Submitted:
12 April 2026
Posted:
14 April 2026
You are already at the latest version
Abstract
Whether copper fundamentally alters Mo-centered redox thermodynamics or mainly tunes hydrogen adsorption in Ni–Mo electrocatalysts under alkaline hydrogen evolution reaction (HER) conditions remains unresolved. Density functional theory calculations combined with a field-corrected computational hydrogen electrode framework are used to evaluate the thermodynamic stability of H3Mo, H3MoOH, H2Mo(OH)2, and MoO(OH)3 on Cu(111) and Ni(111) and to construct surface Pourbaix diagrams under electrochemical conditions. The results show that substrate identity reorganizes the redox stabilization hierarchy of these Mo intermediates. Across the examined conditions, at least one of H3Mo, H3MoOH, or MoO(OH)3 is thermodynamically favored over H2Mo(OH)2 on both surfaces. However, only Cu(111) exhibits measurable pH-dependent free-energy shifts, reaching 0.28 eV on the reversible hydrogen electrode scale. The magnitude of this electrostatic modulation is comparable to the intrinsic substrate-dependent relative Gibbs free-energy differences, suggesting that Cu reshapes Mo redox thermodynamics rather than merely weakening hydrogen binding strength. Electronic structure and vibrational analyses further show that Cu(111) preferentially weakens Mo–O interactions, whereas Ni(111) more strongly perturbs Mo–H bonding in hydrogen-rich complexes. Overall, these results establish that substrate identity governs the electrostatic modulation of Mo redox thermodynamics under alkaline HER conditions and provide a mechanistic insight into substrate effects relevant to Cu-containing Ni–Mo systems.
Keywords:
hydrogen evolution reaction
; molybdenum hydride/oxyhydride species
; Cu(111)
; Ni(111)
; density functional theory method
; field-corrected computational hydrogen electrode
; surface Pourbaix diagrams
; redox thermodynamics
; interfacial charge transfer
1. Introduction
The hydrogen evolution reaction (HER) is a fundamental half-reaction in water splitting and underpins the production of green hydrogen from renewable electricity [1,2,3]. Among available electrolysis technologies, alkaline water electrolysis is particularly attractive for large-scale deployment because of its low materials cost, long-term durability, technological maturity, and compatibility with earth-abundant electrocatalysts under relatively mild operating conditions [4,5]. However, HER in alkaline media is typically slower than in acidic electrolytes, owing in part to the additional water dissociation (Volmer) step required to generate adsorbed hydrogen and to the influence of hydroxyl adsorption and interfacial water structure on surface kinetics [4,6,7,8,9,10,11]. Achieving high activity therefore requires catalysts capable of overcoming these kinetic limitations [4]. Although platinum remains the benchmark HER catalyst, its scarcity and cost constrain large-scale implementation [1,2,3,4].
Among non-precious alternatives, nickel–molybdenum (Ni–Mo) alloys have emerged as leading candidates for alkaline HER because of their high activity, stability, and low cost [4,12]. Their performance has often been attributed to synergistic interactions between Ni and Mo, although the precise mechanistic origin remains debated [2,4,13,14]. In particular, the identity of the active Mo species remains unresolved. While some interpretations assign primary activity to metallic Ni with Mo acting mainly in a supportive role [4,13], recent spectroscopic and electrochemical evidence suggests that oxidized Mo species, including Mo3+, may directly participate in hydride formation and H2 evolution [1,13,14]. Under reaction conditions, Mo has also been reported to undergo structural and compositional evolution, indicating that the active surface may differ substantially from the as-prepared alloy [5,12]. These observations have motivated a Mo3+–hydride-centered framework in which cathodic polarization promotes redox-active Mo species capable of forming reactive hydrides [13,14,15]. Within this picture, four surface intermediates, H3Mo, H3MoOH, H2Mo(OH)2, and MoO(OH)3, define a redox-coupled thermodynamic landscape governing activity in Ni–Mo systems [13,14,15].
Incorporating copper into Ni–Mo catalysts has been reported to further enhance alkaline HER performance [1,12,16]. Most interpretations attribute this improvement to hydrogen-binding modulation and electronic effects, including d-band shifts that weaken hydrogen adsorption [3,16,17], although cooperative interactions between oxidized Cu and Mo species have also been proposed under certain conditions [1]. However, whether Cu merely tunes hydrogen adsorption strength or instead reorganizes the Mo-centered redox stabilization hierarchy under cathodic bias remains unclear. Clarifying this distinction is important because, if Cu perturbs the relative stability of oxidized and hydride-containing Mo species, its role extends beyond simple hydrogen-binding optimization.
Previous theoretical evaluations of these MoOmHn intermediates were performed on Ni(111), representing Ni-rich alloy environments [13]. To isolate substrate-dependent electronic effects without introducing the added complexity of explicit alloy models, here the same intermediates are examined on pure Cu(111). Because Ni and Cu adopt the face-centered cubic structure and exhibit closely related (111) surface geometries, this comparison minimizes geometric differences while enabling direct assessment of substrate effects on Mo redox thermodynamics. In this way, the Cu(111) model is used to isolate electronic stabilization effects rather than to reproduce the full structural complexity of ternary Ni–Mo–Cu catalysts.
Prior theoretical treatments of Ni–Mo HER systems have relied on the conventional computational hydrogen electrode (CHE) formalism [18,19], which neglects explicit interfacial electric field effects. While this approximation is often adequate for weakly polar adsorbates such as atomic hydrogen, it can misrepresent more polar MoOmHn intermediates, whose thermodynamic stability depends on coupling between their dipole moments and the interfacial electric field under cathodic bias. Neglecting this field–dipole interaction may lead to incorrect identification of the thermodynamically preferred Mo oxidation state under operating conditions. To address this limitation, a field-corrected CHE framework [20,21] is employed here to construct surface Pourbaix diagrams for MoOmHn intermediates on Ni(111) and Cu(111) under electrochemically relevant conditions.
Field- and pH-dependent free-energy analysis reveals that substrate identity reorganizes the Mo redox stabilization hierarchy. Whereas Ni(111) exhibits comparatively weak pH sensitivity, Cu(111) shows measurable free-energy shifts of up to 0.28 eV on the reversible hydrogen electrode (RHE) scale, demonstrating significant electrostatic modulation of Mo-centered intermediates. The magnitude of this field-induced stabilization is comparable to intrinsic substrate-dependent Gibbs free-energy differences, suggesting that Cu reshapes Mo redox thermodynamics rather than simply tuning hydrogen binding strength. Electronic structure analysis further reveals contrasting bond-softening tendencies, with Cu preferentially weakening Mo–O interactions and Ni selectively softening Mo–H bonds. Together, these results establish substrate-dependent electrostatic control over Mo redox thermodynamics under alkaline HER conditions.
2. Computational Details
Spin-polarized density functional theory (DFT) calculations were carried out using the Vienna Ab initio Simulation Package (VASP 6.4.2) [22,23]. The revised Perdew–Burke–Ernzerhof (RPBE) functional within the generalized gradient approximation was employed to describe exchange–correlation effects [24,25]. RPBE generally yields less exothermic adsorption energies than PBE for adsorbates on transition-metal surfaces and is widely used in heterogeneous catalysis to mitigate the well-documented overbinding tendency of PBE [19,25,26,27].
The projector augmented-wave method was used to represent the interaction between core and valence electrons [28,29]. A plane-wave cutoff energy of 520 eV was applied throughout all calculations. Electronic self-consistency was achieved with an energy convergence criterion of 10⁻6 eV, while atomic positions were relaxed until residual forces on each atom were below 0.02 eV/Å. To account for electron localization in MoOmHn complexes, the DFT+U approach was applied to the Mo 4d states within the Dudarev formalism [30,31,32], in which the effective Hubbard parameter is given by . Throughout this work, was set to 2.0 eV, consistent with the value used by Bau et al. [13] in their analysis of Mo-centered HER intermediates on Ni-rich surfaces, thereby enabling direct comparison with previous thermodynamic results.
