Submitted:
26 May 2026
Posted:
27 May 2026
You are already at the latest version
Abstract
limiting systemic exposure. We assessed whether co-therapy with the selective cyclooxygenase-2 (COX-2) inhibitor etoricoxib and the corticosteroid betamethasone provides antinociceptive and anti-edema activity in complete Freund’s adjuvant–induced arthritis (AIA) in rats. Methods: Male Wistar rats (n = 10/group) were allocated to seven groups: intact, AIA disease control, indomethacin 5 mg/kg, etoricoxib 8 mg/kg, betamethasone 0.022 mg/kg, low-dose combination (4 + 0.011 mg/kg) and full-dose combination (8 + 0.022 mg/kg), administered orally once daily from Day 4 to Day 28. Paw edema, von Frey withdrawal thresholds and clinical arthritis score were assessed longitudinally as area-under-the-curve (AUC). Terminal joint tissues were profiled for cytokines, prostaglandin pathway mediators and immune cell markers. Results: Both combinations reduced edema and improved mechanical thresholds versus disease control. The full-dose combination produced the greatest restoration of integrated mechanical sensitivity and arthritis index, exceeding either monotherapy, consistent with additive activity of two mechanistically complementary agents. The low-dose combination achieved improvements equivalent to full-dose monotherapies, a pattern consistent with a dose-reduction effect. Biomarker shifts indicated attenuated prostaglandin signaling and a pro-resolving cytokine balance, with macrophage-associated markers trending toward intact levels. Conclusions: These findings support further evaluation of etoricoxib–betamethasone co-therapy for acute inflammatory conditions.
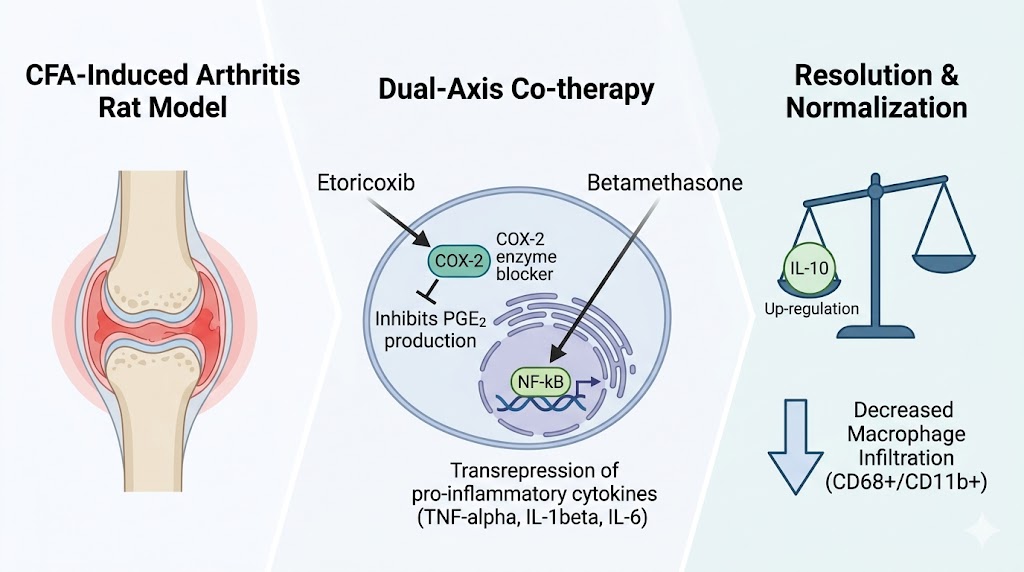
Keywords:
1. Introduction
2. Results
2.1. Induction of AIA-CFA Produced a Robust and Sustained Inflammatory Phenotype
2.2. Etoricoxib–Betamethasone Combinations Reduce Inflammatory Paw Edema
2.3. The Full-Dose Combination Achieves Superior Restoration of Mechanical Withdrawal Threshold
2.4. The Full-Dose Combination Produces the Greatest Reduction in Clinical Arthritis Severity
2.5. Terminal Mediator Profiling Supports Coordinated Dual-Axis Cytokine and Prostaglandin Modulation
2.6. Exploratory Flow Cytometry Suggests Macrophage-Associated Modulation by Betamethasone-Containing Regimens
3. Discussion
3.1. Integrated Efficacy and the Additive-vs-Dose-Reduction Interpretation
3.2. Mechanistic Substrate: Dual-Axis Cytokine and Prostaglandin Modulation
3.3. Translational Implications for Clinical Practice
3.4. Limitations
4. Materials and Methods
4.1. Study Design and Reporting
4.2. Animals and Housing
4.3. Ethical Approval
4.4. Induction of Adjuvant-Induced Arthritis
4.5. Treatments and Dose Selection
4.6. Paw Edema (Plethysmometry)
4.7. Mechanical Allodynia (Von Frey)
4.8. Clinical Arthritis Score
4.9. Enzyme-Linked Immunosorbent Assay (ELISA)
4.10. Flow Cytometry
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leask, M.P.; Crișan, T.O.; Ji, A.; Matsuo, H.; Köttgen, A.; Merriman, T.R. The pathogenesis of gout: molecular insights from genetic, epigenomic and transcriptomic studies. Nat. Rev. Rheumatol. 2024, 20, 510–523. [Google Scholar] [CrossRef]
- Poulsen, R.; Dalbeth, N. Gout and NLRP3 inflammasome biology. Arthritis Rheumatol. 2025, 77, 1317–1326. [Google Scholar] [CrossRef]
- Dalbeth, N.; Merriman, T.R.; Stamp, L.K. Gout. Lancet 2016, 388, 2039–2052. [Google Scholar] [CrossRef] [PubMed]
- Richette, P.; Doherty, M.; Pascual, E.; et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann. Rheum. Dis. 2017, 76, 29–42. [Google Scholar] [CrossRef]
