Submitted:
01 June 2026
Posted:
03 June 2026
You are already at the latest version
Abstract
Small extracellular vesicles (sEVs), operationally referred to as exosomes, where strict biogenesis-based classification cannot be confirmed from the primary literature, are pivotal mediators of intercellular communication in cancer, transferring a diverse epigenetic payload that encompasses noncoding RNAs (ncRNAs), DNA fragments, chromatin-modifying enzymes, and metabolic effectors, which reprogram the recipient cell's chromatin architecture without altering the underlying DNA sequence. sEVs originating from tumors facilitate epigenetic modifications throughout the tumor microenvironment (TME), engaging stromal, immune, and vascular elements to support malignant development. This narrative review synthesizes mechanistic, preclinical, and translational evidence on sEVs-driven epigenetic regulation in cancer, focusing on how distinct cargo classes, including microRNAs (miRNAs), long noncoding RNAs (lncRNAs), circular RNAs (circRNAs), DNA methyltransferases (DNMTs), and histone-modifying enzymes, drive chromatin remodeling, aberrant DNA methylation, transcriptional reprogramming, and acquired drug resistance in recipient cells. We further examine exosomal cargo signatures as minimally invasive liquid biopsy biomarkers and appraise engineered sEVs platforms for precision delivery of miRNA mimics, siRNAs, and small-molecule epigenetic inhibitors. Key methodological challenges, including EV isolation standardization, cargo validation, loading efficiency, in vivo biodistribution, and physiological concentration relevance, are critically evaluated, and a translational roadmap is proposed to guide reproducible clinical implementation of sEVs-mediated epigenetic cancer therapeutics.
Keywords:
exosomes
; extracellular vesicles
; epigenetic regulation
; noncoding RNA
; DNA methylation
; chromatin‑modifying enzymes
; tumor microenvironment
; therapy resistance
; liquid biopsy
; cancer biomarkers
1. Introduction
Analyses of extracellular vesicles (EVs) have turned into a key area of academic interest owing to their vital participation in cellular signaling across both healthy and disease-related scenarios [1,2]. Once considered cellular debris, sEVs are now defined as lipid bilayer-enclosed vesicles secreted by virtually all cell types and are commonly classified by size and biogenesis into exosomes (30-150 nm), microvesicles (100-1000 nm), and apoptotic bodies (>1000 nm) [3,4]. Their cargo, selectively packaged proteins, RNAs, and lipids, travels via regulated intracellular pathways and can modulate recipient cell behavior and gene expression [5,6]. This conceptual framework outlines sEVs as vital components of intercellular communication, which entail considerable consequences for immune responses, tumor progression, and tissue regeneration [7,8]. Over the past two decades, advancements in isolation and characterization techniques have enhanced EV research, moving it from basic cell biology to translational applications, and establishing sEVs as promising biomarkers and therapeutic agents [9,10]. The international consequences of illnesses tied to sEVs like tumors and cardiovascular ailments emphasize the necessity of fully comprehending the biology of EVs [11]. A thorough understanding of the biogenesis of EVs and the selection of their cargo is thus vital for the creation of successful therapeutic interventions, the progression of EV-focused treatments, and the enhancement of diagnostic precision. Notably, the ability of EVs to cross biological barriers, including the blood-brain barrier, is of great interest, as it expands their utility in the field of neurodegenerative diseases, acting as effective delivery systems and as biomarkers that signify disease progression [12]. These dual capabilities heighten interest in EVs while also highlighting the need for standardized isolation methods and regulatory frameworks to ensure safe clinical translation [12].
Despite extensive study of EV biogenesis and cargo composition, important knowledge gaps persist regarding the molecular mechanisms that govern selective cargo sorting and the functional consequences of EV-mediated communication within complex microenvironments [12,13,14]. Continued discourse exists concerning the exact mechanisms that dictate the formation of extracellular vesicles (EVs), the variability of their cargo, and the selectivity with which they connect with recipient cells [15,16]. The contradictory functions of sEVs in facilitating both the advancement of disease and the regeneration of tissue present additional challenges for their therapeutic application [17,18]. These persistent challenges limit the complete actualization of the diagnostic and therapeutic capabilities of sEVs and highlight the necessity for an exhaustive synthesis and critical evaluation of existing research outcomes [19,20]. Since their discovery, insights into exosome biogenesis and cargo selection have evolved to reveal key roles in modulating the tumor microenvironment (TME), maintaining cancer stem cells (CSCs), and facilitating immune evasion [21,22,23]. The clinical relevance is stark: cancers that develop therapy resistance and relapse account for substantial mortality, with exosomal components increasingly recognized as biomarkers and therapeutic targets [24]. For instance, breast cancer was diagnosed in approximately 2.26 million women in 2020, making it the most commonly diagnosed cancer globally, and was responsible for an estimated 685,000 deaths worldwide that year, representing the fifth leading cause of cancer mortality worldwide, underscoring the critical need to clarify the mechanisms of resistance [25,26,27]. Studies on the epigenetic influence on cancer have surfaced as a significant field of research, as it modulates gene expression without changing the core DNA sequences, consequently affecting tumor development, progression, and resistance to therapy [28,29]. Since the initial identification of atypical DNA methylation patterns in neoplasia during the 1980s, the domain has broadened to include histone alterations, chromatin restructuring, and non-coding RNA as pivotal epigenetic processes [30,31]. These reversible alterations represent promising therapeutic avenues: various epigenetic agents have received approval for hematologic cancers, and ongoing endeavors aim to broaden their effectiveness to solid neoplasms [32,33]. Given the global cancer epidemic, enhancing epigenetic diagnostic methods and therapeutic approaches is of paramount importance [34,35,36]. Nonetheless, obstacles persist in precisely identifying the distinct epigenetic modifications that underpin various cancers and in comprehending their contributions to drug resistance and metastatic processes [37,38,39]. While DNA methylation and histone modifications are well studied, the interplay with non-coding RNAs and chromatin remodeling is less well defined [40,41]. The distinct epigenetic shifts identified in separate tumor categories, together with the varied patient outcomes post-epigenetic treatment, sustain a lively discourse in the scientific field [42,43]. Furthermore, the pathways through which epigenetic dysregulation facilitates immune evasion and the modulation of the tumor microenvironment necessitate additional elucidation [44,45]. Confronting these gaps is fundamental for the development of precise and resilient oncological solutions [46,47,48]. Significantly, the convergence of EV biology and epigenetics constitutes a swiftly advancing domain with immediate implications for therapeutic resistance. sEVs miRNAs and lncRNAs have been implicated in reprogramming gene expression and chromatin states in recipient cells, yet the precise pathways and cargo interactions that drive resistance and relapse remain incompletely defined [49,50,51]. Controversy persists over the relative contributions of different sEV cargoes such as chromatin-modifying enzymes versus non-coding RNAs, and their cell-type–specific effects within the TME [52,53,54]. The elaborate interactions involving sEVs produced by tumors and stromal elements, such as cancer-associated fibroblasts (CAFs), create additional layers of complexity in the mechanisms that regulate resistance [55,56]. Conceptually, sEVs function as vehicles for the horizontal transfer of epigenetic regulators, miRNAs, lncRNAs, and chromatin-modifying enzymes, thereby promoting phenotypic plasticity and CSC maintenance that underpin therapeutic resistance [21,57,58]. sEVs cargo can rewire recipient-cell epigenetic landscapes without altering DNA sequence, enabling tumor cells to evade therapy and sustain relapse [50,59]. The collaboration of exosome creation, cargo transportation, and epigenetic modulation establishes a well-organized framework for understanding the dynamics through which cell communication enhances tumor growth while obstructing therapeutic outcomes [60,61,62,63]. This comprehensive review scrutinized the processes through which sEVs originating from tumors facilitate epigenetic reprogramming in malignancies, emphasizing the specific ways in which various classes of cargo alter the epigenetic framework of recipient cells situated within the tumor microenvironment. In addition, we examine the consequences of this intercellular epigenetic dialogue, which alters therapeutic resistance, immune evasion, and the pathways of metastasis, across various cancer types. The translational implications of this finding are evaluated in the context of sEVs cargo as a liquid biopsy biomarker and engineered exosome platform as a tool for epigenetic cancer therapy. Finally, we critically assess the key methodological challenges that remain to be resolved before this insight can be meaningfully translated into clinical practice. Throughout this review, the term exosome is used in the operational sense to encompass a small extracellular vesicle (30 -150 nm) isolated by differential ultracentrifugation or size exclusion chromatography, acknowledging that strict biogenesis-based classification, as defined by MISEV2023[64], is not always possible to confirm from the primary literature cited.
2. Search Strategy
We conducted a structured narrative literature search to compile mechanistic, preclinical, and clinical evidence on exosome-mediated epigenetic regulation in cancer. This review is explicitly a narrative synthesis and not a systematic review; it was not prospectively registered and does not include a PRISMA flow diagram[65]. Eligibility decisions were made by the authors based on mechanistic relevance and methodological quality, and selection bias toward studies supporting the conceptual framework presented cannot be excluded. Databases investigated include: PubMed/MEDLINE, Web of Science, Scopus, and ClinicalTrials.gov for both ongoing and concluded exosome-associated cancer studies. Temporal scope: January 2000-April 2026. Language restriction: English. The principal Boolean string was: ("exosome*" OR "extracellular vesicle*" OR "sEV" OR "small extracellular vesicle*") AND ("epigenetic*" OR "DNA methylation" OR "histone" OR "chromatin" OR "non-coding RNA" OR "epitranscriptom*") AND ("cancer" OR "tumor" OR "oncology*" OR "malignant*"). Inclusion criteria: primary mechanistic studies (in vitro and in vivo) with EV characterization meeting MISEV2018/2023 minimum reporting standards [64,66], translational/clinical biomarker studies, and peer-reviewed reviews that directly address exosomal cargo and epigenetic outcomes. Exclusion criteria: studies lacking EV characterization, reports analyzing cell-free nucleic acids without explicit EV separation, and non-peer-reviewed material unless explicitly cited as preprints and labeled provisional. Readers should interpret the mechanistic synthesis herein as reflecting the current weight of published evidence, subject to publication bias and the rapidly evolving nature of this field.
3. Exosome Biogenesis and Epigenetic Cargo Packaging
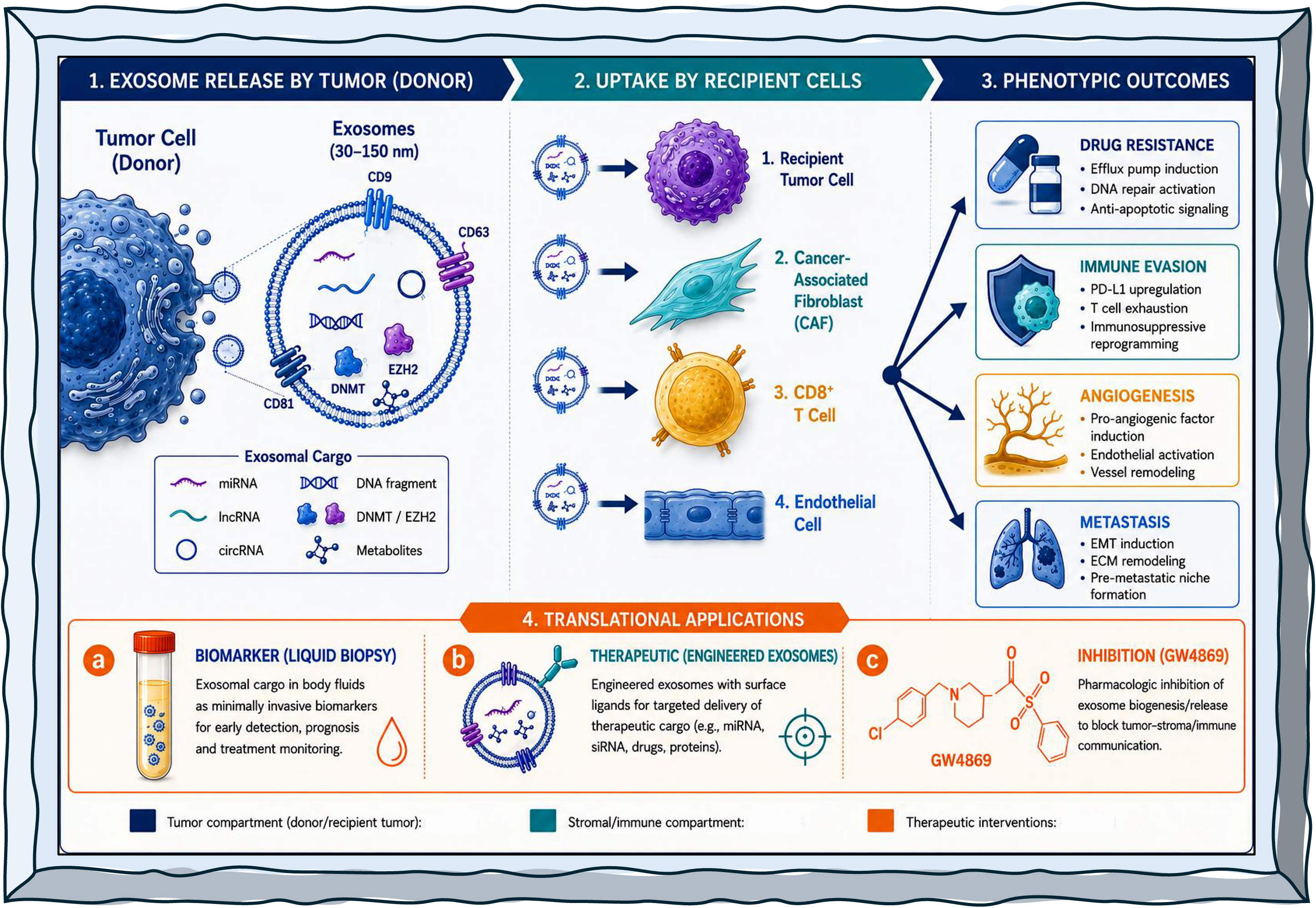
The biogenesis of exosomes is initiated by the invagination of the plasma membrane to form early sorting endosomes, which mature into late endosomes and ultimately give rise to multivesicular bodies (MVBs) [67]. The intraluminal vesicles contained within MVBs are selectively loaded with molecular cargo, including proteins, lipids, and nucleic acids, and are released as exosomes following fusion of MVBs with the plasma membrane [68]. Cargo sorting into exosomes is orchestrated by endosomal sorting complexes required for transport (ESCRT) as well as ESCRT-independent sphingolipid-mediated pathways, with RNA-binding proteins such as heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1), YBX1, and AGO2 directing the selective packaging of miRNAs [69]. The epigenetic payload of exosomes is particularly consequential. Non-coding RNAs includes miRNAs, lncRNAs, and circRNAs, represent the predominant epigenetic cargo and can regulate post-transcriptional gene silencing, chromatin remodeling, and transcription factor activity in recipient cells [68]. Beyond non-coding RNAs, sEVs also transport DNA methyltransferases (DNMT1, DNMT3A, DNMT3B) and histone-modifying enzymes that can reprogram the epigenome of recipient cells, a mechanism with profound relevance for cancer progression and drug resistance [67]. Although DNMTs, EZH2, and histone-modifying enzymes have been detected in exosomal proteomes, this detection does not establish that these proteins are present in catalytically competent form, that they are delivered intact to recipient cell cytoplasm or nucleus, or that they produce heritable chromatin changes in recipient cells. Intact multi-subunit complexes such as PRC2 (EZH2/SUZ12/EED) face particular challenges with respect to intraluminal stability, vesicle loading stoichiometry, and post-endocytic delivery. Direct mechanistic validation of enzymatic activity in recipient cells, not merely cargo detection, is required before these mechanisms can be stated as established. Claims in this review involving protein-mediated epigenetic reprogramming are qualified accordingly as discussed in Section 8 and Section 12.
It is essential to note that the quantity and molecular makeup of sEVs serve as indicators of the physiological and pathological conditions of the source cell, thereby rendering their constituents valuable and informative candidates for diagnostic applications [67]. The RNA content within sEVs is distinctive, safeguarded against degradation by nucleases, proteases, and oxidative stressors, thereby facilitating effective transfer to target cells [70]. This protection makes sEVs cargo a concentrated source of information that can alter the function of neighboring and distant cells, carrying a wealth of information about transcriptomic and epitranscriptomic changes that occur during disease conditions [70] Figure 1.
4. Exosomal Cargo Relevant to Epigenetic Regulation
A specialized subclass of small extracellular vesicles (sEVs) mediate intercellular communication by transferring biologically active macromolecules, including RNAs, proteins, and DNA, between cells, thereby shaping tumor biology at both local and systemic levels [71,72]. In cancer, this communication is increasingly interpreted through an epigenetic lens. Epigenetic regulation, encompassing DNA methylation, histone and chromatin modification, and ncRNA-mediated gene-expression control, is fundamental to malignant transformation, progression, and treatment response [72,73]. A conceptual distinction is essential before describing individual cargo classes. In this review, the term epigenetic cargo refers strictly to molecules that produce heritable, chromatin-level alterations in recipient cells: covalent DNA modifications (5-methylcytosine; 5-hydroxymethylcytosine), post-translational histone modifications (acetylation, methylation, phosphorylation, ubiquitination), or chromatin-remodeling complex-mediated changes in nucleosome positioning [74,75]. sEVs cargo that produces post-transcriptional silencing, for example, miRNA-mediated mRNA destabilization through RNA-induced silencing complex (RISC) loading without a concurrent chromatin-state change, is classified as post-transcriptional regulation and not as epigenetic regulation sensu stricto, even where it influences transcriptional outputs. This distinction is maintained throughout Section 4, Section 5 and Section 6. Cargo classes are described as epigenetic, where direct evidence for chromatin-level changes in recipient cells exists [76]; where such evidence is absent or indirect, claims are explicitly framed as hypotheses or candidate mechanisms. Epigenetic control is organized around chromatin states and the enzymatic writing, erasing, and reading of chromatin marks, implemented by DNA methyltransferases (DNMTs), TET family dioxygenases, histone acetyltransferases (HATs), histone deacetylases (HDACs), and histone methylation enzymes [77,78,79,80]. In cancer, chromatin modifiers including KDM6A, KMT2D, and EZH2 are frequently mutated or overexpressed, reshaping tumor epigenomes and driving aggressive behavior and immune evasion [81,82], while in hematologic malignancies, epigenetic dysregulation is central to pathogenesis and represents a target for therapy [83]. Against this backdrop, sEVs cargo classes can be organized into two functional categories: (i) direct epigenetic cargo; molecules that themselves constitute or transfer epigenetic information (methylated DNA fragments, chromatin-modifying enzymes); and (ii) indirect epigenetic modulators; molecules that alter the expression or activity of the host epigenetic machinery (ncRNAs targeting DNMT or HDAC mRNAs; metabolic intermediates that serve as obligate co-substrates for chromatin enzymes). Both categories are reviewed below, with mechanistic certainty calibrated to the available evidence.
4.1. Exosomal ncRNA Cargo Classes as an Epigenetic Regulator
4.1.1. Exosomal miRNA Cargo
miRNAs regulate gene expression primarily post-transcriptionally; however, in the context of sEVs transfer, their functional consequences extend to chromatin-level alterations, warranting their inclusion as indirect epigenetic cargo. Epigenetic mechanisms and miRNA expression are bidirectionally coupled: promoter hypermethylation silences tumor-suppressive miRNAs, while miRNAs reciprocally target DNMT and polycomb-group mRNAs to alter DNA methylation and histone modification states [75,84]. Crucially, miRNA loading into sEVs is not passive but is governed by selective sorting machinery. The RNA-binding protein SYNCRIP controls miRNA sorting in hepatocyte-derived exosomes through sequence-specific recognition motifs, providing a molecular basis for the selective enrichment of individual miRNA species in secreted vesicles [75,76]. hnRNPA2B1 and AGO2 perform analogous sorting roles in other cell types [68]. This selectivity means that tumor-derived sEVs carry a biased, functionally coherent miRNA payload rather than a random sample of cellular miRNAs. Once delivered to recipient cells, sEVs miRNAs engage the endogenous RISC machinery to suppress target mRNAs. Where targets include DNMT3A, DNMT3B, EZH2, or HDAC isoforms, the downstream consequence is a measurable epigenetic state change, the criterion distinguishing indirect epigenetic modulation from purely post-transcriptional regulation [73,74]. For example, miR-29 family members suppress DNMT3A and DNMT3B, producing genome-wide hypomethylation [85]. miR-21 activates STAT3 signaling that in turn recruits DNMT3A to silence effector immune-cell loci [86]. The evidence for miRNA-mediated DNMT suppression downstream of sEVs transfer reaches Level 3 of the evidence hierarchy (measurable chromatin/methylation change in recipient cells), making it one of the better-supported indirect epigenetic mechanisms in the field. In hepatocellular carcinoma (HCC), sEVs ncRNAs, including miRNAs, have been characterized as contributors to disease biology and candidate non-invasive biomarkers [87]. More broadly, tumor-derived sEVs propagate oncogenic signaling, modulate the tumor microenvironment (TME), and engage epigenetic regulatory programmes linked to progression and therapy resistance [73,74]. sEVs miRNAs are therefore both mechanistic drivers capable of triggering chromatin-state transitions in recipient cells and translational targets, warranting continued investigation as therapeutic and biomarker candidates across tumor types.