Cu(111) and Ni(111) surfaces were modeled as 4 × 4 supercells consisting of three atomic layers, separated by a 17 Å vacuum region along the surface normal to avoid interactions between periodic images (Figure S1). Comparison with a corresponding five-layer slab showed no meaningful changes in optimized adsorption geometries, with adsorption energies differing by less than 0.10 eV. The top two layers and the adsorbates were allowed to relax, while the bottom layer was kept fixed. The Brillouin zone was sampled using a 3 × 3 × 1 Monkhorst–Pack k-point mesh [33] for structural relaxations. For electronic structure analysis and vibrational frequency calculations, a denser 5 × 5 × 1 k-point mesh was used to improve electronic convergence. Gas-phase species were calculated using only the Γ-point in reciprocal space. First-order Methfessel–Paxton smearing [34] with a width of 0.20 eV was used to ensure stable electronic convergence for the metallic slab models. Because adsorption energies were evaluated as differences between similar metallic states, finite-smearing contributions are expected to largely cancel and thus should not affect the relative energetic trends discussed here. Dispersion interactions were included using Grimme’s D3 correction with Becke–Johnson damping [35], and solvent effects were treated with the VASPSol implicit solvent approach [36,37], using a dielectric constant of 78.4 [38] to represent water at 298.15 K. External electric fields were applied along the surface normal using the dipole-layer method implemented in VASP. A dipole correction was included to remove spurious electrostatic interactions between periodic slab images and to ensure a well-defined potential drop across the vacuum region. Field strengths between −0.8 and 0.8 V Å-1 were considered, following the range reported by Kelly et al. [20]. Charge distributions and bond orders were analyzed using the 6th version of the Density Derived Electrostatic and Chemical (DDEC6) method [39,40]. Density-of-states analyses and thermal corrections to the Gibbs free energy were performed using VASPKIT (version 1.3.5) [41]. Thermal corrections were evaluated at 1 atm for gas-phase hydrogen and 0.0317 atm for water. These pressures correspond to the standard-state hydrogen pressure and the equilibrium vapor pressure of water at 298.15 K, respectively [42,43].
The adsorption energy was calculated as
where , , and are the electronic total energies of the gas-phase adsorbate, the pristine slab, and the slab–adsorbate system, respectively. Negative values indicate exothermic adsorption.
The thermodynamic stability of molybdenum complexes on metallic surfaces under HER-relevant electrochemical conditions was evaluated through the Gibbs free energy of the following reaction [13]:
where * denotes an adsorption site on the respective metal surface.
At equilibrium, the electrochemical potential condition yields [44], where is the reaction Gibbs free energy, is the number of electrons involved in the redox process, is the elementary charge in atomic units, and is the electrode potential referenced to the RHE (V). Following the thermodynamic framework previously applied to surface-bound molybdenum complexes by Bau et al. [13,14], the relative Gibbs free energy of formation of each complex on the metal surface was defined as
Here, , , and correspond to the Gibbs free energies of adsorbed , adsorbed gas-phase water, and gas-phase hydrogen, respectively. Negative values indicate thermodynamically favorable formation of the complex on the metal surface, whereas positive values indicate that the reverse reaction is favored.
Following recent extensions of the CHE that explicitly account for interfacial electric-field effects, the Gibbs free energies entering (3) were corrected to include the response of adsorbed species to the electrode field. In this framework, the field-dependent free energy of an adsorbed complex is written as
where is the zero-field relative Gibbs free energy, E is the interfacial electric field, and μ and α are the adsorption-induced dipole moment and polarizability, respectively. Electronic energies of the slab–adsorbate systems were computed at different externally applied electric fields along the surface normal, and was then evaluated for each value of . The parameters μ and α were obtained by plotting – as a function of the externally applied electric field and fitting the data to a second-order polynomial, following the approach introduced by Kelly et al. [20], and subsequently adopted for RHE-dependent surface Pourbaix analyses [21]. The interfacial electric field, , was estimated using the parallel-plate capacitor model, in which the field depends on the electrode potential relative to the potential of zero charge () and on the Helmholtz capacitance (in F m−2):
where ε0 denotes the permittivity of free space (8.85 × 10−12 F m−1), and ε is the dielectric constant. When expressed on the RHE scale, this relation makes E explicitly dependent on both the applied potential U and the pH through the conversion between RHE and SHE potentials, thereby introducing pH-dependent adsorption energetics even at fixed URHE:
Consistent with prior studies, CH was set equal to 0.25 F m−2, and zero-point energy, entropic, and solvation corrections were assumed to be independent of the applied field [20,21]. The UPZC values of Cu and Ni were set to -0.70 V [45] and -0.30 V [46], respectively. In VASP, the externally applied electric field is expressed in V Å-1. Consequently, μ and α, obtained from quadratic fitting of the field-dependent energies are reported in units of eÅ and eÅ2 V-1, respectively. For consistency between the capacitor-model field defined in (6), which is expressed in V m-1, and the VASP convention used in (4), the field values were converted from V m-1 to V Å-1 prior to evaluating the field-dependent free energies.
For Raman spectra, vibrational modes were obtained using density functional perturbation theory calculations [47,48,49], and Raman tensors were evaluated from finite differences of dielectric tensors for positive and negative displacements along each normal mode, using the postprocessing script developed by Fonari and Stauffer [50].
An eigenvector overlap analysis, inspired by the work of Lazzeri et al. [51], was employed to quantitatively compare vibrational modes of adsorbates on different metal surfaces. Prior to comparison, adsorbate geometries were aligned using the Kabsch algorithm [52], and their displacement vectors were rotated accordingly. An iterative atom-index permutation procedure was employed to minimize the root-mean-square deviation between adsorbate geometries on different metal surfaces, thereby accounting for possible atom reordering while preserving chemical equivalence. Only adsorbate-atom displacements were considered in the analysis. The overlap matrix between mode i of system A and mode j of system B was computed as the absolute value of the dot product of the corresponding normalized eigenvectors:
where and are unit-normalized eigenvectors. This metric ranges from 0, corresponding to orthogonal modes, to 1, indicating identical modes, and quantifies the similarity of atomic displacement patterns independently of vibrational frequency. Vibrational modes were visualized using the postprocessing script of Dardzinski [53] to verify mode similarity.
3. Results and Discussion
3.1. Substrate-Dependent Adsorbate–Metal Hybridization Governs Structure and Energetics of Mo Complexes on Cu(111) and Ni(111)
Mo and its hydrogenated and oxygenated complexes (H3Mo, H3MoOH, H2Mo(OH)2, and MoO(OH)3) were adsorbed on Cu(111) and Ni(111) surfaces. Figure 1 shows the most stable adsorption geometries on the two surfaces. The bare Mo adatom preferentially occupies a bridge site on Cu(111) and a threefold hollow site on Ni(111) (Figure 1a and Figure 1f, Table S1). The hollow configuration provides a more symmetric coordination environment than the bridge geometry, consistent with differences in the electronic structure of the two metals and the resulting adsorbate–metal hybridization. Upon ligand coordination, the preferred adsorption configuration changes with the coordination environment of the Mo center, reflecting progressive saturation of Mo-centered bonding interactions by the ligands, which alters the extent and geometry of metal–adsorbate coupling. H3Mo, H3MoOH, and H2Mo(OH)2 adsorb at threefold hollow sites on both metals. However, the Mo–metal distances are unequal, with one bond shorter than the other two (Tables S2–S4), indicating lateral displacement from the ideal C3v hollow position. Such deviations from ideal adsorption-site symmetry are consistent with uneven hybridization between adsorbate orbitals and the three surface atoms, since adsorption-site symmetry governs the symmetry-allowed coupling between adsorbate states and surface d-states [54].
The electronic consequences of this distortion are reflected in the distribution of adsorbate–surface bond orders. As shown in Figure S2, the DDEC6 bond orders between adsorbate atoms and the surface are generally more uneven on Cu(111) than on Ni(111). For example, in H2Mo(OH)2, the two hydroxyl oxygens interact asymmetrically with the Cu surface atoms (bond orders of 0.28 and < 0.05), whereas the corresponding interactions on Ni(111) are nearly equivalent (0.06 and 0.07). A similar trend is observed for MoO(OH)3 and H3MoOH, where O–metal and H–metal bond orders are more broadly distributed on Cu but more uniform on Ni. These observations suggest that bonding on Cu is more localized to individual surface atoms, whereas bonding on Ni is distributed more evenly across the adsorption site, consistent with stronger adsorbate–metal hybridization.
In contrast, the most oxygenated complex, MoO(OH)3, preferentially binds at atop sites on both surfaces and forms a single dominant Mo–metal bond of 2.35 Å on Ni(111) and 2.34 Å on Cu(111) (Table S5). This behavior is consistent with the high oxygen coordination of the Mo center, which strengthens internal Mo–O bonding and consequently reduces Mo-centered interaction with multiple surface atoms.
