Submitted:
21 April 2026
Posted:
22 April 2026
You are already at the latest version
Abstract
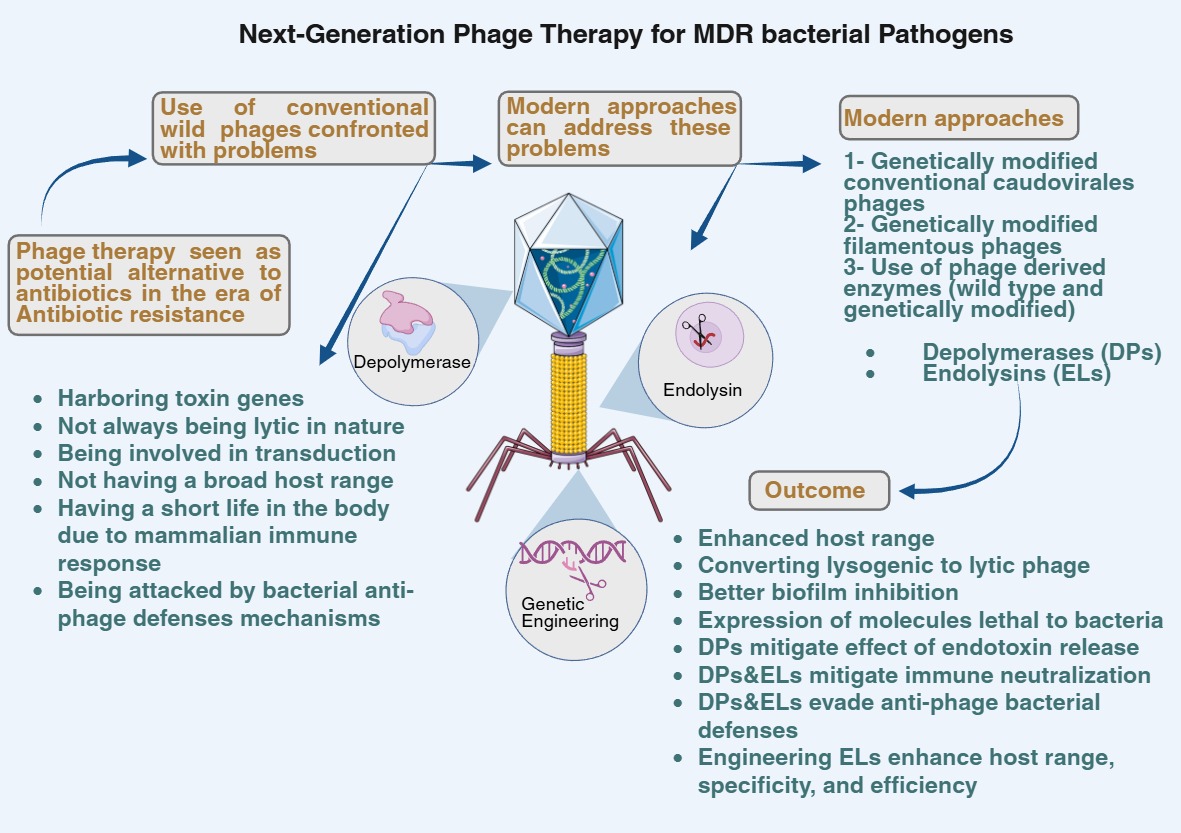
Bacteriophages are viruses that attack bacterial cells, resulting in their lysis. Since the emergence of antibiotic resistance, phages have gained attention from the scientific community and are being explored for their potential use as anti-microbial agents. Since their discovery, naturally occurring wild-type phages have been used as anti-microbial agents. Though usage of antibiotics is confronted with problems, antibiotic resistance being the most pronounced, phages also have limitations, such as harboring toxin genes, not always lytic in nature, being involved in transduction, not having a broad host range, having a short life in the body due to mammalian immune response, and being attacked by bacterial anti-phage defense mechanisms. Modern approaches to phage therapy can address these problems associated with conventional phage therapy. These modern approaches include genetic phage engineering and using phage-derived enzymes, such as depolymerases and endolysins, in their wild-type form as well as in genetically engineered form. Genetic engineering has been performed on lytic, lysogenic, and filamentous phages for desired targeted results. In this review, we discuss the previous studies that have investigated these modern approaches for phage therapy against bacterial infections. Advantages and disadvantages of phage therapy are being discussed, and it is discussed how the use of modern approaches can affect different aspects of phage therapy.
Keywords:
multidrug-resistant bacteria
; phage engineering
; endolysins
; depolymerases
; genetic engineering
; modern phage therapy
; filamentous phages
; bacteriophages
Introduction
In the year 1896, Ernest Hanbury Hankin reported that phages were present in the waters of the rivers Ganges and Yamuna in India for their potent killing effect against Vibrio Cholerae [1]. Phages were discovered by Fredrick William Twort and Felix d’Herelle in 1915 and 1917, respectively [2]. Immediately, following their discovery, these were used to treat bacterial infections, and they were commercially marketed for this purpose during the 1940s. In the meantime, the discovery of antibiotics and their commercial production led to a waning interest in phages for their use for therapeutic purposes [3]. Though antibiotic therapy has largely been successful in treating bacterial infections, the emergence of antibiotic resistance and its spread among pathogenic bacteria have prompted the scientific community to look for alternative ways to combat bacterial infections. Phages can infect and kill bacteria, irrespective of the antibiotic resistance status of bacteria, because these uses different mechanism for eradicating bacteria as compared to antibiotics.
Phages have gained the attention of scientific community and are being explored for their potential use as anti-microbial agents. However, phages do have limitations, such as harboring toxin genes, not always being lytic in nature, being involved in transduction which can lead to unwanted spread of toxin and antibiotic resistance genes, not having a broad host range, having short life span in the body due to the mammalian immune response, and being attacked by bacterial anti-phage defense mechanism. Modern approaches to phage therapy can address some of these problems associated with conventional phage therapy. These modern approaches include, genetic phage engineering, using phage-derived enzymes such as depolymerases and endolysins in their wild-type form as well as in genetically engineered form. Genetic engineering has been performed on lytic, lysogenic and filamentous phages to achieve desired targeted results. Depolymerases can dissolve the bacterial capsule, while endolysins can dissolve the cell wall, whether used in wild-type form or genetically modified form, both types of enzyme can eradicate bacteria. In this review, we summarize the progress regarding the usage of these modern approaches in phage therapy against bacterial infections.
Advantages and limitations of phage therapy
Using phages to control and treat bacterial infections is known as phage therapy. Advantages of using phages include their lytic nature compared to the bacteriostatic properties of some of the antibiotics [4]. They can act against all forms of bacteri i.e., sessile, planktonic and biofilm-forming bacteria [5]. Other advantages include their self-replicating nature, meaning they can auto dose themselves and continue replicating at the site of infection until all bacteria are eliminated. These are inherently low in toxicity as they are composed of natural molecules i.e., proteins and nucleic acid [6]. These can be applied to patients using diverse methods such as inhalation, parenteral routes and topically in the form of creams. There is less propensity for the development of resistance against phages and phage-derived enzymes as they target vital molecules that are indispensable for sustaining bacteria such as peptidoglycan molecules in the cell wall of bacteria [4].
Certain criteria need to be met for a phage to be considered a potential therapeutic agent, including its strict lytic nature, absence of genes responsible for toxins, virulence and spreading antibiotic resistance and a further desired property is to not be involved in transduction. Phages should be well characterized at the genomic level and have a wide host range against pathogenic bacteria, be stable under storage conditions as well as inside the mammalian host and be able to counter defense mechanisms from bacteria and mammalian immune system [7,8,9]. Despite a huge number of phages present naturally, few phage are able to fulfill all of these criteria. In contrast to the desired properties described above, many phages display lysogenic nature, harbor bacterial virulence, harbour toxin determinants and other pathogenic factors [10,11]. Additionally, both temperate and lytic phages participate in transduction, transferring bacterial DNA from one bacterium to other, thus facilitating the spread of pathogenicity-determining genes [12,13,14]. Even when a phage is able to infect a large number of bacteria, the target bacteria can evade the attack of the phage by changing its receptors used by the phage and bacteria can also employ the anti-phage defense mechanisms [15,16,17,18,19]. This makes phage therapy ineffective due to the development of resistance in bacteria against the employed phages, which is a major problem in phage therapy. Numerous anti-phage defense mechanisms are activated after phage DNA is injected inside the bacterial cell, including restriction modification systems and the CRISPR-CAS system; furthermore, bacteria can also activate a suicidal host response to prevent further phage infection spread [19,20]. Most of these disadvantages related to phage therapy can be addressed using strategies discussed in this review, such as 1) using genetically modified phages, and 2) using protein products derived from phages, such as phage-associated hydrolases or endolysins, either in their wild type form or modified form through genetic engineering. Each of these approaches is being discussed in following sections.