4.1.2. Exosomal lncRNA Cargo
Transcripts >200 nucleotides with limited protein-coding potential regulate gene expression at epigenetic, transcriptional, and post-transcriptional levels, primarily through interactions with chromatin-modifying complexes, transcription factors, and other RNA species [88]. As scaffolds or guides for PRC2, DNMT, or KAT/HDAC family proteins, lncRNAs directly nucleate chromatin-state changes at specific genomic loci, qualifying them as direct epigenetic regulators when delivered to recipient cells via sEVs [88,89]. sEVs provide a biologically relevant vehicle for lncRNA transfer across cellular compartments. Dedicated reviews document the biological functions and translational significance of sEVs lncRNAs in tumor progression and drug resistance [73,90]. Disease-specific evidence in colorectal cancer (CRC) is instructive: serum-exosome-protected lncRNA NNT-AS1 has been characterized as a candidate oncogenic biomarker acting through a defined miR-496/RAP2C axis [47]; differential profiling of tumor-tissue versus serum-exosome lncRNA patterns confirms selective lncRNA release into the circulation, reflecting complex extracellular regulation rather than passive leakage [91,92]. Computational ceRNA axis analyses in CRC further map the lncRNA/circRNA-miRNA-mRNA networks carried by circulating sEVs, providing mechanistic plausibility for their downstream gene-regulatory effects [93]. The epigenetic significance of sEVs lncRNA transfer is most directly demonstrated by HOTAIR. HOTAIR recruits the PRC2 complex (EZH2, SUZ12, EED) to silence tumor-suppressor loci via H3K27 trimethylation (H3K27me3) [94,95,96]. Evidence that cisplatin-resistant cells secrete HOTAIR-enriched sEVs, and that HOTAIR transfer propagates H3K27me3-mediated silencing of pro-apoptotic loci (DAPK, PTEN) in cisplatin-sensitive recipient cells, places this mechanism at Level 3-4 of the evidence hierarchy among the strongest available for any sEVs lncRNA cargo. It is important to note, however, that the relative contributions of (i) lncRNA-templated PRC2 nucleation within recipient cells versus (ii) direct transfer of pre-assembled PRC2 complexes remain experimentally unresolved. Transfer of intact multi-subunit complexes requires orthogonal validation (proximity ligation, co-immunoprecipitation within isolated vesicles, single-vesicle proteomics) that has not yet been rigorously provided; this represents an open mechanistic question, not an established pathway. Beyond HOTAIR, sEVs lncRNA transfer between tumor and stromal/immune compartments reshapes therapy-response epigenetic states. Transfer of cargo between cancer cells and tumor-associated macrophages (TAMs) modulates phenotypic reprogramming consistent with chromatin remodeling [97]. These observations support a model in which sEV-mediated lncRNA trafficking constitutes a direct epigenetic communication axis, while cautioning that chromatin-level readouts in recipient cells, not just lncRNA detection in EVs, are required to establish mechanistic completeness.
4.1.3. Exosomal circRNA Cargo
Covalently closed RNA structures generated by back-splicing are increasingly recognized as regulators of gene expression in cancer through two principal mechanisms: (i) competitive endogenous RNA (ceRNA) activity, sponging miRNAs away from their mRNA targets; and (ii) direct interactions with chromatin-regulatory proteins that alter transcriptional programmes [98]. Their relevance spans hematological and solid malignancies [99,100]. Within the exosomal compartment, circRNAs have been identified as functional cargo in breast cancer, pancreatic cancer, and lung cancer contexts [88,101,102,103]. The best-characterized sEVs-circRNA with direct epigenetic consequences is circNSUN2: N6-methyladenosine (m6A) modification of circNSUN2 by the METTL3/METTL14 complex facilitates its YTHDC1-dependent selective loading into sEVs; following uptake by recipient cells, circNSUN2 stabilizes HMGA2, an architectural chromatin protein that remodels nucleosome positioning at metastasis-associated loci, thereby promoting hepatic metastatic colonization [104,105]. This example provides Level 3-4 evidence for sEVs circRNA-mediated epigenetic remodeling. In pancreatic cancer, the circRNA cargo of irradiation-conditioned sEVs is reshuffled relative to untreated cells, consistent with therapy-induced reprogramming of sEVs content [102]. More broadly, sEVs ncRNA biology is synthesised through lncRNA/circRNA-miRNA-mRNA axis models in CRC and breast cancer [93,101]. While these network models provide mechanistic plausibility, the majority of cited circRNA axis evidence rests on in silico ceRNA interaction predictions and correlation analyses rather than chromatin-level functional validation in recipient cells. Cross-cancer generalization of specific circRNA mechanisms should therefore be treated as a testable hypothesis pending experimental confirmation of chromatin-state changes in relevant cancer-type-specific cellular and stromal contexts.
However, evidence for circRNA-specific epigenetic mechanisms in cancer-derived sEVs remains largely descriptive and cancer-type specific; mechanistic validation at the chromatin level is available for only a subset of reported circRNA-cancer associations. The mechanisms and clinical implications remain under active investigation, and cross-cancer extrapolation from in silico axis models requires experimental validation.
4.2. Exosomal DNA Fragments as Epigenetic Regulators
sEVs carry double-stranded DNA fragments, including genomic and mitochondrial sequences, as part of their molecular cargo repertoire [106,107]. Because cancer genomes are characterized by focal promoter hypermethylation at tumor-suppressor CpG islands and by global hypomethylation [108]. DNA transferred via sEVs carries methylation imprints that can, in principle, influence epigenetic states in recipient cells, constituting direct epigenetic cargo in the strictest sense. Evidence that cancer cells transfer hypermethylated promoter fragments to neighboring cells via sEVs, leading to transcriptional silencing of recipient-cell tumor suppressors, has been documented [73,107]. The translational relevance of EV-associated DNA methylation is particularly well established in the liquid-biopsy context: 5-hydroxymethylcytosine (5hmC) signatures in circulating cell-free DNA and sEVs DNA fragments serve as sensitive, tissue-of-origin-specific diagnostic and prognostic biomarkers across multiple cancer types [171]. In lung cancer, integrating circulating DNA methylation profiles with ncRNA measurements enhances diagnostic and therapeutic-prediction accuracy [109]. Single-vesicle nanoscale epigenetic profiling of CRC-derived exosomes using photo-induced force microscopy (PiFM) has successfully distinguished CpG island methylator phenotype (CIMP)-high from CIMP-negative vesicles, revealing intra-population epigenetic heterogeneity that requires single-vesicle resolution for clinically meaningful characterization [110]. An important mechanistic caveat applies transfer of methylated DNA fragments into recipient cells does not automatically reprogram the recipient epigenome. For such fragments to alter gene expression, they would need to integrate into, or otherwise influence, the recipient chromatin landscape, a mechanism that has not been demonstrated in the context of sEVs DNA delivery. The demonstrated translational utility of sEVs DNA methylation, therefore, currently resides in its biomarker value rather than in functional epigenetic reprogramming of recipient cells.
4.3. Exosomal Protein Cargo as Relevant to Epigenetic Regulators
Beyond ncRNA and DNA, sEVs cargo includes proteins, among which chromatin-modifying enzymes represent mechanistically consequential candidates. sEVs DNMT1 transfer has been directly demonstrated in ovarian cancer, where recipient cells acquire elevated DNMT activity and cisplatin resistance, one of the clearest examples of protein-mediated epigenetic reprogramming via sEVs, with evidence reaching Level 3-4 [111]. Similarly, EZH2 has been detected in tumor-derived EVs, and its candidate delivery to recipient cells could deposit H3K27me3 marks at pro-metastatic or immune-evasion loci[112] However, a fundamental unresolved question constrains how strongly these mechanisms can be stated: can single-subunit epigenetic enzymes (e.g., DNMT3A, EZH2 monomers) remain catalytically competent through the intraluminal EV milieu and be delivered in an enzymatically active form to recipient cells? For single-subunit enzymes, folding fidelity during vesicle biogenesis and uptake is the primary requirement, making their functional delivery biologically plausible. For multi-subunit complexes, including PRC2 (EZH2/SUZ12/EED/RBBP7, requiring 4+ subunits for full activity) and NuRD (6-8 subunits)-co-packaging of the complete assembled complex in a single vesicle and its reassembly or maintenance of activity in the recipient cytoplasm have not been demonstrated [113]. Direct transfer of intact PRC2 or NuRD should therefore be regarded as a hypothesis pending proximity ligation assays, co-immunoprecipitation within isolated EVs, and single-vesicle proteomics that establish stoichiometry and activity status of transferred chromatin regulators. Exosome-mediated transfer of epigenetic protein cargo between tumor and stromal cells (cancer-associated fibroblasts, macrophages) modulates chemosensitivity and resistance through epigenetic events that include HDAC activity changes and altered DNA methylation landscapes [73,84]. This crosstalk is particularly relevant in the context of tumor-immune interactions: tumor-derived EVs carry mRNAs and proteins that regulate HDAC activity in T cells [114], potentially reshaping T-cell effector gene accessibility through histone modification. The protein cargo dimension of exosomal epigenetics remains comparatively under-characterized relative to ncRNA cargo, and systematic single-vesicle proteomics studies focused on epigenetic enzyme enrichment and activity represent a high-priority methodological gap.
4.4. Metabolic Regulators as Exosomal Cargo Linked to Epigenetic Regulation
The intersection of sEVs-driven metabolite transfer and epigenetic regulation is mechanistically grounded in the obligate dependency of chromatin-modifying enzymes on specific metabolic intermediates. HATs require acetyl-CoA; DNA and histone methyltransferases depend on S-adenosylmethionine (SAM); and TET family dioxygenases utilize alpha-ketoglutarate (alpha-KG) as an essential cofactor for iterative 5-methylcytosine oxidation [115]. sEVs secreted by metabolically reprogrammed cancer cells can transfer these substrates or the enzymes that generate them to stromal and immune recipient cells, thereby altering the recipient-cell epigenetic landscape independently of genetic instruction. Oncometabolites provide the clearest mechanistic examples. Succinate and fumarate, accumulated through loss-of-function mutations in succinate dehydrogenase (SDH) and fumarate hydratase (FH), competitively inhibit alpha-KG-dependent TET dioxygenases and KDM demethylases, inducing global DNA hypermethylation and H3K methylation accumulation, a hypermethylated epigenetic phenotype demonstrated in paraganglioma and potentially applicable to other SDH/FH-mutant cancers[116].2-Hydroxyglutarate (2-HG), produced by gain-of-function IDH1/IDH2 mutations, similarly inhibits TET activity in recipient stromal and immune cells when transferred via tumor-derived exosomes, inducing hypermethylation programmes that promote immune exclusion [117]. These oncometabolite-mediated mechanisms are supported by direct biochemical evidence (competitive enzyme inhibition assays; DNA methylome profiling), placing them at Level 3 of the evidence hierarchy. The cell-non-autonomous dimension of oncometabolite transfer via EVs is biologically significant: epigenetic silencing of immune-recognition genes in tumor-infiltrating lymphocytes has been documented in hypoxic, metabolite-replete TMEs [116,118], and sEVs delivery of oncometabolite-laden cargo could extend this silencing program beyond the primary tumor niche. Metabolic regulators are best regarded as an emerging integrative cargo class whose contribution to recipient-cell epigenetic states converges with ncRNA-mediated and protein-mediated pathways during therapy response, metastasis, and microenvironmental adaptation [73,75,84,115,118].
4.5. Epitranscriptomic Modifications as an Exosomal Epigenetic Cargo
sEVs RNAs carry covalent chemical modifications collectively the epitranscriptome that influence their biological function in recipient cells and serve as sorting signals that bias which RNAs are packaged into vesicles. N6-methyladenosine (m6A), the most prevalent internal modification on eukaryotic mRNA and lncRNAs, is installed co-transcriptionally by the METTL3/METTL14 complex and removed by the demethylases FTO and ALKBH5 [119]. In cancer, METTL3 is frequently amplified or overexpressed, and oncogenic m6A deposition reprograms RNA stability, translation efficiency, and alternative splicing to promote tumor progression [120]. The role of m6A as a cargo-sorting signal in exosome biogenesis is mechanistically supported: m6A modifications on miRNA precursors facilitate processing via YTHDF2-mediated DGCR8/DROSHA recruitment, altering the mature miRNA repertoire packaged into EVs [121].The m6A-modified circRNA circNSUN2 is selectively exported in sEVs via YTHDC1-dependent recognition; following uptake by recipient cells, it stabilizes HMGA2 and promotes hepatic metastatic colonization, a pathway with Level 3-4 mechanistic evidence[104,105].In gastric and colorectal cancer, tumor-derived EVs enriched for m6A-modified lncRNAs recruit YTHDF1 in recipient macrophages, reprogramming macrophage polarization toward an immunosuppressive, pro-tumorigenic phenotype a mechanism bridging epitranscriptomic and epigenetic regulation within the TME [105].METTL3 protein itself has been detected by single-vesicle mass spectrometry in colorectal- and gastric-cancer-derived EVs, raising the possibility that the m6A writer machinery is transferable and could directly reprogram the epitranscriptome of recipient cells [104]. This represents a mechanistically distinct pathway from ncRNA-mediated DNMT or HDAC modulation: rather than altering chromatin enzyme expression, transferred METTL3 would alter the RNA modification landscape and thereby change the post-transcriptional regulation environment of the recipient cell. Formal demonstration of catalytically active METTL3 delivery as opposed to mere protein detection requires activity assays in recipient cells and has not yet been provided; this is therefore a candidate mechanism. The epitranscriptomic dimension of exosomal biology constitutes a critical mechanistic frontier warranting dedicated investigation. Although m6 A-modified RNA cargo is not yet represented as a row in Table 1 due to the nascent state of direct mechanistic evidence, the epitranscriptomic layer constitutes a functionally important extension of the exosomal epigenetic cargo repertoire described therein, and its systematic incorporation into future summary frameworks is recommended as the field matures.
Throughout this review, we distinguish, where the published evidence permits, four levels of mechanistic evidence for sEVs-mediated epigenetic effects that collectively constitute a mechanistically complete causal chain: level (i) demonstration of cargo enrichment within isolated EVs; defined as detection of a given cargo molecule in purified EV fractions meeting MISEV2018/2023 minimum characterization criteria; level(ii) confirmed delivery of cargo to recipient cells; confirmed by cargo internalization assays such as fluorescent labeling and reporter transfer level(iii) measurable change in recipient-cell chromatin state or DNA methylation pattern; defined as quantifiable alterations in DNA methylation, histone modification, or chromatin accessibility attributable to the transferred cargo; and level (iv) causal contribution of the transferred cargo to a disease-relevant malignant phenotype; demonstrated through functional rescue, genetic complementation, or in vivo validation. Many cited studies provide compelling evidence at levels (i) and (ii), while evidence at levels (iii) and (iv) is available for a more limited subset of cargo-mechanism pairs. Where only levels (i)-(ii) evidence exists, mechanistic claims are qualified as 'consistent with,' 'suggestive of,' or 'hypothesized to involve' epigenetic remodeling, rather than 'demonstrating' it. We further distinguish between indirect modulation of chromatin states via post-transcriptional regulation of chromatin modifier mRNAs, which is mechanistically plausible but not equivalent to direct epigenetic remodeling and bona fide transfer of epigenetic information or enzymatic activity that produces heritable chromatin changes in recipient cells.
5. Exosome-Mediated Epigenetic Alterations in Recipient Cells
The preceding section catalogued the classes of epigenetic cargo carried by sEVs. This section describes how that cargo produces functional epigenetic changes in recipient cells, with attention to the level of mechanistic evidence available for each pathway. Four levels of evidence are used to calibrate claims: Level 1-cargo detected in purified sEV fractions; Level 2- cargo internalization by recipient cells confirmed; Level 3 -measurable chromatin or methylation change in recipient cells attributable to the transferred cargo; Level 4 -causal contribution to a malignant phenotype demonstrated through functional rescue, genetic complementation, or in vivo validation. The majority of studies reviewed below provide strong evidence at Levels 1-2, with growing Level 3 evidence, while Level 4 causal validation remains sparse outside of a subset of miRNA and lncRNA examples. Tumor-derived sEVs (TEXs) reprogram recipient stromal and immune cells through horizontal transfer of ncRNAs, proteins, and metabolic factors that converge on chromatin-modifying pathways [47,62,122,124].TEXs silence tumor suppressors, activate oncogenes, and induce epithelial-to-mesenchymal transition (EMT) in recipient cells via mechanisms that include: (i) miRNA-mediated suppression of DNMT and HDAC mRNAs, altering DNA methylation and histone acetylation patterns; (ii) lncRNA-scaffolded PRC2 recruitment leading to H3K27me3 deposition at pro-apoptotic and tumor-suppressor loci; (iii) direct DNMT enzyme transfer enabling de novo CpG methylation; and (iv) metabolite-mediated inhibition of TET dioxygenases, inducing hypermethylation [47,125,126,127,128]. These mechanisms, individually and in combination, underpin cancer-cell adaptation to the TME and contribute to resistance to chemotherapeutics including tamoxifen, cisplatin, and tyrosine-kinase inhibitors [47,126].
5.1. Exosomal miRNAs as Portable Post-Transcriptional Regulators That Reshape Cell State
sEVs miRNAs produce post-transcriptional silencing of target mRNAs in recipient cells via RISC loading. When targets include chromatin regulators DNMTs, EZH2, HDACs, or their upstream signaling effectors, the downstream effect is a measurable epigenetic state change. This section focuses on miRNA-cargo interactions supported by at least Level 3 evidence (chromatin or methylation readout in recipient cells). Mechanistically, evidence that exosomes lack key miRNA-biogenesis components and Argonaute proteins [129] confirms that sEVs function as passive miRNA carriers rather than active biogenesis compartments; internalized miRNAs must therefore engage the recipient cell's pre-existing RISC machinery to exert their effects. This model is supported by multiple studies demonstrating that cell-line or patient-derived exosomal miRNA profiles can be recapitulated in recipient cells following sEVs uptake, and that the transcriptional consequences mirror those produced by miRNA mimics [129,130,131]. Cancer-associated fibroblast (CAF)-derived sEVs miR-29b suppresses DNMT3B in recipient tumor cells, relieving methylation-mediated repression of target gene promoters and altering transcriptional profiles [85]. This represents a Level 3 mechanism: miRNA detection in EVs (Level 1), uptake confirmed (Level 2), and recipient-cell DNMT3B suppression with downstream promoter demethylation documented (Level 3). Similarly, sEVs miR-21 targets PTEN and PDCD4, activating PI3K/AKT signaling that converges on epigenetic reprogramming through STAT3-mediated DNMT3A recruitment to immune-effector gene loci [98]. sEVs miR-155 downregulates E-cadherin and upregulates mesenchymal markers in recipient non-CSC cells, triggering EMT, a process accompanied by broad chromatin remodeling at epithelial-program loci [132]. Beyond individual tumor types, tumor- and stromal-derived sEVs miRNAs couple distant tissue compartments and shift recipient cells toward phenotypes that favor tumor growth and metastasis [131,133,134]. The systemic dimension of sEVs miRNA signaling reflected in circulating biofluid profiles from cancer patients supports models in which sEV-borne miRNAs coordinate epigenetic state transitions across the tumor, vasculature, immune infiltrate, and stroma. However, it is important to note that most studies demonstrate miRNA transfer and downstream phenotypic effects; the chromatin-level mechanism linking RISC-mediated mRNA silencing to durable epigenetic state change in recipient cells requires explicit validation (e.g., ChIP-seq or ATAC-seq) and is not uniformly established across the cited examples.
5.2. Exosomal Long Noncoding RNAs and Other ncRNAs as Regulators of Gene-Expression Programs
sEVs lncRNAs and circRNAs exert epigenetic effects in recipient cells primarily through two non-mutually exclusive mechanisms: (i) scaffold-directed recruitment of PRC2 or other chromatin-modifying complexes to specific genomic loci; and (ii) competitive endogenous RNA (ceRNA) activity, sponging tumor-suppressive miRNAs away from their DNMT or HDAC targets, thereby indirectly altering chromatin states. Both mechanisms are reviewed here with explicit evidence-level annotation. The clearest example of lncRNA-mediated epigenetic reprogramming via exosomes is HOTAIR. Delivered to recipient cells via cisplatin-resistant cell-derived sEVs, HOTAIR scaffolds the PRC2 complex to deposit H3K27me3 at DAPK, PTEN, and related pro-apoptotic loci, propagating cisplatin resistance across previously sensitive tumor cells [94,95,96]. Separately, HOTAIR sequesters miR-138-5p, relieving miR-138-5p-mediated suppression of EZH2 and SIRT1 and amplifying H3K27me3 deposition [135,136]. These convergent mechanisms constitute Level 3-4 evidence for sEVs lncRNA-driven epigenetic reprogramming. In HCC, sEVs ncRNAs, including lncRNAs, regulate pathophysiological processes in recipient cells and remodel the TME [137]. Pan-cancer reviews acknowledge that mechanistic details in recipient cells remain incompletely defined for most lncRNA cargo beyond HOTAIR [138]. In the ceRNA dimension, computational analyses in CRC and breast cancer map lncRNA/circRNA-miRNA-mRNA networks in circulating sEVs [93,101]; however, the majority of these axis models are based on in silico interaction predictions rather than chromatin-level functional validation in recipient cells, and cross-cancer generalisation of specific lncRNA mechanisms should be treated as a hypothesis until experimental chromatin-state evidence is provided. For circRNAs, the mechanistically best-supported sEVs example with epigenetic consequences is m6A-modified circNSUN2, described in Section 4.5. Beyond this, sEVs circRNA content is reshuffled in therapy-conditioned pancreatic cancer cells [102], suggesting a mechanism by which treatment pressure alters the epigenetic signaling capacity of secreted vesicles, though chromatin-level consequences in recipient cells remain to be characterized in most contexts.
