Submitted:
01 June 2026
Posted:
02 June 2026
You are already at the latest version
Abstract
Photosystem II (PS II) is a water-plastoquinone oxidoreductase. This photosystem has a short ½ life (<1hr) which is driven by repair of oxidative damage to the D1 protein. Reactive oxygen species (ROS) produced by the photosystem under stress conditions oxidatively modify key residues which trigger the sequential disassembly of the photosystem. We hypothesize that the principal target for ROS damage of D1 is 332His. This residue is a ligand to Mn1 of the Mn4CaO5cluster and is the first residue exhibiting oxidative modification during photoinhibition. Recently, cryo-EM studies indicate that PS II monomers containing oxidatively modified 332His lose their manganese clusters and the extrinsic proteins associated with the photosystem, both hallmarks of the early stages of PS II turnover and repair.
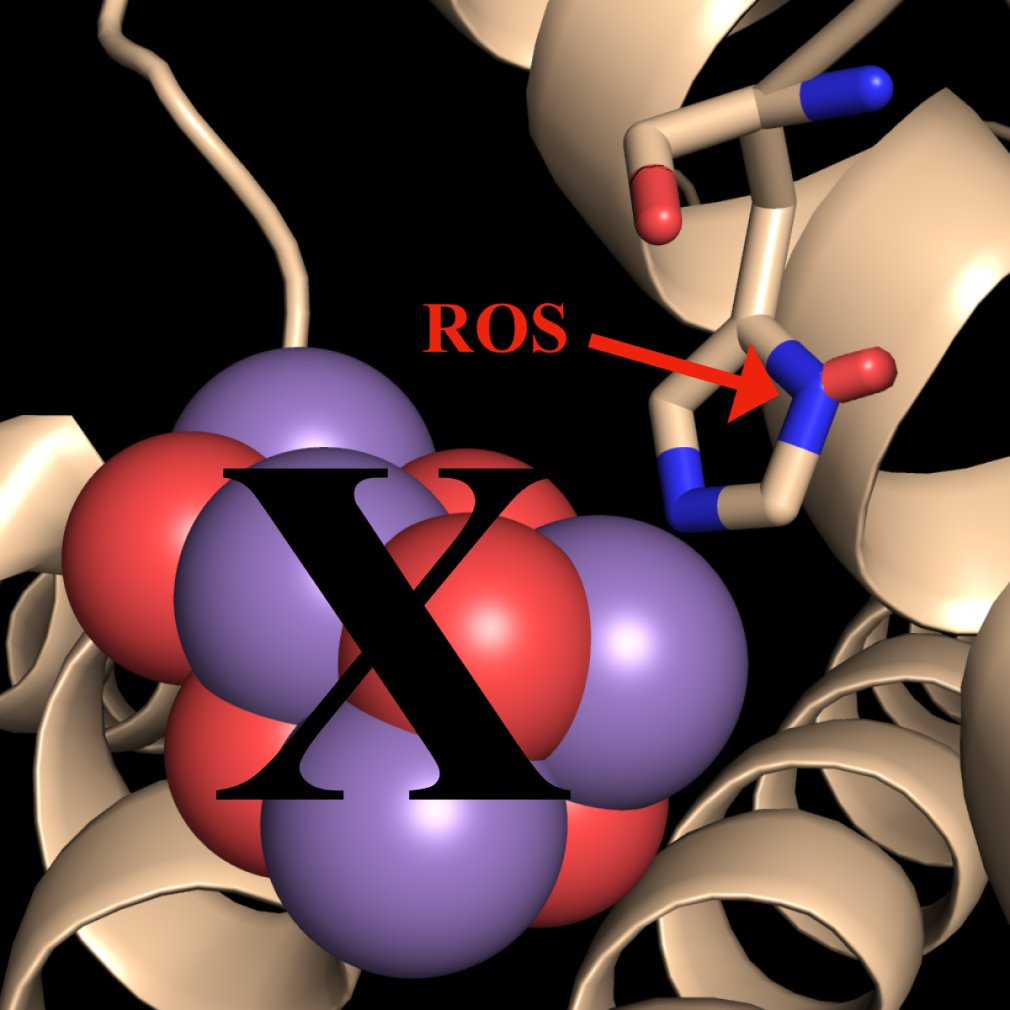
Keywords:
Photosystem II
; Photoinhibition
; ROS
; D1:332His
Introduction
PS II is a water-plastoquinone oxidoreductase and is the source of virtually all O2 in the biosphere. Functionally, excitation energy from the light-harvesting apparatus (phycobilisomes in cyanobacteria and light-harvesting chlorophyll proteins in green plants and algae) is transferred to the reaction center of the photosystem. Initial charge separation occurs between ChlD1 and PheoD1 which yields ChlD1+PheoD1- [1]. This charge separation is stabilized by electron transfer first to QA and then to QB. The accumulation of two electrons on QB leads to protonation and its subsequent release as plastoquinol. ChlD1+ is reduced by YZ, the residue D1:161Y, yielding YZ• with the release of a proton. The subsequent reduction of YZ• by the Mn₄CaO₅ cluster occurs via proton-coupled electron transfer and leads to the accumulation of an oxidizing equivalent at the metal cluster. The accumulation of four oxidizing equivalents, in a manner consistent with the Kok Cycle [2], ultimately leads to the oxidation of two water molecules and the release of dioxygen [3,4]. The photosystem is highly susceptible to photoinhibition and is continuously being repaired. It has been estimated that photoinhibition can reduce biomass yield by as much as 30% in some plant species [5]. In this communication we propose that oxidative modification of D1:332His is the principal residue damaged during photoinhibition and that this triggers the PS II repair cascade (Figure 1).
