Submitted:
02 June 2026
Posted:
03 June 2026
You are already at the latest version
Abstract
Inflammatory bowel diseases (IBD) are increasingly recognized as disorders in which epithelial dysfunction and maladaptive regeneration are as important as immune dysregulation. Tumor necrosis factor (TNF), a key mediator of intestinal inflammation and a therapeutic target, plays a dual role in both immune activation and epithelial repair by regulating progenitor cell expansion, lineage plasticity, and chemokine signaling. During acute injury, TNF-driven responses are adaptive, promoting crypt repair, barrier restitution, and secretory remodeling pathways. However, in chronic disease, persistent TNF exposure, amplified and stabilized by type I interferons (IFN-I), locks epithelial cells into sustained regenerative pathways. IFN-I signaling reinforces chemokine networks and transcription-al imprinting, converting physiological repair into a maladaptive state of “regenerative inflammation,” in which epithelial-derived signals perpetuate immune recruitment and tissue remodeling. Such TNF-IFN imprinted epithelial states may sustain pathology independently of classical inflammatory networks, offering a mechanistic basis for anti-TNF treatment failure in certain patients. By integrating mechanistic, organoid-based, and clinical evidence, we propose that chronic TNF-IFN crosstalk establishes a self-sustaining regenerative inflammatory circuit, redefining therapeutic response in IBD and highlighting new opportunities to target epithelial-immune interactions.
Keywords:
inflammatory bowel disease
; TNF
; type I interferons
; epithelial regeneration
; therapeutic resistance
; epithelial memory
; organoids
; regenerative inflammation
1. Introduction
Inflammatory bowel diseases (IBD), including Crohn’s disease (CD) and ulcerative colitis, involve chronic intestinal inflammation, impaired mucosal repair and barrier function [1]. Although anti-TNF therapies such as infliximab and adalimumab, have significantly improved disease management, therapy fails for approximately 30-40% of patients, and responsiveness is often lost over time [2]. This therapeutic gap has prompted a re-evaluation of disease mechanisms beyond classical immune dysregulation, with increasing attention on the intestinal epithelium as a critical determinant of disease outcome [3]. The epithelium functions not only as a physical barrier but also as an active regulator of immune responses, integrating cytokine signals and coordinating tissue repair [3].
The intestinal epithelium maintains a remarkable capacity for regeneration, driven by a dynamic interplay between stem cells, niche cells, and the surrounding microenvironment [4]. This regenerative capacity is essential for effective epithelial repair following injury and restoration of barrier integrity. Tissue-resident stem cells at the crypt base respond to epithelial injury by initiating proliferation and differentiation pathways, while the niche secretome molecules, including Wingless related integration site (Wnt), R-spondin, and Notch ligands, coordinate spatial and temporal aspects of repair [4]. Regeneration is not solely a stem cell-intrinsic process; rather, it is comprehensively shaped by the surrounding mesenchymal, immune, and microbial cues in the microenvironment [5], which collectively determine whether repair proceeds toward homeostasis or becomes maladaptive. Signals such as inflammatory cytokines, microbial metabolites, and extracellular matrix components dynamically modulate epithelial fate decisions [5]. In physiological contexts, this plasticity allows the intestine to recover from transient insults [6]. However, in chronic inflammatory settings such as IBD, persistent immune cell activation disrupts the regenerative niche [4,6].
While TNF blockade remains central to IBD therapy, a significant subset of patients exhibits refractory disease, with anti-TNF non-response frequently associated with amplified IFN-I activity [7,8,9]. Together, TNF and IFN-I are increasingly implicated in driving a state of “regenerative inflammation,” in which tissue repair programs become reprogrammed to sustain inflammatory signaling rather than restore epithelial homeostasis [10,11,12,13,14]. In this context, elevated epithelial IFN-I signaling has been linked to persistent stress responses and impaired repair capacity [15,16,17], supporting a model in which TNF-IFN-I crosstalk within the epithelium and its niche shapes stem cell behavior and disrupts coordinated regeneration. This basis is being refined through emerging data that maps how dysregulated repair processes evolve into chronic inflammation, highlighting alterations in cytokine crosstalk, epithelial state plasticity, and stromal support [18,19]. Advances in single-cell transcriptomics and tissue-targeted in vivo and in vitro models have revealed fundamental alterations in epithelial states in IBD, including the emergence of regenerative, progenitor-like and inflammatory cell populations that persist during chronic disease [20,21,22,23,24,25]. These findings suggest that epithelial dysfunction is not merely a consequence of inflammation but a driver of disease chronicity. The central question remains: why do regenerative pathways fail to resolve in IBD? We suggest that sustained TNF-IFN-I signaling locks intestinal epithelial cells in a continuously activated state, shifting repair away from homeostatic mechanisms toward maladaptive pathways that reinforce chronic inflammation, defining regenerative inflammation. Understanding these mechanisms will be critical for identifying patients at risk of persistent disease and for developing therapies that restore mucosal homeostasis.
2. Epithelial Regeneration in Homeostasis and Inflammation
The intestinal epithelium undergoes rapid cellular turnover driven by Lgr5⁺ stem cells located at the crypt base [5,6]. Under homeostatic conditions, these stem cells give rise to transit amplifying (TA) progenitor cells, which proliferate rapidly and subsequently differentiate into absorptive enterocytes and secretory lineages, including goblet, Paneth (small intestine), M cells, tuft cells and enteroendocrine cells. TA cells form a key amplification zone linking rapidly dividing stem cells to a committed differentiated progeny, enabling high epithelial turnover while preserving stem cell integrity [5,6]. Importantly, the TA cell compartment not only functions as a proliferative intermediate in homeostasis but also represents a key site where features of regenerative inflammation emerge, contributing also to the cellular pathways that support the formation of wound-associated epithelium (WAE) during injury-driven repair [5,6,9].