5.3. Exosomal mRNAs, Proteins, and Transcriptional Factors as Drivers of Recipient-Cell Program Shifts
sEVs transport mRNAs and proteins between cells, providing a mechanism to alter recipient-cell gene expression and signaling states independent of ncRNA-mediated pathways [3,138,139]. Transcription factors have been explicitly listed among the pathogenic components carried by cancer-derived EVs, and their paracrine delivery contributes to pre-metastatic niche formation and tumor progression [131,133]. The diverse molecular composition of sEVs nucleic acids, proteins, lipids, and metabolites, reflecting the donor-cell state, enables multifaceted reprogramming of recipient-cell transcriptional outputs [140]. In the epigenetic context, the most mechanistically specific protein-mediated pathway remains DNMT enzyme transfer (described in Section 4.3). For transcription factor cargo, the epigenetic consequence depends on whether the delivered transcription factor recruits chromatin-modifying complexes to its target loci, a plausible but incompletely characterized mechanism in most published studies. Where such recruitment has been demonstrated (e.g., STAT3-directed DNMT3A recruitment following sEVs PD-L1-mediated STAT3 activation [86,98]), the pathway qualifies as an indirect but mechanistically complete epigenetic reprogramming axis. In other cases, transcription factor delivery produces transcriptional changes through direct DNA binding without documented chromatin-state alterations, and these effects are therefore classified as signaling-mediated rather than epigenetic.
5.4. Exosomal DNA Transfer and Implications for Regulatory Heterogeneity
Double-stranded genomic DNA has been detected in circulating sEVs from cancer patients, and cancer-derived vesicles can carry mutant oncogene sequences capable of transferring functional information to recipient cells [140,141]. In hematological cancers, exosome-mediated DNA transfer has been proposed to contribute to tumor heterogeneity by distributing oncogenic elements across subclonal populations [141]. In the epigenetic context, it is important to distinguish between two distinct claims: (i) that transferred DNA carries methylation imprints (a well-supported observation reviewed in Section 4.2); and (ii) that such transfer functionally reprogram recipient-cell chromatin (a hypothesis without rigorous experimental support to date). Most evidence positions sEVs DNA primarily as a diagnostic substrate reflecting the epigenetic state of the donor cell rather than as a functional epigenetic reprogram of recipient cells. Until recipient-cell chromatin integration or methylation-pattern alteration downstream of sEVs DNA uptake is demonstrated, this mechanism should be characterized as a candidate pathway rather than an established one.
5.5. Metabolic Reprogramming as a Parallel Axis of State Remodeling That Intersects with Gene Regulation
sEVs-mediated metabolic reprogramming of recipient cancer and stromal cells contributes to angiogenesis, metastasis, drug resistance, immunosuppression, and TME remodeling [126]. The mechanistic link between sEVs metabolite cargo and recipient-cell epigenetics operates through the obligate metabolic dependency of chromatin enzymes described in Section 4.4: when tumour-derived sEVs deliver oncometabolites or metabolic reprogramming signals that deplete alpha-KG or elevate succinate/fumarate/2-HG in recipient cells, TET and KDM enzyme activity is inhibited, inducing global DNA hypermethylation and altered histone methylation states [115,116,123]. Hypoxia is a universal hallmark of solid tumors and a major driver of sEVs biogenesis and cargo composition: hypoxia-conditioned sEVs carry oncogenic ncRNAs, mutant proteins, and metabolites that reprogram stromal cells and prime pre-metastatic niches [142]. Multiple TME stresses hypoxia, inflammation, and nutrient deprivation modulate ncRNA expression, and sEVs externalize those stress-adapted ncRNA programmes to propagate adaptive states across cell populations [143,144]. Metabolic rewiring in PDAC is associated with mesenchymal plasticity and immune evasion through a convergent epigenetic-metabolic program [145], and mTOR-centered metabolic integration in immune cells provides a pathway-level rationale for why metabolically loaded sEVs signals can exert immune-epigenetic consequences at the tissue scale [145]. Collectively, Section 5.1, Section 5.2, Section 5.3, Section 5.4 and Section 5.5 support a multifaceted model in which sEVs deliver ncRNAs, proteins, DNA, and metabolic factors that converge on chromatin-modifying pathways in recipient cells. Through indirect epigenetic modulation (ncRNA-mediated suppression of chromatin enzymes), direct enzyme transfer, and metabolic co-substrate manipulation, sEV-mediated communication promotes phenotypic plasticity, tumor progression, and therapy resistance. The mechanistic completeness of these pathways, particularly the transition from post-transcriptional regulation to durable chromatin-state change, varies across cargo classes and requires explicit experimental validation in each context (Figure 2).
6. Functional Outcomes of Exosome-Driven Regulatory Rewiring Across Cancers
The molecular mechanisms described in Section 4 and Section 5 manifest as tissue-scale phenotypic outcomes that define the biology of cancer progression and therapy resistance. This section maps the key emergent phenotypes, angiogenesis, immune rewiring, stromal activation, metabolic adaptation, and therapy resistance to the sEV-mediated epigenetic signaling axes that drive them. A critical interpretive caveat is maintained throughout mechanistic principles that appear conserved across cancer types are distinguished from cancer-specific evidence that should not be generalized without independent validation in the relevant cellular, stromal, and genomic context. EV diversity further complicates cross-study comparison: some studies operationally analyze sEVs while referring to them as sEVs, and mechanistic conclusions about cargo-sorting or recipient-targeting specificity are contingent on the vesicle identity confirmed by each study [147,148].
A critical interpretive caveat applies throughout this section: while sEVs-mediated epigenetic signaling is emerging as a pan-cancer phenomenon, the mechanistic details, including cargo selection specificity, recipient-cell uptake efficiency, stromal context-dependency, and chromatin-state prerequisites, can differ substantially across tumor types. The evidence reviewed here supports several mechanistic principles that appear to be conserved across malignancies: (i) ESCRT- and ceramide-dependent selective miRNA sorting into exosomes; (ii) lncRNA-scaffolded PRC2 recruitment leading to H3K27me3 deposition; (iii) oncometabolite-mediated TET/KDM inhibition inducing DNA hypermethylation; and (iv) sEVs PD-L1-driven T-cell exhaustion with DNMT3A-dependent effector gene silencing. In contrast, cancer-specific mechanisms, such as HOTAIR-EZH2 cisplatin resistance in ovarian cancer, miR-365-mediated gemcitabine resistance in PDAC, or miR-141-3p-driven JAK/STAT3 angiogenesis in ovarian cancer, should not be generalized to other tumor types without independent validation in the relevant cellular and stromal contexts. This distinction is maintained throughout the following sections.
6.1. Exosomes as Mediators of Angiogenesis and Vascular Remodeling
Tumor-derived sEVs are established mediators of angiogenesis through the transfer of pro-angiogenic ncRNAs and proteins to endothelial recipient cells. In epithelial ovarian cancer, sEVs enriched for miR-141-3p activate JAK/STAT3 and NF-kappaB signaling pathways in endothelial cells, driving neovascularization within the tumor microenvironment [148,149]. JAK/STAT3 and NF-kappaB are canonical inflammatory-angiogenic hubs; their activation by sEVs miRNA constitutes a signaling-level rewiring event that, when sustained, converges on chromatin-state changes at angiogenic gene loci through STAT3-mediated co-recruitment of chromatin-modifying complexes pathway with Level 2-3 evidence in the ovarian cancer context [150].In colorectal cancer (CRC), sEVs are established mediators of tumor angiogenesis and immune evasion, illustrating how vesicle-driven signals produce coordinated, multi-compartment outputs rather than isolated single-cell effects [150,151]. VEGF/VEGFR-targeted therapies and immune checkpoint combinations that engage the tumor vascular compartment are therapeutically relevant downstream of these sEVs signaling axes in NSCLC and gastrointestinal cancers [149].An important distinction applies: the evidence that sEVs miRNAs activate JAK/STAT3 signaling in endothelial cells is well supported (Level 2-3). The inference that this produces durable epigenetic reprogramming of the endothelial transcriptome rather than transient signaling activation requires chromatin-level readouts (ChIP-seq for H3K27ac or H3K4me3 at angiogenic gene enhancers in sEVs -treated endothelial cells) that have not been systematically provided. The angiogenic outcome is therefore classified as primarily signaling-mediated with epigenetic consequences that remain to be quantified.
6.2. Exosomes as Mediators of Immune Rewiring
sEVs-mediated immune rewiring in the TME operates through multiple epigenetic and post-transcriptional mechanisms that collectively establish immunosuppressive chromatin states in infiltrating lymphocytes and reprogram macrophage polarization. Macrophage reprogramming: TME-cargo modulates tumor-associated macrophage (TAM) phenotype via at least two mechanisms: (i) exosomal miRNAs delivered to macrophages engage RISC and suppress anti-inflammatory target mRNAs, promoting M2 polarization [152,153]; and (ii) m6A-modified lncRNAs in tumor-derived EVs recruit YTHDF1 in recipient macrophages, altering polarization state through post-transcriptional and potentially chromatin-level mechanisms [105]. In HCC, knockdown of tumor-secreted exosomal PSMA5 reverses macrophage polarization and restrains disease progression by blocking JAK2/STAT3 signaling [154], providing Level 4 evidence for the macrophage-remodeling function of specific sEVs cargo. TAMs are the most abundant immune cell type in many solid tumors, and their exosome-mediated epigenetic reprogramming represents a therapeutically accessible axis: Wnt/beta-catenin signaling in dendritic cells [155] and mTOR-centered metabolic regulation in T cells [146] provide parallel pathway nodes through which sEV cargo can alter anti-tumor immunity. T-cell exhaustion: sEVs PD-L1, membrane-anchored on the outer leaflet of tumor-derived EVs, suppresses T-cell receptor signaling and induces T-cell exhaustion in draining lymph nodes and the TME, constituting a systemic immunosuppressive mechanism that actively antagonizes PD-1 blockade [156]. Mechanistically, sEVs PD-L1-mediated STAT3 activation recruits DNMT3A to silence granzyme B, perforin, and IFN-gamma loci in CD8+ T cells, establishing a heritable transcriptional silencing program that cannot be reversed by checkpoint antibody monotherapy [86]. This represents Level 3-4 evidence for exosome-driven immune-epigenetic reprogramming with direct clinical relevance, providing a mechanistic rationale for combining exosome biogenesis inhibitors (e.g., GW4869) with DNMT inhibitors and PD-1 blockade. Broader immunosuppressive architecture: Tumor exosomes transfer miRNAs that promote M2 polarization, expand regulatory T cells (Tregs), and suppress natural killer (NK) cell function [157]. sEVs crosstalk between tumor cells, CAFs, endothelial cells, and myeloid-derived suppressor cells (MDSCs) creates a self-reinforcing epigenetic ecosystem that amplifies tumor-promoting signals while systematically suppressing anti-tumor immunity [158].
6.3. Exosomes as Mediators of Stromal Activation and ECM/Mechanics
sEVs-driven stromal rewiring manifests as cancer-associated fibroblast (CAF) activation, extracellular matrix (ECM) remodelling, and altered tissue mechanics that collectively support invasion, immune exclusion, and therapy resistance. CAFs are critical TME components that exert their influence through canonical TGF-beta, Wnt, Notch, Hedgehog, Hippo, and PI3K/AKT/mTOR pathways [159]; sEVs cargo that activates these signaling nodes in fibroblasts constitutes an upstream epigenetic remodeling trigger in the stromal compartment. A direct exosome-to-stroma activation example is provided by a provisional report on salivary adenoid cystic carcinoma, where exosomal S100A9 promotes lung metastasis by activating CAFs [160]. Although this preprint is provisional pending peer review, it exemplifies the mechanistic template in which tumor-derived vesicles carry cargo that induces stable fibroblast activation programmes. In pancreatic ductal adenocarcinoma (PDAC), activated pancreatic stellate cells (PSCs) mediate paracrine signaling, metabolic reprogramming, and onco-immunology through mechanisms that overlap substantially with exosome-mediated stromal communication [161,162]. The epigenetic dimension of CAF activation, including TGF-beta-induced H3K4me3 and H3K27ac changes at fibroblast activation gene enhancers [163] provides a chromatin-level readout framework for future studies of exosome-driven stromal reprogramming. It is important to note that the PDAC literature cited here does not attribute stromal remodeling exclusively to exosomes; rather, paracrine communication and ECM mechanics are dominant organizing principles in tissue-scale tumor behavior for which exosomes represent one, not the only, communication modality [147,150,161,162].
6.4. Exosomes as Mediators of Metabolic and Stress Adaptation Rewiring
Hypoxia, a universal hallmark of solid tumors, directly drives exosome biogenesis, alters cargo composition, and shapes the epigenetic signaling landscape of secreted vesicles: hypoxia-conditioned sEVs carry oncogenic ncRNAs, mutant proteins, and oncometabolites that reprogram stromal cells and prime pre-metastatic niches [142]. This creates a feed-forward mechanism in which tumor microenvironmental stress amplifies sEVs epigenetic signaling to adapt both the tumor and its surrounding stroma. Multiple TME stresses hypoxia, inflammation, and nutrient deprivation modulate ncRNA expression through stress-responsive transcriptional programmes, and sEVs externalize those ncRNA programmes to propagate stress-adapted states across cell populations [143,144]. The epigenetic consequences in recipient cells are mediated through the pathways described in Section 4.4 and Section 5.5: oncometabolite-mediated TET/KDM inhibition, altered HAT/HDAC cofactor availability, and ncRNA-directed chromatin enzyme suppression. mTOR centered metabolic regulation in immune cells provides a pathway-level rationale for why metabolically loaded sEVs signals exert immune-epigenetic consequences at the tissue scale [146].
6.5. Exosomes as Mediators of Therapy Resistance and Adaptive Evolution
EVs are explicitly framed as modulators of cancer-cell adaptive responses to therapy, indicating that vesicle-mediated communication participates in non-genetic adaptation processes that undermine treatment [164]. This adaptive dimension is conceptually distinct from the intrinsic epigenetic differences between drug-resistant and drug-sensitive cells: rather, exosome-mediated resistance operates through the propagation of resistance-associated epigenetic programmes from resistant minority subpopulations to the broader tumor [165,166]. Cross-species proteomics in tumor xenografts demonstrates that PI3K inhibitor treatment differentially reshapes signaling in cancer and stromal cells [167], underscoring that therapeutic pressure acts on multiple compartments simultaneously. EV exchange can plausibly distribute and stabilize these drug-adapted signaling states across tumor and stromal compartments, promoting collective resistance and adaptive evolution [150,165,167]. Drug-induced remodeling of exosome secretion and cargo composition provides a mechanistic route for this distribution: 5-azacytidine (DNMT inhibitor) treatment alters the miRNA repertoire of leukaemic cell-derived exosomes and reprogrammed recipient-cell transcriptional profiles in ways that can circumvent drug action[168,169]; EZH2 inhibitor treatment reshapes the exosomal lncRNA landscape, enabling treated cells to re-establish H3K27me3 in adjacent untreated cells via vesicle-mediated transfer [170]; and HDAC inhibitor exposure enriches vesicles for acetylated histone fragments and heat-shock proteins that activate oncogenic programmes in neighboring cells [171]. These observations support a therapeutic rationale for co-targeting EV biogenesis, for example, with neutral sphingomyelinase inhibitors such as GW4869 alongside epigenetic agents, to interrupt intercellular propagation of adaptive resistance-associated chromatin states. Specific mechanistic pathways of exosome-mediated resistance (drug efflux, DNA damage repair, EMT, and immunosuppressive remodeling) are detailed in Section 9.
7. Exosome-Mediated DNA Methylation and Demethylation in Cancer
An expanding corpus of research characterizes exosomes not solely as inert byproducts of cellular metabolism but as dynamic facilitators of epigenetic reprogramming within the tumor microenvironment (TME). Fundamentally, DNA methylation, the covalent addition of a methyl group to the 5-carbon position of cytosine, predominantly at CpG dinucleotides, is the most extensively studied epigenetic modification and is critically important for the initiation, proliferation, and metastasis of many tumors [74,89,172]. In the realm of oncology, irregularities in DNA methylation profiles, characterized by widespread hypomethylation and localized hypermethylation at the promoters of tumor suppressor genes, serve as defining features of neoplastic progression [172,173]. sEVs are engaged in the modulation of DNA methylation through at least two significant mechanisms. Firstly, exosomes have the capability to transport methylated DNA fragments directly from donor cancer cells to recipient cells, thereby facilitating the transfer of the methylation "imprint" from the originating neoplasm. Qian et al. explicitly describe that extracellular vesicles can transmit proteins and nucleic acids that participate in DNA methylation, and that factors transmitted by EVs reflect the donor cell status [74]. This is supported by findings indicating that neoplastic cells disseminate hypermethylated DNA segments to adjacent cells through exosomes, resulting in the repression of tumor suppressor genes in the recipient cells [87]. Moreover, exosomes have the capacity to transport DNA methyltransferases (DNMTs) and regulatory RNAs that influence the methylation processes within target cells. Hu et al. note that exosomes serve as vehicles for delivering methyltransferases to recipient cells, and that exosomes derived from diverse tissues and cells exhibit varying quantities and sizes of DNA methyltransferases and small RNAs, signifying their distinct roles in epigenetic regulation [67].
Yet, a comprehensively described mechanism entails exosomal microRNAs that specifically engage DNA methyltransferases, thereby facilitating the process of DNA demethylation within recipient cellular populations. Several research investigations indicate that miR-29 family members and other miRNAs can inhibit mRNAs for DNA methyltransferases, resulting in global hypomethylation and overall activation of the genome, a hallmark of cancer [85]. In the context of cancer-associated fibroblasts (CAFs), a critical stromal component of the TME, CAF exosomal miR-29b has been shown to suppress DNA methyltransferase 3B (DNMT3B) in recipient tumor cells, relieving methylation-mediated repression and altering transcriptional profiles [174]. This process delineates a direct relationship between exosome-facilitated intercellular signaling and the epigenetic reconfiguration of the tumor transcriptome. Milk exosome research provides a translational parallel: bovine milk-derived exosomes contain miRNAs (notably miR-148a and miR-29b) that target DNMTs and influence the demethylation of promoter regions of metabolic regulators such as the fat mass and obesity-associated protein (FTO)[175,176]. While this was initially characterized in developmental biology, the same DNMT-targeting miRNA mechanism is directly relevant to cancer, where sEVs miRNA-mediated DNMT suppression can result in DNA demethylation and consequent activation of oncogenic or tumor-suppressive gene programs [85,175]. A complementary and mechanistically distinct axis of exosome-mediated DNA methylation control involves the TET family of dioxygenases, such as TET1, TET2, and TET3, which catalyze iterative oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), and ultimately to unmodified cytosine, thereby executing active DNA demethylation [177]. In the sphere of oncogenesis, TET enzymes are often susceptible to mutations or transcriptional inhibition, producing a notable decrease in the aggregate levels of 5-hydroxymethylcytosine, which is acknowledged as a nearly universal epigenetic indicator of neoplastic transformation [117]. sEVs cargo can modulate this axis bidirectionally. On one hand, tumor-derived exosomes enriched for oncometabolites such as 2-hydroxyglutarate (2-HG), produced by mutant IDH1/2 enzymes, can competitively inhibit TET activity in recipient stromal and immune cells, inducing hypermethylation programs that promote immune exclusion [178]. The translational relevance of this mechanism is underscored by studies demonstrating that 5hmC levels in circulating cell-free DNA and in sEVs DNA fragments serve as sensitive diagnostic and prognostic biomarkers across multiple cancer types, with tissue-of-origin specificity sufficient for multi-cancer early detection [179]. Future studies should characterize how sEVs cargo shapes the TET-5hmC landscape in recipient cells, particularly in immune and stromal compartments, where demethylation-associated transcriptional reactivation could determine the balance between immune activation and suppression within the TME.
8. Exosome-Mediated Histone Modification in Cancer
Changes in histone post-translational modifications, such as acetylation, methylation, phosphorylation, ubiquitination, and sumoylation, emphasize a vital aspect of epigenetic control that affects chromatin structure and the availability of genes [74,89,176]. Histone acetylation, modulated by the enzymatic activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs), mitigates the positive charge of histone proteins, consequently facilitating the relaxation of compact chromatin architecture to enhance the process of gene transcription. The neoplastic microenvironment may consequently exert an influence on the epigenetic modulation of oncogene expression through the mechanism of histone modification [74]. However, as elucidated by Qian et al., the role of extracellular vesicles in the mechanism of histone modification continues to be a topic of significant contention [74]. Sharma et al. used bioinformatic analysis and observed an impressive overlap between genes relevant to transgenerational epigenetic inheritance and the cargo of exosomes released by different cell types, including cancer cells, but direct mechanistic proof of exosome-mediated histone modification transfer remains an active area of investigation [114]. In light of the persistent discourse, a mounting collection of evidence spanning various cancer types confirms multiple mechanistic routes by which exosomes impact histone modification dynamics in recipient cellular structures. Exosomes possess the capability to transport or affect the functionality of histone-modifying enzymes within recipient cellular environments. In the context of T cell dysfunction in the TME, studies indicate that many mRNAs and proteins contained in EVs are involved in epigenetic regulation of immune cell functions, including histone modification [112]. Histone acetylation is acknowledged as a significant area of investigation within post-translational modifications, shaped by the conflicting activities of HATs and HDACs [112]. HDACs exhibit aberrant regulation in numerous malignancies, positioning them as viable therapeutic targets; notably, HDAC3 is indispensable for T cell maturation, whereas HDAC1 and HDAC2 are instrumental in facilitating appropriate thymic development [112]. Tumor-derived EVs that modulate HDAC expression or activity in immune cells could therefore reshape the immune landscape of the TME through histone modification-dependent mechanisms.
