Submitted:
09 April 2026
Posted:
13 April 2026
Read the latest preprint version here
Abstract
N-Methyl-D-aspartate (NMDA) receptors are ligand- and voltage-gated ion channels that are critical for synaptic plasticity, learning, and memory. These receptors are variously composed of GluN1, GluN2A–D, and GluN3A/B subunits. They are widely expressed in the central nervous system and are implicated in several neurological, neurodegenerative, and psychiatric disorders. The GluN2B subunit has garnered particular interest due to its high expression in the forebrain and spinal cord, role in pathophysiological processes, and potential as a therapeutic target. Consequently, there is continuing strong interest in developing radioligands for imaging brain NMDA receptors with positron emission tomography (PET). We report the synthesis of nineteen 3-alkylaryl derivatives of 7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol and some of their enantiomers as prospective GluN2B PET radioligands. The absolute configuration of one core ligand was determined with vibrational circular dichroism and infrared spectroscopy, allowing the determinations of the absolute configurations of enantiomers of other ligands derived from this parent ligand. The GluN2B binding pocket showed generally broad tolerance for alkyl tether chain length and for alterations of both bulk and substitution in the terminal aryl group. No relationship between GluN2B affinity and computed compound lipophilicity was observed. Enantiomers of two prepared ligands, L3 (NR2B-SMe) and L6 (NR2B-Me), have desirably strong affinity for GluN2B, moderate lipophilicity, and amenability for labeling with carbon-11 (t1/2 = 20.4 min). In subsequent and separate PET studies, [S-methyl-11C](S)-L3 and [C-methyl-11C](R)-L6 have shown strong specific binding to GluN2B in animal brain. Synthesis methods and other data from this study can guide and support the further development of candidate GluN2B PET radioligands for clinical applications.
Keywords:
GluN2B
; RADIOLIGAND
; structure–activity relationship (SAR)
; PET imaging
; NMDA receptor
Introduction
N-Methyl-D-aspartate (NMDA) receptors are both ligand- and voltage- gated ion channels that facilitate the synaptic influx of Ca²⁺, Na⁺, and K⁺ ions.(1) NMDA receptors are widely expressed throughout the central nervous system (CNS) and play essential roles in synaptic plasticity, learning, and memory. They are also implicated in the pathophysiology of numerous CNS disorders and have become potential therapeutic targets.(2-4)
NMDA receptors are heterotetrameric complexes composed of subunits that are assembled from seven distinct subunit classes, now named GluN1, GluN2 (A–D), and GluN3 (A or B) (but formerly known as NRB subtypes). This subunit variety gives rise to a rich diversity of NMDA receptor subtypes, each with unique structural features that shape their physiological functions and pharmacological profiles. In general, NMDA receptors contain distinct binding sites for multiple ligands, including L-glutamate, glycine, D-serine, polyamines, Mg²⁺, Zn²⁺, and phencyclidine. Ligands are categorized into four types: glutamate-site ligands, glycine-site ligands, channel blockers, and N-terminal domain ligands. Some NMDA-targeting ligands have approved therapeutic uses. For example, memantine, is approved for treating Alzheimer’s disease.(5,6)
The NMDA receptor GluN2B subunit has gained especially strong interest as a therapeutic target for disorders such as schizophrenia, stroke, neurodegeneration, and neuropathic pain.(7,8) GluN2B-enriched NMDA receptors are predominantly found in the forebrain and the dorsal horn of the spinal cord. They contribute significantly to prefrontal cortex function but also show vulnerability from environmental stressors and psychiatric risk factors.(9) The drug targeting of GluN2B rather than the full range of NMDA receptors has potential to limit debilitating side-effects such as hallucinations, drowsiness, and confusion.
Positron emission tomography (PET) is a uniquely powerful molecular imaging technique that can be used with suitably designed radiotracers to provide information on the abnormal distribution or dysregulation of specific proteins in the brains of living animals and humans. As such, PET has potential for unveiling pathophysiology in neurological (10-12) and psychiatric disorders (13) and for assisting in the development of novel drug therapies (14,15). PET radioligands for brain imaging must be labeled with a positron-emitter, either carbon-11 (t1/2 = 20.4 min) or fluorine18 (t1/2 = 109.8 min). Additionally, they must fulfill a wide range of pharmacological, physicochemical, and other criteria for efficacy.(16-18) Efforts to develop PET radioligands for imaging NMDA receptors in living human brain have been pursued for decades but have met with limited success.(19-21)
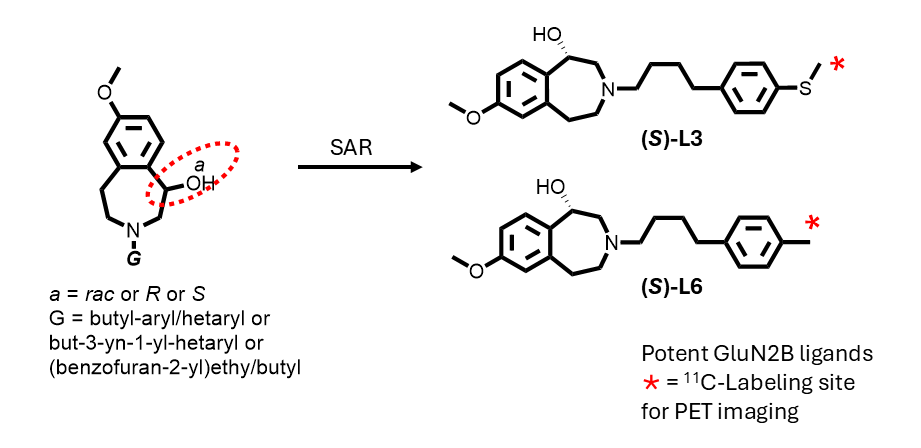
PET-based quantification of GluN2B in vivo could aid in both biomedical research and drug development.(20) To date, only radioligands derived from (±)-7-methoxy-3-(4-(4-(methylthio)phenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (Chart 1) have demonstrated displaceable GluN2B PET signals (22), with one radioligand, [11C](R)-Me-NB-1, evaluated in humans.(23) We have developed and reported structurally similar radioligands, namely enantiomers of [11C]NR2B-SMe (24) and enantiomers of [11C]NR2B-Me (25) (Chart 1). These radioligands emerged out of the present medicinal chemistry effort reported herein. We found that NR2B-SMe (L6) and NR2B-Me (L3) and their enantiomers displayed nanomolar affinity for GluN2B, favorable lipophilicity, and suitability for facile labeling with carbon-11. In separate studies, enantiomers of [11C]L3 showed strong binding to GluN2B in rat brain (24), and [¹¹C](R)-L6 showed the highest binding potential (BPND) of three tested PET radioligands in nonhuman primates.(26)
Here, as part of a broader medicinal chemistry campaign to create GluN2B-specific PET radioligands with high specific signal and minimal background noise, we report the synthesis and pharmacological evaluation of several 7-methoxy-3-substituted-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol derivatives, including NR2B-SMe (L3) and NR2B-Me (L6) and their enantiomers. This study expands knowledge of structure-activity relationships, increases the pool of GluN2B ligands, and contributes to optimizing PET tracers for imaging GluN2B.
