Submitted:
20 May 2026
Posted:
20 May 2026
You are already at the latest version
Abstract
Type 2 diabetes mellitus (T2DM) is a highly prevalent and devastating chronic metabolic disease worldwide, with its pathogenesis centrally characterized by insulin resistance and pancreatic β-cell dysfunction. Accumulating evidence has demonstrated that glucose metabolic reprogramming represents an adaptive metabolic shift from oxidative phosphorylation to aerobic glycolysis in cells in response to a hyperglycemic microenvironment, which acts as an upstream core event driving the initiation and progression of T2DM. This review systematically elaborates the characteristics of glucose metabolic reprogramming in insulin-sensitive target organs under T2DM conditions, including the liver, skeletal muscle, adipose tissue and pancreatic β-cells, and further dissects four major downstream effector mechanisms: mitochondrial energy metabolism disturbance, augmented oxidative stress, disruption of mitochondria-associated endoplasmic reticulum membranes (MAMs) coupled with calcium homeostasis imbalance, and systemic inflammatory response. On this basis, we summarize the intervention strategies targeting the above signaling pathways, including antioxidant therapy, restoration of MAMs integrity and calcium homeostasis, systemic anti-inflammatory intervention, and multi-target regulatory effects of traditional Chinese medicine. Current studies have indicated that early intervention in downstream stress events induced by glucose metabolic reprogramming, particularly the preservation of MAMs integrity, restoration of calcium homeostasis, and inhibition of NLRP3 inflammasome activation, is expected to block or delay the progression from prediabetes to clinical T2DM. Nevertheless, substantial gaps still remain in the understanding of the dynamic regulatory mechanisms of MAMs, tissue-specific therapeutic targets, and relevant clinical translational research. Future integration of multi-omics technologies will provide novel therapeutic strategies and theoretical foundations for the early prevention and treatment of T2DM.
Keywords:
type 2 diabetes mellitus
; glucose metabolic reprogramming
; insulin resistance
; mitochondria‐associated endoplasmic reticulum membranes
; oxidative stress
1. Introduction
Diabetes mellitus (DM) is one of the most severe non-communicable chronic diseases that seriously endanger human health. According to the latest statistics released by the International Diabetes Federation (IDF), the global prevalence of DM reached approximately 589 million cases in 2024, and this figure is projected to rise to 853 million by 2050, with type 2 diabetes mellitus (T2DM) accounting for over 90% of all diabetic cases [1]. Emerging clinical evidence has demonstrated that patients with DM exhibit an increased susceptibility to cancers related to the digestive and reproductive systems. Additionally, these individuals face a higher risk of respiratory tract infections, cutaneous infections, and even systemic soft-tissue infections. Given its high mortality and disability rates, T2DM imposes a substantial economic burden on individual families and the entire healthcare system [2].
As a prevalent chronic metabolic disorder, T2DM has a sophisticated pathogenesis predominantly characterized by disrupted glucose homeostasis and insulin resistance. The core pathological mechanisms mainly include two aspects: the impaired insulin secretion capacity of pancreatic β-cells that disrupts systemic glucose regulation, and the diminished insulin responsiveness of insulin-sensitive target organs, which collectively exacerbate disease progression [3], and abnormal metabolic signal transduction invariably impairs the regulatory function of this axis. Meanwhile, sustained glucolipid metabolic disturbance triggers glucotoxicity and lipotoxicity, which act as central pathological stimuli to initiate glucose metabolic reprogramming in insulin-sensitive organs, including the liver, skeletal muscle, adipose tissue, and pancreatic β-cells. Such metabolic alterations occur in multiple biological processes, such as glucose uptake, glycolysis, and oxidative phosphorylation. Mechanistically, the dysregulated glucose metabolism disrupts cellular energy homeostasis and further provokes a cascade of downstream pathological abnormalities, including chronic inflammatory responses, excessive oxidative stress, mitochondrial dysfunction, activated endoplasmic reticulum stress, and aberrant mitochondria-associated endoplasmic reticulum membranes (MAMs). These detrimental pathological changes synergistically aggravate insulin resistance and deteriorate pancreatic β-cell function, ultimately accelerating the onset and progression of T2DM.
The concept of metabolic reprogramming was initially proposed in cancer therapy. It refers to the metabolic rewiring of signaling pathways in tumor cells, which remodels the flux and distribution of nutrients to satisfy energy demands and anabolic requirements, thereby sustaining tumor cell survival and proliferation [4], Emerging evidence has revealed that cellular metabolic pathways and metabolites undergo adaptive modulation under pathological conditions of cardiovascular, central nervous system, renal, and respiratory diseases [5,6,7,8,9]. This metabolic remodeling is characterized by impaired mitochondrial biogenesis, enhanced glycolysis, and dysregulated lipid and amino acid metabolism, which has been recognized as a pivotal driver facilitating the development and progression of diabetes.
2. Glucose Metabolic Reprogramming: A Core Metabolic Signature of T2DM
Dysregulated feedback modulation between insulin secretion and peripheral insulin action leads to persistent hyperglycemia and continuously promotes the pathophysiological progression of T2DM. On the one hand, the progressive deterioration of pancreatic β-cell function causes insulin secretory defects, impairing the intrinsic capacity for blood glucose homeostasis. On the other hand, insulin resistance markedly inhibits glucose uptake and utilization in the liver, skeletal muscle, and adipose tissue, while concurrently enhancing hepatic gluconeogenesis, thereby further exacerbating metabolic dysregulation. This review systematically summarizes the regulatory roles of organ-specific mediators derived from major insulin-sensitive tissues (e.g., liver, skeletal muscle, and adipose tissue), as well as insulin-secreting regulatory organs including the pancreas and intestine, in the maintenance of glucose metabolic homeostasis.
Diabetes mellitus is tightly correlated with the progression of multiple liver diseases, covering a spectrum of pathological stages ranging from fatty liver and non-alcoholic fatty liver disease (NAFLD) to hepatic fibrosis, cirrhosis, and even hepatocellular carcinoma. Under physiological conditions, insulin effectively suppresses hepatic gluconeogenesis and modulates lipolysis. Nevertheless, in the diabetic pathological microenvironment, hepatic insulin sensitivity is markedly diminished, accompanied by disrupted insulin signaling. Such abnormalities contribute to excessive hepatic gluconeogenesis and sustained hyperglycemia, alongside dysregulated lipolysis that triggers excessive lipid accumulation in hepatocytes. Aberrant intracellular lipid deposition further induces oxidative stress, thereby causing hepatocellular dysfunction and injury. Damaged hepatocytes trigger the activation of Kupffer cells, which secrete abundant pro-inflammatory cytokines and initiate persistent low-grade chronic inflammation. In response to hepatic tissue damage, hepatic stellate cells synthesize and accumulate excessive extracellular matrix, ultimately resulting in progressive liver fibrosis and irreversible hepatic impairment [10].
Skeletal muscle constitutes one of the predominant insulin-sensitive tissues in the human body and serves as the central effector tissue mediating insulin-dependent glucose uptake; accordingly, skeletal muscle health directly determines systemic glucose homeostasis [11]. As critical myogenic progenitor cells responsible for the maintenance of muscle tissue integrity, skeletal muscle satellite cells exert essential functions in muscle fiber repair, remodeling, and the modulation of tissue plasticity [12,13]. Accumulating evidence has validated that diabetic conditions markedly impair the abundance and functional activity of skeletal muscle satellite cells, thereby triggering the onset and progression of diabetic myopathy and accelerating the deterioration rate of diabetic comorbidities [14].
Adipose tissue acts as a pivotal organ for systemic energy storage. Subcutaneous adipose tissue is abundant in mitochondria with thermogenic capacity [15,16], and its biological function is tightly linked to the improvement of metabolic homeostasis and insulin sensitivity. Once energy intake exceeds the storage threshold of subcutaneous adipose tissue, excessive lipids undergo ectopic deposition in multiple tissues and organs, including the liver, vascular walls, and skeletal muscle, which ultimately facilitates the initiation and progression of insulin resistance [17,18,19]. Beyond energy storage, adipose tissue also exerts crucial endocrine functions and secretes adiponectin, diverse cytokines, and bioactive lipid molecules. Both adipose tissue and tissue-infiltrating macrophages can robustly release pro-inflammatory mediators, such as interleukin-6, tumor necrosis factor-α, resistin, visfatin, retinol-binding protein 4, and leptin [20,21]. The expression levels of the above factors are positively correlated with systemic fat mass, which aggravates insulin resistance and accelerates T2DM pathological progression by mediating persistent systemic low-grade chronic inflammation. Collectively, T2DM is characterized by systematic glucose metabolic dysregulation. Specific glucose metabolic reprogramming occurs in key target organs, including the liver, skeletal muscle, adipose tissue, and pancreatic β-cells, which jointly constitute the core metabolic phenotype driving disease progression.
3. Core Downstream Mechanisms of Glucose Metabolic Reprogramming
3.1. Imbalanced Energy Metabolism
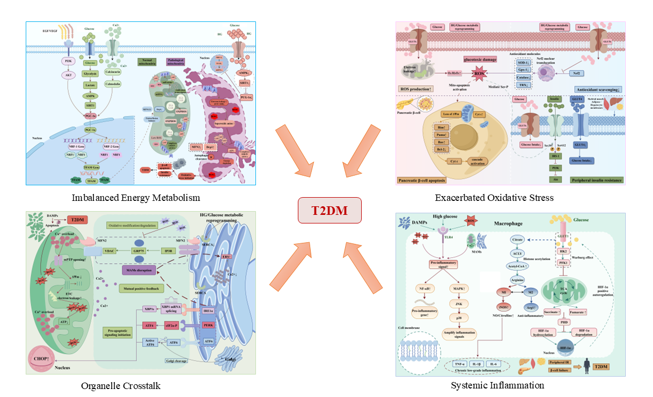
Glucose metabolic reprogramming is initially manifested as decreased mitochondrial oxidative phosphorylation efficiency and disrupted mitochondrial dynamics [22]. The core of mitochondrial dysfunction lies in the systematic imbalance of nutrient signal transduction, energy production, and redox metabolism, ultimately leading to inadequate adenosine triphosphate (ATP) synthesis and subsequent metabolic crisis [23,24]. In response to systemic metabolic disorders, mitochondria undergo a spectrum of pathological alterations, including aberrant substrate oxidation, impaired calcium buffering, disrupted iron transport, morphological dynamic remodeling, reactive oxygen species (ROS) burst, and activation of cellular apoptotic signaling cascades [25,26]. These pathological changes are intuitively illustrated in Figure 1.
Mitochondrial biogenesis and dynamic regulation serve as two indispensable pillars for the maintenance of mitochondrial functional homeostasis. Mitochondria originate from healthy eukaryotic cells and expand their quantity through the fission of fully mature and genetically replicated mitochondrial organelles. As a master regulator of mitochondrial biogenesis, peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) responds to the stimulation of tyrosine kinase receptors and natriuretic peptide receptors as well as nitric oxide. Subsequent post-translational modifications include sirtuin 1 (SIRT1)-mediated deacetylation and activation triggered by alterations in the adenosine monophosphate/adenosine triphosphate (AMP/ATP) ratio via adenosine monophosphate-activated protein kinase (AMPK) [27] Combined with the modulation of calmodulin signaling, PGC-1α sequentially activates downstream nuclear transcription factors, such as nuclear respiratory factor 1/2 (NRF1/2) and estrogen-related receptor-α (ERR-α), thereby initiating de novo mitochondrial biogenesis [28,29]。Meanwhile, AMPK and SIRT1 form a metabolic sensing circuit that further promotes PGC-1α phosphorylation and enhances its transcriptional activity [30,31]. At the dynamic regulatory level, the physiological balance between mitochondrial fusion and fission is precisely modulated by multiple GTPases. Mitofusin 1/2 (MFN1/2) mediates the fusion of the mitochondrial inner and outer membranes [32],while optic atrophy 1 (OPA1) anchors to the inner mitochondrial membrane to facilitate inner membrane remodeling [33]. As a crucial GTPase fission mediator, dynamin-related protein 1 (DRP1) translocates from the cytoplasm to the outer mitochondrial membrane to drive mitochondrial fission, which guarantees the selective autophagic clearance of damaged mitochondria and maintains cellular homeostasis under stress conditions [34].
Under hyperglycemic conditions, this tightly regulated mitochondrial network pathologically deviates toward a detrimental phenotype. Hyperglycemia suppresses PGC-1α expression, downregulates the fusion protein MFN2, and abnormally activates the fission protein DRP1. These alterations result in excessive mitochondrial fragmentation and impaired autophagic clearance. Additionally, the electron leakage of mitochondrial respiratory chain complex I and III is markedly increased, leading to massive superoxide anion generation. Such pathological changes exert dual detrimental consequences: insufficient ATP synthesis induces cellular energy deprivation, accompanied by robust overproduction of mitochondrial-derived ROS. Accordingly, mitochondrial dysfunction acts as an initiating event among all downstream pathological cascades triggered by glucose metabolic reprogramming. Mitochondria-derived ROS further initiate successive oxidative stress and organelle stress cascades, ultimately facilitating T2DM progression [35]。Clinical evidence has demonstrated that the dysregulated glucolipid microenvironment in T2DM patients markedly inhibits the AMPK-SIRT1-PGC-1α signaling axis [36], thereby repressing lipid oxidation and exacerbating lipid-induced insulin resistance [37]. In vivo animal models have revealed that hyperglycemia induces pathological mitochondrial abnormalities through epigenetic modifications and post-translational regulation. On the one hand, enhanced H4K12 lactylation activates the Foxo1 signaling pathway, aggravating mitochondrial oxidative stress and promoting mitochondrial fragmentation alongside defective autophagic clearance in the hippocampus, which contributes to diabetes-associated cognitive impairment [38]. On the other hand, impaired SIRT1-mediated PGC-1α deacetylation suppresses PGC-1α transcriptional activity, causing insufficient hepatic ATP synthesis and subsequently driving hepatic steatosis. Functional intervention experiments have further validated that SIRT1 activation or Foxo1 inhibition can partially reverse these pathological abnormalities [39]. In vitro experiments using high-glucose-treated 3T3-L1 adipocytes have further verified that hyperglycemia reduces mitochondrial mass and decreases the protein levels of mitochondrial respiratory chain complex III and IV. Hyperglycemia also inhibits AMPK phosphorylation, thereby reducing PGC-1α abundance and accelerating mitochondrial fragmentation in adipocytes [40].