Upon injury, the epithelium activates highly plastic regenerative pathways that drive the formation of WAE, a transient repair state that emerges at sites of epithelial disruption to restore barrier integrity [6,12]. WAE is characterized by a flattened, migratory, de-differentiated epithelial phenotype enriched for repair and stress response genes, including Keratin 6 (KRT6), Keratin 17 (KRT17), Clusterin (CLU), Stem cell antigen 1 (SCA1), Tumor associated calcium signal transducer 2 or Trophoblast cell surface antigen 2 (TACSTD2 or TROP2), and Annexin A1 (ANXA1), alongside chemokines such as chemokine (C-X-C motif) ligand 1 (CXCL1), 10 (CXCL10) and 11 (CXCL11) [12,19,26]. These features reflect a transient epithelial program that prioritizes migration, survival, and barrier reconstitution over absorptive or secretory function. This state is coordinated by transcriptional regulatory pathways such as Yes-associated protein/ Transcriptional coactivator with PDZ-binding motif (YAP/TAZ), and inflammatory inputs including TNF and interleukin (IL)6 family cytokines, and IFN-I signaling in the intestine that can vary in magnitude and function depending on tissue context, injury stage, and local cytokine milieu. These molecules collectively shape epithelial plasticity, stress adaptation, and regenerative capacity during injury-driven repair [12,18,19,26].
Lineage tracing in vivo and murine intestinal organoid studies have demonstrated that TA cells in response to injury expand and contribute directly to the regenerative response, often acquiring fetal-like or stress induced transcriptional states [12,20,27,28,29]. Injury can trigger reversion of differentiated epithelial cells into fetal Sca1+ and Clu+ dedifferentiated epithelial states that contribute to crypt repopulation [27,28,29,30]. TA cells display adaptive fate plasticity under stress, with the capacity to transiently adopt stem-like features that enhance proliferative and regenerative capacity [27,30,31,32]. In human and murine inflamed colons, the balance between TA and WAE states shifts toward a broader regenerative axis, characterized by elevated proliferative activity but impaired terminal differentiation, with reduced mature absorptive and secretory cell populations, compromising epithelial barrier integrity [6,12,27,28,29,30,31,32].
In human IBD, single cell transcriptomic studies have identified an expansion of undifferentiated intestinal epithelial populations exhibiting heightened activity of regenerative, inflammatory, and stress response pathways. These epithelial subsets include secretory progenitors marked by Regenerating islet derived family member 4 (REG4), stress responsive epithelial cells expressing Dual oxidase 2 (DUOX2) and cytokine stimulated genes, suggesting injury associated reprogramming within transit amplifying compartments is coupled to innate immune signaling pathways to drive tissue remodeling [20,22]. In inflamed IBD tissue, lineage-restricted epithelial molecular signatures become aberrantly expressed across absorptive, secretory, and progenitor populations, indicating a breakdown in epithelial cell type specificity and impaired differentiation during chronic inflammation [20,22,33,34,35]. Genes such as C-C motif chemokine ligand 20 (CCL20), Gliomedin (GLDN), DUOX2, CXCL1, Olfactomedin 4 (OLFM4), Mucin 1 (MUC1), REG4, Tumor Necrosis Factor Alpha-Induced Protein interacting protein 3 (TNIP3), S100 calcium binding protein P (S100P), and Dedicator of cytokinesis 4 (DOCK4) mark immature TA progenitor populations, consistent with widespread immune driven epithelial remodeling [20,33,34]. These states are associated with activation of TNF responsive pathways, IFN-I signaling, IL22 signaling, oxidative stress programs, and chemokine networks, suggesting that chronic immune activation drives persistence of wound associated and regenerative epithelial pathways [26,35]. Collectively, these findings support a model in which sustained inflammatory signaling disrupts epithelial differentiation, driving a dysregulated regenerative state that perpetuates inflammation and impairs healing.
3. TNF as a Modulator of Epithelial Regeneration and Remodeling
Recent organoid-based and in vivo injury studies demonstrate that TNF, beyond its pro-inflammatory role, is increasingly recognized as a critical regulator of epithelial regeneration and mucosal healing, coordinating multiple signaling pathways that control epithelial survival, proliferation, stem cell plasticity, migration, and restoration of barrier integrity following intestinal injury. Studies in murine colitis show that TNF receptor 2 (TNFR2) expression peaks in crypt epithelial cells at wound margins following Sca1 or Ly6A induction, and loss of TNFR2 prolongs Ly6a expression, sustains hyperproliferation, and delays goblet and secretory lineage differentiation, effects that are also reproduced in organoid systems [11]. Consistent with this, TNF promoted expansion of Reg4⁺ secretory progenitors in murine and human colitis directly linking inflammatory signaling to epithelial lineage remodeling, demonstrating how regenerative pathways can be co-opted in chronic disease [20,22,32]. Notably, DSS-induced colonic injury was more severe in Tnf knockout mice, indicating a role for TNF in epithelial repair processes [36]. In intestinal organoids and the murine gut, TNF activated Wnt signaling, supporting epithelial regeneration through engagement of canonical Wnt dependent repair pathways. This occurred in part through coordinated nuclear factor kappa B (NFKB) and PI3K signaling, which cooperated with Wnt signaling to enhance β catenin activation and drive regenerative epithelial responses [37]. TNF enhanced epithelial wound healing by upregulating platelet activating factor receptor (PAFR), which activated EGFR (epidermal growth factor receptor), Src (proto-oncogene tyrosine-protein kinase Src), and Rac1 (Rac family small GTPase 1) signaling pathways to drive epithelial migration and wound closure [38]. Inhibition of TNF or loss of PAFR delayed mucosal healing, highlighting a regenerative role for TNF dependent PAFR signaling during intestinal injury.