Life cycle of phages and window for genetic modifications
Bacteriophages are viruses that are natural predators of bacteria. Phages contain genetic material, which is either DNA or RNA, and are enclosed in an icosahedral protein capsid head. The tail is attached to the head of a phage, and the tail is composed of a spiral contractile sheath surrounding a core pipe, which is attached to a baseplate containing tail fibers. Tail fibers also act as surface receptors that recognize different molecules for the purpose of adsorption and attachment to the bacterial surface in the first step of the infectious cycle [21]. At the initial stage of the infection cycle, different enzymes attached to the phage tail fiber help to overcome barriers posed by the outer layers of the bacterial cell. Virion-associated lysins (VALs) and depolymerases are enzymes, which are responsible for degradation of cell surface barriers. Depolymerases are responsible for the degradation of polysaccharide molecules such as capsules, lipopolysaccharide (LPS) or biofilm, while VALs are responsible for peptidoglycan degradation, thus creating holes in the cell wall and allowing for the injection of phage genetic material, encased inside the phage capsid, into the bacterial cell [21].
Phage genetic material, when inside the host bacterium, controls host metabolism machinery and leads to synthesis of new virion particles [22]. The phage replicates its genetic material, produces structural and non-structural proteins including different enzymes required for the release of the phage progeny. Numerous newly synthesized copies of phage genetic material are packed into newly assembled phage head capsids. Newly synthesized phage tails are attached to the phage heads, creating a complete phage virion. To release newly assembled phage particles, the holin-endolysin system of enzymes come into action at the final stage. Holin proteins insert themselves into the inner bacterial cell membrane, creating holes in the membrane. Through these holes, endolysins access the cell wall and enzymatically digest the cell wall due to their muralytic capabilities, leading to bacterial cell lysis and the release of phage progeny [22].
The ability of phages to inject their genetic material can be utilized for various possibilities, and phage genome modifications can be done for a targeted desired purpose for to address drawbacks related to conventional phages. The details of such modifications are described in next sections. See Figure 1, for the depiction of the phage lytic cycle and the possible target of genetic modifications.
Genetic engineering of non-filamentous phages for use in phage therapy
One of the strategies to increase the spectrum of host range of phage is to use a cocktail of phages in which different phages have different lytic spectra against different strains of pathogenic bacteria and cover the phage sensitivity profile against target bacteria [23]. Still, such phage cocktails are not able to universally target all strains of a given bacterial species [24,25]. Isolating new natural phages to cover the specificity spectrum against target bacteria is tedious and time consuming. Another strategy is to adapt the phage to resistant bacteria by selecting for mutant phages that are able to lyse the target bacteria, but this strategy is also very tedious and time consuming and may not always be successful [26]. Previous studies have successfully shown that host range of a phage can be affected by manipulating genes responsible for tail fiber through genome engineering techniques [27,28,29,30]. Furthermore, the lytic spectrum of phages with desirable properties against a given pathogen can be increased by converting a lysogenic phage to a variant with lytic capabilities. This can be especially helpful when an appropriate lytic phage is unavailable but a lysogenic phage of given specificity is available that can be converted to lytic one [31,32,33]. A previous study described the first case of human phage therapy using genetically modified phages, wherein lysogenic phages were converted to lytic phage for treating infection with Mycobacterium abscessus bacteria [34]. A patient who had received a lung transplant to cure a condition of cystic fibrosis, was infected with Mycobacterium abscessus bacteria. Only one lytic phage could be found in the phage bank that was able to efficiently lyse the target bacteria. The other two phages were lysogenic and were able to infect the bacterium. Two lytic phages were derived from these lysogenic phages through genetic engineering and mutation, and these two modified phages displayed high lytic potential. The cocktail containing three phages successfully treated the infection [34]. See Table 1 for an overview of all these studies.
In other instances of phage engineering, enhanced bacterial killing and efficient removal of bacterial biofilm were noted when depolymerase enzyme were expressed in phages [35,36]. Biofilm inhibition was also achieved through the expression of enzymes inhibiting quorum sensing [37]. The side effects of toxin release upon bacterial lysis by phage had been mitigated by engineering phage to become non-lytic by deletion of genes responsible for the expression of endolysin [38], holin [39] or deletion of other proteins involved in bacterial lysis [40]. In a study it was investigated whether phages can enter mammalian cells and kill intracellular bacteria through the expression of the GFP protein from the phage genome and imaging it during infection studies [41]. See Table 1 for an overview of all these studies.
Genetic engineering and potential use of filamentous phages for phage therapy
Filamentous phages have been well characterized genomically and possess smaller genomes, containing11 genes and simpler structures [42,43]. Their simpler structure is in contrast to phages belonging to Caudovirales, which are usually used in phage therapy, display lytic properties in nature, possess complex structures, larger genomes and encode genes for 20 to 300 different functions. Therefore, filamentous phages are an attractive choice for phage engineering for the purpose of phage therapy. Furthermore, they do not lyse bacteria while replicating inside a bacterial host; instead, they secrete their progeny from infected bacteria constantly, thus minimizing the release of endotoxins.
Receptors of filamentous phages, which bind to the ligand proteins present on the surface of bacteria, are present on pili and have a narrow host range. Receptor binding proteins (RBPs), used for attaching to surface of bacterial host cells, of filamentous phages can be engineered to make them fit for adsorption to a wide variety of bacteria. Different proteins can be displayed on the surface of the phages, through phage display technology [44]. Libraries of recombinant M13 phages expressing RBPs against a wide variety of bacterial phyla were created. Out of these libraries, phage variants (M13PAB) were identified that bound to several clinical P. aeruginosa isolates. Colistin was cross-linked to the M13 PAB, which showed a lowered minimum inhibitory concentration (MIC) of antibiotics by 1–2 orders of magnitude [45]. Previous studies have shown that the expression of the antimicrobial peptides (AMPs) or toxins in filamentous phages can effectively kill bacteria [46,47]. To lower the release of endotoxins from bacterial lysis, export proteins in their genome have been replaced with other genes such as those for restriction endonucleases [40,48]. Inactivation of the SOS DNA damage response resulted in increased bacterial killing when combined with DNA-damaging antibiotics such as quinolones [49]. Enhanced intracellular killing of intracellular bacteria (Chlamydia trachomatis) was observed when two peptides were expressed in the coat proteins of the filamentous phage. The first peptide was an integrin binding peptide, RGD which helped in the internalization of the phage to the mammalian cell while the other protein was Ct polymorphic membrane protein (Pmp) D peptide which led to the killing of the bacteria [50]. See Table 1 for an overview of all these studies.
Phage-derived enzymes as potential alternatives to antibiotics
Elicitation of immune response, as well as rapid clearance of phages from the body by phagocytic cells, is a major disadvantage when treating bacterial infections using phage therapy [51,52,53]. This problem, presented by neutralizing antibodies against phages, can be tackled by: 1) repeated administration of phages, 2) increasing concentration of phage in a dose or 3) using a different phage targeting the same bacterial strain in repeated future doses. The problem of rapid clearance phage can also be tackled by two other approaches. The first approach is by modifying the phage capsid using genetic engineering techniques (as described in the previous section). The second approach uses enzymes encoded by phages for lysing bacteria which include polysaccharide depolymerases and endolysins [54]. There are fewer chances of these enzymes being neutralized by induced antibodies. Other advantages of using these enzymes for phage therapy include cost effectiveness and quick therapeutic development compared to antibiotics, the development of which requires high cost and more time [55,56]. Additionally, using these enzymes for phage therapy will also bypass the anti-phage defense mechanisms exhibited by bacteria [57]. Furthermore phages display complex biology and pharmacological features which are hurdles in their approval as drugs. Therefore, phage-derived enzymes present an interesting viable alternative to treat bacterial infections rather than using antibiotics or whole phage virion. In the following sections, each class of these enzymes, endolysins and depolymerases, is being discussed one by one for their role in treating bacterial infections.
