Submitted:
12 April 2026
Posted:
14 April 2026
You are already at the latest version
Abstract
Classical subatomic particles simulations were applied to protons, neutrons and electrons to simulate a complete atom using pseudo potentials to simulate nucleus and electrons distributed around nucleus. Molecular dynamics algorithms were applied to subatomic particles to simulate a complete atom for Hydrogen, Carbon and Uranium atoms that were reported in previous studies. Further analysis of energies of Carbon atom reveals that atom has quantized discrete energy values rather than continuous energies as expected in classical simulations. Changes in atom's total energy were observed to be in step changes going to higher/lower energy states and energy values are observed to be in quantized rather than continuous. Electrons energy profiles was observed during the atom's quantum energy shifts and only one electron got affected that has step change in its energies, for short duration, leaving other electrons unaffected. To observe similar phenomenon in other atoms, Fluorine, Magnesium and Chlorine atom were considered in this study and found to have similar quantized discrete energy values properties. This study implements subatomic particles simulations using classical mechanics that explains why atom's energies are quantized in nature as observed in discrete lines in atomic emission/absorption spectrum for atoms.
Keywords:
subatomic particle simulations
; classical physics energies quantized
; atomic emission spectra
; atom’s energies quantized
1. Introduction
In 19th century, atomic emission/absorption spectrum lines [1,2,3,4,5,6] were observed in elements showing atom energies are quantized. In the beginning of 20th century, scientists explore the atomic science on both classical and quantum perspectives. Around the year 1920 many theories like Niels Bohr postulates [7] on stationary orbits, Heisenberg uncertainty principle [8] and Schrödinger’s wave mechanics [9] studies lead to reasonable explanations and lead to the path of exploring quantum studies in atomic physics. Since there was no evidences/reasonable explanation for classical approaches that results in halt of exploring classical approach on subatomic particles that happened around 1930 (Figure 1). Quantum perspective in atomic physics leads to development of Quantum Field Theory [10] in 1930’s. In later decades, many-particle system [11] and solid state systems [12] in quantum approaches emerged and now, in 2026, humanity is in the stage of developing quantum computers [13] and quantum communications systems [14]. In this study, we explore classical approaches by developing subatomic particle simulations for an individual atom by applying molecular dynamics algorithms [15,16,17] to subatomic particles, to form nucleus and electrons distributed around it.
Subatomic particle simulations on atoms were conducted for Carbon atom, Fluorine atom, Magnesium atom and Chlorine atom. Analysis of atom’s total energy (sum of kinetic and potential energies) were observed to be quantized/discrete and are not continuous at all values. This observation, in classical simulations of subatomic particles on atom, explains the phenomenon of atomic emission/absorption spectral lines [18] where atom emits or absorbs energy in discrete manner rather than continuous at all energy values.
Coloumb interactions [19] introduced in the 18th century that guides in modeling electrostatics in molecular simulations. It plays pivotal role in modern applications in Carbon nanotubes [20] and Bilayer Graphene [21] systems. The standard perspective on protons and electrons is that protons bear a positive charge while electrons carry a negative charge and opposite charges attract while the same charges repel each other. In our previous studies [22,23], a new perspective on looking into subatomic particles was presented. Protons, electrons, and neutrons are treated as conventional particles in inside an atom, exhibiting attraction and repulsion between them, akin to the way atoms attract and repel each other in atomic simulations. This new approach leads to development of pseudo potentials for subatomic particles to model the subatomic particle interactions in an individual atom.
