Submitted:
09 June 2026
Posted:
10 June 2026
You are already at the latest version
Abstract
Contemporary radiobiology explains tissue response to ionizing radiation through the linear-quadratic model and the four Rs of fractionation—tools that falter on FLASH sparing, the non-local responses of spatially fractionated radiotherapy (SFRT), the supra-linear control of stereotactic body radiotherapy (SBRT), and reversible non-genetic resistance, each currently explained by its own single-timescale literature. We propose that these are perturbations of one multi-scale adaptive state, organized by the timescale of adaptation into four coupled layers: fast redox chemistry (ms–s), collective organization (s–days), persistent epigenetic memory (days–weeks), and clonal evolution (weeks–months). The framework's claim is not these bands but their dynamical coupling through shared substrates, so that a perturbation entering one propagates to another—made concrete by the recent identification of an Activator Protein-1 (AP-1) –mediated write converting transient signals into heritable states. We formalize this as a coupled, spatially-resolved dynamical system in which every mechanism enters cell kill through a single modulation factor, recovering the linear-quadratic model as its frozen-state limit. Simulations reproduce the defining behaviors of spatially fractionated delivery and adaptive therapy, and show that closing the cross-band loop yields a bistable, hysteretic persistent state, a memory-writing geometry distinct from the dose-optimal one, and history-dependence at fixed dose. The framework generates falsifiable, cross-modality predictions absent from conventional radiobiology—notably that modality interactions are selective, additive when two modalities perturb the same state variable and non-additive when they perturb different coupled ones—and reframes treatment planning as a game against a largely latent, adapting state, played through multiple handles of which dose is only one. Because that state is only partially observable and its parameters are not generally identifiable from clinical data, the planner must act against a posterior rather than a known opponent, making the clinical problem one of Bayesian, latent-state adaptive control rather than fixed calibration.
Keywords:
radiobiology
; linear-quadratic model
; FLASH radiotherapy
; spatially fractionated radiotherapy
; non-genetic resistance
; epigenetic memory
; multi-scale coupling
1. Introduction
Modern radiobiology rests on a small set of durable ideas. The linear-quadratic (LQ) model relates cell survival to absorbed dose through two coefficients, and the “four Rs”—repair, repopulation, redistribution, and reoxygenation—organize how fractionated treatment is understood and planned. Within their domain of validity, these tools are reliable and clinically indispensable.
A number of phenomena, however, have proven difficult to accommodate. FLASH radiotherapy, in which a therapeutic dose is delivered at ultra-high dose rates within milliseconds, produces a reduction in normal-tissue toxicity while tumor control is largely preserved, an effect not predicted by dose alone (Vozenin et al., 2019; Favaudon et al., 2014). Spatially fractionated radiotherapy (SFRT), which delivers a spatially non-uniform dose with high-dose peaks and low-dose valleys, produces tumor responses larger than the directly irradiated fraction would predict, implicating communication between irradiated and unirradiated regions (Asur et al., 2015; Griffin et al., 2020). Stereotactic body radiotherapy (SBRT), which delivers very high doses per fraction, achieves tumor control rates that have been argued to exceed LQ-based expectations, prompting proposals of additional vascular and immune mechanisms (Song et al., 2021; Brown et al., 2014). And treatment resistance increasingly appears to involve not only selection on genetic variants but reversible, non-genetic adaptation that precedes and enables stable resistance (Sharma et al., 2010; Shaffer et al., 2017; Pisco and Huang, 2015).
Each of these phenomena has generated its own mechanistic literature, and each literature is, in isolation, productive. FLASH is debated in terms of radiolytic oxygen depletion and radical chemistry; SFRT in terms of bystander signaling, vascular response, and immune activation; SBRT in terms of endothelial damage and immunogenic cell death; resistance in terms of persister states and clonal evolution. What is largely absent is a structure within which these explanations relate to one another rather than competing for the role of “the” mechanism.
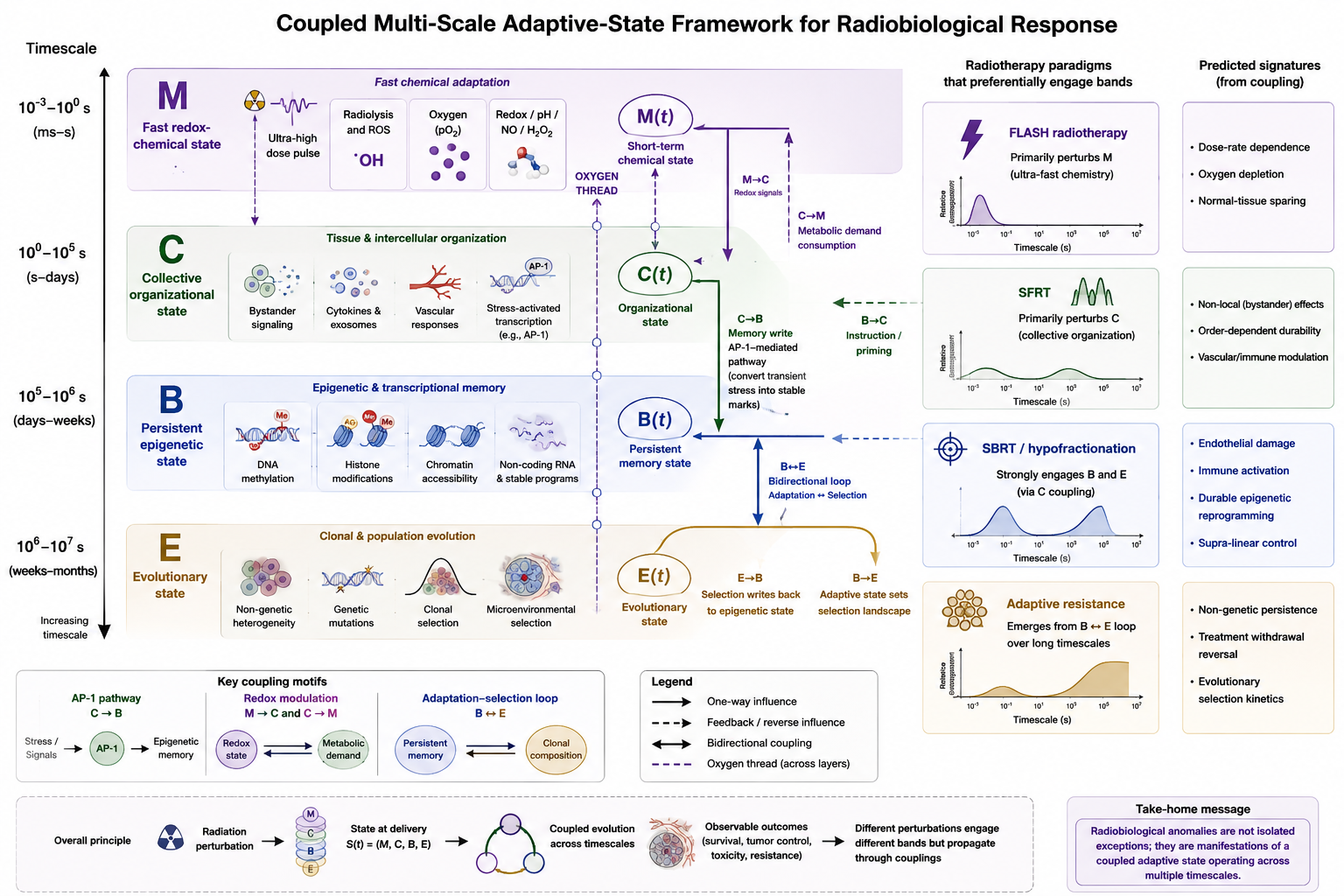
This article proposes such a structure. We suggest that radiobiological response depends not only on the dose delivered but on an evolving internal state of the irradiated tissue—tumor and normal tissue alike—and that this state is naturally organized by timescale. Biological adaptation does not occur at a single rate. Redox chemistry resolves within milliseconds to seconds; intercellular signaling and tissue reorganization unfold over seconds to days; epigenetic reprogramming persists over days to weeks; and clonal evolution operates over weeks to months. We propose that these four timescale bands constitute four coupled state layers, that radiation perturbs all of them but engages each to a different degree depending on how it is delivered, and that the radiobiological anomalies above correspond to perturbations that preferentially probe different bands.
We emphasize at the outset what the framework is and is not. It is an organizing hypothesis, not a mechanistic claim about any single phenomenon. It does not assert that the layers are biologically separable into discrete modules, and it does not propose new chemistry or new molecular pathways. Its contribution is to organize existing, independently supported biology into a coupled multi-scale picture, to make explicit that the coupling between timescale bands may itself be a determinant of treatment response, and to formalize that coupling as an explicit dynamical system whose behavior can be simulated and whose consequences can be tested.
The idea that radiotherapy response should be modeled across coupled biological scales is not itself new. Multiscale models linking gene, cell, and tissue levels were introduced two decades ago by Ribba, Colin and Schnell (2006), who coupled a Boolean genetic network, a cell-cycle model, and tissue mechanics and showed—on a timescale axis explicitly drawn in their schematic—that treatment scheduling can alter outcome at fixed total dose. A substantial computational lineage has since coupled cell-cycle dynamics, oxygen heterogeneity, and bystander signaling within hybrid models of radiation response (Powathil et al., 2012; Powathil et al., 2016), and, on a different substrate, recent work has coupled epigenetic-state dynamics to evolutionary population dynamics to explain non-genetic drug tolerance (Wang, Lei, et al., 2025). Each, however, operates within a single timescale regime: the first couples gene, cell, and tissue scales within an hours-to-days window; the second couples epigenetic and evolutionary dynamics in drug resistance, without radiation, FLASH, or spatial fractionation. The present framework differs not by adding bands but by coupling across the full timescale span—carrying downward causation onto persistent and evolutionary layers that the radiotherapy-modeling tradition does not contain, closing the resulting loop, and reducing to the linear-quadratic model in its frozen-state limit. Because this positioning is what most sharply defines the framework’s contribution, we defer its full treatment to the Discussion (Section 10.2) and note here only that it is the coupling across timescale bands, rather than multiscale modeling as such, that the framework proposes—made possible because the slow-layer biology it requires has only lately been characterized.
The framework presented here is developed at greater length, and situated within a broader argument about adaptation and information in radiation oncology, in a companion volume (Vaitheeswaran, 2026a). The present paper extracts from that wider treatment a single, focused claim—that radiobiological response is the behaviour of a coupled, multi-timescale adaptive state—and develops it in the specific, testable form set out below: a formal dynamical system (Section 4 and Methods), its simulated behavior (Results), and the predictions that distinguish it from conventional radiobiology (Predictions).
2. Adaptation Operates Across Distinct Timescales
The central organizing principle of the framework is that biological adaptation to perturbation is not a single process but a set of processes separated by their characteristic timescales. This separation is not an assumption imposed on the biology; it is a feature of the biology itself, and it is what motivates a layered description rather than a single state variable.
Fast redox and radical chemistry resolves on the scale of milliseconds to seconds. Oxygen consumption, radical recombination, and the chemical fixation of radiation damage occur essentially within and immediately after a radiation pulse (Pratx and Kapp, 2019; Labarbe et al., 2020). Intercellular communication and tissue-level reorganization—bystander signaling, vascular response, and the propagation of stress signals between cells—occur over seconds to days (Asur et al., 2015; Sheth and Esfandiari, 2022). Epigenetic reprogramming that persists after the initiating stimulus has been removed operates over days to weeks (Sharma et al., 2010; Li et al., 2026). And clonal selection, in which the composition of a heterogeneous cell population shifts under treatment pressure, plays out over weeks to months (Greaves and Maley, 2012; Gatenby and Brown, 2020).
These bands are not arbitrary divisions. Each corresponds to a body of experimental evidence describing adaptive behavior at that scale, and the gaps between them—several orders of magnitude in each case—are wide enough that the processes can, to a first approximation, be treated as operating on separate clocks. It is this separation of timescales that licenses a layered description. A single state variable cannot simultaneously capture a millisecond redox transient and a months-long evolutionary shift; the natural representation is a set of coupled variables, each carrying the adaptive history relevant to its own band.