3.2. Exacerbated Oxidative Stress
Excessive mitochondrial-derived reactive oxygen species (ROS) generation coupled with dysregulated endogenous antioxidant defense systems collectively renders oxidative stress a core executor of glucotoxic damage, as depicted in Figure 2. In terms of T2DM-related pathological mechanisms, hyperglycemia is tightly correlated with markedly elevated oxidative stress levels. Reactive substances including hydrogen peroxide (H₂O₂) and superoxide anion (O₂⁻) have been recognized as pivotal driving factors in the pathogenesis of diabetic complications [41,42]. Acute hyperglycemia facilitates the massive influx of glucose into endothelial cells (ECs) via glucose transporters (GLUTs). On the one hand, excessive glucose triggers mitochondrial dysfunction through the polyol pathway; on the other hand, hyperglycemia induces endothelial nitric oxide synthase (eNOS) uncoupling, which synergistically amplifies ROS overproduction [43,44]. Accumulating studies have confirmed that pancreatic β-cells inherently exhibit low expression levels of endogenous antioxidant enzymes, rendering them highly susceptible to oxidative stress. Accordingly, the hyperglycemic state triggers ROS burst accompanied by the depletion of multiple antioxidant molecules, including SOD-1, Gpx-1, catalase, PON, and TRX [45,46]. Mechanistically, ROS constitute the metabolic basis for oxidative stress responses. In insulin-sensitive target tissues, ROS directly participate in the initiation of insulin resistance by oxidatively modifying key molecules involved in insulin signaling cascades. It has been validated that ROS can induce the phosphorylation of insulin receptor substrate-1 (IRS-1) at the Ser307/Ser612 sites, thereby inhibiting the downstream PI3K/Akt signaling pathway and ultimately triggering peripheral insulin resistance [47].
In mammalian cells, the extrinsic death receptor pathway and the intrinsic mitochondria-dependent (Bcl-2-regulated) pathway serve as the central mechanisms underlying ROS-mediated β-cell injury. Excessive ROS accumulation and persistent oxidative stress exert critical pathogenic roles in DM progression. Mechanistically, ROS exacerbate oxidative stress and trigger pancreatic β-cell apoptosis by modulating the expression of the Bcl-2-associated athanogene 3 (BAG3)/nuclear factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) signaling axis, thereby aggravating diabetic complications [48]. In vivo and in vitro intervention experiments have further demonstrated that the overexpression of anti-apoptotic Bcl-2 proteins, as well as the deficiency of the BH3-only proteins (Bim and Puma) and their downstream apoptotic effector Bax, can markedly alleviate high glucose or ribose-induced pancreatic cell death [49,50]. Molecularly, ROS facilitate the loss of mitochondrial outer membrane potential, cytochrome c release, and subsequent activation of the caspase cascade. Clinical and animal model studies have verified that the mRNA levels of pro-apoptotic BH3-only molecules are significantly upregulated in the pancreatic islets of T2DM patients and diabetic mice, indicating that hyperglycemia integrates endoplasmic reticulum stress and mitochondrial oxidative stress to activate the Bim/Puma signaling axis and induce β-cell apoptosis [51]. As a pivotal signaling amplification hub, oxidative stress exerts detrimental effects beyond mitochondrial dysfunction. It further propagates pathological damage to the endoplasmic reticulum and MAMs, laying a critical pathological foundation for subsequent organelle crosstalk dysfunction.
3.3. Organelle Crosstalk
Organelles such as mitochondria and the endoplasmic reticulum are not independently distributed within the cytoplasm but structurally coupled with cell membranes. Accumulating evidence has confirmed that mitochondria can physically interact with the endoplasmic reticulum (ER), Golgi apparatus, and lipid droplets [52]. Mitochondria-associated endoplasmic reticulum membranes (MAMs) represent a specialized structural contact region between mitochondria and the ER. Transmission electron microscopy observations have revealed an intermembrane distance of approximately 10 nm between mitochondria and the smooth ER, and around 25 nm between mitochondria and the rough ER [53], The two organelles are physically interconnected by coupling structures composed of MAM proteins and polymeric complexes, which exert indispensable biological functions in calcium exchange, mitophagy, endoplasmic reticulum stress, and glucolipid metabolism [53,54,55]. Numerous resident proteins are enriched in MAMs, including mitofusin 2 (MFN2), glucose-regulated protein 75 (Grp75), protein kinase R-like endoplasmic reticulum kinase (PERK), and inositol 1,4,5-trisphosphate receptor (IP3R) [56,57,58]. Abnormal expression and dysfunction of these MAM-resident proteins contribute to the initiation and progression of multiple diseases, particularly T2DM-related pathological alterations, as intuitively illustrated in Figure 3.
Under physiological conditions, MAMs facilitate the transport of calcium ions (Ca²⁺) from the ER to mitochondria to maintain mitochondrial calcium homeostasis. Most enzymes involved in glucolipid biosynthesis are anchored on the ER membrane, while a small proportion are located on the mitochondrial membrane [59,60]. Therefore, various lipid intermediates rely on MAMs-mediated trafficking to complete biosynthetic pathways. Meanwhile, MAMs initiate the unfolded protein response to resolve endoplasmic reticulum stress and sustain cell survival [61]. When oxidative stress signals propagate to the endoplasmic reticulum, they trigger endoplasmic reticulum stress (ERS) and disrupt MAMs integrity. Pancreatic β-cells are inherently vulnerable to ERS due to their heavy burden of insulin synthesis and secretion, and this susceptibility can be further aggravated under hyperglycemic conditions. As an indispensable trafficking platform in insulin signaling, the MAMs interface harbors key insulin signaling proteins including GSK3β and AKT; AKT phosphorylation markedly inhibits MAMs formation and subsequently induces insulin resistance (IR). Mechanistically, sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) actively pumps cytoplasmic Ca²⁺ into the ER lumen for storage [62], Under oxidative stress, SERCA activity is suppressed, leading to ER calcium depletion and the activation of three canonical ERS sensor pathways, namely PERK, IRE1 and ATF6 [63], which further upregulate the pro-apoptotic factor CHOP. These pathological signals interact with MAMs-associated proteins MFN2, IP3R and Grp75, thereby inducing disordered Ca²⁺ translocation [64,65]. The resultant mitochondrial calcium overload opens the mitochondrial permeability transition pore (mPTP), causing mitochondrial membrane potential collapse, arrested ATP synthesis and excessive ROS production [66]. Notably, a bidirectional positive feedback loop exists between ERS and MAMs disruption: abnormal MAMs exacerbate ER Ca²⁺ dyshomeostasis and amplify ERS, while ERS further degrades MAMs-related proteins in an IRE1α-dependent manner to compromise MAMs integrity. This vicious inter-organelle cycle amplifies intracellular stress responses, which ultimately transmit to immune cells via the release of ROS and damage-associated molecular patterns (DAMPs), thereby triggering systemic inflammatory responses.
Clinical evidence has demonstrated that mutations in presenilin 1/2 (PS1/PS2) associated with familial Alzheimer’s disease (AD) not only alter protein processing, but also directly impair SERCA pump function and the kinetics of endoplasmic reticulum Ca²⁺ leakage, ultimately activating the pro-apoptotic PERK-CHOP signaling axis [67]. Similarly, in the context of diabetic nephropathy, reduced Mfn2 expression has been observed in high glucose-treated human podocytes, which disrupts the interaction between Mfn2 and PERK and impairs ER–mitochondria contact [68]. Moreover, knockdown of Grp75 in HuH7 cells suppresses the interactions of IP3R-VDAC and IP3R-Grp75, thereby disrupting ER–mitochondria communication [69]. Collectively, these findings indicate that disrupted ER–mitochondrial crosstalk may serve as a common mechanism underlying the aberrant activation of the unfolded protein response in multiple metabolic and neurodegenerative diseases. In pancreatic β-cells, ERS activates the PERK-ATF4-CHOP pathway and upregulates TRB3, a negative regulator of Akt signaling, which further reduces insulin sensitivity and promotes β-cell apoptosis. Meanwhile, excessive Ca²⁺ release from the endoplasmic reticulum into the cytoplasm activates the NLRP3 inflammasome, directly linking ERS to the characteristic chronic low-grade inflammation observed in T2DM [70].
3.4. Systemic Inflammation
Macrophages are widely distributed throughout various tissues. Tissue-resident macrophages originate from embryonic development and circulating monocytes, serving as essential components of the innate immune system [71]. They are capable of recognizing and responding to stress signals derived from damaged organelles, such as ROS and DAMPs, and act as the first line of defense against microorganisms and other endogenous or exogenous threats to homeostasis, thereby initiating inflammatory responses [72,73]. The inflammatory response of macrophages is highly dependent on metabolic reprogramming, especially the enhancement of glycolytic flux, which supports the synthesis and secretion of inflammatory mediators. Once the threat is eliminated, the metabolic pattern of macrophages shifts from glycolysis to oxidative phosphorylation to facilitate tissue repair and inflammation resolution. Macrophages exhibit high phenotypic plasticity and are primarily polarized into two functional subtypes under pathophysiological conditions. Classically activated M1 macrophages rely predominantly on aerobic glycolysis for energy supply and secrete abundant pro-inflammatory cytokines, whereas alternatively activated M2 macrophages are characterized by oxidative phosphorylation and fatty acid oxidation [74], participating in anti-inflammatory processes and tissue remodeling. A growing body of domestic and international consensus has confirmed that the activation status of macrophages is closely implicated in the onset and progression of T2DM, as illustrated in Figure 4 [75].
Under hyperglycemic conditions, macrophages undergo intrinsic glucose metabolic reprogramming, which drives their polarization toward the pro-inflammatory M1 phenotype. Mechanistically, high glucose enhances glycolytic flux in macrophages, enabling cells to preferentially generate ATP through glycolysis even under normoxia, a phenomenon known as the Warburg effect. Hypoxia-inducible factor-1α (HIF-1α) acts as a master transcriptional regulator of glycolysis and anabolic metabolism. It upregulates the expression of key glycolytic molecules, including glucose transporter 1 (GLUT1), hexokinase 2 (HK2), and phosphofructokinase 1 (PFK1), thereby promoting glucose uptake and glycolytic flux to compensate for energy deficiency caused by impaired tricarboxylic acid (TCA) cycle function. Massive glycolysis drives the conversion of accumulated pyruvate toward lactate production. Meanwhile, the TCA cycle is blocked at the α-ketoglutarate dehydrogenase and succinate dehydrogenase sites, leading to substantial accumulation of intermediate metabolites such as succinate and fumarate. As a structural analog of α-ketoglutarate, succinate competitively inhibits prolyl hydroxylase (PHD) activity, blocks proline hydroxylation and von Hippel-Lindau (VHL)-mediated degradation of HIF-1α, stabilizes HIF-1α protein, and consequently upregulates the gene expression of IL-1β and NF-κB [76,77]. Accumulating evidence has shown that following TLR4 activation, macrophages convert glucose-derived citrate into acetyl-CoA via ATP citrate lyase (ACLY), which promotes histone hyperacetylation and further modulates the transcription of inflammatory genes [78]. In M1-polarized macrophages, upregulated inducible nitric oxide synthase (iNOS) catalyzes arginine catabolism into citrulline and nitric oxide to facilitate the clearance of intracellular pathogens, whereas M2 macrophages exhibit elevated arginase-1 (Arg1) expression. Glycolysis-derived pyruvate is further metabolized to generate citrate and acetyl-CoA, which provide substrates for histone acetylation, open the chromatin structure of M1 signature genes such as TNF-α, IL-6 and iNOS, and enhance their transcriptional activity. Meanwhile, upstream ROS and Ca²⁺ signals synergize with TLR4-mediated cascades to co-activate the NF-κB and MAPK signaling pathways, further amplifying pro-inflammatory responses. Polarized M1 macrophages robustly secrete pro-inflammatory cytokines including TNF-α, IL-1β and IL-6, establishing a metabolically driven chronic low-grade inflammatory state. This process serves as a critical hub linking intracellular metabolic stress to systemic tissue injury. Released inflammatory factors act on the liver, skeletal muscle, adipose tissue and pancreatic islets through endocrine and paracrine manners, ultimately inducing peripheral insulin resistance and pancreatic β-cell apoptosis as the terminal pathological outcomes.。
Meanwhile, accumulating in vivo and in vitro studies have further demonstrated that anti-inflammatory pharmacological intervention can effectively inhibit the PI3K/AKT/mTORC1 signaling pathway and glycolysis in mouse macrophages, thereby blocking M1 macrophage polarization [79], Such treatment also markedly suppresses lipocalin-2 expression in LPS-stimulated macrophages and microglia, reduces nitric oxide production as well as the expression levels of pro-inflammatory mediators including iNOS and COX-2, and alleviates neuroinflammation, ROS generation and phagocytic activity [80], In addition, anti-inflammatory agents promote M2 macrophage polarization and upregulate the expression of endogenous antioxidants [81].
