Submitted:
29 April 2026
Posted:
05 May 2026
You are already at the latest version
Abstract
In protein solutions, an additive that increases protein-protein attractive interactions is expected to decrease protein crystal solubility and raise temperature of liquid-liquid phase separation (LLPS). In contrast, addition of 0.10-M 4-(2-hydroxyethyl)-1-piperazineethanesulfonate (HEPES) to lysozyme-NaCl aqueous solutions at constant pH (7.4) and ionic strength (0.20 M) decreases solubility but lowers LLPS temperature. This leads to a broadening of LLPS metastability gap in the phase diagram and an enhancement of protein crystallization yield from LLPS. We theoretically examine the effect of HEPES on both solubility and LLPS boundaries using a colloid model. Under the hypothesis that HEPES stabilizes protein-protein contacts in the crystal lattice by physical cross-linking, we apply cell theory to describe the thermodynamic behavior of the crystalline phase and use solubility data to show that HEPES increases protein-protein attraction energy by 2.7%. Since an increase in attraction incorrectly predicts a raise in LLPS temperature, we consider that HEPES also enhances the anisotropic character of protein-protein interactions. To describe the thermodynamic behavior of the solution phase, we start from Barker-Henderson second-order perturbation theory on the hard-sphere reference fluid with square-well potential and local-compressibility approximation. We modify this model so that it can reproduce the correct mathematical expression of the second virial coefficient. This also leads to a better agreement with Monte Carlo simulations. We then approximately incorporate anisotropy by assuming that the square-well attraction energy is a temperature-dependent average over all particle surface with a given fractional coverage of attractive spots. The attraction energy of the attractive spots is set to be the same as that of protein-protein contacts in the crystal. Only fractional coverage (anisotropy) was varied to successfully fit the effect of HEPES on the LLPS boundary.
Keywords:
lysozyme
; HEPES
; NaCl
; metastability
; anisotropy
; Barker-Henderson
1. Introduction
Understanding and controlling condensation of globular proteins in aqueous media is fundamental for comprehending cell compartmentalization,[1,2,3] protein-aggregation diseases,[4,5] developing stable pharmaceutical formulations,[6,7] preparing protein-based materials,[8,9] and generating protein crystals[10,11,12] with applications in structural biology[13] and separation science[14,15].
An interesting phenomenon of protein aqueous mixtures is the reversible formation of metastable protein-rich liquid microdroplets through liquid-liquid phase separation (LLPS), typically induced by lowering temperature.[16,17,18] Since LLPS is a metastable phase transition, the protein-rich phase can act as an intermediate for the formation of other protein condensed phases such as crystals[17,19,20] and aggregates[4,21,22]. This phenomenon was initially considered for just few protein cases: eye-lens crystallins[16,23,24] and lysozyme[17,25]. However, it has recently attracted considerably more attention as it is believed to drive the formation of membraneless organelles in the cytosol and can promote formation of pathological protein aggregates.[1,2,3,4,5] It has also been investigated in the contest of protein pharmaceutical formulations, where LLPS is described to negatively impact the stability and efficacy of monoclonal antibodies.[8] Finally, LLPS is also known to enhance the rate of protein crystallization,[19,26,27,28] which is beneficial not only for the characterization of protein 3D structure[29,30] but also for protein purification in downstream processing[14,15].
From a molecular point of view, all protein condensation processes are driven by solvent-mediated protein-protein attractive interactions.[31] In principle, it is extremely difficult to describe protein-protein interactions as they ultimately depend on the surface composition and spatial orientation of amino-acid groups. These are responsible for the formation of a highly heterogenous distribution of charged chemical groups, hydrophilic moieties and hydrophobic patches. Moreover, additives, which are invariably present in protein solutions, can significantly alter protein-protein interactions.[32] These cosolvents modify these interactions through mechanisms such as electrostatic screening,[33,34] preferential hydration or binding,[35,36] and molecular crowding[18,37,38,39,40,41]. Nevertheless, some key features associated with the collective behavior of globular proteins can be theoretically described by employing a simple one-component colloid model using hard spheres as a reference system.[42,43,44] For example, LLPS metastability with respect to protein crystallization is successfully explained by assuming that the range of protein-protein interactions is relatively short compared to particle diameter.[42]
From a thermodynamic point of view, the phase behavior of a protein aqueous system is typically described by employing a temperature-composition phase diagram. As schematized in Figure 1A, a typical phase diagram shows a protein-crystal solubility curve with the LLPS boundary positioned in the crystal supersaturated domain, at relatively high protein concentrations. Note that the LLPS boundary can be experimentally characterized because protein crystallization kinetics is usually slow.[6,16,42,45]
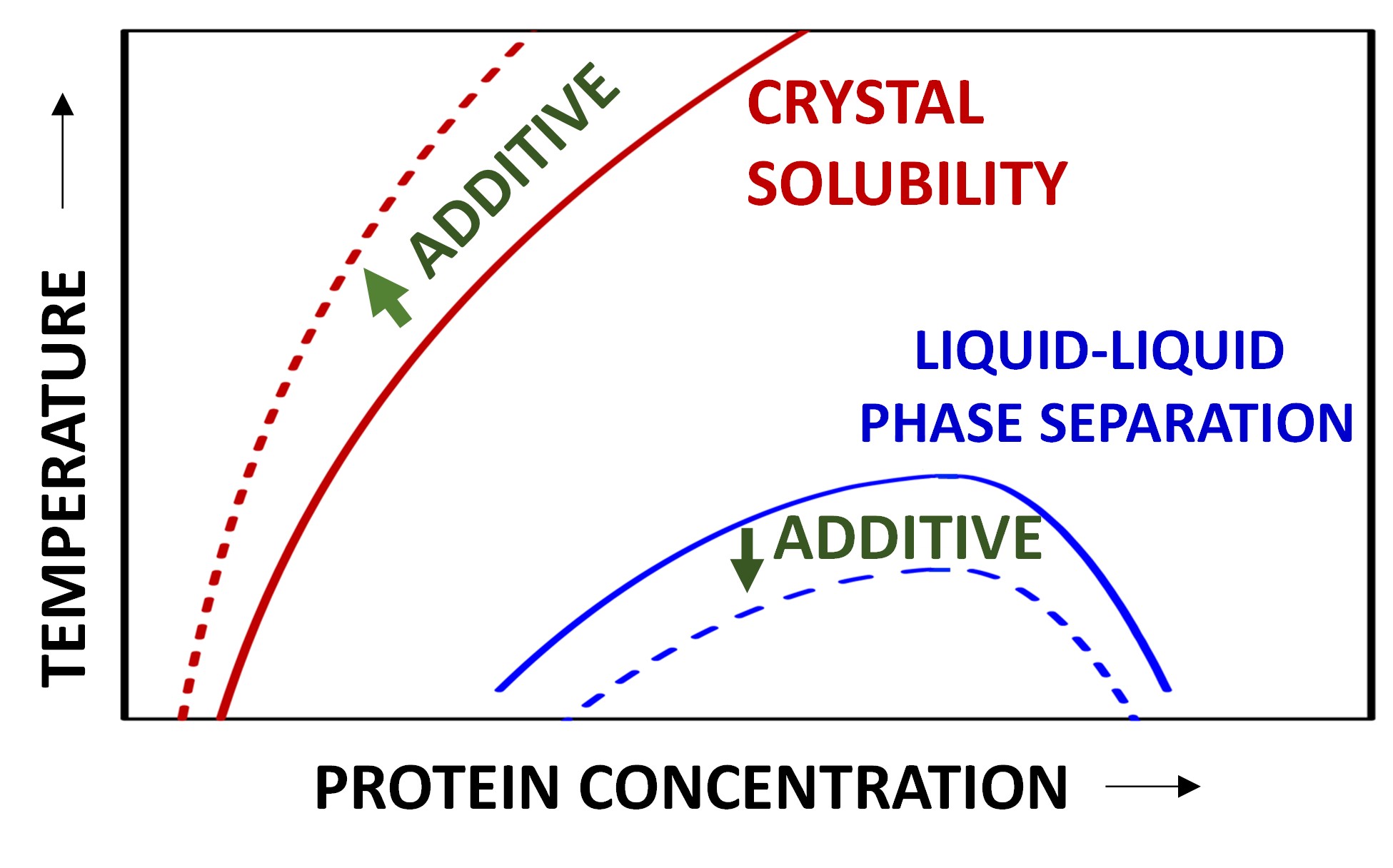