We therefore propose four state layers, defined primarily by timescale and secondarily by the biology that populates each band: a fast adaptive layer M (milliseconds to seconds), a collective organizational layer C (seconds to days), a persistent adaptive layer B (days to weeks), and an evolutionary layer E (weeks to months). The number four reflects the number of well-separated timescale bands the current evidence supports, not a derivation from first principles. We make no claim that this is the only possible decomposition, and we return below to the ways in which the layers overlap.
3. The Four Adaptive Layers and Their Evidence
3.1. The Fast Adaptive Layer (M): Redox and Radical Chemistry
The fastest layer concerns the chemical environment in which radiation damage is fixed or repaired, on the timescale of the radiation delivery itself. The best-characterized variable in this band is molecular oxygen. Oxygen fixes radiation-induced damage, and its local concentration changes during irradiation as it is consumed by radical reactions and replenished by diffusion and perfusion. Treated as a dynamic quantity rather than a static modifier, oxygen has the properties of a fast state variable: it is depleted by the perturbation, recovers between perturbations, and carries the recent history of irradiation forward in its concentration (Pratx and Kapp, 2019; Cao et al., 2021).
Computational and physicochemical models have formalized this dynamic behavior. Pratx and Kapp (2019) modeled transient radiolytic oxygen depletion as a coupled process of consumption, diffusion, and metabolic uptake. Labarbe et al. (2020) developed a reaction-kinetics account centered on radical recombination. More comprehensively, the UNIVERSE model couples oxygen depletion, reoxygenation, and DNA-damage-repair kinetics into a single time-dependent description and reproduces in vitro data on the fast kinetics of the oxygen effect (Liew et al., 2021; Liew et al., 2022). These models establish that response to the fine temporal structure of delivery can arise from dynamic state variables evolving on the millisecond-to-second scale.
We adopt this established dynamic-oxygen description as the substrate of the M layer rather than proposing a new one. We note, however, that the sufficiency of oxygen alone remains debated: recent reviews and measurements indicate that the magnitude of radiolytic oxygen depletion achievable in vivo may be too small to account fully for the observed sparing, and that additional radical, metabolic, or mitochondrial contributions are likely involved (Boscolo et al., 2024; Jin et al., 2025). The M layer is therefore best understood as a fast chemical-state band whose dominant but not exclusive variable is dynamic oxygen.
3.2. The Collective Organizational Layer (C): Intercellular Communication and Tissue Structure
The second layer concerns the organization of interactions among cells, rather than the state of any cell in isolation. Radiation response is not confined to directly irradiated cells: signals propagate to cells that received little or no dose, altering their behavior. These radiation-induced bystander effects are mediated by reactive oxygen species, cytokines, and other secreted factors, and operate over seconds to days (Asur et al., 2015; Casteloes et al., 2025).
This collective behavior is the operative biology of SFRT. In the low-dose valleys of a spatially fractionated field, cells respond as though they had received more dose than they did, an effect attributed to signals propagating from the high-dose peaks (Griffin et al., 2020; Asur et al., 2015). Recent three-dimensional co-culture models have directly demonstrated that clonogenic survival following GRID irradiation is lower than the directly irradiated fraction alone would predict, providing controlled evidence of a bystander contribution (Casteloes et al., 2025). The kinetics of such signaling have been formalized: McMahon et al. (2013) developed a model of radiation-induced intercellular signaling in which a diffusible signal generated by irradiated cells propagates to and modifies the response of neighboring cells, providing a mechanistic description of how the C layer transmits a perturbation beyond the directly irradiated region. Such models describe the intercellular delivery of the signal in detail, but treat the recipient cell’s response as essentially memoryless—the arriving signal maps to an immediate effect, with no representation of whether it leaves a durable internal trace. This gap, at the recipient cell’s membrane, is where the coupling to the persistent layer enters, as developed in Section 4 and the Methods.
That collective organization can causally drive whole-population behavior, rather than merely accompany it, is shown by evidence that minority subpopulations can dictate the growth and behavior of an entire tumor through non-cell-autonomous signaling (Marusyk et al., 2014). This establishes that the organizational state of a cell population is not a passive sum of its members but an active determinant of their collective behavior—the defining property the C layer requires.
The notion that tissue carries an organizational state distinct from the sum of its cells is supported independently by work on bioelectric signaling. Membrane potentials, coordinated across cells through ion channels and gap junctions, encode tissue-level patterning information and are dysregulated in cancer; cancer has been characterized in part as a disorder of tissue organization rather than solely of individual cells (Levin, 2021; Sheth and Esfandiari, 2022). This body of work supports the proposition that a collective organizational state exists, is biologically consequential, and is altered by perturbation—the properties the C layer requires.
3.3. The Persistent Adaptive Layer (B): Epigenetic Memory
The third layer concerns adaptive states that persist after the initiating perturbation has been removed, encoded without genetic change. The clearest evidence comes from the study of drug-tolerant persister (DTP) cells. These are subpopulations of cancer cells that survive otherwise lethal therapy through reversible, non-genetic adaptation, maintained by chromatin remodeling and transcriptional reprogramming, and capable of reverting to a drug-sensitive state after treatment withdrawal (Sharma et al., 2010; Wang, Wang, et al., 2025; Li et al., 2025). A 2026 scoping review spanning 343 studies found this reversible, epigenetically maintained tolerant state to be a convergent feature across cancer types and experimental systems (Lopez-Gonzalez et al., 2026).
The molecular basis of such persistence has been characterized in detail for at least one pathway. Li et al. (2026) demonstrated that the transcription factor AP-1 mediates the formation of cellular memory during the acquisition of therapy resistance: the transcriptional state of a cell at the time of stress is encoded in cis through AP-1 activity and propagated forward, such that cells exposed to a low dose of therapy adapt to survive a higher dose than selection alone would predict. This provides a concrete, demonstrated mechanism by which a transient perturbation becomes a persistent, heritable adaptive state—the defining property of the B layer.
The capacity of biological substrates to encode and act on prior experience extends beyond cancer, and recent work establishes it at several levels of organization. At the level of intracellular biochemistry, Eckert et al. (2024) showed that simple, biochemically plausible reaction networks can reproduce the defining features of habituation—including frequency sensitivity and spontaneous recovery—without any specialized memory machinery, demonstrating that adaptive memory can be an emergent property of ordinary regulatory chemistry. At the level of the whole single cell, Doan et al. (2026) provided experimental evidence of associative learning in the ciliate Stentor coeruleus, in which temporally paired stimuli produce a transiently enhanced response that depends systematically on the temporal structure of conditioning and is well described by a model combining associative learning with habituation. At the level of regulatory networks, associative conditioning has been demonstrated in gene regulatory network models, where training increases the integrative causal emergence of the network (Pigozzi et al., 2025). Together this work supports the broader proposition that biological substrates—from reaction networks to single cells to regulatory architectures—can store and act on aspects of prior experience, of which the AP-1 mechanism in cancer is a specific, experimentally grounded instance directly relevant to therapy resistance.
3.4. The Evolutionary Layer (E): Clonal Selection
The slowest layer concerns changes in the composition of a heterogeneous cell population under selective pressure, operating over weeks to months. That cancer is an evolutionary disease, and that treatment imposes a selective pressure favoring resistant subpopulations, is well established (Greaves and Maley, 2012). The clinical relevance of treating this explicitly has been demonstrated: adaptive therapy, which modulates treatment to manage rather than maximally suppress the resistant population, prolonged tumor control relative to standard dosing in a prospective trial in metastatic castration-resistant prostate cancer (Zhang et al., 2017; Gatenby and Brown, 2020).
The evolutionary layer is conceptually distinct from the persistent adaptive layer, and the distinction matters. The B layer concerns adaptation within cells—reversible, induced, encoded epigenetically. The E layer concerns selection among cells—a shift in population composition. Recent work has emphasized that these interact: the reversible persister state of the B layer acts as a staging ground from which stable, heritable resistance subsequently emerges (Wang, Wang, et al., 2025). Some authors have characterized the induced, heritable component of this process in explicitly Lamarckian terms, as the induction of non-genetic resistance traits that are then inherited, operating alongside Darwinian selection (Valcz et al., 2025). The framework represents both: induced adaptation in B, selection in E, and their coupling between the two layers.
4. Coupled Dynamics: The Structural Core of the Framework
The layers are not independent, and the coupling between them is where the framework makes its distinctive claim. A description that merely partitioned radiobiology into four timescale bands would add little to the existing modality-specific literatures. The proposal here is stronger: that the bands are dynamically coupled, that the coupling is in general bidirectional, and that a single physical variable may participate in several bands through its dynamics on different timescales. It is this coupling that generates consequences no single-band model produces, and it is therefore the part of the framework on which its value rests. The Methods section formalizes this coupling as an explicit dynamical system; here we set out its structure.
4.1. A Minimal Representation
We represent the system by a state composed of the four layers,
evolving under an external input U(t) that encodes the treatment—its dose, dose rate, temporal structure, and spatial distribution. The evolution of each layer is written as a function of the full state and the input,
The presence of every layer in the argument of every function is the formal statement that the layers are coupled. We make no claim to know the functional forms fM, fC, fB, fE; different biological hypotheses correspond to different choices of these functions, and the framework’s purpose is to specify the coupling structure rather than to prescribe a mechanism. The well-separated timescales of the four layers mean that, to a first approximation, each function carries an intrinsic rate set by its band, with the cross-layer arguments acting as slower or faster modulations.
To connect this state to an observable outcome, we posit that the rate at which lethal damage accumulates depends on the current state and on the instantaneous dose rate. Writing the surviving fraction SF, the natural form that preserves the dose and dose-squared structure of classical radiobiology is
where D(t) is the cumulative dose delivered up to time t, Ḋ(t) is the instantaneous dose rate, and the state-dependent coefficients α(X) and β(X) represent, respectively, the single-track and two-track lethality of the tissue in its current configuration. This response functional is the bridge between the state dynamics and the measurable outcome: the state evolves according to the coupled equations above, and the damage it sustains at each instant is read out through α(X) and β(X). We do not claim specific forms for α(X) and β(X); we claim only that lethality is a function of the evolving state rather than a fixed constant, which is the minimal departure from classical radiobiology that the framework requires. Writing the radiosensitivity coefficients as state-dependent functions α(X) and β(X), rather than as the fixed constants α and β of the classical model, is the formal core of the framework.
A feature of this functional, developed in the Methods, is worth drawing out here. Both coefficients are modulated by the same state-dependent factor—each layer rescales the lethality of a track rather than adding lethality of its own—so the entire influence of the multi-scale state on kill can be written through a single dimensionless modulation Φ(X), with α(X) = α0·Φ(X) and β(X) = β0·Φ(X). The state therefore acts on radiosensitivity in an essentially one-dimensional way: it rescales the survival curve through Φ. We treat this single-factor form as a deliberate idealization, not an established fact—oxygen is known to act more strongly on the quadratic term, so α/β is not strictly invariant—and its testable content is that the pattern by which a perturbation changes α relative to β diagnoses which component of lethality it acts on, a point developed in the Methods and Predictions.
4.2. Oxygen as a Vertical Thread (Bottom-Up and Top-Down Through All Bands)
The clearest illustration that the layers share variables is oxygen, which participates in every band through its dynamics on the corresponding timescale.
Bottom-up, the fast oxygen state shapes the slower layers. On the M timescale oxygen is depleted and replenished within and between pulses (Pratx and Kapp, 2019). The resulting oxygenation, sustained over longer periods, drives hypoxia-induced transcriptional and epigenetic reprogramming in the B layer (Sheth and Esfandiari, 2022), and acts as one of the strongest selective pressures shaping clonal composition in the E layer (Greaves and Maley, 2012). A fast chemical variable thus has consequences that propagate upward to the slowest band.