Results and Discussion
Ligand design.N-Substituted 7-methoxy and 7-hydroxy derivatives of 2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol have featured prominently among potent GluN2B ligands and candidate PET radioligands, such as [11C]Me-NB-1 (23,27-29), [18F]PF-NB1 (30,31), [18F]OF-NB1 (26,32-37) and their enantiomers (Chart 1). The chemical, pharmacokinetic, and pharmacodynamic characterization of the GluN2B antagonist, 3-(4-phenylbutyl)2,3,4,5-tetrahydro-1H-3-benazepine-1,7-diol (WMS-1410), has been well reviewed as a starting point for the development of radioligands for PET imaging of GluN2B.(35,38) WMS-1410 was developed by conformational constraint of ifenprodil, a prototypical GluN2B ligand that lacked good selectivity, especially towards GluN2A, GluN2C, or GluN2D subunits of NMDA receptors (Chart 2). The 7-hydroxy group of WMS-1410 is at a chiral center mimicking one of the two chiral centers in ifenprodil. Ifenprodil shows stereoselectivity for binding to GluN2B receptors with the eutomer identified as the (1R,2R)-enantiomer. This enantiomer has 2.5-fold higher affinity (Ki = 5.8 nM) than the (1S,2S)-enantiomer.(39) WMS-1410 likewise shows stereoselectivity for binding to GluN2B with the R-enantiomer being the eutomer with a Ki value of 30 nM; the S-enantiomer has a 25-fold greater Ki value, measured as 740 nM.(40) X-ray crystallography has elucidated the binding interactions of ifenprodil with GluN2B NMDA receptors.(41,42) Ifenprodil binds at the interface between GluN1A and GluN2B in a heterodimer, rather than within a cleft of GluN2B. The phenolic hydroxy group forms a hydrogen bond with the terminal carboxylate group of glutamate-236 in GluN2B. Another hydrogen bond forms between the protonated amino group of ifenprodil and the terminal carbamoyl group of the glutamine-210 residue in GluN2B. The terminal aryl groups of ifenprodil have p-p interactions with tyrosine-109 (GluN1A), phenylalanine-114 (GluN2B) and phenylalanine-176 (GluN2B) and hydrophobic interactions with phenyalanine-113 and leucine-135 in GluN1A and with isoleucine-111 and proline-177 in GluN2B. Docking studies indicate that WMS-1410 binds at the same site as ifenprodil and with a similar binding pose.(41) Importantly, the 2,3,4,5-tetrahydro-1H-benzo[d]azepine-1,7-diol group confers high binding affinity for GluN2B with high selectivity over GluN2A, GluN2C, and GluN2D NMDA receptors, in addition to high selectivity over more than 100 receptor, enzyme, transporter, and ion channel proteins.(43,44)
The phenolic hydroxy group of WMS-1410 underdoes glucuronidation in vivo limiting its bioavailability.(40) Replacement of the 7-hydroxy group in WMS-1410 with a methoxy group, as in WMS-1405 (Chart 1), blocks glucuronidation. Two reports indicated that WMS-1405 had low GluN2B affinity (Ki, ~700 nM) (40,45) with very modest retention of stereoselectivity in favor of the R-enantiomer (40) whereas others have reported a Ki value of 5.4 nM for the racemate.(43,46) With regards to PET radioligand development, the 7-methoxy group of WMS-1405 usefully provides a site for 11C-methylation. In fact, racemic [methoxy-11C]WMS-1405, dubbed 11C-Me-NB1 (Chart 1), has been produced as a PET radioligand for imaging brain GluN2B receptors in rodents (47) and as the R-enantiomer in humans.(23,27,29) Thus, the methoxy group in WMS-1405 is not detrimental to high affinity for GluN2B in vivo.
In this study, we aimed to explore variations in the alkylaryl substituents at the 3-position of 7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol. Specifically, we aimed to investigate: i) the effect of para substitution on the terminal phenyl group of WMS-1405, ii) the effect of alkyl linker chain length, and iii) the effect of unsaturation in the alkyl chain (Chart 2).
Ligand Syntheses
The overall strategy for ligand synthesis was to couple a requisite alkyl tosylate with 7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol. Tosylates were prepared from corresponding alcohols that were either purchased directly or obtained by various methods from commercially available starting materials (Scheme 1).
Syntheses of alcohols. Reduction of 4-(p-iodophenyl)butyric acid with diborane in dry tetrahydrofuran (THF) gave alcohol 1 (Scheme 2).(48) Use of a new unopened bottle of diborane was critical for achieving a clean and high yield reaction. Even the use of a large excess of diborane did not compensate for this need. Temperature control was also critical. The reaction mixture must be kept below 40 °C but above 0 °C to ensure complete conversion while minimizing side-product formation and maintaining sufficient reaction rates. A palladium-catalyzed reaction in the presence of chlorotri(methyl)stannane successfully substituted the iodo substituent in compound 1 with methylthiolate, probably through trimethyl(methylthio)stannane (Me3SnSMe) as an intermediate, to give 2 (Scheme 2).
Reagents, conditions, and yields: (i) BH3, THF, < 40 °C, 3 d; (ii) MeOH, RT, 1 h. (86% over 2 steps); (iii) NaSMe, (Me)3SnCl, [1,1’-bis(diphenylphosphino)ferrocene]dichloropalladium(II)●CH2Cl2, DMSO, MW (110 °C, 30 min, 50 W, 250 psi). (48%).
Substitution of the iodo substituent in 4-iodo-trifluoromethylbenzene by but-3-yn-1-ol under catalysis by copper(I) and palladium(II) complexes, gave 3, probably through a copper-alkyne intermediate. For the synthesis of the p-trifluoromethyl substituted alcohol 4, various catalysts, including Pd/CF3Cu, Ru/CF3Cu, and CF3Cu, failed to replace the iodo substituent in 1 with a trifluoromethyl group. Nonetheless, hydrogenation of the triple bond in 3 by hydrogen in the presence of Pd/C readily generated 4 (Scheme 3).
Reagents, conditions, and yields: (i) but-3-yn-1-ol, CuI, Pd(PPh3)2Cl2●CH2Cl2, Et2NH, 16 h, 60 °C. (62%); (ii) H2, Pd/C, MeOH, 40 °C, 8 h. (78%).
Reaction conditions for 5 (Scheme 4) mirrored those of 3 with the addition of PPh3. Protection of the hydroxy group in but-3-yn-1-ol reagent was unnecessary.
Reagents, conditions, and yields: (i) but-3-yn-1-ol, CuBr, (PPh3)2PdCl2, PPh3, Et2NH, RT for 1 h then 88 °C for 24 h. (88%).
Catalysis with copper(I) salt and palladium complex readily converted 2-iodophenol p-substituted derivatives into the hydroxyalkyl-benzofurans 6 to 11 (Scheme 5). These syntheses probably occur through substitution of the iodo substituent by the alkyne followed by intramolecular cyclization under the basic conditions.