4. Tissue-Specific Heterogeneity of Targeted Insulin Resistance
Insulin resistance constitutes the core pathological hallmark of T2DM and exhibits pronounced heterogeneity across different tissues [82]. The liver, skeletal muscle, and adipose tissue display distinct tissue-specific molecular mechanisms in terms of impaired insulin signaling transduction, aberrant glucolipid metabolism, and inflammatory responses, as illustrated in Figure 5.
4.1. Hepatic Steatosis-Driven Insulin Resistance
Under physiological conditions, insulin promotes glucose uptake mediated by glucose transporters in hepatocytes, myocytes and adipocytes. However, in the setting of hepatic steatosis, the glucose uptake capacity of hepatocytes is impaired, and physiological levels of insulin fail to efficiently reduce blood glucose [83]. Accordingly, disordered hepatic glucolipid metabolism triggers oxidative stress and hepatocellular injury, which further activates Kupffer cells to secrete abundant inflammatory cytokines, ultimately resulting in the development of hepatic insulin resistance and subsequent T2DM progression.
Figure 5.
Tissue-Specific Heterogeneity of Targeted Insulin Resistance. FFA: free fatty acid; NEFA: non-esterified fatty acid; BCAA: branched-chain amino acid; SCFA: short-chain fatty acid; IR: insulin resistance; DAG: diacylglycerol; PKCα/θ/ζ: protein kinase C α/θ/ζ; IRK: insulin receptor kinase; IRS1/2: insulin receptor substrate 1/2; PI3K: phosphatidylinositol 3-kinase; AKT: protein kinase B; PP2A: protein phosphatase 2A; aPKCλ/ζ: atypical protein kinase C λ/ζ; ROS: reactive oxygen species; JNK: c-Jun N-terminal kinase; FoxO1: forkhead box protein O1; PEPCK: phosphoenolpyruvate carboxykinase; G6Pase: glucose-6-phosphatase; GLUT4: glucose transporter 4; ERS: endoplasmic reticulum stress; TCA cycle: tricarboxylic acid cycle; OXPHOS: oxidative phosphorylation; mtROS: mitochondrial reactive oxygen species; mtDNA: mitochondrial DNA; NLRP3: NLR family pyrin domain containing 3; HIF-α: hypoxia-inducible factor α; mTOR: mechanistic target of rapamycin; M1: classically activated macrophage; TNF-α: tumor necrosis factor-α; IL-1β: interleukin-1β; IL-6: interleukin-6; UCP2: uncoupling protein 2; MD: mitochondrial dysfunction; ATP: adenosine triphosphate; GSIS: glucose-stimulated insulin secretion; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; T2DM: type 2 diabetes mellitus.
Figure 5.
Tissue-Specific Heterogeneity of Targeted Insulin Resistance. FFA: free fatty acid; NEFA: non-esterified fatty acid; BCAA: branched-chain amino acid; SCFA: short-chain fatty acid; IR: insulin resistance; DAG: diacylglycerol; PKCα/θ/ζ: protein kinase C α/θ/ζ; IRK: insulin receptor kinase; IRS1/2: insulin receptor substrate 1/2; PI3K: phosphatidylinositol 3-kinase; AKT: protein kinase B; PP2A: protein phosphatase 2A; aPKCλ/ζ: atypical protein kinase C λ/ζ; ROS: reactive oxygen species; JNK: c-Jun N-terminal kinase; FoxO1: forkhead box protein O1; PEPCK: phosphoenolpyruvate carboxykinase; G6Pase: glucose-6-phosphatase; GLUT4: glucose transporter 4; ERS: endoplasmic reticulum stress; TCA cycle: tricarboxylic acid cycle; OXPHOS: oxidative phosphorylation; mtROS: mitochondrial reactive oxygen species; mtDNA: mitochondrial DNA; NLRP3: NLR family pyrin domain containing 3; HIF-α: hypoxia-inducible factor α; mTOR: mechanistic target of rapamycin; M1: classically activated macrophage; TNF-α: tumor necrosis factor-α; IL-1β: interleukin-1β; IL-6: interleukin-6; UCP2: uncoupling protein 2; MD: mitochondrial dysfunction; ATP: adenosine triphosphate; GSIS: glucose-stimulated insulin secretion; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; T2DM: type 2 diabetes mellitus.

Epidemiological and multi-omics studies have revealed that elevated circulating levels of triglycerides, phosphatidylethanolamine, ceramides and certain branched-chain amino acids are closely associated with the typical insulin resistance phenotype of T2DM [84,85,86]. During the development of insulin resistance, the concentrations of branched-chain amino acids rise rapidly, accompanied by sustained increases in major unsaturated non-esterified fatty acids (NEFAs). NEFAs and their metabolites not only act as allosteric regulators of hepatic gluconeogenesis but also serve as inhibitory signaling molecules in peripheral tissues [87,88]. Clinical studies have further demonstrated that the abundance of sn-1,2-diacylglycerol (DAG) is markedly elevated in the hepatic plasma membrane of insulin-resistant patients [89]. In vivo and in vitro experiments have confirmed that plasma membrane DAG accumulation in adipocytes, cardiomyocytes and hepatocytes primarily originates from fatty acid re-esterification. Accumulated DAG activates novel protein kinase C (PKC) isoforms, which catalyze inhibitory phosphorylation of upstream molecules in the insulin signaling cascade [90,91]. Under the hyperglycemic and hyperlipidemic microenvironment, the liver uptakes excessive NEFAs, which are re-esterified to generate DAG. Subsequent DAG accumulation in the hepatocyte plasma membrane activates PKCε. Activated PKCε induces phosphorylation of insulin receptor kinase (IRK) at the Thr1160 site, thereby blocking IRK autophosphorylation at Tyr1162 and disrupting proximal insulin signaling. Impaired insulin signaling relieves the inhibition of the transcription factor FoxO1, which undergoes enhanced nuclear translocation and transcriptionally upregulates phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase), ultimately driving excessive hepatic gluconeogenesis [92]. Meanwhile, ceramides suppress distal insulin signaling via two distinct mechanisms: on the one hand, ceramides allosterically activate protein phosphatase 2A (PP2A) to induce dephosphorylation of AKT at the Thr308/Ser473 sites; on the other hand, ceramides stimulate atypical PKC isoforms (aPKCλ and aPKCζ) to hinder the plasma membrane recruitment and activation of AKT.
Excessively elevated free fatty acid (FFA) levels in hepatocytes markedly enhance mitochondrial β-oxidation [93]. Long-term overloading of this metabolic pathway readily disrupts fatty acid homeostasis and subsequently induces mitochondrial dysfunction. Incomplete fatty acid oxidation synergizes with endoplasmic reticulum stress to promote hydrogen peroxide (H₂O₂) release, alter cellular redox status, and trigger robust ROS overproduction [94], which initiates lipid peroxidation and may eventually cause mitochondrial DNA damage [95]. Excessive ROS further impairs electron transport chain function through redox-sensitive kinases such as ASK1-JNK, thereby aggravating the impairment of insulin signaling transduction [10]. Meanwhile, hepatocytes overproduce pro-inflammatory cytokines, including TNF-α, IL-1β and IL-6, which amplify local inflammatory responses via paracrine and autocrine pathways. The synergistic effect of the above mechanisms blocks downstream insulin signaling cascades, promotes excessive hepatic glucose output, and ultimately drives the development of hepatic insulin resistance.
4.2. Skeletal Muscle Insulin Resistance: Core Mechanisms of Impaired Glucose Uptake
Skeletal muscle is one of the largest metabolic organs in the human body, accounting for approximately 30% of body weight in females and 40% in males. It is mainly composed of multinucleated contractile muscle fibers. Beyond its fundamental roles in locomotion, physical support and thermoregulation, skeletal muscle maintains continuous intertissue communication by secreting a variety of polypeptide factors and occupies a central position in the regulation of systemic glucose homeostasis [96]. Under insulin stimulation, skeletal muscle takes up nearly 85% of circulating glucose mainly via glucose transporter 4 (GLUT4), serving as the primary site for postprandial glucose disposal. The hallmark feature of skeletal muscle insulin resistance is a marked reduction in insulin-stimulated glucose uptake and glycogen synthesis [97].
Upon binding to its cell membrane receptor, insulin initiates intracellular signaling cascades involving insulin receptor substrate 1 (IRS1), phosphoinositide 3-kinase (PI3K), and protein kinase B (Akt). This signaling cascade ultimately promotes the translocation of glucose transporter 4 (GLUT4) from the cytoplasm to the plasma membrane, thereby mediating glucose uptake [98]. Multiple pathological mechanisms can disrupt this process, including the accumulation of fatty acid metabolites, inflammatory responses, and mitochondrial dysfunction [99]. Skeletal muscle in T2DM patients frequently presents mitochondrial defects and impaired GLUT4 translocation [100]. A clinical study induced acute skeletal muscle insulin resistance by lipid infusion in lean healthy volunteers with serial muscle biopsies, and confirmed that insulin resistance was temporally highly correlated with transient diacylglycerol (DAG) accumulation, particularly membrane-associated and cytoplasmic C18:2-containing DAG species [101], Such lipid accumulation activates protein kinase Cθ (PKCθ) and increases IRS1 phosphorylation at the Ser1101 site, thereby inhibiting downstream insulin signaling transduction [102]. Impaired insulin signaling disrupts the protein phosphorylation cascade, reduces PI3K activity, hinders GLUT4 translocation to the cell membrane, and consequently diminishes cellular glucose uptake [103]. In T2DM mouse models, GLUT4 expression is significantly downregulated in the gastrocnemius muscle, accompanied by suppressed membrane translocation [104]. Consistently, in vitro experiments have further verified that palmitate-induced insulin-resistant myotubes exhibit decreased GLUT4 content and reduced AMPK phosphorylation, along with impaired glucose uptake capacity [105]. Moreover, declined skeletal muscle mitochondrial function is closely linked to incomplete fatty acid oxidation. Increased fatty acid flux exacerbates mitochondrial dysfunction and further impairs insulin signaling, creating a self-amplifying vicious cycle between mitochondrial impairment and lipid deposition. Distinct from hepatic insulin resistance, the core defect of skeletal muscle insulin resistance lies primarily in impaired glucose transporter trafficking and phosphorylation signaling, which accounts for reduced insulin-stimulated glycogen synthesis and the development of systemic insulin resistance in T2DM patients [88].
4.3. Adipose Tissue Insulin Resistance: A Key Link in Systemic Insulin Resistance
As a central sensor and signaling hub for whole-body energy status, adipose tissue systematically transmits metabolic signals across the body through multidirectional crosstalk with the liver, skeletal muscle, pancreatic islets and other target organs. Adipose tissue dysfunction is recognized as an essential initiating event in the pathogenesis of insulin resistance and T2DM. Such dysfunction originates from pathological remodeling of the adipose microenvironment, mainly manifested as adipocyte apoptosis and macrophage infiltration, which further reshapes the functional characteristics and secretory profile of adipose tissue. Beyond storing excess energy in the form of lipids, adipose tissue also serves as a critical site for the initiation and regulation of local inflammatory responses. Under obese conditions, the remodeled adipose microenvironment drives macrophage polarization toward the pro-inflammatory M1 phenotype, which secretes abundant inflammatory cytokines, disrupts insulin signaling, and accelerates the progression of insulin resistance. Notably, the immune microenvironment of adipose tissue is highly complex. In addition to macrophages, adipose tissue harbors a variety of immune cells, including T cells (CD4, CD8, Tregs), B cells, natural killer T (NKT) cells, dendritic cells, neutrophils, eosinophils, and mast cells [106]. Experimental studies have further revealed phenotypic switching in adipose tissue macrophages (ATMs). Compared with lean mice, obese mice exhibit a marked increase in macrophage abundance in adipose tissue [107], accompanied by elevated expression of M1-like classical activation-related genes such as TNF-α, Nos2 and Ccr2 [108]. Furthermore, in vitro experiments demonstrate that bone marrow-derived macrophages (BMDMs) exposed to palmitate adopt a phenotype highly similar to ATMs from high-fat diet-fed mice, characterized by robust upregulation of pro-inflammatory cytokines including TNF-α, IL-6 and IL-1β.
Under conditions of nutrient excess, local hypoxia in adipose tissue induces the expression of hypoxia-inducible factor-1α (HIF-1α). Upon activation under hypoxic conditions, HIF-1α reprograms cellular energy metabolism from oxidative phosphorylation (OXPHOS) toward glycolysis, and directly promotes macrophage polarization toward the pro-inflammatory M1 phenotype. Excessive uptake of free fatty acids by adipose tissue macrophages (ATMs) shifts their metabolic profile away from OXPHOS toward the synthesis of triglycerides, phospholipids and ceramides, thereby triggering intracellular lipotoxicity in macrophages. Fatty acids induce endoplasmic reticulum (ER) structural disruption and ER stress, further leading to mitochondrial calcium overload and mitochondrial dysfunction [109]. Impaired autophagy mediated by ceramides, palmitate and saturated fatty acids results in abnormal accumulation of dysfunctional mitochondria. Meanwhile, lipotoxic substances such as excess fatty acids and ceramides directly impair mitochondrial function in adipocytes and macrophages, triggering the release of mitochondrial reactive oxygen species (mtROS) and mitochondrial DNA (mtDNA) as damage-associated molecular patterns (DAMPs). These mediators activate the NLRP3 inflammasome and further amplify local inflammatory signaling. The synergistic action of the above signals drives robust M1 pro-inflammatory macrophage polarization and excessive secretion of TNF-α, IL-6 and IL-1β, establishing a chronic low-grade inflammatory state within adipose tissue. Such inflammatory signals subsequently disseminate via the bloodstream to target organs including the liver, skeletal muscle and pancreatic islets [110].