Additives such as salts, buffer components and polyethylene glycols are typically present in protein aqueous solutions. They are used to stabilize proteins against unfolding, favor protein crystallization and mimic physiological conditions. The thermodynamic effect of additive concentration has often been described using the theory of preferential interaction.[46] Accordingly, an additive that is preferentially excluded from the vicinity of protein surface (preferential hydration) reduces protein solubility (e.g., NaCl for lysozyme, sulfates). In contrast, an additive that accumulates near protein surface (preferential binding) increases protein solubility (e.g., thiocyanates, urea).[47,48] A one-component colloid model may be still used to describe the phase behavior of these multicomponent protein solutions provided that the additive has a low molecular weight and is therefore considered as integral part of the solvent. The effect of additive concentration is then implicitly taken into account by considering a corresponding change in the solvent-mediated protein-protein interactions.[44] For example, attraction energy should increase with additive concentration in the presence of preferential hydration.[49] As illustrated in Figure 1B, this salting-out effect leads to a shift of both crystal solubility and LLPS boundary towards higher temperatures, with crystal solubility shifting toward lower protein concentrations. This behavior has been experimentally demonstrated for lysozyme in the presence of NaCl and other additives.[17,50,51] Interestingly, shifts in the LLPS boundary appear to be stronger than those of crystal solubility. This results in an increase in the metastability gap between LLPS boundary and solubility curve.[51]
In our previous studies, we have shown that addition of 4-(2-hydroxyethyl)-1-piperazineethanesulfonate (HEPES) to lysozyme-NaCl solutions at constant ionic strength and pH produces even a more complex effect on the phase diagram.[52,53] As qualitatively schematized in Figure 1C, HEPES moves the solubility curve toward higher temperatures (as in Figure 1B) while moving the LLPS boundary in the opposite direction. This leads to a significant increase in the metastability gap between the two phase boundaries. Remarkably, addition of HEPES was also found to significantly increase yield of protein crystallization, especially in the presence of LLPS.[28] Clearly, understanding the multifaceted effect of this type of additives the phase behavior of protein solutions, especially in relation to the metastability gap, is important for controlling protein crystallization and other aggregation processes.
To explain the effect of HEPES on lysozyme phase diagram, partitioning experiments were also carried out. It was found that HEPES accumulates in the protein-rich liquid phase showing that HEPES preferentially binds to lysozyme.[52] Furthermore, dynamic-light-scattering experiments showed that HEPES weakens protein-protein attraction energy.[52] While these results are consistent with HEPES suppressing lysozyme LLPS, they cannot explain HEPES salting-out effect on lysozyme solubility.
The lysozyme 3D structure obtained from crystals grown in HEPES buffer also shows that his organic molecules act as a ligand. Indeed, it is found in the lysozyme catalytic site, with the hydroxyethyl and sulfonate groups pointing inward and outward, respectively.[54] The sulfonate group is close to a cationic arginine group of a neighboring protein. It is therefore reasonable to assume that the electrostatic attraction between these two ionic groups may strengthen one of the crystal contacts.[53] In other words, the effect of HEPES on lysozyme solubility may be qualitatively explained by considering that this organic molecule acts as a physical cross-linker enhancing attractive interactions between neighboring protein units in the crystal lattice. This thermodynamically stabilizes the crystalline phase. Our hypothesis aligns with the general belief that multi-functional organic molecules are beneficial in protein crystallography.[55]
In this paper, we examine the ability of a colloid model to quantitatively describe the multifaceted effects of HEPES on lysozyme phase behavior assuming that it does not alter protein conformational state. We will specifically model the effect of this additive by assuming that it causes an increase in protein-protein attractive interactions accompanied with an increase in their anisotropic character. It is also assumed that the additive does not alter protein conformational state.
2. Methods
A protein aqueous solution is described as a one-component compressible fluid of colloidal particles provided that this aqueous medium is regarded as incompressible and low-molecular weight additives such as salts and buffer components are implicitly described as a part of the background solvent. These additives are modulators of solvent-mediated protein-protein interactions. Correspondingly, the protein osmotic pressure, , becomes a “gas pressure” and LLPS is equivalent to a “gas-liquid” phase transition. Furthermore, protein chemical potential, , must describe particle insertion accompanied with isochoric removal of solvent.[31] It is then appropriate to start from the Helmholtz free energy, , of the colloid fluid. It is also practically convenient to introduce the corresponding reduced unitless quantity, , where , is the Boltzmann constant, the absolute temperature, the protein volume, and is the volume of the fluid phase. We shall set so that is the same as temperature, and energy parameters adopt temperature units. The composition of the fluid phase is described by the volume fraction, , where is the number of colloidal particles. An experimental volume fraction of lysozyme solution is readily calculated by multiplying lysozyme mass concentration by the known[56] specific volume of this protein (0.713 cm3·g-1).
The protein phase diagram[42] shows protein volume fraction () and temperature () on the x- and y-axis, respectively. The LLPS boundary is described by the protein volume fractions, and , of the two coexisting of protein-diluted (“gas”, I) and -concentrated (“liquid”, II) fluid phases. These two volume fractions become equal at the critical point, which is described by the critical coordinates, .To determine and , the expressions of chemical potential and osmotic pressure are extracted from the Helmholtz free energy using
where and . At any sufficiently high value of , the protein volume fractions, and , of the two coexisting phases are determined by numerically solving the chemical-equilibrium conditions:
Critical coordinates, , are extracted using the conditions: .