Top-down, the slower layers set the conditions under which the fast oxygen state operates. The spatial distribution of oxygen is determined by the vascular and perfusion structure that constitutes much of the C layer, and is reshaped by radiation-induced vascular change and inter-fraction reoxygenation. The metabolic phenotype encoded in the B layer modulates oxygen consumption. And the clonal composition of the E layer—through differences in angiogenic and metabolic phenotype—reshapes the oxygen environment that in turn selects it. The same physical variable therefore threads through all four bands in both directions. This is not a category error but the natural consequence of representing a multi-scale system: a mechanism that operates across timescales is represented in each band by its behavior on that band’s clock, and the cross-band influence of that mechanism is precisely a coupling.
4.3. The C→B Coupling: Transient Signal to Durable State
The coupling for which the framework has the strongest mechanistic support is from the collective layer to the persistent layer, and it is best understood as supplying a module that the standard bystander models structurally omit. Kinetic models of intercellular signaling describe how the bystander signal is generated and delivered to a recipient cell, but stop at the recipient’s membrane: the signal maps to an immediate effect, with no account of the cell’s internal response or whether that response persists (McMahon et al., 2013). The coupling concerns precisely what happens past the membrane. Bystander signals propagating through the C layer—reactive oxygen species, cytokines, and stress mediators—can, in recipient cells, activate the stress-responsive transcriptional machinery that the B layer encodes. The AP-1 pathway provides a demonstrated molecular route for this intracellular step: AP-1 is activated by reactive oxygen species and stress signaling, and through CBP/p300 it writes durable chromatin marks that are propagated across cell divisions (Li et al., 2026). AP-1 thus furnishes exactly the recipient-cell response dynamics that the bystander models leave unspecified—the conversion of a transient organizational signal into a durable heritable state, which is the operation required to couple an intermediate layer into a slow, persistent one. We note that this mechanism has been demonstrated in the setting of induced drug resistance rather than in the radiation setting itself; that the specific radiation-bystander signal engages AP-1 in this way is the proposed coupling, and an explicit extension of the kinetic-bystander model to include such an intracellular epigenetic-memory step has been developed separately (Vaitheeswaran, 2026b). The concrete form of this coupling—a bistable, autoregulatory write—is specified in the Methods.
Because the same write machinery is redox-sensitive, the framework anticipates that the fast oxygen/redox layer may feed the same integrator—an M→B coupling in which the redox excursion of a fast delivery, rather than a bystander signal, supplies the input. This is a hypothesis, not a demonstrated coupling: AP-1’s redox sensitivity and the redox excursion of ultra-high-dose-rate delivery make the linkage plausible, but it has not been shown that fast delivery drives AP-1 memory. We flag it here as the second target for mechanistic test, and treat AP-1 as a candidate convergent entry point into the B layer that may be fed by more than one upstream band.
4.4. The B↔E Coupling: Induced State and Selection
The persistent and evolutionary layers are coupled in both directions, and recent work makes the relationship concrete. Bottom-up, the induced persister state of the B layer acts as a staging ground from which stable, heritable resistance subsequently emerges: cells held in a reversible drug-tolerant state provide the substrate on which selection in the E layer then operates (Wang, Wang, et al., 2025). The induced, heritable component of this process has been described in explicitly Lamarckian terms—as the induction of non-genetic resistance traits that are subsequently inherited—operating alongside Darwinian selection rather than instead of it (Valcz et al., 2025). Top-down, the evolutionary layer reshapes the persistent layer: as selection changes the clonal composition of the population, it changes the distribution of epigenetic programs available to be induced, so that the B-layer states reachable by a future perturbation depend on the E-layer history. The coupling thus runs induced-state → selection → altered-inducible-states in a loop, which the Methods closes explicitly.
4.5. The Remaining Couplings
The couplings not detailed above—M↔C beyond the oxygen thread, and the top-down constraint of organizational state on fast dynamics—are biologically plausible but least developed. The reactive oxygen species of the M layer are themselves among the bystander mediators of the C layer, so a fast redox excursion is simultaneously a C-layer signal; conversely, tissue organization constrains where and how fast redox events occur. We include these in the coupling structure for completeness but do not assign them concrete dynamics, and we count them among the framework’s open problems rather than its established claims.
4.6. Treatment as Perturbation of a Coupled System
Within this picture, radiation is not simply a source of damage but a perturbation applied to a coupled adaptive system. The same dose may produce different outcomes depending on the configuration of the state at the time of delivery, because the response integrates the perturbation against an evolving, coupled internal state rather than mapping dose to outcome through fixed coefficients. This reframing provides a natural interpretation of the radiobiological anomalies: each corresponds to a mode of delivery that preferentially engages a different timescale band, while the coupling between bands determines the downstream and cross-modality consequences. The framework’s central and most testable claims, developed in the Predictions section, concern these couplings rather than the individual bands.
5. The Radiobiological Anomalies as Band-Specific Perturbations
5.1. FLASH as a Fast-State Perturbation
FLASH delivers a therapeutic dose within milliseconds, on the timescale of the M layer. Its defining signature—reduced normal-tissue toxicity with preserved tumor control—is a property of the differential response between normal and tumor tissue, and the leading mechanistic accounts locate that difference in the fast oxygen and radical chemistry of the M band (Vozenin et al., 2019; Boscolo et al., 2024). The dependence of the effect on dose rate, pulse structure, and delivery time follows naturally from treating oxygen as a dynamic state variable: closely spaced pulses do not allow oxygen to recover between them, whereas widely spaced pulses do, so that the same total dose produces a different outcome depending on its temporal structure (Pratx and Kapp, 2019; Liew et al., 2022).
We note that, on this account, FLASH does not require a novel memory mechanism; its temporal phenomenology is reproduced by the dynamic chemistry of the M band as described by existing models, and the differential between tumor and normal tissue reflects their different oxygenation states. The contribution of the framework here is not to explain FLASH—the dynamic-oxygen models do that—but to situate FLASH as the temporal-axis probe of the M layer, and to make explicit that the oxygen state it perturbs is coupled to the slower layers through the mechanisms described above.
5.2. SFRT As a Collective-Organizational Perturbation
SFRT delivers a spatially structured dose, engaging the C layer directly. Its signature—response in regions that received little dose—is the operative definition of a collective effect, and is attributed to bystander signaling propagating from high-dose to low-dose regions (Asur et al., 2015; Griffin et al., 2020; Casteloes et al., 2025). Within the framework, SFRT is the spatial-axis probe of the C layer: it perturbs the organizational state of the tissue and reads out the consequence through the response of cells that were not directly hit.
The framework adds to the modality-specific account in one respect. If the persistent consequences of SFRT depend on the coupling from C into B—on whether the transient bystander signal is stabilized into a durable epigenetic state through pathways such as AP-1—then the durability of SFRT effects across fractions becomes a question about inter-layer coupling, not about the signaling event alone. This is a consequence available only to a coupled multi-layer description, and the spatially-resolved model in the Methods makes it concrete: the durable signature is predicted to be displaced toward the low-dose valleys.
5.3. SBRT As a High-Magnitude Perturbation
SBRT delivers very high doses per fraction. Its tumor control has been argued to exceed LQ-based predictions, and additional mechanisms have been proposed, prominently endothelial damage above a dose threshold of roughly 8–10 Gy, leading to vascular collapse and secondary tumor cell death, together with enhanced immune activation (Song et al., 2021; Sheth and Esfandiari, 2022). It is important to record that this remains contested: other analyses argue that the clinical results are consistent with increased direct cell killing at high dose per fraction and do not require new biology (Brown et al., 2014; Kirkpatrick et al., 2008).
The framework does not adjudicate this debate, and should not be read as endorsing the vascular account over the direct-killing account. Its more limited claim is that the high-magnitude regime engages threshold and reorganization behavior in the slower layers—vascular and organizational change in C, and downstream consequences in B and E—rather than introducing a distinct layer of its own. SBRT is thus interpreted as a magnitude-axis perturbation that drives the existing layers across nonlinear thresholds, an interpretation consistent with both the vascular and direct-killing accounts insofar as both locate the supra-linear behavior in the response of an already-represented part of the system.
5.4. Resistance as a Persistent–Evolutionary Perturbation
Treatment resistance engages the two slowest layers. The reversible, non-genetic persister state is a B-layer phenomenon: induced by treatment stress, maintained epigenetically, and reversible on treatment withdrawal (Sharma et al., 2010; Wang, Wang, et al., 2025). The subsequent emergence of stable, heritable resistance is an E-layer phenomenon: selection acting on a population whose composition has been shaped by the persister state (Greaves and Maley, 2012; Gatenby and Brown, 2020). The framework’s interpretation of resistance is that it is the joint output of these two layers and their coupling—induced adaptation in B providing the substrate on which selection in E subsequently acts—rather than a single process. This is consistent with recent characterizations of the persister state as a staging ground for stable resistance (Wang, Wang, et al., 2025) and with the explicitly induced-and-inherited framing some authors have adopted (Valcz et al., 2025).
6. Existing Models as Limiting Cases
The framework is intended to contain, not replace, existing radiobiological models, and the linear-quadratic model emerges from it explicitly in a well-defined limit. Suppose the state does not evolve appreciably over the course of a single delivery—the frozen-state condition X(t) ≈ X0. Then the state-dependent coefficients α(X(t)) and β(X(t)) in the response functional of Section 4 reduce to the constants α(X0) and β(X0), and the accumulated log-kill can be integrated directly. Using that the cumulative dose rises from 0 to its total value D as the integral of the dose rate, the two terms integrate to
which is exactly the linear-quadratic form: the classical α corresponds to α(X0) and the classical β to ½ β(X0), the radiosensitivity of the tissue evaluated at its fixed configuration X0. The LQ coefficients are thus revealed as the lethality of the tissue in a fixed state, and the LQ model as the frozen-state limit of the coupled dynamics. Its success in conventional fractionation corresponds to regimes in which state evolution is slow relative to the delivery, so that the frozen-state condition holds to good approximation. Its documented difficulties in the FLASH, SFRT, and SBRT regimes correspond, on this account, to regimes in which one or another layer evolves on a timescale comparable to the perturbation, so that the coefficients can no longer be treated as constant during delivery and the frozen-state limit fails.
This derivation also clarifies what the framework adds. The classical model treats α and β as fixed tissue constants; here they are values of a state-dependent lethality evaluated at a particular configuration. When the configuration is stable, the two descriptions coincide and the LQ model is recovered exactly. When the configuration evolves during or between deliveries—because a fast layer is perturbed faster than it relaxes, or a slow layer has been altered by prior fractions—the effective α and β change, and the departure from LQ behavior is the signature of that state evolution. The same single-factor structure that produces this also allows the separate within-band models to be combined into one expression, and a non-additive cross-modality interaction term to be derived from it; both are developed in the Methods.
Similarly, kinetic models of bystander signaling—such as the radiation-induced intercellular signaling model of McMahon et al. (2013)—can be read as descriptions of the C layer in isolation, and population-dynamic models of resistance as descriptions of the E layer in isolation. Each is a valid description of its own band; what the framework adds is the proposition that the bands are coupled, so that a model restricted to one band omits contributions from the others. On this view the coexistence of multiple successful but incompatible mechanistic models is not paradoxical: each correctly describes a different band of the same coupled system.
7. Methods: The Coupled System as an Explicit Dynamical Model
This section formalizes the framework as a coupled, spatially-resolved dynamical system. We show that the mainstream accounts of the anomalies enter cell kill through a single shared quantity, collapse into one master equation, and—once spatial structure, the evolutionary layer, and downward couplings are included—form a closed cross-band system. The construction is illustrative: parameters are chosen to expose structure, not fitted to data, and the resulting claims are conditional on the modelling assumptions made explicit below. All symbols are defined in the Glossary (Table 2).