Lipid storage and mobilization in adipose tissue are intrinsically regulated in a highly coordinated manner, characterized by minute-level regulation and rapid fluctuations in metabolic flux, as typically observed during the fasting-postprandial metabolic transition [111]. Adipose tissue plays a pivotal role in modulating circulating non-esterified fatty acid (NEFA) levels and metabolic flux under both fasting and postprandial conditions, functioning analogously to the liver and skeletal muscle in the regulation of glucose homeostasis and glucose flux [112]. Meanwhile, elevated plasma NEFA levels mainly result from unrestrained lipolysis in insulin-resistant adipose tissue, which constitutes the primary source of systemic lipotoxic burden. Beyond serving as energy substrates, NEFAs also participate in the regulatory mechanism of the glucose-fatty acid cycle [113]. When blood glucose is depleted, NEFAs are utilized as alternative energy fuels; however, ectopic deposition of NEFAs in the liver and skeletal muscle leads to the generation of lipotoxic intermediates such as diacylglycerol (DAG) and ceramides. These metabolites activate PKC and atypical PKC signaling cascades, directly impairing insulin transduction and inducing peripheral insulin resistance [114]. In addition, excessive NEFAs exert direct lipotoxic effects on pancreatic β-cells, exacerbating β-cell injury and accelerating functional exhaustion.
4.4. Pancreatic β-Cell Dysfunction
In addition to being predominantly located within the pancreatic islets of Langerhans, pancreatic β-cells also exist as single cells or small clusters scattered throughout the acinar parenchyma, adjacent to capillaries and other endocrine cell populations. β-cell dysfunction represents the critical turning point in the progression from insulin resistance to overt hyperglycemia in T2DM, and its underlying mechanisms mainly involve endoplasmic reticulum stress, mitochondrial dysfunction and apoptosis induced by glucolipotoxicity. Abnormal cellular cholesterol deposition, islet amyloid polypeptide accumulation, lipotoxicity, glucotoxicity, and the persistent accumulation of pro-inflammatory cytokines all contribute to the initiation and progression of β-cell dysfunction [115]. Furthermore, impaired β-cell function further exacerbates oxidative stress and endoplasmic reticulum stress, and promotes the development of inflammatory disorders and T2DM pathogenesis by activating inflammatory signaling pathways [116].
Circulating free fatty acids (FFAs) consist of saturated and unsaturated fatty acids, which are mainly derived from dietary intake, while FFAs synthesized de novo from glucose are predominantly produced in the form of palmitate. Palmitate can be converted into other FFA species through chain elongation and desaturation. Long-term excessive intake of carbohydrates and lipids elevates circulating FFA concentrations. FFAs undergo β-oxidation in mitochondria and peroxisomes, and those with lipotoxic potential preferentially target pancreatic β-cells. Under lipotoxic conditions, lipid droplet deposition in the form of triglycerides is a common phenomenon in multiple mammalian cell types such as hepatocytes and cardiomyocytes, and this pattern also occurs in pancreatic β-cells. Lipotoxicity disturbs cellular metabolism via multiple pathways, including inflammation, autophagy and endoplasmic reticulum stress [117,118].
Insulin receptor substrate (IRS) molecules serve as pivotal mediators in insulin signaling pathways and play essential roles in maintaining fundamental cellular functions such as cell growth, proliferation and metabolism [119]. NEFAs activate downstream serine kinases including JNK, IKKβ, p38 and RIP1, which directly phosphorylate inhibitory serine sites on IRS1/2. This modification promotes IRS1/2 degradation and suppresses the binding of transcription factors MafA, PDX1 and NeuroD to the insulin promoter, thereby downregulating insulin gene transcription. Meanwhile, the deficiency of upstream scaffold proteins directly disrupts the PI3K/Akt signaling cascade and blocks intracellular insulin signal transduction. In vitro experiments using STZ-treated INS-1 cells have demonstrated that the IRS1/PI3K/Akt/FOXO1 pathway is markedly inhibited, accompanied by decreased cell viability, enhanced apoptosis, impaired insulin secretion, elevated oxidative stress and altered glycolysis, ultimately facilitating β-cell apoptosis [120]. In vivo studies further reveal that IL-1β substantially induces the release of mitochondrial DNA (mtDNA) from mitochondria via uncoupling protein 2 (UCP2) in β-cells. Under physiological conditions, pancreatic β-cells maintain high expression of glucokinase (GCK) and low expression of lactate dehydrogenase (LDH), ensuring that glucose-derived pyruvate preferentially enters the mitochondrial oxidative phosphorylation pathway to generate ATP and trigger insulin exocytosis. Under lipotoxic conditions, fatty acids upregulate UCP2 expression, leading to oxidative phosphorylation uncoupling, reduced ATP production, and subsequent impairment of glucose-stimulated insulin secretion (GSIS). In addition, inflammatory signals derived from adipose tissue and pancreatic islets, such as HIF-1α, MCP1/CCL2 and CXCL1, promote the infiltration of pro-inflammatory M1 macrophages into pancreatic islets. Infiltrated M1 macrophages and damaged β-cells activate the NLRP3 inflammasome via the TLR4 pathway, which further cleaves caspase-1 to drive the maturation and release of IL-1β and IL-18. These local inflammatory cytokines exert feedback effects on β-cells, further activating the NF-κB and JNK pathways and forming a self-sustaining vicious cycle.
5. Intervention Strategies Targeting Downstream Glucose Metabolic Pathways
5.1. Antioxidant Strategies
Hyperglycemia is the core driver of excessive ROS overproduction and oxidative stress. Abnormal glucose metabolism, such as enhanced glucose uptake and accelerated glycolysis, induces massive ROS generation through multiple mechanisms, including mitochondrial electron transport chain leakage, the hexosamine pathway, and advanced glycation end products (AGEs) formation. Excessive ROS not only damages lipids, proteins and DNA, but also further suppresses oxidative phosphorylation [121]. Therefore, direct ROS scavenging or activation of the endogenous antioxidant system represents one of the key strategies for intervening in downstream glucose metabolic disorders. Clinical trials have demonstrated that hypoglycemic agents such as nuclear factor erythroid 2-related factor 2 (Nrf2) activators, sodium-glucose cotransporter 2 (SGLT2) inhibitors, and glucagon-like peptide-1 receptor agonists (GLP-1RAs) can effectively retard T2DM progression by mitigating oxidative stress. In addition, other antioxidants, including vitamins, lipoic acid, NADPH oxidase (Nox) inhibitors, epigenetic regulators and complement inhibitors, offer promising therapeutic alternatives for T2DM treatment. Accumulating evidence indicates that alleviating oxidative stress reduces the levels of MDA, ROS and JNKs, while upregulating the expression of antioxidant molecules such as Nrf2, SOD, NQO1, HO-1 and GSH-Px, thereby ameliorating the pathological progression of T2DM.
As a first-line therapeutic agent for T2DM, metformin markedly alleviates oxidative stress injury and tissue fibrosis in high-fat diet/streptozotocin (HFD/STZ)-induced T2DM mice [122]. Its underlying mechanism involves activation of the AMPK/SIRT1-FoxO1 signaling pathway, which effectively relieves oxidative stress and modulates autophagy [123]. Meanwhile, metformin acts as a PGC-1α activator and exerts therapeutic effects against T2DM by maintaining mitochondrial integrity and inhibiting the NLRP3 inflammasome [124]. Beyond hypoglycemic efficacy, sodium-glucose cotransporter 2 (SGLT2) inhibitors also possess definite anti-inflammatory and antioxidant properties [125]. In diabetic rat models, dapagliflozin, either used alone or combined with metformin, improves glycemic control, upregulates organic anion transporter 3 expression and promotes autophagy [126]. It also suppresses IL-1β secretion in macrophages via the ROS-NLRP3-Caspase-1 signaling cascade [127]. As a member of the insulin hormone family, glucagon-like peptide-1 (GLP-1) enhances insulin secretion and inhibits glucagon release, thereby lowering blood glucose. GLP-1 activates autophagy through regulation of the GLP-1R-AMPK-mTOR-autophagy-ROS axis to ameliorate T2DM pathogenesis. In addition, exenatide treatment effectively reduces macrophage infiltration [128], and alleviates oxidative stress as well as NF-κB activation independent of glycemic changes in diabetic models [129]. Dimethyl fumarate (DMF) exerts immunomodulatory, anti-inflammatory and antioxidant effects by activating Nrf2 and agonizing hydroxycarboxylic acid receptor 2 (HCAR2) [130], Studies have demonstrated that DMF modulates macrophage subpopulation distribution in mouse skeletal muscle, significantly reduces adipocyte abundance, and inhibits apoptotic and pro-inflammatory pathways, thereby exerting anti-inflammatory, anti-fibrotic and anti-lipotoxic actions [130]. In summary, ROS-mediated tissue damage can be alleviated by upregulating antioxidant enzymes. Therapeutic intervention can also be achieved by reducing ROS production, blocking specific enzymatic reactions, or preventing the occurrence of mitochondrial dysfunction.
5.2. Repair of Organelle Dysfunction
Increasing attention has been paid to the pivotal roles of mitochondrial dysfunction and endoplasmic reticulum stress in cellular injury and apoptosis during T2DM pathogenesis [131]. In podocytes from STZ-induced diabetic rats, abnormal mitochondrial morphology and reduced MAMs are observed, accompanied by downregulated Mfn2 expression and activation of the unfolded protein response (UPR) pathways, including IRE1, ATF6 and PERK. As a GTPase, Mfn2 mediates outer mitochondrial membrane (OMM) fusion by forming homodimers or heterodimers to bridge adjacent mitochondria [132]. Loss of Mfn2 function markedly impairs mitochondrial fusion efficiency and induces mitochondrial fragmentation. Further studies have shown that Mfn2 is highly enriched in MAMs and regulates multiple biological functions of the endoplasmic reticulum and mitochondria, including calcium homeostasis, protein synthesis, mitochondrial fusion and fission, mitophagy, and inflammatory responses [133,134].
Mfn2 expression is also decreased in the skeletal muscle of T2DM patients. High glucose (HG)-induced mitochondrial dysfunction, reduced MAMs and increased apoptosis are accompanied by Mfn2 downregulation and PERK pathway activation. Mechanistically, Mfn2 physically interacts with PERK, and the HG microenvironment weakens their binding affinity [68]. Other studies have shown that the expression of Mfn2 and Mfn1 is markedly reduced, while Fis1 and Drp1 levels are elevated in the renal tissue of diabetic rats. Vitamin D receptor (VDR) agonists can partially restore mitophagy through the Mfn2-MAMs-FUNDC1 axis, alleviate mitochondrial swelling, reduce mitochondrial ROS production, and exert beneficial regulatory effects on ERS. Further experiments have confirmed that mitochondrial calcium levels are significantly decreased in HK-2 cells under HG/TGF-β conditions, which may be attributed to impaired MAM integrity and reduced calcium transfer from the endoplasmic reticulum to mitochondria. Treatment with paricalcitol or VDR overexpression plasmid can partially rescue mitochondrial function [135]. These findings directly reveal the intrinsic association among MAM integrity, mitochondrial Ca²⁺ homeostasis and ATP production.
Early studies have established that the mitochondrial outer membrane voltage-dependent anion channel 1 (VDAC1) physically interacts with endoplasmic reticulum IP3 receptor 1 (IP3R1) via the MAM-associated chaperone GRP75, which directly modulates calcium transfer from the endoplasmic reticulum to mitochondria [136]. Either calcium depletion in the ER or MAM-mediated mitochondrial calcium overload can enhance ROS production and trigger ER dysfunction, further opening the mitochondrial permeability transition pore (mPTP) and ultimately inducing cell death [137]. Recent studies have confirmed that the IP3R1–GRP75–VDAC1 complex mediates the critical crosstalk between ER stress and mitochondrial oxidative stress in the pathogenesis of T2DM. Pharmacological blockade of ERS with the chemical chaperone 4-phenylbutyric acid (4-PBA) [138], or silencing GRP75 via small interfering RNA (siRNA) in HL-1 cardiomyocytes and Hspa9 conditional knockout mouse models, can both restrain ER-to-mitochondria calcium trafficking and thereby mitigate mitochondrial oxidative stress and calcium overload [139]. In addition, animal experiments demonstrate that compared with the model group, tea polyphenol (TP) intervention significantly reduces the protein levels of p-IP3R1 (Ser-1756), GRP75, VDAC1 and MCU in T2DM rats, accompanied by a marked decrease in the proportion of MAM contact length relative to mitochondrial perimeter. These findings indicate that TP ameliorates memory impairment in T2DM rats by regulating GRP75-targeted MAM homeostasis in hippocampal neurons, providing a novel mechanistic insight into the neuroprotective effects of TP in T2DM-associated complications [140].