Reagents, conditions, and yields: (i) but-3-yn-1-ol, CuBr, (PPh3)2PdCl2, PPh3, Et2NH, 90 °C, 4–5 h and then overnight. (6, 67%; 7, 76%; 8; 81%; 9, 78%); (ii) hex-5-yn-1-ol, CuBr, (PPh3)2PdCl2, Et2NH, 90 °C, 4–5 h and then overnight. (10, 84%; 11, 67%).
Treatment of the synthesized alcohols with tosyl chloride in the presence of base gave the tosylates, X1–X18 in yields ranging from 13 to 77% (Scheme 6). Some reactions did not go to completion even with excess reagent. A long reaction time risked reduced yield from substitution of the product tosylate group by the chloride anion generated from the tosyl chloride. Normally, overnight reactions gave the best compromise between yield and purity.
Reagents, conditions, and yields: (i) TsCl, Et3N, DCM, RT, overnight–24 h (X1, 77%; X2, 61%; X3, 47%; X4, 74%; X5, 21%; X6, 24%; X7, 58% after 5 d stirring; X8, 52%; X9, 22%; X10, 21%; X11, 22%; X12, 13%; X13, 45%; X14, 54%; X15, 72%; X16, 39%; X17, 76%; X18, 56%.
For the syntheses of the prospective GluN2B ligands, treatment of the prepared tosylates with 7-methoxy-2,3,4,5-terahydro-1H-benzo[d]azepin-1-ol was generally successful (Scheme 7), except in two cases, namely L10 and L12, which resulted in competing cyclization reactions.
Reagents, conditions and yields: (i) ROTs (X1), K2CO3, MeCN, 6 d at reflux. (L1, 72%). (ii) ROTs (X2), K2CO3, MeCN, 5 d at 75 °C. (L2, 73%). (iii) ROTs (X3 or X4), K2CO3, MeCN, 2 d at 75 °C. (L3, 75%; L4, 71%). (iii) ROTs (X5, X6, or X8), K2CO3, MeCN, 2 d at 90 °C. (L5, 87%; L6, 37%; L8, 79%). (iv) ROTs (X7, X9, or X11), K2CO3, MeCN, 5 d at 90 °C. (L7, 62%; L9, 74%; L11, 77%). (iv) ROTs (X12, X14, X15, X16, X17, or X18) MW (135 °C, 10 min, 60 W, 250 psi). (L12, 12%; L14, 51%; L15, 59%; L16, 64%; L17, 71%; L18, 68%). (v) ROTs (X13), Na2HPO4, MeCN, 22 d at 90 °C. (L13, 71%).
When X10 (R = 4-(pyridin-2-yl)butyl) was used in the synthesis, only a cyclized product was observed (Scheme 8). Changing the concentration of X10, including the use of pure X10 as solvent, did not generate any of the desired ligand (L10). No further attempt was made to synthesize L10.
Reagents, conditions and yields: (i) Na2HPO4, DMSO, MW (80 °C, 10 min, 30 W, 250 psi, or 120 °C, 10 min, 50 W, 250 psi, or 150 °C, 10 min, 60 W, 250 psi). (no L10, only the shown cyclized product); (ii) Na2HPO4, MeCN, 90 °C for 22 d. (no L10, only the shown cyclized product).
When X12 was used to synthesize L12 in the presence of disodium hydrogen phosphate as base, a cyclized product was also obtained in addition to L12 (Scheme 9), probably from the intramolecular cyclization of the product through substitution of the 6-fluoro group by the remote hydroxy group. The use of other bases, such as sodium carbonate or bicarbonate, did not improve the yield of L12.
Reagents, conditions, and yields: (i) MW (135 °C, 10 min, 60 W, 250 psi). (12% of L12 plus 8.1% of cyclized product).
Precursors for 11C-labeling
The methyl arylthiopropanoate ester, L19, and its enantiomers served as precursors for labeling L3 and its enantiomers, respectively, with carbon-11.(24) The enantiomers of L19 were obtained and purified by preparative chiral HPLC.
L20, the precursor for 11C-labeling of L6, was synthesized by substitution of either the iodo or bromo substituent in L2 or L7, respectively, with Bpin, using DPPF-PdCl2●CH2Cl2 as catalyst under microwave heating conditions (Scheme 10). The enantiomers were obtained by chiral resolution.
Reagents, conditions, and yields: (i) 4,4,4’,4’,5,5,5’,5’-octamethyl-2,2’-bi(1,3,2-dioxaborolane), DPPF-PdCl2●CH2Cl2, DMSO, MW (80 °C, 60 min, 60 W, 250 psi). (37% from L2). (ii) 4,4,4’,4’,5,5,5’,5’-octamethyl-2,2’-bi(1,3,2-dioxaborolane), DPPF-PdCl2●CH2Cl, DMSO, MW (110 °C, 30 min, 50 W, 250 psi). (54% from L7).
Absolute configuration correlation. We used vibrational circular dichroism/infrared (VCD/IR) plus quantum calculations to determine the absolute configuration of the enantiomers of ligand L3 (see Supporting Information).(24) Several homochiral ligands (enantiomers of L2, L6, L19, and L20) were related to (S)-L3 through chemical reactions that did not alter stereochemistry, thereby allowing unequivocal assignment of their absolute configurations (Scheme 11). In each case, we observed that the early eluting enantiomers of these ligands on chiral HPLC analysis with the (S,S)-Whelk column had the same absolute configuration, which we determined to be S-configuration. We also observed that each enantiomer of L3 has opposite specific optical rotation sign in ethanol and chloroform (see Experimental), which had to be considered in the VCD/IR analysis.
Reagents, conditions, and yields: (i) methyl 3-((trimethylstannyl)thio)propanoate, Pd(DPPF)Cl2●CH2Cl2, Et3N, MeCN, MW (90 °C, 20 min, 120 W, 250 psi). 81% (ii) MeI, TBAOH, DCM, rt, 20 min. 75%. (iii) bis(pinacolato)diboron, KOAc, Pd(DPPF)Cl2, MeCN, MW (80 °C. 20 min, 150 W, 250 psi). 31%. (iv) MeI, CsF, Pd(PPh3)4, MeOH/MeCN, MW (90 °C. 20 min, 80 W, 250 psi). 87%.
It is noteworthy that the chemical transformations shown in Scheme 11 did not cause any appreciable racemization of the products. Steps ii and iv are reactions which were adapted for labeling the enantiomers of S3 (24) and L6 (25) with carbon-11 utilizing [11C]methyl iodide as a labeling agent to give the homochiral radioligands, thereby supplanting the need for chiral chromatography.
Ligand structure versus affinity for GluN2B. 20 ligands were evaluated through various in vitro binding assays, including both in-house binding assays with rat brain homogenate and those in use at the Psychoactive Drug Screening Program (PDSP) (Table 1 and Table 2).