Despite these local differences, global structural descriptors indicate broadly similar adsorption geometries on the two substrates. Figure 2a shows the distance between the adsorbate centroid and the average z-coordinate of the top-layer slab atoms (centroid height). For all complexes, the centroid height is slightly larger on Ni(111) than on Cu(111), although the differences remain modest across the series: 0.02, 0.13, 0.13, 0.18, and 0.29 Å for Mo, H3Mo, H3MoOH, H2Mo(OH)2, and MoO(OH)3, respectively. Even with these modestly larger centroid heights, Ni nevertheless exhibits stronger adsorption, indicating that electronic factors outweigh the small geometric differences between the two substrates. A complementary descriptor is the relative variation in the radius of gyration () between the two adsorbed states, normalized by the gas-phase value: (Figure 2b). H3Mo exhibits a larger expansion on Ni (6.4%) relative to Cu, consistent with elongation of the Mo–H bonds by 0.16 Å relative to the gas phase, whereas H3MoOH and H2Mo(OH)2 show only minor expansion (< 3.0%) on Cu. MoO(OH)3 displays essentially no size difference between the two surfaces. Overall, these descriptors confirm that geometric differences between the two substrates are modest.
Electronic and energetic descriptors, however, reveal systematic differences. All complexes exhibit net electron donation to the substrate upon adsorption, with Ni(111) consistently accepting more charge than Cu(111), despite the slightly larger centroid heights noted above. The computed charge-transfer values (in units of e) are −0.57 (Cu) versus −1.10 (Ni) for H3Mo, −0.15 versus −0.43 for H3MoOH, −0.32 versus −0.52 for H2Mo(OH)2, and −0.12 versus −0.14 for MoO(OH)3 (Figure 2c). Within the d-band model of chemisorption [55,56,57,58], adsorption strength is correlated with the position of the metal d-band center relative to the Fermi level. Because the d-band center of Ni lies closer to the Fermi level than that of Cu (−1.84 eV versus −2.75 eV for clean slabs, Figure S3a), stronger adsorbate–metal hybridization and enhanced charge transfer are expected on Ni. Consistently, adsorption on Ni(111) is more exothermic for all complexes considered, with adsorption energy differences of −0.67 eV for H3Mo, −0.35 eV for H3MoOH, −0.52 eV for H2Mo(OH)2, and −0.45 eV for MoO(OH)3. Increasing oxygen coordination is accompanied by reduced charge transfer and weaker adsorption (Figure 2d), consistent with strong internal Mo–O bonding and diminished Mo-centered interaction with the surface.
The stronger interaction with the Ni surface also alters the internal bonding of the complexes. Enhanced adsorbate–metal hybridization on Ni is consistent with redistribution of electron density from the Mo–ligand framework toward the surface, thereby weakening Mo–H and O–H bonds relative to Cu. These electronic effects are reflected in the vibrational spectra discussed below. Because net atomic charge analysis assigns negative partial charge to Mo–H hydrogens and positive partial charge to O–H hydrogens in both isolated and adsorbed complexes (Tables S6–S12), the former are referred to as hydride hydrogens and the latter as protic hydrogens.
3.2. Substrate-Dependent Weakening and Reorganization of Mo–H Bonding in H3Mo on Cu(111) and Ni(111)
On both Ni(111) and Cu(111), the hydride hydrogens in H3Mo adopt similar geometries, remaining closer to Mo than to the surface atoms (Figure 1b and Figure 1g). The H–metal distances exceed the H–Mo bond lengths by an average of 0.32 Å, and the H–Ni separations are 0.11 Å longer than the corresponding H–Cu distances (Table S2). Despite these larger distances, the summed DDEC6 H–metal bond orders remain comparable on both substrates (Figures S2a and S2b), indicating similar direct hydride–surface interactions.
The main difference between Cu and Ni lies in the Mo–H bonding framework. Greater charge transfer to Ni(111) is accompanied by an average 19.9% reduction in Mo–H bond order relative to Cu (range: 19.7–20.1%; Figure 3a). Consistent with this trend, the partial charge on Mo increases from +1.40 on Cu(111) to +1.67 on Ni(111), while hydride charges become less negative (−0.28 to −0.19; Tables S8 and S9), indicating reduced electron density in the Mo–H bonding framework.
These changes are reflected in the vibrational spectra. Eigenmode overlap analysis (Figure S4a) shows only moderate correspondence between the Mo–H stretching manifolds on the two surfaces, despite their broadly similar adsorption geometries. The Cu(111) modes at 1347, 1344, and 1341 cm-1 show high overlap with lower-frequency modes on Ni(111) at 763, 782, and 733 cm-1, with projection scores of 0.83, 0.71, and 0.65, respectively. The reduced Mo–H bond order on Ni is consistent with the observed red shifts in the Mo–H stretching modes. However, the large frequency difference (Δν ≈ 590 cm-1), together with only moderate eigenvector overlap, suggests appreciable reorganization of the Mo–H stretching manifold rather than simple preservation of mode character.
The Raman response is consistent with this interpretation (Figure 4a). The Mo–H stretching features paired based on the overlap analysis display large Cu/Ni Raman intensity contrasts: peak 0 versus 4 (1:80), peak 1 versus 3 (1:13), and peak 2 versus 5 (1:20). These intensity differences indicate substantial modification of the vibrational mode character on Ni, likely reflecting increased mixing between Mo–H stretching and H–metal motions.
3.3. Substrate-Dependent Redistribution of Mo–H Bonding and Weakly Perturbed O–H Modes in H3MoOH on Cu(111) and Ni(111)
A related but distinct pattern is observed for H3MoOH, which contains three hydride hydrogens and one protic hydrogen. The hydrides adopt similar geometries on both surfaces, remaining closer to Mo than to the surface (Figure 1c and Figure 1h). The H–metal separations exceed the H–Mo bond lengths by 0.56 Å on Ni and 0.72 Å on Cu. In contrast to H3Mo, the average H–Ni distance is 0.10 Å shorter than the corresponding H-Cu distance (Table S3), indicating closer hydride approach to Ni. The protic hydrogen shows the opposite trend, lying 1.0 Å farther from the surface on Ni(111) than on Cu(111).
The summed H–metal bond orders indicate slightly stronger hydride–surface interactions on Ni than on Cu (Figures S2a and S2b). In contrast, Mo–H bond orders decrease on Ni by an average of 12.9% relative to Cu (Figure 3b), consistent with redistribution of electron density toward H–Ni interactions. Accordingly, the partial charge on Mo increases from +0.95 on Cu(111) to +1.13 on Ni(111), while the hydride charges become slightly less negative (Tables S10 and S11), indicating reduced electron density in the Mo–H bonding framework.
Vibrational spectra further show pronounced Mo–H softening and Mo-H-metal coupling on Ni. Modes at 1487, 1429, and 1406 cm-1 on Cu correspond to modes at 902, 1027, and 952 cm-1 on Ni (Figure S4b), with projection scores of 0.77, 0.88, and 0.74. Displacement patterns suggest that these modes change from predominantly Mo–H stretches on Cu to coupled Mo–H–Ni vibrations on Ni. The modest Raman intensity ratios Cu/Ni (≈1:1–3:1; Figure 4b) together with the appreciable projection scores indicate partial preservation of mode character on both substrates.
The O–H stretching mode of the protic hydrogen remains largely unaffected by the surface because the proton resides relatively far from the slab (3.66 Å on Cu(111) and 4.66 Å on Ni(111); Table S3). The O–H bond order decreases only slightly on Ni relative to Cu (2.5%), as reflected by the modest red shift from 3544 cm-1 on Cu(111) to 3511 cm-1 on Ni(111) (Figure 3b and Figure 4b). The partial charge on the protic hydrogen increases slightly (+0.45 → +0.48, Tables S10 and S11), while the oxygen becomes marginally more negative, indicating only minor polarization changes within the O–H bond. The eigenmode projection score of 0.86 indicates substantial, though not complete, conservation of normal-mode character. Inspection of the adsorption geometries reveals that the O–H bond lies approximately parallel to the surface on Cu(111) but aligns more closely with the surface normal on Ni(111), providing a plausible origin for the reduced overlap. The Raman intensity ratio remains close to unity (≈1:1; Figure 4b), further supporting the assignment of analogous surface-distal O–H stretching modes on the two substrates.
3.4. Substrate-Dependent Symmetry Breaking and Loss of Vibrational Correspondence in H2Mo(OH)2 on Cu(111) and Ni(111)
H2Mo(OH)2 adopts markedly different adsorption geometries on the two substrates (Figure 1d and Figure 1i). On Cu(111), the hydroxyl hydrogens lie on opposite sides of the Mo center, producing inequivalent hydroxyl groups. One oxygen interacts strongly with the surface (bond order 0.28), while the second interacts weakly (bond order < 0.05) (Figure S2c). On Ni(111), both hydroxyl groups lie on the same side of the complex, and both OH oxygens lie at similar heights above the surface, interacting weakly and symmetrically with the slab (< 0.10; Figure S2d). The centroid heights capture this structural contrast (Figure 2a), with the Cu/Ni separation increasing to 0.18 Å compared with 0.13 Å for the preceding complexes.