Depolymerases as anti-infective agents
At the adsorption stage of phage to bacteria, the counter defense mechanisms employed by bacteria include the production of structures with thick layers of polysaccharides such as capsules, slime, and biofilm matrix, all of which prevent adsorption of phages to the bacterial surface [58]. Phages have also evolved strategies to overcome these obstacles. Polysaccharide depolymerases are enzymes located on tail fibers of phages, responsible for degrading of structures present on the outer surface of the bacteria, such as capsules containing capsular polysaccharide (CPS), exopolysaccharide (EPS), LPS, and biofilm matrix, thus exposing cell surface receptors for binding to phage tail fibers. VALs are responsible for the degradation of PG layers of bacterial cell wall. Both of these classes of enzymes help the virion to inject its genetic material inside a bacterial cell at the start of infection cycle [59]. Because CPS, EPS and LPS play an important role in the formation of bacterial biofilms, therefore depolymerases are being investigated in the prevention and treatment of biofilm related infections. Biofilm formation is a characteristic exhibited by a large number of gram-negative bacteria, including important pathogens such as ESKAPE pathogens [60]. The ESKAPE pathogens (Enterococcus faecium, S. aureus, Klebsiella pneumoniae, A. baumannii, P. aeruginosa, and Enterobacter species) are the leading cause of nosocomial opportunistic infections. These bacteria are continuously developing antibiotic resistance mechanisms, and all of them are MDR bacteria and among the list of top priority pathogens of WHO regarding issue of antibiotic resistance [61]. The capsule is an important virulence factor in this group of pathogens, and depolymerases can play a crucial role in combating infections arising from this group of pathogens. A wide range of bacteria-specific depolymerases have been documented in phages [62]. Due to their ability to disrupt vital structures in bacterial cells, phage-derived depolymerases have been proposed as anti-infective agents and can augment the action of antibiotics in combating resistant bacteria. Indeed, experimental in vivo studies using animals have shown promising therapeutic results against bacterial infections [63,64]. In contrast to antibiotics, which kill or halt the growth of bacteria, capsular depolymerases disarm bacteria from their virulence abilities, i.e. capsule, which protects bacteria from immune defense mechanisms such as phagocytosis and antibodies. Depolymerases may be a preferred choice as these would not lyse the bacteria and thus also limit the release of endotoxins that are released when bacteria are lysed using phages or bactericidal antibiotics [65]. Recombinant capsular depolymerases have been shown to protect mice against lethal systemic bacterial infections [54,66,67]. An in vitro study showed that depolymerase DA7 was able to remove both dynamic and static biofilms present on polystyrene and glass surfaces [68]. Dep 42 depolymerase was able to remove K. pneumoniae biofilms alone as well as in combination with Polymyxin B [69]. A previous study demonstrated that KP34p57 depolymerase could reduce biofilms by itself alone and also increased the biofilm removal abilities of phages that did not possess a depolymerase enzyme by itself [70]. For an overview of previous studies investigating depolymerase, see Table 2.
Engineered endolysins as anti-infective
Towards the end of the lytic cycle, phages use a lytic mechanism to lyse bacterial cells to release their progeny (Figure 1 for details). This is achieved by using two types of enzymes: amurins, which inhibit the synthesis of PGs [71], or endolysins which dissolve the cell wall using PG hydrolases attacking from the inside. These enzymes are assisted by another protein called holins, which pierce holes in the inner cell membrane and allow the endolysin to access the PG layer of the bacterial cell wall and together this constitutes Holin-endolysin system [56,72,73]. The dissolution of the PG layer results in decreased cell wall strength, leading to the leakage of cytoplasmic contents and ultimately killing of the bacteria through bursting. The advantages of using endolysins for to combat bacterial infections include rapid killing of bacteria, extremely low to zero levels of resistance development, their specific nature resulting in minimal disruption of normal microflora and lower chance of developing neutralizing antibodies compared to using the entire phage. In the following paragraphs, we discuss endolysins with targeted genetic modifications to improve their therapeutic properties.
Endolysins possess lytic activity against both gram-positive and gram-negative bacteria. Under natural conditions the holin-lysin system of kills bacteria from inside of bacterial cell during infection cycle has also been referred to as “lysis from within” [72]. The simpler cell wall structure of gram-positive bacteria allows for the therapeutic application of purified recombinant lysins when applied from the outside as the access to the cell wall is not hindered by the outer cell membrane for enzymatic hydrolysis of the PG cell wall. This phenomenon is referred to as “lysis from without”. In contrast, this action of lysis by endolysin is hindered by the outer cell membrane in the case of gram-negative bacteria, and only a few lysins have been reported to date that can lyse bacteria without the help of membrane permeants when applied from outside. This concept of “lysis from without” opens up the possibility of using recombinant lysins for the treatment of bacterial infections, mostly concerning gram-positive bacteria [74]. However, for using these against Gram-negative bacteria, lysins have been fused with another peptide using genetic engineering to help lysin cross the outer cell membrane and access the PG. Such lysins have been termed as “Artificial lysins” or “Artilysins” [75]. A study reported that the fusion of albumin binding domain with an endolysin resulted in extended stability of the endolysin circulating in blood without affecting its lytic capabilities, using a mice model [76]. The fusion of membrane penetrating peptides (MPPs) has been used for targeting E. coli in the case of Lysep3 lysin and LoGT-0377 [77,78], for P. aeruginosa and A. baumannii in case of LoGT-001 and LoGT-008 lysins respectively [78], for Streptomyces aureofaciens in case of lysin LytAmfi [79]. Furthermore, this study showed that fusion also resulted in broadening of lytic activity against other bacterial species such as Bacillus immobilis and Citrobacter fumigatus [79]. The rationale for the fusion of such peptides is their amphipathic or hydrophilic properties, which enhance their interaction with the bacterial cell membrane allowing endolysins to pass through the membrane. Such properties can also be introduced by changing amino acid compositions through mutations as described below. The fusion of MPPs with endolysins in all these cases enhanced the bactericidal activities compared to wild type endolysins. Fusion of AMP cecropin A, which is generally known to cause perturbations and create holes in bacterial cell membrane, to the 5′ end of mutant EC340 endolysin resulted in the creation of the engineered endolysin LNT113 with improved outer membrane permeability and increased activity against E. coli, A. baumannii, P. aeruginosa, and K. pneumoniae [80]. In another study, cecropin A was fused to the N-terminus of AbEndolysin, which increased its activity by at least 2–8 fold against MDR A. baumannii. It also rescued the mice from systemic A. baumannii infection and displayed synergism with different antibiotics [81]. In another study cecropin A was fused to its N terminus via a linker of three Ala-Gly repeats creating LysMK34, which displayed improved antibacterial activity against A. baumannii compared to that of the parental lysin [82]. Endolysin KZ144 was fused with the sheep myeloid 29-amino acid peptide (SMAP), resulting in the formation of Artilysin-175 and -085, both of which could efficiently kill multidrug-resistant strains of P. aeruginosa. Artilysin-175 was reported to be highly refractory to resistance development due to natural mutations [83]. Holin was fused at the N terminus of endolysin (RL_Lys) targeting P. aeruginosa, and the engineered endolysin not only gained increased activity but was also able to lyse K. pneumoniae, Salmonella sp. and Methicillin-Resistant S. aureus (MRSA) [84]. Another strategy for enhancing the penetrability of endolysins for crossing the outer membrane of bacteria, is the fusion of lysins with domains or peptides interacting with outer membrane transport proteins or transport systems. For example, pyocin S2 (PyS2), a bacteriocin that can use the FpvAI protein channel for passing through the bacterial OM, was fused with GN4 lysin for targeting P. aeruginosa, resulting in successful killing and disruption of biofilm [85]. Pesticin, another bacteriocin that can use the FyuA protein channel for passing through the bacterial OM, was fused with T4 lysozyme for targeting both Yersinia pestis and E. coli. The fusion not only resulted in efficient bacterial killing but also led to evasion over pesticin immunity protein, produced by bacteria as defense against pesticin, which is a clear advantage against using pesticin alone [86]. Receptor binding domains of colicin A, another bacteriocin that uses the BtuB receptor on the bacterial outer membrane, were fused with lysin Lysep3 for targeting E. coli while applying the endolysin from the outside [87]. For an overview of these previous studies, see Table 3.
Endolysins from phages of gram-positive and gram-negative bacteria display different structures. Endolysins from phages of gram-positive bacteria contain two domains: an enzymatically active domain (EAD) on the N-terminal which hydrolyzes PG through its enzymatic activity, and the cell wall binding domain (CBD) on the C-terminal, which specifically recognizes the cell wall structures peculiar to each bacterial species [88]. With a few exceptions, the majority of endolysins from gram-negative bacteria possess a globular structure and only one domain, the catalytic domain [89,90]. This modular nature of the structure of lysins for gram-positive bacteria allows for swapping domains from two different lysins using genetic engineering techniques [75]. Such lysins have been termed as “Chimeric lysins” or “Chimeolysins” [75]. EAD can attack most of the bonds present in the PG of bacteria, whereas CBD is responsible for recognizing the substrate in PG and confers specificity for recognizing the specific host bacterial cell. Because endolysins from different bacteria exhibit different properties, regarding enzymatic action and specificity, swapping the domains among endolysins can result in changing their properties toenhance their therapeutic efficiency. A number of previous studies have shown that such swapping of domains has resulted in: 1) different specificity directed against different bacteria [91,92,93,94], 2) improved catalytic efficiency resulting in enhanced bacterial killing [95] and 3) altered desired physical properties, such as increased solubility [94]. Domain shuffling of the 1,4-beta-N-acetylmuramidase lysin encoded by Gardnerella prophages led to endolysin PM-477 with 10-fold higher bactericidal activity than any wild-type enzyme against different Gardnerella strains and effectively disrupted biofilms [96]. Several investigations applied engineered lysins through domain swapping against clinically important bacteria such as S. aureus [91,92,93,94,95,97,98,99,100,101], Streptococcus pyogenes [92,93], Streptococcus pneumoniae [91,92], Streptococcus agalactiae [98], Pseudomonas aeruginosa [83], E. coli [102], A. baumannii [78], Bacillus amyloliquefaciens [102], Enterococcus [94] and Gardnerella [96]. For an overview of these previous studies, see Table 3.