In this work, substituents on the terminal aryl group and the alkyl linker were varied. Among ligands having a terminal p-substituted phenylbutyl group (L2–L8), the electronic property of the p-phenyl substituent had very limited effect on ligand affinity with Ki, only varying by a factor of 2 from the electron-donating methoxy group in L5 to the electron-withdrawing fluoro group in L8 (Table 1). The size of the aryl p-substituent also has limited effect (compare L1 with L2). Likewise, the bulky naphthalenyl group in L11 was well tolerated. A terminal hydrogen bond acceptor, such as the 2-pyridinyl group in L11 and L12, dramatically reduced binding affinity, reinforcing the conclusion that the binding pocket is hydrophobic for interaction with this terminus of the ligands. From comparisons of L13 with L17 and of L16 with L18, the length of the tether appears to play an appreciable role in the binding affinity. The optimal tether is a flexible four carbon straight chain with low space requirements, as in L17 and L18. The addition of a 5-bromo (L14), 5-fluoro (L15) or 5-trifluoromethyl (L16) substituent to the terminal 2-benzofuranylethyl of L13 enhanced binding potency. Likewise, addition of a 5-trifluoromethyl group (L18) to the 2-benzofuranylbutyl group of L17, appreciably improved binding affinity. Finally, L19 was designed as a precursor for labeling L3 with carbon-11 rather than as prospective GluN2B ligand. Notably, L19 also showed remarkably high binding affinity, and further underscores the tolerance of the GluN2B binding pocket for bulky p-substituents on the terminal phenyl group of L1.
A plot of ligand pKi (from PDSP) versus clogD (from Pallas software) for the ligands shown in Table 1 (Figure S1) showed almost no correlation (r2 = 0.0285; n = 17) and therefore suggests that the GluN2B binding pocket is very tolerant of hydrophobic moieties.
PET radioligands. From a total of twenty two new ligands prepared in this study, we selected two ligands and their enantiomers for labeling with carbon-11 and evaluation as GluN2B PET radioligands in animals, namely L3 (NRB-SMe) (24) and L6 (NR2B-Me) (25). Our choice of these two ligands was based on their high affinities for GluN2B, acceptable lipophilicities (moderate clogD values), and amenability to labeling with carbon-11 at a methyl group (Table 1), in addition to low affinity for s1 receptors (Table 2).
We have previously reported the labeling of L3 and its enantiomers in the S-methyl position with carbon-11 for their evaluation for PET imaging of brain GluN2B in rat brain.(24) Labeling was achieved readily by treatment of a methyl thiopropanoate precursor (L19 or an enantiomer) with [11C]methyl iodide, as fully described in the Supporting Information of reference 24. Both enantiomers of [11C]L3 performed well as radioligands for PET imaging in rats, by showing both high brain uptake and specific signals uncontaminated by radiometabolites.(24)
[11C]L6 and its enantiomers have been labeled by palladium-mediated treatment of the boronic ester precursor L21 or enantiomer with [11C]methyl iodide, as fully described in the Supporting Information of reference 25. These radioligands also entered rat brain avidly to give receptor-specific signals that were uncontaminated by radiometabolites. Unexpectedly high binding seen in cerebellum with these radioligands was determined not to be off-site binding to s1 receptors.(25)
Conclusions
In this study, we synthesized and evaluated more than twenty-two 3-alkylaryl derivatives of 7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol and its enantiomers as GluN2B ligands. We explored variation in the alkyl group linking the head 2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol moiety to a terminal aryl group, and the nature of the terminal aryl group. The measured Ki values correlated with clogD values and demonstrated that the binding affinity derives strongly from hydrophilic interactions between the ligand and protein binding pocket. Ligands emerged with properties desirable in candidate PET radioligands, namely L3 and L6, and their enantiomers. These properties included high affinity, high selectivity, moderate lipophilicity, stereoselectivity, and amenability to labeling with carbon-11. We have labeled these ligands with carbon-11 and in separate studies have evaluated their PET radioligand performance. [11C](S)-L3 ([11C](S)-NR2B-Me) gave high specific PET signal in rhesus monkey brain and the highest binding potentials (BPND) values across brain regions in comparison with those of two other radioligands, [18F](R)-OF-Me-NB1 (Chart 1) and [18F](S)-OF-NB1 (Chart 1) (26) showing that lower binding radioligands are preferred based on faster washout for better mathematical model of pharmacokinetics.
Materials and Methods
Starting Materials
7-Methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol was obtained from Ambeed Inc. (Arlington Heights, Il). 4-Phenyl-1-butanol, 4-(4-methoxyphenyl)-1-butanol, 4-tolylbutan-1-ol, 4-(4-bromophenyl)butan-1-ol, 4-(4-fluorophenyl)butan-1-ol, 4-(p-iodophenyl)butyric acid, 3-butyn-1-ol, 5-hexyn-1-ol, 2-iodophenol, 4-iodobenzotrifluoride, diborane, triethylamine, bis(triphenylphosphine)palladium(II) dichloride, [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II)·CH2Cl2), and 4-(4-nitrophenyl)-1-butanol, were obtained from Aldrich (St Louis, MO). 2-Naphthalenebutanol was obtained from AstaTech (Bristol, PA). (Pyridin-2-yl)but-3-yn-1-ol, 2-bromo-6-fluoropyridine, 4-bromo-2-iodophenol, 4-fluoro-2-iodophenol, 2-iodo-4(trifluoromethyl)-phenol were obtained from Combi-Blocks (San Diego, CA).
Instruments and General Methods
A microwave apparatus (Discover CP-D; CEM; Matthews, NC) was used for microwave promoted reactions where indicated. A Combi-Flash apparatus (Teledyne Labs, Lincoln, NE) with a silica gel cartridge (4–80 g) was used in compound purifications, as later indicated. A Centrifan PE apparatus (Fisher Scientific; Waltham, MA) was used for drying many isolated compounds. Melting points were measured with a glass capillary on a digital melting point apparatus (SMP 20; Cole-Parmer Ltd, Stone, UK). Preparative, analytical, and chiral HPLC methods are detailed in supporting information (general methods A-F). Purities were calculated as area percentage of all chromatogram peaks. HRMS (ESI or EI) were acquired from Mass Spectrometry Lab (University of Illinois Urbana-Champaign, https://illinois.edu/). Optical rotation measurements were performed on Jasco P-1010 Polarimeter.
LC-MS was performed on an Acquity UPLC M-Class System (Waters; Milford, MA), using a UPLC BEH column (1.7 mm, 2.1 × 50 mm) eluted at 0.4 mL/min with 0.1% formic acid in H2O (A)/0.1% formic acid in acetonitrile (B) (B from 5 to 90% over 7 min).
1 H-NMR spectra were obtained at 400 MHz and 13C-NMR spectra at 101 MHz at room temperature (RT) on a multinuclear instrument (Bruker Biospin) in deuterated solvent. TMS (δ = 0 ppm) was used as an internal standard for 1H and 13C-NMR spectroscopy. The abbreviations s, d, t, m, q, quint, dd, dt, brs, vs, vt, and AB denote singlet, doublet, triplet, multiplet, quartet, quintet, double doublet, double triplet, broad singlet, virtual singlet, virtual triplet, and AB coupling, respectively.