A principal mechanism through which exosomes exert influence on histone modification is via the transfer of non-coding RNAs, especially long non-coding RNAs (lncRNAs), which serve to recruit chromatin-modifying complexes to designated genomic sites. The varied assortment of long non-coding RNAs contains transcripts that facilitate fundamental cellular functions through their engagement with proteins, chromatin, and RNA molecules themselves [173]. An important regulatory aspect of lncRNAs is their association with the epigenetic machinery and the recruitment of its regulatory apparatus to specific loci, resulting in DNA methylation and/or post-translational modifications of histones [173]. Well-known representatives include the Polycomb Repressive Complex 2 (PRC2) and its catalytic component Enhancer of Zeste Homolog 2 (EZH2), which promote H3K27 methylation, and the Lysine Acetyltransferase (KAT) and HDAC families that mediate histone acetylation and deacetylation, respectively [173]. Several lncRNAs that epigenetically regulate cancer cells through chromatin modification mechanisms are part of the sEVs cargo secreted from tumors [173]. sEVs lncRNAs have been deemed important regulators of gene expression owing to their associations with transcription factors or enzymes that modify chromatin [89]. In gastric cancer specifically, histone deacetylases (HDACs) can repress the expression of tumor suppressor lncRNAs and microRNAs, while histone methyltransferases and demethylases modulate the expression of oncogenic lncRNAs and microRNAs, influencing cancer progression [89]. The bidirectional relationship where histone modifications regulate sEVs ncRNA content, and sEVs ncRNAs in turn modulate histone marks in recipient cells, creates a self-reinforcing epigenetic communication loop. A distinctive example of histone changes associated with exosomes in cancer contexts pertains to the epigenetic repression orchestrated by EZH2. In the context of tumor-derived exosomes and pre-metastatic niche formation, trimethylation of H3K27 and the inhibition of PTEN expression via EZH2-mediated epigenetic silencing have been described [180].
While the lncRNA-scaffolding model whereby sEVs lncRNAs delivered to recipient cells recruit the Polycomb Repressive Complex 2 (PRC2) to specific genomic loci, leading to H3K27me3 deposition, is mechanistically well-supported by studies of HOTAIR and MALAT1 function in cancer cells [95,98], the direct transfer of assembled, catalytically competent PRC2 or NuRD complexes via exosomes remains an unresolved and important question. Intact multi-subunit complexes require the assembly of 3-7 protein subunits for catalytic activity [113], and whether such assemblies can survive the intraluminal EV milieu and be delivered in enzymatically competent form to recipient cells has not been rigorously demonstrated. The relative quantitative contributions of (i) lncRNA-templated PRC2 nucleation within recipient cells versus (ii) direct transfer of EZH2 monomer as a single-subunit cargo are experimentally unresolved. Single-subunit epigenetic enzymes such as DNMT3A or EZH2 in isolation represent mechanistically more plausible EV cargo candidates, as their functional delivery requires only protein folding fidelity rather than multi-protein complex reassembly in the recipient cytoplasm. Future studies employing proximity ligation assays, co-immunoprecipitation within isolated EVs, and single-vesicle proteomics should prioritize resolving this hierarchy. Until such validation is provided, exosomal transfer of intact PRC2 should be regarded as a hypothesis rather than an established mechanism.
9. Exosome-Mediated Epigenetic Therapy Resistance
The reality of resistance to chemotherapeutic agents and targeted therapeutic modalities continues to pose a major clinical impediment within the sphere of oncology, with exosome-mediated epigenetic mechanisms increasingly recognized as critical players in the shortcomings of therapeutic interventions [48]. sEVs cargo orchestrates resistance through interconnected pathways: drug efflux, DNA damage repair, metabolic reprogramming, apoptosis evasion, and maintenance of cancer stem cell properties. Among these cargo classes, sEVs miRNAs are especially influential in modulating gene expression to diminish therapeutic efficacy [132]. An adaptive dimension of resistance involves drug induced remodeling of exosome secretion and cargo composition: exposure to the DNMT inhibitor 5 azacytidine (5 AZA) alters the miRNA repertoire of leukemic cell derived exosomes and thereby reprograms recipient cell transcriptional profiles in ways that may circumvent drug action [168,169]. Likewise, treatment with EZH2 inhibitors (for example, tazemetostat) can provoke compensatory upregulation of alternative PRC2 subunits and reshape the sEVs lncRNA landscape, enabling treated cells to reestablish H3K27me3 marks in adjacent, untreated cells via vesicle-mediated transfer a form of epigenetic contagion that propagates resistance across tumor subpopulations [170]. HDAC inhibitor exposure has similarly been reported to increase exosome secretion and to enrich vesicles for acetylated histone fragments and heat shock proteins that activate oncogenic programs in neighboring cells [171]. Collectively, these observations support a therapeutic rationale for co targeting EV biogenesis (for example, with neutral sphingomyelinase inhibitors such as GW4869) alongside epigenetic agents to interrupt intercellular propagation of adaptive, resistance associated states.
9.1. Drug Efflux and Metabolic Resistance
sEVs miR-1246 secreted by ovarian cancer cells inhibit Caveolin-1 (Cav1) and upregulates ABCB1 (P-glycoprotein) expression in recipient cells, conferring a drug-resistant phenotype by enhancing efflux of chemotherapeutic agents[132,181]. Conversely, exosome-transmitted miR-128-3p downregulates MDR5 expression, thereby decreasing oxaliplatin efflux and restoring chemosensitivity in oxaliplatin-resistant colorectal cancer cells, illustrating the bidirectional regulatory potential of sEV miRNAs on transporter-mediated resistance [182]. Tumor-associated macrophage (TAM)-derived exosomes transfer miR-365 to pancreatic ductal adenocarcinoma (PDAC) cells, where it increases triphosphate nucleotide (NTP) levels that compete with phosphorylated gemcitabine for DNA incorporation and upregulates cytidine deaminase (CDA) to inactivate gemcitabine, generating gemcitabine resistance through a coordinated epigenetic and metabolic mechanism[183].
9.2. DNA Damage Repair and Apoptosis Evasion
sEVs miR-151a, transferred from temozolomide (TMZ)-resistant glioblastoma multiforme (GBM) cells to sensitive recipient cells, target XRCC4, a key mediator of non-homologous end joining (NHEJ) repair, and its downregulation activates DNA repair, conferring TMZ resistance in recipient cells. When researchers restored miR-151a in TMZ-resistant exosomes, resistance in recipient GBM cells was significantly attenuated, validating sEVs miRNA as both a mechanistic driver and a potential therapeutic target for glioblastoma chemoresistance [132]. Cancer-associated adipocyte (CAA) and CAF-derived sEVs miR-21 isomiRNAs bind to APAF1 in ovarian cancer cells, suppressing apoptosome formation and activation of caspase-9 and caspase-3, thereby conferring paclitaxel resistance, revealing a stromal-tumor epigenetic crosstalk that shapes therapeutic outcome in the TME [132].
9.3. Epithelial-Mesenchymal Transition, and Cancer Stem Cells
Epigenetic differences between cancer stem cells (CSCs) and non-CSCs, driven in significant part by epithelial-to-mesenchymal transition (EMT), underlie the plastic, therapy-resistant phenotype of CSCs [132]. sEVs miR-32-5p promotes multidrug resistance in HCC by activating the PI3K/AKT pathway to drive EMT and angiogenesis, while sEVs miR-155, enriched in vesicles secreted by both CSCs and drug-resistant cells, downregulates E-cadherin and upregulates mesenchymal biomarkers in recipient non-CSC cells, triggering the EMT process and conferring drug resistance upon previously sensitive cells [132]. In the framework of pancreatic carcinoma, gemcitabine-resistant cancer stem cells (CSCs) facilitate the transfer of miR-210-enriched exosomes to gemcitabine-sensitive cellular entities, thereby promoting the dissemination of chemoresistance among the neoplastic cell population [133]. The epigenetic mechanisms mediated by exosomes exemplify a phenomenon of intercellular "resistance contagion," in which a minority of cells exhibiting drug resistance possess the capability to epigenetically reconfigure the larger tumor microenvironment.
9.4. Immunosuppressive Epigenetic Remodeling by Tumor-Derived Exosomes
Resistance to anti-PD-1/PD-L1 immune checkpoint therapy remains a major unresolved clinical problem because exosomal PD-L1, when membrane-anchored on the outer leaflet of tumor-derived EVs, suppresses T-cell receptor signaling and induces T-cell exhaustion in the tumor-draining lymph nodes and TME, thereby constituting a systemic immunosuppressive mechanism that actively antagonizes PD-1 blockade [156]. Mechanistically, this effect is accompanied by epigenetic silencing of T-cell effector genes: exosomal PD-L1 activates STAT3 in CD8+ T cells, which recruits DNMT3A to silence granzyme B, perforin, and IFN-γ loci, thereby establishing a heritable transcriptional silencing program in exhausted T cells that cannot be reversed by checkpoint antibody monotherapy [86] These findings provide a mechanistic rationale for combining exosome biogenesis inhibitors (e.g. GW4869) with epigenetic reprogramming agents (such as DNMT inhibitors, TET activators) alongside PD-1 blockade to address both the extrinsic immunosuppressive EV signal and the cell-intrinsic epigenetic silencing it induces. Additionally, tumor-derived exosomes are active exploiters of epigenetic mechanisms that remodel the immune landscape of the TME in favor of immune evasion: exosomal PD-L1 is a prototypic immune checkpoint molecule that induces resistance to paclitaxel by activating the STAT3/miR-21/PTEN/Akt signaling axis in esophageal cancer cells, while simultaneously suppressing cytotoxic T-lymphocyte (CTL) activity by engaging PD-1 on T cells, a dual epigenetic and immunosuppressive effect[184,185]. Tumor exosomes transfer miRNAs that promote M2 macrophage polarization, expand regulatory T cells (Tregs), and suppress natural killer (NK) cell function, collectively establishing an immunosuppressive microenvironment that facilitates cancer progression and attenuates immunotherapy responses [157]. The intercellular transfer of epigenetic cargo within the TME extends to exosomal crosstalk between tumor cells and cancer-associated fibroblasts (CAFs), endothelial cells, and myeloid-derived suppressor cells (MDSCs), which creates a self-reinforcing epigenetic ecosystem in which tumor-promoting signals are amplified and propagated throughout the tumor stroma while anti-tumor immunity is systematically suppressed [158].
9.5. Radiotherapy Resistance and Epigenetic Bystander Effects via Exosomes
sEVs have been implicated in radiation-induced bystander effects (RIBE), whereby irradiated cells transmit epigenetic signals to non-irradiated neighboring cells via sEVs miRNA and lncRNA cargo, resulting in radiotherapy resistance and DNA damage responses in distant cells [186]. This mechanism holds considerable relevance in the discourse of tumor heterogeneity, where a designated population of radioresistant cells exhibits the capability to epigenetically transmit resistance to the broader tumor community through exosomal communication, thereby thwarting the intended therapeutic efficacy of ionizing radiation.
9.6. Cisplatin and Platinum-Based Resistance via Exosomal Epigenetic Crosstalk
Platinum-based chemotherapy (cisplatin, oxaliplatin, carboplatin) forms the backbone of treatment across multiple major cancers, and cisplatin resistance remains a leading cause of treatment failure in ovarian, bladder, head-and-neck, and non-small cell lung cancers. Exosome-mediated epigenetic resilience to platinum-based therapeutics functions through multiple converging mechanisms. Cancer-associated fibroblast (CAF)-derived exosomes enriched for miR-21-5p have been shown to silence PTEN and PDCD4 in epithelial ovarian cancer cells, activating AKT/PI3K signaling and conferring cisplatin resistance through inhibition of caspase-3-dependent apoptosis [187]. Additionally, cisplatin-resistant ovarian cancer cells overexpress HOTAIR, which sequesters miR-138-5p and thereby relieves miR-138-5p-mediated suppression of EZH2 and SIRT1; the resulting elevation of EZH2 catalytic activity deposits H3K27me3 at pro-apoptotic loci, including DAPK and PTEN, propagating cisplatin resistance across previously sensitive tumor cells [135,136]. Knockdown of HOTAIR restores miR-138-5p expression, suppresses EZH2 and SIRT1, and re-sensitizes ovarian cancer cells to cisplatin in vitro and in vivo, providing a pharmacological rationale for combining EZH2 inhibitors (such as tazemetostat, GSK126) with platinum-based chemotherapy in HOTAIR-high tumors [135,136].
10. Translational Landscape
10.1. Therapeutic Strategies Targeting Exosome-Mediated Epigenetic Pathways
The clinical advancement of engineered exosome therapies requires rigorous characterization of pharmacokinetic (PK) and pharmacodynamic (PD) parameters that remain poorly defined for most EV-based modalities. Electroporation is the most widely studied active loading method for nucleic acid cargo, achieving loading efficiencies for miRNAs of approximately 10-25% per loading cycle, with significant batch-to-batch variability driven by RNA aggregation on the exosomal surface and disruption of vesicle integrity at high field strengths [188]. Co-incubation (passive loading) achieves substantially lower efficiencies (~2-5%) but preserves membrane integrity and is more scalable. Novel membrane-active loading approaches including saponin-mediated transient permeabilization and click chemistry-based surface conjugation, are emerging as alternatives with improved efficiency and reproducibility [189]. In terms of pharmacokinetics, intravenously administered exosomes demonstrate rapid hepatic and splenic clearance (half-life: 15-30 minutes in murine models), with preferential accumulation in Kupffer cells and sinusoidal endothelium rather than tumor tissue in the absence of surface targeting modifications [190]. Surface decoration with tumor-homing peptides (such as iRGD, GE11, anti-EGFR nanobodies) significantly improves tumor biodistribution but adds manufacturing complexity and potential immunogenicity. Regarding safety, the inherent miRNA pleiotropy, individual miRNAs regulate hundreds of target mRNAs, which raises the risk of off-target epigenetic effects in non-tumor tissues; systematic off-target profiling using RNA sequencing and ATAC-seq in tissues of accumulation (liver, spleen, lung) should be standard in preclinical IND-enabling packages for any exosome-miRNA therapeutic candidate[191]. The immunogenicity of allogeneic exosome preparations, particularly those derived from non-autologous mesenchymal stem cells (MSCs), may trigger complement activation or adaptive immune responses that accelerate clearance and reduce therapeutic efficacy, necessitating immunological profiling as part of quality control [192]. The varied and sophisticated functions of exosomes in the epigenetic control of cancer have disclosed a vast range of treatment possibilities. These can be broadly categorized into: (a) inhibition of exosome biogenesis and secretion, (b) engineering of exosomes as drug delivery platforms, (c) loading of tumor-suppressive miRNAs, siRNAs, and epigenetic reprogramming agents, and (d) exosome-based cancer immunotherapy. Figure 3.
10.1.1. Inhibition of Exosome Biogenesis and Release
Given that cancer cells secrete exosomes at markedly elevated levels compared to normal cells, pharmacological inhibition of exosome biogenesis and release represents an attractive strategy for blocking the intercellular epigenetic communication that sustains tumor progression [69]. The neutral sphingomyelinase 2 (nSMase2) inhibitor GW4869 impedes both the biosynthesis and release of exosomes in multiple cancer cell types, including ovarian cancer and prostate cancer, and has been shown to enhance sensitivity to trastuzumab in HER2-overexpressing breast cancer cells and to reverse paclitaxel resistance conferred by exosomal PD-L1 in esophageal cancer [193]. GW4869 has additionally been shown to enhance the efficacy of PD-L1 checkpoint blockade by stimulating CTL activity in a mouse melanoma model, highlighting the potential for combining exosome inhibitors with immune checkpoint therapies [69]. RAB GTPases, which regulate exosome biogenesis, represent additional pharmacological targets. Tipifarnib, a pharmacological substance aimed at RAB27A, effectively diminishes the exosome count and the expression of PD-L1 across various cancer cell lines, thereby enhancing susceptibility to sunitinib [194]. Knockout of RAB27A in B-cell lymphoma enhances sensitivity to chemo-immunotherapy regimens, validating the utility of targeting exosome secretory machinery as a means of overcoming resistance[194].
10.1.2. Engineered sEVs as Delivery Vehicles for Epigenetic Cargo
Exosomes exhibit intrinsic properties that make them uniquely suited as drug delivery vehicles: they display low toxicity and immunogenicity, excellent biodegradability, efficient tissue-targeting capabilities, the ability to cross physiological barriers, including the blood-brain barrier, and a double-membrane structure that protects encapsulated cargo from enzymatic degradation[69]. These properties are not confined to neural applications: the capacity of exosomes to traverse restrictive anatomical barriers, including the corneal epithelium, blood-retinal barrier, and conjunctival membrane, further validates their universal potential as delivery platforms for epigenetic cargo in compartmentalized disease sites where conventional nanoparticles fail to achieve therapeutic concentrations[195]. These properties position engineered exosomes as a compelling alternative to synthetic nanoparticle delivery systems, particularly for the delivery of nucleic acid-based epigenetic therapeutics that are vulnerable to degradation in the systemic circulation[69]. A range of techniques has been established for the incorporation of therapeutic agents into exosomes, encompassing co-incubation, electroporation, sonication, extrusion, and endogenous loading achieved via genetic alteration of donor cells [69]. Electroporation has demonstrated particular utility for loading miRNAs and siRNAs, with expanded natural killer cells (eNK-EXO) derived exosomes loaded with cisplatin via electroporation enhance its cytotoxic effect against drug-resistant ovarian cancer (OC) cells, and umbilical cord blood-derived macrophage exosomes loaded with cisplatin exhibiting greater potency against cisplatin-resistant ovarian cancer cells than free cisplatin[196,197]. BMSC-derived exosomes loaded with doxorubicin via extrusion show enhanced efficacy against osteosarcoma with fewer side effects than the free drug, while exosome-delivered cisplatin evades endosomal trapping through clathrin-independent endocytosis, achieving more uniform cytosolic distribution [198].
10.1.3. Delivery of Tumor-Suppressive miRNAs and Epigenetic Reprogramming Agents
The delivery of tumor-suppressive miRNAs via engineered exosomes represents one of the most actively investigated therapeutic strategies in cancer epigenetics. Exosomes derived from bone marrow stem cells, which are encapsulated with miR-22-3p, impede the proliferation of human melanoma cells by specifically targeting galectin-1 (LGALS1) and subsequently inhibiting epithelial-mesenchymal transition (EMT) [199]. NSCLC serum-derived exosomes loaded with miR-126 suppress tumor growth by reducing integrin α-6 expression, while lung cancer-derived exosomes carrying miR-563 inhibit cancer cell proliferation, invasion, and apoptosis with acceptable tissue-level safety profiles[200,201]. HEK293T-derived exosomes carrying miR-34a suppress oral squamous carcinoma cell proliferation by reducing SATB2 expression, and exosomes loaded with miR-317b-5b suppress lung cancer proliferation and induce apoptosis [202,203]. Exosomal siRNA delivery has also demonstrated strong therapeutic potential. Glioma cell-derived exosomes loaded with PDGFRβ siRNA suppress glioma cell proliferation via the PI3K/Akt/EZH2 pathway and display no appreciable toxicity with high targeting specificity [69]. NSCLC-derived exosomes loaded with PD-L1 siRNA via electrostatic interaction induce apoptosis with enhanced PD-L1 downregulation and low toxicity compared to controls, while BMSC-derived exosomes loaded with galectin-9 siRNA suppress pancreatic cancer proliferation by enhancing CTL activity and inhibiting Treg function through M1 macrophage polarization [204,205]. Of particular significance, MSC-derived exosomes carrying KrasG12D siRNA are currently being evaluated in a Phase I clinical trial (NCT03608631) in patients with metastatic pancreatic cancer harboring the KrasG12D mutation, with overall survival and progression-free survival as primary outcomes [206].
10.1.4. Exosome-Based Cancer Immunotherapy
The immunomodulatory characteristics of exosomes have been strategically employed in the formulation of exosome-derived cancer vaccines and immune-enhancing therapeutic interventions. Exosomes originating from dendritic cells (DCs), especially those activated by interferon-gamma (IFN-γ-Dex), facilitate natural killer (NK) cell-mediated anti-tumor immune responses via the activation of the NKp30 signaling pathway. A Phase II clinical trial of IFN-γ-Dex in 22 patients with NSCLC demonstrated disease stabilization in 64% of participants and partial response to platinum-based chemotherapy in 36% following treatment cessation, validating the therapeutic potential of exosome-based immunotherapy [207]. Chimeric exosomal tumor vaccines constructed from antigen-presenting cell (APC)-tumor chimeric cells are currently under evaluation in a Phase I trial in bladder cancer patients (NCT05559177), measuring clinical response rate, overall survival, and safety. A Phase I trial of plant exosome-encapsulated curcumin (NCT01294072) in 35 colon cancer patients is further evaluating the safety, tolerability, and immune-modulatory effects of this natural epigenetic agent delivered via the exosomal platform. Curcumin is generally regarded as a deterrent of DNMT enzymatic processes and a facilitator of histone modifications, and the incorporation of this compound within exosomes is expected to ameliorate its historically poor bioavailability while augmenting its epigenetic impacts in neoplastic tissues.
Figure 3.