Other genetic engineering strategies for improving the properties of endolysins include domain deletion, domain addition and changing the amino acid composition of endolysin. For example truncation of the CBD plus 13 amino acids at the C-terminal of the lysin PlyGBS increased the enzymatic activity by 28-fold against group B Streptococci [103]. The C-terminal protein truncation without disturbing the EAD of lysin CD27L resulted in increased lytic activity against Clostridium difficile along with a broadened lytic spectrum [104]. A truncated protein harboring the EAD of endolysin LysSAP26 displayed effective killing of Clostridioides difficile in a murine model [105]. It has been hypothesized that deletion of CBD could result in imparting a net positive charge to the remaining intact EAD domain, which interacts more strongly with the negatively charged cell wall of the gram-positive bacteria, thus increasing enzymatic activity [106]. Conversely, the addition of the CBD domain has been shown to increase activity for some endolysins. A previous study showed that the addition of an extra copy of CBD to endolysin Ply500 resulted in increased affinity by 50-fold and also increased the ability to act at high salt concentrations against Listeria [107]. The addition of lysostaphin CBD (SH3b) to lysin HydH5 of S. aureus resulted in increased activity and higher thermostability [108]. It has been hypothesized that the addition of CBD for some lysins may optimize their ability to interact and recognize the bacterial cell wall [106]. Mutating 15 amino acids from the Cpl-7 lysin resulted in increased activity and expanded spectrum against S. pneumoniae and S. pyogenes [109]. The hypothesized reason for the increased activity was the reversal of the net charge of CBD to be positive which facilitated its interaction with the cell wall [109]. Mutating the 88th amino acid glutamate of lysin LysF1 to other hydrophobic amino acids such as Leucine, Phenylalanine and Methionine respectively, resulted in improved thermal stability and lytic activity against E. coli [110]. A point mutation that prevented N-linked glycosylation enhanced the killing effect against S. pneumoniae [111]. For an overview of these previous studies, see Table 3.
In a unique fashion, an engineered endolysin was expressed from probiotic bacteria Lactobacillus johnsonii, targeting C. perfringens in the gastrointestinal tract [112]. L. lactis was engineered to express endolysin and virion-associated peptidoglycan hydrolase enzyme after fusing with the SPK1 signal peptide for secretion. The engineered bacteria were able to lyse all MRSA strains tested and S. epidermidis [113]. In another unique fashion, nanoparticles of endolysin were created due to self-assembling peptide (P114) in response to pH reduction. P128 nanoparticles consisting of endolysin demonstrated efficient bacterial killing even at lower concentrations against MRSA. The nanoparticles also demonstrated enhanced thermal stability (up to 65 °C) and storage stability [114]. For an overview of these previous studies, see Table 3.
Conclusions
Phage therapy is showing promising results for the treatment of antibiotic-resistant bacteria, but still, a lot of progress is to be made to make it a regular choice of physicians in clinical practice. It is confronted with technical issues as well as regulatory issues. Modern genetic engineering approaches are making it possible to address some of the technical issues faced by phage therapy. Such approaches can help modify already discovered and characterized phages and their enzymes regarding the desired properties rather than isolating new ones, which is a time-consuming process. These desired properties pertain to altering their host range, mitigating undesirable effects when lytic phages are used, such as the release of bacterial toxins upon bacterial lysis, evading bacterial defense mechanisms, evading mammalian immune response, helping with the direct removal of biofilms, and allowing phages to directly deliver toxins to target bacteria. Such techniques are definitely improving the results of phage therapy in laboratory experiments and can definitely improve the outcome of phage therapy in clinical settings.
Author Contributions
SN: Conceptualization, Investigation, Writing—original draft. RK: Conceptualization, Investigation, Writing—original draft. MAA: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Supervision. All authors have read and approved the submitted version.
Funding
Authors acknowledge the financial, practical and logistical support extended by the Institute of
Microbiology, University of Agriculture, Faisalabad, Punjab, Pakistan.
Acknowledgments
Authors acknowledge the contribution from Sohaib H. Mazhar for creating hight quality Figures used in this paper.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Abbreviations
| AMP | antimicrobial peptides |
| CBD | wall binding domain |
| CPS | Capsular polysaccharide |
| EAD | enzymatically active domain |
| EPS | Exopolysaccharide |
| LAB | Lactic acid bacteria |
| LPS | Lipopolysaccharide |
| MRSA | Methicillin Resistant S. aureus |
| Psa | Pseudomonas syringae pv. actinidiae |
| RBPs | Receptor binding proteins |
| SMPA | sheep myeloid 29-amino acid peptide |
| VALs | Virion-associated lysins |
References
- Elfadadny A, Ragab RF, Abou Shehata MA, Elfadadny MR, Farag A, Abd El-Aziz AH, et al. Exploring bacteriophage applications in medicine and beyond. Acta microbiol hell. 2024;69:167-79. [CrossRef]
- Sahoo K, Meshram S, Sahoo Jr K. The Evolution of Phage Therapy: A Comprehensive Review of Current Applications and Future Innovations. Cureus. 2024;16:e70414. [CrossRef]
- Grigson SR, Giles SK, Edwards RA, Papudeshi B. Knowing and naming: phage annotation and nomenclature for phage therapy. Clin Infect Dis. 2023;77:S352-S9. [CrossRef]
- Guo Z, Yuan M, Chai J. Mini review advantages and limitations of lytic phages compared with chemical antibiotics to combat bacterial infections. Heliyon. 2024;10:e34849. [CrossRef]
- Mobarezi Z, Esfandiari AH, Abolbashari S, Meshkat Z. Efficacy of phage therapy in controlling staphylococcal biofilms: a systematic review. Eur J Med Res. 2025;30:605. [CrossRef]
- Diallo K, Dublanchet A. Benefits of combined phage–antibiotic therapy for the control of antibiotic-resistant bacteria: A literature review. Antibiotics. 2022;11:839. [CrossRef]
- Merabishvili M, Pirnay J-P, De Vos D. Guidelines to compose an ideal bacteriophage cocktail. Bacteriophage Therapy: From Lab to Clinical Practice. 2018;1693:99-110. [CrossRef]
- Fernández L, Gutiérrez D, García P, Rodríguez A. The perfect bacteriophage for therapeutic applications—a quick guide. Antibiotics. 2019;8:126. [CrossRef]
- Nayab S, Idrees K, Aslam MA. Synergism of phages and antimicrobial peptides for treating multidrug resistant bacterial pathogens. Explor Drug Sci. 2025;3:1008133. [CrossRef]
- Keen EC, Dantas G. Close encounters of three kinds: bacteriophages, commensal bacteria, and host immunity. Trends Microbiol. 2018;26:943-54. [CrossRef]
- Ingmer H, Gerlach D, Wolz C. Temperate phages of Staphylococcus aureus. Microbiol Spectr. 2019;7:10.1128/microbiolspec. gpp3-0058-2018. [CrossRef]
- Schicklmaier P, Schmieger H. Frequency of generalized transducing phages in natural isolates of the Salmonella typhimurium complex. Appl Environ Microbiol. 1995;61:1637-40. [CrossRef]
- Penadés JR, Chen J, Quiles-Puchalt N, Carpena N, Novick RP. Bacteriophage-mediated spread of bacterial virulence genes. Curr Opin Microbiol. 2015;23:171-8. [CrossRef]