5.3. Anti-Systemic Inflammation Strategies
Hyperglycemia-induced abnormal glucose metabolism serves not only as the primary source of oxidative stress but also provides the metabolic foundation for the chronic inflammatory cascade. Enhanced glycolysis, activation of the hexosamine biosynthetic pathway (HBP), and reprogramming of the pentose phosphate pathway (PPP) can directly or indirectly regulate NLRP3 inflammasome activation, macrophage polarization, and pro-inflammatory cytokine release through the accumulation of metabolic intermediates and modification of signaling molecules [141]. Among these events, abnormally elevated glycolytic flux represents a hallmark metabolic reprogramming feature of inflammatory cells, particularly M1 macrophages. Such metabolic reprogramming occurs dynamically at different stages of inflammation and is precisely modulated by the type, abundance, and competitive interaction of regulatory factors such as IL-4 and IFN-γ in the local microenvironment. Furthermore, hypoxia and lactate accumulation within the microenvironment can independently trigger metabolic reprogramming in macrophages [142].
Pharmacological intervention targeting glucose metabolic reprogramming has demonstrated cardiorenal protective effects beyond simple hypoglycemia. Sodium-glucose cotransporter 2 inhibitors (SGLT2is) suppress sympathetic overactivation, a common complication in T2DM patients, by alleviating oxidative stress and inflammation [143]. A meta-analysis covering six major cardiovascular and renal outcome trials in T2DM confirmed that SGLT2is reduced the composite renal endpoint by 36%, independent of baseline atherosclerotic cardiovascular disease status [144]. Mechanistic studies have shown that empagliflozin ameliorates endothelial dysfunction and arterial stiffness in diabetic rats and delays the progression of nephropathy [145]. Moreover, empagliflozin reduces adipose tissue mass in normoglycemic animals and lowers blood pressure in diabetic patients [146,147]. Dapagliflozin has also been shown to significantly decrease the risk of renal events in cardiorenal protection trials [148]. Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) improve glycemic control independent of insulin signaling by enhancing endothelial function, optimizing body weight and blood pressure regulation, and exerting direct anti-inflammatory and anti-fibrotic effects on renal tissues. Combined administration of SGLT2is and GLP-1 RAs achieves augmented protection against cardiovascular and renal deterioration through synergistic modulation of hemodynamic, metabolic, and inflammatory pathways [149].
Beyond the above broad intervention strategies, precise targeting of key rate-limiting enzymes involved in glycolysis has gradually become a cutting-edge research direction for T2DM therapy. As a pivotal rate-limiting enzyme of glycolysis, upregulated PFKFB3 expression enhances glycolytic activity in endothelial cells and macrophages, and promotes the production of pro-inflammatory cytokines such as IL-1β and TNF-α. Pharmacological inhibition of PFKFB3 markedly reduces glycolytic flux and alleviates vascular inflammation in diabetic atherosclerosis models [150]. Accumulating evidence indicates that PFKFB3 mediates diabetic organ damage by regulating immune inflammatory responses, fibrosis, vascular dysfunction, as well as pancreatic β-cell function and survival, suggesting that PFKFB3 represents a promising therapeutic target for diabetic complications [150]. As a functionally complementary molecule, PKM2 acts as a core linker between glycolysis and inflammatory signaling. In addition to catalyzing the conversion of phosphoenolpyruvate to pyruvate, PKM2 can translocate into the nucleus and cooperate with HIF-1α to initiate the transcription of NLRP3 inflammasome-related genes. Studies have verified that PKM2 activators can reverse LPS-induced M1 macrophage polarization and suppress NLRP3-dependent caspase-1 activation as well as IL-1β secretion [151]. Notably, elevated levels of succinate stabilize HIF-1α to sustain the M1 macrophage phenotype, which is closely correlated with exacerbated inflammatory responses [152]. In the context of diabetic encephalopathy, the glycolytic status of central microglia determines their inflammatory phenotype. A high-glucose microenvironment drives a metabolic shift from oxidative phosphorylation to glycolysis in microglia, accompanied by PKM2 nuclear translocation and NLRP3 inflammasome activation. This process triggers the release of IL-1β and TNF-α, ultimately leading to neuronal injury and cognitive dysfunction [153]. Previous studies have demonstrated that citrus-derived polyphenols, including hesperidin, naringenin, and nobiletin, can systematically remodel the glucose metabolic network via multi-target regulation of the AMPK, PI3K/Akt, NF-κB and Nrf2 signaling pathways, thereby ameliorating insulin resistance and metabolic inflammation [154].
5.4. Traditional Chinese Medicine and Natural Products
In recent years, in addition to continuous breakthroughs in Western medicine represented by SGLT2 inhibitors and GLP-1 receptor agonists, bioactive components derived from traditional Chinese medicine (TCM) have exhibited unique advantages in regulating macrophage metabolic reprogramming and immune homeostasis. Compared with synthetic chemical drugs, which are often limited to single-target action or accompanied by potential adverse reactions, these natural products exert direct antioxidant and anti-inflammatory effects. More importantly, they can remodel macrophage metabolism through epigenetic regulation and modification of metabolic enzyme activity, thereby ameliorating insulin resistance and chronic inflammation in T2DM.
Berberine induces aerobic glycolysis by blocking the tricarboxylic acid cycle and modulates cytokine responses in bone marrow-derived macrophages (BMDMs) isolated from mouse and human peripheral blood mononuclear cells (PBMCs). Berberine gradually upregulates GLUT1 expression and enhances total cellular hexokinase activity in BMDMs. Moreover, in LPS-injected mice, berberine treatment attenuates the elevation of serum TNF-α and the reduction of blood glucose [155]. Puerarin activates the AMPK-mTOR and PPARγ-NF-κB signaling pathways, modulates insulin signaling and glycolipid metabolism, and alleviates inflammatory injury [156], Meanwhile, puerarin reduces macrophage infiltration in adipose tissue, downregulates TNF-α expression, and improves obesity-associated inflammation and dyslipidemia [157]. Resveratrol is a polyphenolic compound widely present in various plants and synthesized mainly under pathogen-induced stress. It possesses multiple biological activities, including antioxidant, anti-inflammatory, anti-carcinogenic, anti-aging, anti-diabetic and neuroprotective effects [158]. Under high-glucose conditions, resveratrol markedly inhibits NO production and pro-inflammatory interleukin secretion in LPS-stimulated macrophages. Existing evidence indicates that resveratrol effectively suppresses the release of pro-inflammatory cytokines from macrophages and remodels the extracellular metabolic microenvironment under hyperglycemic conditions [159]. In vitro, astragalus polysaccharides inhibit the differentiation of LPS/HG-stimulated THP-1 cells into pro-inflammatory M1 macrophages, accompanied by reduced ROS generation and decreased secretion of pro-inflammatory cytokines. Meanwhile, astragalus polysaccharides promote M2 polarization and the release of anti-inflammatory factors (IL-4, IL-10 and Arg-1) by activating the Nrf2/HO-1 signaling pathway in THP-1-derived macrophages. Such regulatory effects can be abolished by Nrf2 siRNA interference. In addition, astragalus polysaccharides enhance endothelial migration and angiogenesis in the co-culture system of HUVECs and macrophages under high-glucose conditions [160]. Collectively, these studies suggest that natural products can regulate macrophage glycolysis and polarization through multi-target mechanisms, providing novel therapeutic strategies for T2DM. Nevertheless, current research on TCM interventions targeting the upstream event of macrophage metabolic reprogramming remains relatively limited, and further in-depth investigations are warranted in the future.
6. Conclusions
In summary, glucose metabolic reprogramming acts as an upstream core event in the occurrence and progression of type 2 diabetes mellitus (T2DM). The hyperglycemic microenvironment drives metabolic cells such as macrophages to shift their energy metabolism from oxidative phosphorylation to aerobic glycolysis. This metabolic transition not only meets the energy demands for rapid cell proliferation and functional activation, but also initiates and amplifies downstream pathological cascades through the accumulation of metabolic intermediates and the modification of signaling molecules. As a central hub in this pathological process, oxidative stress triggers excessive ROS production via multiple pathways, including mitochondrial electron transport chain leakage, hexosamine pathway activation, and advanced glycation end product (AGEs) formation. Mitochondrial-endoplasmic reticulum dysfunction further exacerbates cellular injury and apoptosis through the disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity, calcium homeostasis imbalance, and abnormal activation of the unfolded protein response (UPR). The synergistic interplay of these downstream effectors ultimately leads to pancreatic β-cell dysfunction, insulin resistance and chronic inflammation, thereby facilitating the progression of T2DM and its related complications.
Although the pivotal role of glucose metabolic reprogramming and its downstream mechanisms in T2DM has been widely recognized, several limitations still remain. First, the regulatory mechanisms of MAMs in T2DM have not been fully elucidated. As critical structural and functional interfaces mediating crosstalk between mitochondria and the endoplasmic reticulum, MAMs are essential for calcium signal transmission, lipid metabolism, mitophagy and inflammatory responses. Nevertheless, the dynamic alterations of MAM structural integrity and functional status under T2DM pathological conditions remain poorly defined. Second, clinical translational research is insufficient. Current evidence is predominantly derived from in vitro cell experiments and in vivo animal studies, while large-scale, multicenter clinical cohort studies are still lacking to validate the therapeutic efficacy of interventions targeting glucose metabolic reprogramming and downstream stress pathways. Meanwhile, the research and development of targeted drugs against MAM integrity, mitochondrial dynamics and endoplasmic reticulum stress are still in the early stage, with a long way remaining before clinical application.
Future research can be carried out from the following perspectives. Firstly, targeting mitochondria-endoplasmic reticulum crosstalk represents a promising novel therapeutic target for T2DM. Strategies aimed at preserving MAM integrity and regulating its key functional proteins are expected to restore calcium homeostasis, inhibit mitochondrial permeability transition pore opening, and alleviate both oxidative and endoplasmic reticulum stress. Secondly, the prediabetic stage serves as a critical window for T2DM prevention. At this stage, abnormal glucose metabolism has not yet become irreversible, and oxidative as well as endoplasmic reticulum stress remain reversible. Early intervention targeting downstream effectors of glucose metabolic reprogramming may block or delay the progression from prediabetes to overt T2DM. Finally, integrating metabolomics, transcriptomics, proteomics and single-cell sequencing technologies can systematically reveal the dynamic characteristics of glucose metabolic reprogramming across different T2DM stages, as well as its crosstalk with downstream stress signaling pathways. The anticipated findings will provide novel therapeutic targets and theoretical evidence for the early prevention and clinical management of T2DM.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure 1: Imbalanced Energy Metabolism; Figure 2: Exacerbated Oxidative Stress; Figure 3: Organelle Crosstalk; Figure 4: Systemic Inflammation; Figure 5: Tissue-Specific Heterogeneity of Targeted Insulin Resistance.
Author Contributions
Conceptualization, C.W., M.W. and A.L.; Data curation, C.W., Q.Z., J.G., A.S. and X.G.; Formal analysis, C.W. and Y.G.; Writing—original draft preparation, all authors; Writing—review and editing, all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Administration of Traditional Chinese Medicine Young Qi Huang Scholars Support Project (National Traditional Chinese Medicine Human Education Development [2020] No.7), the Fundamental Research Funds for the Central Universities (2023-JYB-JBZD-010), the Postdoctoral Fellowship Program of China Postdoctoral Science Foundation (GZC20230324), the China Postdoctoral Science Foundation (2024M750263), and the Independent Research Project for Postgraduates of Dongzhimen Hospital, Beijing University of Chinese Medicine (No. DZMYJS2026006).