Reactive Oxygen Species (ROS) and Photoinhibition
Excitation energy transfer and electron transport within PS II are unavoidably associated with production of ROS when the absorption of light by the chlorophyll antenna exceeds the capacity for energy utilization or when the Mn₄CaO₅ cluster is damaged. PS II appears particularly susceptible to damage by ROS. A variety of different ROS species have been proposed to oxidatively damage PS II components, including 1O2, O2-•, H2O2 (and cofactor-bound peroxides) and HO•; with a variety of mechanisms being proposed for their possible production both on the oxidizing- and reducing-side of the photosystem (22,23). ROS can oxidatively damage proteins, lipids and DNA [6]. PS II photoinhibition occurs when ROS oxidatively modifiy PS II component proteins, including D1, D2, CP47, and CP43. Many studies support a two-step model of PSII photoinhibition in which the earliest detectable photodamage occurs at the oxygen-evolving Mn₄CaO₅ cluster, followed by secondary reaction center damage occurring when the impaired oxygen-evolving complex can no longer efficiently reduce P680+ [7,8,9]. This model is supported by the observation that Mn is released during the early stages of photoinhibition, by experiments showing that visible light can directly damage the oxygen-evolving complex prior to detectable reaction-center damage [10], and the observation that the earliest detectable oxidative modification events occur in the vicinity of the Mn₄CaO₅ cluster of the D1 protein. Subsequent oxidative damage occurs in the vicinity of PheoD1, QA, non-heme iron, and QB [11,12].
PS II Repair
Photoinhibitory damage to PS II by ROS occurs at all light levels [13,14], necessitating that the photosystem be continuously repaired [15,16]. Briefly, during the repair process in cyanobacteria, functional dimeric PS II becomes oxidatively damaged during photoinhibition, leading to loss of the extrinsic proteins and subsequent monomerization of the complex. Subsequently, the damaged PS II core at least partially disassembles, yielding a CP43 repair module and a damaged CP43-less reaction center module, both of which are likely associated with suites of repair-specific proteins. These would include proteins that stabilize these dissociated sub-complexes and facilitate their interaction with other repair components. The damaged D1 protein is degraded by the FtsH and Deg proteases and is subsequently replaced by co-translational insertion of a nascent D1 chain. In Synechocystis, membrane integration of D1 depends on the Oxa1/Alb3/YidC homolog Slr1471 [17], while YCF48/HCF136 stabilizes newly synthesized pD1/iD1 [18] in supporting efficient PSII assembly and repair. Concomitantly, D1 chls are probably inserted as the nascent D1 elongates and associates with D2 within a repaired CP43-less reaction center module. C-terminal processing of D1 (and assembly of the Mn4O5Ca cluster) may occur prior to re- association of the undamaged CP43-less reaction center complex with CP43, yielding PS II monomers. Subsequently, dimerization of the PS II monomers and reassociation of the extrinsic proteins with the dimeric complex and re-association with the PS II antennae completes the repair cycle. In higher plants and cyanobacteria, these repair processes are thought to be quite similar, although there are differences with respect to the specific accessory proteins functioning in both systems. So the question is: how are these events triggered? What subset of oxidative modifications within the photosystem are necessary and sufficient to induce this repair cascade?