In this study, molecular dynamics algorithms [15,16,17] were applied to protons, neutrons and electrons to simulate a complete atom called as "Subatomic Particle Simulations". [22,23] Simulations of Carbon atom, Fluorine atom, Magnesium atom and Chlorine atom were considered in this study to study the atom’s energy pattern that evolve with time. Using classical mechanics, subatomic particle simulations on individual atoms provide insights on behavior of quantum effects on atom’s energies that observed in atomic emission/absorption spectrum. Conventional perspective in classical mechanics is that atom’s energies are continuous rather than discrete quantized energy values. Analysis made in this study show different perspective that change of energies of atom, in these simulations, happens only by step change giving discrete energy values rather than continuous increase or decrease in atom’s energies. This study and our previous studies [22,23] provides explanation to atom’s energies is having quantization effects using classical mechanics in subatomic particle simulations. Energy quantization property was already enclosed in classical mechanics and this property was observed in this study.
In the remainder of this paper, simulation details were presented for subatomic particle simulations followed by results and discussion section. In the results and discussion section, atom’s energies at every time steps (up to 1 time steps) were presented to study the discrete quantum effects for Carbon, Fluorine, Magnesium and Chlorine atoms. In conclusion section, observations and finding made in this study were summarized.
2. Simulation Details
Simulation code was developed in Fortran for subatomic particles in an atom using molecular dynamics (MD) algorithm using Velocity Verlet integrator [24]. A dimensionless time step of 0.02 was used in MD simulations and the time step was calculated, from dimensionless variables, and found to be 1.67 attoseconds. The initial configuration was created by placing protons and neutrons, randomly, in the centre of the atom (nucleus) and the electrons placed randomly around the nucleus. Carbon, Fluorine, Magnesium and Chlorine atoms were considered in this study to analyze atoms total energy behavior over time, like quantization, in subatomic particle simulations. Pseudo potentials, introduced in previous study, [22,23] were used to calculate force and potential acting on the subatomic particles at each time step. Boundary conditions and cut-off were not applied in the simulation box for calculating the force/potential acting on particles. All MD simulations were run for 16.7 picoseconds (1 x time steps) and first 10,000 time steps were neglected in analysis. MD simulation video for Carbon atom were captured using the VMD package [25].
Pseudo potentials for subatomic particles were developed [22,23] on the principle of exhibiting repulsion (at smaller distances) and attraction (at larger distances) between them. Considering this behavior for subatomic particles in MD code gives stable individual atom simulations.
Following pseudo potentials were used for subatomic particle simulation:
for electron to proton:
for proton to proton, proton to neutron, neutron to neutron,
for electron to electron,
where V(r) is the potential acting on particles, r is the distance between particles, and are pseudo potential parameters as shown in Table 1.
The total potential energy of atom (force field potential function) was considered as sum of potential functions of electron to proton (eq. 1), electron to electron (eq. 3), proton to proton, proton to neutron and neutron to neutron (eq. 2).
3. Results and Discussion
3.1. Carbon Atom Energy Analysis
Subatomic particle simulations for Carbon, Hydrogen and Uranium atoms were developed and analyzed in previous studies [22,23] using pseudo potentials. Monte Carlo and molecular dynamics simulations were developed for subatomic particles to simulate a full Carbon atom and properties like radius, energies of atom were reported. [22,23] Figure 2 (taken from previous studies) shows subatomic particles trajectories of Carbon atom using molecular dynamics algorithm for 5000 time steps, as reported in previous studies. Simulation video of Carbon atom using subatomic particle simulations was shown in supplementary material video 1.
To analyze the total energy of Carbon atom, sum of kinetic energy and potential energy of protons, neutrons and electrons were considered. Potential energy was calculated between subatomic particles using pseudo potentials and kinetic energy was calculated from particle velocities as shown in equation 4.