TNF directly impaired LGR5+ intestinal stem cell function in CD-derived organoids by reducing self-renewal and organoid-forming capacity. This change was associated with disrupted Wnt/β-catenin signaling and activation of stress and inflammatory pathways, indicating that TNF-driven inflammation can compromise epithelial regeneration by targeting the stem cell compartment [39]. This loss of stem cell function in CD organoids was partially rescued by exogenous prostaglandin E2 (PGE2), which restored stem cell expansion and organoid forming efficiency, and synergized with low dose TNF blockade to improve epithelial regeneration. However, studies in azoxymethane/dextran sulfate sodium (AOM/DSS) colitis associated cancer models, TNFΔARE mice, and IL10/TNF double deficient mice demonstrate that TNF can promote IBD associated tumorigenesis by sustaining chronic inflammation, epithelial hyperproliferation, and NFKB-mediated survival signaling, thereby driving hyperplasia, dysplasia, and progression to colitis associated cancer [40]. These studies identify TNF as an injury associated cytokine that regulates epithelial repair and intestinal stem cell plasticity, coordinating regenerative pathways that promote wound healing, progenitor expansion, and barrier restoration following injury, while chronic TNF signaling may sustain dysfunctional regenerative states [11,12,20,26,35,38].
4. Type I Interferons and Intestinal Epithelial Homeostasis and Repair
IFN-Is are a family of cytokines, including multiple IFNα subtypes and IFNβ, that are rapidly induced in response to viral infection, cellular stress, and tissue injury to coordinate antimicrobial defense, inflammatory signaling, and immune regulation [10,14]. IFN-Is have highly context dependent roles in epithelial health, functioning as both protective regulators of tissue homeostasis and drivers of chronic inflammatory pathology depending on the timing, magnitude, and persistence of signaling during inflammation [10,14,16,17,41]. Under homeostatic and acute injury conditions, IFN-I signaling supports epithelial barrier integrity, antimicrobial defense, and controlled epithelial turnover by promoting tight junction formation, regulating proliferation, restricting excessive apoptosis, and regulating immune tolerance pathways towards commensal microbes. Within the intestinal epithelium, IFN-I signaling regulates stem and progenitor cell dynamics through pathways involving Signal transducer and activator of transcription 1 (STAT1), cyclin dependent kinase inhibitor 1A (p21/CDKN1A), tumor protein p53 (TP53/p53), and Wnt associated signaling networks, helping balance proliferation with differentiation and barrier restoration [14,42]. However, persistent or excessive IFN-I signaling can become pathological in chronic inflammatory settings [8,9,16,43]. IFN-Is promoted gut epithelial turnover and tissue repair during chronic viral infection by enhancing regenerative responses through immune-epithelial crosstalk [44]. Sustained IFN-I signaling acted on macrophages to induce regenerative factors, including apolipoprotein L9a and L9b (Apol9a/b), which subsequently activated extracellular signal regulated kinase (ERK) signaling in epithelial cells to enhance proliferation, turnover, and wound repair. These findings highlight a role for IFN-I in regulation of immune-epithelial crosstalk that supports tissue restitution during injury [44]. Sustained IFN-I activity amplifies inflammatory transcriptional pathways, promoting interferon stimulated gene expression that reinforces NFKB driven cytokine and apoptotic signaling, and can impair epithelial plasticity and regenerative resolution [44]. Persistent IFN-I exposure has been linked with prolonged epithelial stress states, de-differentiation, stem cell depletion and altered states, and hyperplasia that disrupted effective mucosal healing in chronic inflammation [42,43,44,45]. The outcome of IFN-I signaling therefore appears highly dependent on context, where transient signaling may support epithelial protection and repair, whereas prolonged activation may promote chronic inflammation and impaired mucosal recovery.
Genetic models reveal the nuanced roles of IFN-I in epithelial homeostasis. Intestinal epithelial cell (IEC)-specific Interferon alpha and beta receptor subunit 1 (Ifnar1) deficient mice had increased Paneth cell numbers and epithelial hyperproliferation relative to wildtype littermates [45]. While these Ifnar1 deficient mice did not develop spontaneous inflammation or heightened DSS colitis severity, they showed increased tumor burden in the AOM/DSS model. Notably, both hyperproliferation and tumor promotion were microbiota-dependent, as differences between genotypes were abrogated by co-housing. These findings show that epithelial IFN-I signaling acts as a homeostatic regulator of Paneth cell function and epithelial renewal, while also indirectly controlling microbial ecology. Disruption of this axis may shift host-microbiota interactions toward a state that promotes epithelial hyperproliferation and increases susceptibility to colitis-associated tumorigenesis [45].
In an in vivo murine model of Casein kinase 1 alpha (CK1α) deletion, which regulates β-catenin and Ifnar1 turnover, IFN-I signaling prevented uncontrolled epithelial proliferation despite constitutive β-catenin activity [46], IFN-I contributed to activation of the p53 pathway, apoptosis, and senescence in the CK1α-deficient gut, whereas concurrent ablation of CK1α and Ifnar1 resulted in marked intestinal hyperplasia, impaired apoptosis, and rapid loss of barrier function, demonstrating the capacity of IFN-I to restrain epithelial proliferation in contexts of oncogenic signaling.
Studies of acute injury models further highlight regenerative roles of IFN-Is. In a DSS model of acute colitis, mice lacking both type I and III IFN receptors exhibited worsened epithelial injury, including increased barrier disruption, goblet cell depletion, and reduced epithelial proliferation compared to control-DSS subjected mice [42]. This impaired mucosal repair was associated with diminished amphiregulin (AREG) expression, normally induced by IFN signaling in epithelial and hematopoietic compartments to promote epithelial regeneration and wound healing [42]. These findings demonstrate that IFN-Is support mucosal regeneration and recovery following acute epithelial injury.
5. TNF-IFN Crosstalk and the Emergence of Regenerative Inflammation
Most studies investigating TNF and IFN-I crosstalk have been performed in vitro and in murine models of immune cells, particularly macrophages [reviewed in 46], and comparatively little is known about how these synergistic pathways operate within the intestinal epithelium. In murine primary macrophages in vitro, TNF signaling initiated a sequential inflammatory amplification loop in which early NFKB and mitogen activated protein kinase (MAPK) activation induced Interferon regulatory factor 1 (IRF1) expression, leading to low level IFN-β production [47,48]. Autocrine IFN-β signaling subsequently activated JAK-STAT pathways, particularly STAT1, which cooperated with TNF induced signaling to sustain chemokine expression, amplified inflammatory transcriptional mediators, and enhanced expression of additional IFN-I responsive signaling genes including IKKε, IRF7, and STAT1 [47,48,49]. Collectively, these pathways sustain prolonged inflammatory activation and biased cellular responses toward persistent immune signaling. These findings provide a mechanistic framework for how TNF-IFN-I synergy may similarly perpetuate chronic inflammatory and dysregulated regenerative programs within the intestinal epithelium.