Earlier, we have hypothesized that amino acid residues in the vicinity of the sites of ROS production would be particularly susceptible to ROS modification [19,20]. Consequently, the identification of such oxidatively modified residues should serve to identify the site(s) of ROS generation within PS II and, perhaps, assist in the identification of residues whose modification elicit PS II turnover. The PS II cofactors required for water splitting are primarily associated with the D1 protein and this component is the primary site for photodamage in PS II [21]. Due to this damage, D1 is replaced rapidly (a t1/2 of <1 hr in Synechocystis [22]) during repair, which is much faster than observed for other PS II core components (D2, 3.3 hrs., CP43, 6.5 hrs, CP47, 11.5 hrs [22]). Modules containing these other PS II components have been demonstrated to be reused during the repair process [13,23]. Consequently, this necessitates that ROS targeted residues in D1 are the primary trigger for initiating the repair cascade, while the other, more stable PS II core components serve as a scaffold for reassembly of the repaired photosystem. If the initiatial triggering oxidative modification(s) resided on other PS II core proteins, removal and replacement of damaged D1 would lead to a newly repaired PS II complex already containing the triggering oxidative modification (due to reuse of the modules). This would result in a futile cycle of D1 degradation and replacement.
Modification of D1 by ROS
So, which residues of D1 are oxidatively modified? In early work, Sharma et al. [24] identified a number of D1 peptides which exhibited oxidative modifications in a higher plant PS II core preparation isolated from pea. Because of instrumental limitations in 1997 the individual residues modified were not identified. These oxidized peptides included 130E-136R (+16 amu), which lies in the vicinity of PheoD1 (7-16 Å), and oxidized peptides in the vicinity of the Mn4CaO5 cluster (4-24 Å); 184Ile-199Hse (+48 amu), 313Val-323Arg (+32 amu), and 332His-344Ala (+32 amu). We confirmed a number of these observations, identifying several 1natively oxidized amino acid residues (D1:130E, 133L and 135F) present in the D1: 130E-136R peptide [20] and in the domain 313Val-344Ala (316T, 317W, 319D, 328M, 331M and 333E, [12]). Additionally, other natively oxidized residues (D1:239F, 241Q, 242E) were in close proximity to the non-heme iron [20]. Barry and coworkers identified an oxidatively modified tryptophan residue, D1:317W, which appears to be a target for oxidizing-side ROS modification [25]. Kato et al. [26] identified D1:317W and D1:14W as accumulating in Chlamydomonas mutants lacking the FtsH protease which degrades damaged D1, speculating that D1:14W may be involved in triggering D1 degradation. This latter residue has not been identified in experiments mapping oxidative modifications in response to light treatments in either cyanobacteria or higher plants [11,12]. These studies indicate that residues in the vicinity of the Mn4CaO5 cluster, PheoD1, and the non-heme iron, are natively oxidized; however, the relative importance of these sites during the photoinhibition time-course was not determined. It should be noted that several studies [20,25] were performed on a standard PS II preparation (BBYs, [27]) isolated from field grown spinach which could arguably be challenged by a variety of environmental insults prior to harvesting (high light intensity, high temperatures, drought, wound damage during harvesting, etc.), all of which could elicit ROS production and protein damage. Consequently, while the accumulation of oxidative modifications at specific residues which are adjacent to various PS II cofactors suggest possible sites of ROS production, these studies do not demonstrate any direct correlation to serious photodamage which would elicit the repair cascade, nor do they necessarily address the temporal order that these oxidative modifications were produced.
More recently two studies addressed these limitations [11,12]. In Synechocystis 6803, Weisz et al. [11] examined the differential accumulation of oxidized residues occurring either in the absence or presence of illumination. They identified numerous oxidative modifications in the D1, D2 and CP43 proteins, some of which were present only in illuminated samples ([11], Supplementary Information, Table S2). The D1 residues which were modified only during illumination were 235Y (near QA), 249V, and 257R (near QB). Additionally, two residues, 332H and 333E, which are in close proximity to the Mn4O5Ca cluster were oxidatively modified. These results indicated that a relatively long-term exposure (6 hr) to modest light intensities (50 μmoles photons/m-2/s-1) elicited significant oxidative modification of the proteins examined.
In our laboratory we examined the time-course of the appearance of oxidatively modified residues during photoinhibition [12]. In these studies, prior to the isolation of PS II membranes, the spinach leaves were incubated overnight under dim light conditions (<5 μmoles photons/m-2/s-1) to facilitate repair of the photosystem. This pre-treatment markedly decreased the abundance of the natively oxidized residues that we had observed in earlier studies [19,20]. Illumination at 1,400 μmoles photons/m-2/s-1 was carried out for 30 min, with mass spectrometry analysis performed on samples collected after 0, 15 and 30 min of ilumination. Photoinhibition, as monitored by oxygen evolution, exhibited a t1/2 of ~20 min. Only oxidized residues in the vicinity of the manganese cluster were observed at 0 and 15 min, with extensive modification of residues in the vicinity of PheoD1, QA, the non-heme iron and QB appearing only after 30 min of illumination ([12], Supplementary Information, Table S1). At 0 min the native oxidative modification of the D1 residues 316T, 317W, 319D, 328M, 331M and 333E were observed. After 15 min of illumination only a single additional residue was observed to be oxidatively modified, D1:332His.