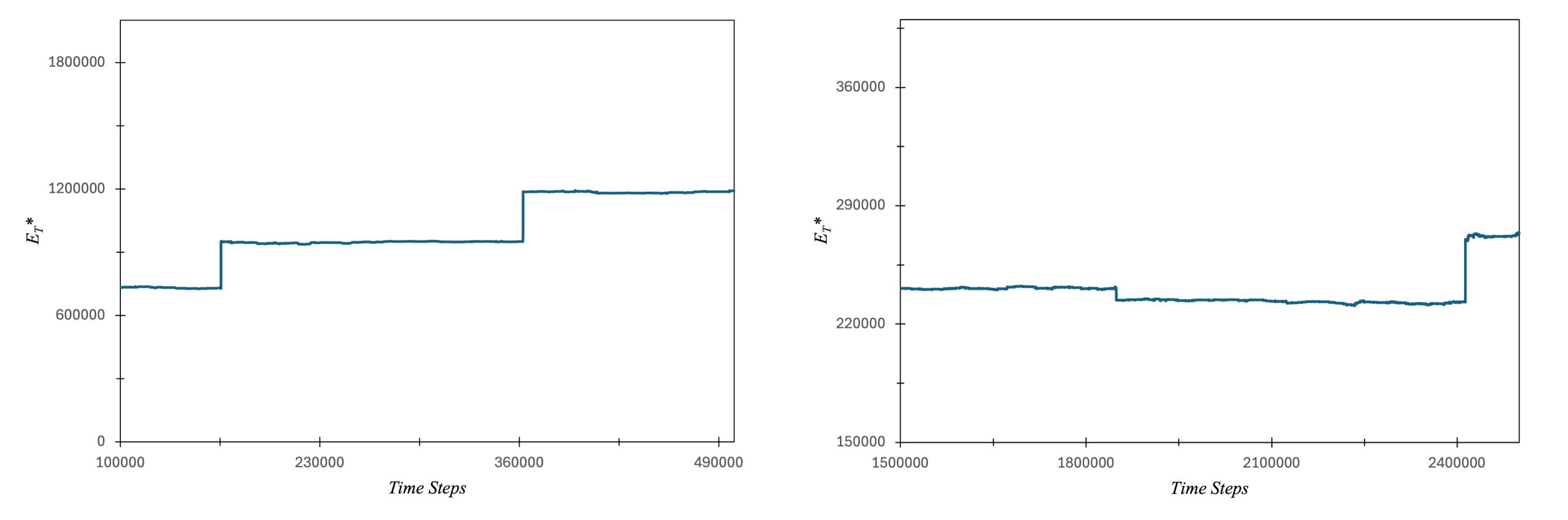
To study the behavior of atom’s energy like quantization effects, analysis of total energy of Carbon atom at each time step in the subatomic particle simulations were considered. Figure 3 and Figure 4 shows atom’s total energy (, dimensionless) at each time step in the simulation of 16.7 picoseconds ( time steps). It was observed that Carbon atom’s energy was not continuous at all values rather than have discrete values and exhibits quantization property over time. Sudden step change of total energy, in duration of few time steps, was observed showing energy quantization property of Carbon atom energies. First step change of atom’s energy was observed in Figure 3(a) around 140,000 time step (at 0.23 picoseconds) with energy values changed from 39,700 to 59,300 () energy units within duration of few time steps. Few smaller step changes in energies were observed in Figure 3(b) that too in discrete behavior rather than continuous energy pattern. Second major energy quantization shift (step change) happened in Figure 3(c) around 2,350,000 time step (at 3.9 picoseconds). with energy changes from 73,900 to 116,000 () units. In other words, Carbon atom total energy exists either at 73,900 units or at 116,000 units and doesn’t have continuous values exhibting behavior of quantum effects of energies over time. Third energy quantization phenomenon was observed in Figure 4(b) around 6,802,000 time step (at 11.3 picoseconds) with energy change from 129,400 to 156,000 units.
Figure 3 and Figure 4 shows that atom’s doesn’t have continuous increase or decrease in energy values rather it have discrete quantized energy values as observed in atomic emission/absorption spectrum of atoms. This total energy analysis over each time step for Carbon atom in subatomic particle simulation explains quantum effects of atom energies using classical theory. It is expected that in classical mechanics, atom tends to have continuous energy values at all levels, but on contrary, classical subatomic particle simulations shows energy quantization property in real time. It also shows that atom’s energies is only possible for certain discrete quantized values and does not exist for all energy values as shown in Figure 3 and Figure 4. This behavior in energy patterns in classical simulations can provide explanation of quantum effects of atomic emission/absorption spectrum behavior where classical models failed to explain till date.