From studies reviewed so far, it becomes clear that TNF can promote early regenerative responses by stimulating epithelial proliferation, survival, and chemokine production, whereas IFN-I signaling appears to amplify and stabilize these inflammatory repair programs. Together, these pathways may form a feed forward inflammatory circuit that sustains epithelial activation. Recent experimental studies in human IBD organoid systems support this model, showing that combined TNF and IFN-I stimulation induces greater chemokine production, epithelial stress responses, and cytotoxicity than either cytokine alone [50].
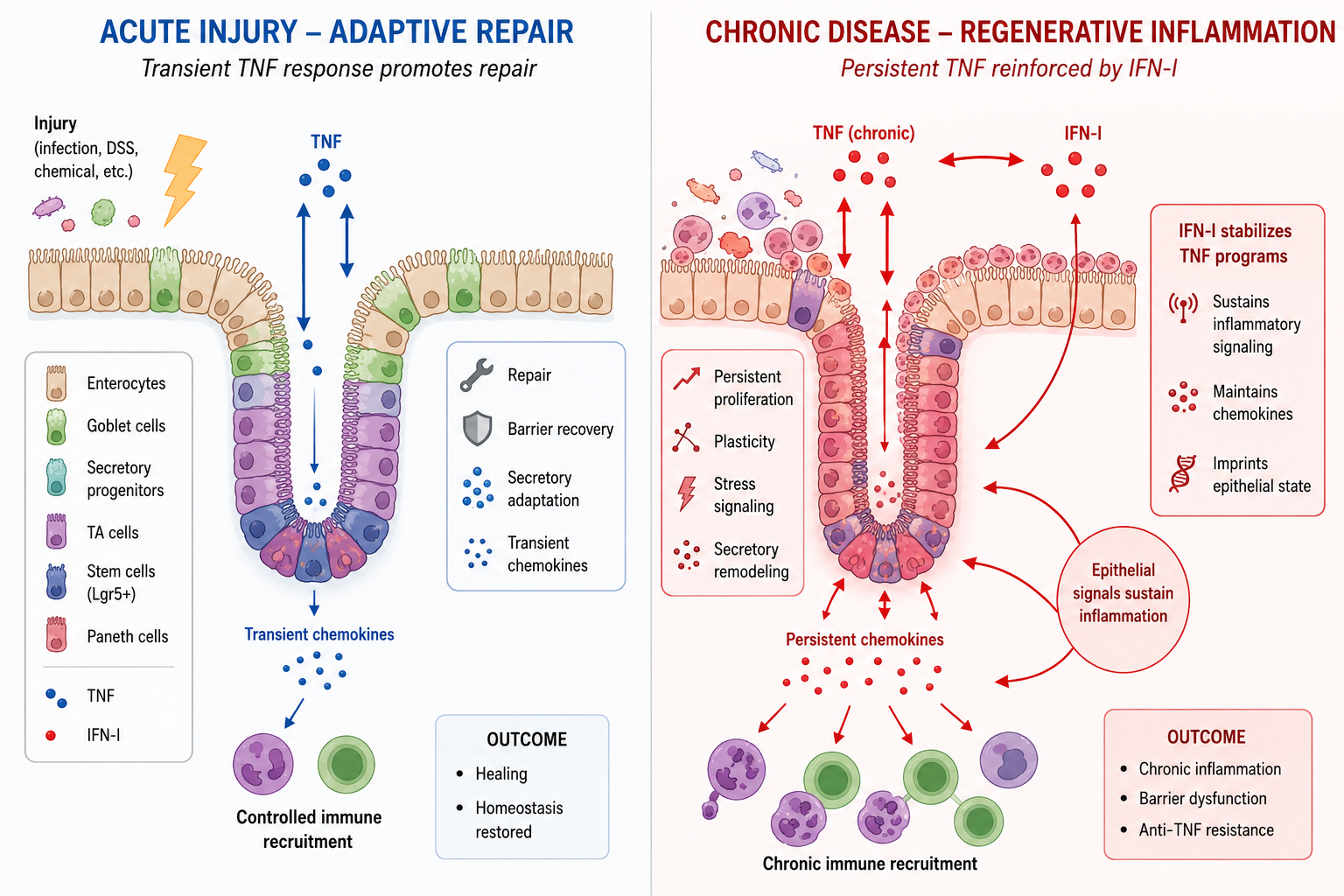
We propose a model in which TNF and IFN-I signaling are dynamically reprogrammed across phases of epithelial injury and inflammation. During acute injury, coordinated TNF and IFN-I signaling supports mucosal repair and restoration of epithelial barrier integrity (Figure 1). In contrast, under chronic inflammatory conditions, persistent TNF and IFN-I activity may impair resolution by sustaining inflammatory chemokine expression, altering apoptotic signaling pathways, and reinforcing regenerative inflammatory programs that perpetuate a self-sustaining cycle of inflammation. Consequently, epithelial cells remain locked in activated and poorly differentiated states associated with continuous immune cell recruitment and chronic tissue remodeling. We refer to this persistent injury response as regenerative inflammation, in which repair pathways become dysregulated and perpetuate disease rather than restore homeostasis. As these regenerative programs become progressively stabilized, they may also become less dependent on TNF signaling alone, potentially contributing to therapeutic resistance in IBD.
