V. E.; Landreth, G. E. TREM2 in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 56. [Google Scholar] [CrossRef]
- Lampl, Y.; et al. Minocycline treatment in acute stroke. Neurology 2007, 69, 1404–1410. [Google Scholar] [CrossRef]
- Kohler, E.; et al. Intravenous Minocycline in Acute Stroke. Stroke 2013, 44, 2493–2499. [Google Scholar] [CrossRef]
- Sierra, S.; et al. Statins as Neuroprotectants: A Comparative In Vitro Study of Lipophilicity, Blood-Brain-Barrier Penetration, Lowering of Brain Cholesterol, and Decrease of Neuron Cell Death. J. Alzheimer’s Dis. 2011, 23, 307–318. [Google Scholar] [CrossRef]
- Westover, M. B.; Bianchi, M. T.; Eckman, M. H.; Greenberg, S. M. Statin Use Following Intracerebral Hemorrhage. Arch. Neurol. 2011, 68. [Google Scholar] [CrossRef]
- Fu, Y.; et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc. Natl. Acad. Sci. 2014, 111, 18315–18320. [Google Scholar] [CrossRef]
- Liesz, A.; et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef]
- Nguyen, H.; et al. Stem cell therapy for neurological disorders: A focus on aging. Neurobiol. Dis. 2019, 126, 85–104. [Google Scholar] [CrossRef]
- Jolkkonen, J.; et al. Neuroprotection by the α2-adrenoceptor agonist, dexmedetomidine, in rat focal cerebral ischemia. Eur. J. Pharmacol. 1999, 372, 31–36. [Google Scholar] [CrossRef]
- Manwani, B.; et al. Differential effects of aging and sex on stroke induced inflammation across the lifespan. Exp. Neurol. 2013, 249, 120–131. [Google Scholar] [CrossRef]
- McCullough, L. D.; Zeng, Z.; Blizzard, K. K.; Debchoudhury, I.; Hurn, P. D. Ischemic Nitric Oxide and Poly (ADP-Ribose) Polymerase-1 in Cerebral Ischemia: Male Toxicity, Female Protection. J. Cereb. Blood Flow Metab. 2005, 25, 502–512. [Google Scholar] [CrossRef]
- Quinn, T. J.; Dawson, J.; Walters, M. R.; Lees, K. R. Functional Outcome Measures in Contemporary Stroke Trials. Int. J. Stroke 2009, 4, 200–205. [Google Scholar] [CrossRef]
- Sena, E. S.; van der Worp, H. B.; Bath, P. M. W.; Howells, D. W.; Macleod, M. R. Publication Bias in Reports of Animal Stroke Studies Leads to Major Overstatement of Efficacy. PLoS Biol. 2010, 8, e1000344. [Google Scholar] [CrossRef]
- Clayton, J. A.; Collins, F. S. Policy: NIH to balance sex in cell and animal studies. Nature 2014, 509, 282–283. [Google Scholar] [CrossRef]
- Pollock, A.; St George, B.; Fenton, M.; Firkins, L. Top 10 Research Priorities Relating to Life after Stroke – Consensus from Stroke Survivors, Caregivers, and Health Professionals. Int. J. Stroke 2014, 9, 313–320. [Google Scholar] [CrossRef]
Figure 1.
Temporal trajectory of post-stroke cognitive impairment. Post-stroke cognitive impairment (PSCI) evolves dynamically across acute (days-weeks), subacute (weeks-months), and chronic (>6 months-years) phases. The schematic depicts cognitive trajectories over time with their symptoms and projected outcomes as impairments persist into the chronic stage.
Figure 1.
Temporal trajectory of post-stroke cognitive impairment. Post-stroke cognitive impairment (PSCI) evolves dynamically across acute (days-weeks), subacute (weeks-months), and chronic (>6 months-years) phases. The schematic depicts cognitive trajectories over time with their symptoms and projected outcomes as impairments persist into the chronic stage.


Table 1.
Overview of Preclinical Stroke Models and Their Lesion Features, Cognitive Effects, Strengths, and Limitations. This table summarizes commonly used preclinical stroke models, including lesion location, associated cognitive impairments, experimental advantages, and limitations. Middle Cerebral Artery Occlusion: MCAO; Endothelin-1: ET-1; N5-(1 iminoethyl)-L-ornithine: L-NIO.
Table 1.
Overview of Preclinical Stroke Models and Their Lesion Features, Cognitive Effects, Strengths, and Limitations. This table summarizes commonly used preclinical stroke models, including lesion location, associated cognitive impairments, experimental advantages, and limitations. Middle Cerebral Artery Occlusion: MCAO; Endothelin-1: ET-1; N5-(1 iminoethyl)-L-ornithine: L-NIO.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