7.1. The Shared Kill Kernel
All accounts ultimately modify the rate of log-kill, so we begin from the state-dependent response functional of Section 4, with Σ ≡ −ln SF,
Each mechanism modifies the radiosensitivity coefficients. We adopt—as a deliberate idealization, not an established fact—the assumption that oxygenation and the persistent state scale both coefficients multiplicatively and in the same proportion, because each rescales the lethality of a track rather than adding lethality of its own,
This equal-proportion form is known to be violated for oxygen, which modulates the quadratic term more strongly than the linear one; the single-factor structure is therefore the simplest coupling ansatz, adopted for analytical transparency. Because both coefficients carry the same factor, it can be named—the master modulation function Φ(X)—and the kernel factors accordingly,
7.2. Spatial Structure: The Collective and Persistent Layers as Fields
A spatially uniform state cannot represent spatially fractionated radiotherapy, which is defined by the spatial pattern of peaks and valleys and by signal propagating between them. We therefore promote the collective signal and the persistent state to fields over position x. The signal obeys a reaction–diffusion equation—generated where the delivered dose pattern σ(x) is high, diffusing, and decaying, with its generation additionally modulated top-down by the persistent and evolutionary states (the χB and χE factors introduced in 7.4)—
The persistent state is written locally by the local signal. Here we use the molecular identity of the write to constrain its form: AP-1 is autoregulatory—it drives its own transcription—so the write is self-reinforcing and switch-like rather than graded. We therefore include a positive-feedback (Hill) term in B itself, giving a bistable write,
where the term in Bh/(Khh+B^h) is the AP-1 self-activation. This is consequential: numerical solution shows the write becomes bistable, latching into a high state once a signal threshold is crossed and remaining there after the signal decays (the steady B differs depending on whether it is approached from low or high B—genuine hysteresis), whereas a graded write relaxes whenever the signal falls. This is what makes “write-and-hold” actually hold—the persistent state is a latched memory, not a decaying trace—and it yields a falsifiable signature: the persistent state should respond to the collective signal with a threshold and hysteresis, not gradually. The spatially resolved survival integrates the kernel over dose and space,
7.3. Recovering the Conventional Models
If the state does not move over a delivery and space is uniform, Φ = Φ0 is constant and the master equation reduces to Σ = Φ0(α0D + ½β0D2)—the linear-quadratic model, with α = Φ0α0 and β = Φ0β0. Switching off individual couplings recovers each mechanistic account: with only ω(p) dynamic, an oxygen-depletion (FLASH) model; with only the signal field active, a bystander (SFRT) model; with only V dynamic, a threshold-vascular (SBRT) model; with only B dynamic, an induced-resistance model. The system contains all four as limits and the linear-quadratic model as its fully frozen, spatially-uniform limit.
7.4. The Evolutionary Layer and the Closed Loop
The persistent state B is the substrate on which selection acts. We model the evolutionary layer following two established ideas. From the adaptive-therapy literature, resistance carries a fitness cost and sensitive and resistant cells compete for shared resources, so withholding treatment lets sensitive cells suppress resistance (Gatenby and Brown, 2020). From the induced-resistance literature, the resistant state is induced from the reversible reservoir and then heritably fixed (Valcz et al., 2025). We therefore use two competing compartments—sensitive Ns and resistant Nr—with radiation kill on each, a maturation flux m(B) carrying cells from the B-reservoir into fixed resistance, and logistic competition under a shared carrying capacity,
The maturation flux m(B)·Ns is implemented as a stochastic birth process, the one place stochasticity is scientifically necessary rather than decorative, following recent epigenetic-instability persister models (Wang, Lei, et al., 2025).
Direction of coupling and top-down constraint. Coupling runs in both directions across the hierarchy. Information flows upward (fast to slow): the signal writes B, and B supplies the reservoir m(B) that matures into resistance. It also flows downward (slow to fast)—the top-down constraint that distinguishes a coupled hierarchy from a bottom-up cascade—and we implement downward coupling where it is best motivated, while noting that other downward arrows are admissible but left unspecified. This is an instance of genuine top-down causation in the sense of Ellis (2012) and Noble’s biological relativity (2012): the higher (evolutionary) layer does not merely influence the lower population dynamics but constrains their accessible state space, so that no single level holds privileged causal status.
Three downward couplings are included: the persistent state lowers radiosensitivity (B in the kill kernel through Φ); vascular damage lowers oxygenation (V in the oxygen dynamics); and the persistent and evolutionary states modulate collective signal generation (the χB and χE factors in the signal-field equation). In addition, the resistant fraction reduces the carrying capacity, K(E) = K0(1 − cK·E), an instance of genuine top-down constraint rather than mere influence: the higher layer reshapes the accessible state space of the lower population dynamics. The χE and K(E) terms close the cycle C → B → E → C. In the regimes we simulated these downward terms are structurally important—they close the causal hierarchy and are required for the loop to exist—but quantitatively second-order, shifting outcomes modestly rather than dominating; their role is to make the hierarchy genuinely bidirectional. The fast layer M (through p) is coupled into the loop as a driving input—conditioning C through ω(p) and writing B through εr(p)—but is not a member of the closed cycle, because its relaxation is fast relative to the loop and the slow layers cannot feed back to it on its timescale. The full coupled state is
7.5. Cross-Modality Interaction: An Elementary Identity With Non-Elementary Consequences
Comparing a treatment in which only oxygenation varies, one in which only the persistent state varies, and one in which both vary, an exact expansion of Φ gives a residual equal to −κB·δω·δB, where δω and δB are the departures of the two factors from baseline. As pure algebra this is elementary—the statement that a product is not additive—and we do not present it as a discovery. Its interest lies in two non-elementary consequences. First, selectivity: the cross term is bilinear in different state variables, so two modalities perturbing the same variable (FLASH and SBRT, both acting through oxygen) combine additively with no interaction, while only modalities perturbing different, coupled variables interact. The framework predicts a definite pattern of which pairs interact, fixed by which variable each modality moves. Second, the surviving interaction enters the master equation as a term proportional to κB that vanishes unless both an oxygen-moving and a resistance-writing mechanism are present—predicting that spatially fractionated delivery at FLASH dose rates is non-additive, while two oxygen-only modalities are not.
8. Results
This section reports the behaviors we were able to compute from the coupled system of the Methods. We state at the outset what these calculations are and are not. They are theoretical: each is a numerical solution of the illustrative model, whose parameters are chosen to expose structure rather than fitted to any dataset, so the results demonstrate what behaviors the coupled architecture can generate, not the magnitude with which they occur in tissue. They are deliberately falsification-oriented: their value is that each produces a qualitative signature—an inversion, a threshold, an interaction sign, an order-dependence—that is absent from conventional band-separated radiobiology, so that an experiment finding the signature supports the framework and one failing to find it bounds or refutes the relevant coupling. Common assumptions and limitations apply throughout and are stated once here: the model is one-dimensional in space where spatial; cell populations are treated as continuous except for the stochastic maturation step; the single-factor modulation Φ is an idealization that the oxygen effect is known to violate (Section 7.1); the coupling constants are unmeasured; and absolute timescales are nominal. Item-specific caveats are noted with each result.
8.1. The Persistent State is Written in the Unirradiated Valleys
Solving the spatial field equations of Section 7.2 under a spatially fractionated dose pattern, the bystander signal generated at the high-dose peaks diffuses into the low-dose valleys and writes the persistent state there, where the delivered dose is zero. In this solution the valley persistent state reaches approximately 36% of its peak value despite receiving no direct dose, and the B profile is markedly broader than the delivered-dose pattern (Figure 1), so the durable molecular signature is displaced from the dose map toward the valleys. Assumptions and limitations: the signal-generation, diffusion, and decay constants are illustrative; the degree of valley writing depends on the ratio of diffusion length to peak spacing, so the 36% figure is regime-specific and not a tissue prediction. The robust, falsifiable claim is qualitative—that durable marks appear in unirradiated valleys at all—not the specific fraction.
8.2. The Evolutionary Layer Reproduces The Adaptive-Therapy Trade-Off
Integrating the two-compartment evolutionary layer of Section 7.4 under two delivery policies reproduces the established signature of adaptive therapy without it being imposed (Figure 2). Continuous maximum-dose delivery drives the total burden down but drives the sensitive compartment to extinction, leaving a surviving population that is essentially entirely resistant (resistant fraction → 1.0). Adaptive delivery, which withholds dose when burden is low, maintains a larger but controlled burden in which sensitive cells competitively suppress the resistant compartment, leaving a resistant fraction of roughly 0.14. Assumptions and limitations: the fitness cost of resistance, the competition under shared carrying capacity, and the maturation rate are illustrative parameters drawn from the adaptive-therapy modelling tradition rather than measured here; the result demonstrates that the coupled layer is structurally capable of the trade-off, and its reproduction of a clinically established behavior (Zhang et al., 2017; Gatenby and Brown, 2020) is a consistency check on the evolutionary layer rather than a new prediction.
8.3. Closing the Loop Produces History-Dependence at Fixed Dose
Integrating the closed stochastic C→B→E system for two schedules that deliver identical total dose in identical fraction number, differing only in whether the signal-generating deliveries fall early or late, produces significantly different final resistant fractions (Figure 3): in this calculation early delivery yields a resistant fraction of about 0.35 against about 0.27 for late delivery, a separation of roughly eleven times the run-to-run spread across stochastic realizations. Early signal-generating delivery writes the persistent reservoir sooner, the resistant population then feeds back through the return path to amplify subsequent signalling, and the difference compounds. Assumptions and limitations: the magnitude of the separation depends on the return-coupling strength and the maturation rate, both illustrative; the robust claim is that a difference exists at matched dose, which a feed-forward or dose-only description forbids, not its specific size. This is the calculation that most directly distinguishes a closed loop from a cascade.
8.4. The Persistent Write is a Bistable Switch with Threshold and Hysteresis
Because the C→B write carries the AP-1 autoregulatory (self-activating) term, the steady persistent state is a bistable function of the collective signal rather than a graded one. Sweeping the signal upward, the steady state jumps abruptly from a low to a high branch at a critical value—in this calculation a rise of about 0.5 in B across a signal increment near 0.06—rather than rising smoothly (Figure 4, left). Sweeping the signal back down, the high state is retained: increasing-signal and decreasing-signal paths differ by up to about 0.6 over an intermediate signal range, a hysteresis loop (Figure 4, right). Assumptions and limitations: the existence of bistability requires the self-activation strength to exceed the reversal rate, which holds for the illustrative parameters but is itself an experimental question; the location of the threshold is regime-specific. The falsifiable signatures are qualitative—an abrupt threshold and a hysteresis loop in the persistent response, as opposed to graded adaptation—and are accessible by measuring AP-1–associated marks against signal delivered in increasing versus decreasing sequence.
8.5. Spatially Fractionated Geometry Has an Optimum for Memory Writing
Because the durable signature is written by the diffusing collective signal rather than by local dose, the geometry that maximizes persistent memory per unit delivered dose is distinct from the geometry that maximizes peak or valley dose. Sweeping peak spacing in the spatial model, the memory written per unit dose rises and then saturates, with efficiency increasing from about 0.95 to about 1.5 (arbitrary units) and reaching a plateau beyond an intermediate spacing (Figure 5): tightly packed peaks waste signal in already-written tissue, while widely spaced peaks leave valleys unwritten. Assumptions and limitations: the optimum is a saturating plateau rather than a sharp peak in this calculation, and its location depends on the diffusion-length-to-spacing ratio; the result should be read as identifying memory-writing geometry as the relevant design variable, not as specifying an optimal spacing in tissue. The qualitative claim—that the memory-optimal geometry differs from the dose-optimal one—is the falsifiable content.
8.6. FLASH and SFRT Interact Non-Additively
The selectivity result of Section 7.5 predicts that two modalities perturbing different coupled state variables interact, while those perturbing the same variable do not. Computing the change in log-kill for FLASH alone, SFRT alone, and both together in the illustrative model, the combined effect (about 2.48) falls below the sum of the separate effects (about 2.97), the deficit of roughly 0.49 being the cross-modality interaction term −κB·δω·δB (Figure 6). FLASH acts through oxygen (ω) and SFRT through the collective signal that writes B; because Φ is bilinear in these, their combination is non-additive. Assumptions and limitations: the sign and magnitude of the interaction depend on the coupling constant κB and on the assumed departures δω and δB, none measured; the calculation demonstrates that a non-additive term of definite structure exists and is computable, and predicts that two oxygen-only modalities would show no such term, but does not predict the tissue magnitude. The robust, falsifiable claim is the pattern—interaction between different-variable modalities, additivity between same-variable ones—not the numerical deficit.