6. Regenerative Inflammation and Anti-TNF Response
A persistent TNF-IFN-I crosstalk in the intestinal epithelium under chronic inflammatory conditions establishing a self-reinforcing state of “regenerative inflammation,” may provide a mechanistic explanation for a lack of anti-TNF therapy response. While TNF blockade effectively neutralizes inflammation in many patients, persistent IFN-I activity can stabilize or amplify regenerative inflammatory pathways and maintain chemokine-driven immune recruitment independently of TNF. Clinically, such epithelial states may correspond to non-responder profiles, where inflammation persists despite apparent TNF inhibition. Importantly, regenerative inflammation is not solely a marker of disease severity, it actively drives tissue remodeling, alters differentiation trajectories, and sensitizes epithelium to apoptotic and senescence cues, creating a permissive environment for IBD-associated fibrosis or tumorigenesis. Patient-derived organoids and single-cell analyses indicate that epithelial cells in non-responders exhibit signatures of both proliferative plasticity and stress adaptation, suggesting that anti-TNF response may be an epithelial-intrinsic phenomenon reinforced by IFN-I signaling [20,22,27,39].
7. Therapeutic Implications and Conclusions
Recognition of regenerative inflammation as a central feature of IBD reframes therapeutic strategies beyond conventional immunosuppression. Targeting interferon signaling, for example via JAK-STAT pathway inhibition, offers a promising avenue to disrupt epithelial memory, restore repair-resolving mechanisms, and potentially re-sensitize patients to anti-TNF therapy. Combination approaches that simultaneously modulate TNF and IFN-I-dependent pathways may be particularly effective in interrupting self-perpetuating inflammatory circuits. Epithelial-derived biomarkers, including REG4, DUOX2, CXCL1, and CXCL10, could serve as precision tools for patient stratification, enabling identification of individuals at risk for therapeutic non-response and guiding personalized treatment. Targeting IFN-I signaling, either alone or in combination with TNF blockade, may release epithelial cells from maladaptive regenerative loops, restore proper differentiation, and improve barrier function in refractory patients. Furthermore, organoid-based stratification of regenerative inflammation could enable early identification of individuals unlikely to respond to anti-TNF therapy, guiding precision interventions and combination strategies. Recognizing regenerative inflammation as a mechanistic driver of anti-TNF resistance could reframe IBD management, emphasizing the need to integrate epithelial biology into therapeutic decision-making alongside classical immune modulation. Future strategies should prioritize restoring the dynamic balance between regeneration and resolution, rather than merely suppressing inflammation, which will require a deeper mechanistic understanding of epithelial-immune interactions and the regenerative niche.
In conclusion, IBD pathogenesis encompasses both immune dysregulation and fundamental alterations in epithelial regeneration. TNF-driven repair pathways, while essential for acute mucosal healing, can be driven into maladaptive regenerative inflammation when reinforced by sustained interferon signaling. This chronic epithelial state, characterized by persistent activation, chemokine production, and therapeutic resistance, underscores the importance of epithelial-immune crosstalk in disease progression. Integrating experimental, organoid, and clinical insights provides a roadmap for novel interventions that target epithelial memory, offering the potential to improve outcomes for patients with refractory IBD and to advance precision medicine approaches in the field.
Author Contributions
Safina Gadeock (Study conceptualization, writing; editorial revisions, acquired funding). Emelia Hinton, (writing; editorial revisions). Roslyn A Kemp (Editorial revisions, acquired funding). .
Funding
This research was funded by the Health Research Council of New Zealand [#22/262] to RK, Health Research Council of New Zealand Emerging Researcher Award 2024 [24/659/A] to SG.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| IFN-I | Type I Interferon |
| TNFR | Tumor Necrosis Factor Receptor |
| TNF | Tumor Necrosis Factor |
| IBD | Inflammatory Bowel Disease |
| RNA-Seq | RNA Sequencing |
| CD | Crohn’s Disease |
| NR-CD | Non-Responsive CD |
| R-CD | Responsive CD |
| IEC | Intestinal epithelial cells |
| IFNs | Interferons |
| scRNA-Seq | Single cell RNA Sequencing |
| TNFR1 | TNF receptor 1 |
| Wnt | Wingless related integration site |
| TA cells | Transit amplifying cells |
| WAE | Wound-associated epithelium |
| KRT6 | Keratin 6 |
| KRT17 | Keratin 17 |
| CLU | Clusterin |
| SCA1 | Stem cell antigen 1 |
| TACSTD2 / TROP2 | Tumor associated calcium signal transducer 2 / Trophoblast cell surface antigen 2 |
| ANXA1 | Annexin A1 |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 |
| CXCL10 | Chemokine (C-X-C motif) ligand 10 |
| CXCL11 | Chemokine (C-X-C motif) ligand 11 |
| YAP/TAZ | Yes-associated protein / Transcriptional coactivator with PDZ-binding motif |