Recently, a critically important cryo-EM study by Zhao et al. [28] has provided strong evidence that D1:332His is directly involved in PS II remodeling in response to photoinhibition. In the cyanobacterium Thermosynechococcus vestitus grown under moderate light conditions (60 μmoles photons/m-2/s-1), three types of PS II dimers were isolated: fully active (PDB:9G6H), containing two active monomers; semi-active (PDB: 9G6G), containing one active and one inactive monomer; and fully inactive (PDB: 9G6F), containing two inactive monomers. These inactive monomers were likely the result of constitutive photodamage occurring under standard growth light conditions [14,29]. The inactive monomers exhibited loss of the Mn4CaO5 cluster, increased flexibility of the C-terminus of D1 (such that it is no longer resolved in cryo-EM maps), structural alterations in CP43 and CP47, and loss of PsbO, PsbU and PsbV. Monomerization of the inactive dimers was not observed. Importantly, all inactive monomers also exhibited oxidative modification of D1:332His (+16 amu, Figure 1). The authors speculated that oxidative damage to D1:332His was an early event in photoinactivation of the photosystem, echoing proposals in earlier studies [11,12]. Importantly, this was the only oxidized residue identifiable in their cryo-EM maps. Mass spectrometry analysis indicated that D1:328M and 331M are also oxidized (+16 amu), however the cryo-EM maps associated with these residues do not show any oxidative modification in the inactive monomers, indicating low occupancy of any of these oxidized species in their cryo-EM structures. Earlier, both D1:328M and D1:338M had been identified as being natively oxidized [11,12]. The authors hypothesized that the oxidative modification to D1:332His is the addition of a hydroxyl group at the ring Nδ1 position, forming a 2Nδ1-hydroxy-L-histidine residue, a highly unusual, and if correct, novel oxidative protein modification. Better documented oxidized histidine ring modifications include 2-oxo-histidine [30] and possibly 2-hydroxy-histidine [31]. The latter has also been proposed as a possible short-lived intermediate or transient product in histidine oxidation pathways, but is not well established as a stable protein modification. It should be pointed out that the authors did not report examining rotomers of 2-oxo-histidine (or 2-hydroxy histidine) for possible fit to their observed cryo-EM maps.
While these and earlier results are highly suggestive, an important question persists: why would oxidative modification of 332His lead to disassembly of PS II? The core observation in this regard is that 332His is an inner shell ligand to the Mn4CaO5 cluster [32], with the histidyl Nε2 nitrogen coordinating Mn1 of the cluster. Oxidative modification of this residue would, with high probability, modify this interaction. For instance, if the modification is indeed Nδ1-hydroxy-L-histidine, it is estimated that the approximate pK of the Nε2 nitrogen would shift from ~6.9 to ~4.9 (pK estimates using the MolGpkA online resource [33]). Earlier, mutagenesis studies replacing 332His with the acidic residues Glu (pK ~4.5) and Asp (pK ~4.1) indicated that these substitutions had a dramatic effect on the structure/function of the active site [34]. For instance, replacement of 332His with 332Glu led to profoundly altered electronic and geometric structure of the manganese cluster, with the authors stating, “The substantial structural changes provide an explanation not only for the altered properties of the D1-H332E mutant but also the importance of the histidine ligand for proper assembly of the Mn4Ca cluster [35].” It should be noted that if the oxidative modification were to 2-oxo-histidine, similar defects might be expected. The pKs of the Nδ1 and Nε2 nitrogens are estimated to be 12.3 and 12.8, respectively [15]. The protonation of these nitrogens at physiological pH would effectively preclude their coordination to Mn1 [34]. Although the 2-keto oxygen of 2-oxo-histidine could, in principle, interact with Mn1, it would not be expected to substitute effectively for the native histidine imidazole nitrogen ligand. Mutants which substitute 332His with 332Gln or 332Asn, both of which contain amide carbonyl substituents, exhibit very low oxygen evolution rates, assemble fewer PS II reaction centers and exhibit very rapid fluorescence relaxation kinetics, all of which are consistent with a defective Mn4CaO5 cluster [34]. Importantly, in active PS II monomers, the 332His Nδ1 atom lies within van der Waals contact with the backbone carbonyl oxygen of D1-329Glu (~2.7 Å). This close native contact leaves little space for addition of an Nδ1-hydroxy substituent. Formation of Nδ1-hydroxy-L-histidine would therefore be expected to generate a severe steric conflict with the D1-329Glu backbone carbonyl unless relieved by local rearrangement. Comparison of the active and inactive monomers [28] suggests that such relief could involve an approximately 180° rotation of the 332His imidazole ring about the Cβ–Cγ bond, which corresponds to a χ2 rotamer change. This rotation would increase the Nε2–Mn1 distance from ~2.2 Å to ~2.8 Å and introduce an angular distortion of >45°, changes that are consistent with severe weakening or functional loss of the normal Mn–imidazole coordination interaction. A qualitative estimate based on bond-valence distance dependence and simple angular-overlap considerations suggests that the effective coordination interaction could be reduced to roughly ~10% of that in the active monomer [36,37].