- FitzGerald, J.D.; Dalbeth, N.; Mikuls, T.; et al. 2020 American College of Rheumatology guideline for the management of gout. Arthritis Care Res. 2020, 72, 744–760. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewé, R.B.M.; Bergstra, S.A.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann. Rheum. Dis. 2023, 82, 3–18. [Google Scholar] [CrossRef]
- Fraenkel, L.; Bathon, J.M.; England, B.R.; et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 1108–1123. [Google Scholar] [CrossRef] [PubMed]
- National Institute for Health and Care Excellence. Rheumatoid arthritis in adults: management. NICE guideline [NG100]; published July 2018, last updated 12 October 2020; NICE: London, UK, 2020; Available online: https://www.nice.org.uk/guidance/ng100 (accessed on 13 May 2026).
- Schumacher, H.R., Jr.; Boice, J.A.; Daikh, D.I.; et al. Randomised double blind trial of etoricoxib and indometacin in treatment of acute gouty arthritis. BMJ 2002, 324, 1488. [Google Scholar] [CrossRef]
- Matsumoto, A.K.; Melian, A.; Mandel, D.R.; et al. A randomized, controlled, clinical trial of etoricoxib in the treatment of rheumatoid arthritis. J. Rheumatol. 2002, 29, 1623–1630. [Google Scholar]
- Laine, L.; Curtis, S.P.; Cryer, B.; Kaur, A.; Cannon, C.P.; MEDAL Steering Committee. Assessment of upper gastrointestinal safety of etoricoxib and diclofenac in patients with osteoarthritis and rheumatoid arthritis in the Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL) programme: a randomised comparison. Lancet 2007, 369, 465–473. [Google Scholar] [CrossRef]
- Strehl, C.; Spies, C.M.; Buttgereit, F. Pharmacodynamics of glucocorticoids. Clin. Exp. Rheumatol. 2011, 29 (Suppl. 68), S13–S18. [Google Scholar] [PubMed]
- Stahn, C.; Löwenberg, M.; Hommes, D.W.; Buttgereit, F. Molecular mechanisms of glucocorticoid action and selective glucocorticoid receptor agonists. Mol. Cell. Endocrinol. 2007, 275, 71–78. [Google Scholar] [CrossRef]
- Janssens, H.J.E.M.; Janssen, M.; van de Lisdonk, E.H.; van Riel, P.L.C.M.; van Weel, C. Use of oral prednisolone or naproxen for the treatment of gout arthritis: a double-blind, randomized equivalence trial. Lancet 2008, 371, 1854–1860. [Google Scholar] [CrossRef]
- Rainer, T.H.; Cheng, C.H.; Janssens, H.J.E.M.; et al. Oral prednisolone in the treatment of acute gout: a pragmatic, multicenter, double-blind, randomized trial. Ann. Intern. Med. 2016, 164, 464–471. [Google Scholar] [CrossRef]
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-induced inflammation: relevance of prostaglandin E receptors. Biochim. Biophys. Acta 2015, 1851, 414–421. [Google Scholar] [CrossRef]
- Diaz-Jimenez, D.; Kolb, J.P.; Cidlowski, J.A. Glucocorticoids as Regulators of Macrophage-Mediated Tissue Homeostasis. Front. Immunol. 2021, 12, 669891. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Zimmermann, G.R.; Avery, W.; Finelli, A.L.; Farwell, M.; Fraser, C.C.; Borisy, A.A. Selective amplification of glucocorticoid anti-inflammatory activity through synergistic multi-target action of a combination drug. Arthritis Res. Ther. 2009, 11, R12. [Google Scholar] [CrossRef]
- Laboratorios Silanes S.A. de C.V. Efficacy and Safety Study for the Combination of Etoricoxib/Betamethasone Compared to Etoricoxib for the Treatment of Patients Diagnosed With Acute Gout Arthritis. ClinicalTrials.gov Identifier NCT06863701; Phase III, randomized, double-blind, multicenter trial; status: recruiting; first posted 2025. Available online: https://clinicaltrials.gov/study/NCT06863701 (accessed on 13 May 2026).