- Chiang YN, Penadés JR, Chen J. Genetic transduction by phages and chromosomal islands: the new and noncanonical. PLoS Pathog. 2019;15:e1007878. [CrossRef]
- Bertozzi Silva J, Storms Z, Sauvageau D. Host receptors for bacteriophage adsorption. FEMS Microbiol Lett. 2016;363:fnw002. [CrossRef]
- Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018;359:eaar4120. [CrossRef]
- Ofir G, Sorek R. Contemporary phage biology: from classic models to new insights. Cell. 2018;172:1260-70. [CrossRef]
- Bernheim A, Sorek R. The pan-immune system of bacteria: antiviral defence as a community resource. Nature Reviews Microbiology. 2020;18:113-9. [CrossRef]
- Hampton HG, Watson BN, Fineran PC. The arms race between bacteria and their phage foes. Nature. 2020;577:327-36. [CrossRef]
- Rostøl JT, Marraffini L. (Ph) ighting phages: how bacteria resist their parasites. Cell Host and Microbe. 2019;25:184-94. [CrossRef]
- Leprince A, Mahillon J. Phage adsorption to gram-positive bacteria. Viruses. 2023;15:196. [CrossRef]
- Samir S. Molecular machinery of the triad Holin, Endolysin, and Spanin: key players orchestrating bacteriophage-induced cell lysis and their therapeutic applications. Protein Peptide Lett. 2024;31:85-96. [CrossRef]
- Pirnay J-P, De Vos D, Verbeken G, Merabishvili M, Chanishvili N, Vaneechoutte M, et al. The phage therapy paradigm: prêt-à-porter or sur-mesure? Pharm Res. 2011;28:934-7. [CrossRef]
- Cui Z, Guo X, Feng T, Li L. Exploring the whole standard operating procedure for phage therapy in clinical practice. J Transl Med. 2019;17:1-7. [CrossRef]
- Gibson SB, Green SI, Liu CG, Salazar KC, Clark JR, Terwilliger AL, et al. Constructing and characterizing bacteriophage libraries for phage therapy of human infections. Front Microbiol. 2019;10:2537. [CrossRef]
- Friman VP, Soanes-Brown D, Sierocinski P, Molin S, Johansen HK, Merabishvili M, et al. Pre-adapting parasitic phages to a pathogen leads to increased pathogen clearance and lowered resistance evolution with Pseudomonas aeruginosa cystic fibrosis bacterial isolates. J Evol Biol. 2016;29:188-98. [CrossRef]
- Mahichi F, Synnott AJ, Yamamichi K, Osada T, Tanji Y. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS Microbiol Lett. 2009;295:211-7. [CrossRef]
- Ando H, Lemire S, Pires DP, Lu TK. Engineering modular viral scaffolds for targeted bacterial population editing. Cell systems. 2015;1:187-96. [CrossRef]
- Yehl K, Lemire S, Yang AC, Ando H, Mimee M, Torres MDT, et al. Engineering phage host-range and suppressing bacterial resistance through phage tail fiber mutagenesis. Cell. 2019;179:459-69. e9. [CrossRef]
- Yoichi M, Abe M, Miyanaga K, Unno H, Tanji Y. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157: H7. J Biotechnol. 2005;115:101-7. [CrossRef]
- Lynch KH, Seed KD, Stothard P, Dennis JJ. Inactivation of Burkholderia cepacia complex phage KS9 gp41 identifies the phage repressor and generates lytic virions. J Virol. 2010;84:1276-88. [CrossRef]
- Kilcher S, Studer P, Muessner C, Klumpp J, Loessner MJ. Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc Natl Acad Sci. 2018;115:567-72. [CrossRef]
- Zhang H, Fouts D, DePew J, Stevens R. Genetic modifications to temperate Enterococcus faecalis phage ϕEf11 that abolish the establishment of lysogeny and sensitivity to repressor, and increase host range and productivity of lytic infection. Microbiology. 2013;159:1023-35. [CrossRef]
- Dedrick RM, Guerrero-Bustamante CA, Garlena RA, Russell DA, Ford K, Harris K, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019;25:730-3. [CrossRef]
- Lu TK, Collins JJ. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci. 2007;104:11197-202. [CrossRef]
- Born Y, Fieseler L, Thöny V, Leimer N, Duffy B, Loessner MJ. Engineering of bacteriophages Y2:: dpoL1-C and Y2:: luxAB for efficient control and rapid detection of the fire blight pathogen, Erwinia amylovora. Appl Environ Microbiol. 2017;83:e00341-17. [CrossRef]
- Pei R, Lamas-Samanamud GR. Inhibition of biofilm formation by T7 bacteriophages producing quorum-quenching enzymes. Appl Environ Microbiol. 2014;80:5340-8. [CrossRef]
- Paul VD, Sundarrajan S, Rajagopalan SS, Hariharan S, Kempashanaiah N, Padmanabhan S, et al. Lysis-deficient phages as novel therapeutic agents for controlling bacterial infection. BMC Microbiol. 2011;11:1-9. [CrossRef]
- Matsuda T, Freeman TA, Hilbert DW, Duff M, Fuortes M, Stapleton PP, et al. Lysis-deficient bacteriophage therapy decreases endotoxin and inflammatory mediator release and improves survival in a murine peritonitis model. Surgery. 2005;137:639-46. [CrossRef]
- Hagens S, Habel A, Von Ahsen U, Von Gabain A, Bläsi U. Therapy of experimental Pseudomonas infections with a nonreplicating genetically modified phage. Antimicrob Agents Chemother. 2004;48:3817-22. [CrossRef]
- Møller-Olsen C, Ho SFS, Shukla RD, Feher T, Sagona AP. Engineered K1F bacteriophages kill intracellular Escherichia coli K1 in human epithelial cells. Sci Rep. 2018;8:17559. [CrossRef]
- Mai-Prochnow A, Hui JGK, Kjelleberg S, Rakonjac J, McDougald D, Rice SA. Big things in small packages: the genetics of filamentous phage and effects on fitness of their host. FEMS Microbiol Rev. 2015;39:465-87. [CrossRef]
- Rakonjac J, Russel M, Khanum S, Brooke SJ, Rajič M. Filamentous phage: structure and biology. Recombinant antibodies for infectious diseases. 2017;1053:1-20. [CrossRef]
- Yacoby I, Shamis M, Bar H, Shabat D, Benhar I. Targeting antibacterial agents by using drug-carrying filamentous bacteriophages. Antimicrob Agents Chemother. 2006;50:2087-97. [CrossRef]
- Yang Y, Kang D, Mihalache B, Vexler S, Jain S, Peng H, et al. Metagenome-inspired libraries to engineer phage M13 for targeted killing of Gram-negative bacterial species. Nucleic Acids Res. 2025;53:gkaf984. [CrossRef]
- Krom RJ, Bhargava P, Lobritz MA, Collins JJ. Engineered phagemids for nonlytic, targeted antibacterial therapies. Nano Lett. 2015;15:4808-13. [CrossRef]
- Westwater C, Kasman LM, Schofield DA, Werner PA, Dolan JW, Schmidt MG, et al. Use of genetically engineered phage to deliver antimicrobial agents to bacteria: an alternative therapy for treatment of bacterial infections. Antimicrob Agents Chemother. 2003;47:1301-7. [CrossRef]
- Hagens S, Bläsi U. Genetically modified filamentous phage as bactericidal agents: a pilot study. Lett Appl Microbiol. 2003;37:318-23. [CrossRef]
- Lu TK, Collins JJ. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci. 2009;106:4629-34. [CrossRef]
- Bhattarai SR, Yoo SY, Lee S-W, Dean D. Engineered phage-based therapeutic materials inhibit Chlamydia trachomatis intracellular infection. Biomaterials. 2012;33:5166-74. [CrossRef]
- Smith HW, Huggins MB, Shaw KM. Factors influencing the survival and multiplication of bacteriophages in calves and in their environment. Microbiology. 1987;133:1127-35. [CrossRef]
- Dabrowska K, Switała-Jelen K, Opolski A, Weber-Dabrowska B, Gorski A. Bacteriophage penetration in vertebrates. J Appl Microbiol. 2005;98:7-13. [CrossRef]
- Cisek AA, Dąbrowska I, Gregorczyk KP, Wyżewski Z. Phage therapy in bacterial infections treatment: one hundred years after the discovery of bacteriophages. Curr Microbiol. 2017;74:277-83. [CrossRef]
- Maciejewska B, Olszak T, Drulis-Kawa Z. Applications of bacteriophages versus phage enzymes to combat and cure bacterial infections: an ambitious and also a realistic application? Appl Microbiol Biotechnol. 2018;102:2563-81. [CrossRef]
- Lewis K. Platforms for antibiotic discovery. Nat Rev Drug Discov. 2013;12:371-87. 10.1038/nrd3975.