The hard-sphere model is employed as a reference model. The reduced Helmholtz free energy, , is then given by
where is the hard-sphere contribution and is a residual term describing deviation from reference model. The hard-sphere term, which describes both particle translational motion (ideal contribution) and hard-sphere excluded-volume interactions (steric repulsion), is given by
where is the uninfluential standard chemical potential, the second term represents the ideal-gas contribution and the third term describes steric repulsion according to Carnahan-Starling equation of state.[57] It follows from Eqs. 1a,b that the hard-sphere chemical potential and pressure are given by and , respectively. The residual term in Eq. 3 characterizes protein-protein attraction energy and vanishes in the limit of high temperatures ().[58] This residual term will be further discussed in the next section.
The crystalline ordered phase, which has a very high protein volume fraction (typically more than 50%), is assumed to be incompressible.[42,59] In this case, the protein chemical potential in the solid crystalline phase, , coincides with the Helmholtz free energy of one particle. The crystal solubility boundary is described by the protein volume fraction of the coexisting fluid phase, . At any given , this is determined by numerically solving the chemical-equilibrium condition:
where . A simple expression for was obtained from cell theory as discussed in the next section.
3. Results and Discussion
To examine the effect of HEPES on the phase diagram of lysozyme aqueous solutions, we have previously considered two systems. The first system contains HEPES (0.10 M) and NaCl (0.15 M) at pH 7.4, while the second system is a reference system (denoted as REF) and mostly contains NaCl (0.183 M; Tris buffer, 0.020 M) at the same pH and ionic strength (0.20 M).[52,53] In this way, replacement of NaCl with HEPES is carried out in conditions in which long-range electrostatic repulsive interactions between proteins remain the same. Note the presence of Tris buffer is REF, which was employed to improve buffer capacity. Although its contribution to the total ionic strength of 0.20 was taken into account, its concentration is fairly low and Tris-specific effects are not expected to be significant.
In the following subsections, we shall first examine the effect of HEPES on lysozyme crystal solubility and then the corresponding effect on the LLPS boundary.
3.1. Crystal Solubility and Thermodynamics of Protein Crystal
To theoretically describe crystal solubility, expressions for the protein chemical potential in the crystal and fluid phases in Eq. 5 are needed. According to cell theory,[42,59] , may be written as:
where is the same as in Eq. 4. The second term represents crystal internal energy, with loosely representing the number of interactions “sites” or “contacts” between a central protein and its neighboring surrounding proteins. The factor “2” considers that a protein-protein interaction involves two proteins; i.e., the range of interactions is sufficiently short that one interaction cannot extend to more than two proteins. In Eq. 6, represents the average attraction energy of a contact. In principle, the number of contacts could be defined as involving interactions between single amino acids (atomic contacts) or as relatively big regions on the protein surface encompassing multiple functional groups on the protein surface (region contacts). Thus, the value of depends on the choice of and only their product, , can be unambiguously defined. The last term in Eq. 6 is an entropic contribution related to both the residual translational and orientational motion of a protein in the crystal lattice, with representing the phase volume[59] (as a multiple of particle volume, ) accessible to both particle center of mass and particle rotation. It will be further discussed at the end of this subsection.
The highest experimental[53] are 0.014 and 0.027 for the HEPES and REF system, respectively. We assume that these values are sufficiently low that the protein chemical potential of the fluid phase is approximated by its ideal-dilute approximation, , neglecting contribution of protein-protein interactions in the fluid phase. We will later validate this assumption by verifying that inclusion of steric repulsion and protein-protein attraction energy terms do not significantly alter solubility boundary. Insertion of Eq. 6 into Eq. 5 yields the following Van’t Hoff equation:
Van’t Hoff plots for lysozyme solubility in the HEPES and REF systems are shown in Figure 2. Here, we can see that solubility values in the HEPES system are appreciably lower than those in the REF system. Application of the method of least squares yields: (10.4±0.5)103 K (86.6 kJ·mol-1) and -(12.9±1.0) for the HEPES system and (10.1±1.0)103 K (84.0 kJ·mol-1) and -(12.9±1.8) for the REF system. These values are essentially the same within the experimental errors due to correlation between slope and intercept parameters. To further examine these solubility data, we assume that addition of HEPES has a negligible effect on crystal entropy. This assumption is motivated by the hypothesis that HEPES energetically stabilizes region contacts through physical crosslinking while not appreciably altering crystal lattice, consistent with available crystal structures.[54,60,61] Accordingly, we define as a region contact and assume it is the same for both systems. We then set -12.9 so that we can attribute solubility variations entirely to . We can now determine appreciably different values of . These are (10.39±0.02)×103 K and (10.12±0.03)×103 K for the HEPES and REF systems, respectively. The corresponding linear fits are also shown in Figure 2. These share the same intercept at =0.
To determine , we need to identify a reasonable value of . It is known[61,62] that there are eight lysozyme molecules interacting with every lysozyme molecule in the tetragonal crystal structure. Correspondingly, there are eight region contacts on each protein involved in the formation of multiple interatomic bonds with neighboring proteins. However, the interactions occurring with the two proteins immediately above and below the reference protein (along the crystallographic c axis) are weak and can be neglected.[62] The proteins involved in the interactions with a reference protein (M) are represented on the (0,0,1) crystallographic plane in Figure 3. Here, we can see that there are two neighboring proteins, F (at ) and G (at ), related to M by twofold screw symmetry. There are also two B (at and ) and two C (at and ) proteins with different positions along the c-axis interacting with M. These are related by fourfold screw symmetry to M.[61,62] The catalytic site (cleft) of protein M, which can be occupied by HEPES, is near the protein B at . It is therefore reasonable to attribute the increase in attraction energy caused by HEPES to this specific contact region.
In summary, there are six main region contacts on the protein that can be identified as binding sites. We therefore set 6 and determine that the average attraction energy of an interacting region is 1732 K and 1687 K for the HEPES and REF system, respectively. In summary, our data analysis shows that HEPES causes an increase of 2.7% in the value of inside the crystal.
We conclude this subsection by examining the extracted describing crystallization entropy. Although its accurate interpretation should factor in protein shape and changes in solvent entropy, it is important to examine whether the value of phase volume, , yields physically acceptable geometric parameters. We specifically assume that is the product of a translational factor, , and an orientational factor, . The translational factor is given by , where is the center-of-mass excursion volume. If proteins are assumed to be spherical particles, we can use unit cell volume of tetragonal lysozyme (238 nm3), number of proteins inside unit cell (8)[61] and protein volume (16.9 nm3) to determine that protein volume fraction in the crystal is 0.57. For spherical particles, translational motion is lost when volume fraction reaches the classical close-packing value of 0.7405. Since an increase of 1.091 in particle diameter is needed to increase experimental volume fraction to close-packing value, the diameter of the excursion sphere is estimate to be , where is the diameter of the protein. This implies that from which we calculate: . To estimate angular degree of freedom, we may set: , where represents the maximum angular phase corresponding to a freely-rotating particle, and is the geometric mean of the three Euler angles accessible through trough particle rotation in the crystal.[59,63] We then extract 18º, which is a physically acceptable[59] angular degree of freedom.