8.7. Consolidated View and What the Results Imply
Table 1 collects the six calculations, the equation or mechanism each exercises, the qualitative signature each produces, and the conventional expectation each violates. Read together, they make a single point: the behaviors that distinguish the framework from conventional radiobiology are not artefacts of any one equation but recur across independent calculations on the same coupled system. Three of the six (8.1, 8.2, 8.3) confirm that the coupled construction behaves as intended—signal reaches unirradiated tissue, the evolutionary layer reproduces a clinically established trade-off, and closing the loop makes outcome depend on history at fixed dose. The other three (8.4, 8.5, 8.6) expose properties that conventional radiobiology does not represent at all: a bistable, hysteretic persistent state in place of graded adaptation; a memory-writing geometry distinct from the dose-optimal geometry; and a structured, selective non-additivity between modalities. We stress that every one of these is a theoretical result on an uncalibrated model, offered so that the framework can be falsified: each names a specific qualitative observation—valley-displaced durable marks, an abrupt threshold and hysteresis in the persistent response, a memory-optimal spacing, a sign-definite cross-modality interaction, an order effect at matched dose—whose absence in experiment would bound or refute the corresponding coupling, and whose presence is not anticipated by the linear-quadratic model or by any single-band account. It is on this falsifiable ground, rather than on the calculations’ present quantitative values, that we rest their significance.
9. Predictions
The predictions below are not accompanied by calculations, and we are explicit about why each is excluded from the Results: in every case the prediction requires a variable, coupling, or regime that the present illustrative model does not contain or cannot constrain, so any number we attached would be invented rather than computed. They are nonetheless stated specifically enough to be tested, and we regard developing the model to the point where each can be simulated as the principal direction for future work. We group them by what would be needed to promote them into a calculation.
9.1. Predictions Requiring Couplings the Model Contains but Cannot Yet Constrain
Sequence-order optimization of clinical schedules. The order-dependence demonstrated in Section 8.3 implies that, among the many schedules sharing a fixed biologically effective dose, some minimize the final resistant fraction and others maximize it. The model can show that the spread exists; it cannot yet identify the optimal schedule, because doing so requires the true write and reversal rates of the persistent layer, which are unmeasured. We predict that front-loaded or clustered signal-generating fractions will outperform uniform schedules at matched dose, and flag schedule optimization as the most clinically reachable extension once those rates are known. This is excluded from Results because the optimization target depends entirely on unmeasured rate constants.
Critical timing of resistance emergence. Because the maturation flux from the persistent reservoir to fixed resistance is rate-limited, the model implies that the same cumulative persistent burden delivered as a short intense episode versus a slow sustained one may yield different resistance. We predict timing-dependence of resistance at matched cumulative B. This is excluded from Results because a meaningful calculation requires the maturation kinetics to be specified beyond the illustrative value, which would make any computed difference an artefact of the assumed rate.
Memory-erasure windows. The competition between write and reversal in the persistent layer implies that an intervention timed to a particular window could disproportionately reduce long-term resistance. Exploratory runs suggested the effect is small and highly parameter-sensitive, so we decline to present a calculated window and instead predict, qualitatively, that such windows exist. This is excluded from Results precisely because our calculations did not produce a robust, parameter-independent effect.
9.2. Predictions Requiring Structures Not yet in the Model
Multiple attractors and irreversible resistance. Because the closed C→B→E loop carries the bistable write of Section 8.4, the loop sits in two coexisting states across a substantial parameter range—a low-memory, low-resistance state and a latched high-memory, high-resistance one—so that the system as a whole inherits the write’s bistability. What we have not established is the stronger and more consequential claim: that the loop’s feedback itself creates genuinely emergent multistability, with a tipping point beyond which the closed dynamics drive an irreversible transition to a self-sustaining resistant state even after the perturbation is withdrawn. In our simulations the return coupling shifts the coexisting states only marginally, so the bistability we observe is essentially that of the isolated write propagated through the loop, not a new attractor structure created by closure. We regard the emergent, irreversible-tipping version as potentially the deepest prediction of the architecture, and explicitly leave it as a prediction: establishing it requires a dedicated bifurcation analysis of the closed system—mapping where feedback strength qualitatively changes the attractor landscape—which is beyond the present illustrative model and would justify a separate study.
Cross-modality memory transfer. Because all modalities act on a shared state, the framework predicts that writing the persistent state with one modality should alter the response to a later, different modality: spatially fractionated delivery that writes B should change how conventional or stereotactic delivery subsequently behaves, and vice versa. We exclude this from Results because a credible calculation requires modelling two modalities in sequence with a persistent state that carries between them at calibrated rates, which compounds several unmeasured couplings; the qualitative prediction, however, follows directly from the shared-state architecture and is distinctive to it.
The full modality-interaction matrix. Section 8.6 computes one entry (FLASH × SFRT) of a matrix whose structure the framework predicts in general: interaction strength set by whether two modalities perturb shared or distinct coupled variables. We predict, for example, weak FLASH × SBRT interaction (both largely oxygen-mediated), strong SFRT × resistance-inducing-schedule interaction (distinct variables meeting in B), and moderate SBRT × resistance interaction. We present only the single computed entry as a result and leave the matrix as a prediction, because the remaining entries require modality-specific couplings we have not parameterized.
9.3. Predictions Requiring New External Variables
Hyperthermia: order effects and convergence with SFRT on memory. Hyperthermia perturbs the fast layer (oxygen kinetics, radical chemistry, protein damage) and plausibly the collective layer (heat-shock signalling). The framework predicts, beyond the conventional radiosensitizer role, that heat-then-radiation need not equal radiation-then-heat at matched thermal and radiation dose, and that hyperthermia combined with spatially fractionated delivery should interact more strongly than additivity implies—because the two perturb different variables that meet in the persistent layer, by the same selectivity mechanism as FLASH × SFRT. We exclude this from Results because the model contains no temperature variable; adding a thermal coupling to M and C, with its own parameters, is required before the predicted order effect or interaction can be computed rather than asserted. The associated experiment measures AP-1 activity, epigenetic persistence, and later resistance rather than immediate survival, the framework predicting the largest differences appear weeks to months later through the slow layers.
Bioelectric modulation as a non-cytotoxic write. Placing bioelectric signalling in the collective layer, and noting that C→B already exists, the framework predicts that a purely bioelectric perturbation routed through C can write persistent adaptive memory—altering future radiotherapy response without delivering cytotoxic dose—and, through the closed loop, can alter resistance trajectories with little direct cell kill. Concretely: pre-conditioning tissue with externally applied bioelectric or impedance modulation should change the subsequent spatially fractionated response even when radiation delivery is unchanged. We exclude this from Results because the model contains no bioelectric variable; its addition to the C layer, and measurement of its coupling, are prerequisites for any calculation. We flag this as the most speculative prediction, and also the one that most directly tests the framework’s largest implied claim—that radiation is one of several handles on a shared adaptive state.
Radiotherapy–immunotherapy trade-off. Treating adaptive immune memory as a second instance of the persistent-memory abstraction in a separate host compartment, the framework predicts that one radiation delivery writes both a host-protective immune memory and a tumour-protective resistance memory through the same collective signal, so that delivery parameters optimized for durable immune activation may carry a concomitant resistance-writing cost, and an intervention on the shared write mechanism would move both. We exclude this from Results because representing it requires a second persistent compartment with its own dynamics and an immune cross-coupling, none of which the present single-compartment model contains. The prediction is distinctive because single-purpose models of the radiation–immune interaction, representing only the immune arm, cannot express a coupled trade-off. A single observation unifies the predictions of Section 9.3. If radiation is merely one way of moving the coupled state, then temperature, bioelectric field, and immune intervention are alternative handles on the same state space, differing only in which variable each perturbs—a view whose planning consequences are developed in Section 10.3, and whose calculation awaits a model extended to carry these additional variables.
10. Discussion
The framework proposed here is an organizing hypothesis, now given explicit dynamical form. Its purpose is to provide a structure within which the diverse anomalies of contemporary radiobiology can be related to one another, by recognizing that each engages biological adaptation on a different timescale and that these timescales are coupled. Its principal claims are individually grounded in current evidence: that adaptation operates across separable timescale bands; that each band is populated by experimentally characterized biology; that radiation delivered in different ways preferentially engages different bands; and that the bands are coupled, with at least one coupling—from the collective to the persistent layer, via AP-1—now supported by a demonstrated molecular mechanism.
10.1. What the Framework Adds Beyond Conventional Radiobiology
It is fair to ask what this framework provides that the linear-quadratic model, the classical four Rs, and the existing single-mechanism models do not. We state the answer sharply, because the framework’s contribution is narrower and more specific than its scope might suggest: its distinctive claim is not the existence of four adaptive layers but the coupling between them, and the cross-modality predictions that coupling generates.
It is worth being explicit about why this direction is available now rather than earlier. The behavioral facts the framework organizes are not themselves new—evolutionary models of resistance and the bystander phenomenology of non-targeted effects have been developed over two decades (Gatenby and Brown, 2020; Greaves and Maley, 2012). What was missing was a demonstrated molecular mechanism by which a transient signal becomes a durable, heritable cellular state—the very operation a coupling between a fast or intermediate layer and a persistent one requires. The recent characterization of AP-1 as a mediator of cellular adaptation and memory formation supplies precisely this write-and-inherit step (Li et al., 2026), and parallel work has reframed induced, non-genetic resistance as a heritable trait that is selected upon (Valcz et al., 2025). At the same time, evidence that ordinary biological substrates can store and act on prior experience has accumulated across several systems—associative conditioning in gene regulatory network models (Pigozzi et al., 2025), demonstrated learning in a single-celled organism (Doan et al., 2026), and biochemically plausible models of habituation in simple reaction networks (Eckert et al., 2024)—making a persistent adaptive layer a credible general assumption rather than an exotic one. These developments do not establish the radiobiological couplings, which remain to be tested; but they are what make the couplings concrete enough to state and to falsify, and in that sense they are the proximate reason the framework can be formulated now.
Two of the framework’s apparent contributions, by contrast, do not survive scrutiny as genuine additions, and we say so plainly. The decomposition into timescale bands is, in part, a reorganization of structure the field already possesses: the four Rs are themselves a timescale-ordered description, and recasting repair, reoxygenation, and repopulation alongside non-genetic resistance as layers M, C, B, and E relabels as much as it discovers. Likewise, the reinterpretation of the radiosensitivity coefficients as state-dependent quantities α(X) and β(X)—while conceptually correct, and a genuine departure from treating α and β as fixed tissue constants—adds no predictive power in its general form: until the functional dependence on state is specified, “α and β depend on state” cannot be falsified, because any departure from fixed-LQ behavior can be attributed post hoc to a change in state. We regard the state-dependent reinterpretation as a necessary reframing that the coupling claims require, not as an independent result.
The genuine addition lies in what no single-band description contains. Each existing mechanistic model—dynamic-oxygen models of FLASH, kinetic models of bystander signaling, evolutionary models of resistance—operates within a single timescale band, and the four Rs treat their components as independent, additive corrections. None represents coupling between bands. The framework’s central and distinctive claim is that these bands are dynamically coupled through shared biological substrates, so that a perturbation applied through one phenomenon propagates to another. This is not left as an assertion: as the Methods show, once the state-dependent radiosensitivity is written in its natural form, every mechanism enters cell kill through a single shared modulation factor, and the four mainstream models compose into one master equation that reduces to each of them when the other couplings are removed and to the linear-quadratic model when the state is frozen. The coupling is therefore a structural property of the combined system, not a narrative imposed on it.