- Drulis-Kawa Z, Majkowska-Skrobek G, Maciejewska B. Bacteriophages and phage-derived proteins–application approaches. Curr Med Chem. 2015;22:1757-73. [CrossRef]
- Olszak T, Shneider MM, Latka A, Maciejewska B, Browning C, Sycheva LV, et al. The O-specific polysaccharide lyase from the phage LKA1 tailspike reduces Pseudomonas virulence. Sci Rep. 2017;7:16302. [CrossRef]
- Zou X, Xiao X, Mo Z, Ge Y, Jiang X, Huang R, et al. Systematic strategies for developing phage resistant Escherichia coli strains. Nat commun. 2022;13:4491. [CrossRef]
- Latka A, Maciejewska B, Majkowska-Skrobek G, Briers Y, Drulis-Kawa Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl Microbiol Biotechnol. 2017;101:3103-19. [CrossRef]
- Chen Y, Wang S, Wang Y, Zhangxiang L, Chen H, Li X, et al. Complete genome sequence of the novel phage vB_EcoS_PHB17, which infects Shiga-toxin-producing Escherichia coli. Arch Virol. 2019;164:3111-3. [CrossRef]
- Ma YX, Wang CY, Li YY, Li J, Wan QQ, Chen JH, et al. Considerations and caveats in combating ESKAPE pathogens against nosocomial infections. Advanced Science. 2020;7:1901872. [CrossRef]
- Pires DP, Oliveira H, Melo LD, Sillankorva S, Azeredo J. Bacteriophage-encoded depolymerases: their diversity and biotechnological applications. Appl Microbiol Biotechnol. 2016;100:2141-51. [CrossRef]
- Lin H, Paff ML, Molineux IJ, Bull JJ. Antibiotic therapy using phage depolymerases: robustness across a range of conditions. Viruses. 2018;10:622. [CrossRef]
- Chen Y, Sun E, Yang L, Song J, Wu B. Therapeutic application of bacteriophage PHB02 and its putative depolymerase against Pasteurella multocida capsular type A in mice. Front Microbiol. 2018;9:1678. [CrossRef]
- Lin H, Paff ML, Molineux IJ, Bull JJ. Therapeutic application of phage capsule depolymerases against K1, K5, and K30 capsulated E. coli in mice. Front Microbiol. 2017;8:2257. [CrossRef]
- Lin T-L, Hsieh P-F, Huang Y-T, Lee W-C, Tsai Y-T, Su P-A, et al. Isolation of a bacteriophage and its depolymerase specific for K1 capsule of Klebsiella pneumoniae: implication in typing and treatment. J Infect Dis. 2014;210:1734-44. [CrossRef]
- Chen Y, Li X, Wang S, Guan L, Li X, Hu D, et al. A novel tail-associated O91-specific polysaccharide depolymerase from a podophage reveals lytic efficacy of Shiga toxin-producing Escherichia coli. Appl Environ Microbiol. 2020;86:e00145-20. [CrossRef]
- Olsen NM, Thiran E, Hasler T, Vanzieleghem T, Belibasakis GN, Mahillon J, et al. Synergistic removal of static and dynamic Staphylococcus aureus biofilms by combined treatment with a bacteriophage endolysin and a polysaccharide depolymerase. Viruses. 2018;10:438. [CrossRef]
- Wu Y, Wang R, Xu M, Liu Y, Zhu X, Qiu J, et al. A novel polysaccharide depolymerase encoded by the phage SH-KP152226 confers specific activity against multidrug-resistant Klebsiella pneumoniae via biofilm degradation. Front Microbiol. 2019;10:2768. [CrossRef]
- Latka A, Drulis-Kawa Z. Advantages and limitations of microtiter biofilm assays in the model of antibiofilm activity of Klebsiella phage KP34 and its depolymerase. Sci Rep. 2020;10:20338. [CrossRef]
- Husain FM, Zahra A, Ali A, Kamthan M, Al-Shabib NA, Farooqui Z, et al. Bacteriophages and Their Enzymes: Allies Against Microbial Biofilms. Pharmaceuticals. 2025;18:1771. [CrossRef]
- Wang I-N, Smith DL, Young R. Holins: the protein clocks of bacteriophage infections. Annu Rev Microbiol. 2000;54:799-825. [CrossRef]
- Loessner MJ. Bacteriophage endolysins—current state of research and applications. Curr Opin Microbiol. 2005;8:480-7. [CrossRef]
- Abedon ST. Lysis from without. Bacteriophage. 2011;1:46-9. [CrossRef]
- Yang H, Yu J, Wei H. Engineered bacteriophage lysins as novel anti-infectives. Front Microbiol. 2014;5:542. [CrossRef]
- Seijsing J, Sobieraj AM, Keller N, Shen Y, Zinkernagel AS, Loessner MJ, et al. Improved biodistribution and extended serum half-life of a bacteriophage endolysin by albumin binding domain fusion. Front Microbiol. 2018;9:2927. [CrossRef]
- Yan G, Yang R, Fan K, Dong H, Gao C, Wang S, et al. External lysis of Escherichia coli by a bacteriophage endolysin modified with hydrophobic amino acids. Amb Express. 2019;9:106. [CrossRef]
- Briers Y, Walmagh M, Van Puyenbroeck V, Cornelissen A, Cenens W, Aertsen A, et al. Engineered endolysin-based “Artilysins” to combat multidrug-resistant gram-negative pathogens. MBio. 2014;5:10.1128/mbio. 01379-14. [CrossRef]
- Mancoš M, Šramková Z, Peterková D, Vidová B, Godány A. Functional expression and purification of tailor-made chimeric endolysin with the broad antibacterial spectrum. Biologia. 2020;75:2031-43. [CrossRef]
- Hong H-W, Kim YD, Jang J, Kim MS, Song M, Myung H. Combination effect of engineered endolysin EC340 with antibiotics. Front Microbiol. 2022;13:821936. [CrossRef]
- Islam MM, Kim D, Kim K, Park S-J, Akter S, Kim J, et al. Engineering of lysin by fusion of antimicrobial peptide (cecropin A) enhances its antibacterial properties against multidrug-resistant Acinetobacter baumannii. Front Microbiol. 2022;13:988522. [CrossRef]
- Abdelkader K, Gutierrez D, Tames-Caunedo H, Ruas-Madiedo P, Safaan A, Khairalla AS, et al. Engineering a Lysin with Intrinsic Antibacterial Activity (LysMK34) by Cecropin A Fusion Enhances Its Antibacterial Properties against Acinetobacter baumannii. Appl Environ Microbiol. 2022;88:e0151521. [CrossRef]
- Briers Y, Walmagh M, Grymonprez B, Biebl M, Pirnay J-P, Defraine V, et al. Art-175 is a highly efficient antibacterial against multidrug-resistant strains and persisters of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2014;58:3774-84. [CrossRef]
- Basit A, Qadir S, Qureshi S, Rehman SU. Cloning and expression analysis of fused holin-endolysin from RL bacteriophage; Exhibits broad activity against multi drug resistant pathogens. Enzyme Microb Technol. 2021;149:109846. [CrossRef]
- Heselpoth RD, Euler CW, Schuch R, Fischetti VA. Lysocins: bioengineered antimicrobials that deliver lysins across the outer membrane of Gram-negative bacteria. Antimicrob Agents Chemother. 2019;63:10.1128/aac. 00342-19. [CrossRef]
- Lukacik P, Barnard TJ, Keller PW, Chaturvedi KS, Seddiki N, Fairman JW, et al. Structural engineering of a phage lysin that targets Gram-negative pathogens. Proc Natl Acad Sci. 2012;109:9857-62. [CrossRef]
- Yan G, Liu J, Ma Q, Zhu R, Guo Z, Gao C, et al. The N-terminal and central domain of colicin A enables phage lysin to lyse Escherichia coli extracellularly. Antonie Van Leeuwenhoek. 2017;110:1627-35. [CrossRef]
- Oliveira H, Melo LD, Santos SB, Nóbrega FL, Ferreira EC, Cerca N, et al. Molecular aspects and comparative genomics of bacteriophage endolysins. J Virol. 2013;87:4558-70. [CrossRef]
- Walmagh M, Briers Y, Santos SBd, Azeredo J, Lavigne R. Characterization of modular bacteriophage endolysins from Myoviridae phages OBP, 201φ2-1 and PVP-SE1. PLoS ONE. 2012;7:e36991. [CrossRef]
- Briers Y, Schmelcher M, Loessner MJ, Hendrix J, Engelborghs Y, Volckaert G, et al. The high-affinity peptidoglycan binding domain of Pseudomonas phage endolysin KZ144. Biochem Biophys Res Commun. 2009;383:187-91. [CrossRef]
- Díez-Martínez R, De Paz HD, García-Fernández E, Bustamante N, Euler CW, Fischetti VA, et al. A novel chimeric phage lysin with high in vitro and in vivo bactericidal activity against Streptococcus pneumoniae. J Antimicrob Chemother. 2015;70:1763-73. [CrossRef]
- Becker SC, Foster-Frey J, Stodola AJ, Anacker D, Donovan DM. Differentially conserved staphylococcal SH3b_5 cell wall binding domains confer increased staphylolytic and streptolytic activity to a streptococcal prophage endolysin domain. Gene. 2009;443:32-41. [CrossRef]
- Dong Q, Wang J, Yang H, Wei C, Yu J, Zhang Y, et al. Construction of a chimeric lysin Ply187N-V12C with extended lytic activity against staphylococci and streptococci. Microbial biotechnology. 2015;8:210-20. [CrossRef]
- Fernandes S, Proença D, Cantante C, Silva FA, Leandro C, Lourenço S, et al. Novel chimerical endolysins with broad antimicrobial activity against methicillin-resistant Staphylococcus aureus. Microb Drug Resist. 2012;18:333-43. [CrossRef]