Data Availability Statement
Data Availability Statement: No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACLY: | ATP-citrate lyase |
| AKT | Protein kinase B |
| AMPK | Adenosine monophosphate-activated protein kinase |
| Arg-1 | Arginase 1 |
| ATF4 | Activating transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| BCAA | Branched-chain amino acid |
| CHOP | C/EBP homologous protein |
| Cyt c | Cytochrome c |
| DAG | Diacylglycerol |
| DAMPs | Damage-associated molecular patterns |
| Drp1 | Dynamin-related protein 1 |
| EGF/VEGF | Epidermal growth factor/vascular endothelial growth factor |
| eIF2α-P | Phosphorylated eukaryotic initiation factor 2α |
| ERS | Endoplasmic reticulum stress |
| ETC | Electron transport chain |
| FFA/NEFA | Free fatty acid/non-esterified fatty acid |
| FoxO1 | Forkhead box protein O1 |
| G6Pase | Glucose-6-phosphatase |
| GLUT1 | Glucose transporter 1 |
| GLUT4 | Glucose transporter 4 |
| GLUTs | Glucose transporters |
| GPx-1 | Glutathione peroxidase 1 |
| GRP75 | Glucose-regulated protein 75 |
| GSH-Px | Glutathione peroxidase |
| HG | High glucose |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HK2 | Hexokinase 2 |
| HO-1 | Heme oxygenase-1 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| iNOS | Inducible nitric oxide synthase |
| IP3R | Inositol 1,4,5-trisphosphate receptor |
| IRE1α | Inositol-requiring enzyme 1α |
| IRK | Insulin receptor kinase |
| IRS1/2 | Insulin receptor substrate 1/2 |
| JNK | c-Jun N-terminal kinases |
| M1 | Classically activated macrophage |
| M2 | Alternatively activated macrophage |
| MAPK | Mitogen-activated protein kinase |
| MAMs | Mitochondria-associated endoplasmic reticulum membranes |
| MDA | Malondialdehyde |
| MFN1/2 | Mitofusin 1/2 |
| mPTP | Mitochondrial permeability transition pore |
| mtDNA | Mitochondrial DNA |
| mtROS | Mitochondrial reactive oxygen species |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NLR family pyrin domain containing 3 |
| NQO1 | NADPH quinone oxidoreductase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NRF1/2 | Nuclear respiratory factor 1/2 |
| OXPHOS | Oxidative phosphorylation |
| p38 | p38 mitogen-activated protein kinase |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PFK1 | Phosphofructokinase 1 |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PHD | Prolyl hydroxylase |
| PI3K | Phosphatidylinositol 3-kinase |
| PKCα/θ/ζ | Protein kinase C α/θ/ζ |
| PP2A | Protein phosphatase 2A |
| ROS | Reactive oxygen species |
| SCFA | Short-chain fatty acid |
| SERCA | Sarco/endoplasmic reticulum calcium ATPase |
| SIRT1 | Sirtuin 1 |
| SOD | Superoxide dismutase |
| SOD-1 | Superoxide dismutase 1 |
| T2DM | Type 2 diabetes mellitus |
| TCA cycle | Tricarboxylic acid cycle |
| TFAM | Mitochondrial transcription factor A |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor-α |
| TRX | Thioredoxin |
| UCP2 | Uncoupling protein 2 |
| VDAC | Voltage-dependent anion channel |
| XBP1 | X-box binding protein 1 |
| XBP1s | Spliced X-box binding protein 1 |
| ΔΨm | Mitochondrial membrane potential |
References
- Genitsaridi, I.; Salpea, P.; Salim, A.; et al. 11th edition of the IDF Diabetes Atlas: global, regional, and national diabetes prevalence estimates for 2024 and projections for 2050. Lancet Diabetes Endocrinol. 2026, 14, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.I.G.; Cockram, C.S.; Ma, R.C.W.; et al. Diabetes and infection: review of the epidemiology, mechanisms and principles of treatment. Diabetologia 2024, 67, 1168–1180. [Google Scholar] [CrossRef]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef]
- Ye, L.; Jiang, Y.; Zhang, M. Crosstalk between glucose metabolism, lactate production and immune response modulation. Cytokine Growth Factor Rev. 2022, 68, 81–92. [Google Scholar] [CrossRef]
- Russo, S.; Kwiatkowski, M.; Govorukhina, N.; et al. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Front. Immunol. 2021, 12, 746151. [Google Scholar] [CrossRef]
- Kelly, B.; O'Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cao, J.; Li, S.; et al. M2 Macrophage-Derived sEV Regulate Pro-Inflammatory CCR2(+) Macrophage Subpopulations to Favor Post-AMI Cardiac Repair. Adv. Sci. 2023, 10, e2202964. [Google Scholar] [CrossRef]
- Liu, J.; Cao, X. Glucose metabolism of TAMs in tumor chemoresistance and metastasis. Trends Cell Biol. 2023, 33, 967–978. [Google Scholar] [CrossRef]
- Usuba, F.S.; de Medeiros-Ribeiro, A.C.; Novaes, P.; et al. Dry eye in rheumatoid arthritis patients under TNF-inhibitors: conjunctival goblet cell as an early ocular biomarker. Sci. Rep. 2020, 10, 14054. [Google Scholar] [CrossRef]
- Kumar, V.; Mahato, R.I. Delivery and targeting of miRNAs for treating liver fibrosis. Pharm. Res. 2015, 32, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; et al. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef] [PubMed]
- D'Souza, D.M.; Al-Sajee, D.; Hawke, T.J. Diabetic myopathy: impact of diabetes mellitus on skeletal muscle progenitor cells. Front Physiol. 2013, 4, 379. [Google Scholar] [CrossRef]
- Chadt, A.; Al-Hasani, H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflug. Arch. Eur. J. Physiol. 2020, 472, 1273–1298. [Google Scholar] [CrossRef]
- Yeh, C.J.; Sattler, K.M.; Lepper, C. Molecular regulation of satellite cells via intercellular signaling. Gene 2023, 858, 147172. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Mantzoros, C.S. Obesity: seize the day, fight the fat. Metabolism 2019, 92, 1–5. [Google Scholar] [CrossRef]
- Boutari, C.; Perakakis, N.; Mantzoros, C.S. Association of Adipokines with Development and Progression of Nonalcoholic Fatty Liver Disease. Endocrinol. Metab. 2018, 33, 33–43. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Valvano, C.M.; De Nardo, W.; et al. Integrative metabolism in MASLD and MASH: Pathophysiology and emerging mechanisms. J. Hepatol. 2025, 83, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Boutari, C.; Mantzoros, C.S. A 2022 update on the epidemiology of obesity and a call to action: as its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism 2022, 133, 155217. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; et al. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Gasbarrino, K.; Hafiane, A.; Gianopoulos, I.; et al. Relationship between circulating adipokines and cholesterol efflux in subjects with severe carotid atherosclerosis. Metabolism 2023, 140, 155381. [Google Scholar] [CrossRef]
- Van Huynh, T.; Rethi, L.; Rethi, L.; et al. The Complex Interplay between Imbalanced Mitochondrial Dynamics and Metabolic Disorders in Type 2 Diabetes. Cells 2023, 12. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. TEM. 2012, 23, 435–443. [Google Scholar] [CrossRef]
- Cheng, X.; Gu, X.; Wang, F. Mitochondrial Dysfunction During TGF-β1-Induced Epithelial-Mesenchymal Transition in Retinal Pigment Epithelial Cells. Curr. Eye Res. 2025, 50, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Shi, G.; Zhao, L.; et al. Blue light exposure exacerbates obesity in high-fat diet-fed mice by inducing mitochondrial dysfunction in the white adipose tissue. Ecotoxicol. Environ. Saf. 2026, 316, 120111. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; Li, X.; Hall, M.E.; et al. Obesity, Hypertension, and Cardiac Dysfunction: Novel Roles of Immunometabolism in Macrophage Activation and Inflammation. Circ. Res. 2020, 126, 789–806. [Google Scholar] [CrossRef]
- Tábara, L.C.; Segawa, M.; Prudent, J. Molecular mechanisms of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2025, 26, 123–146. [Google Scholar] [CrossRef]
- Larsson, O.; Morita, M.; Topisirovic, I.; et al. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proceedings of the National Academy of Sciences of the United States of America 2012, 109, 8977–8982. [Google Scholar] [CrossRef] [PubMed]
- Wood Dos Santos, T.; Cristina Pereira, Q.; Teixeira, L.; et al. Effects of Polyphenols on Thermogenesis and Mitochondrial Biogenesis. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St-Pierre, J.; et al. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proceedings of the National Academy of Sciences of the United States of America 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; van der Wel, N.N.; Orozco, I.J.; et al. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 2004, 279, 18792–18798. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, Z.; Zhang, S.; et al. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics 2023, 13, 736–766. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria. Cold Spring Harbor perspectives in biology, 2021; 13. [Google Scholar]
- Zhao, X.; Huang, F.; Sun, Y.; et al. Mechanisms of endurance and resistance exercise in type 2 diabetes mellitus: A Narrative review. Biochem. Biophys. Res. Commun. 2025(761), 151731. [CrossRef]
- Zhang, S.; Zhang, S.; Zhang, Y.; et al. Activation of NRF2 by epiberberine improves oxidative stress and insulin resistance in T2DM mice and IR-HepG2 cells in an AMPK dependent manner. J. Ethnopharmacol. 2024, 327, 117931. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, L.; Yu, L.; et al. H4K12 lactylation potentiates mitochondrial oxidative stress via the Foxo1 pathway in diabetes-induced cognitive impairment. J. Adv. Res. 2025, 78, 391–407. [Google Scholar] [CrossRef]
- Pang, J.; Yin, L.; Jiang, W.; et al. Sirt1-mediated deacetylation of PGC-1α alleviated hepatic steatosis in type 2 diabetes mellitus via improving mitochondrial fatty acid oxidation. Cell. Signal. 2024, 124, 111478. [Google Scholar] [CrossRef]
- Sun, J.; Leng, P.; Li, X.; et al. Salvianolic acid A promotes mitochondrial biogenesis and mitochondrial function in 3T3-L1 adipocytes through regulation of the AMPK-PGC1α signalling pathway. Adipocyte 2022, 11, 562–571. [Google Scholar] [CrossRef]
- Iacobini, C.; Vitale, M.; Pesce, C.; et al. Diabetic Complications and Oxidative Stress: A 20-Year Voyage Back in Time and Back to the Future. Antioxidants 2021, 10. [Google Scholar] [CrossRef]
- Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11. [Google Scholar] [CrossRef]
- Tu, Y.; Li, L.; Zhu, L.; et al. Geniposide Attenuates Hyperglycemia-Induced Oxidative Stress and Inflammation by Activating the Nrf2 Signaling Pathway in Experimental Diabetic Retinopathy. Oxidative Med. Cell. Longev. 2021, 2021, 9247947. [Google Scholar] [CrossRef]
- Maruhashi, T.; Higashi, Y. Pathophysiological Association between Diabetes Mellitus and Endothelial Dysfunction. Antioxidants 2021, 10. [Google Scholar] [CrossRef]
- Dunn, L.L.; Buckle, A.M.; Cooke, J.P.; et al. The emerging role of the thioredoxin system in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2089–2098. [Google Scholar] [CrossRef]
- Levy, D.; Reichert, C.O.; Bydlowski, S.P. Paraoxonases Activities and Polymorphisms in Elderly and Old-Age Diseases: An Overview. Antioxidants 2019, 8. [Google Scholar] [CrossRef]
- Araújo, T.G.; Oliveira, A.G.; Vecina, J.F.; et al. Treatment with Parkinsonia aculeata combats insulin resistance-induced oxidative stress through the increase in PPARγ/CuZn-SOD axis expression in diet-induced obesity mice. Mol. Cell. Biochem. 2016, 419, 93–101. [Google Scholar] [CrossRef]
- Chien, C.Y.; Wen, T.J.; Cheng, Y.H.; et al. Diabetes Upregulates Oxidative Stress and Downregulates Cardiac Protection to Exacerbate Myocardial Ischemia/Reperfusion Injury in Rats. Antioxidants 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, M.D.; Jamieson, E.; Jansen, E.S.; et al. Glucose induces pancreatic islet cell apoptosis that requires the BH3-only proteins Bim and Puma and multi-BH domain protein Bax. Diabetes 2010, 59, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Subas Satish, H.P.; Iyer, S.; Shi, M.X.; et al. A novel inhibitory BAK antibody enables assessment of non-activated BAK in cancer cells. Cell Death Differ. 2024, 31, 711–721. [Google Scholar] [CrossRef]
- Wali, J.A.; Rondas, D.; McKenzie, M.D.; et al. The proapoptotic BH3-only proteins Bim and Puma are downstream of endoplasmic reticulum and mitochondrial oxidative stress in pancreatic islets in response to glucotoxicity. Cell Death Dis. 2014, 5, e1124. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, C.; Sun, L. Mitochondria-Associated Membranes (MAMs): A Novel Therapeutic Target for Treating Metabolic Syndrome. Curr. Med. Chem. 2021, 28, 1347–1362. [Google Scholar] [CrossRef]
- Csordás, G.; Renken, C.; Várnai, P.; et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, Q.; Cao, B.; et al. Ca(2+) transfer via the ER-mitochondria tethering complex in neuronal cells contribute to cadmium-induced autophagy. Cell Biol. Toxicol. 2022, 38, 469–485. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Jaña, F.; Urra, H.; et al. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Yoboue, E.D.; Valente, E.M. PINK1 and Parkin: The odd couple. Neurosci. Res. 2020, 159, 25–33. [Google Scholar] [CrossRef]