Therapeutic targeting strategies for exosome-mediated epigenetic pathways. Diagram illustrating proposed intervention points to modulate exosome-driven epigenetic communication between tumor and recipient cells, where exosomes cargos transfer (miRNAs, lncRNAs, DNA, and proteins) are shown influencing epigenetic reprogramming in recipient cells. Panel (a); Biogenesis inhibition: Blocking exosome production/release using agents (e.g., GW4869) or genetic approaches (e.g., RAB27A targeting) to limit downstream effects Panel (b); Cargo interception: Neutralizing oncogenic exosomal contents (e.g., miRNAs) via antisense or RNA interference Panel (c); Engineered delivery: Using modified exosomes to deliver therapeutic cargos (tumor-suppressive miRNAs, epigenetic drugs) to reprogram aberrant states Panel (d); Combination immunotherapy: Integrating exosome-targeting with immune checkpoint blockade to enhance anti-tumor responses.
Figure 3.
Therapeutic targeting strategies for exosome-mediated epigenetic pathways. Diagram illustrating proposed intervention points to modulate exosome-driven epigenetic communication between tumor and recipient cells, where exosomes cargos transfer (miRNAs, lncRNAs, DNA, and proteins) are shown influencing epigenetic reprogramming in recipient cells. Panel (a); Biogenesis inhibition: Blocking exosome production/release using agents (e.g., GW4869) or genetic approaches (e.g., RAB27A targeting) to limit downstream effects Panel (b); Cargo interception: Neutralizing oncogenic exosomal contents (e.g., miRNAs) via antisense or RNA interference Panel (c); Engineered delivery: Using modified exosomes to deliver therapeutic cargos (tumor-suppressive miRNAs, epigenetic drugs) to reprogram aberrant states Panel (d); Combination immunotherapy: Integrating exosome-targeting with immune checkpoint blockade to enhance anti-tumor responses.

10.2. Translational Implications Across Cancers
As of early 2026, more than 100 clinical trials evaluating EV-based diagnostics or therapeutic platforms are registered on ClinicalTrials.gov, reflecting the rapid maturation of this field toward clinical application (ClinicalTrials.gov; accessed April 2026). Among those with explicit epigenetic endpoints, the MSC-KrasG12D siRNA exosome trial (NCT03608631, Phase I) stands as the most advanced therapeutic application, with safety and preliminary efficacy data establishing proof of concept for nucleic acid delivery via vesicle platforms in pancreatic cancer [206]. Regulatory agencies, including the FDA and EMA, are increasingly engaging with EV-based companion diagnostic concepts; however, jurisdiction-specific guidance documents tailored to EV-derived biomarker assays remain absent, and the classification of EV products varies substantially across regulatory regions from biologics in the United States to Advanced Therapy Medicinal Products (ATMPs) in Europe creating significant barriers to multi-regional trial design and commercialization[208], and harmonization of pre-analytical variables (sample type, storage, isolation method) across trial sites remains an unmet need [209]. Table 2.
10.2.1. Exosomal Biomarkers for Cancer Diagnosis and Prognosis
The diagnostic and prognostic utility of exosomal content in cancer, particularly exosomal non-coding RNAs, is one of the most rapidly advancing areas of translational oncology[210]. Because exosomes are shed into all body fluids, including blood, urine, saliva, and bile, and because their molecular cargo reflects the genomic and epigenomic state of their cell of origin, they constitute an accessible, non-invasive source of cancer biomarkers that can be profiled through liquid biopsy approaches [211]. In clinical validation studies, plasma exosomal miR-320d, miR-4479, and miR-6763-5p are significantly decreased in patients with epithelial cancers compared to healthy controls, while serum exosomal miR-122-5p, let-7d-5p, and miR-425-5p reliably distinguish HCC patients from healthy donors [69]. The expression levels of exosomal miR-21, miR-155, miR-182, and miR-373 in the serum of breast cancer patients are markedly elevated compared to healthy controls, and urinary exosomal lncRNAs distinguish bladder cancer patients from controls with high sensitivity by RNA sequencing [69]. The biomarker utility of EV-associated miRNA signatures extends beyond saliva to additional non-invasive biofluids. In the ophthalmological setting, EV-miRNA profiles in tears have been identified as sensitive, non-invasive biomarkers for corneal disorders, a precedent that demonstrates the feasibility of tissue-of-origin-specific EV-miRNA profiling across diverse biological fluids and supports the broader application of this approach to cancer liquid biopsy workflows[212]. For prognostication, low serum exosomal miR-134 predicts shorter overall and relapse-free survival in gastric cancer patients, while high levels of circulating exosomal LINC00963 predict poor prognosis in lung cancer[69]. In cholangiocarcinoma, high bile exosomal miR-200a-3p, miR-200c-3p, and serum exosomal miR-200c-3p levels predict poor prognosis, and low levels of circulating exosomal lncRNA-GC1 predict a favorable response to adjuvant chemotherapy in gastric cancer patients[69]. The collective findings elucidate a foundational structure for panels of exosomal epigenetic biomarkers that have the potential to inform clinical decision-making across a diverse array of cancer types. A particularly innovative advance is the nanoscale epigenetic profiling of individual exosomes derived from colorectal cancer cell lines using photo-induced force microscopy (PiFM), which has successfully distinguished CpG island methylator phenotype (CIMP)-high from CIMP-negative exosomes at the single-vesicle level[110]. This technique reveals heterogeneity among individual exosomes, suggesting the existence of distinct subpopulations with unique epigenetic profiles, a finding that may require single-vesicle resolution for clinically meaningful diagnostic precision [110].
10.2.2. Exosomal miRNAs as Predictors of Therapeutic Response
Together with their diagnostic and prognostic value, the examination of exosomal miRNA is slowly being recognized as an optimistic biomarker for evaluating therapeutic success. In a research investigation involving patients with triple-negative breast cancer (TNBC) who were subjected to neoadjuvant chemotherapy, microarray-based miRNA profiling discerned 16 distinctively expressed exosomal miRNAs differentiating between individuals achieving pathological complete response (pCR) and those not achieving pCR [213]. A combined signature of four miRNAs (miR-4448, miR-2392, miR-2467-3p, and miR-4800-3p) discriminated between pCR and non-pCR patients with an area under the curve (AUC) of 0.7652, establishing exosomal miRNA panels as viable tools for predicting chemotherapy sensitivity in aggressive breast cancer subtypes [213]. Exosomal miR-7-5p enhances the therapeutic efficacy of everolimus by inhibiting the MNK/eIF4E signaling axis, suggesting that high levels of this circulating exosomal miRNA could predict a favorable response to mTOR inhibitor-based regimens [69]. In NSCLC, plasma exosomal lnc-SNAPC5-3:4 is significantly upregulated when anlotinib treatment is effective, positioning it as a pharmacodynamic biomarker for anti-angiogenic therapy response [69]. These empirical findings suggest the potential for real-time epigenetic surveillance of therapeutic responses through the utilization of minimally invasive liquid biopsy techniques. Figure 4.
11. Challenges & Limitations
Several methodological limitations of this narrative review merit explicit acknowledgement. First, as described in the search strategy, this review was not prospectively registered and does not include the PRISMA flow diagram; eligibility decisions were made by the authors based on the mechanistic relevance, introducing the possibility of selection bias towards studies that support the conceptual framework presented. Second, the literature based on sEVs epigenetics is dominated by the positive mechanistic findings, and publication bias is likely inflated. Third, the rapidly evolving nature of the field means that studies published after April 2026 (such cutoffs are not represented). Readers should interpret the mechanistic synthesis herein as reflecting the current weight of the published evidence rather than a stable consensus.
Notwithstanding the substantial potential inherent in exosome-mediated epigenetic regulation for cancer therapies, numerous obstacles persist. The engagement of exosomes in histone modification remains an area of ongoing discussion, emphasizing the importance of conducting additional research to thoroughly clarify the methods by which sEVs contents shape the epigenetic environment of target cells [74]. A limitation is associated with determining the role of drugs in epigenetic mechanisms, issues concerned with specificity and sensitivity, and their role in establishing the prediction of response to treatment [214]. Understanding the role of epigenetic mechanisms and their targeting through drugs is an important area of research in cancer treatment [215]. Due to their fundamental ability for reversibility and dynamic nature, epigenetic modifications offer considerable therapeutic strategies, while particular epigenetic markers are undergoing research as potential biomarkers for early diagnosis and monitoring of therapeutic effectiveness.[216]. The results indicate that further research is imperative to elucidate the mechanisms underlying epigenetic modifications and to examine biomarkers, response rates, and customized treatment combinations [217]. In future developments, epigenetics has the potential to transform into a crucial element of diagnostic techniques, prognostic evaluations, and individualized treatment strategies [217].
Moreover, from a manufacturing standpoint, the limited yield of exosomes from biological sources is insufficient to meet therapeutic application requirements in preclinical and clinical studies, and ultracentrifugation, the current gold-standard isolation method, is labor-intensive, inefficient, and difficult to scale for Good Manufacturing Practice (GMP) grade production [68]. A persistent and under-reported caveat across the exosome-epigenetics literature is the disconnect between the EV doses used in mechanistic in vitro studies and the physiological exosome concentrations measured in patient biofluids. Multiple primary reports of miRNA-mediated phenotype reprogramming use vesicle preparations at 10⁹-10¹¹ particles/mL applied to 10⁵-10⁶ recipient cells [218], concentrations that exceed plasma levels by two to four orders of magnitude, and stoichiometric analyses indicate that most circulating sEVs miRNAs are present at less than one copy per vesicle [218]. Therefore, future mechanistic claims about sEVs miRNA-driven epigenetic reprogramming should be benchmarked against absolute miRNA copy quantification per vesicle and against EV doses calibrated to physiologically attainable concentrations in the recipient compartment. A second, equally consequential dimension of the standardization problem is the variability in how individual studies report or fail to report their EV preparations. The MISEV2023 consensus emphasizes that primary reports should disclose particle counts, protein content, and the presence of at least one transmembrane (such as CD9, CD63, CD81) and one cytosolic (such as TSG101, ALIX, syntenin-1) EV marker, alongside negative markers (such as calnexin, GM130) to exclude intracellular contaminants [64]. Compliance with these criteria across the exosome-epigenetics literature remains uneven, particularly in studies that infer functional cargo transfer from bulk EV preparations without orthogonal single-vesicle validation. Future mechanistic claims about exosomal epigenetic cargo should therefore be benchmarked against MISEV2023 reporting, and meta-analytic syntheses should pre-specify MISEV adherence as a quality criterion[64,66]. Additionally, standardization of exosome isolation, characterization, and cargo detection represents a critical unmet need. Beyond methodological standardization, the absence of unified global regulatory frameworks creates an equally significant translational barrier: regulatory pathways for EV therapeutics currently differ substantially between the FDA (biologics/IND pathway), EMA (ATMP classification), and Asian jurisdictions (source-based categorization), and no single international standard yet governs engineered EVs containing epigenetic cargo, necessitating region-specific regulatory strategies that substantially increase development costs and timelines[208]. The considerable congruence in dimensions between exosomes and alternative extracellular vesicles, coupled with the potential for contamination from lipoprotein aggregates during the production process, undermines the purity and consistency of exosome preparations [69]. Raw exosomes that remain in their native form are promptly eliminated from the bloodstream and demonstrate a tendency for preferential localization within various organ systems, such as the pulmonary, splenic, and hepatic tissues, which can lead to unintended biological consequences, reduced therapeutic efficacy, and the risk of organ-specific toxicity[68]. The surface engineering of exosomes, which entails the conjugation of tumor-specific targeting ligands, antibodies, or peptides, serves as a fundamental enhancement focused on optimizing tumor-targeted biodistribution while concurrently reducing systemic exposure [69]. The loading efficiency of nucleic acid cargo into exosomes using electroporation or sonication remains technically challenging. A parallel and equally consequential standardization gap exists at the analytical level: variability in EV-miRNA quantification arising from differences in isolation method, RNA extraction protocol, normalization strategy, and detection platform introduces significant inter-study variability that undermines the reproducibility of both biomarker discovery studies and functional mechanistic experiments[212].This quantification variability problem is not oncology-specific; it has been documented across EV-miRNA research in multiple disease contexts, and resolving it through universal analytical standards should be treated as a prerequisite for clinical-grade biomarker validation in cancer epigenetics. . Additionally, the inherent promiscuity of miRNAs, which typically regulate hundreds of target mRNAs simultaneously, raises concerns about off-target epigenetic effects and unintended phenotypic consequences when delivered systemically [69]. Future strategies must incorporate robust off-target profiling and miRNA dose optimization to minimize these risks.
12. Discussion & Future Directions
The evidence synthesized in this review establishes that exosome-mediated epigenetic communication is not a peripheral phenomenon in cancer biology but a central mechanism through which tumors coordinate malignant progression across cellular compartments [219]. Several unresolved controversies, however, deserve direct confrontation. First, the question of whether intact multi-subunit chromatin-modifying complexes, such as PRC2 or NuRD, are transferred in a catalytically competent form through exosomes remains an open and critical mechanistic question [113,134]. As we described in Section 8, single-subunit enzymes such as DNMT3A and EZH2 in isolation represent more plausible EV cargo than assembled multi-protein complexes [122,170]. Future studies should employ proximity ligation assays and single-vesicle proteomics specifically designed to assess the stoichiometry and enzymatic activity of transferred chromatin regulators[64,220], rather than inferring function from bulk EV preparations alone. Second, the stoichiometric problem identified in the Challenges section deserves emphasis here; most functional studies reporting miRNA-mediated epigenetic reprogramming use vesicle concentrations that exceed physiological plasma levels by two to four orders of magnitude[218]. Until mechanistic claims are benchmarked against absolute miRNA copy quantification per vesicle and against EV doses calibrated to physiologically attainable concentrations, the translational relevance of these findings remains provisional. We call on the field to adopt stoichiometric reporting as a standard requirement in primary mechanistic publications[64,209]. Third, the functional stratification encompassing the various classes of exosomal cargo, specifically non-coding RNAs, proteins, metabolites, and epitranscriptomic modifications, is yet to be elucidated [123,221]. The m6A epitranscriptome represents an especially underexplored dimension; METTL3-containing EVs and m6A-modified circRNAs (such as circNSUN2) have now been shown to carry functional epigenetic instructions that extend beyond canonical post-transcriptional regulation into chromatin remodeling in recipient cells[104,121]. A comprehensive examination of the manner in which m6A modification status affects cargo sorting, uptake efficiency, and the epigenetic ramifications for recipient cells constitutes a significant research priority.
On the translational frontier, numerous advancements are set to revolutionize the discipline. AI-driven multi-omics integration of exosomal miRNA profiles, cfDNA methylation signatures, and proteomics from liquid biopsies holds genuine promise for early cancer detection and real-time treatment monitoring[110,179], but only if the pre-analytical standardization problems outlined in Section 11 are resolved first. This analytical ambition requires as its foundation the resolution of a more basic technical problem: miRNA quantification variability across EV-miRNA studies remains sufficiently large that biomarker signatures identified in one laboratory frequently fail validation in independent cohorts [212]. AI-driven integration can amplify signal, but it cannot substitute for the upstream analytical harmonization, standardized isolation, extraction, normalization, and detection that must precede any clinically actionable multi-analyte panel. The convergence of CRISPR-dCas9 epigenome editing tools with exosomal delivery platforms represents the most conceptually transformative therapeutic approach on the horizon[171,206], enabling programmable, locus-specific epigenetic reprogramming without permanent genome modification. While this remains preclinical, proof-of-concept studies demonstrating exosomal delivery of catalytically inactive Cas9 fused to DNMT3A or TET1 domains should be prioritized [189,190]. The engineering of hybrid exosome-liposome nanoparticles to improve drug-loading capacity and circulation half-life addresses two of the most persistent barriers to clinical translation and merits accelerated preclinical evaluation. Complementary evidence from exosome-based drug delivery in ophthalmology, where the same low-toxicity, barrier-penetrating properties that make exosomes useful in oncology have been harnessed to overcome restrictive ocular membranes, reinforces the universality of the exosomal delivery platform and suggests that lessons from non-oncological applications should be systematically integrated into cancer epigenetics drug delivery research[195]. The regulatory landscape is evolving, but has not kept pace with scientific progress. Neither the FDA nor the EMA has issued guidance documents specific to EV-derived companion diagnostics, and clear regulatory frameworks for bioengineered EVs remain incomplete across all major jurisdictions[208]. Harmonization of pre-analytical variables, including sample collection, processing, storage, and isolation methods, across clinical trial sites is an unmet need that should be addressed through inter-institutional consortia and standardized biobanking protocols before Phase II/III trials are initiated [208].
This detailed analysis specifies the importance of exosome-mediated epigenetic reprogramming as an essential mechanistic convergence that links tumor biology, microenvironmental interactions, therapeutic resistance, and liquid biopsy diagnostics. Realizing its translational potential will require the field to move from descriptive cataloguing of cargo to functional validation under physiologically relevant conditions, from single-omic to integrated multi-omics analyses [66], and from unengineered to precision-targeted exosomal platforms. The next decade of research at this interface has the potential to yield a fundamentally new class of precision oncology agents.
Additionally, a key conceptual caveat in pan-cancer interpretation is that sEVs cargo selection, recipient cell uptake, stromal and immune context, and chromatin state dependency are all highly tumor-type specific. The cross-cancer mechanistic framework proposed in this review, wherein exosome-mediated transfer of miRNAs, lncRNAs, DNMTs, and metabolites broadly reprograms recipient-cell epigenomes, should be understood as a conceptual scaffold that identifies shared biological principles, not as evidence that identical mechanisms operate across all tumor types. Future mechanistic studies should be stratified by cancer type, stromal composition, and immune context to delineate which elements of this framework are conserved and which are tumor-specific.
13. Conclusions
This examination propounds a cohesive conceptual schema: exosomes originating from tumors operate not solely as inert vehicles for molecular remnants but as proactive orchestrators of the epigenetic milieu within the tumor microenvironment. Through the coordinated transfer of miRNAs, lncRNAs, circRNAs, DNA methyltransferases, histone-modifying enzymes, oncometabolites, and m6A-modified RNAs, exosomes reprogram recipient-cell chromatin states, silence tumor suppressors, induce drug efflux programs, exhaust cytotoxic immune cells, and propagate therapy resistance across heterogeneous tumor populations, all without altering the recipient genome. This mode of intercellular epigenetic communication constitutes a previously underappreciated dimension of cancer biology with direct implications for why tumors that appear genetically identical can behave so differently and respond so variably to treatment.
Three key questions necessitate resolution to actualize the translational efficacy of this discipline. First, can intact, catalytically competent epigenetic enzyme complexes survive the intraluminal exosomal milieu and be functionally delivered to recipient cells, or does epigenetic reprogramming occur primarily through exosomal ncRNA-templated nucleation of recipient-cell chromatin machinery? Second, do the exosomal miRNA concentrations achievable in vivo, which stoichiometric analyses suggest may be sub-copy-per-vesicle for many miRNAs, produce the phenotypic reprogramming reported in vitro at supraphysiological doses? Third, can engineered exosomes carrying CRISPR-dCas9 epigenome editing complexes achieve sufficient tumor biodistribution and on-target epigenetic reprogramming in vivo to warrant clinical investigation?
Realizing the translational potential of this field will require resolving the three foundational mechanistic questions identified above. Until these are answered, exosome-mediated epigenetic cancer therapeutics should be framed as a well-motivated, mechanistically grounded translational hypothesis; one with compelling preclinical foundations and early clinical signals, rather than an emerging therapeutic class approaching readiness. The concrete validation priorities that will determine whether this hypothesis can be translated into the clinic include: (i) dose-relevant functional assays using physiologically calibrated EV concentrations and stoichiometrically quantified cargo; (ii) single-vesicle cargo characterization by proximity ligation assay and sin-gle-vesicle mass spectrometry; (iii) orthogonal EV purification protocols combining immunoaffinity capture and size-exclusion chromatography; (iv) recipient-cell chromatin readouts (ATAC-seq, ChIP-seq, WGBS) directly paired with phenotypic assays; and (v) in vivo biodistribution studies with co-registered epigenetic endpoint measurements in tumor, immune, and stromal compartments. The convergence of single-vesicle analytical technologies, AI-driven multi-omics integration, and precision EV engineering places these validation experiments within near-term reach. It creates an unprecedented opportunity to deliver a new class of therapies that do not merely kill cancer cells but reprogram the epigenetic ecosystem that sustains them.