- Mao J, Schmelcher M, Harty WJ, Foster-Frey J, Donovan DM. Chimeric Ply187 endolysin kills Staphylococcus aureus more effectively than the parental enzyme. FEMS Microbiol Lett. 2013;342:30-6. [CrossRef]
- Landlinger C, Tisakova L, Oberbauer V, Schwebs T, Muhammad A, Latka A, et al. Engineered phage endolysin eliminates Gardnerella biofilm without damaging beneficial bacteria in bacterial vaginosis ex vivo. Pathogens. 2021;10:54. [CrossRef]
- Daniel A, Euler C, Collin M, Chahales P, Gorelick KJ, Fischetti VA. Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2010;54:1603-12. [CrossRef]
- Donovan DM, Dong S, Garrett W, Rousseau GM, Moineau S, Pritchard DG. Peptidoglycan hydrolase fusions maintain their parental specificities. Appl Environ Microbiol. 2006;72:2988-96. [CrossRef]
- Behera M, Singh G, Vats A, Roshan M, Gautam D, Rana C, et al. Expression and characterization of novel chimeric endolysin CHAPk-SH3bk against biofilm-forming methicillin-resistant Staphylococcus aureus. Int J Biol Macromol. 2024;254:127969. [CrossRef]
- Behera M, Singh P, Verma AK, De S, Ghorai SM. Activity of GS-linked chimeric endolysin CHAPk-SH3bk against methicillin-resistant Staphylococcus aureus biofilms: an in-vitro, ex-vivo and in-vivo study. npj Biofilms and Microbiomes. 2025;11:94. [CrossRef]
- Momen S, Soleimani N, Azizmohseni F, Ahmadbeigi Y, Borhani S, Amini-Bayat Z. Characterization and bioinformatic analysis of a new chimeric endolysin against MRSA with great stability. AMB Express. 2024;14:143. [CrossRef]
- Wang S, Gu J, Lv M, Guo Z, Yan G, Yu L, et al. The antibacterial activity of E. coli bacteriophage lysin lysep3 is enhanced by fusing the Bacillus amyloliquefaciens bacteriophage endolysin binding domain D8 to the C-terminal region. J Microbiol. 2017;55:403-8. [CrossRef]
- Cheng Q, Fischetti VA. Mutagenesis of a bacteriophage lytic enzyme PlyGBS significantly increases its antibacterial activity against group B streptococci. Appl Microbiol Biotechnol. 2007;74:1284-91. [CrossRef]
- Mayer MJ, Garefalaki V, Spoerl R, Narbad A, Meijers R. Structure-based modification of a Clostridium difficile-targeting endolysin affects activity and host range. J Bacteriol. 2011;193:5477-86. [CrossRef]
- Dahal RH, Choi Y-J, Kim S, Kim J. CHAPSAP26-161, A Truncated Protein and Enzymatic Active Domain of Endolysin LysSAP26, as a Potential Therapeutic Agent to Combat Clostridioides difficile Infection. J Bacteriol Virol. 2024;54:107-21. [CrossRef]
- Wang Y, Wang X, Liu X, Lin B. Research progress on strategies for improving the enzyme properties of bacteriophage endolysins. J Microbiol Biotechnol. 2024;34:1189. [CrossRef]
- Schmelcher M, Tchang VS, Loessner MJ. Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb Biotechnol. 2011;4:651-62. [CrossRef]
- Rodríguez-Rubio L, Martínez B, Rodríguez A, Donovan DM, García P. Enhanced staphylolytic activity of the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA88 HydH5 virion-associated peptidoglycan hydrolase: fusions, deletions, and synergy with LysH5. Appl Environ Microbiol. 2012;78:2241-8. [CrossRef]
- Díez-Martínez R, de Paz H, Bustamante N, García E, Menéndez M, García P. Improving the lethal effect of Cpl-7, a pneumococcal phage lysozyme with broad bactericidal activity, by inverting the net charge of its cell wall-binding module. Antimicrob Agents Chemother. 2013;57:5355-65. [CrossRef]
- Love MJ, Coombes D, Manners SH, Abeysekera GS, Billington C, Dobson RC. The molecular basis for Escherichia coli O157: H7 phage FAHEc1 endolysin function and protein engineering to increase thermal stability. Viruses. 2021;13:1101. [CrossRef]
- Jansson MK, Nguyen DT, Mikkat S, Warnke C, Janssen MB, Warnke P, et al. Synthetic mRNA delivered to human cells leads to expression of Cpl-1 bacteriophage-endolysin with activity against Streptococcus pneumoniae. Mol Ther Nucleic Acids. 2024;35:102145. [CrossRef]
- Gervasi T, Horn N, Wegmann U, Dugo G, Narbad A, Mayer MJ. Expression and delivery of an endolysin to combat Clostridium perfringens. Appl Microbiol Biotechnol. 2014;98:2495-505. [CrossRef]
- Chandran C, Tham HY, Rahim RA, Lim SHE, Yusoff K, Song AA-L. Lactococcus lactis secreting phage lysins as a potential antimicrobial against multi-drug resistant Staphylococcus aureus. PeerJ. 2022;10:e12648. [CrossRef]
- Dzuvor CK, Shanbhag BK, Younas T, Shen H-H, Haritos VS, He L. Engineering self-assembled endolysin nanoparticles against antibiotic-resistant bacteria. ACS Applied Bio Materials. 2022;5:4993-5003. [CrossRef]
- Nobrega FL, Costa AR, Santos JF, Siliakus MF, Van Lent JW, Kengen SW, et al. Genetically manipulated phages with improved pH resistance for oral administration in veterinary medicine. Sci Rep. 2016;6:39235. [CrossRef]
- Peng H, Borg RE, Dow LP, Pruitt BL, Chen IA. Controlled phage therapy by photothermal ablation of specific bacterial species using gold nanorods targeted by chimeric phages. Proc Natl Acad Sci. 2020;117:1951-61. [CrossRef]
- Gattuboyena N, Tsai Y-C, Lin L-C. Therapeutic and Diagnostic Potential of a Novel K1 Capsule Dependent Phage, JSSK01, and Its Depolymerase in Multidrug-Resistant Escherichia coli Infections. Int J Mol Sci. 2024;25:12497. [CrossRef]
- Napolitano V, Privitera M, Drulis-Kawa Z, Marasco D, Fallarini S, Berisio R, et al. Structural and functional features of Klebsiella pneumoniae capsular degradation by the phage depolymerase KP32gp38: Implications for vaccination against K. pneumoniae. Int J Antimicrob Agents. 2025:107596. [CrossRef]
- Chen Y, Fan Z, Fu T, Li Z, Feng J, Cui X, et al. Characterization of a Phage-Encoded Depolymerase Against Klebsiella pneumoniae K30 Capsular Type and Its Therapeutic Application in a Murine Model of Aspiration Pneumonia. Viruses. 2025;17:1446. [CrossRef]
- Li P, Guo G, Zheng X, Xu S, Zhou Y, Qin X, et al. Therapeutic efficacy of a K5-specific phage and depolymerase against Klebsiella pneumoniae in a mouse model of infection. Vet Res. 2024;55:59. [CrossRef]
- Wang R, Liu Y, Zhang Y, Yu S, Zhuo H, Huang Y, et al. Identification and characterization of the capsule depolymerase Dpo27 from phage IME-Ap7 specific to Acinetobacter pittii. Front Cell Infect Microbiol. 2024;14:1373052. [CrossRef]
- Borzilov AI, Volozhantsev NV, Korobova OV, Kolupaeva LV, Pereskokova ES, Kombarova TI, et al. Bacteriophage and phage-encoded depolymerase exhibit antibacterial activity against K9-type Acinetobacter baumannii in mouse sepsis and burn skin infection models. Viruses. 2025;17:70. [CrossRef]
- Yang P, Shan B, Hu X, Xue L, Song G, He P, et al. Identification of a novel phage depolymerase against ST11 K64 carbapenem-resistant Klebsiella pneumoniae and its therapeutic potential. J Bacteriol. 2025;207:e00387-24. [CrossRef]
- Zhao J, Wang J, Zhang C, Xu S, Ren H, Zou L, et al. Characterization of a Salmonella abortus equi phage 4FS1 and its depolymerase. Frontiers in Veterinary Science. 2024;11:1496684. [CrossRef]
- Zelmer A, Martin MJ, Gundogdu O, Birchenough G, Lever R, Wren BW, et al. Administration of capsule-selective endosialidase E minimizes upregulation of organ gene expression induced by experimental systemic infection with Escherichia coli K1. Microbiology. 2010;156:2205-15. [CrossRef]
- Waseh S, Hanifi-Moghaddam P, Coleman R, Masotti M, Ryan S, Foss M, et al. Orally administered P22 phage tailspike protein reduces Salmonella colonization in chickens: prospects of a novel therapy against bacterial infections. PLoS ONE. 2010;5:e13904. [CrossRef]
- Li M, Li P, Chen L, Guo G, Xiao Y, Chen L, et al. Identification of a phage-derived depolymerase specific for KL64 capsule of Klebsiella pneumoniae and its anti-biofilm effect. Virus Genes. 2021;57:434-42. [CrossRef]
- V. Volozhantsev N, M. Shpirt A, I. Borzilov A, V. Komisarova E, M. Krasilnikova V, S. Shashkov A, et al. Characterization and therapeutic potential of bacteriophage-encoded polysaccharide depolymerases with β galactosidase activity against Klebsiella pneumoniae K57 capsular type. Antibiotics. 2020;9:732. [CrossRef]
- Chen X, Liu M, Zhang P, Xu M, Yuan W, Bian L, et al. Phage-derived depolymerase as an antibiotic adjuvant against multidrug-resistant Acinetobacter baumannii. Frontiers in microbiology. 2022;13:845500. [CrossRef]