The atomic charge distribution follows the same pattern (Tables S12 and S13). On Cu(111), the two hydroxyl groups exhibit distinct charges (O: −0.67 and −0.73; H: +0.44 and +0.41), consistent with the stronger interaction of one oxygen with the surface. In contrast, the corresponding charges on Ni(111) are identical for the two hydroxyl groups (O: −0.71 and −0.71; H: +0.45 and +0.45), reflecting the more symmetric adsorption geometry. In both cases, the Mo center becomes more positively charged on Ni (1.24 → 1.38), indicating greater electron transfer to the Ni surface.
Because of these structural differences, O–H stretching manifolds cannot be consistently mapped between the two substrates. Modes at 3584 and 3546 cm-1 on Cu correspond poorly to modes on Ni at 3599 and 3622 cm-1, with projection scores of 0.19 and 0.53 (Figure S4c). Their Raman activities also differ significantly (Cu/Ni ≈ 1:6; Figure 4c), reflecting the distinct adsorption geometries.
3.5. Substrate-Dependent Redistribution of Mo–OH Bonding and Mode-Dependent O–H Vibrational Response in MoO(OH)3 on Cu(111) and Ni(111)
MoO(OH)3 adsorption geometries remain broadly similar on both substrates (Figure 1e and Figure 1j), although they exhibit the largest centroid-height difference in the series (0.29 Å; Figure 2a). The atomic charge distribution indicates that the overall electronic differences between the two substrates are modest for this complex (Tables S14 and S15). The Mo center becomes only slightly more positively charged on Ni (1.58 → 1.68), while the average oxygen charge changes only marginally (−0.69 → −0.72), consistent with the relatively small difference in charge transfer between Cu and Ni (Figure 2c).
The main distinction arises from the orientation of the O–H groups and the resulting redistribution of electron density within the Mo–OH framework. The surface-proximal O–H groups associated with bonds B1 and B2 in Figure 3d point in opposite directions on Cu, but align in the same direction on Ni (Figure 1e and Figure 1j).
In both adsorption geometries, three of the four oxygen atoms interact with the slab through the hydroxyl groups (bond orders 0.34 and 0.37 on Cu versus 0.47 and 0.47 on Ni), whereas the terminal Mo=O oxygen remains comparatively weakly coupled (0.24 on Cu versus 0.13 on Ni) (Figures S2c and S2d). The summed O–metal bond orders are more uneven on Cu than on Ni, indicating more localized adsorbate–surface interactions on Cu. In contrast, the nearly identical O–metal bond orders on Ni reflect more evenly distributed hybridization between the hydroxyl oxygens and the surface d-states, consistent with stronger adsorbate–metal coupling. The weaker hybridization on Cu is partially compensated by the shorter adsorption distance, as reflected in the smaller centroid height on Cu relative to Ni (Figure 2a).
This redistribution of bonding is reflected in the vibrational behavior of the two surface-proximal O–H bonds. One O–H bond is slightly softened on Cu (4.7%), whereas the other is softened on Ni (2.6%) (Figure 3d). The synchronous surface-proximal O–H stretching mode (3576 cm-1 on Cu and 3586 cm-1 on Ni) involves in-phase motion of these two bonds and exhibits low eigenmode overlap (projection score 0.25; Figure S4d), reflecting its sensitivity to the different orientations of the two O–H bonds. In contrast, the asynchronous stretch (3595 cm-1 on Cu → 3568 cm-1 on Ni) involves out-of-phase motion and retains higher overlap (projection score 0.73). Notably, the synchronous mode exhibits a blue shift whereas the asynchronous mode displays a red shift, consistent with asymmetric softening of the two O–H bonds across the substrates (Figure 3d).
The surface-distal O–H stretch associated with bond B3 shows the strongest correspondence between the two substrates: mode 0 at 3623 cm-1 on Cu corresponds to mode 1 at 3572 cm-1 on Ni with a projection score of 0.97. Despite only minor bond softening on Ni (1.2%), a red shift of 51 cm-1 is nevertheless observed. The Raman intensity ratio for these modes is 3:1 (Figure 4d), consistent with the assignment of analogous surface-distal O–H stretching vibrations.
3.6. Substrate-Dependent Bonding and Vibrational Trends Across Mo Complexes on Cu(111) and Ni(111)
Across the series, combined geometric, electronic, and vibrational descriptors reveal a consistent substrate-dependent bonding pattern. Global structural metrics indicate broadly similar adsorption geometries on Cu(111) and Ni(111), with only modest differences in centroid height and molecular size. The principal distinctions instead emerge from local bonding descriptors: bond orders and atomic charges show stronger and more evenly distributed adsorbate–metal hybridization on Ni, whereas bonding on Cu is more localized and partially compensated by shorter adsorption distances. These electronic differences redistribute bonding within the Mo–ligand framework and are captured directly in the vibrational analysis, where projection scores quantify the conservation of normal modes between substrates and Raman intensities reflect changes in vibrational character associated with these bonding changes. This behavior manifests as preferential softening of Mo–H and O–H bonds on Ni, while Cu more strongly perturbs the Mo–O framework, as illustrated in Figure 3c, where Cu weakens one Mo–O bond of H2Mo(OH)2 more strongly than Ni, and in Figure 3d, where three of the four Mo–O bonds in MoO(OH)3 are more strongly perturbed on Cu.
3.7. Field-Dependent Thermodynamics and Electrochemical Stability of Mo Complexes on Cu(111) and Ni(111)
Relative Gibbs free energies () under HER-relevant potential conditions were evaluated for H3Mo, H3MoOH, H2Mo(OH)2, and MoO(OH)3 adsorbed on Ni(111). The results (Table S16) are in good agreement with those reported by Bau et al. [13], with deviations below 0.06 eV for all species except H3Mo, for which a difference of 0.22 eV is observed. Re-evaluation using both three- and five-layer slab models yields consistent adsorption geometries, with H3Mo adopting a trigonal pyramidal structure in which the hydride H atoms point toward the surface and the Mo center resides at the apex (Figure 1g). The discrepancy with the structure reported by Bau et al. [13], which features one outward-pointing hydride, likely accounts for the energetic difference.
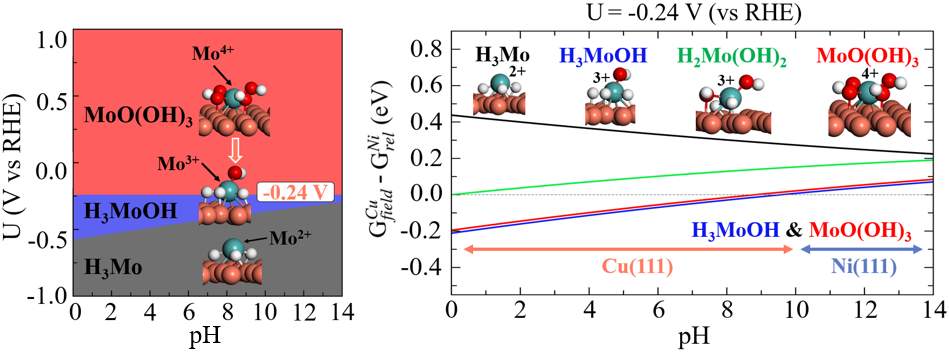
Comparison of surface Pourbaix diagrams for Ni(111) and Cu(111) (Figure 5) establishes the baseline thermodynamic differences between the two substrates. H3MoOH and MoO(OH)3 exhibit nearly identical stability on both surfaces (differences < 0.05 eV), whereas H3Mo and H2Mo(OH)2 are stabilized more strongly on Ni by 0.30 and 0.13 eV, respectively. Across the explored pH–potential space, at least one of H3Mo, H3MoOH, or MoO(OH)3 is thermodynamically preferred over H2Mo(OH)2, effectively restricting the accessible redox manifold to these three species. These baseline differences define a static thermodynamic landscape; however, under electrochemical conditions, interfacial electric fields can further modulate the relative stability of these states.
To assess how these thermodynamic trends evolve under electrochemical conditions, the response of the metal–adsorbate interface to an applied electric field was examined. Charge redistribution was quantified at nine values of the external electric field between −0.8 and 0.8 V Å-1 as
where is the sum of the DDEC6 atomic charges on the metal slab. Thus, quantifies the field-induced change in slab charge relative to the zero-field configuration.