| IL6 | Interleukin 6 |
| REG4 | Regenerating islet derived family member 4 |
| DUOX2 | Dual oxidase 2 |
| CCL20 | C-C motif chemokine ligand 20 |
| GLDN | Gliomedin |
| OLFM4 | Olfactomedin 4 |
| MUC1 | Mucin 1 |
| TNIP3 | Tumor Necrosis Factor Alpha-Induced Protein interacting protein 3 |
| S100P | S100 calcium binding protein P |
| DOCK4 | Dedicator of cytokinesis 4 |
| TNFR2 | TNF receptor 2 |
| PAFR | Platelet activating factor receptor |
| EGFR | Epidermal growth factor receptor |
| Src | Proto-oncogene tyrosine-protein kinase Src |
| Rac1 | Rac family small GTPase 1 |
| PGE2 | Prostaglandin E2 |
| AOM/DSS | Azoxymethane/dextran sulfate sodium |
| STAT1 | Signal transducer and activator of transcription 1 |
| p21/CDKN1A | Cyclin dependent kinase inhibitor 1A |
| TP53/p53 | Tumor protein p53 |
| Apol9a/b | Apolipoprotein L9a and L9b |
| ERK | Extracellular signal regulated kinase |
| IEC | Intestinal epithelial cell |
| Ifnar1 | Interferon alpha and beta receptor subunit 1 |
| CK1α | Casein kinase 1 alpha |
| AREG | Amphiregulin |
| NF-κB | Nuclear factor kappa B |
| MAPK | Mitogen activated protein kinase |
| IRF1 | Interferon regulatory factor 1 |
References
- Otte; Tamang, M.L.; Papapanagiotou, R.L.; Ahmad, J.; Dhawan, R.; P. Singh, A.B. Mucosal healing and inflammatory bowel disease: Therapeutic implications and new targets. World J. Gastroenterol. 2023, 21, 1157–1172. [Google Scholar] [CrossRef]
- Marsal; Barreiro-de Acosta, J.; Blumenstein, M.; Cappello, I.; Bazin, M.; T. Sebastian, S. Management of Non-response and Loss of Response to Anti-tumor Necrosis Factor Therapy in Inflammatory Bowel Disease. Front Med. 2022, 15, 897936. [Google Scholar] [CrossRef] [PubMed]
- Martini; Krug, E.; Siegmund, S.M.; Neurath, B.; M.F. Becker, C. Mend Your Fences: The Epithelial Barrier and its Relationship With Mucosal Immunity in Inflammatory Bowel Disease. Cell Mol. Gastroenterol. Hepatol. 2017, 23, 33–46. [Google Scholar] [CrossRef]
- Zhang; Chan, M.J.; Jia, S.X.; Lv, Z.G.; Chen, C.; J.J. Hong, S.C. Roles of intestinal stem cells in inflammatory bowel disease pathogenesis. World J. Stem Cells 2025, 26, 107639. [Google Scholar] [CrossRef]
- Hageman; Heinz, J.H.; Kretzschmar, M.C.; van der Vaart, K.; Clevers, J.; Snippert, H.H.J.G. Intestinal Regeneration: Regulation by the Microenvironment. Dev. Cell 2020, 54, 435–446. [Google Scholar] [CrossRef]
- de Sousa E Melo; F. de Sauvage, F.J. Cellular Plasticity in Intestinal Homeostasis and Disease. Cell Stem Cell 2019, 24, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.H.; Colgan, S.P. Mucosal responses to type II interferon in IBD. Inflamm. Bowel Dis. 2025, 31, 2584–2592. [Google Scholar] [CrossRef]
- Mavragani; Nezos, C.P.; Dovrolis, A.; Andreou, N.; Legaki, N.P.; Sechi, E.; Bamias, L.A.; G. Gazouli, M. Type I and II interferon signatures can predict the response to anti-TNF agents in inflammatory bowel disease donors: involvement of the microbiota. Inflamm. Bowel Dis. 2020, 26, 1543–1553. [Google Scholar] [CrossRef]
- van Baarsen; Wijbrandts, L.G.; Rustenburg, C.A.; Cantaert, F.; van der Pouw Kraan, T.; Baeten, T.C.; Dijkmans, D.L.; Tak, B.A.; P.P. Verweij, C.L. Regulation of IFN response gene activity during infliximab treatment in rheumatoid arthritis is associated with clinical response to treatment. Arthritis Res. Ther. 2010, 12, R11. [Google Scholar] [CrossRef] [PubMed]
- King; Kemp, M.R.; Gadeock, R.A.S. Type I interferons in inflammatory bowel diseases: balancing barrier integrity, repair and inflammation in the intestinal epithelium. Front Med. 2025, 12, 1713424. [Google Scholar] [CrossRef]
- Sharifkhodaei; Liu, Z.; Girish, C.Y.; Huang, N.; Punit, Y.; Washington, S.; M.K. Polk, D.B. Colitis-induced upregulation of tumor necrosis factor receptor-2 (TNFR2) terminates epithelial regenerative signaling to restore homeostasis. iScience 2023, 26, 107829. [Google Scholar] [CrossRef]
- Liu; Cham, C.Y.; C.M. Chang, E.B. Epithelial wound healing in inflammatory bowel diseases: the next therapeutic frontier. Transl. Res. 2021, 236, 35–51. [Google Scholar] [CrossRef]
- Sato; Y. Yu, J. Jump-starting IFN-I responses and epithelial regeneration. Sci. Signal 2025, 18, eaeb6151. [Google Scholar] [CrossRef]
- Kotredes; Thomas, K.P.; B. Gamero, A.M. The Protective Role of Type I Interferons in the Gastrointestinal Tract. Front Immunol. 2017, 8, 410. [Google Scholar] [CrossRef] [PubMed]
- Sprooten, J.; Garg, A.D. Type I interferons and endoplasmic reticulum stress in health and disease. Int. Rev. Cell Mol. Biol. 2020, 350, 63–118. [Google Scholar] [PubMed]
- Andreou; Legaki, N.P.; E. Gazouli, M. Inflammatory bowel disease pathobiology: the role of the interferon signature. Ann. Gastroenterol. 2020, 33, 125–133. [Google Scholar] [CrossRef]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Hausmann; Steenholdt, A.; Nielsen, C.; O.H. Jensen, K.B. Immune cell-derived signals governing epithelial phenotypes in homeostasis and inflammation. Trends Mol. Med. 2024, 30, 239–251. [Google Scholar] [CrossRef]
- Crifo, B.; MacNaughton, W.K. Cells and mediators of inflammation as effectors of epithelial repair in the inflamed intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 322, G169–G182. [Google Scholar] [CrossRef]