- Liu Y, Mi Z, Mi L, Huang Y, Li P, Liu H, et al. Identification and characterization of capsule depolymerase Dpo48 from Acinetobacter baumannii phage IME200. PeerJ. 2019;7:e6173. [CrossRef]
- Rice CJ, Kelly SA, O’Brien SC, Melaugh EM, Ganacias JC, Chai ZH, et al. Novel phage-derived depolymerase with activity against Proteus mirabilis biofilms. Microorganisms. 2021;9:2172. [CrossRef]
- Tung CW, Thapa K, Phan A, Mohapatra A, Hashmi M, Bleich K, et al. Antibacterial Activity of a Fused Endolysin ENDO-1252/KL9P Against Multiple Serovars of Salmonella enterica. Microbiol Biotechnol. 2025;18:e70237. [CrossRef]
- Kogawa M, Yoda T, Matsuhashi A, Matsushita A, Otsuka Y, Shibagaki S, et al. Development of chimera AMP–Endolysin with wider spectra against Gram-Negative Bacteria using High-Throughput assay. Viruses. 2025;17:200. [CrossRef]
- Kim D, Kim J, Kim M. Potential of antimicrobial peptide-fused endolysin LysC02 as therapeutics for infections and disinfectants for food contact surfaces to control Cronobacter sakazakii. Food Control. 2024;157:110190. [CrossRef]
- Lim J, Myung H, Lim D, Song M. Antimicrobial peptide thanatin fused endolysin PA90 (Tha-PA90) for the control of Acinetobacter baumannii infection in mouse model. J Biomed Sci. 2024;31:36. [CrossRef]
- Antonova N, Grigoriev I, Lendel A, Usacheva O, Klimova A, Usachev E, et al. Engineering of recombinant endolysin LysSi3 to increase its antibacterial properties. Appl Biochem Microbiol. 2024;60:802-11. [CrossRef]
- Vasina DV, Antonova NP, Gushchin VA, Aleshkin AV, Fursov MV, Fursova AD, et al. Development of novel antimicrobials with engineered endolysin LysECD7-SMAP to combat Gram-negative bacterial infections. J Biomed Sci. 2024;31:75. [CrossRef]
- Antonova NP, Abdullaeva SD, Vasina DV, Grigoriev IV, Usachev EV, Usacheva OV, et al. Modifying Pharmacokinetic Properties of the Gram-Negative Bacteria Targeting Endolysin ML06 Without Affecting Antibacterial Activity. Int J Mol Sci. 2025;26:4376. [CrossRef]
- Wang Y, Li H, Cui Y, Li Y, Yu Y, Zhang X, et al. Enhancing food system protection: synergistic action of engineered endolysin LYSMP and berberine hydrochloride against Streptococcus suis on pork. Food Res Int. 2025;221:117214. [CrossRef]
- Li X, Shangguan W, Yang X, Hu X, Li Y, Zhao W, et al. Influence of lipopolysaccharide-interacting peptides fusion with endolysin LysECD7 and fatty acid derivatization on the efficacy against Acinetobacter baumannii infection in vitro and in vivo. Viruses. 2024;16:760. [CrossRef]
- Rani R, Poria V, Jaglan AB, Kapoor P, Bhutani K, Sharma P, et al. Improved lytic action of engineered phage-encoded endolysin and docking insights into its action on bacterial peptidoglycan. Int J Biol Macromol. 2025:146105. [CrossRef]
- Amiryan A, Sannikova E, Serkina A, Gubaidullin I, Bulushova N, Kozlov D. Optimization of the Structure of the Staphylococcus aureus Phage K Endolysin CHAP Domain to Increase Lytic Activity. Appl Biochem Microbiol. 2024;60:1565-74. [CrossRef]
- Easwaran M, Govindaraj RG, Naderi M, Brylinski M, De Zoysa M, Shin H-J. Evaluating the antibacterial activity of engineered phage ФEcSw endolysin against multi-drug-resistant Escherichia coli strain Sw1. Int J Antimicrob Agents. 2025;65:107395. [CrossRef]
- Mortazavi N, Aliakbarlu J, Imani M. The antibacterial effects of engineered Salmonella phage PVP-SE1 endolysin on the planktonic cells and biofilms of food-borne pathogens and its antibacterial activity in milk. Food Bioscience. 2025:107274. [CrossRef]
- Park W, Park M, Chun J, Hwang J, Kim S, Choi N, et al. Delivery of endolysin across outer membrane of Gram-negative bacteria using translocation domain of botulinum neurotoxin. Int J Antimicrob Agents. 2024;64:107216. [CrossRef]
- Chen W, Han L-M, Chen X-Z, Yi P-C, Li H, Ren Y-Y, et al. Engineered endolysin of Klebsiella pneumoniae phage is a potent and broad-spectrum bactericidal agent against “ESKAPEE” pathogens. Front Microbiol. 2024;15:1397830. [CrossRef]
- Krishnan M, Tham HY, Wan Nur Ismah WAK, Yusoff K, Song AA-L. Effect of Domain Manipulation in the Staphylococcal Phage Endolysin, Endo88, on Lytic Efficiency and Host Range. Mol Biotechnol. 2025;67:2536-44. [CrossRef]
- Warring SL, Sisson HM, Randall G, Grimon D, Dams D, Gutiérrez D, et al. Engineering an antimicrobial chimeric endolysin that targets the phytopathogen Pseudomonas syringae pv. actinidiae. J Biol Chem. 2025:110224. [CrossRef]
- Munetomo S, Uchiyama J, Takemura-Uchiyama I, Wanganuttara T, Yamamoto Y, Tsukui T, et al. Examination of yield, bacteriolytic activity and cold storage of linker deletion mutants based on endolysin S6_ORF93 derived from Staphylococcus giant bacteriophage S6. PLoS ONE. 2024;19:e0310962. [CrossRef]
- Lee J, Cho H, Kim K-s. Surface-displaying protein from Lacticaseibacillus paracasei–derived extracellular vesicles: Identification and utilization in the fabrication of an endolysin-displaying platform against Staphylococcus aureus. Chem Eng J. 2025;512:162196. [CrossRef]
- Sáez Moreno D, Cunha J, Melo LDR, Tanaka K, Bamba T, Hasunuma T, et al. CRISPR-Cas9 engineered Saccharomyces cerevisiae for endolysin delivery to combat Listeria monocytogenes. Appl Microbiol Biotechnol. 2025; 109(1), 81. [CrossRef]
Figure 1.
Schematic illustration of phage-induced bacteriolysis through the lytic cycle. (1) Adsorption of phage to the bacterial body and DNA injection. (2) Circularization of phage DNA to avoid enzymatic degradation. (3) Replication of phage DNA as well as transcription of phage DNA to produce phage proteins. (4) DNA packaging and assembly of the head and tail into a complete phage. (5) Disruption of the cell wall through the endolysin-holin system. (6) Release of the phage progeny. (A) Genetic modifications in tail fiber (receptor binding proteins) of lytic phage can affect host range. Virion associated lysins and polysaccharide depolymerases (DPs), responsible for degradation of cell layers, present in tail fibers can be used in pure form after expressing recombinantly. (B) Genetic modifications in any pertinent important features can affect the desirable phage properties i.e., expressing depolymerases, inhibiting quorum sensing, deletion of genes resulting in dampening of bacterial killing for mitigating effects of toxin release upon bacterial killing and conversion of lysogenic to lytic phage. Deletion of endolysin/holin genes or export protein gens resulted in slow bacterial killing. Disruption of phage repressors or lysogeny gene module converted lysogenic to lytic phage (C) Endolysins can be used in pure form after recombinantly expressing either in wild type form or after genetic modifications.
Figure 1.
Schematic illustration of phage-induced bacteriolysis through the lytic cycle. (1) Adsorption of phage to the bacterial body and DNA injection. (2) Circularization of phage DNA to avoid enzymatic degradation. (3) Replication of phage DNA as well as transcription of phage DNA to produce phage proteins. (4) DNA packaging and assembly of the head and tail into a complete phage. (5) Disruption of the cell wall through the endolysin-holin system. (6) Release of the phage progeny. (A) Genetic modifications in tail fiber (receptor binding proteins) of lytic phage can affect host range. Virion associated lysins and polysaccharide depolymerases (DPs), responsible for degradation of cell layers, present in tail fibers can be used in pure form after expressing recombinantly. (B) Genetic modifications in any pertinent important features can affect the desirable phage properties i.e., expressing depolymerases, inhibiting quorum sensing, deletion of genes resulting in dampening of bacterial killing for mitigating effects of toxin release upon bacterial killing and conversion of lysogenic to lytic phage. Deletion of endolysin/holin genes or export protein gens resulted in slow bacterial killing. Disruption of phage repressors or lysogeny gene module converted lysogenic to lytic phage (C) Endolysins can be used in pure form after recombinantly expressing either in wild type form or after genetic modifications.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