The inactive monomers observed by Zhao et al. [28] were isolated from T. vestitis cells, grown under standard light conditions which had apparently lost their Mn4CaO5 clusters due to constitutive photodamage. Earlier cryo-EM studies in Synechocystis indicated that removal of the Mn4CaO5 cluster by chemical treatment [38] also led to increased flexibility of the C-terminus of D1 (such that it was no longer resolved in cryo-EM maps), structural alterations of CP43 and CP47, and loss of PsbO, PsbU and PsbV. In this case, dimers were not observed, only inactive monomers. We speculate that in both systems the Mn2 ligand 344Ala (via its C-terminal carboxylate) normally locks the D1 C-terminus in position in active monomers. Removal of the Mn4O5Ca either during photoinactivation as a result of 332His oxidation or after chemical treatment disrupts this association and allows for much greater mobility of the D1 C-terminus, precluding its identification in these cryo-EM maps [28,38]. This enhanced mobility may predispose the photosystem for the structural modifications of CP43 and CP47 and loss of the extrinsic subunits and, ultimately, monomerization of the PS II dimers.
Testing the Hypothesis
The studies outlined above provide a mechanistic framework which supports the hypothesis that oxidative modification of D1:332His triggers the disassembly of photodamaged PS II (Figure 3). Hypotheses, however, must necessarily be tested. Additional cryo-EM and mass spectrometry studies performed on cyanobacterial and higher plant samples which initially are allowed to repair in the dark and then subsequently collected during a photoinhibitory time-course would be most welcomed. These studies could potentially confirm or reject the hypothesis. While cryo-EM studies capture protein structures which, by definition, are frozen in time, the damage repair process is highly dynamic and can potentially be studied using molecular dynamic simulation. Atomistic molecular dynamic simulations examining the stability of the Mn4CaO5 cluster in the presence of D1:332Nδ1-hydroxy-L-histidine or D1:3322-oxo-histidine could provide direct evidence that oxidation of this residue results in lower Mn4CaO5 cluster stability (simulated on ns-μs timescales). Additionally, course-grained molecular dynamic simulations would allow the examination of the flexibility of the D1 C-terminus, possible CP43/CP47 remodeling, and potentially, the release of the extrinsic proteins (simulated on 1-10 μs timescales), in addition to possible dimer monomerization. These molecular dynamic studies are ongoing.
Figure 2.
Orientation of 332His in the presence and absence of the Mn4CaO5 cluster (PDB:9G6G, [28]) A. Active monomer, D1 is shown in light brown, the Mn4CaO5 cluster is shown as spheres and color coded by atom type with the manganese atoms (Mn1-Mn4) labeled. 332His is shown as sticks and color-coded by atom type with the imidazole nitrogens shown in blue. Nδ1 and Nε2 are labeled. B. Inactive monomer, D1 is shown in yellow, 332Nδ1-hydroxy histidine is shown as sticks and color-coded by atom type, with the imidazole nitrogens shown in blue, Nδ1 and Nε2 are labeled. Some regions of D1 have been omitted for clarity.
Figure 2.
Orientation of 332His in the presence and absence of the Mn4CaO5 cluster (PDB:9G6G, [28]) A. Active monomer, D1 is shown in light brown, the Mn4CaO5 cluster is shown as spheres and color coded by atom type with the manganese atoms (Mn1-Mn4) labeled. 332His is shown as sticks and color-coded by atom type with the imidazole nitrogens shown in blue. Nδ1 and Nε2 are labeled. B. Inactive monomer, D1 is shown in yellow, 332Nδ1-hydroxy histidine is shown as sticks and color-coded by atom type, with the imidazole nitrogens shown in blue, Nδ1 and Nε2 are labeled. Some regions of D1 have been omitted for clarity.