On Cu(111), exhibits a quasi-linear dependence on the applied field within the weak-field regime (−0.4 to 0.4 V Å-1), spanning −0.33 to 0.35 e for Mo (minimum–maximum values within this window), while pronounced nonlinearity emerges beyond ±0.6 V Å-1 (Figure 6a and Figure 6b). In contrast, on Ni(111), remains effectively zero over the entire field range (−0.8 to 0.8 V Å-1) for all systems, with variations within the numerical resolution of the charge analysis (|ΔQ| < 0.01 e) [40].
This contrast reflects fundamental differences in adsorbate–surface bonding. Within the d-band framework [55,58], the Ni(111) d-band center lying closer to the Fermi level than that of Cu(111) suggests enhanced adsorbate–surface hybridization. The resulting larger adsorption-induced charge transfer on Ni (Figure 2c) yields a more strongly polarized interface and, consequently, diminished sensitivity to external perturbations. This insensitivity may further be reinforced by a more delocalized electronic coupling on Ni, contributing to field screening. In contrast, the less polarized, more localized bonding on Cu may result in greater susceptibility to field-induced charge redistribution. This interpretation remains qualitative, as the present work does not directly resolve the microscopic origin of the screening response.
Consistent with this contrasting charge response, two distinct energetic regimes emerge in the field-dependent free energies. On Ni(111), the relative Gibbs free energies remain invariant with applied field, yielding μ and α that are effectively zero within numerical precision. On Cu(111), in contrast, displays a clear field dependence.
Quadratic fitting of the field-dependent energies using (4) over the full ±0.8 V Å-1 field range yields μ values of 0.15–0.24 eÅ for all Cu-supported complexes. The corresponding α values are approximately 0.12 eÅ2V-1 for H3MoOH, H2Mo(OH)2, and MoO(OH)3, and lower (0.09 eÅ2V-1) for H3Mo (insets of Figure 6c and Figure 6d). Notably, H3Mo exhibits dipole and polarizability values of opposite sign relative to the other complexes, defining two distinct electrostatic response regimes. These parameters, extracted from the field-dependent form of represent effective descriptors of interfacial electrostatic coupling rather than intrinsic molecular properties.
To assess the robustness of the field-correction procedure, additional fits were performed over the narrower ±0.4 and ±0.6 V Å-1 field ranges (Figure S5), motivated by the quasi-linear charge response on Cu(111) within ±0.4 V Å-1. Although the magnitudes of μ and α vary modestly, the resulting 2D Pourbaix diagrams (Figure S6) and cross-metal trends remain unchanged (Figures S7 and S8). Similarly, varying the Helmholtz capacitance CH from 0.20 to 0.60 F/m2 [20] alters the magnitude of the field-induced shifts by up to 0.45 eV but does not affect their direction or relative ordering (Table S17). The field-induced shifts in relative Gibbs free energy discussed in the present work were computed using CH = 0.25 F/m2 following Kelly et al. [20].
The thermodynamic impact of electrostatic coupling is illustrated in Figure 7, which compares zero-field relative Gibbs free energies (dotted lines) with field-corrected profiles (solid lines) on Cu(111). shifts reach up to 0.28 eV across the examined pH range, stabilizing H3Mo and destabilizing oxygenated species under alkaline conditions. In contrast, Ni(111) exhibits negligible field-induced changes, reflecting its screened interfacial response.
The magnitude of these field-induced shifts on Cu is comparable to the intrinsic substrate-dependent differences observed at zero field (e.g., 0.30 eV for H3Mo), demonstrating that electrostatic effects constitute a thermodynamic contribution on par with substrate identity. Thus, the relative stability of Mo intermediates on Cu is not fixed but evolves dynamically with pH and potential.
The resulting changes in relative stability are reflected in the 2D Pourbaix diagram (Figure S6). At pH 0, the stability sequence with increasing potential is H3Mo → H3MoOH → MoO(OH)3, while H2Mo(OH)2 exhibits no stability domain despite negative values. Across all conditions, at least one of the three dominant species is more stable than H2Mo(OH)2, indicating that this complex is not thermodynamically preferred, although its potential kinetic accessibility cannot be ruled out.
With increasing pH, the stability region of H3Mo expands, whereas that of H3MoOH contracts. The H3Mo/H3MoOH boundary shifts with both pH and potential, whereas the H3MoOH/MoO(OH)3 boundary remains horizontal, indicating negligible pH sensitivity of that transition. Within the double-layer capacitor framework (6), the pH dependence of the stability boundaries arises from pH-induced modulation of the interfacial electric field and its coupling to the adsorption-induced dipole moment and polarizability (4). At the H3MoOH/MoO(OH)3 boundary, the condition leads to a quadratic expression in the potential U and pH through the electric field terms E and E2, whose coefficients are Δµ and Δα, respectively. Because the fitted dipole moments and polarizabilities of the two complexes are very similar (μ ≈ 0.23 eÅ, α ≈ 0.12 eÅ2V-1, Figure 6c), the coefficients associated with the field-dependent, and therefore pH-dependent, terms are close to zero. As a result, the boundary effectively reduces to a function of potential only, producing the observed horizontal line. In contrast, the distinct electrostatic responses of H3Mo and H3MoOH generate finite field-coupling terms, leading to the oblique, pH-dependent H3Mo/H3MoOH boundary.
Overall, these results demonstrate that Cu introduces a field-sensitive thermodynamic response absent on Ni, enabling electrostatic modulation of Mo-centered redox equilibria under operating conditions. The magnitude of this effect is comparable to intrinsic substrate-dependent stabilization, establishing electrostatic coupling as a key factor in determining the thermodynamic landscape of Mo-based HER intermediates.
3.8. Contrasting Roles of Ni and Cu in Governing Mo-Centered Redox Thermodynamics and Reactivity in Ni–Mo HER
In the Mo3+–hydride framework proposed by Bau et al. [13,14,15], HER proceeds through interconversion of oxygenated Mo4+ precursors and reduced hydride-containing Mo3+ species under cathodic bias, with the metallic substrate acting primarily as a modulator of relative state stability rather than as an independent catalytic center. In the following paragraphs, reactivity within this framework is discussed indirectly through trends in thermodynamic stabilization, bond softening, and field response, rather than through explicit transition-state calculations.
Under zero-field conditions, adsorption energetics establish a clear baseline distinction between the two substrates. Hydrogen-rich intermediates are systematically more stable on Ni(111), as reflected in adsorption-energy differences of −0.67 eV for H3Mo, −0.35 eV for H3MoOH, and −0.52 eV for H2Mo(OH)2 relative to Cu(111) (Figure 2d), and in the greater stability of H3Mo and H2Mo(OH)2 by 0.30 and 0.13 eV, respectively, in relative Gibbs free energy (Figure 5). While these trends are primarily electronic in origin, structural factors modulate their magnitude: H3Mo, which exhibits a 6.4% expansion on Ni (Figure 2b), and H2Mo(OH)2, which adopts distinct symmetries on Ni and Cu (Figure 1d and Figure 1i), show the largest adsorption-energy differences, whereas H3MoOH and MoO(OH)3, which retain similar geometries on both surfaces (Figure 1e and Figure 1j), exhibit smaller gaps (−0.35 and −0.45 eV, respectively). This stronger stabilization is accompanied by enhanced bond softening: all three Mo–H bonds in H3Mo are weakened by approximately 20% on Ni relative to Cu, those in H3MoOH by approximately 13%, and those in H2Mo(OH)2 by approximately 2%, with O–H bonds following a similar but smaller trend (Figure 3a–c). These results indicate that Ni stabilizes hydrogen-rich intermediates while simultaneously increasing Mo–H lability within the reduced manifold, demonstrating that stronger adsorption does not imply a more rigid hydride framework.
In contrast, Cu perturbs the oxygen-coordinated manifold more strongly. Bond-softening analysis shows that three of the four Mo–O bonds in MoO(OH)3 are weakened more strongly on Cu than on Ni, and that in H2Mo(OH)2 the Mo–O response is asymmetric, with one bond weakened more strongly on Cu (20.6%) and the other on Ni (12.5%) (Figure 3c and Figure 3d), consistent with stronger destabilization of oxygen-bound configurations on Cu. This effect is reflected in adsorption energetics: MoO(OH)3 is less stable than H3MoOH by 0.82 eV on Cu, compared to 0.71 eV on Ni, indicating a slightly larger energetic separation between oxidized and reduced states on Cu. Despite this difference, the relative Gibbs free energies of both species remain nearly identical across the two substrates (Figure 5), with cross-metal differences below 0.03 eV. This apparent insensitivity arises from the definition of , which references adsorbed Mo and therefore cancels part of the substrate-dependent baseline stabilization. Adsorption energies thus more clearly capture the substrate effect, revealing that Cu perturbs the internal thermodynamic spacing of the Mo4+/Mo3+ couple more strongly than Ni, consistent with the Cu–Mo redox cooperation proposed by Nishimoto et al. [1].