Further investigation on changes in kinetic and potential energy patterns of an atom in energy quantization effects will provide some insights in understanding quantum effects in classical simulations. Figure 5 shows comparison of kinetic and potential energy of Carbon atom with its total energy that has step change as observed in Figure 4(b) at around 6,802,000 time step (at 11.3 picoseconds). During total energy quantum step change, kinetic and potential energy pattern doesn’t show any quantum effects but rather shows continuous variation within the range. Step change in kinetic energy of atom during quantization was observed and later tends to have normal trend as before quantization. One observation from Figure 5 is that before and after energy quantization, kinetic and potential energy fluctuate at higher frequency at higher energy value. It shows that frequency of atom increases with increase in quantum discrete energy values. Correlation between atom frequency and its discrete energy values can be developed and will be considered in future studies.
3.1.1. Study of Electrons Energy Patterns during Atom’s Energy Quantization Shift
To know whether electrons exhibit similar energy quantization property in an individual Carbon atom, energy profiles of individual electrons (six electrons of Carbon atom) were analyzed during the step change of atom total energy occurred. Figure 6 shows total energy of an individual electron with electron ID 3 during the atom made step change as shown in Figure 4(b) at 6,802,000 time step. All electrons except one (electron ID 5) shows usual energy profile with no observed phenomenon during the atom’s energy quantization. Only electron ID 5 has step change in total energy during the atom’s quantum effect, as shown in Figure 7(b). This step change in electron ID 5 is also temporary only during the quantum energy shift and usual electron energy pattern prevails in long term as similar to other electrons as shown in Figure 6. Only one electron out of six electrons of Carbon atom have step change in energy profile during atom’s quantum energy shift that too for a short duration of time. One can increase the kinetic energy of a particular electron in an atom, imitating electron excitation to higher energy levels by photon absorption, and study the quantum effects of total energy of an atom. This approach is considered in future studies.
3.2. Quantum Effects of Fluorine, Magnesium and Chlorine Atom in Subatomic Particle Simulations
To study this phenomenon of energy quantization in atoms, other atoms are considered that has different number of protons, neutrons, and electrons. Fluorine, Magnesium, and Chlorine atom were considered to investigate whether quantum effects applies to all atoms or not. Supplementary material Figure S1 to S6 show energy trends for these individual atoms, at every time step in subatomic particle simulations. For Fluorine atom, step change or quantum discrete energy values were observed in supplementary material Figure S1 (a), (b), (c) and (d). In Figure S1 (c), total energy of Fluorine atom had decreased energy state with step change. Both decrease and increase in atom’s total energy () are following an instant step change in energy levels that has explains phenomenon of atomic emission spectrum and atomic absorption spectrum, respectively. For Magnesium atom, step change in energy levels was observed in supplementary material Figure S3 (b) and (c). For Chlorine atom, discrete energy levels was observed in supplementary material Figure S5 (a) and (d). Energy quantization property was observed in classical subatomic particle simulations for all four atoms considered here in this study.