Chemistry
4. -(4-Iodophenyl)butan-1-ol (1).(48) 4-(p-Iodophenyl)butyric acid (11.87 g, 40.9 mmol) was dissolved in THF (100 mL) from a SealSure bottle (Sigma Aldrich, Burlington, MA). The solution was stirred while BH3·THF (1 M, 100 mL) was added slowly, resulting in gas release. The addition was controlled and the reaction temperature was maintained below 40 oC. A clear, colorless solution formed after 5 min. Reversed phase HPLC (general method A) showed complete conversion of the starting acid to the alcohol. Extending the reaction time to 72 h showed no observable change. Methanol (100 mL) was then added slowly to quench any remaining borane, resulting in hydrogen gas evolution. Once gas evolution had ceased, water (100 mL) was added gradually; no additional gas or heat was generated. The reaction mixture was then stirred at RT for 1 h. A saturated solution of ammonium chloride (100 mL) was then added. The product was extracted with DCM (3 × 100 mL) and the combined organic layers were dried (MgSO₄) and the solvent removed under vacuum. The resultant oil was redissolved in acetonitrile, transferred, and then dried with Centrifan to give 1 as a pale yellow oil (14.87 g, yield 86%). 1H-NMR (CDCl3): δ 7.59 (d, 2H, 3JHH = 12 Hz, Ar-H), 6.93 (d, 2H, 3JHH = 8.0 Hz, Ar-H), 3.64 (t, 2H, 3JHH = 6.0 Hz, CH2OH), 2.58 (t, 2H, 3JHH = 8.0 Hz, CH2OH), 1.68 (m, 2H, CH2), 1.59 (m, 2H, CH2). 13C-NMR (CDCl3): δ 142.07 (CAr), 137.44 (CAr), 130.69 (CAr), 90.90 (CAr-I), 62.71 (CH2OH), 36.24 (CH2), 32.26 (CH2), 27.49 (CH2).
4-(4-Methylthiophenyl)butan-1-ol (2).(49) 4-(p-Iodophenyl)butan-1-ol (1; 2.01 g, 7.3 mmol), NaSMe (0.60 g, 8.6 mmol), Me3SnCl (1.67 g, 8.4 mmol), and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II)·CH2Cl2 (0.82 g, 1.1 mmol) were dissolved in DMSO (10 mL) in a 35-mL microwave tube. The reaction mixture was then heated in a microwave reactor (110 °C, 30 min, 50 W, 250 psi). Reversed phase HPLC (general method A) showed complete conversion. The reaction mixture was then dissolved in DCM (200 mL) and washed with water. The organic layer was then dried over MgSO4, filtered, and then added to silica gel (200 mesh, 50 mL). The solvent was removed from the silica gel slurry under vacuum and the reaction mixture was purified (Combi-Flash; silica, hexanes/ethyl acetate). LC-MS analysis confirmed product isolation. The product fractions were dried under vacuum. The resulting residue was then redissolved in acetonitrile, and passed through a 0.2 µm syringe filter, and then dried with Centrifan to give 2 as a brown oil (0.69 g, yield 48%). 1H-NMR (CDCl3): δ 7.20 (d, 2H, 3JHH = 8.0 Hz, Ar-H), 7.11 (d, 2H, 3JHH = 8.0 Hz, Ar-H), 3.66 (t, 2H, 3JHH = 6.0 Hz, CH2OH), 2.61 (t, 2H, 3JHH = 8.0 Hz, CH2OH), 2.47 (s, 3H, CH3), 1.68 (m, 2H, CH2), 1.59 (m, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 139.69 (CAr), 135.41 (CAr), 129.17 (CAr), 127.39 (C Ar-I), 63.01 (CH2OH), 35.28 (CH2), 32.47 (CH2), 27.72 (CH2), 16.59 (SCH3). HRMS calcd for C11H17O2S [M + OH]+: m/z = 213.0952, found 213.0949; error: 1.4 ppm.
4-(4-(Trifluoromethyl)phenyl)but-3-yn-1-ol (3).(50) 1-Iodo-4-(trifluoromethyl)benzene (20.0 g, 73.5 mmol), CuI (2.1 g, 11.0 mmol), Pd(PPh3)2Cl2·CH2Cl2 (2.9 g, 3.7 mmol), and but-3-yn-1-ol (10.3 g, 147.1 mmol) were added to triethylamine (300 mL) under nitrogen. The mixture was heated at 60 °C for 16 h. Reversed phase HPLC (general method B) showed near complete consumption of the starting material. The reaction solution was cooled to RT, ethyl acetate (550 mL) was added, stirred for 20 min, and then filtered. The filter cake was then washed with ethyl acetate (200 mL). The combined ethyl acetate washings were then dried under vacuum to give a black oil, which was purified (Combi-Flash; silica gel, petroleum ether/ethyl acetate, from 50: 1 to 3: 1 v/v) to give 3 as a light-yellow liquid (9.7 g, yield 62.0%). 1H-NMR (CDCl3): δ 7.55 (d, 2H, 3JHH = 8.0 Hz, Ar-H), 7.50 (d, 2H, 3JHH = 8.0 Hz, Ar-H), 3.84 (t, 2H, 3JHH = 6.0 Hz, CH2OH), 2.72 (t, 2H, 3JHH = 8.0 Hz, CH2CH2OH). 13C{1H}-NMR (CDCl3): δ 132.12 (CH), 129.92 (q, 2JCF = 32.3 Hz), 127.41, 125.39 (q, 2JCF = 4.0 Hz, CH), 124.14 (q, 1JCF = 270 Hz, CF3), 89.41 (C2), 81.43 (C2), 61.22 (CH2O), 23.99. HRMS calcd for C11H10OF3 [M + H]+: m/z = 215.0684, found 215.0681; error: –0.4 ppm.
4-(4-(Trifluoromethyl)phenyl)butan-1-ol (4).(50) Compound 3 (9.7 g, 45.3 mmol) was dissolved in methanol (160 mL) and Pd/C (1.0 g, 10% wt) was added. The reaction flask was then evacuated and filled with hydrogen gas to 1 atmosphere. The reaction mixture was then stirred at 40 ℃ for 8 h. TLC showed consumption of the starting material. The reaction mixture was then cooled to RT and filtered. After solvent removal under vacuum, the reaction mixture was purified (Combi-Flash; silica gel, hexane/ethyl acetate) to give 4 as a pale yellow liquid (7.7 g, yield 78%). 1H-NMR (CDCl3): δ 7.53 (d, 2H, 3JHH = 8.0 Hz, Ar-H), 7.29 (d, 2H, 3JHH = 8.0 Hz, Ar-H), 3.67 (t, 2H, 3JHH = 6.0 Hz, CH2OH), 2.71 (t, 2H, 3JHH = 6.0 Hz, CH2CH2OH), 1.72 (quint, 2H, 3JHH = 6.0 Hz, CH2CH2OH), 1.62 (quint, 2H, 3JHH = 6.0 Hz, CH2CH2OH). 13C{1H}-NMR (CDCl3): δ 146.62, 128.90 (CH), 128.38 (q, 2JCF = 33.0 Hz, CCF), 127.41, 125.45 (q, 2JCF = 4.0 Hz, CHCF), 124.57 (q, 1JCF = 270 Hz, CF3), 62.88 (CH2O), 35.68, 32.37, 27.52.