Figure 3.
Proposed Sequence at the Mn4CaO5 Cluster Binding Site. A. Active monomer, D1 is shown in light brown, 332His and 344Ala and the Mn4CaO5 cluster are shown as spheres and color- coded by atom type. B. After exposure to ROS, Nδ1-hydroxy 332His is formed. The Nδ1-hydroxy modification is highlighted by the arrow. C. Loss of effective ligation of Mn1 by the Nδ1 immidazole nitrogen leads to destabilization and loss of the Mn4CaO5 cluster, which is highlighted by the arrow. D. Loss of the Mn4CaO5 cluster allows increased flexibility of the D1 C-terminus, including 344Ala, which precludes structure determination in cryo-EM. Sphere color code: carbon, light brown; nitrogen, blue; oxygen, red and manganese, purple.
Figure 3.
Proposed Sequence at the Mn4CaO5 Cluster Binding Site. A. Active monomer, D1 is shown in light brown, 332His and 344Ala and the Mn4CaO5 cluster are shown as spheres and color- coded by atom type. B. After exposure to ROS, Nδ1-hydroxy 332His is formed. The Nδ1-hydroxy modification is highlighted by the arrow. C. Loss of effective ligation of Mn1 by the Nδ1 immidazole nitrogen leads to destabilization and loss of the Mn4CaO5 cluster, which is highlighted by the arrow. D. Loss of the Mn4CaO5 cluster allows increased flexibility of the D1 C-terminus, including 344Ala, which precludes structure determination in cryo-EM. Sphere color code: carbon, light brown; nitrogen, blue; oxygen, red and manganese, purple.

Acknowledgements
This work was supported by the US Department of Energy, Office of Basic Energy Sciences Grant DE-FG02-09ER20310 (to T.M.B. and L.K.F.).
| 1 | Oxidized residues which are present in PS II even in the absence of experimental photoinhibitory irradiation. These accumulate due to constitutive photodamage occurring under growth light conditions. |
| 2 | In the SI for this paper, the authors identify the modified residue as 3-hydroxy-L-histidine, in this communication we prefer the designation Nδ1-hydroxy-L-histidine. |
References
- Cardona, T.; Sedoud, A.; Cox, N.; Rutherford, A.W. Charge separation in Photosystem II: A comparative and evolutionary overview. Biochim Biophys. Acta 2012, 1817, 26–43. [Google Scholar] [CrossRef]
- Kok, B.; Forbush, B.; McGloin, M. Cooperation of charges in photosynthetic O2 evolution-I. A linear four step mechanism. Photochem Photobiol. 1970, 11, 457–475. [Google Scholar] [CrossRef]
- Vinyard, D.J.; Ananyev, G.M.; Dismukes, G.C. Photosystem II: The reaction center of oxygenic photosynthesis. Annu Rev. Biochem 2013, 82, 577–606. [Google Scholar] [CrossRef]
- Yano, J.; Yachandra, V. Mn4Ca cluster in photosynthesis: Where and how water is oxidized to dioxygen. Chem. Rev. 2014, 114, 4175–4205. [Google Scholar] [CrossRef]
- Murchie, E.H.; Ali, A.; Herman, T. Photoprotection as a trait for rice yield improvement: Status and prospects. Rice 2015, 8, 31. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef]
- Ohnishi, N.; Allakhverdiev, S.I.; Takahashi, S.; Higashi, S.; Watanabe, M.; Nishiyama, Y.; Murata, N. Two-step mechanism of photodamage to photosystem II: Step 1 occurs at the oxygen-evolving complex and step 2 occurs at the photochemical reaction center. Biochemistry 2005, 44, 8494–8499. [Google Scholar] [CrossRef] [PubMed]
- Hakala, M.; Tuominen, I.; Keränen, M.; Tyystjärvi, T.; Tyystjärvi, E. Evidence for the role of the oxygen-evolving manganese complex in photoinhibition of Photosystem II. Biochem Biophys. Acta 2005, 1706, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Tyystjärvi, E. Photoinhibition of Photosystem II and photodamage of the oxygen evolving manganese cluster. Coord. Chem. Rev. 2008, 252, 361–376. [Google Scholar] [CrossRef]
- Zavafer, A.; Cheah, M.H.; Hillier, W.; Chow, W.S.; Takahashi, S. Photodamage to the oxygen evolving complex of photosystem II by visible light. Sci. Rep. 2015, 5, 16363. [Google Scholar] [CrossRef]