That single-factor structure has consequences conventional radiobiology cannot reproduce. The first is a constraint on radiosensitivity itself: because the state rescales the linear and quadratic terms together, the ratio α/β should be comparatively conserved under changing treatment history—exactly so where each mechanism acts as a pure rescaling—so that the degree of α/β conservation becomes a direct test against existing oxygen-effect data. The second, and most distinctive, is that cross-modality interaction is predicted to be selective: because each modality acts by perturbing a particular state variable, two modalities that perturb the same variable combine additively and do not interact, whereas only modalities perturbing different, coupled variables produce a residual interaction term. Delivering spatially fractionated radiotherapy at FLASH dose rates, for example, should yield an effect that is not the sum of the two delivered separately, while combining two modalities that both act through tumour oxygenation should show no such interaction. This is a definite, falsifiable pattern—a prediction of precisely which combinations interact and which do not—that is absent from any account in which modality effects simply superpose.
The most concrete of the couplings is mechanistically grounded: the AP-1–mediated epigenetic write that converts a transient collective signal into a persistent state—the C→B coupling—is, on this account, the same substrate operating in both the durability of spatially fractionated treatment and the acquisition of non-genetic resistance (Li et al., 2026). Because this coupling has a definite, bistable form, it yields sharp entailments developed in the Predictions: that the durable component of spatially fractionated treatment should depend on the temporal ordering of delivery at fixed total dose; that the durable molecular signature should invert the dose map, peaking in the low-dose valleys; and that a single intervention on the persistent layer should move both SFRT durability and the kinetics of resistance acquisition. A correlation of this last, cross-modality form—one molecular lever shifting two phenomena conventionally regarded as unrelated—together with the selectivity pattern, is the framework’s sharpest empirical signature.
We state plainly that these predictions have not yet been tested in the radiation context. The framework’s present status is therefore hypothesis-generating, and its value will be settled by whether the cross-layer couplings are borne out experimentally. If they are, the framework will have identified links that the conventional, band-separated picture cannot represent; if they are not, it remains a suggestive reorganization. We prefer to rest the framework’s claim to novelty on this falsifiable ground rather than on the more easily granted observation that radiobiology can be organized by timescale.
That the framework also accommodates phenomena beyond the four it was built on is supporting evidence that the coupling structure captures something general rather than fitted. Tumor hypoxia and inter-fraction reoxygenation—already among the four Rs—emerge as the inter-layer dynamics of a single shared variable: oxygen as a fast M-layer quantity whose spatial distribution is set by C-layer vasculature, whose chronic form drives B-layer epigenetic reprogramming, and which acts as an E-layer selective pressure, so that reoxygenation is recovered not as a separate “R” but as coupling among layers. The abscopal effect and the broader radiotherapy–immunity interaction are similarly accommodated: out-of-field responses are a collective (C) signal propagating beyond the treated volume, whose durable consequences depend on coupling into the persistent (B) and evolutionary (E) layers through immune memory and clonal selection.
The framework also offers a specific, if speculative, vantage on the combination of radiotherapy with immunotherapy. Adaptive immune memory is itself a persistent, heritable adaptive state written by a transient signal—structurally the same kind of object as the B-layer tumor state, though residing in a separate host compartment. The framework therefore accommodates it as a second instance of the persistent-memory abstraction, and in doing so makes an observation that single-purpose models of the radiation–immune interaction do not: that one radiation delivery engages two write-and-hold processes at once—a host-protective immune memory and a tumor-protective resistance memory—both driven by the collective-layer signal and both set by the same delivery parameters. This predicts a structural coupling between them: the fractionation and dose-rate choices that maximize the durable immune response may also be those that most strongly write tumor-resistance memory, so that the two cannot in general be optimized independently. We raise this only as an illustration of the framework’s reach; it rests on treating immune memory and tumor persistence as instances of one abstraction, and the radiation-context mechanisms on both sides remain to be established.
Equally important is what the framework does not improve upon, which we record so that its contribution is not overstated. It does not provide a better account of the FLASH effect than existing dynamic-oxygen models, which already reproduce its temporal phenomenology; it only situates FLASH as a probe of the fast layer. It does not resolve the contested mechanism of SBRT, which it explicitly declines to adjudicate. And it does not revise the established behavioral facts of treatment resistance—directional adaptation, the advantage of non-stationary over fixed treatment policies—which derive from evolutionary dynamics; the framework’s contribution there is to supply a candidate molecular substrate for the induced component and to couple it to the other layers, not to alter the behavior itself. The framework’s novelty, in short, is concentrated entirely in the couplings: remove them, and what remains is a timescale-ordered relabeling of known radiobiology.
10.2. Relation to Existing Multi-Scale Mechanistic Models
Radiobiology already contains sophisticated multi-scale models, and it is important to be precise about how the present framework relates to them, since it neither competes with nor improves upon them on their own terms. Mechanistic DNA-damage-and-repair models such as Medras trace the response from the spatial distribution of double-strand breaks through repair kinetics and misrepair to survival, mutation, and chromosome aberration (McMahon et al., 2016; McMahon and Prise, 2021). Multi-process models of the FLASH effect, such as UNIVERSE, couple radiolytic oxygen depletion, reoxygenation, and time-dependent damage repair within a single computational description (Liew et al., 2021; Liew et al., 2022). These are genuine multi-scale models, quantitative and predictive in a way the present framework is not.
The distinction is not that the present framework resolves finer detail—it resolves less, and deliberately so—but that it is organized along a different axis and makes a different kind of claim. Existing multi-scale models are predominantly vertical and bottom-up along the spatial and causal axis, linking microscopic energy deposition to a cellular or tissue endpoint, and they operate within a single phenomenon and a single broad timescale regime. Neither couples the millisecond redox regime to the weeks-to-months evolutionary regime, because each was constructed to explain one anomaly rather than to relate several. The present framework is organized instead along the temporal axis of adaptation, and its central object is the lateral coupling between timescale bands that those models, by design, do not represent.
Two consequences follow. First, the framework contains these models rather than replacing them, and the Methods make this exact: written in its natural form, the response routes every mechanism through a single shared modulation factor, so the within-band models compose into one master equation that returns each of them when the other couplings are switched off and the linear-quadratic model when the state is frozen. UNIVERSE or Medras can thus be read as detailed descriptions of the fast and damage-repair components the framework treats, at coarser grain, as the M layer; its contribution is the embedding and the coupling, not the within-band mechanism, which these models supply far more rigorously. Second, existing multi-scale models treat the slow biological variables—hypoxic fraction, repair capacity, the effective radiosensitivity coefficients—as fixed parameters specified for a given exposure. The framework’s defining move is to promote these to dynamical, coupled state variables with feedback onto the fast layers. It is this promotion that makes the radiosensitivity coefficients state-dependent, and that produces predictions a fixed-parameter model cannot express: not merely that modalities interact, but that they interact selectively. We note, in fairness, that evolutionary tumor-control models already treat the slowest variables dynamically (Gatenby and Brown, 2020); the framework’s novelty is not dynamics within any single band but the coupling across bands, and the specific interaction pattern it forces.
A second tradition is more directly comparable, since it couples scales rather than resolving one in detail. Beginning with Ribba et al. (2006), who linked a Boolean genetic network, a cell-cycle model, and tissue mechanics to show that scheduling can alter outcome at fixed total dose, this lineage has grown into sophisticated hybrid models coupling cell-cycle state, oxygen dynamics, and bystander signaling, reproducing behavior such as low-dose hyper-radiosensitivity that the linear-quadratic model misses (Powathil et al., 2012; Powathil et al., 2016). More recently, and on a different substrate, multiscale models have coupled epigenetic instability to evolutionary population dynamics to account for drug-tolerant persistence and to derive optimal intermittent schedules (Wang, Lei, et al., 2025). The present framework is positioned precisely between these two bodies of work, and claims neither’s territory as its own: it concedes to the first the coupling of fast redox, oxygen, and collective-signaling processes within the hours-to-days regime—including microenvironmental feedback onto cell-cycle and repair state, which these models already represent—and to the second the coupling of persistent epigenetic state to clonal selection.
Its contribution is to join them, and the join is not merely additive. Treating the fast and collective layers of the radiotherapy-modeling tradition and the persistent and evolutionary layers of the resistance-modeling tradition as bands of one coupled system brings FLASH, spatially fractionated, and stereotactic delivery, along with non-genetic resistance, under a single account that neither tradition alone provides; but the substantive novelty is in the coupling itself. The downward couplings the framework adds—the persistent state lowering radiosensitivity, and the evolutionary state feeding back to the collective layer—reach layers the radiotherapy-modeling tradition does not contain, so its established microenvironmental feedback onto cell-cycle state cannot express them; the cross-band loop they close produces a history-dependence whose origin is a persistent memory feeding selection, distinct in mechanism and in timescale from the cell-cycle-timing scheduling effects of the earlier models. We make no claim to exceed either tradition in mechanistic completeness within its own regime—Ribba’s and Powathil’s within-band machinery is more detailed than ours, and we use it rather than replace it. The contribution is the span and the coupling across regimes, and the structural consequences that follow only from coupling: the single-factor reduction that routes every modality through one quantity and yields the linear-quadratic model as its frozen-state limit; the selectivity of cross-modality interactions; and the bistable, hysteretic persistent state whose durable signature is displaced from the delivered-dose map—none of which a model lacking the persistent and evolutionary layers, or the couplings onto them, can generate.
10.3. Implications for Radiotherapy Planning and Delivery
If radiobiological response is the behaviour of a coupled adaptive state rather than a function of dose alone, then treatment planning is no longer the problem of choosing a dose distribution but the problem of steering a dynamical system. This reframing has both a corrective component—how the conventional planning quantities must be modified—and a constructive component—what new control handles become available, and how they should be chosen against a system that adapts in response. We take these in turn.
The conventional planning quantities become history-dependent. The biologically effective dose, tumour control probability, and normal-tissue complication probability are all built on the frozen-state assumptions of the linear-quadratic model, and each acquires a specific correction in the framework. Because α and β are values of a state-dependent lethality rather than constants, the biologically effective dose, BED = nd [1 + d/(α/β)], loses its order-independence: two schedules with identical nominal BED can differ in outcome if they drive the underlying state differently, so rearranging the temporal sequence at fixed BED is predicted to change the biological effect wherever a persistent layer is engaged (Fowler, 1989). Tumour control probability, conventionally TCP = exp(−N·SF) with a single fixed surviving fraction, will systematically overestimate control whenever treatment itself induces a less sensitive persister compartment during the course, because the clonogen population is heterogeneous in state and that state evolves. And normal-tissue complication probability, currently handled by summing delivered dose and applying empirical recovery factors (Lyman, 1985; Kutcher et al., 1991), is reframed: the carried-forward injury is not a static dose-bookkeeping quantity but a dynamical persistent state that is written by prior treatment, decays on its own timescale, and couples back to raise the damage produced by subsequent dose. This predicts that effective tolerance depends on the temporal pattern and recency of prior dose rather than its total alone, that reirradiation tolerance and consequential late effects should co-vary because they share a substrate, and that injury memory may be modifiable by intervention rather than fixed. In each case the conventional quantity is recovered exactly in the frozen-state limit, and departs from it in precisely the regimes where the linear-quadratic model itself departs from observation; the departures are measurable as dependences on treatment history that the conventional, history-independent formulations forbid.
The available control handles. The framework’s more constructive implication is that planning has more degrees of freedom than dose distribution alone. Each state variable can in principle be addressed by a distinct external handle: dose and dose distribution set the magnitude and spatial pattern of the perturbation; dose rate and pulse microstructure address the fast layer M through the oxygen and redox excursion (the FLASH regime); spatial fractionation geometry addresses the collective layer C and, through the C→B coupling, the geometry of memory writing (Section 9.3); temporal sequence and inter-fraction interval address the integrating dynamics of the persistent layer B and the order-dependence it produces (Section 9.4); temperature, through hyperthermia, addresses M and C jointly (Section 9.6); and bioelectric field modulation, applied externally, addresses the organizational layer C directly and upstream of both memory and evolution (Section 9.7). The significance of organizing planning this way is that it makes explicit which handle reaches which part of the adaptive state, and therefore which combinations should interact: the selectivity result predicts that handles addressing different coupled variables (dose rate and spatial geometry, say, or temperature and fractionation) interact, while handles addressing the same variable do not, so the space of useful combinations is structured rather than arbitrary. Several of these handles—temperature, sequence, bioelectric field—are not represented at all in dose-based planning, yet on this account they are levers on the same state the dose acts on.