- Kjærgaard, J.; Stocks, B.; Henderson, J.; et al. Personalized molecular signatures of insulin resistance and type 2 diabetes. Cell 2025, 188, 4106–4122.e4116. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Y.; Li, Y.; et al. FAM134B-Mediated ER-Phagy in Mg(2+)-Free Solution-Induced Mitochondrial Calcium Homeostasis and Cell Death in Epileptic Hippocampal Neurons. Neurochem. Res. 2021, 46, 2485–2494. [Google Scholar] [CrossRef]
- Wu, S.; Lu, Q.; Ding, Y.; et al. Hyperglycemia-Driven Inhibition of AMP-Activated Protein Kinase α2 Induces Diabetic Cardiomyopathy by Promoting Mitochondria-Associated Endoplasmic Reticulum Membranes In Vivo. Circulation 2019, 139, 1913–1936. [Google Scholar] [CrossRef]
- Gong, Y.; Lu, X.; Wang, X.; et al. Mitochondrial Tumor Suppressor 1A Attenuates Myocardial Infarction Injury by Maintaining the Coupling Between Mitochondria and Endoplasmic Reticulum. Circulation 2025, 152, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Casellas-Díaz, S.; Larramona-Arcas, R.; Riqué-Pujol, G.; et al. Mfn2 localization in the ER is necessary for its bioenergetic function and neuritic development. EMBO Rep. 2021, 22, e51954. [Google Scholar] [CrossRef]
- Chambers, P.J.; Juracic, E.S.; Fajardo, V.A.; et al. Role of SERCA and sarcolipin in adaptive muscle remodeling. Am. J. Physiol. Cell Physiol. 2022, 322, C382–C394. [Google Scholar] [CrossRef]
- Gawlak-Socka, S.; Kowalczyk, E.; Wiktorowska-Owczarek, A. Unfolded Protein Response at the Crossroads: Integrating Endoplasmic Reticulum Stress with Cellular Stress Networks. Int. J. Mol. Sci. 2026, 27. [Google Scholar] [CrossRef]
- Ye, L.; Zeng, Q.; Ling, M.; et al. Inhibition of IP3R/Ca2+ Dysregulation Protects Mice From Ventilator-Induced Lung Injury via Endoplasmic Reticulum and Mitochondrial Pathways. Front. Immunol. 2021, 12, 729094. [Google Scholar] [CrossRef]
- Clarke, W.R.; Amundadottir, L.; James, M.A. CLPTM1L/CRR9 ectodomain interaction with GRP78 at the cell surface signals for survival and chemoresistance upon ER stress in pancreatic adenocarcinoma cells. Int. J. Cancer 2019, 144, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tian, S.; Ruan, S.; et al. Neuroprotective effects of cordycepin on MPTP-induced Parkinson's disease mice via suppressing PI3K/AKT/mTOR and MAPK-mediated neuroinflammation. Free Radic. Biol. Med. 2024, 216, 60–77. [Google Scholar] [CrossRef] [PubMed]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, Z.; Hu, J.; et al. Mfn2 Regulates High Glucose-Induced MAMs Dysfunction and Apoptosis in Podocytes via PERK Pathway. Front. Cell Dev. Biol. 2021, 9, 769213. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, Y.; Jin, X.; et al. PDZD8 Augments Endoplasmic Reticulum-Mitochondria Contact and Regulates Ca2+ Dynamics and Cypd Expression to Induce Pancreatic β-Cell Death during Diabetes. Diabetes Metab. J. 2024, 48, 1058–1072. [Google Scholar] [CrossRef]
- Li, C.; Guo, N.; Han, S.; et al. Impact of positive CD4 cells on event-free survival in follicular lymphoma patients. Cancer Med. 2024, 13, e70117. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Kikuta, J.; Matsui, T.; et al. Periportal macrophages protect against commensal-driven liver inflammation. Nature 2024, 629, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Galli, G.; Saleh, M. Immunometabolism of Macrophages in Bacterial Infections. Front. Cell. Infect. Microbiol. 2020, 10, 607650. [Google Scholar] [CrossRef]
- Kadomoto, S.; Izumi, K.; Mizokami, A. Macrophage Polarity and Disease Control. Int. J. Mol. Sci. 2021, 23. [Google Scholar] [CrossRef]
- Devericks, E.N.; Carson, M.S.; McCullough, L.E.; et al. The obesity-breast cancer link: a multidisciplinary perspective. Cancer Metastasis Rev. 2022, 41, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Niven, J.; Haas, R.; et al. The sedoheptulose kinase CARKL controls T-cell cytokine outputs and migration by promoting metabolic reprogramming. Discov. Immunol. 2024, 3, kyae016. [Google Scholar] [CrossRef]
- Gomes, M.T.R.; Guimarães, E.S.; Marinho, F.V.; et al. STING regulates metabolic reprogramming in macrophages via HIF-1α during Brucella infection. PLoS Pathog. 2021, 17, e1009597. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 2019, 51, 997–1011.e1017. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, S.; Jin, J.; et al. Cassiaside C Inhibits M1 Polarization of Macrophages by Downregulating Glycolysis. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Tsai, C.F.; Chen, G.W.; Chen, Y.C.; et al. Regulatory Effects of Quercetin on M1/M2 Macrophage Polarization and Oxidative/Antioxidative Balance. Nutrients 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Shu, A.; Chen, J.; Shao, S.; et al. Breviscapine enhances angiogenesis in diabetic wound healing by regulating macrophage polarization via mitochondrial metabolic reprogramming. Phytomed. Int. J. Phyther. Phytopharm. 2026, 151, 157794. [Google Scholar] [CrossRef]
- Suzuki, K.; Hatzikotoulas, K.; Southam, L.; et al. Genetic drivers of heterogeneity in type 2 diabetes pathophysiology. Nature 2024, 627, 347–357. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Hornburg, D.; Wu, S.; Moqri, M.; et al. Dynamic lipidome alterations associated with human health, disease and ageing. Nat. Metab. 2023, 5, 1578–1594. [Google Scholar] [CrossRef] [PubMed]
- Markova, I.; Hüttl, M.; Šťastný, J.; et al. Skeletal muscle insulin resistance in prediabetes: a lipidomic perspective on diacylglycerols, ceramides, and phospholipids. Sci. Rep. 2025, 15, 38784. [Google Scholar] [CrossRef]
- O'Sullivan, S.; Qi, L.; Zalloua, P. From omics to AI-mapping the pathogenic pathways in type 2 diabetes. FEBS Lett. 2025, 599, 3244–3280. [Google Scholar] [CrossRef]
- Goto, H.; Takamura, T. Metabolic Dysfunction-Associated Steatotic Liver Disease Complicated by Diabetes: Pathophysiology and Emerging Therapies. J. Obes. Metab. Syndr. 2025, 34, 224–238. [Google Scholar] [CrossRef]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef]
- Lyu, K.; Zhang, Y.; Zhang, D.; et al. A Membrane-Bound Diacylglycerol Species Induces PKCϵ-Mediated Hepatic Insulin Resistance. Cell Metab. 2020, 32, 654–664.e655. [Google Scholar] [CrossRef]
- James, D.E.; Stöckli, J.; Birnbaum, M.J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell Biol. 2021, 22, 751–771. [Google Scholar] [CrossRef] [PubMed]
- Gancheva, S.; Jelenik, T.; Álvarez-Hernández, E.; et al. Interorgan Metabolic Crosstalk in Human Insulin Resistance. Physiol. Rev. 2018, 98, 1371–1415. [Google Scholar] [CrossRef]
- Unterman, T.G. Regulation of Hepatic Glucose Metabolism by FoxO Proteins, an Integrated Approach. Curr. Top. Dev. Biol. 2018, 127, 119–147. [Google Scholar]
- Pleiner, J.; Schaller, G.; Mittermayer, F.; et al. FFA-induced endothelial dysfunction can be corrected by vitamin C. J. Clin. Endocrinol. Metab. 2002, 87, 2913–2917. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bi, Y.; Sowers, J.R.; et al. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Shum, M.; Ngo, J.; Shirihai, O.S.; et al. Mitochondrial oxidative function in NAFLD: Friend or foe? Mol. Metab. 2021, 50, 101134. [Google Scholar] [CrossRef]
- Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal Muscle Extracellular Matrix - What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front Physiol. 2020, 11, 253. [Google Scholar] [CrossRef]
- Aleksandrowicz, R.; Strączkowski, M. Link between insulin resistance and skeletal muscle extracellular matrix remodeling. Endocr. Connect 2023, 12. [Google Scholar] [CrossRef]
- Xie, X.; Shu, R.; Yu, C.; et al. Mammalian AKT, the Emerging Roles on Mitochondrial Function in Diseases. Aging Dis. 2022, 13, 157–174. [Google Scholar] [CrossRef]
- Stefanowicz, M.; Nikołajuk, A.; Matulewicz, N.; et al. Skeletal muscle RUNX1 is related to insulin sensitivity through its effect on myogenic potential. Eur. J. Endocrinol. 2022, 187, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Wang, D.; Zhou, J.; et al. Diabetes Pharmacotherapy and its effects on the Skeletal Muscle Energy Metabolism. Mini Rev. Med. Chem. 2024, 24, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Ritter, O.; Jelenik, T.; Roden, M. Lipid-mediated muscle insulin resistance: different fat, different pathways? J. Mol. Med. 2015, 93, 831–843. [Google Scholar] [CrossRef]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proceedings of the National Academy of Sciences of the United States of America 2014, 111, 9597–9602. [Google Scholar] [CrossRef]
- Mucinski, J.M.; Salvador, A.F.; Moore, M.P.; et al. Histological improvements following energy restriction and exercise: The role of insulin resistance in resolution of MASH. J. Hepatol. 2024, 81, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wu, S.; Gong, C.; et al. Neuregulin-1β increases glucose uptake and promotes GLUT4 translocation in palmitate-treated C2C12 myotubes by activating PI3K/AKT signaling pathway. Front. Pharmacol. 2022, 13, 1066279. [Google Scholar] [CrossRef]
- Yao, X.; Liu, L.; Shao, W.; et al. Tectorigenin targets PKACα to promote GLUT4 expression in skeletal muscle and improve insulin resistance in vitro and in vivo. Int. J. Biol. Sci. 2023, 19, 1579–1596. [Google Scholar] [CrossRef]
- Ivanov, S.; Merlin, J.; Lee, M.K.S.; et al. Biology and function of adipose tissue macrophages, dendritic cells and B cells. Atherosclerosis 2018, 271, 102–110. [Google Scholar] [CrossRef]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; et al. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef]
- Hinojosa Vera, K.F.; Hemakumar, C.; Bilachi, R.S.; et al. Macrophage Phenotypic Switch and Obesity-Associated Metabolic Risk: Mechanisms and Targets. Oxidative Med. Cell. Longev. 2025(2025), 6710641. [CrossRef]
- Caslin, H.L.; Bhanot, M.; Bolus, W.R.; et al. Adipose tissue macrophages: Unique polarization and bioenergetics in obesity. Immunol. Rev. 2020, 295, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yan, X.; Zhao, Y.; et al. Macrophage Polarization Mediated by Mitochondrial Dysfunction Induces Adipose Tissue Inflammation in Obesity. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Coppack, S.W.; Patel, J.N.; Lawrence, V.J. Nutritional regulation of lipid metabolism in human adipose tissue. Exp. Clin. Endocrinol. Diabetes 2001, 109, S202–S214. [Google Scholar] [CrossRef]
- Frayn, K.N. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002, 45, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; et al. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Yuan, S.; Merino, J.; Larsson, S.C. Causal factors underlying diabetes risk informed by Mendelian randomisation analysis: evidence, opportunities and challenges. Diabetologia 2023, 66, 800–812. [Google Scholar] [CrossRef]
- Vilas-Boas, E.A.; Almeida, D.C.; Roma, L.P.; et al. Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress. Cells 2021, 10. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J. Endoplasmic Reticulum (ER) Stress and Its Role in Pancreatic β-Cell Dysfunction and Senescence in Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- von Hanstein, A.S.; Tsikas, D.; Lenzen, S.; et al. Potentiation of Lipotoxicity in Human EndoC-βH1 β-Cells by Glucose is Dependent on the Structure of Free Fatty Acids. Mol. Nutr. Food Res. 2023, 67, e2200582. [Google Scholar] [CrossRef] [PubMed]
- Bugliani, M.; Mossuto, S.; Grano, F.; et al. Modulation of Autophagy Influences the Function and Survival of Human Pancreatic Beta Cells Under Endoplasmic Reticulum Stress Conditions and in Type 2 Diabetes. Front. Endocrinol. 2019, 10, 52. [Google Scholar] [CrossRef]
- Metz, H.E.; Kargl, J.; Busch, S.E.; et al. Insulin receptor substrate-1 deficiency drives a proinflammatory phenotype in KRAS mutant lung adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America 2016, 113, 8795–8800. [Google Scholar] [CrossRef]
- Cui, F.; He, X. IGF-1 ameliorates streptozotocin-induced pancreatic β cell dysfunction and apoptosis via activating IRS1/PI3K/Akt/FOXO1 pathway. Inflamm. Res. 2022, 71, 669–680. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, C. Oxidative Stress: A Culprit in the Progression of Diabetic Kidney Disease. Antioxidants 2024, 13. [Google Scholar] [CrossRef]
- Han, Y.C.; Tang, S.Q.; Liu, Y.T.; et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 2021, 12, 925. [Google Scholar] [CrossRef]
- Ren, H.; Shao, Y.; Wu, C.; et al. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol. Cell. Endocrinol. 2020, 500, 110628. [Google Scholar] [CrossRef]
- Nam, B.Y.; Jhee, J.H.; Park, J.; et al. PGC-1α inhibits the NLRP3 inflammasome via preserving mitochondrial viability to protect kidney fibrosis. Cell Death Dis. 2022, 13, 31. [Google Scholar] [CrossRef]
- Mima, A. Mitochondria-targeted drugs for diabetic kidney disease. Heliyon 2022, 8, e08878. [Google Scholar] [CrossRef]
- Jaikumkao, K.; Thongnak, L.; Htun, K.T.; et al. Dapagliflozin and metformin in combination ameliorates diabetic nephropathy by suppressing oxidative stress, inflammation, and apoptosis and activating autophagy in diabetic rats. Biochim. Et. Biophys. Acta Mol. Basis Dis. 2024, 1870, 166912. [Google Scholar] [CrossRef]