4-(6-Fluoropyridin-2-yl)but-3-yn-1-ol (5). 2-Bromo-6-fluoropyridine (6.11 g, 34.7 mmol), but-3-yn-1-ol (2.61g, 37.2 mmol), CuBr (509.6 mg, 3.6 mmol), Pd(PPh3)2Cl2 (1.2 g, 1.7 mmol), and PPh3 (0.96 g, 3.7 mmol) were dissolved in Et2NH (10 mL) resulting in a green solution and white precipitate. This mixture was stirred at RT for 1 h and then heated at 88 oC for 24 h. The solution turned brown after stirring at RT for 5 min, forming an off-white precipitate. HPLC showed complete conversion after heating for 3 h, however, heating continued for the full 24 h. The next day, solvent was removed under vacuum and the residue was redissolved in DCM (200 mL). Silica gel (80 mL) was added, the solvent was removed under vacuum, and the reaction mixture was purified (Combi-Flash; silica gel, hexane/ethyl acetate) to give 5 as a colorless oil (0.50 g, yield 88%). 1H-NMR (CDCl3): δ 7.73 (dd, 1H, 3JHH = 8.0 Hz, Ar-H), 7.27 (dd, 1H, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, Ar-H), 6.88 (dd, 1H, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, Ar-H), 3.86 (t, 2H, 3JHH = 6.0 Hz, CH2OH), 2.71 (t, 2H, 3JHH = 8.0 Hz, CH2OH), 3.20 (brs, 1H, OH). 13C{1H}-NMR (CDCl3): δ 163.03 (d, 1JCF = 241.5 Hz, Fpy, CF), 141.52 (d, 4JCF = 8.0 Hz, Fpy, CH), 141.37 (CH), 124.55 (d, 4JCF = 4.0 Hz, Fpy, CH), 89.31 (C2), 80.76 (C2), 60.85 (CH2O), 23.90. HRMS calcd for C9H9NOF [M + H]+: m/z = 166.0668, found 166.0665; error: –1.8 ppm.
2-(Benzofuran-2-yl)ethan-1-ol (6).(51,52) 2-Iodophenol (1.069 g, 4.86 mmol), but-3-yn-1-ol (0.340 g, 4.85 mmol), CuBr (67.8 mg, 0.47 mmol), (Ph3P)2●PdCl2 (172.4 mg, 0.246 mmol), and Ph3P (152.7 mg, 0.582 mmol) were dissolved in Et2NH (10 mL). The reaction mixture was heated at 90 o C for 4 h. Reversed phase HPLC (general method B) showed starting material consumption. The reaction mixture was left to stir overnight. The following morning DCM (200 mL) was added and this was stirred for 5 min. Silica gel (20 mL) was then added. After removal of the solvent under vacuum, the reaction mixture was purified (Combi-Flash, silica gel, ethyl acetate/hexane) to give 6 as a colorless oil (0.531 g, yield 67%). 1H-NMR (CDCl3): δ 7.50 (dd, 3JHH = 8.0 Hz, 4JHH = 2.0 Hz, 1H, Ar-H), 7.42 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 7.24 (dt, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, 1H, Ar-H), 7.19 (dt, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, 1H, Ar-H), 6.50 (s, 1H, Ar-H), 3.98 (t, 3JHH = 6.0 Hz, 2H, CH2O), 3.04 (t, 3JHH = 6.0 Hz, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 156.14, 155.01, 128.88, 123.74 (CH), 122.84 (CH), 120.63 (CH), 111.06 (CH), 103.89 (CH), 60.95 (CH2O), 32.26 (CH2).
2-(5-Bromobenzofuran-2-yl)ethan-1-ol (7).(53) Use of the method for 6 in same molar proportions to 4-bromo-2-iodophenol (2.12 g, 7.10 mmol) gave 7 as a colorless oil (1.26 g, yield 76%). 1H-NMR (CDCl3): δ 7.61 (s, 1H, Ar-H), 7.32 (d, 3JHH = 8.9 Hz, 1H, Ar-H), 7.28 (d, 3JHH = 8.6 Hz, 1H, Ar-H), 6.45 (s, 1H, Ar-H), 3.98 (t, 3JHH = 6.2 Hz, 2H, CH2O), 3.03 (t, 3JHH = 6.2 Hz, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 157.74, 153.76, 130.92, 126.58 (CH), 123.29 (CH), 115.88, 112.49 (CH), 103.41 (CH), 60.76 (CH2O), 32.20 (CH2).
2-(5-Fluorobenzofuran-2-yl)ethan-1-ol (8). Use of the method for 6 in same molar proportions to 4-fluoro-2-iodophenol (2.29 g, 9.61 mmol) gave 8 as a colorless oil (1.41 g, yield 81%). 1H-NMR (CDCl3): δ 7.33 (dd, 3JHF = 8.0 Hz, 4JHF = 4.0 Hz, 1H, Ar-H), 7.14 (dd, 3JHH = 8.0 Hz, 4JHF = 2.0 Hz, 1H, Ar-H), 6.94 (dt, 3JHF = 3JHH = 8.0 Hz, 4JHF = 2.0 Hz, 1H, Ar-H), 6.47 (s, 1H, Ar-H), 3.98 (t, 3JHH = 6.0 Hz, 2H, CH2O), 3.02 (t, 3JHH = 6.0 Hz, 2H, CH2). 13C{1H}-NMR (CDCl3)3: δ 159.36 (d, 1JCF = 236.0 Hz, CF), 158.18, 151.21, 129.71 (d, C, JCF = 11.0 Hz), 111.55 (d, CH, JCF = 9.0 Hz), 111.23 (d, CH, JCF = 26 Hz), 106.20 (d, CH, JCF = 25.0 Hz), 104.11 (d, CH, JCF = 4.0 Hz), 60.78 (CH2O), 32.28 (CH2).
2-(5-(Trifluoromethyl)benzofuran-2-yl)ethan-1-ol (9). Use of method for 6 in same molar proportions to 2-iodo-4-(trifluoromethyl)phenol (1.06 g, 3.68 mmol) gave 9 as a colorless oil (661 mg, yield 78%). 1H-NMR (CDCl3): δ 7.79 (s, 1H, Ar-H), 7.50 (s, 2H, Ar-H), 6.59 (s, 1H, Ar-H), 4.02 (t, 3JHH = 6.2 Hz, 2H, CH2O), 3.08 (t, 3JHH = 6.2 Hz, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 158.34, 156.36, 129.02, 126.25, 125.58 (q, 2JCF = 31.9 Hz, CCF3), 123.55, 120.94 (q, 3JCF = 3.6 Hz, CHCCF3), 118.30 (q, 3JCF = 3.6 Hz, CHCCF3), 111.40 (CH), 104.10 (CH), 60.74 (CH2O), 32.22 (CH2).