3.2. Thermodynamic Model for the Fluid Phase
In order to describe the thermodynamic behavior of protein solutions, an expression for in Eq. 3 is needed. To enable LLPS, this expression must incorporate protein-protein attraction energy. According to our solubility results, HEPES is responsible for an increase in protein-protein attraction energy. This increase, however, should also cause a corresponding increase in LLPS temperature, in contrast with experimental results showing that HEPES causes a ≈5% decrease[52,53] in LLPS temperature. To further examine the effect of HEPES on protein-protein interactions, we examine previous measurements of lysozyme diffusion coefficient[52] as a function of protein concentration for the HEPES and REF models at 298 K. Assuming that the hydrodynamic factor[64] is the same for both systems, these diffusion data show that HEPES causes an increase of in the second virial coefficient[43] ( in ). This confirms that HEPES decreases protein-protein attraction energy in the fluid phase. It is in apparent conflict with HEPES being able to increase protein-protein attraction energy in in the crystalline phase.
We have previously mentioned that HEPES can stabilize lysozyme crystals by physical cross-linking interactions. This type of interaction can also occur in the fluid phase. However, their highly directional character makes them entropically unfavored, i.e., crosslinking can occur only if the relative orientation between two proteins is appropriate. In the crystalline phase, this entropy cost is marginal because protein orientation is essentially fixed by the lattice structure. In contrast, protein-protein attraction energy in the fluid phase can be weaker on average due to free rotation. In other words, it is the anisotropic character of protein-protein interactions that may explain the opposite effects of HEPES on lysozyme solubility and LLPS.
To theoretically derive LLPS boundaries, we need to discuss in Eq. 3. There are several Monte-Carlo studies accurately describing the Helmholtz free energy of model fluid systems appropriate for protein solutions.[31,63,65,66,67,68,69] Nonetheless, it remains practically convenient to use thermodynamic-perturbation theories[44,58,59,70,71,72] that provide approximate analytical expression of the Helmholtz free energy. In our case, we shall consider a model that depends on just three parameters, describing attraction energy, range of interactions and degree of anisotropy. These are linked to the three main topological features of a dome-shaped LLPS boundary: the two critical point coordinates, and the boundary width.
In this work, we shall consider the Barker-Henderson perturbation theory of square-well fluids (BHPT)[58,72] to describe the effect of HEPES on lysozyme phase behavior. Note that BHPT treats particle-particle interactions as isotropic. However, anisotropy may be incorporated in BHPT by introducing temperature-dependent energy parameter that depends on the fraction of protein surface engaging in protein-protein interactions.[63] Our revised BHPT model will be further discussed below. It is important to note that Wertheim perturbation theory of associating spheres (WPT)[44,59,70,73] has also been also applied to examine the phase behavior of protein solutions. WPT explicitly describes anisotropic interactions by considering a specified number of sites that can link two spherical particles. However, we chose to use BHPT instead of WPT as there is a noticeable difference in between WPT and computer simulation data[31,69] on spherical particles with same number of sites. Moreover, it also yields a relatively large difference in second virial coefficient between HEPES and REF systems. Nonetheless, application of WPT with the same number of parameters employed in our revised BHPT is discussed in Supplementary Material for completeness.
The residual Helmholtz free energy for the original BHPT model is written as
where rigorously represents a mean-field first-order correction to the hard-sphere reference model. In the case of isotropic square-well potential, we have:[58]
where is the magnitude of the square-well attraction energy. Attraction occurs when the distance between the centers of two particles, , is such that while no interactions occur if , where is the particle diameter and specifies the range of attractive interactions. In Eq. 9, is the average number of contacts that each particle makes within the range , evaluated using the pair distribution function of the reference hard-sphere fluid, with . We specifically write:
with represents in the limit of and is the Carnahan-Staling contact-value of evaluated at the particle volume fraction, . The following Pade` approximant of is available: [72,74,75]
where with and the matrix of coefficients is given by [-3.1649, 13.3501, -14.8057, 5.7029; 43.0042, -191.6623, 273.8968, -128.9334; 65.0419, -266.4627, 361.0431, -162.6996].
The second contribution in Eq. 8, , takes into account higher order terms in BHPT and can be evaluated only approximately. The most successful approximation is the local compressibility approximation,[58,72] where is the second-order perturbation term of the Helmholtz free energy. The term, , is approximately obtained by examining the radial profile of fluctuations of particle density around a central particle.[58] At a given distance from this particle, these fluctuations are locally described by the isothermal compressibility of the hard-sphere reference model. This leads to[58,72]
where and represents the second-order term in the series expansion of the Mayer function, . We can numerically calculate from Eq. 10 while we can use Carnahan-Starling equation of state[57,72] to determine that. It is important to note that the BHPT model is applicable for moderately short interaction ranges; and higher.[75] This is appropriate for protein solution with and less.[31] It should not be used when particle-particle interactions are very short ().
The range of interaction, , is the only parameter that affects the value of the critical volume fraction, . In our application of the BHPT model, we should consider as a fitting parameter that is just needed to reproduce experimental values of . It is also important to note that the use of protein specific volume to convert concentrations into volume fractions is debatable as the diameter of a protein is not straightforwardly connected to protein specific volume. This adds uncertainty in the comparison between theoretical and experimental values of . In some studies,[44,73] the protein diameter has also been proposed as an extra fitting parameter to be varied in order to enhance model accuracy.
It is known that the range of interactions specifies the value of , with corresponding to .[31] Thus, we focus on the LLPS boundary extracted using Eqs. 8-12 with . For comparison, the critical point extracted from related Monte Carlo simulations[31] is also included. We can appreciate that the values of and calculated using BHTP are both ≈6% higher than those extracted from simulations. For completeness, we also included the boundary calculated after setting 0. This exhibits relatively larger deviations from simulation results as expected.
It is important to note that Eq. 12 does not yield the correct expression of the second-virial coefficient, , for the square-well fluid.[75] Specifically, it yields instead of . Since the second-virial coefficient is a fundamental thermodynamic property of fluids, it is important that the expression of reproduces the correct expression of . We therefore replace Eq. 12 with
which is the same as Eq. 12 to second order in . As shown in Figure 4A, Eq. 13 significantly improves agreement with Monte Carlo Simulations. Specifically, the values of and are now found to be just ≈2% higher than those extracted from simulations. Hence, we will use Eq. 13 instead of Eq. 12 in our data analysis. In Figure 4B, we show the LLPS boundaries calculated at different values of . Here, we can see that increases as decreases as expected from theory and Monte Carlo simulations.[31]
We now need to incorporate anisotropy in the BHPT model. One way to approximately describe anisotropy in the square-well fluid is to replace with a temperature-dependent effective energy, , given by the following free-energy expression:[63]
where the factor “2” considers that a protein-protein interaction involves two proteins, is a degeneracy factor representing the fraction of particle surface occupied by binding sites with energy , and represents the fraction of particle surface with zero binding energy. If , we recover the isotropic case: . As decreases, the area covered by binding sites decreases and the anisotropic character of protein-protein interactions increases.