- Gadeock; Girish, S.; Liu, N.; Huang, C.Y.; Grikscheit, Y.; T.C. Polk, D.B. Tumor necrosis factor signaling promotes differentiation of colonic deep crypt secretory cells. bioRxiv 2025. [Google Scholar] [CrossRef]
- Arijs; Li, I.; Toedter, K.; Quintens, G.; Van Lommel, R.; Van Steen, L.; Leemans, K.; De Hertogh, P.; Lemaire, G.; Ferrante, K.; Schnitzler, M.; Thorrez, F.; Ma, L.; Song, K.; Marano, X.Y.; Van Assche, C.; Vermeire, G.; Geboes, S.; Schuit, K.; Baribaud, F.; F. Rutgeerts, P. Mucosal gene signatures to predict response to infliximab in ulcerative colitis. Gut 2009, 58, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Smillie; Biton, C.S.; Ordovas-Montanes, M.; Sullivan, J.; Burgin, K.M.; Graham, G.; Herbst, D.B.; Rogel, R.H.; Slyper, N.; Waldman, M.; Sud, J.; Andrews, M.; Velonias, E.; Haber, G.; Jagadeesh, A.L.; Vickovic, K.; Yao, S.; Stevens, J.; Dionne, C.; Nguyen, D.; Villani, L.T.; Hofree, A.C.; Creasey, M.; Huang, E.A.; Rozenblatt-Rosen, H.; Garber, O.; Khalili, J.J.; Desch, H.; Daly, A.N.; Ananthakrishnan, M.J.; Shalek, A.N.; Xavier, A.K.; R.J. Regev, A. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 2019, 178, 714–730. [Google Scholar] [CrossRef]
- VanDussen; Stojmirović, K.L.; Li, A.; Liu, K.; Kimes, T.C.; Muegge, P.K.; Simpson, B.D.; Ciorba, K.F.; Perrigoue, M.A.; Friedman, J.G.; Towne, J.R.; Head, J.E.; R.D. Stappenbeck, T.S. Abnormal small intestinal epithelial microvilli in Crohn’s disease. Gastroenterology 2018, 155, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Noble; Abbas, C.L.; Lees, A.R.; Cornelius, C.W.; Toy, J.; Modrusan, K.; Clark, Z.; Arnott, H.F.; Penman, I.D.; Satsangi, I.D.; J. Diehl, L. Characterization of intestinal gene expression profiles in Crohn’s disease by genome-wide microarray analysis. Inflamm. Bowel Dis. 2010, 16, 717–1728. [Google Scholar] [CrossRef]
- Verstockt; Verstockt, B.; Veny, S.; Dehairs, M.; Arnauts, J.; Van Assche, K.; De Hertogh, G.; Vermeire, G.; Salas, S.; A. Ferrante, M. Expression Levels of 4 Genes in Colon Tissue Might Be Used to Predict Which Donors Will Enter Endoscopic Remission After Vedolizumab Therapy for Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2020, 18, 1142–1151.e10. [Google Scholar] [CrossRef]
- Brazil; Quiros, J.C.; Nusrat, M.; A. Parkos, C.A. Innate immune cell-epithelial crosstalk during wound repair. J. Clin. Invest 2019, 129, 2983–2993. [Google Scholar] [CrossRef] [PubMed]
- Liu; Girish, C.Y.; Gomez, N.; Kalski, M.L.; Bernard, M.; Simons, J.K.; B.D. Polk, D.B. Wound-healing plasticity enables clonal expansion of founder progenitor cells in colitis. Dev. Cell 2023, 58, 2309–2325.e7. [Google Scholar] [CrossRef]
- Maimets; Nikolaev, M.; Lövkvist, M.; Bertillot, C.; Larsen, F.; Bressan, H.L.; Georgantzoglou, R.B.; Gjorevski, A.; Filidou, N.; Zøylner, E.; Hansen, M.; Baattrup, S.L.; Banjac, A.M.; Guiu, I.; Wickström, J.; Lutolf, S.A.; Jensen, M.P.; B., K. Engineered intestinal crypt geometry uncovers YAP1-dependent fetal-to-adult transition. Cell Stem Cell 2026, 33, 487–501.e7. [Google Scholar] [CrossRef]
- Guiu; Hannezo, J.; Yui, E.; Demharter, S.; Ulyanchenko, S.; Maimets, S.; Jørgensen, M.; Perlman, A.; Lundvall, S.; Mamsen, L.; Larsen, L.S.; Olesen, A.; Andersen, R.H.; Thuesen, C.Y.; Hare, L.L.; Pers, K.J.; Khodosevich, T.H.; Simons, K.; Jensen, B.D.; B., K. Tracing the origin of adult intestinal stem cells. Nature 2019, 570, 107–111. [Google Scholar] [CrossRef]
- Nusse; Savage, Y.M.; Marangoni, A.K.; Rosendahl-Huber, P.; Landman, A.K.M.; de Sauvage, T.A.; Locksley, F.J.; R.M. Klein, O.D. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 2018, 559, 109–113. [Google Scholar] [CrossRef]
- Buczacki; Zecchini, S.J.; Nicholson, H.I.; Russell, A.M.; Vermeulen, R.; Kemp, L.; R. Winton, D.J. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 2013, 495, 65–9. [Google Scholar] [CrossRef]
- Sasaki; Sachs, N.; Wiebrands, N.; Ellenbroek, K.; Fumagalli, S.I.; Lyubimova, A.; Begthel, A.; van den Born, H.; van Es, M.; Karthaus, J.H.; Li, W.R.; López-Iglesias, V.S.; Peters, C.; van Rheenen, P.J.; van Oudenaarden, J.; A. Clevers, H. Reg4+ deep crypt secretory cells function as epithelial niche for Lgr5+ stem cells in colon. Proc. Natl. Acad. Sci. U S A 2016, 113, E5399-407. [Google Scholar] [CrossRef]
- Serigado; Foulke-Abel, J.M.; Hines, J.; Hanson, W.C.; In, J.A.; J. Kovbasnjuk, O. Ulcerative Colitis: Novel Epithelial Insights Provided by Single Cell RNA Sequencing. Front Med. 2022, 9, 868508. [Google Scholar] [CrossRef]
- Balasubramanian; Patel, B.; Gall, S.; Hannan, L.; Dalleywater, N.R.F.; Huelsken, W.; Pin, J.; Moran, C.; G.W. Ordóñez-Morán, P. Intestinal stem and progenitor cells exhibit distinct adaptive responses to inflammatory stress in IBD. Stem Cell Res. Ther. 2025, 17, 71. [Google Scholar] [CrossRef]
- Andrews; McLean, C.; M.H. Durum, S.K. Cytokine Tuning of Intestinal Epithelial Function. Front Immunol. 2018, 9, 1270. [Google Scholar] [CrossRef]
- Naito; Takagi, Y.; Handa, T.; Ishikawa, O.; Nakagawa, T.; Yamaguchi, S.; Yoshida, T.; Minami, N.; Kita, M.; Imanishi, M.; J. Yoshikawa, T. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J. Gastroenterol. Hepatol. 2003, 18, 560–9. [Google Scholar] [CrossRef]