- Weisz, D.A.; Gross, M.L.; Pakrasi, H.B. Reactive oxygen species leave a damage trail that reveals water channels in Photosystem II. Sci. Adv. 2017, 3, eaao3013. [Google Scholar] [CrossRef]
- Kale, R.; Hebert, A.E.; Frankel, L.K.; Sallans, L.; Bricker, T.M.; Pospisil, P. Amino acid oxidation of the D1 and D2 proteins by oxygen radicals during photoinhibition of Photosystem II. Proc. Natl. Acad. Sci. U S A 2017, 114, 2988–2993. [Google Scholar] [CrossRef]
- Theis, J.; Schroda, M. Revisiting the photosystem II repair cycle. Plant Signal. Behav. 2016, 11, e1218587. [Google Scholar] [CrossRef]
- Sundby, C.; McCaffery, S.; Anderson, J.M. Turnover of the Photosystem II D l Protein in Higher Plants under Photoinhibitory and Nonphotoinhibitory Irradiation. J. Biol. Chem. 1993, 268, 25476–25482. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.M.; Pakrasi, H.B. Advances in the Understanding of the Lifecycle of Photosystem II. Microorganisms 2022, 10, 836. [Google Scholar] [CrossRef]
- Komenda, J.; Sobotka, R.; Nixon, P.J. The biogenesis and maintenance of PSII: Recent advances and current challenges. Plant Cell 2024, 36, 3997–4013. [Google Scholar] [CrossRef]
- Ossenbühl, F.; Inaba-Sulpice, M.; Meurer, J.; Soll, J.; Eichacker, L.A. The Synechocystis sp. PCC 6803 Oxa1 homolog is essential for membrane integration of reaction center precursor protein pD1. Plant Cell 2006, 18, 2236–2246. [Google Scholar] [CrossRef]
- Komenda, J.; Nickelsen, J.; Tichý, M.; Prášil, O.; Eichacker, L.A. The cyanobacterial homologue of HCF136/YCF48 is a component of an early photosystem II assembly complex and is important for both the efficient assembly and repair of photosystem II in Synechocystis sp. PCC 6803. J. Biol. Chem. 2008, 283, 22390–22399. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.K.; Sallans, L.; Limbach, P.A.; Bricker, T.M. Identification of oxidized amino acid residues in the vicinity of the Mn(4)CaO(5) cluster of Photosystem II: implications for the identification of oxygen channels within the photosystem. Biochemistry 2012, 51, 6371–6377. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.K.; Sallans, L.; Limbach, P.A.; Bricker, T.M. Oxidized amino acid residues in the vicinity of Q(A) and Pheo(D1) of the Photosystem II reaction center: putative generation sites of reducing-side reactive oxygen species. PLoS ONE 2013, 8, e58042. [Google Scholar] [CrossRef]
- Keren, N.; Berg, A.; van Kan, P.J.M.; Levanon, H.; Ohad, I. Mechanism of photosystem II photoinactivation and D1 protein degradation at low light: the role of back electron flow. Proc. Natl. Acad. Sci. U S A 1997, 94, 1579–1584. [Google Scholar] [CrossRef]
- Yao, D.C.I.; Brune, D.C.; Vermaas, W.F.J. Lifetimes of photosystem I and II proteins in the cyanobacterium Synechocystis sp. PCC 6803. FEBS Lett. 586(212), 169–173. [CrossRef]
- Nixon, P.J.; Michoux, F.; Yu, J.; Boehm, M.; Komenda, J. Recent advances in understanding the assembly and repair of photosystem II. Ann. Bot. 2010, 106, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Panico, M.; Shipton, C.A.; Nilsson, F.; H.R. Morris, B. J. Primary Structure Characterization of the Photosystem II D1 and D2 Subunits. J. Biol. Chem. 1997, 272, 33158–33166. [Google Scholar] [CrossRef]
- Dreaden-Kasson, T.M.; Rexroth, S.; Barry, B.A. Light-induced oxidative stress, N-formylkynurenine, and oxygenic photosynthesis. PLoS ONE 2012, 7, e42220. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kuroda, H.; Ozawa, S.-I.; Saito, K.; Dogra, V.; Scholz, M.; Zhang, G.; de Vitry, C.; Ishikita, I.; Kim, C.; Hippler, M.; Takahashi, Y.; Sakamoto, W. Characterization of tryptophan oxidation affecting D1 degradation by FtsH in the photosystem II quality control of chloroplasts. eLife 2023, 88822.88822. [Google Scholar]
- Berthold, D.A.; Babcock, G.T.; Yocum, C.F. A highly resolved, oxygen-evolving Photosystem II preparation from spinach thylakoid membranes: EPR and electron-transport properties. FEBS Lett. 1981, 134, 231–234. [Google Scholar] [CrossRef]