A game against an adapting state, played under uncertainty. Because the persistent and evolutionary layers respond to treatment, each intervention changes the state the next one will face, so maximizing immediate kill is not generally optimal—it is the continuous-maximum-dose policy that the Results show selects a purely resistant population (Zhang et al., 2017; Gatenby and Brown, 2020). The natural formulation, developed in the companion volume (Vaitheeswaran, 2026a), is game-theoretic: planner and adapting state are players in a sequential game, and the optimal strategy anticipates the state’s response—timing interventions to the writing and decay windows of the persistent layer, and choosing among the control handles above to move variables the tumour cannot cheaply counter—rather than playing the myopically strongest move. Conventional dose-maximization is the open-loop limit of this closed-loop problem, recovered, like BED, TCP, and NTCP, in the frozen-state limit.
This control picture is actionable only if the underlying state can be inferred and its couplings estimated, and here the framework meets a structural obstacle that we treat in detail separately (Vaitheeswaran, 2026c): the adaptive state is largely latent—the persistent and evolutionary layers are not directly observable in the clinic—and the model’s parameters, spanning widely separated timescales and entering nonlinearly, are in general neither observable nor structurally identifiable from feasible measurements. This is the well-characterized problem of structural and practical non-identifiability in systems-biology models, where distinct parameter combinations produce indistinguishable observable trajectories and fitting returns confident but unconstrained point estimates (Chis et al., 2011; Raue et al., 2009; Gutenkunst et al., 2007). What remains robustly estimable is not the parameter vector but the qualitative features the Results emphasize: the reversal rate from how the durable SFRT component decays with inter-fraction interval; the write threshold and hysteresis from AP-1 marks assayed against increasing versus decreasing signal; the sign of the cross-modality interaction from a single matched-kill FLASH-versus-conventional comparison. These feature-level observables can be fixed in preclinical models before any patient calibration (Vaitheeswaran, 2026b).
Even then, the clinic forecloses point identification: per-patient data are sparse and irregularly sampled, the slow layers are seen at only a few timepoints, and the perturbations that would cleanly excite individual parameters are constrained. The realistic target is therefore a posterior over a partially-observed, latent state rather than a fixed parameter vector, and the appropriate route is Bayesian—priors set from preclinical and population data, updated per patient as longitudinal data accrue, with decisions made by propagating the posterior through the control problem (Gelman et al., 2013). This is precisely the game played under uncertainty about the opponent: the planner acts against a distribution over the state’s parameters, hedging against the posterior rather than best-responding to an assumed value, with Bayesian experimental design choosing the few feasible measurements that most reduce decision-relevant uncertainty. Framed this way, the framework’s clinical translation is not a calibration yielding fixed numbers but a latent-state adaptive-control problem under explicit identifiability limits, in which uncertainty is carried through to the treatment decision (Vaitheeswaran, 2026c).
10.4. Limitations
The decomposition into four layers reflects the number of well-separated timescale bands the evidence currently supports; it is not derived from first principles, and other decompositions are possible. In other words, the framework should be interpreted as a coarse-graining of a continuous adaptive hierarchy. Four layers are the minimal decomposition currently supported by available evidence. The layers are not cleanly separable: the same mechanism, such as oxygen dynamics, may contribute to more than one band, and we have argued that this overlap is a feature of a multi-scale representation rather than an inconsistency, but it does mean the layers are defined by timescale rather than by exclusive molecular identity. The couplings between layers are, with the noted exception of C→B, proposed on physiological grounds rather than measured; formalizing and testing them is the principal direction for future work. The dynamical model of the Methods is illustrative—its parameters are chosen to expose structure, not fitted to data—so its simulations demonstrate that the framework can produce the claimed behaviors, not that those behaviors occur with any particular magnitude in tissue. And the framework deliberately does not adjudicate the mechanistic debates internal to each band, most notably the contested role of vascular damage in SBRT.
The framework does not propose new molecular mechanisms; every mechanism invoked is drawn from existing literature. It does not claim that the layered state is directly or fully measurable, nor that it is the unique correct description—only that organizing radiobiological adaptation by timescale, and coupling the bands explicitly, yields a coherent picture in which otherwise disparate phenomena and their separate mechanistic literatures find a common place, and from which falsifiable cross-modality predictions follow.
The value of such a structure, if it holds, is twofold. It suggests that the proliferation of competing mechanistic explanations for radiobiological anomalies may reflect that each describes a different band of a single coupled system. And it directs attention to the couplings between bands—particularly the conversion of transient organizational signals into persistent epigenetic states—as a locus where treatment might intervene, not by delivering more dose, but by altering how the irradiated tissue’s adaptive state evolves.
Figure 7.
An illustration of the coupled multi-time scale adaptive state framework of radiobiology.


Table 2.
Master glossary of symbols used throughout the paper and Methods.
| Symbol | Meaning |
| D, Ḋ | cumulative dose; instantaneous dose rate |
| SF, Σ | surviving fraction; log-kill, Σ ≡ −ln SF |
| X | full multi-scale state vector, X = (M, C, B, E) and, in the Methods, its resolved variables |
| M, C, B, E | the four layers: fast redox (M), collective (C), persistent epigenetic (B), evolutionary (E) |
| U(t) | treatment input (dose, dose rate, temporal and spatial structure) |
| fM, fC, fB, fE | layer evolution functions |
| α0, β0 | intrinsic (baseline) linear and quadratic radiosensitivity |
| α(X), β(X) | state-dependent radiosensitivity coefficients |
| Φ(X) | master modulation function, Φ = ω(p)(1 − κB B) |
| p, ω(p) | oxygen tension; oxygen sensitivity factor |
| r(p) | redox excursion driving the persistent-state write |
| s(x, D) | bystander signal field (collective layer, C) |
| σ(x) | delivered spatial dose pattern (high at peaks, zero in valleys) |
| Ds, μs, gs | signal diffusion coefficient; decay rate; generation rate |
| V | vascular-damage variable (collective layer, C) |
| B(x, D) | persistent epigenetic state field (B layer) |
| kw, kr | write rate; reversal (forgetting) rate of B |
| kfb, Kh, h | AP-1 self-activation strength; half-saturation constant; Hill exponent |
| Ε | strength of the redox (M→B) write contribution |
| Ns, Nr | sensitive and resistant cell populations (evolutionary layer, E) |
| E | resistant fraction, E = Nr/(Ns + Nr) |
| Φs, Φr | modulation factors for the sensitive and resistant compartments |
| rs, rr | proliferation rates of sensitive and resistant cells |
| C | fitness cost of resistance, rr = (1 − c) rs |
| m(B) | maturation flux from induced reservoir to fixed resistance, m(B) = sE B |
| K(E) | carrying capacity, reduced by the resistant fraction, K0(1 − cK E) |
| χB, χE | strengths of the B→C and E→C downward (top-down) couplings |
| cK | strength of the E→carrying-capacity constraint |
| κB | suppression of radiosensitivity by the persistent state |
| δω, δB | departures of ω and B from baseline (interaction analysis) |
Table 3.
The four adaptive layers of the framework, defined by timescale, with representative biology, current evidence level and key supporting studies..
Table 3.
The four adaptive layers of the framework, defined by timescale, with representative biology, current evidence level and key supporting studies..
| Layer | Timescale | Representative biology | Evidence level | Key examples |
| M -fast adaptive | Milliseconds–seconds | Radiolytic oxygen depletion and recovery, radical recombination, fast damage fixation | Established biology (as dynamic oxygen/radical chemistry); the novel-layer status beyond known chemistry is unestablished | Pratx & Kapp (2019) ROD model; Labarbe et al. (2020) radical recombination; UNIVERSE model (Liew et al. 2021, 2022) |
| C — collective organizational | Seconds–days | Bystander signaling, intercellular communication, vascular/perfusion structure, bioelectric tissue organization | Established biology (bystander effects, non-cell-autonomous behavior); collective-state framing is strongly supported synthesis; intracellular epigenetic-memory through AP-1 (proposed, not established) | McMahon et al. (2013) kinetic signaling model; Marusyk et al. (2014) non-cell-autonomous growth; SFRT valley response (Casteloes et al. 2025); Levin (2021) bioelectric organization; Vaitheeswaran (2026b) |
| B — persistent adaptive (epigenetic memory) | Days–weeks | Reversible non-genetic drug-tolerant states, transcriptional/chromatin conditioning, AP-1 cis-memory | Strongly supported synthesis; the C→B write mechanism rests on demonstrated molecular biology (AP-1) | Sharma et al. (2010) chromatin-mediated tolerance; Li et al. (2026) AP-1 memory; Lopez-Gonzalez et al. (2026) DTP scoping review; Eckert et al. (2024), Doan et al. (2026), Pigozzi et al. (2025) for substrate-general memory |
| E — evolutionary | Weeks–months | Clonal selection, competition, ecological/fitness dynamics under treatment pressure | Established biology | Greaves & Maley (2012) clonal evolution; Gatenby & Brown (2020) and Zhang et al. (2017) adaptive therapy; Valcz et al. (2025) induced-and-inherited framing |
References
- Asur, R., Butterworth, K. T., Penagaricano, J. A., Prise, K. M., & Griffin, R. J. (2015). High dose bystander effects in spatially fractionated radiation therapy. Cancer Letters, 356(1), 52–57. [CrossRef]
- Boscolo, D., Scifoni, E., Carlino, A., et al. (2024). The oxygen puzzle in FLASH radiotherapy: A comprehensive review and experimental outlook. Clinical and Translational Radiation Oncology, 49, 100863.
- Brown, J. M., Carlson, D. J., & Brenner, D. J. (2014). The tumor radiobiology of SRS and SBRT: Are more than the 5 Rs involved? International Journal of Radiation Oncology, Biology, Physics, 88(2), 254–262. [CrossRef]
- Cao, X., Zhang, R., Esipova, T. V., et al. (2021). Quantification of oxygen depletion during FLASH irradiation in vitro and in vivo. International Journal of Radiation Oncology, Biology, Physics, 111(1), 240–248. [CrossRef]
- Casteloes, N., House, C. D., & Tambasco, M. (2025). A 3D co-culture scaffold approach to assess spatially fractionated radiotherapy bystander and abscopal immune effects on clonogenic survival. International Journal of Molecular Sciences, 26(9), 4436. [CrossRef]
- Chis, O.-T., Banga, J. R., & Balsa-Canto, E. (2011). Structural identifiability of systems biology models: A critical comparison of methods. PLoS ONE, 6(11), e27755. [CrossRef]
- Doan, V., et al. (2026). Associative learning in the single-celled organism Stentor coeruleus. bioRxiv, 2026.02.25.708045.
- Eckert, L., et al. (2024). Habituation as an emergent property of biochemical reaction networks. Current Biology, 34(24), 5646–5658. [CrossRef]
- Ellis, G. F. R. (2012). Top-down causation and emergence: some comments on mechanisms. Interface Focus 2(1), 126–140. [CrossRef]
- Favaudon, V., Caplier, L., Monceau, V., et al. (2014). Ultrahigh dose-rate FLASH irradiation increases the differential response between normal and tumor tissue in mice. Science Translational Medicine, 6(245), 245ra93. [CrossRef]
- Fowler, J. F. (1989). The linear-quadratic formula and progress in fractionated radiotherapy. British Journal of Radiology, 62(740), 679–694. [CrossRef]
- Gatenby, R. A., & Brown, J. S. (2020). Integrating evolutionary dynamics into cancer therapy. Nature Reviews Clinical Oncology, 17(11), 675–686. [CrossRef]
- Gelman, A., Carlin, J. B., Stern, H. S., Dunson, D. B., Vehtari, A., & Rubin, D. B. (2013). Bayesian Data Analysis (3rd ed.). Chapman and Hall/CRC.