Author Contributions
Conceptualization, N.V.; writing—original draft preparation, N.V.; Investigation, N.V; writing—review and editing, S.A.; data curation, S.A.; visualization, S.A and K.C.; supervision, K.C.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
Swati Arora was employed by LP1 TSMS, Eli Lilly and Company. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript
| EV | Extracellular Vesicles |
| sEV | Small Extracellular Vesicles |
| MVBs | Multivesicular Bodies |
| TME | Tumor Microenvironment |
| EMT | Epithelial to Mesenchymal Transition |
| ECM | Extracellular Matrix |
| GMP | Good Manufacturing Practice |
| FDA | Food and Drug Administration |
| EMA | European Medicines Agency |
| IND | Investigational New Drug |
| TET | Ten Eleven Translocation proteins |
| CAF | Cancer Associated Fibroblasts |
| DNMT | DNA methyltransferase |
| mRNA | Messenger Ribonucleic Acid |
| miRNA | MicroRNA |
| lncRNA | Long noncoding RNA |
| MISEV | Minimal Information for Studies of Extracellular Vesicles |
| SYNCRIP | Synaptotagmin binding cytoplasmic RNA interacting protein |
| TEXs | Tumor derived exosomes |
| ncRNA | Noncoding RNA |
| VEGF | Vascular Endothelial Growth Factor |
| HAT | Histone Acetyltransferase |
| PRC2 | Polycomb Repressive Complex 2 |
| HDAC | Histone Deacetylase |
| ESCRT | Endosomal Sorting Complexes Required for Transport |
| CSCs | Caner Stem Cells |
| ESCRT | Endosomal Sorting Complexes Required for Transport |
| RISC | RNA Induced Silencing Complex |
References
- Yeat, N.Y.; Chen, R.H. Extracellular vesicles: biogenesis mechanism and impacts on tumor immune microenvironment. J Biomed Sci 2025, 32, 85. [CrossRef]
- Li, Q.; Wang, H.; Peng, H.; Huyan, T.; Cacalano, N.A. Exosomes: Versatile Nano Mediators of Immune Regulation. Cancers (Basel) 2019, 11, 1557. [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 2013, 200, 373-383. [CrossRef]
- Stoorvogel, W.; Kleijmeer, M.J.; Geuze, H.J.; Raposo, G. The biogenesis and functions of exosomes. Traffic 2002, 3, 321-330. [CrossRef]
- Qian, F.; Huang, Z.; Zhong, H.; Lei, Q.; Ai, Y.; Xie, Z.; Zhang, T.; Jiang, B.; Zhu, W.; Sheng, Y.; et al. Analysis and Biomedical Applications of Functional Cargo in Extracellular Vesicles. ACS Nano 2022, 16, 19980-20001. [CrossRef]
- Wei, H.; Chen, Q.; Lin, L.; Sha, C.; Li, T.; Liu, Y.; Yin, X.; Xu, Y.; Chen, L.; Gao, W.; et al. Regulation of exosome production and cargo sorting. Int J Biol Sci 2021, 17, 163-177. [CrossRef]
- Porcu, C.; Dobrowolny, G.; Scicchitano, B.M. Exploring the Role of Extracellular Vesicles in Skeletal Muscle Regeneration. Int J Mol Sci 2024, 25, 5811. [CrossRef]
- Cerrotti, G.; Buratta, S.; Latella, R.; Calzoni, E.; Cusumano, G.; Bertoldi, A.; Porcellati, S.; Emiliani, C.; Urbanelli, L. Hitting the target: cell signaling pathways modulation by extracellular vesicles. Extracell Vesicles Circ Nucl Acids 2024, 5, 527-552. [CrossRef]
- Di Bella, M.A. Overview and Update on Extracellular Vesicles: Considerations on Exosomes and Their Application in Modern Medicine. Biology 2022, 11, 804.
- Ragni, E. Extracellular Vesicles: Recent Advances and Perspectives. Front Biosci (Landmark Ed) 2025, 30, 36405. [CrossRef]
- Zhang, X.; Wu, Y.; Cheng, Q.; Bai, L.; Huang, S.; Gao, J. Extracellular Vesicles in Cardiovascular Diseases: Diagnosis and Therapy. Front Cell Dev Biol 2022, 10, 875376. [CrossRef]
- Han, Q.F.; Li, W.J.; Hu, K.S.; Gao, J.; Zhai, W.L.; Yang, J.H.; Zhang, S.J. Exosome biogenesis: machinery, regulation, and therapeutic implications in cancer. Mol Cancer 2022, 21, 207. [CrossRef]
- A. Alli, A. Mechanisms of Extracellular Vesicle Biogenesis, Cargo Loading, and Release. In Extracellular Vesicles - Role in Diseases, Pathogenesis and Therapy, Paul, M.K., Ed.; Physiology; IntechOpen: London, 2022.
- Lee, Y.J.; Shin, K.J.; Chae, Y.C. Regulation of cargo selection in exosome biogenesis and its biomedical applications in cancer. Exp Mol Med 2024, 56, 877-889. [CrossRef]
- Gurung, S.; Perocheau, D.; Touramanidou, L.; Baruteau, J. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Commun Signal 2021, 19, 47. [CrossRef]
- Russell, A.E.; Sneider, A.; Witwer, K.W.; Bergese, P.; Bhattacharyya, S.N.; Cocks, A.; Cocucci, E.; Erdbrugger, U.; Falcon-Perez, J.M.; Freeman, D.W.; et al. Biological membranes in EV biogenesis, stability, uptake, and cargo transfer: an ISEV position paper arising from the ISEV membranes and EVs workshop. J Extracell Vesicles 2019, 8, 1684862. [CrossRef]
- Li, M.; Fang, F.; Sun, M.; Zhang, Y.; Hu, M.; Zhang, J. Extracellular vesicles as bioactive nanotherapeutics: An emerging paradigm for regenerative medicine. Theranostics 2022, 12, 4879-4903. [CrossRef]
- Chang, W.-H.; Cerione, R.A.; Antonyak, M.A. Extracellular Vesicles and Their Roles in Cancer Progression. In Cancer Cell Signaling: Methods and Protocols, Robles-Flores, M., Ed.; Springer US: New York, NY, 2021; pp. 143-170.
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Extracellular vesicles: masters of intercellular communication and potential clinical interventions. J Clin Invest 2016, 126, 1139-1143. [CrossRef]
- Rezaie, J.; Akbari, A.; Rahbarghazi, R. Inhibition of extracellular vesicle biogenesis in tumor cells: A possible way to reduce tumorigenesis. Cell Biochem Funct 2022, 40, 248-262. [CrossRef]
- Sonar, S.; Nyahatkar, S.; Kalele, K. Tumor and cancer stem cells-derived exosomes interplay: A secret of cancer complications. Clinical and Translational Discovery 2024, 4, e325. [CrossRef]
- Yildirim, D.T.; Baki Yildirim, A.; Salzet, M.; Bertelli, M.; Beccari, T.; Prakash, S.; Pascucci, L.; Dundar, M. Messaging malignancy: Tumour-derived exosomes at the nexus of immune escape, vascular remodelling and metastatic competence. The EuroBiotech Journal 2025, 9, 216-237. [CrossRef]
- Sundararajan, V.; Sarkar, F.H.; Ramasamy, T.S. The multifaceted role of exosomes in cancer progression: diagnostic and therapeutic implications [corrected]. Cell Oncol (Dordr) 2018, 41, 223-252. [CrossRef]
- Wenxuan, P.; Qun, M.; Wenqian, Y.; Xiaobo, L.; Wencai, Y.; Dongmei, Z.; Lijuan, D.; Junqiu, Z.; Minfeng, C. The role and clinical applications of exosomes in cancer drug resistance. Cancer Drug Resistance 2024, 7, 43.
- Rezaee, M.; Mohammadi, F.; Keshavarzmotamed, A.; Yahyazadeh, S.; Vakili, O.; Milasi, Y.E.; Veisi, V.; Dehmordi, R.M.; Asadi, S.; Ghorbanhosseini, S.S.; et al. The landscape of exosomal non-coding RNAs in breast cancer drug resistance, focusing on underlying molecular mechanisms. Front Pharmacol 2023, 14, 1152672. [CrossRef]
- Ye, S.; Chen, S.; Yang, X.; Lei, X. Drug resistance in breast cancer is based on the mechanism of exocrine non-coding RNA. Discov Oncol 2024, 15, 138. [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209-249. [CrossRef]
- Gökalp, E.E.; Işık, S.; Artan, S. Introduction to Cancer Epigenetics. In Cancer Epigenetics, Kalkan, R., Ed.; Epigenetics and Human Health; Springer International Publishing: Cham, 2023; pp. 77-134.
- Oluwaseyi Ajibola, F.; Damilola Samuel, O.-o.; Samuel Imeh, B.; Festus Ikechukwu, O.; Onyeyili Ikemefuna, N.; Nyerovwo Charity, O.; Sunday, K. Epigenetic therapies in cancer treatment: Opportunities and challenges. World Journal of Biology Pharmacy and Health Sciences 2024, 20, 454-478. [CrossRef]
- Feng, S.; De Carvalho, D.D. Clinical advances in targeting epigenetics for cancer therapy. FEBS J 2022, 289, 1214-1239. [CrossRef]
- Jin, N.; George, T.L.; Otterson, G.A.; Verschraegen, C.; Wen, H.; Carbone, D.; Herman, J.; Bertino, E.M.; He, K. Advances in epigenetic therapeutics with focus on solid tumors. Clin Epigenetics 2021, 13, 83. [CrossRef]
- Karami Fath, M.; Azargoonjahromi, A.; Kiani, A.; Jalalifar, F.; Osati, P.; Akbari Oryani, M.; Shakeri, F.; Nasirzadeh, F.; Khalesi, B.; Nabi-Afjadi, M.; et al. The role of epigenetic modifications in drug resistance and treatment of breast cancer. Cell Mol Biol Lett 2022, 27, 52. [CrossRef]
- Pathak, S.; Tomar, S.; Pathak, A. Epigenetics and Cancer: A Comprehensive Review. Asian Pacific Journal of Cancer Biology 2023, 8, 75-89. [CrossRef]
- Yu, B.; Yu, X.; Xiong, J.; Ma, M. Methylation Modification, Alternative Splicing, and Noncoding RNA Play a Role in Cancer Metastasis through Epigenetic Regulation. Biomed Res Int 2021, 2021, 4061525. [CrossRef]
- Sadida, H.Q.; Abdulla, A.; Marzooqi, S.A.; Hashem, S.; Macha, M.A.; Akil, A.S.A.; Bhat, A.A. Epigenetic modifications: Key players in cancer heterogeneity and drug resistance. Transl Oncol 2024, 39, 101821. [CrossRef]
- Wang, N.; Ma, T.; Yu, B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct Target Ther 2023, 8, 69. [CrossRef]
- Rubatto, M.; Borriello, S.; Sciamarrelli, N.; Pala, V.; Tonella, L.; Ribero, S.; Quaglino, P. Exploring the role of epigenetic alterations and non-coding RNAs in melanoma pathogenesis and therapeutic strategies. Melanoma Res 2023, 33, 462-474. [CrossRef]
- Yang, J.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Epigenetic regulation in the tumor microenvironment: molecular mechanisms and therapeutic targets. Signal Transduct Target Ther 2023, 8, 210. [CrossRef]
- Prabhakaran, R.; Thamarai, R.; Sivasamy, S.; Dhandayuthapani, S.; Batra, J.; Kamaraj, C.; Karthik, K.; Shah, M.A.; Mallik, S. Epigenetic frontiers: miRNAs, long non-coding RNAs and nanomaterials are pioneering to cancer therapy. Epigenetics Chromatin 2024, 17, 31. [CrossRef]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat Rev Clin Oncol 2020, 17, 91-107. [CrossRef]
- Chaudhary, A.; Orchard, K.J.A.; Salani, F.; Partsou, T.; Eccleston, M.; Bocci, G.; Italiano, A.; Crea, F. Precision epigenetic therapies in oncology. Cancer Metastasis Rev 2025, 44, 71. [CrossRef]
- Liu, M.; Zhou, J.; Chen, Z.; Cheng, A.S. Understanding the epigenetic regulation of tumours and their microenvironments: opportunities and problems for epigenetic therapy. J Pathol 2017, 241, 10-24. [CrossRef]
- Andreescu, M. Epigenetic Alterations That Are the Backbone of Immune Evasion in T-cell Malignancies. Cureus 2024, 16, e51662. [CrossRef]
- Raz, D.J. Targeting Epigenetic Regulators in Cancer to Overcome Targeted Therapy Resistance. In Targeted Therapies for Lung Cancer, Salgia, R., Ed.; Current Cancer Research; Springer International Publishing: Cham, 2019; pp. 217-232.
- Hamid, F.F.H. Epigenetic Dysregulation and Cancer Progression: How Epigenetic Modifications Shape Tumor Fate. Health Innovation Reports 2025, 1, 55-59. [CrossRef]
- Ying, Z.; Wenjing, S.; Jing, B.; Songbin, F.; Kexian, D. Advances in long non-coding RNA regulating drug resistance of cancer. Gene 2023, 887, 147726. [CrossRef]
- Ren, B.; Li, X.; Zhang, Z.; Tai, S.; Yu, S. Exosomes: a significant medium for regulating drug resistance through cargo delivery. Front Mol Biosci 2024, 11, 1379822. [CrossRef]
- Khan, M.I.; Alsayed, R.; Choudhry, H.; Ahmad, A. Exosome-Mediated Response to Cancer Therapy: Modulation of Epigenetic Machinery. Int J Mol Sci 2022, 23, 6222. [CrossRef]
- Zhang, H.; Wu, B.; Wang, Y.; Du, H.; Fang, L. Extracellular Vesicles as Mediators and Potential Targets in Combating Cancer Drug Resistance. Molecules 2025, 30, 498.
- Shchegolev, Y.Y.; Sorokin, D.V.; Scherbakov, A.M.; Andreeva, O.E.; Salnikova, D.I.; Mikhaevich, E.I.; Gudkova, M.V.; Krasil'nikov, M.A. Exosomes are involved in the intercellular transfer of rapamycin resistance in the breast cancer cells. Bioimpacts 2023, 13, 313-321. [CrossRef]
- Ferrer-Diaz, A.I.; Sinha, G.; Petryna, A.; Gonzalez-Bermejo, R.; Kenfack, Y.; Adetayo, O.; Patel, S.A.; Hooda-Nehra, A.; Rameshwar, P. Revealing role of epigenetic modifiers and DNA oxidation in cell-autonomous regulation of Cancer stem cells. Cell Commun Signal 2024, 22, 119. [CrossRef]
- Masoudi-Khoram, N.; Soheilifar, M.H.; Ghorbanifar, S.; Nobari, S.; Hakimi, M.; Hassani, M. Exosomes derived from cancer-associated fibroblasts mediate response to cancer therapy. Crit Rev Oncol Hematol 2023, 185, 103967. [CrossRef]
- Nedaeinia, R.; Najafgholian, S.; Salehi, R.; Goli, M.; Ranjbar, M.; Nickho, H.; Haghjooy Javanmard, S.; G, A.F.; Manian, M. The role of cancer-associated fibroblasts and exosomal miRNAs-mediated intercellular communication in the tumor microenvironment and the biology of carcinogenesis: a systematic review. Cell Death Discov 2024, 10, 380. [CrossRef]
- Gou, Z.; Li, J.; Liu, J.; Yang, N. The hidden messengers: cancer associated fibroblasts-derived exosomal miRNAs as key regulators of cancer malignancy. Front Cell Dev Biol 2024, 12, 1378302. [CrossRef]
- Wandrey, M.; Jablonska, J.; Stauber, R.H.; Gul, D. Exosomes in Cancer Progression and Therapy Resistance: Molecular Insights and Therapeutic Opportunities. Life (Basel) 2023, 13, 2033. [CrossRef]
- Zhang, H.D.; Jiang, L.H.; Hou, J.C.; Zhong, S.L.; Zhu, L.P.; Wang, D.D.; Zhou, S.Y.; Yang, S.J.; Wang, J.Y.; Zhang, Q.; et al. Exosome: a novel mediator in drug resistance of cancer cells. Epigenomics 2018, 10, 1499-1509. [CrossRef]
- Hu, X.; Wen, Y.; Tan, L.Y.; Wang, J.; Tang, F.; Wang, Y.T.; Zheng, C.X.; Zhang, Y.Q.; Gong, T.J.; Min, L. Exosomal Long Non-Coding RNA ANCR Mediates Drug Resistance in Osteosarcoma. Front Oncol 2021, 11, 735254. [CrossRef]
- Cao, J.; Feng, B.; Xv, Y.; Yu, J.; Cao, S.; Ma, C. Continued attention: The role of exosomal long non-coding RNAs in tumors over the past three years. Int Immunopharmacol 2025, 144, 113666. [CrossRef]
- Salehi, M.; Kamali, M.J.; Arab, D.; Safaeian, N.; Ashuori, Z.; Maddahi, M.; Latifi, N.; Jahromi, A.M. Exosomal microRNAs in regulation of tumor cells resistance to apoptosis. Biochem Biophys Rep 2024, 37, 101644. [CrossRef]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol Cancer 2019, 18, 75. [CrossRef]
- Khan, N.; Umar, M.S.; Haq, M.; Rauf, T.; Zubair, S.; Owais, M. Exosome-encapsulated ncRNAs: Emerging yin and yang of tumor hallmarks. Front Genet 2022, 13, 1022734. [CrossRef]
- Xu, C.; Xu, P.; Zhang, J.; He, S.; Hua, T.; Huang, A. Exosomal noncoding RNAs in gynecological cancers: implications for therapy resistance and biomarkers. Front Oncol 2024, 14, 1349474. [CrossRef]
- Wang, S.; Jin, S.; Zhang, J.; Wang, X. Exosomal miRNAs: Key Regulators of the Tumor Microenvironment and Cancer Stem Cells. Int J Mol Sci 2025, 26, 9323. [CrossRef]
- Welsh, J.A.; Goberdhan, D.C.I.; O'Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrugger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J Extracell Vesicles 2024, 13, e12404. [CrossRef]
- Rethlefsen, M.L.; Kirtley, S.; Waffenschmidt, S.; Ayala, A.P.; Moher, D.; Page, M.J.; Koffel, J.B.; Group, P.-S. PRISMA-S: an extension to the PRISMA Statement for Reporting Literature Searches in Systematic Reviews. Syst Rev 2021, 10, 39. [CrossRef]
- Zhang, Y.; Lan, M.; Chen, Y. Minimal Information for Studies of Extracellular Vesicles (MISEV): Ten-Year Evolution (2014-2023). Pharmaceutics 2024, 16. [CrossRef]
- Hu, S.; Feng, L.; Yang, Z.; Fan, X.; Gao, H.; Yang, T. A recognition of exosomes as regulators of epigenetic mechanisms in central nervous system diseases. Front Mol Neurosci 2024, 17, 1370449. [CrossRef]
- Zhang, Y.; Zhang, S.; Li, Y.; Jin, W.; Zhou, L.; Lu, J. Role and relevance of exosome-mediated epigenetic regulation in the pathogenesis, diagnosis and treatment of cardiovascular diseases (Review). Mol Med Rep 2026, 33. [CrossRef]
- Jo, H.; Shim, K.; Jeoung, D. Exosomes: Diagnostic and Therapeutic Implications in Cancer. Pharmaceutics 2023, 15. [CrossRef]
- Padmasekar, M.; Savai, R.; Seeger, W.; Pullamsetti, S.S. Exposomes to Exosomes: Exosomes as Tools to Study Epigenetic Adaptive Mechanisms in High-Altitude Humans. Int J Environ Res Public Health 2021, 18. [CrossRef]
- Bhattacharya, B.; Dhar, R.; Mukherjee, S.; Gorai, S.; Devi, A.; Krishnan, A.; Alexiou, A.; Papadakis, M. Exosome DNA: An untold story of cancer. Clinical and Translational Discovery 2023, 3, e218. [CrossRef]
- Ng, C.T.; Azwar, S.; Yip, W.K.; Zahari Sham, S.Y.; Faisal Jabar, M.; Sahak, N.H.; Mohtarrudin, N.; Seow, H.F. Isolation and Identification of Long Non-Coding RNAs in Exosomes Derived from the Serum of Colorectal Carcinoma Patients. Biology (Basel) 2021, 10, 918. [CrossRef]
- Mezher, M.; Abdallah, S.; Ashekyan, O.; Shoukari, A.A.; Choubassy, H.; Kurdi, A.; Temraz, S.; Nasr, R. Insights on the Biomarker Potential of Exosomal Non-Coding RNAs in Colorectal Cancer: An In Silico Characterization of Related Exosomal lncRNA/circRNA-miRNA-Target Axis. Cells 2023, 12, 1081. [CrossRef]
- Qian, Z.; Shen, Q.; Yang, X.; Qiu, Y.; Zhang, W. The Role of Extracellular Vesicles: An Epigenetic View of the Cancer Microenvironment. Biomed Res Int 2015, 2015, 649161. [CrossRef]
- Chen, Y.Y.; Jiang, M.J.; Tian, L. Analysis of exosomal circRNAs upon irradiation in pancreatic cancer cell repopulation. BMC Med Genomics 2020, 13, 107. [CrossRef]
- Wang, Z.; Yang, B.; Zhang, M.; Guo, W.; Wu, Z.; Wang, Y.; Jia, L.; Li, S.; Cancer Genome Atlas Research, N.; Xie, W.; et al. lncRNA Epigenetic Landscape Analysis Identifies EPIC1 as an Oncogenic lncRNA that Interacts with MYC and Promotes Cell-Cycle Progression in Cancer. Cancer Cell 2018, 33, 706-720 e709. [CrossRef]
- Zhang, Q.; Li, H.; Liu, Y.; Li, J.; Wu, C.; Tang, H. Exosomal Non-Coding RNAs: New Insights into the Biology of Hepatocellular Carcinoma. Current Oncology 2022, 29, 5383-5406.