- Zhao, Z.; Vercellino, I.; Whitelegge, J.P.; Maghlaoui, K.; Nixon, P.J.; Sazanov, L.A. Cryo-EM structures of naturally occurring dimeric photosystem II complexes lacking the Mn4CaO5 cluster. BioRxiv 2025. [Google Scholar] [CrossRef]
- Tyystjärvi, T.; Herranen, M.; Aro, E.-M. Regulation of translation elongation in cyanobacteria: membrane targeting of the ribosome nascent-chain complexes controls the synthesis of D1 protein. Mol. Microbiol. 2001, 40, 476–484. [Google Scholar] [CrossRef]
- Uchida, K.; Kawakishi, S. 2-Oxo-histidine as a novel biological marker for oxidatively modified proteins. FEBS Lett. 1993, 332, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Torbjörnsson, M.; Hagemann, M.M.; Ryde, U.; Hedegård, E.D. Histidine oxidation in lytic polysaccharide monooxygenase. J. Biol. Inorg. Chem. 2023, 28, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Yachandra, V.K. Mn₄Ca cluster in photosynthesis: where and how water is oxidized to dioxygen. Chem. Rev. 2014, 114, 4175–4205. [Google Scholar] [CrossRef]
- Pan, X.; Wang, H.; Cuiyu, L.; Zhang, J.Z.H.; Changge, J. MolGpka: a web server for small molecule pKa prediction using a graph-convolutional neural network. J. Chem. Info Model 2021, 61, 3159–3165. [Google Scholar] [CrossRef]
- Chu, H.A.; Nguyen, A.P.; Debus, R.J. Amino acid residues that influence the binding of manganese or calcium to photosystem II. 2. The carboxy-terminal domain of the D1 polypeptide. Biochemistry 1995, 34, 5859–5882. [Google Scholar] [CrossRef]
- Yano, J.; Walker, L.M.; Strickler, M.A.; Service, R.J.; Yachandra, V.K.; Debus, R.J. Altered Structure of the Mn₄Ca Cluster in the Oxygen-evolving Complex of Photosystem II by a Histidine Ligand Mutation. J. Biol. Chem. 2011, 286, 9257–9267. [Google Scholar] [CrossRef]
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystall Sec B 1985, 41, 244–247. [Google Scholar] [CrossRef]
- P. Comba, Strains and stresses in coordination compounds. Coord. Chem. Rev. 1999, 182, 343–371. [CrossRef]
- Gisriel, C.J.; Zhou, K.F.; Huang, H.L.; Debus, R.J.; Xiong, Y.; Brudvig, G.W. Cryo-EM structure of monomeric photosystem II from Synechocystis sp. PCC 6803 lacking the water-oxidation complex. Joule 2020, 4, 2131–2148. [Google Scholar] [CrossRef]
Figure 1.
The Hypothesis. Intact PS II dimers are subject to photodamage by ROS which oxidatively modify D1:332His (shown in red), leading to the loss of the Mn4CaO5 cluster. This leads to remodeling of the protein scaffold which includes loss of the interaction of 344Ala with the metal cluster, increased structural flexibility of the C-terminus of D1, structural reorientation of D2, CP43 and CP47 and loss of the extrinsic proteins (PsbO, PsbU and PsbV). Subsequently, the PS II dimers monomerize and the ROS-damaged monomers dissociate, yielding a CP43 repair module and a damaged CP43-less reaction center module along with their suites of accessory proteins. Subsequently, the oxidatively modified D1 protein is removed from the damaged CP43-less reaction center module and replaced by a nascent D1 subunit.
Figure 1.
The Hypothesis. Intact PS II dimers are subject to photodamage by ROS which oxidatively modify D1:332His (shown in red), leading to the loss of the Mn4CaO5 cluster. This leads to remodeling of the protein scaffold which includes loss of the interaction of 344Ala with the metal cluster, increased structural flexibility of the C-terminus of D1, structural reorientation of D2, CP43 and CP47 and loss of the extrinsic proteins (PsbO, PsbU and PsbV). Subsequently, the PS II dimers monomerize and the ROS-damaged monomers dissociate, yielding a CP43 repair module and a damaged CP43-less reaction center module along with their suites of accessory proteins. Subsequently, the oxidatively modified D1 protein is removed from the damaged CP43-less reaction center module and replaced by a nascent D1 subunit.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