- Greaves, M., & Maley, C. C. (2012). Clonal evolution in cancer. Nature, 481(7381), 306–313. [CrossRef]
- Griffin, R. J., Ahmed, M. M., Amendola, B., et al. (2020). Understanding high-dose, ultra-high dose-rate, and spatially fractionated radiotherapy. International Journal of Radiation Oncology, Biology, Physics, 107(4), 766–778. [CrossRef]
- Gutenkunst, R. N., Waterfall, J. J., Casey, F. P., Brown, K. S., Myers, C. R., & Sethna, J. P. (2007). Universally sloppy parameter sensitivities in systems biology models. PLoS Computational Biology, 3(10), e189. [CrossRef]
- Jin, J., et al. (2025). Re-examining the oxygen depletion hypothesis in FLASH radiotherapy. Radiotherapy and Oncology (in press).
- Kirkpatrick, J. P., Meyer, J. J., & Marks, L. B. (2008). The linear-quadratic model is inappropriate to model high dose per fraction effects in radiosurgery. Seminars in Radiation Oncology, 18(4), 240–243. [CrossRef]
- Kutcher, G. J., Burman, C., Brewster, L., Goitein, M., & Mohan, R. (1991). Histogram reduction method for calculating complication probabilities for three-dimensional treatment planning evaluations. International Journal of Radiation Oncology, Biology, Physics, 21(1), 137–146. [CrossRef]
- Labarbe, R., Hotoiu, L., Barbier, J., & Favaudon, V. (2020). A physicochemical model of reaction kinetics supports peroxyl radical recombination as the main determinant of the FLASH effect. Radiotherapy and Oncology, 153, 303–310. [CrossRef]
- Levin, M. (2021). Bioelectric signaling: Reprogrammable circuits underlying embryogenesis, regeneration, and cancer. Cell, 184(8), 1971–1989. [CrossRef]
- Li, B., et al. (2025). Drug-tolerant persister cell states in cancer (review). Cell Reports Medicine (in press).
- Li, X., & Raj, A., et al. (2026). AP-1 mediates the formation of cellular memory during the acquisition of therapy resistance. Nature Communications, 17, 4265. [CrossRef]
- Liew, H., Mein, S., Dokic, I., Haberer, T., Debus, J., Abdollahi, A., & Mairani, A. (2021). Deciphering time-dependent DNA damage complexity, repair, and oxygen tension: A mechanistic model for FLASH-dose-rate radiotherapy. International Journal of Radiation Oncology, Biology, Physics, 110(2), 574–586. [CrossRef]
- Liew, H., Mein, S., Tessonnier, T., Abdollahi, A., Debus, J., Dokic, I., & Mairani, A. (2022). The impact of sub-millisecond damage fixation kinetics on the in vitro sparing effect at ultra-high dose rate in UNIVERSE. International Journal of Molecular Sciences, 23(6), 2954. [CrossRef]
- Lopez-Gonzalez, J. S., Perez-Medina, M., Galicia-Velasco, M., & Aguilar-Cazares, D. (2026). Drug-tolerant persister cells in cancer: a scoping review of definitions, models, and molecular mechanisms. Frontiers in Oncology, 16, 1771061. [CrossRef]
- Lyman, J. T. (1985). Complication probability as assessed from dose-volume histograms. Radiation Research Supplement, 8, S13–S19.
- Marusyk, A., Tabassum, D. P., Altrock, P. M., Almendro, V., Michor, F., & Polyak, K. (2014). Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature, 514(7520), 54–58. [CrossRef]
- McMahon, S. J., Butterworth, K. T., McGarry, C. K., et al. (2013). A kinetic-based model of radiation-induced intercellular signalling. PLOS ONE, 8(1), e54526. [CrossRef]
- McMahon, S. J., Schuemann, J., Paganetti, H., & Prise, K. M. (2016). Mechanistic modelling of DNA repair and cellular survival following radiation-induced DNA damage. Scientific Reports, 6, 33290. [CrossRef]
- McMahon, S. J., & Prise, K. M. (2021). Mechanistic modelling of radiation responses. Cancers, 13(9), 2154. [CrossRef]
- Noble, D. (2012). A theory of biological relativity: no privileged level of causation. Interface Focus 2(1), 55–64. [CrossRef]
- Pigozzi, F., et al. (2025). Associative conditioning in gene regulatory networks increases integrated causal emergence. Communications Biology, 8, 1027. [CrossRef]
- Pisco, A. O., & Huang, S. (2015). Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse. British Journal of Cancer, 112(11), 1725–1732. [CrossRef]
- Powathil, G. G., Gordon, K. E., Hill, L. A., & Chaplain, M. A. J. (2012). Modelling the effects of cell-cycle heterogeneity on the response of a solid tumour to chemotherapy: Biological insights from a hybrid multiscale cellular automaton model. Journal of Theoretical Biology, 308, 1–19. [CrossRef]
- Powathil, G. G., Munro, A. J., Chaplain, M. A. J., & Swat, M. (2016). Bystander effects and their implications for clinical radiation therapy: Insights from multiscale in silico experiments. Journal of Theoretical Biology, 401, 1–14. [CrossRef]
- Pratx, G., & Kapp, D. S. (2019). A computational model of radiolytic oxygen depletion during FLASH irradiation and its effect on the oxygen enhancement ratio. Physics in Medicine & Biology, 64(18), 185005. [CrossRef]
- Raue, A., Kreutz, C., Maiwald, T., Bachmann, J., Schilling, M., Klingmüller, U., & Timmer, J. (2009). Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics, 25(15), 1923–1929. [CrossRef]
- Ribba, B., Colin, T., & Schnell, S. (2006). A multiscale mathematical model of cancer, and its use in analyzing irradiation therapies. Theoretical Biology and Medical Modelling, 3, 7. [CrossRef]
- Shaffer, S. M., Dunagin, M. C., Torborg, S. R., et al. (2017). Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature, 546(7658), 431–435. [CrossRef]
- Sharma, S. V., Lee, D. Y., Li, B., et al. (2010). A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell, 141(1), 69–80. [CrossRef]
- Sheth, M., & Esfandiari, L. (2022). Bioelectric dysregulation in cancer initiation, promotion, and progression. Frontiers in Oncology, 12, 846917. [CrossRef]
- Song, C. W., Glatstein, E., Marks, L. B., et al. (2021). Biological principles of stereotactic body radiation therapy (SBRT) and stereotactic radiation surgery (SRS): Indirect cell death. International Journal of Radiation Oncology, Biology, Physics, 110(1), 21–34. [CrossRef]
- Vaitheeswaran, R. (2026a). The Ghost in the Cancer: Towards a New Science of Radiation Oncology. Notion Press. ISBN 979-8903629169.
- Vaitheeswaran, R. (2026b). Extending the kinetic-bystander model for spatially fractionated radiotherapy with intracellular epigenetic memory. Preprints. [CrossRef]
- Vaitheeswaran, R. (2026c). On the observability and structural identifiability of radiobiological models and the case for latent-state adaptive control. Preprints. [CrossRef]
- Valcz, G., Gatenby, R. A., Újvári, B., Buzás, E. I., & Molnár, B. (2025). Adaptive cancer therapy: can non-genetic factors become its Achilles heel? Oncogene, 44(42), 3999–4005. [CrossRef]
- Vozenin, M.-C., De Fornel, P., Petersson, K., et al. (2019). The advantage of FLASH radiotherapy confirmed in mini-pig and cat-cancer patients. Clinical Cancer Research, 25(1), 35–42. [CrossRef]
- Wang, Z., Wang, M., Dong, B., Wang, Y., Ding, Z., & Shen, S. (2025). Drug-tolerant persister cells in cancer: Bridging the gaps between bench and bedside. Nature Communications, 16, 10048. [CrossRef]
- Wang, S., Lei, J., Zou, X., & Jin, S. (2025). Integrating multiscale mathematical modeling and multidimensional data reveals the effects of epigenetic instability on acquired drug resistance in cancer. PLOS Computational Biology, 21(2), e1012815. [CrossRef]
- Zhang, J., Cunningham, J. J., Brown, J. S., & Gatenby, R. A. (2017). Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nature Communications, 8, 1816. [CrossRef]
Figure 1.
Numerical solution of the spatial field equations under a spatially fractionated dose pattern (grey). The bystander signal (teal) is generated at the peaks and diffuses; the persistent state B (amber) is written across a region substantially broader than the delivered dose, reaching into the unirradiated valleys.
Figure 1.
Numerical solution of the spatial field equations under a spatially fractionated dose pattern (grey). The bystander signal (teal) is generated at the peaks and diffuses; the persistent state B (amber) is written across a region substantially broader than the delivered dose, reaching into the unirradiated valleys.

Figure 2.
The evolutionary layer reproduces the adaptive-therapy signature. Continuous dosing (left) drives sensitive cells to extinction and selects a purely resistant survivor population; adaptive dosing (right) preserves a competing sensitive population that suppresses resistance, at the cost of a higher controlled burden.
Figure 2.
The evolutionary layer reproduces the adaptive-therapy signature. Continuous dosing (left) drives sensitive cells to extinction and selects a purely resistant survivor population; adaptive dosing (right) preserves a competing sensitive population that suppresses resistance, at the cost of a higher controlled burden.

Figure 3.
Closed-loop history dependence. Two schedules deliver identical total dose and fraction number, differing only in the temporal order of signal-generating deliveries; the final resistant fraction differs significantly (shaded bands: spread over stochastic realizations).
Figure 3.
Closed-loop history dependence. Two schedules deliver identical total dose and fraction number, differing only in the temporal order of signal-generating deliveries; the final resistant fraction differs significantly (shaded bands: spread over stochastic realizations).

Figure 4.
Bistable write. Left: the steady persistent state jumps abruptly above a critical signal (shaded), not gradually. Right: increasing-signal and decreasing-signal paths differ (shaded region), a hysteresis loop indicating that the write-and-hold mechanism stores history.
Figure 4.
Bistable write. Left: the steady persistent state jumps abruptly above a critical signal (shaded), not gradually. Right: increasing-signal and decreasing-signal paths differ (shaded region), a hysteresis loop indicating that the write-and-hold mechanism stores history.

Figure 5.
Memory written per unit delivered dose as a function of peak spacing, showing a saturating optimum rather than a monotonic dependence on dose.
Figure 5.
Memory written per unit delivered dose as a function of peak spacing, showing a saturating optimum rather than a monotonic dependence on dose.

Figure 6.
FLASH × SFRT interaction in the illustrative model. The combined effect (right) falls below the sum of the separate effects (grey); the deficit is the cross-modality interaction term. Same-variable modality pairs show no such deviation.
Figure 6.
FLASH × SFRT interaction in the illustrative model. The combined effect (right) falls below the sum of the separate effects (grey); the deficit is the cross-modality interaction term. Same-variable modality pairs show no such deviation.

Table 1.
Consolidated view of the computed results. All are numerical solutions of the illustrative, uncalibrated model of the Methods; magnitudes are regime-specific and the falsifiable content is qualitative.
Table 1.
Consolidated view of the computed results. All are numerical solutions of the illustrative, uncalibrated model of the Methods; magnitudes are regime-specific and the falsifiable content is qualitative.
| # | Result | Mechanism / equation exercised | Qualitative signature | Conventional expectation it violates |
| 8.1 | Valley memory writing | Spatial reaction–diffusion of s; local B write | Durable marks in unirradiated valleys; B broader than dose | Durable effect tracks local dose |
| 8.2 | Adaptive-therapy trade-off | Two-compartment E layer; cost + competition | Continuous → all-resistant; adaptive → low resistance | Maximal dose is always optimal |
| 8.3 | Order-dependence at fixed dose | Closed C→B→E loop; stochastic maturation | Different resistant fraction, same total dose | BED / LQ are order-independent |
| 8.4 | Bistable threshold + hysteresis | AP-1 autoregulatory write | Abrupt switch; escalation ≠ de-escalation path | Graded, history-independent response |
| 8.5 | SFRT geometric optimum | Spatial signal field + threshold write | Memory-optimal geometry ≠ dose-optimal | Optimize peak/valley dose or PVDR |
| 8.6 | FLASH × SFRT non-additivity | Bilinear Φ; interaction term −κB·δω·δB | Combined ≠ sum; selective by shared variable | Modality effects superpose additively |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