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat Rev Genet 2016, 17, 487-500. [CrossRef]
- Baniahmad, A.; Ozen, M. Editorial: Epigenetics in prostate cancer. Front Oncol 2023, 13, 1268519. [CrossRef]
- Jakob, F.; Hesse, E.; Bischof, O.; Kornak, U.; Ebert, R.; Taipaleenmäki, H. Epigenetics and Noncoding RNA – Principles and Clinical Impact. Osteologie 2021, 30, 201-210. [CrossRef]
- Liu, X.; Ding, G.; Liu, Y.; Yan, X.; Zhao, Y.; Lv, H.; Xu, X. Epigenetic regulation of bladder cancer in the context of aging. Front Pharmacol 2025, 16, 1617452. [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol Cancer 2020, 19, 79. [CrossRef]
- Bhalla, K.N. Epigenetic Dysregulation in Lymphoid Malignancies. Blood 2010, 116, SCI-29-SCI-29. [CrossRef]
- Pajares, M.J.; Alemany-Cosme, E.; Goni, S.; Bandres, E.; Palanca-Ballester, C.; Sandoval, J. Epigenetic Regulation of microRNAs in Cancer: Shortening the Distance from Bench to Bedside. Int J Mol Sci 2021, 22, 7350. [CrossRef]
- Redzic, J.S.; Balaj, L.; van der Vos, K.E.; Breakefield, X.O. Extracellular RNA mediates and marks cancer progression. Semin Cancer Biol 2014, 28, 14-23. [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142-157 e119. [CrossRef]
- Papakonstantinou, E.; Dragoumani, K.; Chrousos, G.P.; Vlachakis, D. Exosomal Epigenetics. EMBnet J 2024, 29. [CrossRef]
- Li, S.P.; Lin, Z.X.; Jiang, X.Y.; Yu, X.Y. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharmacol Sin 2018, 39, 542-551. [CrossRef]
- Jiang, C.; Zhang, J.; Wang, W.; Shan, Z.; Sun, F.; Tan, Y.; Tong, Y.; Qiu, Y. Extracellular vesicles in gastric cancer: role of exosomal lncRNA and microRNA as diagnostic and therapeutic targets. Front Physiol 2023, 14, 1158839. [CrossRef]
- Bortoluzzi, S.; Lovisa, F.; Gaffo, E.; Mussolin, L. Small RNAs in Circulating Exosomes of Cancer Patients: A Minireview. High Throughput 2017, 6, 13. [CrossRef]
- Bonizzato, A.; Gaffo, E.; Te Kronnie, G.; Bortoluzzi, S. CircRNAs in hematopoiesis and hematological malignancies. Blood Cancer J 2016, 6, e483. [CrossRef]
- Wang, Y.; Zhang, M.; Zhou, F. Biological functions and clinical applications of exosomal long non-coding RNAs in cancer. J Cell Mol Med 2020, 24, 11656-11666. [CrossRef]
- Arun, G.; Diermeier, S.D.; Spector, D.L. Therapeutic Targeting of Long Non-Coding RNAs in Cancer. Trends Mol Med 2018, 24, 257-277. [CrossRef]
- Guil, S.; Soler, M.; Portela, A.; Carrere, J.; Fonalleras, E.; Gomez, A.; Villanueva, A.; Esteller, M. Intronic RNAs mediate EZH2 regulation of epigenetic targets. Nat Struct Mol Biol 2012, 19, 664-670. [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311-1323. [CrossRef]
- Hannafon, B.N.; Ding, W.Q. Intercellular communication by exosome-derived microRNAs in cancer. Int J Mol Sci 2013, 14, 14240-14269. [CrossRef]
- Yin, H.; Hu, J.; Ye, Z.; Chen, S.; Chen, Y. Serum long non-coding RNA NNT-AS1 protected by exosome is a potential biomarker and functions as an oncogene via the miR-496/RAP2C axis in colorectal cancer. Mol Med Rep 2021, 24, 585. [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689-693. [CrossRef]
- Suzuki, H.; Maruyama, R.; Yamamoto, E.; Kai, M. Epigenetic alteration and microRNA dysregulation in cancer. Front Genet 2013, 4, 258. [CrossRef]
- Ashekyan, O.; Abdallah, S.; Shoukari, A.A.; Chamandi, G.; Choubassy, H.; Itani, A.R.S.; Alwan, N.; Nasr, R. Spotlight on Exosomal Non-Coding RNAs in Breast Cancer: An In Silico Analysis to Identify Potential lncRNA/circRNA-miRNA-Target Axis. Int J Mol Sci 2022, 23, 8351. [CrossRef]
- Wang, M.; Zhao, X.; Huang, F.; Wang, L.; Huang, J.; Gong, Z.; Yu, W. Exosomal proteins: Key players mediating pre-metastatic niche formation and clinical implications (Review). Int J Oncol 2021, 58, 4. [CrossRef]
- Zhao, Z.; Zhang, L.; Ocansey, D.K.W.; Wang, B.; Mao, F. The role of mesenchymal stem cell-derived exosome in epigenetic modifications in inflammatory diseases. Front Immunol 2023, 14, 1166536. [CrossRef]
- Kerachian, M.A.; Azghandi, M. Identification of long non-coding RNA using single nucleotide epimutation analysis: a novel gene discovery approach. Cancer Cell Int 2022, 22, 337. [CrossRef]
- Chen, R.X.; Chen, X.; Xia, L.P.; Zhang, J.X.; Pan, Z.Z.; Ma, X.D.; Han, K.; Chen, J.W.; Judde, J.G.; Deas, O.; et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun 2019, 10, 4695. [CrossRef]
- Alarcon, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299-1308. [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet 2019, 20, 675-691. [CrossRef]
- Hussain, M.Z.; Haris, M.S.; Rizwan, M.; Ashraf, N.S.; Arshad, M.; Mahjabeen, I. Deregulation of exosomal miRNAs in rheumatoid arthritis patients. PLoS One 2023, 18, e0289301. [CrossRef]
- Gahan, P.B.; Schwarzenbach, H. Liquid biopsies in lung cancer—a narrative review. ExRNA 2022, 4.
- Ali Syeda, Z.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int J Mol Sci 2020, 21, 1723. [CrossRef]
- Kang, M.; Lee, E.S.; Lee, E.S.; Yu, H.; Lee, T.G.; Suh, Y.D.; Na, H.K.; Park, K.D.; Jahng, J. Nanoscale Epigenetic Profiling of Colorectal Cancer Cell-Derived Exosomes via Single-Vesicle Nanoscopy. Small Methods 2025, 9, e00919. [CrossRef]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res 2014, 24, 766-769. [CrossRef]
- Ma, F.; Vayalil, J.; Lee, G.; Wang, Y.; Peng, G. Emerging role of tumor-derived extracellular vesicles in T cell suppression and dysfunction in the tumor microenvironment. J Immunother Cancer 2021, 9. [CrossRef]
- O'Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol 2020, 21, 585-606. [CrossRef]
- Benites, B.D.; Alvarez, M.C.; Saad, S.T.O. Small Particles, Big Effects: The Interplay Between Exosomes and Dendritic Cells in Antitumor Immunity and Immunotherapy. Cells 2019, 8. [CrossRef]
- Santangelo, L.; Giurato, G.; Cicchini, C.; Montaldo, C.; Mancone, C.; Tarallo, R.; Battistelli, C.; Alonzi, T.; Weisz, A.; Tripodi, M. The RNA-Binding Protein SYNCRIP Is a Component of the Hepatocyte Exosomal Machinery Controlling MicroRNA Sorting. Cell Rep 2016, 17, 799-808. [CrossRef]
- Letouze, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739-752. [CrossRef]
- Haffner, M.C.; Chaux, A.; Meeker, A.K.; Esopi, D.M.; Gerber, J.; Pellakuru, L.G.; Toubaji, A.; Argani, P.; Iacobuzio-Donahue, C.; Nelson, W.G.; et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011, 2, 627-637. [CrossRef]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481-484. [CrossRef]
- Kovaleva, O.; Sorokin, M.; Egorova, A.; Petrenko, A.; Shelekhova, K.; Gratchev, A. Macrophage - tumor cell interaction beyond cytokines. Front Oncol 2023, 13, 1078029. [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 2014, 10, 93-95. [CrossRef]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millan-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 2017, 552, 126-131. [CrossRef]
- Cao, Y.L.; Zhuang, T.; Xing, B.H.; Li, N.; Li, Q. Exosomal DNMT1 mediates cisplatin resistance in ovarian cancer. Cell Biochem Funct 2017, 35, 296-303. [CrossRef]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56-69. [CrossRef]
- Behbahani, G.D.; Khani, S.; Hosseini, H.M.; Abbaszadeh-Goudarzi, K.; Nazeri, S. The role of exosomes contents on genetic and epigenetic alterations of recipient cancer cells. Iran J Basic Med Sci 2016, 19, 1031-1039.
- Ge, R.; Tan, E.; Sharghi-Namini, S.; Asada, H.H. Exosomes in Cancer Microenvironment and Beyond: have we Overlooked these Extracellular Messengers? Cancer Microenviron 2012, 5, 323-332. [CrossRef]
- Costa, P.; Sales, S.L.A.; Pinheiro, D.P.; Pontes, L.Q.; Maranhao, S.S.; Pessoa, C.D.O.; Furtado, G.P.; Furtado, C.L.M. Epigenetic reprogramming in cancer: From diagnosis to treatment. Front Cell Dev Biol 2023, 11, 1116805. [CrossRef]
- Conigliaro, A.; Cicchini, C. Exosome-Mediated Signaling in Epithelial to Mesenchymal Transition and Tumor Progression. J Clin Med 2018, 8. [CrossRef]
- Jafari, A.; Babajani, A.; Abdollahpour-Alitappeh, M.; Ahmadi, N.; Rezaei-Tavirani, M. Exosomes and cancer: from molecular mechanisms to clinical applications. Med Oncol 2021, 38, 45. [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev Cell 2019, 49, 347-360. [CrossRef]
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct Target Ther 2020, 5, 242. [CrossRef]
- Mathieu, M.; Nevo, N.; Jouve, M.; Valenzuela, J.I.; Maurin, M.; Verweij, F.J.; Palmulli, R.; Lankar, D.; Dingli, F.; Loew, D.; et al. Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9. Nat Commun 2021, 12, 4389. [CrossRef]
- Guo, Q.R.; Wang, H.; Yan, Y.D.; Liu, Y.; Su, C.Y.; Chen, H.B.; Yan, Y.Y.; Adhikari, R.; Wu, Q.; Zhang, J.Y. The Role of Exosomal microRNA in Cancer Drug Resistance. Front Oncol 2020, 10, 472. [CrossRef]
- Li, K.; Chen, Y.; Li, A.; Tan, C.; Liu, X. Exosomes play roles in sequential processes of tumor metastasis. Int J Cancer 2019, 144, 1486-1495. [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428-445 e418. [CrossRef]
- Tang, M.; Wang, Y.; Xie, Y.; Li, J.; Ding, D.; Li, C.; Chen, Q.; Han, F. Targeting epigenetic networks to overcome cisplatin resistance in ovarian cancer: from mechanisms to clinical translation. J Ovarian Res 2026, 19. [CrossRef]
- Zhang, Y.; Ai, H.; Fan, X.; Chen, S.; Wang, Y.; Liu, L. Knockdown of long non-coding RNA HOTAIR reverses cisplatin resistance of ovarian cancer cells through inhibiting miR-138-5p-regulated EZH2 and SIRT1. Biol Res 2020, 53, 18. [CrossRef]
- Fujita, Y.; Yoshioka, Y.; Ochiya, T. Extracellular vesicle transfer of cancer pathogenic components. Cancer Sci 2016, 107, 385-390. [CrossRef]
- Lu, Y.; Liu, D.; Feng, Q.; Liu, Z. Diabetic Nephropathy: Perspective on Extracellular Vesicles. Front Immunol 2020, 11, 943. [CrossRef]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther 2020, 5, 145. [CrossRef]
- van Niel, G.; D'Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 2018, 19, 213-228. [CrossRef]
- Thind, A.; Wilson, C. Exosomal miRNAs as cancer biomarkers and therapeutic targets. J Extracell Vesicles 2016, 5, 31292. [CrossRef]
- Zhao, Z.; Li, J.; Li, H.; Yuan Wu, N.Y.; Ou-Yang, P.; Liu, S.; Cai, J.; Wang, J. Integrative Bioinformatics Approaches to Screen Potential Prognostic Immune-Related Genes and Drugs in the Cervical Cancer Microenvironment. Front Genet 2020, 11, 727. [CrossRef]
- Herrera, M.; Galindo-Pumarino, C.; Garcia-Barberan, V.; Pena, C. A Snapshot of The Tumor Microenvironment in Colorectal Cancer: The Liquid Biopsy. Int J Mol Sci 2019, 20. [CrossRef]
- Giordano, A.; Rucci, N.; Falone, S. Editorial: Extracellular vesicles as modulators of cancer cell adaptive responses linked to therapy resistance. Front Oncol 2022, 12, 1101103. [CrossRef]
- Wang, L.; Li, J.; Mei, N.; Chen, H.; Niu, L.; He, J.; Wang, R. Identifying subtypes and developing prognostic models based on N6-methyladenosine and immune microenvironment related genes in breast cancer. Sci Rep 2024, 14, 16586. [CrossRef]
- Fang, Z.; Meng, Q.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Zhao, Y.; Yu, X.; et al. Signaling pathways in cancer-associated fibroblasts: recent advances and future perspectives. Cancer Commun (Lond) 2023, 43, 3-41. [CrossRef]
- Kalluri, R.; LeBleu, V.S. Discovery of Double-Stranded Genomic DNA in Circulating Exosomes. Cold Spring Harb Symp Quant Biol 2016, 81, 275-280. [CrossRef]
- Yao, J.; Chen, Y.; Lin, Z. Exosomes: Mediators in microenvironment of colorectal cancer. Int J Cancer 2023, 153, 904-917. [CrossRef]
- Zhu, N.; Yang, Y.; Wang, H.; Tang, P.; Zhang, H.; Sun, H.; Gong, L.; Yu, Z. CSF2RB Is a Unique Biomarker and Correlated With Immune Infiltrates in Lung Adenocarcinoma. Front Oncol 2022, 12, 822849. [CrossRef]
- Rojas, A.; Lindner, C.; Schneider, I.; Gonzalez, I.; Araya, H.; Morales, E.; Gomez, M.; Urdaneta, N.; Araya, P.; Morales, M.A. Diabetes mellitus contribution to the remodeling of the tumor microenvironment in gastric cancer. World J Gastrointest Oncol 2021, 13, 1997-2012. [CrossRef]
- Zhu, L.; Liu, J.; Zhou, G.; Liu, T.M.; Dai, Y.; Nie, G.; Zhao, Q. Remodeling of Tumor Microenvironment by Tumor-Targeting Nanozymes Enhances Immune Activation of CAR T Cells for Combination Therapy. Small 2021, 17, e2102624. [CrossRef]
- Sikorski, H.; Zmijewski, M.A.; Piotrowska, A. Tumor Microenvironment in Melanoma-Characteristic and Clinical Implications. Int J Mol Sci 2025, 26. [CrossRef]
- Srinivasan, G.; Le, M.K.; Azher, Z.; Liu, X.; Vaickus, L.; Kaur, H.; Kolling, F.t.; Palisoul, S.; Perreard, L.; Lau, K.S.; et al. Histology-Based Virtual RNA Inference Identifies Pathways Associated With Metastasis Risk in Colorectal Cancer. Mod Pathol 2025, 38, 100866. [CrossRef]
- Masoumi-Dehghi, S.; Babashah, S.; Sadeghizadeh, M. microRNA-141-3p-containing small extracellular vesicles derived from epithelial ovarian cancer cells promote endothelial cell angiogenesis through activating the JAK/STAT3 and NF-kappaB signaling pathways. J Cell Commun Signal 2020, 14, 233-244. [CrossRef]
- Sempere, L.F.; Powell, K.; Rana, J.; Brock, A.A.; Schmittgen, T.D. Role of non-coding RNAs in tumor progression and metastasis in pancreatic cancer. Cancer Metastasis Rev 2021, 40, 761-776. [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382-386. [CrossRef]
- Sheta, M.; Taha, E.A.; Lu, Y.; Eguchi, T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology (Basel) 2023, 12. [CrossRef]
- Garcia-Gomez, A.; Rodriguez-Ubreva, J.; Ballestar, E. Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment. Clin Immunol 2018, 196, 64-71. [CrossRef]
- Atanaki, F.F.; Mirsadeghi, L.; Manesh, M.R.; Kavousi, K. Integrative analysis of single-cell transcriptomic and multilayer signaling networks in glioma reveal tumor progression stage. Front Genet 2024, 15, 1446903. [CrossRef]
- Suryawanshi, A.; Hussein, M.S.; Prasad, P.D.; Manicassamy, S. Wnt Signaling Cascade in Dendritic Cells and Regulation of Anti-tumor Immunity. Front Immunol 2020, 11, 122. [CrossRef]
- Cheng, B.; Chen, C.; zhang, S.; Yan, Y.; Pan, K.; Ou, F.; Su, K. Exosomal S100A9 Promotes Lung Metastasis of Adenoid Cystic Carcinoma via Activating Cancer-Associated Fibroblasts. Research Square 2025. [CrossRef]
- Curtale, G. MiRNAs at the Crossroads between Innate Immunity and Cancer: Focus on Macrophages. Cells 2018, 7. [CrossRef]
- Ning, W.; Marti, T.M.; Dorn, P.; Peng, R.W. Non-genetic adaptive resistance to KRAS(G12C) inhibition: EMT is not the only culprit. Front Oncol 2022, 12, 1004669. [CrossRef]
- Wade, J.D.; Lun, X.K.; Zivanovic, N.; Voit, E.O.; Bodenmiller, B. Mechanistic Model of Signaling Dynamics Across an Epithelial Mesenchymal Transition. Front Physiol 2020, 11, 579117. [CrossRef]
- Kochumon, S.; Al-Sayyar, A.; Jacob, T.; Bahman, F.; Akhter, N.; Wilson, A.; Sindhu, S.; Hannun, Y.A.; Ahmad, R.; Al-Mulla, F. TGF-beta and TNF-alpha interaction promotes the expression of MMP-9 through H3K36 dimethylation: implications in breast cancer metastasis. Front Immunol 2024, 15, 1430187. [CrossRef]
- Mitsuyasu Barbosa, B.; Todorovic Fabro, A.; da Silva Gomes, R.; Rainho, C.A. Deciphering the Heterogeneity of Pancreatic Cancer: DNA Methylation-Based Cell Type Deconvolution Unveils Distinct Subgroups and Immune Landscapes. Epigenomes 2025, 9. [CrossRef]
- Bungaro, C.; Guida, M.; Apollonio, B. Spatial proteomics of the tumor microenvironment in melanoma: current insights and future directions. Front Immunol 2025, 16, 1568456. [CrossRef]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep 2017, 18, 2162-2174. [CrossRef]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113, 6411-6418. [CrossRef]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat Med 2016, 22, 128-134. [CrossRef]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.J.; Liu, B.; Gao, Y.; Chen, W.; Liu, F.; Sun, W.; Li, X.F.; et al. Exosome-Transmitted lncARSR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell 2016, 29, 653-668. [CrossRef]
- Mannavola, F.; D'Oronzo, S.; Cives, M.; Stucci, L.S.; Ranieri, G.; Silvestris, F.; Tucci, M. Extracellular Vesicles and Epigenetic Modifications Are Hallmarks of Melanoma Progression. Int J Mol Sci 2019, 21. [CrossRef]
- Begolli, R.; Sideris, N.; Giakountis, A. LncRNAs as Chromatin Regulators in Cancer: From Molecular Function to Clinical Potential. Cancers (Basel) 2019, 11. [CrossRef]
- Gao, H.; Nishikubo, H.; Ma, D.; Pan, J.; Sano, T.; Imanishi, D.; Sakuma, T.; Fan, C.; Yashiro, M. Significance of Epigenetic Alteration in Cancer-Associated Fibroblasts on the Development of Carcinoma. Int J Mol Sci 2025, 26. [CrossRef]
- Melnik, B.C.; Schmitz, G. Milk's Role as an Epigenetic Regulator in Health and Disease. Diseases 2017, 5. [CrossRef]
- Mitsis, T.; Papakonstantinou, E.; Dragoumani, K.; Chrousos, G.; Vlachakis, D. Influence of epigenetics and microbiota in early-life development: A possible role for exosomes (Review). International Journal of Epigenetics 2024, 4, 3. [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930-935. [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739-744. [CrossRef]
- Li, W.; Zhang, X.; Lu, X.; You, L.; Song, Y.; Luo, Z.; Zhang, J.; Nie, J.; Zheng, W.; Xu, D.; et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res 2017, 27, 1243-1257. [CrossRef]
- Nicolini, A.; Ferrari, P.; Biava, P.M. Exosomes and Cell Communication: From Tumour-Derived Exosomes and Their Role in Tumour Progression to the Use of Exosomal Cargo for Cancer Treatment. Cancers (Basel) 2021, 13. [CrossRef]
- Kanlikilicer, P.; Bayraktar, R.; Denizli, M.; Rashed, M.H.; Ivan, C.; Aslan, B.; Mitra, R.; Karagoz, K.; Bayraktar, E.; Zhang, X.; et al. Exosomal miRNA confers chemo resistance via targeting Cav1/p-gp/M2-type macrophage axis in ovarian cancer. EBioMedicine 2018, 38, 100-112. [CrossRef]
- Liu, T.; Zhang, X.; Du, L.; Wang, Y.; Liu, X.; Tian, H.; Wang, L.; Li, P.; Zhao, Y.; Duan, W.; et al. Correction to: Exosome-transmitted miR-128-3p increase chemosensitivity of oxaliplatin-resistant colorectal cancer. Mol Cancer 2020, 19, 89. [CrossRef]
- Binenbaum, Y.; Fridman, E.; Yaari, Z.; Milman, N.; Schroeder, A.; Ben David, G.; Shlomi, T.; Gil, Z. Transfer of miRNA in Macrophage-Derived Exosomes Induces Drug Resistance in Pancreatic Adenocarcinoma. Cancer Res 2018, 78, 5287-5299. [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414-427 e413. [CrossRef]
- Wang, H.; Qi, Y.; Lan, Z.; Liu, Q.; Xu, J.; Zhu, M.; Yang, T.; Shi, R.; Gao, S.; Liang, G. Exosomal PD-L1 confers chemoresistance and promotes tumorigenic properties in esophageal cancer cells via upregulating STAT3/miR-21. Gene Ther 2023, 30, 88-100. [CrossRef]
- Jassi, C.; Kuo, W.W.; Kuo, C.H.; Chang, C.M.; Chen, M.C.; Shih, T.C.; Li, C.C.; Huang, C.Y. Mediation of radiation-induced bystander effect and epigenetic modification: The role of exosomes in cancer radioresistance. Heliyon 2024, 10, e34460. [CrossRef]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun 2016, 7, 11150. [CrossRef]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A comprehensive overview of exosomes as drug delivery vehicles - endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta 2014, 1846, 75-87. [CrossRef]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J Control Release 2015, 205, 35-44. [CrossRef]
- Wiklander, O.P.; Nordin, J.Z.; O'Loughlin, A.; Gustafsson, Y.; Corso, G.; Mager, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles 2015, 4, 26316. [CrossRef]
- Li, X.; Corbett, A.L.; Taatizadeh, E.; Tasnim, N.; Little, J.P.; Garnis, C.; Daugaard, M.; Guns, E.; Hoorfar, M.; Li, I.T.S. Challenges and opportunities in exosome research-Perspectives from biology, engineering, and cancer therapy. APL Bioeng 2019, 3, 011503. [CrossRef]
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.S.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; Perle, K.; et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J Extracell Vesicles 2017, 6, 1324730. [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244-1247. [CrossRef]
- Datta, A.; Kim, H.; Lal, M.; McGee, L.; Johnson, A.; Moustafa, A.A.; Jones, J.C.; Mondal, D.; Ferrer, M.; Abdel-Mageed, A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett 2017, 408, 73-81. [CrossRef]
- Verma, N.; Arora, S.; Singh, A.K.; Ahmed, J. Unlocking the potential of exosomes ‘extracellular vesicles’: drug delivery advancements and therapeutics in ocular diseases. RSC Pharmaceutics 2025, 2, 1201-1226. [CrossRef]
- Zhang, X.; Liu, L.; Tang, M.; Li, H.; Guo, X.; Yang, X. The effects of umbilical cord-derived macrophage exosomes loaded with cisplatin on the growth and drug resistance of ovarian cancer cells. Drug Dev Ind Pharm 2020, 46, 1150-1162. [CrossRef]
- Luo, H.; Zhou, Y.; Zhang, J.; Zhang, Y.; Long, S.; Lin, X.; Yang, A.; Duan, J.; Yang, N.; Yang, Z.; et al. NK cell-derived exosomes enhance the anti-tumor effects against ovarian cancer by delivering cisplatin and reactivating NK cell functions. Front Immunol 2022, 13, 1087689. [CrossRef]
- Wang, J.; Li, M.; Jin, L.; Guo, P.; Zhang, Z.; Zhanghuang, C.; Tan, X.; Mi, T.; Liu, J.; Wu, X.; et al. Exosome mimetics derived from bone marrow mesenchymal stem cells deliver doxorubicin to osteosarcoma in vitro and in vivo. Drug Deliv 2022, 29, 3291-3303. [CrossRef]
- Chen, Y.; Fang, Y.; Li, L.; Luo, H.; Cao, T.; Tu, B. Exosomal miR-22-3p from Mesenchymal Stem Cells Inhibits the Epithelial-Mesenchymal Transition (EMT) of Melanoma Cells by Regulating LGALS1. Front Biosci (Landmark Ed) 2022, 27, 275. [CrossRef]
- Gao, W.; Yang, N.; Yin, C.; Zeng, Y.; Zhu, X. Engineered Exosomes Loaded with miR-563 Inhibit Lung Cancer Growth. J Oncol 2022, 2022, 6141857. [CrossRef]
- Li, M.; Wang, Q.; Zhang, X.; Yan, N.; Li, X. Exosomal miR-126 blocks the development of non-small cell lung cancer through the inhibition of ITGA6. Cancer Cell Int 2020, 20, 574. [CrossRef]
- Xue, Q.; Yang, Y.; Yang, L.; Yan, X.; Shen, Z.; Liu, J.; Xue, J.; Zhao, W.; Liu, X. miR-371b-5p-Engineered Exosomes Enhances Tumor Inhibitory Effect. Front Cell Dev Biol 2021, 9, 750171. [CrossRef]
- Deng, W.; Meng, Y.; Wang, B.; Wang, C.X.; Hou, C.X.; Zhu, Q.H.; Tang, Y.T.; Ye, J.H. In vitro experimental study on the formation of microRNA-34a loaded exosomes and their inhibitory effect in oral squamous cell carcinoma. Cell Cycle 2022, 21, 1775-1783. [CrossRef]
- Lin, X.; Lin, L.; Wu, J.; Jiang, W.; Wu, J.; Yang, J.; Chen, C. A targeted siRNA-loaded PDL1-exosome and functional evaluation against lung cancer. Thorac Cancer 2022, 13, 1691-1702. [CrossRef]
- Zhou, W.; Zhou, Y.; Chen, X.; Ning, T.; Chen, H.; Guo, Q.; Zhang, Y.; Liu, P.; Zhang, Y.; Li, C.; et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials 2021, 268, 120546. [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498-503. [CrossRef]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [CrossRef]
- Verma, N.; Arora, S. Navigating the Global Regulatory Landscape for Exosome-Based Therapeutics: Challenges, Strategies, and Future Directions. Pharmaceutics 2025, 17. [CrossRef]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 2014, 3, 26913. [CrossRef]
- Maurya, A.K.; Maurya, A.K.; Muntane, J.; Kumar, V.B.S. Exosomal MicroRNAs in Cancer: Mechanisms, Clinical Applications, and Challenges in Biomarker Discovery. Preprints 2025. [CrossRef]
- Jiang, L.; Gu, Y.; Du, Y.; Liu, J. Exosomes: Diagnostic Biomarkers and Therapeutic Delivery Vehicles for Cancer. Mol Pharm 2019, 16, 3333-3349. [CrossRef]
- Verma, N.; Arora, S.; Singh, A.K.; Kumar, A. Extracellular Vesicle-Associated miRNAs in Cornea Health and Disease: Diagnostic Potential and Therapeutic Implications. Targets 2025, 3, 32.