4. Conclusions
Implementation of molecular dynamics algorithm to subatomic particles to simulate a full atom leads to development of "Subatomic Particle Simulations" reported in previous studies. Analyzing Carbon atom’s total energy pattern with every time step reveals the property of quantum discrete energy values property in classical simulations. Investigation on individual electron energy patterns reveal that only one electron has step change in its total energy, for short period, leaving other electrons unaffected. Considering Fluorine atom, Magnesium atom, and Chlorine atom shows the same property of discrete energy values rather than continuous values. It was observed in all these case studies that there is no trend observed in energy patterns of decreasing or increasing trend over time that leads to large differences in atom’s total energy, as shown in Figure 3, Figure 4 and in supplementary material Figure S1 to S6. Atom’s total energy patterns strictly follows horizontal lines (parallel to x-axis) and to change its energies, atom follows instant step changes, showing the property of discrete quantum energy levels in classical simulations. Conventional perspective in classical mechanics for having nucleus in center and electrons distributed around nucleus is to have continuous energy values. This was not observed in classical subatomic particle simulations rather discrete quantum energy levels of atom was observed in this study. Based on these understanding of classical mechanics explaining quantum effects of atom’s energies, one can propose theoretical models explaining quantum effects of individual atoms using classical physics without the need of subatomic particle simulations reported in this study.
Acknowledgments
I thank Gomathi, Punya Koti and Dr Delli Ganesh for their guidance in this project. I thank Dr Subhash Patri for his guidance in this project. I thank Mississippi State University (MSU) where part of the Hydrogen atom calculations were performed on MSU computer. I thank Dr. Neeraj Rai for his valuable discussions on Lennard-Jones potentials and on molecular simulations.
References
- Wollaston, W.H. XII. A Method of Examining the Refractive and Dispersive Powers of Different Substances, by Prismatic Reflection. Philosophical Transactions of the Royal Society of London 1802, 92, 365–380. [CrossRef]
- Fraunhofer, J.v. Bestimmung des Brechungs- und Farbenzerstreuungs-Vermögens verschiedener Glasarten, in Bezug auf die Vervollkommnung achromatischer Fernröhre. Denkschriften der Königlichen Akademie der Wissenschaften zu München 1817, 5, 193–226.
- Kirchhoff, G. Über die Fraunhofer’schen Linien. Annalen der Physik 1860, 185, 148–150.
- Kirchhoff, G.; Bunsen, R. Chemische Analyse durch Spectralbeobachtungen. Annalen der Physik und Chemie 1860, 110, 161–189. [CrossRef]
- Ångström, A.J. Optiska Undersökningar. Kongl. Svenska Vetenskaps-Akademiens Handlingar 1853.
- Thomsen, V. A Timeline of Atomic Spectroscopy. Spectroscopy 2006, 21, 32–38.
- Bohr, N. I. On the Constitution of Atoms and Molecules. The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science 1913, 26, 1–25. [CrossRef]
- Heisenberg, W. Über den anschaulichen Inhalt der quantentheoretischen Kinematik und Mechanik. Zeitschrift für Physik 1927, 43, 172–198. [CrossRef]
- Schödinger, E. Quantisierung als eigenwertproblem.(erste mitteilung). Annalen der Physik 1926, 79, 361–376.
- Heisenberg, W.; Pauli, W. Zur Quantentheorie der Wellenfelder. II. Zeitschrift für Physik 1930, 59, 168–190. [CrossRef]
- Heisenberg, W. Mehrkörperproblem und Resonanz in der Quantenmechanik. Zeitschrift für Physik 1926, 38, 411–426. [CrossRef]
- Bloch, F. Über die Quantenmechanik der Elektronen in Kristallgittern. Zeitschrift für Physik 1928, 52, 555–600. [CrossRef]
- Deutsch, D. Quantum Theory, the Church–Turing Principle and the Universal Quantum Computer. Proceedings of the Royal Society of London. A. Mathematical and Physical Sciences 1985, 400, 97–117. [CrossRef]
- Bennett, C.H.; Brassard, G. Quantum Cryptography: Public Key Distribution and Coin Tossing. In Proceedings of the Proceedings of the IEEE International Conference on Computers, Systems and Signal Processing, Bangalore, India, December 1984; pp. 175–179.