- Leng, W.; Ouyang, X.; Lei, X.; et al. The SGLT-2 Inhibitor Dapagliflozin Has a Therapeutic Effect on Atherosclerosis in Diabetic ApoE(-/-) Mice. Mediat. Inflamm. 2016, 2016, 6305735. [Google Scholar] [CrossRef]
- Yang, S.; Lin, C.; Zhuo, X.; et al. Glucagon-like peptide-1 alleviates diabetic kidney disease through activation of autophagy by regulating AMP-activated protein kinase-mammalian target of rapamycin pathway. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E1019–E1030. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Kourakis, S.; Timpani, C.A.; Bagaric, R.M.; et al. Repurposed Nrf2 activator dimethyl fumarate rescues muscle inflammation and fibrosis in an aggravated mdx mouse model of Duchenne muscular dystrophy. Redox Biol. 2025, 84, 103676. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ma, Y.; Yang, Q.; et al. AKAP1 mediates high glucose-induced mitochondrial fission through the phosphorylation of Drp1 in podocytes. J. Cell. Physiol. 2020, 235, 7433–7448. [Google Scholar] [CrossRef]
- Escobar-Henriques, M.; Anton, F. Mechanistic perspective of mitochondrial fusion: tubulation vs. fragmentation. Biochim. Et. Biophys. Acta 2013, 1833, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Gong, M.; Zhang, Z.; et al. Hyperglycemia Induces Endoplasmic Reticulum Stress in Atrial Cardiomyocytes, and Mitofusin-2 Downregulation Prevents Mitochondrial Dysfunction and Subsequent Cell Death. Oxidative Med. Cell. Longev. 2020, 2020, 6569728. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e817. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, H.; Li, A.M.; et al. VDR regulates mitochondrial function as a protective mechanism against renal tubular cell injury in diabetic rats. Redox Biol. 2024, 70, 103062. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- La Rovere, R.M.; Roest, G.; Bultynck, G.; et al. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87. [Google Scholar] [CrossRef]
- Wiersma, M.; Meijering, R.A.M.; Qi, X.Y.; et al. Endoplasmic Reticulum Stress Is Associated With Autophagy and Cardiomyocyte Remodeling in Experimental and Human Atrial Fibrillation. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Yuan, M.; Gong, M.; He, J.; et al. IP3R1/GRP75/VDAC1 complex mediates endoplasmic reticulum stress-mitochondrial oxidative stress in diabetic atrial remodeling. Redox Biol. 2022, 52, 102289. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Cheng, L.; Lv, C.; et al. Tea Polyphenol Modulates Mitochondria-Associated Endoplasmic Reticulum Membrane of Hippocampal Neurons Targeting Grp75 to Ameliorate Memory Impairment in the Aged T2DM Rats. Food Sci. Nutr. 2025, 13, e70776. [Google Scholar] [CrossRef]
- Pomeyie, K.; Abrokwah, F.; Boison, D.; et al. Macrophage immunometabolism dysregulation and inflammatory disorders. Biomed. Pharmacother. Biomed. Pharmacother. 2025(188), 118142. [CrossRef]
- Zhou, H.C.; Xin-Yan, Y.; Yu, W.W.; et al. Lactic acid in macrophage polarization: The significant role in inflammation and cancer. Int. Rev. Immunol. 2022, 41, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.H. SGLT2 Inhibitors and GLP-1 Receptor Agonists in Diabetic Kidney Disease: Evolving Evidence and Clinical Application. Diabetes Metab. J. 2025, 49, 386–402. [Google Scholar] [CrossRef]
- McGuire, D.K.; Shih, W.J.; Cosentino, F.; et al. Association of SGLT2 Inhibitors With Cardiovascular and Kidney Outcomes in Patients With Type 2 Diabetes: A Meta-analysis. JAMA Cardiol. 2021, 6, 148–158. [Google Scholar] [CrossRef]
- Jardine, M.J.; Mahaffey, K.W.; Neal, B.; et al. The Canagliflozin and Renal Endpoints in Diabetes with Established Nephropathy Clinical Evaluation (CREDENCE) Study Rationale, Design, and Baseline Characteristics. Am. J. Nephrol. 2017, 46, 462–472. [Google Scholar] [CrossRef]
- Kaze, A.D.; Zhuo, M.; Kim, S.C.; et al. Association of SGLT2 inhibitors with cardiovascular, kidney, and safety outcomes among patients with diabetic kidney disease: a meta-analysis. Cardiovasc. Diabetol. 2022, 21, 47. [Google Scholar] [CrossRef]
- Anson, M.; Zhao, S.S.; Austin, P.; et al. SGLT2i and GLP-1 RA therapy in type 1 diabetes and reno-vascular outcomes: a real-world study. Diabetologia 2023, 66, 1869–1881. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, S.A.; El-Tanani, M.; Kumar, R.; et al. Repurposing Diabetes Therapies in CKD: Mechanistic Insights, Clinical Outcomes and Safety of SGLT2i and GLP-1 RAs. Pharmaceuticals 2025, 18. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; An, X. 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3: A potential target in the development of diabetes and its complications. Metabolism 2026, 180, 156621. [Google Scholar] [CrossRef]
- Le, S.; Zhang, H.; Huang, X.; et al. PKM2 Activator TEPP-46 Attenuates Thoracic Aortic Aneurysm and Dissection by Inhibiting NLRP3 Inflammasome-Mediated IL-1β Secretion. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 364–376. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Yu, S.; Pei, S.; Zhang, M.; et al. PKM2-mediated STAT3 phosphorylation promotes acute liver failure via regulating NLRP3-dependent pyroptosis. Commun. Biol. 2024, 7, 1694. [Google Scholar] [CrossRef] [PubMed]
- Shimu, S.J.; Mahir, J.U.K.; Shakib, F.A.F.; et al. Metabolic Reprogramming Through Polyphenol Networks: A Systems Approach to Metabolic Inflammation and Insulin Resistance. Med. Sci. 2025, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, H.; Zhang, Y.; et al. Berberine Modulates Macrophage Activation by Inducing Glycolysis. J. Immunol. 2022, 208, 2309–2318. [Google Scholar] [CrossRef]
- Gong, P.; Wang, J.; Wang, S.; et al. Metabolomic analysis of the Puerarin hypoglycemic activity via AMPK-mTOR and PPARγ-NF-κB signaling pathways. Phytomed. Int. J. Phyther. Phytopharm. 2024, 130, 155546. [Google Scholar] [CrossRef]
- Noh, J.W.; Yang, H.K.; Jun, M.S.; et al. Puerarin Attenuates Obesity-Induced Inflammation and Dyslipidemia by Regulating Macrophages and TNF-Alpha in Obese Mice. Biomedicines 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.T.; Li, X.; Xie, M.L.; et al. Resveratrol: Review on its discovery, anti-leukemia effects and pharmacokinetics. Chem.-Biol. Interact. 2019, 306, 29–38. [Google Scholar] [CrossRef]
- Manríquez-Núñez, J.; Mora, O.; Villarroya, F.; et al. Macrophage Activity under Hyperglycemia: A Study of the Effect of Resveratrol and 3H-1,2-Dithiole-3-thione on Potential Polarization. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Sha, W.; Zhao, B.; Wei, H.; et al. Astragalus polysaccharide ameliorates vascular endothelial dysfunction by stimulating macrophage M2 polarization via potentiating Nrf2/HO-1 signaling pathway. Phytomed. Int. J. Phyther. Phytopharm. 2023, 112, 154667. [Google Scholar] [CrossRef]
Figure 1.
Imbalanced Energy Metabolism. EGF/VEGF, epidermal growth factor/vascular endothelial growth factor; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; Ca²⁺, calcium ion; Calcineurin, serine/threonine protein phosphatase 2B; Calmodulin, calcium-modulated protein; HG, high glucose; AMPK, AMP-activated protein kinase; SIRT1, sirtuin 1; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; NRF1/2, nuclear respiratory factor 1/2; TFAM, mitochondrial transcription factor A; mtDNA, mitochondrial DNA; ETC, electron transport chain; OXPHOS, oxidative phosphorylation; TCA Cycle, tricarboxylic acid cycle; MFN1/2, mitofusin 1/2; Drp1, dynamin-related protein 1; ROS, reactive oxygen species; ATP, adenosine triphosphate; β-cell, pancreatic β cell.
Figure 1.
Imbalanced Energy Metabolism. EGF/VEGF, epidermal growth factor/vascular endothelial growth factor; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; Ca²⁺, calcium ion; Calcineurin, serine/threonine protein phosphatase 2B; Calmodulin, calcium-modulated protein; HG, high glucose; AMPK, AMP-activated protein kinase; SIRT1, sirtuin 1; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; NRF1/2, nuclear respiratory factor 1/2; TFAM, mitochondrial transcription factor A; mtDNA, mitochondrial DNA; ETC, electron transport chain; OXPHOS, oxidative phosphorylation; TCA Cycle, tricarboxylic acid cycle; MFN1/2, mitofusin 1/2; Drp1, dynamin-related protein 1; ROS, reactive oxygen species; ATP, adenosine triphosphate; β-cell, pancreatic β cell.

Figure 2.
Exacerbated Oxidative Stress. GLUTs: glucose transporters; HG: high glucose; ROS: reactive oxygen species; O₂/H₂O₂: oxygen/hydrogen peroxide; SOD-1: superoxide dismutase 1; Gpx-1: glutathione peroxidase 1; TRX: thioredoxin; Nrf2: nuclear factor erythroid 2-related factor 2; ΔΨm: mitochondrial membrane potential; Cyt c: cytochrome c; Bim: Bcl-2-interacting mediator of cell death; Puma: p53 upregulated modulator of apoptosis; Bax: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma 2; IRS-1: insulin receptor substrate 1; PI3K: phosphatidylinositol 3-kinase; Akt: protein kinase B; GLUT4: glucose transporter 4.
Figure 2.
Exacerbated Oxidative Stress. GLUTs: glucose transporters; HG: high glucose; ROS: reactive oxygen species; O₂/H₂O₂: oxygen/hydrogen peroxide; SOD-1: superoxide dismutase 1; Gpx-1: glutathione peroxidase 1; TRX: thioredoxin; Nrf2: nuclear factor erythroid 2-related factor 2; ΔΨm: mitochondrial membrane potential; Cyt c: cytochrome c; Bim: Bcl-2-interacting mediator of cell death; Puma: p53 upregulated modulator of apoptosis; Bax: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma 2; IRS-1: insulin receptor substrate 1; PI3K: phosphatidylinositol 3-kinase; Akt: protein kinase B; GLUT4: glucose transporter 4.

Figure 3.
Organelle Crosstalk. DAMPs: damage-associated molecular patterns; T2DM: type 2 diabetes mellitus; Ca²⁺: calcium ion; mPTP: mitochondrial permeability transition pore; ΔΨm: mitochondrial membrane potential; ETC: electron transport chain; ATP: adenosine triphosphate; ROS: reactive oxygen species; MFN2: mitofusin 2; VDAC: voltage-dependent anion channel; GRP75: glucose-regulated protein 75; IP3R: inositol 1,4,5-trisphosphate receptor; MAMs: mitochondria-associated endoplasmic reticulum membranes; SERCA: sarco/endoplasmic reticulum calcium ATPase; ERS: endoplasmic reticulum stress; IRE1α: inositol-requiring enzyme 1α; XBP1: X-box binding protein 1; XBP1s: spliced X-box binding protein 1; PERK: protein kinase R-like endoplasmic reticulum kinase; eIF2α-P: phosphorylated eukaryotic initiation factor 2α; ATF4: activating transcription factor 4; ATF6: activating transcription factor 6; CHOP: C/EBP homologous protein.
Figure 3.
Organelle Crosstalk. DAMPs: damage-associated molecular patterns; T2DM: type 2 diabetes mellitus; Ca²⁺: calcium ion; mPTP: mitochondrial permeability transition pore; ΔΨm: mitochondrial membrane potential; ETC: electron transport chain; ATP: adenosine triphosphate; ROS: reactive oxygen species; MFN2: mitofusin 2; VDAC: voltage-dependent anion channel; GRP75: glucose-regulated protein 75; IP3R: inositol 1,4,5-trisphosphate receptor; MAMs: mitochondria-associated endoplasmic reticulum membranes; SERCA: sarco/endoplasmic reticulum calcium ATPase; ERS: endoplasmic reticulum stress; IRE1α: inositol-requiring enzyme 1α; XBP1: X-box binding protein 1; XBP1s: spliced X-box binding protein 1; PERK: protein kinase R-like endoplasmic reticulum kinase; eIF2α-P: phosphorylated eukaryotic initiation factor 2α; ATF4: activating transcription factor 4; ATF6: activating transcription factor 6; CHOP: C/EBP homologous protein.

Figure 4.
Systemic Inflammation. DAMPs: damage-associated molecular patterns; ROS: reactive oxygen species; TLR4: toll-like receptor 4; MAMs: mitochondria-associated endoplasmic reticulum membranes; Ca²⁺: calcium ion; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; MAPK: mitogen-activated protein kinase; JNK: c-Jun N-terminal kinase; TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1β; IL-6: interleukin-6; GLUT1: glucose transporter 1; HK2: hexokinase 2; PFK1: phosphofructokinase 1; ACLY: ATP-citrate lyase; Acetyl-CoA: acetyl coenzyme A; TCA cycle: tricarboxylic acid cycle; PHD: prolyl hydroxylase; HIF-1α: hypoxia-inducible factor 1-alpha; iNOS: inducible nitric oxide synthase; Arg1: arginase 1; IR: insulin resistance; T2DM: type 2 diabetes mellitus.
Figure 4.
Systemic Inflammation. DAMPs: damage-associated molecular patterns; ROS: reactive oxygen species; TLR4: toll-like receptor 4; MAMs: mitochondria-associated endoplasmic reticulum membranes; Ca²⁺: calcium ion; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; MAPK: mitogen-activated protein kinase; JNK: c-Jun N-terminal kinase; TNF-α: tumor necrosis factor-alpha; IL-1β: interleukin-1β; IL-6: interleukin-6; GLUT1: glucose transporter 1; HK2: hexokinase 2; PFK1: phosphofructokinase 1; ACLY: ATP-citrate lyase; Acetyl-CoA: acetyl coenzyme A; TCA cycle: tricarboxylic acid cycle; PHD: prolyl hydroxylase; HIF-1α: hypoxia-inducible factor 1-alpha; iNOS: inducible nitric oxide synthase; Arg1: arginase 1; IR: insulin resistance; T2DM: type 2 diabetes mellitus.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



