Submitted:
23 April 2026
Posted:
27 April 2026
You are already at the latest version
Abstract
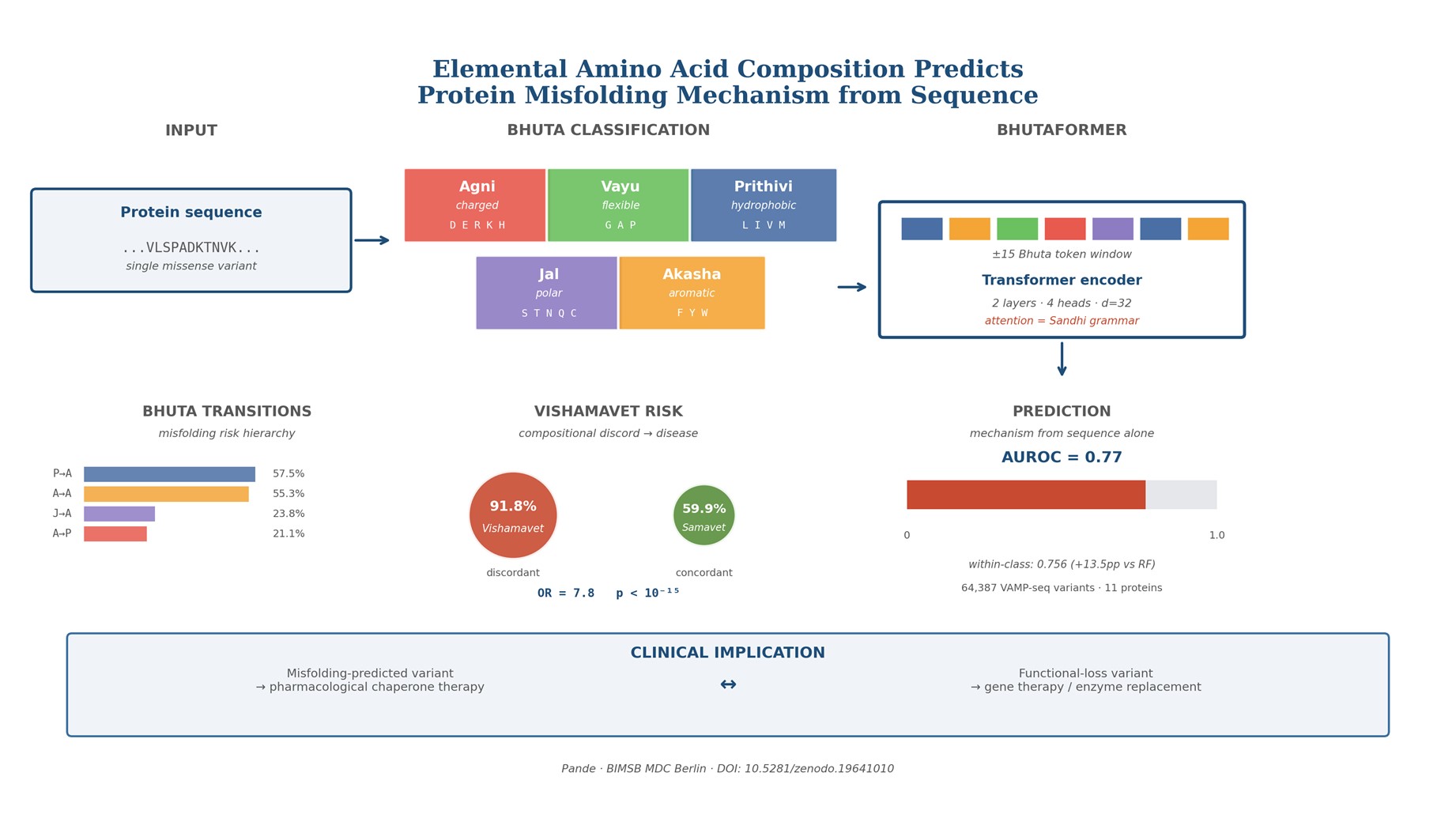
Pathogenic missense variants cause disease through two mechanistically distinct routes: structural destabilization leading to protein misfolding and degradation, or functional disruption of a stably folded protein. Despite the clinical importance of this distinction — pharmacological chaperones rescue misfolded proteins, while functionally defective proteins require gene therapy or enzyme replacement — no existing framework predicts which mechanism underlies a given variant. All current tools, from SIFT and PolyPhen-2 to AlphaMissense, predict pathogenicity but not mechanism.Here we show that the elemental composition of a protein’s amino acids, classified by dominant biophysical property, predicts misfolding mechanism from sequence alone. We map each amino acid to one of five elemental classes: charged residues (Agni), flexible residues (Vayu), hydrophobic residues (Prithivi), polar residues (Jal), and aromatic residues (Akasha). This classification is inspired by and named after the Panchamahabhuta system of classical Indian natural philosophy, whose five-element grouping aligns with the five dominant thermodynamic forces governing protein stability. The elemental composition profile of a domain — its elemental constitution (Prakriti) — explains 36.4% of variance in domain biological function across 14 functional classes (η² = 0.364, F = 18, p = 9.99×10⁻³³, n = 420 domains) without any machine learning.Applying this framework to 64,387 VAMP-seq stability measurements across 11 disease proteins, we derive a substitution risk hierarchy: mutations introducing charged residues into hydrophobic cores (Prithivi→Agni) cause misfolding in 57.5% of cases versus 21.1% for Agni→Prithivi. Secondary structure context reveals a 3.7-fold gradient — hydrophobic and aromatic residues in β-strands misfold at 51–52% when mutated; charged residues in turns at only 14%. Proteins whose domain composition conflicts with biological function — compositional discord (Vishamavet) — carry pathogenic variants at 91.8% versus 59.9% in concordant proteins (OR = 7.8, p < 10⁻¹⁵).Key finding: structured-position hydrophobic/aromatic residues misfold at 3.7× the rate of loop charged residues — mechanistic information absent from all conservation-based predictors.We introduce BhutaFormer, a transformer architecture that encodes sequence context as Bhuta tokens and learns elemental interaction grammar via multi-head self-attention, achieving AUROC = 0.77 overall and 0.76 on within-class (Tanmatra) variants — a 13.5 percentage point improvement over Random Forest. On ProteinGym DMS abundance assays, BhutaFormer achieves Spearman ρ = 0.29 for training-distribution proteins, exceeding the Site-Independent baseline (ρ = 0.175) without evolutionary alignment, structural data, or large language model pre-training.
Keywords:
protein misfolding
; variant mechanism prediction
; Panchamahabhuta
; BhutaFormer
; Tanmatra
; Vishamavet
; VAMP-seq
; elemental constitution
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