- Sueta, A.; Fujiki, Y.; Goto-Yamaguchi, L.; Tomiguchi, M.; Yamamoto-Ibusuki, M.; Iwase, H.; Yamamoto, Y. Exosomal miRNA profiles of triple-negative breast cancer in neoadjuvant treatment. Oncol Lett 2021, 22, 819. [CrossRef]
- Kalani, A.; Kamat, P.K.; Tyagi, S.C.; Tyagi, N. Synergy of homocysteine, microRNA, and epigenetics: a novel therapeutic approach for stroke. Mol Neurobiol 2013, 48, 157-168. [CrossRef]
- Ramazi, S.; Dadzadi, M.; Sahafnejad, Z.; Allahverdi, A. Epigenetic regulation in lung cancer. MedComm (2020) 2023, 4, e401. [CrossRef]
- Wali, A.F.; Ansari, A.R.; Mir, P.A.; El-Tanani, M.; Babiker, R.; Hussain, M.S.; Uppal, J.; Zargar, A.I.; Mir, R.H. Epigenetic Alterations in Hepatocellular Carcinoma: Mechanisms, Biomarkers, and Therapeutic Implications. Pharmaceuticals (Basel) 2025, 18. [CrossRef]
- Sulewska, A.; Pilz, L.; Manegold, C.; Ramlau, R.; Charkiewicz, R.; Niklinski, J. A Systematic Review of Progress toward Unlocking the Power of Epigenetics in NSCLC: Latest Updates and Perspectives. Cells 2023, 12. [CrossRef]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc Natl Acad Sci U S A 2014, 111, 14888-14893. [CrossRef]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol Sci 2016, 37, 606-617. [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 2018, 7, 1535750. [CrossRef]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 2019, 20, 608-624. [CrossRef]
Figure 1.
Exosome biogenesis and selective epigenetic cargo sorting. Schematic overview of multivesicular body (MVB) formation, selective loading of epigenetic regulators into intraluminal vesicles (ILVs), and release of mature exosomes. ESCRT-dependent (HRS, TSG101, CHMP4/VPS4) and ESCRT-independent pathways drive ILV biogenesis (Panel 1). RNA-binding proteins and sequence motifs mediate selective packaging of small RNAs (such as miR-21, miR-155), lncRNAs/circRNAs, and methylated DNA, while epigenetic enzymes (DNMT1/3A/3B, EZH2) are recruited as cargo (Panel 2). Two sorting checkpoints, ceramide-enriched lipid domains and tetraspanin-enriched microdomains (CD9/CD63/CD81) bias cargo selection (Panel 3). MVB–plasma membrane fusion releases exosomes bearing canonical markers (CD63, CD9, CD81, HSP70, TSG101) to modulate recipient-cell epigenomes (Panel 4).
Figure 1.
Exosome biogenesis and selective epigenetic cargo sorting. Schematic overview of multivesicular body (MVB) formation, selective loading of epigenetic regulators into intraluminal vesicles (ILVs), and release of mature exosomes. ESCRT-dependent (HRS, TSG101, CHMP4/VPS4) and ESCRT-independent pathways drive ILV biogenesis (Panel 1). RNA-binding proteins and sequence motifs mediate selective packaging of small RNAs (such as miR-21, miR-155), lncRNAs/circRNAs, and methylated DNA, while epigenetic enzymes (DNMT1/3A/3B, EZH2) are recruited as cargo (Panel 2). Two sorting checkpoints, ceramide-enriched lipid domains and tetraspanin-enriched microdomains (CD9/CD63/CD81) bias cargo selection (Panel 3). MVB–plasma membrane fusion releases exosomes bearing canonical markers (CD63, CD9, CD81, HSP70, TSG101) to modulate recipient-cell epigenomes (Panel 4).

Figure 2.
Mechanistic landscape of exosome-driven epigenetic rewiring in recipient cancer and stromal cells. Schematic overview illustrating both established and proposed mechanisms by which tumor-derived exosomes may transfer epigenetic regulators to recipient cells and reprogram gene expression. Panel A (established mechanism): Exosomal miRNAs (e.g., miR-21, miR-29, miR-148a) alter the expression or activity of DNA methyltransferases (DNMTs) and TET demethylases in recipient cells, shifting CpG methylation patterns at tumor suppressor loci; this pathway is supported by functional cargo delivery and recipient-cell methylation readout studies. Panel B (established mechanism): Exosome-delivered long noncoding RNAs (e.g., HOTAIR, MALAT1) scaffold PRC2 components (EZH2, SUZ12, EED) in recipient cells, promoting H3K27 trimethylation at target loci and facilitating transcriptional programs associated with EMT and invasion; this pathway is supported by recipient-cell chromatin immunoprecipitation evidence. Panel C (established mechanism): Metabolites carried by vesicles (acetyl-CoA, succinate, 2-HG) modulate chromatin-modifying enzyme cofactors, biasing histone acetylation states (HAT/HDAC balance) and favoring oncogenic transcriptional programs; supported by metabolomics and epigenomics data. Panel D (proposed model; not yet rigorously demonstrated): Transfer of DNMT3A protein via exosomes may deposit new CpG methylation marks on previously unmethylated promoters in recipient cells, contributing to immune evasion and stable gene repression. This pathway is mechanistically plausible for single-subunit enzyme transfer but has not been validated by orthogonal single-vesicle proteomics demonstrating enzymatically competent DNMT3A delivery; it is depicted as a hypothetical model pending rigorous experimental confirmation.
Figure 2.
Mechanistic landscape of exosome-driven epigenetic rewiring in recipient cancer and stromal cells. Schematic overview illustrating both established and proposed mechanisms by which tumor-derived exosomes may transfer epigenetic regulators to recipient cells and reprogram gene expression. Panel A (established mechanism): Exosomal miRNAs (e.g., miR-21, miR-29, miR-148a) alter the expression or activity of DNA methyltransferases (DNMTs) and TET demethylases in recipient cells, shifting CpG methylation patterns at tumor suppressor loci; this pathway is supported by functional cargo delivery and recipient-cell methylation readout studies. Panel B (established mechanism): Exosome-delivered long noncoding RNAs (e.g., HOTAIR, MALAT1) scaffold PRC2 components (EZH2, SUZ12, EED) in recipient cells, promoting H3K27 trimethylation at target loci and facilitating transcriptional programs associated with EMT and invasion; this pathway is supported by recipient-cell chromatin immunoprecipitation evidence. Panel C (established mechanism): Metabolites carried by vesicles (acetyl-CoA, succinate, 2-HG) modulate chromatin-modifying enzyme cofactors, biasing histone acetylation states (HAT/HDAC balance) and favoring oncogenic transcriptional programs; supported by metabolomics and epigenomics data. Panel D (proposed model; not yet rigorously demonstrated): Transfer of DNMT3A protein via exosomes may deposit new CpG methylation marks on previously unmethylated promoters in recipient cells, contributing to immune evasion and stable gene repression. This pathway is mechanistically plausible for single-subunit enzyme transfer but has not been validated by orthogonal single-vesicle proteomics demonstrating enzymatically competent DNMT3A delivery; it is depicted as a hypothetical model pending rigorous experimental confirmation.

Figure 4.
Liquid biopsy biomarker pipeline for extracellular vesicle (EV)-based precision oncology. Panel a; Schematic of minimally invasive collection sites (blood, urine, saliva, pleural effusion, ascites, cerebrospinal fluid) used to obtain patient-derived EVs; emphasizes routine, low-burden sampling across cancers stages. Panel b; Overview of common isolation strategies (differential ultracentrifugation, size-exclusion chromatography, precipitation polymers, immunoaffinity capture) and the target EV size range (≈30–150 nm), highlighting trade-offs between yield and purity. Panel c; Workflow for molecular characterization of EV cargo, including small RNA (miRNA) and lncRNA profiling, cell-free DNA and methylation assays, proteomics (DDA/DIA, targeted MS), and metabolomics/lipidomics; illustrates downstream assays used to generate high-dimensional biomarker data. Panel d; Data-processing and machine-learning pipeline for quality control, normalization, feature extraction, multi-omics integration, predictive modeling, and validation; depicts how computational models translate EV signatures into diagnostic, prognostic, and therapeutic predictions. Panel e; Illustration of translational endpoints showing applications in diagnosis (early detection, tumor classification), prognosis (risk stratification, survival modeling), treatment prediction (therapy selection, resistance mechanisms, companion biomarkers), and disease monitoring (minimal residual disease, recurrence surveillance, longitudinal response tracking).
Figure 4.
Liquid biopsy biomarker pipeline for extracellular vesicle (EV)-based precision oncology. Panel a; Schematic of minimally invasive collection sites (blood, urine, saliva, pleural effusion, ascites, cerebrospinal fluid) used to obtain patient-derived EVs; emphasizes routine, low-burden sampling across cancers stages. Panel b; Overview of common isolation strategies (differential ultracentrifugation, size-exclusion chromatography, precipitation polymers, immunoaffinity capture) and the target EV size range (≈30–150 nm), highlighting trade-offs between yield and purity. Panel c; Workflow for molecular characterization of EV cargo, including small RNA (miRNA) and lncRNA profiling, cell-free DNA and methylation assays, proteomics (DDA/DIA, targeted MS), and metabolomics/lipidomics; illustrates downstream assays used to generate high-dimensional biomarker data. Panel d; Data-processing and machine-learning pipeline for quality control, normalization, feature extraction, multi-omics integration, predictive modeling, and validation; depicts how computational models translate EV signatures into diagnostic, prognostic, and therapeutic predictions. Panel e; Illustration of translational endpoints showing applications in diagnosis (early detection, tumor classification), prognosis (risk stratification, survival modeling), treatment prediction (therapy selection, resistance mechanisms, companion biomarkers), and disease monitoring (minimal residual disease, recurrence surveillance, longitudinal response tracking).


Table 1.
Summary table comparing the major exosomal cargo classes by cancer type, epigenetic mechanism, and functional outcome.
Table 1.
Summary table comparing the major exosomal cargo classes by cancer type, epigenetic mechanism, and functional outcome.
| Cargo Class | Cancer Type | Epigenetic Mechanism | Functional Outcome | References |
|---|---|---|---|---|
| miRNA | Breast, NSCLC | miR-21 suppresses PTEN/PDCD4; miR-29 family inhibits DNMT3A/3B mRNA, reducing CpG methylation; exosomal miR-148a targets DNMT1, promoting promoter demethylation; net effect: altered DNA methylation landscape and H3K27me3 at tumor suppressor loci. | Drug resistance (tamoxifen, cisplatin), EMT induction, cancer stem cell maintenance | [96] |
| lncRNA (HOTAIR) | CRC, HCC, ovarian cancer | HOTAIR scaffolds PRC2 (EZH2/SUZ12/EED) to gene promoters, depositing H3K27me3; sequesters miR-138-5p, relieving EZH2 suppression; promotes SIRT1-mediated deacetylation at pro-apoptotic loci. | Invasion, metastasis, cisplatin resistance | [94,95,98] |
| circRNA | Pancreatic ductal adenocarcinoma, Breast cancer (TNBC) | ceRNA sponging of tumor-suppressive miRNAs relieves post-transcriptional repression of chromatin modifiers; chromatin looping via interaction with RNA-binding proteins alters topological domains. | Therapy resistance, lineage plasticity | [106] |
| EV-DNA | Lung, CRC, pancreatic | Transfer of hypermethylated CpG island DNA from donor cancer cells to recipient cells silences tumor suppressor promoters in trans; 5-hydroxymethylcytosine (5hmC) content serves as cancer-state indicator. | TSG silencing in recipient cells; liquid biopsy biomarker | [111] |
| DNMT1/3A/3B proteins | Breast, Gastric, Ovarian | Exosomal DNMT1 transferred to cisplatin-sensitive cells methylates pro-apoptotic gene promoters (e.g., DAPK, RASSF1A), silencing them de novo; DNMT3A deposits new CpG methylation at immune-recognition loci contributing to immune exclusion. | Gene silencing, cisplatin resistance, immune evasion | [122] |
| Metabolites (acetyl-CoA, SAM) | Pan-cancer (IDH-mutant glioma/AML | Acetyl-CoA availability drives HAT activity and H3K27ac deposition at oncogene enhancers; SAM depletion reduces DNMT/HMT activity; alpha-KG is essential TET cofactor for 5mC | Metabolites epigenetic rewiring, immune evasion via epigenetic silencing of immune effector genes | [123] |
Table 2.
Extensive clinical studies demonstrate exosome-mediated epigenetic approaches for oncological treatment, in conjunction with associated biomarkers and diagnostic practices. Recruitment statuses verified at ClinicalTrials.gov accessed (04/05/2026).
Table 2.
Extensive clinical studies demonstrate exosome-mediated epigenetic approaches for oncological treatment, in conjunction with associated biomarkers and diagnostic practices. Recruitment statuses verified at ClinicalTrials.gov accessed (04/05/2026).
| NCT identifier | Source of Exosomes/Cargo class | Cancer type | Phase | Status (As cited) | Epigenetic Target |
|---|---|---|---|---|---|
| Therapeutic Clinical Trials | |||||
| NCT03608631 | MSC-derived exosomes + KrasG12D siRNA | Metastatic pancreatic ductal adenocarcinoma (PDAC) cancer | I | Recruiting | KrasG12D oncogene silencing via RNA interference; siRNA-mediated post-transcriptional gene silencing with downstream epigenetic consequences for MAPK/ERK transcriptional programs |
| NCT01294072 | Plant exosome-encapsulated curcumin (oral delivery) | Colon cancer | I | Unknown status | DNMT inhibition (curcumin is a DNMT enzymatic inhibitor); HAT activation; general epigenetic reprogramming toward tumor-suppressive states |
| NCT05559177 | APC-tumor chimeric exosome cancer vaccine | Bladder cancer | I | Unknown status | Immune epigenetic reprogramming via antigen-presenting cell activation; T cell epigenetic rejuvenation |
| NCT06536712 | MSC-derived Exosome Therapy | Rectal Cancer | I | Not yet recruiting | Not specified; general MSC-derived EV cargo (likely anti-inflammatory ncRNAs and growth factors). Biomarker trials |
| Biomarker and Diagnostic Trials | |||||
| NCT06388967 | Case-Control/ Exosomal miRNAs | Pancreatic ductal adenocarcinoma (PDAC) | Observational | Recruiting | exosomal miRNA signatures, lncRNA panels, or cfDNA methylation profiles as specified by the respective trial protocols |
| NCT06342427 | Case-Control/ Exosomal miRNAs | Gastric cancer (Stomach Cancer) | Observational | Completed | |
| NCT07553754 | Case-Control/ plasma exosomal RNAs | Prostate Cancer | Observational | Recruiting | |
| NCT05705583 | Case-Control/ circulating exosomes as companion diagnostic biomarker | Renal Cell Carcinoma | Observational | Unknown status | |
| NCT04939324 | Molecular profiling pulmonary and peripheral vein exosomes | early-stage non-small-cell lung cancer (NSCLC) | Not Applicable | Unknown status | |
| NCT01840306 | Exosomal and RNAs and Proteins | Breast Cancer | Observational | Completed | |
| NCT07558447 | Analysis of blood extracellular vesicles (lipids and microRNAs) | Cancer patients receiving chemotherapy | Not Applicable | Recruiting | |
| NCT06991452 | Body fluids exosomes | Colorectal cancer | Observational | Not yet recruiting | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