These static trends define a thermodynamic manifold in which the relevant competition is effectively confined to H3Mo, H3MoOH, and MoO(OH)3. Within this landscape, H2Mo(OH)2 occupies an intermediate position on the adsorption-energy ladder between MoO(OH)3 and H3MoOH (Figure 2d) and exhibits mixed bonding characteristics, combining weak Mo–H softening with asymmetric Mo–O perturbation (Figure 3c). At the same time, its relative Gibbs free energy remains nearly potential-independent, appearing as horizontal lines in the 1D Pourbaix diagrams (Figure 5). These features suggest that H2Mo(OH)2 may act as a thermodynamic bridge between oxygenated and hydride states, although its precise role within the operative HER mechanism cannot be established without explicit transition-state analysis.
The distinction between the two substrates becomes more pronounced when electrostatic effects are considered. The thermodynamic separation between Cu(111) and Ni(111) is strongly modulated by potential and pH through the interfacial electric field, with a markedly stronger response on Cu than on Ni. This behavior can be rationalized in terms of adsorbate–surface bonding: Cu exhibits more localized and uneven interactions, whereas Ni displays more distributed bonding and stronger hybridization, which enhances baseline stabilization but reduces sensitivity to external perturbations.
This heightened field sensitivity on Cu strongly influences the accessible Mo redox manifold, which, based on the oxidation-state assignments of Bau et al. [13], includes MoO(OH)3 (Mo4+), H3MoOH and H2Mo(OH)2 (Mo3+), and H3Mo (Mo2+). However, the surface Pourbaix diagram on Cu(111) (Figure S6) narrows this space to MoO(OH)3, H3MoOH, and H3Mo, with H2Mo(OH)2 excluded under relevant conditions. Because the Mo4+/Mo3+ interconversion underpins the Mo3+–hydride mechanism, the analysis centers on the MoO(OH)3/H3MoOH redox couple. Figure S8 shows that this pair undergoes pronounced potential- and pH-dependent stabilization relative to Ni, leading to a crossover in which Cu becomes thermodynamically favored: at −0.40 V versus RHE, this preference is limited to acidic to near-neutral conditions (pH < 7; Figure S8a), whereas at −0.10 V it extends up to pH ≈ 12 (Figure S8d). These trends demonstrate that the Mo4+/Mo3+ thermodynamic balance is field-tunable on Cu. Consistent with this picture, enhanced performance for Ni–Mo(Cu) catalysts under moderately alkaline environments has been reported [1], suggesting that such electrostatic modulation may be relevant under operando conditions.
Although increasing pH stabilizes H3Mo on Cu, the field perturbation is insufficient to overcome the substantial baseline thermodynamic gap (Figures S7 and S8), indicating that electrostatic effects modulate but do not fully override the stronger intrinsic stabilization on Ni. H2Mo(OH)2 follows a similar trend, remaining favored on Ni across most conditions, except within a narrow regime at −0.10 V and pH < 2.4 (Figure S8d), where it becomes more stable on Cu.
Taken together, these results show that Ni and Cu influence the Mo-centered HER manifold through distinct yet complementary mechanisms. Ni primarily stabilizes hydrogen-rich intermediates through strong electronic coupling while enhancing Mo–H bond lability, whereas Cu selectively destabilizes oxygen-coordinated states and enables field-dependent tuning of the Mo4+/Mo3+ thermodynamic balance. Whether these thermodynamic and bonding differences translate into altered hydride-transfer kinetics within the Mo3+-centered HER mechanism [13,14,15], or into changes in hydrogen-evolution rates, cannot be inferred from the present analysis and will require explicit free-energy and transition-state calculations.
4. Conclusions
Mo complexes adsorbed on Cu(111) and Ni(111) exhibit substrate-dependent electronic structure and thermodynamic behavior that extends beyond simple differences in adsorption strength to include electrostatic modulation of redox energetics. Although Ni(111) promotes greater interfacial charge transfer and stronger overall adsorption, the key thermodynamic distinction lies in the electrochemical response of the Mo-centered redox states. Across the examined pH–potential range, at least one of H3Mo, H3MoOH, or MoO(OH)3 remains thermodynamically favored over H2Mo(OH)2 on both substrates. However, only Cu(111) displays measurable pH-dependent field-corrected relative Gibbs free energy shifts on the RHE scale, reaching 0.28 eV. The magnitude of this field-driven modulation is comparable to the intrinsic substrate-induced differences in zero-field relative Gibbs free energy, indicating that Cu incorporation does more than tune hydrogen binding and also reshape the Mo redox landscape through its electrostatic response.
Preferential Mo–O bond weakening on Cu(111) is consistent with greater thermodynamic destabilization of the oxidized MoO(OH)3 state relative to the reduced H3MoOH state, thereby increasing the adsorption-energy separation between these redox states compared to Ni(111). In contrast, preferential softening of Mo–H and O–H bonds on Ni(111) indicates enhanced hydrogen-ligand lability despite stronger adsorption stabilization, reflecting more extensive redistribution of charge within the Mo–ligand framework. Vibrational analysis further shows that even modest geometric differences can reorganize surface-proximal normal modes through substrate-specific bond-order redistribution, whereas surface-distal O–H modes remain largely conserved across the two surfaces.
Collectively, these results establish that Cu modifies Mo-centered redox thermodynamics through electrostatic field effects whose magnitude is comparable to intrinsic substrate differences, while Ni primarily stabilizes hydrogen-rich intermediates and enhances Mo–H bond lability. These findings imply that substrate identity governs not only the static ordering of Mo oxidation states but also their field sensitivity under operating conditions, underscoring the need for explicit free-energy and transition-state analyses to assess the kinetic consequences for hydride transfer and hydrogen-evolution rates in Cu-containing Ni–Mo HER systems.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, D.R. and T.I.; methodology, D.R., E.K., Y.K. and T.I; software, E.K.; validation, D.R., Y.K. and T.I.; formal analysis, E.K.; investigation, E.K.; resources, T.I.; data curation, E.K.; writing—original draft preparation, E.K.; writing—review and editing, D.R.; visualization, E.K.; supervision, T.I.; project administration, T.I.; funding acquisition, T.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Japan Science and Technology Agency (JST) through the GteX (Green Technologies of Excellence) Program, grant number JPMJGX23H2.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| 1D | One-dimensional |
| 2D | Two-dimensional |
| CHE | Computational Hydrogen Electrode |
| DDEC | Density Derived Electrostatic and Chemical (method) |
| DFT | Density Functional Theory (method) |
| HER | Hydrogen Evolution Reaction |
| PBE | Perdew–Burke–Ernzerhof (functional) |
| RHE | Reversible Hydrogen Electrode |
| RPBE | Revised Perdew–Burke–Ernzerhof (functional) |
| SHE | Standard Hydrogen Electrode |
| VASP | Vienna Ab initio Simulation Package |
References
- Nishimoto, T.; Obata, K.; Komiya, H.; Naito, T.; Harada, K.; Yoshida, M.; Takanabe, K. Oxidized Copper and Molybdenum Species Exclusively Boosting Electrocatalytic Hydrogen Evolution in Non-Extreme pH Carbonate Buffer Electrolyte. ACS Catal. 2023, 13, 14725–14736. [Google Scholar] [CrossRef]
- Miao, F.; Cui, P.; Yu, S.; Gu, T. In situ fabrication of a 3D self-supported porous Ni–Mo–Cu catalyst for an efficient hydrogen evolution reaction. Dalton Trans. 2023, 52, 8654–8660. [Google Scholar] [CrossRef]