3.3. Effect of HEPES on Lysozyme LLPS Boundary
We shall now employ the BHTP model with to describe the experimental LLPS boundaries for REF and HEPES systems. These are shown in the phase diagrams of Figure 5 together with the corresponding crystal solubility curves. To make the crystal model consistent with the BHPT model, we set the attraction energy parameter, , for the fluid phase to be the same as extracted from solubility data. We are then left with identifying the values of that best fit the two sets of LLPS data. We determine that describes the LLPS of the REF system well. This value must be then decreased to in order to accurately represent the lower LLPS temperatures of the HEPES system. Noteworthily, these values of also describe the width of the LLPS boundaries fairly well. Specifically, experimental LLPS data show a decrease of ≈4% in LLPS temperature when protein volume fraction changes from ≈0.2 to 0.05. These variations are comparable with those shown for in Figure 4D. In Figure 5, theoretical solubility curves are now obtained by calculating the protein chemical potential using the BHPT model. These theoretical curves, which are also shown in Figure 5, accurately describe solubility data justifying the initial use of . Finally, we can also make predictions on the values of second virial coefficient at 298 K. Specifically, we use Eq. 15 to calculate that 278 K for the REF system is higher than 244 K for the HEPES system. If then use the square-well expression of second virial coefficient with replacing :
we calculate -3.4 for the REF system and the higher value of -2.1 for the REF system. Their difference, +1.3, is essentially the same as that estimated from DLS data at 298 K ().
4. Conclusions
We applied a colloid model to theoretically examine the opposite effects of HEPES on lysozyme crystal solubility and LLPS boundary. We specifically applied cell theory to solubility data to determine that HEPES increases protein-protein attraction energy by 2.7%. To explain the observed decrease in LLPS temperature, we considered that HEPES also enhances the anisotropic character of protein-protein interactions. We developed an analytical model based on BHPT to describe the LLPS boundaries using the same protein-protein attraction energy parameter extracted from solubility data (). Only the parameter, , was decreased in order to increase anisotropy and successfully explain the effect of HEPES on LLPS temperature and change in second virial coefficient. Our work describes a useful analytical model for describing the multifaceted effects of additives on the phase diagram of protein solutions.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
This work was supported by TCU Research and Creative Activity Funds (grant number: 61038).
References
- Patra, S.; Sharma, B.; George, S. Programmable Coacervate Droplets via Reaction-Coupled Liquid-Liquid Phase Separation (LLPS) and Competitive Inhibition. J. Am. Chem. Soc. 2025, 147(19), 16027–16037. [Google Scholar] [CrossRef]
- Nakashima, K.; van Haren, M.; André, A.; Robu, I.; Spruijt, E. Active coacervate droplets are protocells that grow and resist Ostwald ripening. Nat. Commun. 2021, 12(1). [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C. P. Liquid phase condensation in cell physiology and disease. Science 2017, 357(6357). [Google Scholar] [CrossRef]
- Sudhakar, S.; Manohar, A.; Mani, E. Liquid-Liquid Phase Separation (LLPS)-Driven Fibrilization of Amyloid-β Protein. ACS Chem. Neurosci. 2023, 14(19), 3655–3664. [Google Scholar] [CrossRef]
- Carey, J.; Guo, L. Liquid-Liquid Phase Separation of TDP-43 and FUS in Physiology and Pathology of Neurodegenerative Diseases. Front. Mol. Biosci. 2022, 9. [Google Scholar] [CrossRef]
- Rowe, J. B.; Cancel, R. A.; Evangelous, T. D.; Flynn, R. P.; Pechenov, S.; Subramony, J. A.; Zhang, J. F.; Wang, Y. Metastability Gap in the Phase Diagram of Monoclonal IgG Antibody. Biophys. J. 2017, 113(8), 1750–1756. [Google Scholar] [CrossRef]
- Raut, A. S.; Kalonia, D. S. Pharmaceutical Perspective on Opalescence and Liquid-Liquid Phase Separation in Protein Solutions. Mol. Pharm. 2016, 13(5), 1431–1444. [Google Scholar] [CrossRef]
- Liu, J.; Spruijt, E.; Miserez, A.; Langer, R. Peptide-based liquid droplets as emerging delivery vehicles. Nat. Rev. Mater. 2023, 8(3), 139–141. [Google Scholar] [CrossRef]
- Ng, T.; Hoare, M.; Maristany, M.; Wilde, E.; Sneideris, T.; Huertas, J.; Agbetiameh, B.; Furukawa, M.; Joseph, J.; Knowles, T.; et al. Tandem-repeat proteins introduce tuneable properties to engineered biomolecular condensates. Chem. Sci. 2025, 16(23), 10532–10548. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, H.; Qiao, B.; Wang, Y. Review of Liquid-Liquid Phase Separation in Crystallization: From Fundamentals to Application. Cryst. GROWTH Des. 2021, 21(12), 7306–7325. [Google Scholar] [CrossRef]
- McPherson, A. Introduction to the Crystallization of Biological Macromolecules. Membr. Protein Cryst. 2009, 63, 5–23. [Google Scholar] [CrossRef]
- Chen, J.; Sarma, B.; Evans, J. M. B.; Myerson, A. S. Pharmaceutical Crystallization. Cryst. Growth Des. 2011, 11(4), 887–895. [Google Scholar] [CrossRef]
- McPherson, A. Crystallization of Biological Macromolecules; Cold Spring Harbor Lab. Press, 1999. [Google Scholar]
- Hekmat, D.; Breitschwerdt, P.; Weuster-Botz, D. Purification of proteins from solutions containing residual host cell proteins via preparative crystallization. Biotechnol. Lett. 2015, 37(9), 1791–1801. [Google Scholar] [CrossRef]
- Hubbuch, J.; Kind, M.; Nirschl, H. Preparative Protein Crystallization. Chem. Eng. Technol. 2019, 42(11), 2275–2281. [Google Scholar] [CrossRef]
- Broide, M. L.; Berland, C. R.; Pande, J.; Ogun, O. O.; Benedek, G. B. BINARY-LIQUID PHASE-SEPARATION OF LENS PROTEIN SOLUTIONS. Proc. Natl. Acad. Sci. U. S. A. 1991, 88(13), 5660–5664. [Google Scholar] [CrossRef]
- Muschol, M.; Rosenberger, F. Liquid-liquid phase separation in supersaturated lysozyme solutions and associated precipitate formation/crystallization. J. Chem. Phys. 1997, 107(6), 1953–1962. [Google Scholar] [CrossRef]