4-(Benzofuran-2-yl)butan-1-ol (10).(54) 2-Iodophenol (1.363 g, 6.20 mmol), hex-5-yn-1-ol (0.646 g, 6.58 mmol), CuBr (94.5 mg, 0.66 mmol), (Ph3P)2PdCl2 (222.3 mg, 0.32 mmol), and Ph3P (161.6 mg, 0.62 mmol) were dissolved in Et2NH (10 mL). The reaction mixture was heated at 90 oC for 4 to 5 h, turning a deep red. HPLC (general method B) showed consumption of the starting material. The reaction mixture was stirred overnight. DCM (200 mL) was then added, and this was stirred for 5 min. Silica gel (20 mL) was then added and the solvent was removed under vacuum. The reaction mixture was purified (Combi-Flash, silica gel, ethyl acetate/hexane) to give 10 as a colorless oil (986 mg, yield 84%). 1H-NMR (CDCl3): δ 7.49 (d, 3JHH = 7.4 Hz, 1H, Ar-H), 7.42 (d, 3JHH = 7.6 Hz, 1H, Ar-H), 7.23–7.16 (m, 2H, Ar-H), 6.40 (s, 1H, Ar-H), 3.68 (t, 3JHH = 6.4 Hz, 2H, CH2O), 2.81 (t, 3JHH = 6.4 Hz, 2H, CH2), 1.86–1.82 (m, 2H, CH2), 1.69–1.59 (m, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 159.31, 154.81, 129.10, 123.32 (CH), 122.60 (CH), 120.39 (CH), 110.89 (CH), 102.23 (CH), 62.70 (CH2O), 32.29, 28.33, 24.14.
4-(5-(Trifluoromethyl)benzofuran-2-yl)butan-1-ol (11). Use of the method for 10 in same molar proportions to 2-iodo-4-(trifluoromethyl)phenol (1.06 g, 3.68 mmol) gave 11 as a colorless oil (637 mg, yield 67%). 1H-NMR (CDCl3): δ 7.76 (vs, 1H, Ar-H), 7.47 (vs, 2H, Ar-H), 6.46 (s, 1H, Ar-H), 3.70 (t, 3JHH = 8.0 Hz, 2H, CH2O), 2.83 (t, 3JHH = 8.0 Hz, 2H, CH2), 1.89‒1.82 (m, 2H, CH2), 1.71–1.51 (m, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 161.44, 156.22, 129.28, 129.20 (CH), 129.10, 125.34 (q, 2JCF = 31.7 Hz), 124.97 (q, 1JCF = 270 Hz, CF3), 120.56 (q, 2JCF = 3.7 Hz, CH), 110.05 (q, 2JCF = 4.0 Hz, CH), 62.66 (CH2O), 32.26, 28.37, 24.06.
4-Phenylbutyl 4-methylbenzenesulfonate (X1). 4-Phenylbutan-1-ol (6.5 mL, d = 0.984 g/mL, 42.6 mmol), tosyl chloride (15.11 g, 79.3 mmol), and Et3N (21 mL, d = 0.726 g/mL, 15.2 g, 151 mmol) were dissolved in DCM (100 mL) and stirred at RT overnight. A saturated sodium solution (200 mL) was added the following day and this was extracted with DCM . The organic layer was dried (MgSO4), filtered, and then added to silica gel (60 mL). The solvent was removed under vacuum. The reaction mixture was purified (Combi-Flash, silica gel, hexane/ethyl acetate) to give XI as a colorless waxy material (10.0 g, yield 77%). 1H-NMR (CDCl3): δ 7.78 (d, 3JHH = 12 Hz, 2H, Ar-H), 7.33 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.26 (vt, 3JHH = 8.0 Hz, 2H, Ar-H), 7.17 (t, 3JHH = 8.0 Hz, 1H, Ar-H), 7.10 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 4.04 (t, 3JHH = 6.0 Hz, 2H, CH2O), 2.56 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.45 (s, 3H, CH3), 1.66 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 144.88, 141.75, 133.35, 130.02 (CH), 128.54 (CH), 128.06 (CH), 126.10 (CH), 70.58 (CH2O), 35.26, 28.51, 27.26, 21.82 (CH3). HRMS calcd for C17H24NO3 [M + NH4]+: m/z = 322.1477, found 322.1476; error: –0.3 ppm.
4-(4-Iodophenyl)butyl 4-methylbenzenesulfonate (X2). Compound 1 (1.20 g, 4.35 mmol), tosyl chloride (1.07 g, 5.61 mmol), and Et3N (2.0 mL, d = 0.726 g/mL, 14.3 mmol) were dissolved in DCM (20 mL) and stirred at RT for 24 h. Reaction progress was monitored with reversed phase HPLC (general method B). The next day, silica gel (25 mL) was added and the solvent removed under vacuum. The reaction mixture was then purified (Combi-Flash, silica gel, hexane/ethyl acetate) to give X2 as a white crystalline solid (1.13 g. yield 61%). Mp: 58–60 °C. 1H-NMR (CDCl3): δ 7.77 (d, 3JHH = 8.2 Hz, 2H, Ar-H), 7.57 (d, 3JHH = 8.2 Hz, 2H, Ar-H), 7.33 (d, 3JHH = 8.1 Hz, 2H, Ar-H), 6.86 (d, 3JHH = 8.2 Hz, 2H, Ar-H), 4.03 (t, 3JHH = 5.8 Hz, 2H, CH2O), 2.51 (t, 3JHH = 5.8 Hz, 2H, CH2O), 2.45 (s, 3H, CH3), 1.63–1.61 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 144.96, 141.38, 137.61 (CH), 133.32, 130.67 (CH), 130.05 (CH), 128.08 (CH), 91.15, 70.42 (CH2O), 34.78, 28.45, 27.11, 21.86 (CH3). HRMS calcd for C17H23NO3SI [M + NH4]+: m/z = 448.0443, found 448.0441; error: –0.4 ppm.
4-(4-(Methylthio)phenyl)butyl 4-methylbenzenesulfonate (X3). Compound 2 (0.51 g, 2.60 mmol), tosyl chloride (0.56 g, 2.94 mmol), and Et3N (2.1 mL, d = 0.726 g/mL, 15.0 mmol) were dissolved in DCM (20 mL). The reaction mixture was stirred at RT for 24 h. Reaction progress was monitored with reversed phase HPLC (general method B). The next day, silica gel (10 mL) then was added and the solvent removed under vacuum. The reaction mixture was then purified (Combi-Flash, silica gel, hexane/ethyl acetate) to give X3 as a colorless oil (0.43 g, yield 47%). 1H-NMR (CDCl3): δ 7.77 (d, 3JHH = 12 Hz, 2H, Ar-H), 7.33 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.17 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.03 (d, 3JHH = 12 Hz, 2H, Ar-H), 4.03 (t, 3JHH = 6.0 Hz, 2H, CH2O), 2.52 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.46 (s, 3H, CH3), 2.44 (s, 3H, CH3), 1.64 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 144.91, 138.88, 135.70, 133.38, 130.04, 129.11, 128.09, 127.34, 70.54 (CH2O), 34.73, 28.49, 27.27, 21.86 (CH3), 16.51 (SCH3). HRMS calcd for C18H26NO3S2 [M + NH4]+: m/z = 368.1354, found 368.1348; error: –1.6 ppm.