- Wu, X.-F.; Li, Z.-Y.; Wang, H.; Wang, J.-C.; Xi, G.-Q.; Zhao, X.-J.; Zhang, C.-X.; Liao, W.-G.; Ho, J.C. Defect-Engineered Multi-Intermetallic Heterostructures as Multisite Electrocatalysts for Efficient Water Splitting. Advanced Science 2025, 12, 2502244. [Google Scholar] [CrossRef]
- Luo, M.; Yang, J.; Li, X.; Eguchi, M.; Yamauchi, Y.; Wang, Z.-L. Insights into alloy/oxide or hydroxide interfaces in Ni–Mo-based electrocatalysts for hydrogen evolution under alkaline conditions. Chem. Sci. 2023, 14, 3400–3414. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Shi, Y.; Zhou, W.; Yu, Y.; Zhang, B. Unveiling the In Situ Dissolution and Polymerization of Mo in Ni4 Mo Alloy for Promoting the Hydrogen Evolution Reaction. Angew Chem Int Ed 2021, 60, 7051–7055. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Shi, C.; Guo, X.; He, Z.; Pan, L.; Huang, Z.-F.; Zhang, X.; Zou, J.-J. Rational Design of Better Hydrogen Evolution Electrocatalysts for Water Splitting: A Review. Advanced Science 2022, 9, 2200307. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, Y.; Zhao, Z.; Zhang, Q.-H.; Huang, L.-B.; Gu, L.; Lu, G.; Hu, J.-S.; Wan, L.-J. Steering elementary steps towards efficient alkaline hydrogen evolution via size-dependent Ni/NiO nanoscale heterosurfaces. Natl Sci Rev 2020, 7, 27–36. [Google Scholar] [CrossRef]
- Shen, L.; Lu, B.; Li, Y.; Liu, J.; Huang-fu, Z.; Peng, H.; Ye, J.; Qu, X.; Zhang, J.; Li, G.; Cai, W.; Jiang, Y.; Sun, S. Interfacial Structure of Water as a New Descriptor of the Hydrogen Evolution Reaction. Angewandte Chemie International Edition 2020, 59, 22397–22402. [Google Scholar] [CrossRef]
- Liu, E.; Qin, X.; Delmo, E.P.; Shao, M.; Vegge, T.; Drisdell, W.S. Discovering the Catalytic Role of Interfacial Water Towards Hydrogen Evolution/Oxidation Reactions in Aqueous Solutions, Meet. Abstr. MA2024-02. 2024, 2822. [Google Scholar] [CrossRef]
- Sun, M.; Staykov, A.; Yamauchi, M. Understanding the Roles of Hydroxide in CO2 Electroreduction on a Cu Electrode for Achieving Variable Selectivity. ACS Catal. 2022, 12, 14856–14863. [Google Scholar] [CrossRef]
- Guha, A.; Sahoo, M.; Alam, K.; Rao, D.K.; Sen, P.; Narayanan, T.N. Role of water structure in alkaline water electrolysis. iScience 2022, 25, 104835. [Google Scholar] [CrossRef]
- Zhang; Yao, J.; Liu, J.; Zhang, T.; Wan, H.; Wang, H. Reducing the pH dependence of hydrogen evolution kinetics via surface reactivity diversity in medium-entropy alloys. EES Catal. 2023, 1, 1017–1024. [Google Scholar] [CrossRef]
- Bau, J.A.; Kozlov, S.M.; Azofra, L.M.; Ould-Chikh, S.; Emwas, A.-H.; Idriss, H.; Cavallo, L.; Takanabe, K. Role of Oxidized Mo Species on the Active Surface of Ni–Mo Electrocatalysts for Hydrogen Evolution under Alkaline Conditions. ACS Catal. 2020, 10, 12858–12866. [Google Scholar] [CrossRef]
- Bau, J.A.; Ahmad, R.; Cavallo, L.; Rueping, M. A Unified Theory for H2 Evolution on Mo-Based Electrocatalysts. ACS Energy Lett. 2022, 7, 3695–3702. [Google Scholar] [CrossRef]
- Bau, J.A.; Emwas, A.-H.; Nikolaienko, P.; Aljarb, A.A.; Tung, V.; Rueping, M. Mo3+ hydride as the common origin of H2 evolution and selective NADH regeneration in molybdenum sulfide electrocatalysts. Nat Catal 2022, 5, 397–404. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, J.; Zhang, W.; Wu, A. Growth of Ni/Mo/Cu on carbon fiber paper: An efficient electrocatalyst for hydrogen evolution reaction. International Journal of Hydrogen Energy 2021, 46, 35550–35558. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Z.; Oliveira, A.M.; Xi, S.; Zhiani, M.; Zhang, J.; Tu, Z.; Xiao, F.; Wang, S.; Yan, Y.; Xiao, J. Accelerating Hydrogen Desorption of Nickel Molybdenum Cathode via Copper Modulation for Pure-Water-Fed Hydroxide Exchange Membrane Electrolyzer. Advanced Functional Materials 2024, 34, 2313275. [Google Scholar] [CrossRef]
- Hansen, H.A.; Rossmeisl, J.; Nørskov, J.K. Surface Pourbaix diagrams and oxygen reduction activity of Pt, Ag and Ni(111) surfaces studied by DFT. Phys. Chem. Chem. Phys. 2008, 10, 3722–3730. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Kelly, S.R.; Kirk, C.; Chan, K.; Nørskov, J.K. Electric Field Effects in Oxygen Reduction Kinetics: Rationalizing pH Dependence at the Pt(111), Au(111), and Au(100) Electrodes. J. Phys. Chem. C 2020, 124, 14581–14591. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, D.; Wang, Y.; Li, H. Reversible Hydrogen Electrode (RHE) Scale Dependent Surface Pourbaix Diagram at Different pH. Langmuir 2024, 40, 7632–7638. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational Materials Science 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Wellendorff, J.; Silbaugh, T.L.; Garcia-Pintos, D.; Nørskov, J.K.; Bligaard, T.; Studt, F.; Campbell, C.T. A benchmark database for adsorption bond energies to transition metal surfaces and comparison to selected DFT functionals. Surface Science 2015, 640, 36–44. [Google Scholar] [CrossRef]
- Tonigold, K.; Groß, A. Dispersive interactions in water bilayers at metallic surfaces: A comparison of the PBE and RPBE functional including semiempirical dispersion corrections. J Comput Chem 2012, 33, 695–701. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Lutfalla, S.; Shapovalov, V.; Bell, A.T. Calibration of the DFT/GGA+U Method for Determination of Reduction Energies for Transition and Rare Earth Metal Oxides of Ti, V, Mo, and Ce. J. Chem. Theory Comput. 2011, 7, 2218–2223. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA + U framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 2010, 132, 154104. [Google Scholar] [CrossRef]
- Mathew, K.; Sundararaman, R.; Letchworth-Weaver, K.; Arias, T.A.; Hennig, R.G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106. [Google Scholar] [CrossRef] [PubMed]
- Mathew, K.; Kolluru, V.S.C.; Mula, S.; Steinmann, S.N.; Hennig, R.G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 2019, 151, 234101. [Google Scholar] [CrossRef] [PubMed]
- Archer, D.G.; Wang, P. The Dielectric Constant of Water and Debye-Hückel Limiting Law Slopes. Journal of Physical and Chemical Reference Data 1990, 19, 371–411. [Google Scholar] [CrossRef]
- Manz, T.A.; Limas, N.G. DDEC6: A Method for Computing Even-Tempered Net Atomic Charges in Periodic and Nonperiodic Materials. 2015. [Google Scholar] [CrossRef]
- Limas, N.G.; Manz, T.A. Introducing DDEC6 atomic population analysis: part 4. Efficient parallel computation of net atomic charges, atomic spin moments, bond orders, and more. RSC Adv. 2018, 8, 2678–2707. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Computer Physics Communications 2021, 267, 108033. [Google Scholar] [CrossRef]
- Trasatti, S. The Absolute Electrode Potential: An Explanatory Note: (Recommendations 1986). n.d. [Google Scholar] [CrossRef]
- Jerkiewicz, G. Standard and Reversible Hydrogen Electrodes: Theory, Design, Operation, and Applications. ACS Catal. 2020, 10, 8409–8417. [Google Scholar] [CrossRef]
- Singh, A.K.; Zhou, L.; Shinde, A.; Suram, S.K.; Montoya, J.H.; Winston, D.; Gregoire, J.M.; Persson, K.A. Electrochemical Stability of Metastable Materials. Chem. Mater. 2017, 29, 10159–10167. [Google Scholar] [CrossRef]
- Łukomska, A.; Sobkowski, J. Potential of zero charge of monocrystalline copper electrodes in perchlorate solutions. Journal of Electroanalytical Chemistry 2004, 567, 95–102. [Google Scholar] [CrossRef]
- Ohmori, T. MEASUREMENT OF THE POTENTIAL OF ZERO-CHARGE ON NICKEL ELECTRODE BY THE GALVANOSTATIC TRANSIENT METHOD. n.d. [Google Scholar]
- Porezag, D.; Pederson, M.R. Infrared intensities and Raman-scattering activities within density-functional theory. Phys. Rev. B 1996, 54, 7830–7836. [Google Scholar] [CrossRef]
- Baroni, S.; De Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Gonze, X. Perturbation expansion of variational principles at arbitrary order. Phys. Rev. A 1995, 52, 1086–1095. [Google Scholar] [CrossRef]
- Fonari, A.; Stauffer, S. vasp_raman.py. 2013. Available online: https://github.com/raman-sc/VASP/.