- Alder, B.J.; Wainwright, T.E. Phase Transition for a Hard Sphere System. The Journal of Chemical Physics 1957, 27, 1208. [CrossRef]
- Alder, B.J.; Wainwright, T.E. Studies in Molecular Dynamics. I. General Method. The Journal of Chemical Physics 1959, 31, 459–466. [CrossRef]
- Rahman, A. Correlations in the Motion of Atoms in Liquid Argon. Physical Review 1964, 136, A405–A411. [CrossRef]
- Kramida, A.; Ralchenko, Y.; Reader, J.; Team, N. A. NIST Atomic Spectra Database (ver. 5.12). https://physics.nist.gov/asd,2024; National Institute of Standards and Technology, Gaithersburg, MD.
- Coulomb, C.A. Premier mémoire sur l’électricité et le magnétisme. Histoire de l’Academie royale des sciences 1785, 569.
- Kane, C.; Balents, L.; Fisher, M.P. Coulomb interactions and mesoscopic effects in carbon nanotubes. Physical review letters 1997, 79, 5086.
- Cea, T.; Guinea, F. Coulomb interaction, phonons, and superconductivity in twisted bilayer graphene. Proceedings of the National Academy of Sciences 2021, 118, e2107874118.
- Venkatesan, S. Subatomic Particle Simulations using Monte Carlo and Molecular Dynamics Algorithms to Simulate Stable Atom and Model Electronic Structures. Preprints 2025. [CrossRef]
- Subatomic Particle Simulations using Monte Carlo and Molecular Dynamics Algorithms to Simulate Stable Atom and Model Electronic Structures. Innovative Journal of Applied Science 2025, 2, 20. [CrossRef]
- Swope, W.C.; Andersen, H.C.; Berens, P.H.; Wilson, K.R. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters. The Journal of chemical physics 1982, 76, 637–649.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD – Visual Molecular Dynamics. Journal of Molecular Graphics 1996, 14, 33–38.
Figure 1.
Timeline showing halt of exploring classical approach to subatomic particles.

Figure 2.
Trajectories of subatomic particles in Carbon atom simulation for 5000 time steps. Reproduced from previous studies. [22,23]

Figure 3.
Atom’s energy exhibiting quantization property in classical simulation by plotting Carbon atom’s total energy over time steps of the simulation from 10,000 to time steps (Figure (a) to Figure (f)). Atom’s energy quantization was observed in Figure (a), (b) and (c).
Figure 3.
Atom’s energy exhibiting quantization property in classical simulation by plotting Carbon atom’s total energy over time steps of the simulation from 10,000 to time steps (Figure (a) to Figure (f)). Atom’s energy quantization was observed in Figure (a), (b) and (c).

Figure 4.
Atom’s total energy quantization graph showing Carbon atom’s total energy with time steps of the simulation from to time steps. Atom’s energy quantization was observed in Figure (b).
Figure 4.
Atom’s total energy quantization graph showing Carbon atom’s total energy with time steps of the simulation from to time steps. Atom’s energy quantization was observed in Figure (b).

Figure 5.
Atom’s total energy quantization (step change) with time and comparison of changes in kinetic energy and potential energy.
Figure 5.
Atom’s total energy quantization (step change) with time and comparison of changes in kinetic energy and potential energy.

Figure 6.
Electron ID 3 total energy profile with time during subatomic particle simulations. Energy pattern follows random pattern with time, giving no observed phenomenon for quantum effects of atom’s total energy.
Figure 6.
Electron ID 3 total energy profile with time during subatomic particle simulations. Energy pattern follows random pattern with time, giving no observed phenomenon for quantum effects of atom’s total energy.

Figure 7.
Atoms total energy quantization observed in Figure (a) and compared with electron ID 5 energies with time, shown in Figure (b).
Figure 7.
Atoms total energy quantization observed in Figure (a) and compared with electron ID 5 energies with time, shown in Figure (b).


Table 1.
Pseudo potentials parameters, parameterized based on Carbon Atom Simulation
| (Joules) | (picometers) | |
|---|---|---|
| Parameterized based on Carbon atom | 7.837 * | 0.7777 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