- Annunziata, O.; Asherie, N.; Lomakin, A.; Pande, J.; Ogun, O.; Benedek, G. B. Effect of polyethylene glycol on the liquid-liquid phase transition in aqueous protein solutions. Proc. Natl. Acad. Sci. U. S. A. 2002, 99(22), 14165–14170, Article. [Google Scholar] [CrossRef]
- Galkin, O.; Vekilov, P. G. Control of protein crystal nucleation around the metastable liquid-liquid phase boundary. Proc. Natl. Acad. Sci. U. S. A. 2000, 97(12), 6277–6281. [Google Scholar] [CrossRef] [PubMed]
- Pantuso, E.; Mastropietro, T.; Briuglia, M.; Gerard, C.; Curcio, E.; ter Horst, J.; Nicoletta, F.; Di Profio, G. On the Aggregation and Nucleation Mechanism of the Monoclonal Antibody Anti-CD20 Near Liquid-Liquid Phase Separation (LLPS). Sci. Rep. 2020, 10(1). [Google Scholar] [CrossRef] [PubMed]
- Galkin, O.; Chen, K.; Nagel, R.; Hirsch, R.; Vekilov, P. Liquid-liquid separation in solutions of normal and sickle cell hemoglobin. Proc. Natl. Acad. Sci. U. S. A. 2002, 99(13), 8479–8483. [Google Scholar] [CrossRef]
- Babinchak, W.; Surewicz, W. Liquid-Liquid Phase Separation and Its Mechanistic Role in Pathological Protein Aggregation. J. Mol. Biol. 2020, 432(7), 1910–1925. [Google Scholar] [CrossRef]
- Liu, C. W.; Asherie, N.; Lomakin, A.; Pande, J.; Ogun, O.; Benedek, G. B. Phase separation in aqueous solutions of lens gamma-crystallins: Special role of gamma(s). Proc. Natl. Acad. Sci. U. S. A. 1996, 93(1), 377–382. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, O.; Pande, A.; Pande, J.; Ogun, O.; Lubsen, N. H.; Benedek, G. B. Oligomerization and phase transitions in aqueous solutions of native and truncated human beta B1-crystalline. Biochemistry 2005, 44(4), 1316–1328. [Google Scholar] [CrossRef]
- Taratuta, V. G.; Holschbach, A.; Thurston, G. M.; Blankschtein, D.; Benedek, G. B. LIQUID LIQUID-PHASE SEPARATION OF AQUEOUS LYSOZYME SOLUTIONS - EFFECTS OF PH AND SALT IDENTITY. J. Phys. Chem. 1990, 94(5), 2140–2144. [Google Scholar] [CrossRef]
- Jim, A. I.; Goh, L. T.; Oh, S. K. W. Crystallization of IgG(1) by mapping its liquid-liquid phase separation curves. Biotechnol. Bioeng. 2006, 95(5), 911–918. [Google Scholar] [CrossRef]
- Maier, R.; Zocher, G.; Sauter, A.; Da Vela, S.; Matsarskaia, O.; Schweins, R.; Sztucki, M.; Zhang, F. J.; Stehle, T.; Schreiber, F. Protein Crystallization in the Presence of a Metastable Liquid-Liquid Phase Separation. Cryst. Growth Des. 2020, 20(12), 7951–7962. [Google Scholar] [CrossRef]
- Thomas, S.; Dougay, J.; Annunziata, O. Yield of Protein Crystallization from Metastable Liquid-Liquid Phase Separation. MOLECULES 2025, 30(11). [Google Scholar] [CrossRef] [PubMed]
- McPherson, A. Introduction to protein crystallization. Methods 2004, 34(3), 254–265. [Google Scholar] [CrossRef]
- Saridakis, E.; Chayen, N. E. Towards a 'universal' nucleant for protein crystallization. Trends Biotechnol. 2009, 27(2), 99–106. [Google Scholar] [CrossRef]
- Lomakin, A.; Asherie, N.; Benedek, G. B. Monte Carlo study of phase separation in aqueous protein solutions. J. Chem. Phys. 1996, 104(4), 1646–1656. [Google Scholar] [CrossRef]
- Hribar-Lee, B.; Luksic, M. Biophysical Principles Emerging from Experiments on Protein-Protein Association and Aggregation. Annu. Rev. Biophys. 2024, 53, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pellicane, G. Colloidal Model of Lysozyme Aqueous Solutions: A Computer Simulation and Theoretical Study. J. Phys. Chem. B 2012, 116(7), 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kuehner, D.; Heyer, C.; Ramsch, C.; Fornefeld, U.; Blanch, H.; Prausnitz, J. Interactions of lysozyme in concentrated electrolyte solutions from dynamic light-scattering measurements. Biophys. J. 1997, 73(6), 3211–3224. [Google Scholar] [CrossRef]
- Timasheff, S. N. Protein-solvent preferential interactions, protein hydration, and the modulation of biochemical reactions by solvent components. Proceedings of the National Academy of Sciences of the United States of America 2002, 99(15), 9721–9726. [Google Scholar] [CrossRef]
- Record, M. T.; Anderson, C. F. Interpretation of preferential interaction coefficients of nonelectrolytes and of electrolyte ions in terms of a two-domain model. Biophys. J. 1995, 68(3), 786–794. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Annunziata, O. Comparison between protein-polyethylene glycol (PEG) interactions and the effect of PEG on protein-protein interactions using the liquid-liquid phase transition. J. Phys. Chem. B 2007, 111(5), 1222–1230. [Google Scholar] [CrossRef]
- Vergara, A.; Capuano, F.; Paduano, L.; Sartorio, R. Lysozyme mutual diffusion in solutions crowded by poly(ethylene glycol). Macromolecules 2006, 39(13), 4500–4506. [Google Scholar] [CrossRef]
- Bhat, R.; Timasheff, S. N. STERIC EXCLUSION IS THE PRINCIPAL SOURCE OF THE PREFERENTIAL HYDRATION OF PROTEINS IN THE PRESENCE OF POLYETHYLENE GLYCOLS. Protein Sci. 1992, 1(9), 1133–1143. [Google Scholar] [CrossRef]
- Vivares, D.; Belloni, L.; Tardieu, A.; Bonnete, F. Catching the PEG-induced attractive interaction between proteins. Eur. Phys. J. E 2002, 9(1), 15–25. [Google Scholar] [CrossRef]
- Bloustine, J.; Virmani, T.; Thurston, G. M.; Fraden, S. Light scattering and phase behavior of lysozyme-poly(ethylene glycol) mixtures. Phys. Rev. Lett. 2006, 96(8). [Google Scholar] [CrossRef]
- Asherie, N.; Lomakin, A.; Benedek, G. B. Phase diagram of colloidal solutions. Phys. Rev. Lett. 1996, 77(23), 4832–4835. [Google Scholar] [CrossRef]
- Platten, F.; Valadez-Pérez, N.; Castañeda-Priego, R.; Egelhaaf, S. Extended law of corresponding states for protein solutions. J. Chem. Phys. 2015, 142(17). [Google Scholar] [CrossRef]
- Kastelic, M.; Kalyuzhnyi, Y. V.; Hribar-Lee, B.; Dill, K. A.; Vlachy, V. Protein aggregation in salt solutions. Proc. Natl. Acad. Sci. U. S. A. 2015, 112(21), 6766–6770. [Google Scholar] [CrossRef] [PubMed]
- Asherie, N. Protein crystallization and phase diagrams. Methods 2004, 34(3), 266–272. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S. N. Preferential interactions of proteins with salts in concentrated solutions. Biochemistry 1982, 21(25), 6545–6552. [Google Scholar] [CrossRef]