- Lazzeri, M.; Mauri, F. First-Principles Calculation of Vibrational Raman Spectra in Large Systems: Signature of Small Rings in Crystalline ${\mathrm{S}\mathrm{i}\mathrm{O}}_{2}$. Phys. Rev. Lett. 2003, 90, 036401. [Google Scholar] [CrossRef]
- Kabsch, W. A solution for the best rotation to relate two sets of vectors. Acta Cryst A 1976, 32, 922–923. [Google Scholar] [CrossRef]
- Yu, M.; Moayedpour, S.; Yang, S.; Dardzinski, D.; Wu, C.; Pribiag, V.S.; Marom, N. Dependence of the electronic structure of the EuS/InAs interface on the bonding configuration. Phys. Rev. Materials 2021, 5, 064606. [Google Scholar] [CrossRef]
- Ruban, A.; Hammer, B.; Stoltze, P.; Skriver, H.L.; Nørskov, J.K. Surface electronic structure and reactivity of transition and noble metals1. Journal of Molecular Catalysis A: Chemical 1997, 115, 421–429. [Google Scholar] [CrossRef]
- Hammer, J.K. Norskov, Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Hammer; Morikawa, Y.; Nørskov, J.K. CO Chemisorption at Metal Surfaces and Overlayers. Phys. Rev. Lett. 1996, 76, 2141–2144. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Waghmare, U.V.; Lee, S.-C. An improved d-band model of the catalytic activity of magnetic transition metal surfaces. Sci Rep 2016, 6, 35916. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surface Science 1995, 343, 211–220. [Google Scholar] [CrossRef]
Figure 1.
Top and side views of the most stable adsorption configurations of Mo (a, f); H3Mo (b, g); H3MoOH (c, h); H2Mo(OH)2 (d, i); and MoO(OH)3 (e, j) on Cu(111) (a–e) and Ni(111) (f–j). Copper, nickel, molybdenum, oxygen, and hydrogen atoms are shown as orange, blue, teal, red, and white spheres, respectively. Atomic radii are not to scale.
Figure 1.
Top and side views of the most stable adsorption configurations of Mo (a, f); H3Mo (b, g); H3MoOH (c, h); H2Mo(OH)2 (d, i); and MoO(OH)3 (e, j) on Cu(111) (a–e) and Ni(111) (f–j). Copper, nickel, molybdenum, oxygen, and hydrogen atoms are shown as orange, blue, teal, red, and white spheres, respectively. Atomic radii are not to scale.


Figure 2.
(a) Adsorbate-centroid height relative to the average z-coordinate of the top surface layer for Cu(111) and Ni(111); (b) Relative adsorbate expansion upon adsorption on Cu(111) and Ni(111), quantified by the radius of gyration (); (c) Charge transfer to the slab upon adsorption of Mo complexes on Cu(111) and Ni(111); (d) Adsorption energies of Mo complexes on Cu(111) and Ni(111). Orange and blue denote adsorption on Cu(111) and Ni(111), respectively.
Figure 2.
(a) Adsorbate-centroid height relative to the average z-coordinate of the top surface layer for Cu(111) and Ni(111); (b) Relative adsorbate expansion upon adsorption on Cu(111) and Ni(111), quantified by the radius of gyration (); (c) Charge transfer to the slab upon adsorption of Mo complexes on Cu(111) and Ni(111); (d) Adsorption energies of Mo complexes on Cu(111) and Ni(111). Orange and blue denote adsorption on Cu(111) and Ni(111), respectively.

Figure 3.
Changes in DDEC6 bond orders (ΔBO) of internal bonds in Mo complexes upon adsorption on Cu(111) and Ni(111): (a) H3Mo; (b) H3MoOH; (c) H2Mo(OH)2; and (d) MoO(OH)3. Bond “softening” denotes a decrease in bond order upon adsorption. Orange bars indicate greater bond softening on Cu(111), whereas blue bars indicate greater bond softening on Ni(111).
Figure 3.
Changes in DDEC6 bond orders (ΔBO) of internal bonds in Mo complexes upon adsorption on Cu(111) and Ni(111): (a) H3Mo; (b) H3MoOH; (c) H2Mo(OH)2; and (d) MoO(OH)3. Bond “softening” denotes a decrease in bond order upon adsorption. Orange bars indicate greater bond softening on Cu(111), whereas blue bars indicate greater bond softening on Ni(111).

Figure 4.
Computed Raman spectra of Mo complexes adsorbed on Cu(111) (orange) and Ni(111) (blue): (a) H3Mo, (b) H3MoOH, (c) H2Mo(OH)2, and (d) MoO(OH)3. Numerical labels denote the vibrational mode indices corresponding to the peaks, as assigned from the overlap matrices (Figure S4). Intensities are normalized.
Figure 4.
Computed Raman spectra of Mo complexes adsorbed on Cu(111) (orange) and Ni(111) (blue): (a) H3Mo, (b) H3MoOH, (c) H2Mo(OH)2, and (d) MoO(OH)3. Numerical labels denote the vibrational mode indices corresponding to the peaks, as assigned from the overlap matrices (Figure S4). Intensities are normalized.


Figure 5.
One-dimensional (1D) Pourbaix diagrams of Mo complexes adsorbed on Cu(111) (dashed) and Ni(111) (solid). Black, blue, green, and red curves correspond to H3Mo, H3MoOH, H2Mo(OH)2, and MoO(OH)3, respectively.
Figure 5.
One-dimensional (1D) Pourbaix diagrams of Mo complexes adsorbed on Cu(111) (dashed) and Ni(111) (solid). Black, blue, green, and red curves correspond to H3Mo, H3MoOH, H2Mo(OH)2, and MoO(OH)3, respectively.

Figure 6.
(a, b) Charge redistribution under an applied electric field for Mo complexes adsorbed on Cu(111) and Ni(111). (c, d) Changes in relative Gibbs free energy under an applied electric field. Gas-phase H2O and H2 energies, as well as thermal corrections to the Gibbs free energies, are assumed constant. Ni(111) exhibits negligible variation, whereas Cu(111) shows measurable changes in slab charge with increasing field strength. The relative Gibbs free energies follow the same trend.
Figure 6.
(a, b) Charge redistribution under an applied electric field for Mo complexes adsorbed on Cu(111) and Ni(111). (c, d) Changes in relative Gibbs free energy under an applied electric field. Gas-phase H2O and H2 energies, as well as thermal corrections to the Gibbs free energies, are assumed constant. Ni(111) exhibits negligible variation, whereas Cu(111) shows measurable changes in slab charge with increasing field strength. The relative Gibbs free energies follow the same trend.


Figure 7.
One-dimensional (1D) Pourbaix diagrams of Mo complexes adsorbed on Cu(111) at (a) pH = 2; (b) pH = 7; (c) pH = 10; and (d) pH = 14. Black, blue, green, and red curves correspond to H3Mo, H3MoOH, H2Mo(OH)2, and MoO(OH)3, respectively. Stability is evaluated using the relative Gibbs free energy (, dashed) and the electric field-corrected relative Gibbs free energy (, solid). With increasing pH, the blue, green, and red solid curves shift upward, indicating decreased stability of the corresponding complexes, whereas the black solid curve shifts downward, indicating enhanced stability H3Mo at high pH.
Figure 7.
One-dimensional (1D) Pourbaix diagrams of Mo complexes adsorbed on Cu(111) at (a) pH = 2; (b) pH = 7; (c) pH = 10; and (d) pH = 14. Black, blue, green, and red curves correspond to H3Mo, H3MoOH, H2Mo(OH)2, and MoO(OH)3, respectively. Stability is evaluated using the relative Gibbs free energy (, dashed) and the electric field-corrected relative Gibbs free energy (, solid). With increasing pH, the blue, green, and red solid curves shift upward, indicating decreased stability of the corresponding complexes, whereas the black solid curve shifts downward, indicating enhanced stability H3Mo at high pH.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