- Bradford; Ryu, E.M.; Singh, S.H.; Lee, A.P.; Goretsky, G.; Sinh, T.; Williams, P.; Cloud, D.B.; Gounaris, A.L.; Patel, E.; Lamping, V.; Lynch, O.F.; Moyer, E.B.; De Plaen, M.P.; Shealy, I.G.; Yang, D.J.; Barrett, G.Y.; A., T. Epithelial TNF Receptor Signaling Promotes Mucosal Repair in Inflammatory Bowel Disease. J. Immunol. 2017, 199, 1886–1897. [Google Scholar] [CrossRef] [PubMed]
- Birkl; Quiros, D.; García-Hernández, M.; Zhou, V.; Brazil, D.W.; Hilgarth, J.C.; Keeney, R.; Yulis, J.; Bruewer, M.; García, M.; O Leary, A.J.; Parkos, M.N.; C.A. Nusrat, A. TNFα promotes mucosal wound repair through enhanced platelet activating factor receptor signaling in the epithelium. Mucosal Immunol. 2019, 12, 909–918. [Google Scholar] [CrossRef]
- Lee, C. An; Joung, M.; Park, J.G.; Chang, W.Y.; Kim, D.K.; Y.H. Hong, S.N. TNFα Induces LGR5+ Stem Cell Dysfunction In Patients With Crohn's Disease. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Waldner, M.J.; Neurath, M.F. Mechanisms of Immune Signaling in Colitis-Associated Cancer. Cell Mol. Gastroenterol. Hepatol. 2014, 1, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Sato; Y. Yu, J. Jump-starting IFN-I responses and epithelial regeneration. Sci. Signal 2025, 18, eaeb6151. [Google Scholar] [CrossRef]
- McElrath; Espinosa, C.; Lin, V.; Peng, J.D.; Sridhar, J.; Dutta, R.; Tseng, O.; Smirnov, H.C.; Risman, S.V.; Sandoval, H.; Davra, M.J.; Chang, V.; Pollack, Y.J.; Birge, B.P.; Galan, R.B.; Rivera, M.; Durbin, A.; J.E. Kotenko, S.V. Critical role of interferons in gastrointestinal injury repair. Nat. Commun. 2021, 12, 2624. [Google Scholar] [CrossRef]
- Rauch, I.; et al. Type I interferons have opposing effects during the emergence and recovery phases of colitis. Eur. J. Immunol. 2014, 44, 2749–2760. [Google Scholar] [CrossRef]
- Sun; Miyoshi, L.; Origanti, H.; Nice, S.; Barger, T.J.; Manieri, A.C.; Fogel, N.A.; French, L.A.; Piwnica-Worms, A.R.; Piwnica-Worms, D.; Virgin, H.; Lenschow, H.W.; D.J. Stappenbeck, T.S. Type I interferons link viral infection to enhanced epithelial turnover and repair. Cell Host Microbe 2015, 17, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Tschurtschenthaler; Wang, M.; Fricke, J.; Fritz, C.; Niederreiter, T.M.; Adolph, L.; Sarcevic, T.E.; Künzel, E.; Offner, S.; Kalinke, F.A.; Baines, U.; Tilg, J.F.; H. Kaser, A. Type I interferon signalling in the intestinal epithelium affects Paneth cells, microbial ecology and epithelial regeneration. Gut 2014, 63, 1921–31. [Google Scholar] [CrossRef]
- Katlinskaya; Katlinski, Y.V.; Lasri, K.V.; Li, A.; Beiting, N.; Durham, D.P.; Yang, A.C.; Pikarsky, T.; Lengner, E.; Johnson, C.J.; Ben-Neriah, F.B.; Y. Fuchs, S.Y. Type I Interferons Control Proliferation and Function of the Intestinal Epithelium. Mol. Cell Biol. 2016, 36, 1124–35. [Google Scholar] [CrossRef]
- Yarilina; A. Ivashkiv, L.B. Type I interferon: a new player in TNF signaling. Curr. Dir. Autoimmun. 2010, 11, 94–104. [Google Scholar]
- Yarilina; Park-Min, A.; Antoniv, K.H.; Hu, T.; X. Ivashkiv, L.B. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol. 2008, 9, 378–87. [Google Scholar] [CrossRef] [PubMed]
- Sharma; tenOever, S.; Grandvaux, B.R.; Zhou, N.; Lin, G.P.; R. Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–51. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Raichon; Neil, L.; Kim, J.A.; Heaney, K.; Miller, T.; Moon, B.M.; Lubkin, D.; DuMont, A.; Torres, A.L.; Axelrad, V.J.; Matsuzawa-Ishimoto, J.; Y. Cadwell, K. Type I and III interferons synergize with TNF to promote virally-triggered damage to the intestinal epithelium. bioRxiv 2026. [Google Scholar] [CrossRef]
Figure 1.
TNF-IFN crosstalk drives regenerative inflammation and therapeutic resistance in IBD. During acute injury, TNF signaling promotes epithelial regeneration through progenitor expansion, transient chemokine production, and restoration of barrier integrity. In chronic disease, persistent exposure to TNF and type I interferons (IFN-I) amplifies and stabilizes these responses, leading to sustained chemokine expression and altered epithelial differentiation. This establishes a self-reinforcing epithelial-immune circuit, defined here as regenerative inflammation. As these pathways become imprinted, epithelial activation becomes partially independent of TNF signaling, providing a mechanistic basis for resistance to anti-TNF therapy.
Figure 1.
TNF-IFN crosstalk drives regenerative inflammation and therapeutic resistance in IBD. During acute injury, TNF signaling promotes epithelial regeneration through progenitor expansion, transient chemokine production, and restoration of barrier integrity. In chronic disease, persistent exposure to TNF and type I interferons (IFN-I) amplifies and stabilizes these responses, leading to sustained chemokine expression and altered epithelial differentiation. This establishes a self-reinforcing epithelial-immune circuit, defined here as regenerative inflammation. As these pathways become imprinted, epithelial activation becomes partially independent of TNF signaling, providing a mechanistic basis for resistance to anti-TNF therapy.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