- Silanes, Laboratorios; S.A. de, C.V. Efficacy and Safety of Etoricoxib/Betamethasone Combination in Acute Bursitis, Tendinitis and Synovitis. Phase III, multicenter, randomized, double-blind, parallel-group trial of etoricoxib/betamethasone 90 mg/0.25 mg vs etoricoxib 90 mg once daily for 14 days. Available online: https://ctv.veeva.com/study/efficacy-and-safety-of-etoricoxib-betamethasone-combination-in-acute-bursitis-tendinitis-and-synovi (accessed on 13 May 2026).
- Stouten, V.; Westhovens, R.; Pazmino, S.; De Cock, D.; Van der Elst, K.; Joly, J.; Bertrand, D.; Verschueren, P. Five-year treat-to-target outcomes after methotrexate induction therapy with or without other csDMARDs and temporary glucocorticoids for rheumatoid arthritis in the CareRA trial. Ann. Rheum. Dis. 2021, 80, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Boers, M.; Hartman, L.; Opris-Belinski, D.; Bos, R.; Kok, M.R.; Da Silva, J.A.P.; Griep, E.N.; Klaasen, R.; Allaart, C.F.; Baudoin, P.; et al. Low dose, add-on prednisolone in patients with rheumatoid arthritis aged 65+: the pragmatic randomised, double-blind placebo-controlled GLORIA trial. Ann. Rheum. Dis. 2022, 81, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.; Mai, A.; Klaassen-Mielke, R.; Timmesfeld, N.; Trampisch, U.; Rudolf, H.; Baraliakos, X.; Schmitz, E.; Fendler, C.; Klink, C.; et al. The Efficacy of Short-Term Bridging Strategies With High- and Low-Dose Prednisolone on Radiographic and Clinical Outcomes in Active Early Rheumatoid Arthritis: A Double-Blind, Randomized, Placebo-Controlled Trial. Arthritis Rheumatol. 2022, 74, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Kvien, T.K.; Greenwald, M.; Peloso, P.M.; Wang, H.; Mehta, A.; Gammaitoni, A. Do COX-2 inhibitors provide additional pain relief and anti-inflammatory effects in patients with rheumatoid arthritis who are on biological disease-modifying anti-rheumatic drugs and/or corticosteroids? Post-hoc analyses from a randomized clinical trial with etoricoxib. BMC Musculoskelet. Disord. 2015, 16, 26. [Google Scholar] [CrossRef]
- Paglia, M.D.G.; Silva, M.T.; Lopes, L.C.; Barberato-Filho, S.; Mazzei, L.G.; Abe, F.C.; Bergamaschi, C.C. Use of corticoids and non-steroidal anti-inflammatories in the treatment of rheumatoid arthritis: Systematic review and network meta-analysis. PLoS ONE 2021, 16, e0248866. [Google Scholar] [CrossRef]
- Fischer, B.D.; Adeyemo, A.; O’Leary, M.E.; Bottaro, A. Animal models of rheumatoid pain: experimental systems and insights. Arthritis Res. Ther. 2017, 19, 146. [Google Scholar] [CrossRef]
- Mohd Noh, A.S.; Chuan, T.D.; Mohamed Khir, N.A.; Zin, A.A.M.; Ghazali, A.K.; Long, I.; Ab Aziz, C.B.; Ismail, C.A.N. Effects of different doses of complete Freund’s adjuvant on nociceptive behaviour and inflammatory parameters in polyarthritic rat model mimicking rheumatoid arthritis. PLoS ONE 2021, 16, e0260423. [Google Scholar] [CrossRef]
- National Institute for Health and Care Excellence. Gout: diagnosis and management. NICE guideline [NG219]; NICE: London, UK, June 2022; Available online: https://www.nice.org.uk/guidance/ng219 (accessed on 13 May 2026).