- Arakawa, T.; Timasheff, S. N. THEORY OF PROTEIN SOLUBILITY. Methods Enzymol. 1985, 114, 49–77. [Google Scholar]
- Jungwirth, P.; Cremer, P. S. Beyond Hofmeister. Nat. Chem. 2014, 6(4), 261–263. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, O.; Payne, A.; Wang, Y. Solubility of lysozyme in the presence of aqueous chloride salts: Common-ion effect and its role on solubility and crystal thermodynamics. J. Am. Chem. Soc. 2008, 130(40), 13347–13352, Article. [Google Scholar] [CrossRef] [PubMed]
- Retailleau, P.; RiesKautt, M.; Ducruix, A. No salting-in of lysozyme chloride observed at how ionic strength over a large range of pH. Biophys. J. 1997, 73(4), 2156–2163. [Google Scholar] [CrossRef]
- Hansen, J.; Platten, F.; Wagner, D.; Egelhaaf, S. Tuning protein-protein interactions using cosolvents: specific effects of ionic and non-ionic additives on protein phase behavior. Phys. Chem. Chem. Phys. 2016, 18(15), 10270–10280. [Google Scholar] [CrossRef]
- Fahim, A.; Annunziata, O. Effect of a Good buffer on the fate of metastable protein-rich droplets near physiological composition. Int. J. Biol. Macromol. 2021, 186, 519–527. [Google Scholar] [CrossRef]
- Fahim, A.; Pham, J.; Thomas, S.; Annunziata, O. Boosting protein crystallization from liquid-liquid phase separation by increasing metastability gap. J. Mol. Liq. 2024, 398, 124164. [Google Scholar] [CrossRef]
- Camara-Artigas, A.; Salinas-Garcia, M. C.; Plaza-Garrido, M. Crystal structure of Lysozyme in complex with Hepes. [CrossRef]
- McPherson, A.; Cudney, B. Searching for silver bullets: An alternative strategy for crystallizing macromolecules. J. Struct. Biol. 2006, 156(3), 387–406. [Google Scholar] [CrossRef]
- Albright, J. G.; Annunziata, O.; Miller, D. G.; Paduano, L.; Pearlstein, A. J. Precision measurements of binary and multicomponent diffusion coefficients in protein solutions relevant to crystal growth: Lysozyme chloride in water and aqueous NaCl at pH 4.5 and 25 degrees C-perpendicular to. In Journal of the American Chemical Society; Proceedings Paper, 1999; Volume 121, 14, pp. 3256–3266. [Google Scholar] [CrossRef]
- CARNAHAN, N.; STARLING, K. EQUATION OF STATE FOR NONATTRACTING RIGID SPHERES. J. Chem. Phys. 1969, 51(2), 635–&. [Google Scholar] [CrossRef]
- BARKER, J.; HENDERSON, D. PERTURBATION THEORY AND EQUATION OF STATE FOR FLUIDS - SQUARE-WELL POTENTIAL. J. Chem. Phys. 1967, 47(8), 2856–+. [Google Scholar] [CrossRef]
- Sear, R. Phase behavior of a simple model of globular proteins. J. Chem. Phys. 1999, 111(10), 4800–4806. [Google Scholar] [CrossRef]
- Vaney, M.; Maignan, S.; RiesKautt, M.; Ducruix, A. High-resolution structure (1.33 angstrom) of a HEW lysozyme tetragonal crystal grown in the APCF apparatus. Data and structural comparison with a crystal grown under microgravity from SpaceHab-01 mission. ACTA Crystallogr. Sect. D.-Struct. Biol. 1996, 52, 505–517. [Google Scholar] [CrossRef]
- Nadarajah, A.; Pusey, M. Growth mechanism and morphology of tetragonal lysozyme crystals. ACTA Crystallogr. Sect. D.-Biol. Crystallogr. 1996, 52, 983–996. [Google Scholar] [CrossRef]
- Weiss, M.; Palm, G.; Hilgenfeld, R. Crystallization, structure solution and refinement of hen egg-white lysozyme at pH 8.0 in the presence of MPD. ACTA Crystallogr. Sect. D.-Struct. Biol. 2000, 56, 952–958. [Google Scholar] [CrossRef]
- Lomakin, A.; Asherie, N.; Benedek, G. Aeolotopic interactions of globular proteins. Proc. Natl. Acad. Sci. U. S. A. 1999, 96(17), 9465–9468. [Google Scholar] [CrossRef]
- Fine, B. M.; Lomakin, A.; Ogun, O. O.; Benedek, G. B. Static structure factor and collective diffusion of globular proteins in concentrated aqueous solution. J. Chem. Phys. 1996, 104(1), 326–335. [Google Scholar] [CrossRef]
- HENDERSON, D.; SCALISE, O.; SMITH, W. MONTE-CARLO CALCULATIONS OF THE EQUATION OF STATE OF THE SQUARE-WELL FLUID AS A FUNCTION OF WELL WIDTH. J. Chem. Phys. 1980, 72(4), 2431–2438. [Google Scholar] [CrossRef]
- VEGA, L.; DEMIGUEL, E.; RULL, L.; JACKSON, G.; MCLURE, I. PHASE-EQUILIBRIA AND CRITICAL-BEHAVIOR OF SQUARE-WELL FLUIDS OF VARIABLE WIDTH BY GIBBS ENSEMBLE MONTE-CARLO SIMULATION. J. Chem. Phys. 1992, 96(3), 2296–2305. [Google Scholar] [CrossRef]
- LOMBA, E.; ALMARZA, N. ROLE OF THE INTERACTION RANGE IN THE SHAPING OF PHASE-DIAGRAMS IN SIMPLE FLUIDS - THE HARD-SPHERE YUKAWA FLUID AS A CASE-STUDY. J. Chem. Phys. 1994, 100(11), 8367–8372. [Google Scholar] [CrossRef]
- Kern, N.; Frenkel, D. Fluid-fluid coexistence in colloidal systems with short-ranged strongly directional attraction. J. Chem. Phys. 2003, 118(21), 9882–9889. [Google Scholar] [CrossRef]
- Bianchi, E.; Largo, J.; Tartaglia, P.; Zaccarelli, E.; Sciortino, F. Phase diagram of patchy colloids: Towards empty liquids. Phys. Rev. Lett. 2006, 97(16). [Google Scholar] [CrossRef]
- WERTHEIM, M. FLUIDS WITH HIGHLY DIRECTIONAL ATTRACTIVE FORCES.2. THERMODYNAMIC PERTURBATION-THEORY AND INTEGRAL-EQUATIONS. J. Stat. Phys. 1984, 35(1-2), 35–47. [Google Scholar] [CrossRef]
- GilVillegas, A.; Galindo, A.; Whitehead, P.; Mills, S.; Jackson, G.; Burgess, A. Statistical associating fluid theory for chain molecules with attractive potentials of variable range. J. Chem. Phys. 1997, 106(10), 4168–4186. [Google Scholar] [CrossRef]
- López-Picón, J.; Escamilla-Herrera, L.; Torres-Arenas, J. The square-well fluid: A thermodynamic geometric view. J. Mol. Liq. 2022, 368. [Google Scholar] [CrossRef]
- Brudar, S.; Hribar-Lee, B. Effect of Buffer on Protein Stability in Aqueous Solutions: A Simple Protein Aggregation Model. J. Phys. Chem. B 2021, 125(10), 2504–2512. [Google Scholar] [CrossRef]
- Patel, B.; Docherty, H.; Varga, S.; Galindo, A.; Maitland, G. Generalized equation of state for square-well potentials of variable range. Mol. Phys. 2005, 103(1), 129–139. [Google Scholar] [CrossRef]
- GilVillegas, A.; delRio, F.; Benavides, A. Deviations from corresponding-states behavior in the vapor-liquid equilibrium of the square-well fluid. FLUID PHASE Equilib. 1996, 119(1-2), 97–112. [Google Scholar] [CrossRef]
Figure 1.