| Group | Edema AUC (mL·day) | p (vs AIA-CFA) Edema | von Frey AUC (g·day) | p (vs AIA-CFA) von Frey | Arthritis AUC (score·day) | p (vs AIA-CFA) Arthritis |
| Intact | 2.23 (8.33) | <0.0001 | 1431.73 (184.07) | <0.0001 | 0.00 (0.00) | <0.0001 |
| AIA-CFA | 16.37 (3.32) | — | 462.86 (56.86) | — | 43.74 (9.51) | — |
| Indomethacin | 7.45 (2.26) | 0.0001 | 1131.12 (273.04) | <0.0001 | 19.93 (8.01) | <0.0001 |
| Etoricoxib | 8.59 (3.55) | 0.0007 | 937.48 (223.80) | 0.0004 | 30.77 (5.55) | 0.0094 |
| Betamethasone | 8.89 (1.82) | 0.0013 | 884.15 (226.34) | 0.0024 | 34.04 (8.08) | 0.1097 |
| Combo-L | 7.16 (2.06) | <0.0001 | 1028.86 (276.61) | <0.0001 | 27.56 (8.12) | 0.0005 |
| Combo-H | 5.09 (1.33) | <0.0001 | 1252.01 (288.89) | <0.0001 | 18.23 (11.29) | <0.0001 |
| Endpoint | Intact (p vs AIA-CFA) | AIA-CFA | Indomethacin (p vs AIA-CFA) | Etoricoxib (p vs AIA-CFA) | Betamethasone (p vs AIA-CFA) | Combo-L (p vs AIA-CFA) | Combo-H (p vs AIA-CFA) |
| TNF-α (pg/mg protein) | 15.09 ± 2.31 (<0.0001) |
37.09 ± 4.68 (—) |
23.69 ± 3.39 (<0.0001) |
25.60 ± 3.17 (<0.0001) |
18.40 ± 1.86 (<0.0001) |
20.40 ± 2.81 (<0.0001) |
16.50 ± 1.77 (<0.0001) |
| IL-1β (pg/mg protein) | 3.80 ± 0.62 (<0.0001) |
7.90 ± 1.13 (—) |
6.00 ± 0.91 (<0.0001) |
5.20 ± 0.56 (<0.0001) |
4.00 ± 0.55 (<0.0001) |
4.30 ± 0.55 (<0.0001) |
3.90 ± 0.45 (<0.0001) |
| IL-6 (pg/mg protein) | 4.19 ± 0.63 (<0.0001) |
8.30 ± 0.88 (—) |
6.30 ± 0.82 (<0.0001) |
5.90 ± 0.67 (<0.0001) |
6.40 ± 0.60 (<0.0001) |
5.00 ± 0.48 (<0.0001) |
4.50 ± 0.50 (<0.0001) |
| IL-10 (pg/mg protein) | 58.20 ± 7.35 (<0.0001) |
27.40 ± 2.93 (—) |
32.10 ± 3.97 (N.S.) |
36.88 ± 5.65 (0.0101) |
50.19 ± 7.18 (<0.0001) |
46.31 ± 4.67 (<0.0001) |
54.61 ± 7.54 (<0.0001) |
| COX-2 (ng/mg protein) | 2.80 ± 0.43 (<0.0001) |
12.30 ± 1.55 (—) |
7.20 ± 1.03 (<0.0001) |
6.30 ± 0.78 (<0.0001) |
2.90 ± 0.29 (<0.0001) |
3.60 ± 0.50 (<0.0001) |
2.70 ± 0.29 (<0.0001) |
| PGE-2 (ng/mg protein) | 24.00 ± 3.94 (<0.0001) |
62.70 ± 6.65 (—) |
25.30 ± 2.99 (<0.0001) |
34.50 ± 3.42 (<0.0001) |
39.10 ± 3.95 (<0.0001) |
28.20 ± 4.34 (<0.0001) |
25.30 ± 3.01 (<0.0001) |
| CD3+ lymphocytes (%) | 4.60 ± 0.61 (N.S.) |
4.70 ± 0.97 (—) |
5.90 ± 1.17 (N.S.) |
4.70 ± 0.87 (N.S.) |
3.36 ± 1.36 (N.S.) |
4.48 ± 2.55 (N.S.) |
4.52 ± 1.97 (N.S.) |
| CD68+/CD11b+ macrophages (%) | 7.33 ± 2.51 (0.0375) |
17.17 ± 6.77 (—) |
9.61 ± 3.56 (N.S.) |
16.14 ± 6.06 (N.S.) |
7.47 ± 5.21 (0.0312) |
11.10 ± 7.00 (N.S.) |
8.76 ± 4.43 (N.S.) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