(A) Schematic temperature-concentration phase diagram showing crystal solubility (red) and LLPS boundary (blue; critical point, ×). The star symbols indicate two representative states with same protein concentration. Both states are supersaturated with respect to protein crystallization and are expected to generate crystals (diamonds). The state at lower temperature is below the LLPS boundary and is associated with the formation of protein-rich spherical droplets (circles). (B) Schematic temperature-concentration phase diagram showing the effect of an additive that shifts the two phase boundaries toward higher temperatures (e.g., NaCl for lysozyme). (C) Schematic temperature-concentration phase diagram showing the effect of an additive that shifts the two phase boundaries in opposite directions thereby increasing metastability gap (e.g., HEPES for lysozyme).
Figure 1.
(A) Schematic temperature-concentration phase diagram showing crystal solubility (red) and LLPS boundary (blue; critical point, ×). The star symbols indicate two representative states with same protein concentration. Both states are supersaturated with respect to protein crystallization and are expected to generate crystals (diamonds). The state at lower temperature is below the LLPS boundary and is associated with the formation of protein-rich spherical droplets (circles). (B) Schematic temperature-concentration phase diagram showing the effect of an additive that shifts the two phase boundaries toward higher temperatures (e.g., NaCl for lysozyme). (C) Schematic temperature-concentration phase diagram showing the effect of an additive that shifts the two phase boundaries in opposite directions thereby increasing metastability gap (e.g., HEPES for lysozyme).

Figure 2.
Van’t Hoff plot of crystal solubility, fS, as a function of inverse temperature, b, for lysozyme in HEPES (circles) and REF (squares) systems. Solid lines are linear fits through the data based on Eq. 7.
Figure 2.
Van’t Hoff plot of crystal solubility, fS, as a function of inverse temperature, b, for lysozyme in HEPES (circles) and REF (squares) systems. Solid lines are linear fits through the data based on Eq. 7.

Figure 3.
Simplified representation of lysozyme proteins on the (0,0,1) crystallographic plane as described in the literature.[61,62] Dashed box indicates unit cell along the a- and b- crystallographic axis. Each type of protein is labeled using a letter: reference protein (M; black), proteins interacting with M (B,C,G and F; gray) and proteins non-interacting with M (A,D,E; white). Positions (and orientations) of proteins relative to M (x,y,z) are also listed.
Figure 3.
Simplified representation of lysozyme proteins on the (0,0,1) crystallographic plane as described in the literature.[61,62] Dashed box indicates unit cell along the a- and b- crystallographic axis. Each type of protein is labeled using a letter: reference protein (M; black), proteins interacting with M (B,C,G and F; gray) and proteins non-interacting with M (A,D,E; white). Positions (and orientations) of proteins relative to M (x,y,z) are also listed.

Figure 4.
(A) Phase diagram showing normalized temperature, , as a function of protein volume fraction, . LLPS boundaries extracted using 1.3, 1, and calculated using Eq. 12 (dashed curve), Eq. 13 (solid curve) and setting 0 (dotted curve). Diamond indicates critical point extracted from Monte Carlo simulations. (B) Phase diagram showing LLPS boundaries extracted using 1 and 1.2 (dotted curve), 1.3 (solid curve) and 1.4 (dashed curve). (C) Plot describing dependence of on at the critical temperature with 1.3. (D) Phase diagram showing how the width of LLPS boundaries depend on at 1.3. The number associated with each curve is the corresponding values of .
Figure 4.
(A) Phase diagram showing normalized temperature, , as a function of protein volume fraction, . LLPS boundaries extracted using 1.3, 1, and calculated using Eq. 12 (dashed curve), Eq. 13 (solid curve) and setting 0 (dotted curve). Diamond indicates critical point extracted from Monte Carlo simulations. (B) Phase diagram showing LLPS boundaries extracted using 1 and 1.2 (dotted curve), 1.3 (solid curve) and 1.4 (dashed curve). (C) Plot describing dependence of on at the critical temperature with 1.3. (D) Phase diagram showing how the width of LLPS boundaries depend on at 1.3. The number associated with each curve is the corresponding values of .

Figure 5.
Phase diagram showing experimental LLPS data for the HEPES (solid circles) and REF (solid squares) systems. Theoretical LLPS boundaries are calculated from our BHPT model using in both cases. The values of 1732 K and , and 1687 K and are used for the HEPES and REF systems, respectively. Solubility data for the HEPES (open circles) and REF (open squares) systems are also included. Theoretical solubility curves are obtained using the BHPT model for the fluid phase and cell model for the solid phase, with , and .
Figure 5.
Phase diagram showing experimental LLPS data for the HEPES (solid circles) and REF (solid squares) systems. Theoretical LLPS boundaries are calculated from our BHPT model using in both cases. The values of 1732 K and , and 1687 K and are used for the HEPES and REF systems, respectively. Solubility data for the HEPES (open circles) and REF (open squares) systems are also included. Theoretical solubility curves are obtained using the BHPT model for the fluid phase and cell model for the solid phase, with , and .

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