4-(4-(Trifluoromethyl)phenyl)butyl 4-methylbenzenesulfonate (X4). Use of the method for X3 in same molar proportions to 5 (1.0 g, 4.58 mmol) gave X4 as a white crystalline solid (1.26 g, yield 74%). Mp: 52–54 °C. 1H-NMR (CDCl3): δ 7.78 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.51 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.33 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.22 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 4.04 (d, 3JHH = 6.0 Hz, 2H, CH2), 2.65 (d, 3JHH = 6.0 Hz, 2H, CH2), 1.68 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 145.87, 144.99, 133.31, 130.05, 128.85, 128.55 (2JCF = 32.2 Hz), 128.07, 125.50 (3JCF = 4.0 Hz), 124.51 (1JCF = 272.7 Hz), 70.33 (CH2O), 35.11, 28.49, 27.06, 21.82 (CH3). HRMS calcd for C18H23NO3SF3 [M + NH4]+: m/z = 390.1351, found 390.1347; error: –1.0 ppm.
4-(4-Methoxyphenyl)butyl 4-methylbenzenesulfonate (X5). Use of the method for X3 in the same molar proportions to 4-(4-methoxyphenyl)butan-1-ol (1.0 g, 5.5 mmol) gave X5 as a pale yellow oil (0.39 g, yield 21%). 1H-NMR (CDCl3): δ 7.77 (d, 3JHH = 8.1 Hz, 2H, Ar-H), 7.33 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.01 (d, 3JHH = 8.4 Hz, 2H, Ar-H), 7.28 (d, 3JHH = 8.5 Hz, 2H, Ar-H), 4.03 (t, 3JHH = 6.0 Hz, 2H, CH2), 3.78 (s, 3H, OCH3), 2.50 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.44 (s, CH3), 1.65–1.58 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 158.03, 144.87, 133.82, 133.38, 130.01 (CH), 129.42 (CH), 128.07 (CH), 113.96 (CH), 70.63 (CH2O), 55.45 (OCH3), 34.35, 28.47, 27.49, 21.83 (CH3). HRMS calcd for C18H26NO4S [M + NH4]+: m/z = 352.1583, found 352.1581; error: –0.6 ppm.
4-(p-Tolyl)butyl 4-methylbenzenesulfonate (X6). Use of the method for X3 in the same molar proportions to 4-(p-tolyl)butan-1-ol (1.0 g, 6.1 mmol) gave X6 as a colorless waxy solid (0.46 g, yield 24%). 1H-NMR (CDCl3): δ 7.78 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.33 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.07 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 6.99 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 4.03 (t, 3JHH = 6.0 Hz, 2H, CH2O), 2.52 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.44 (s, 3H, CH3), 2.31 (s, 3H, CH3), 1.66‒1.61 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 144.87, 138.67, 135.57, 133.40, 130.02 (CH), 129.24 (CH), 128.43 (CH), 128.09 (CH), 70.63 (CH2O), 34.83, 28.53, 27.38, 21.85 (CH3), 21.19 (CH3). HRMS calcd for C18H26NO3S [M + NH4]+: m/z = 336.1633, found 366.1633; error: 0.0 ppm.
4-(4-Bromophenyl)butyl 4-methylbenzenesulfonate (X7). 4-(4-Bromophenyl)butan-1-ol (1.0 g, 4.4 mmol), tosyl chloride (0.99 g, 5.2 mmol) and Et3N (2 mL, d = 0.726 g/mL, 14.3 mmol) were dissolved in DCM (20 mL). The reaction mixture was stirred at RT for 5 d. Reaction progress was monitored with HPLC(general method A). Next, silica gel (60 mL) was added and the solvent removed under vacuum. The reaction mixture was then purified (Combi-Flash, silica gel, hexane/ethyl acetate) to give X7 as a pale yellow waxy solid (0.97 g, yield 58%). 1H-NMR (CDCl3): δ 7.77 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.36 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.33 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 6.97 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 4.02 (t, 3JHH = 6.0 Hz, 2H, CH2O), 2.51 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.44 (s, 3H, CH3), 1.64–1.62 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 144.93, 140.68, 133.27, 131.56 (CH), 130.28 (CH), 130.01 (Ts, CH), 128.02 (Ts, CH), 119.80, 70.40 (CH2O), 34.63, 28.40, 27.10, 21.81 (CH3). HRMS calcd for C17H23NO3SBr [M + NH4]+: m/z = 400.0582, found 400.0588; error: 1.5 ppm.
4-(4-Fluorophenyl)butyl 4-methylbenzenesulfonate (X8). 4-(4-Fluorophenyl)butan-1-ol (1.0 g, 4.4 mmol), tosyl chloride (1.22 g, 6.4 mmol), and Et3N (2.5 mL, d = 0.726 g/mL, 17.9 mmol) were dissolved in DCM (20 mL). The reaction mixture was stirred at RT for 5 d. Next, silica gel (20 mL) was added, and the solvent was removed under vacuum. The reaction mixture was then purified (Combi-Flash, silica gel, hexane/ethyl acetate) to give X8 as a colorless oil (0.99 g. yield 52%). 1H-NMR (CDCl3): δ 7.78 (d, 3JHH = 12.0 Hz, 2H, Ar-H), 7.33 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.06 (dd, 3JHH = 8.0 Hz, 3JHF = 4.0 Hz, 2H, Ar-H), 6.94 (t, 3JHH = 8.0 Hz, 2H, Ar-H), 4.03 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.54 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.44 (s, CH3), 1.64 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 161.49 (d, 1JCF = 243.5 Hz, Fph, C-F), 144.92, 137.6 (d, 4JCF = 3.0 Hz, Fph, CHCHCHCF), 133.35, 130.03 (CH), 129.85 (d, 3JCF = 8.0 Hz, Fph, CHCHCF), 128.07 (CH), 115.29 (d, 2JCF = 21.1 Hz, Fph, CHCF), 70.49 (CH2O), 34.47, 28.46, 27.42, 21.83 (CH3). HRMS calcd for C17H23NO3SF [M + NH4]+: m/z = 340.1383, found 340.1378; error: –1.5 ppm.
4-(Naphthalen-2-yl)butyl 4-methylbenzenesulfonate (X9). Use of the method for X8 in the same molar proportions to 4-(naphthalen-2-yl)butan-1-ol (1.0 g, 5.0 mmol) gave X9 was as a tan crystalline solid (0.39 g, yield 22%). Mp: 34–36 °C). 1H-NMR (CDCl3): δ 7.81–7.75 (m, 5H, Ar-H), 7.54 (s, 1H, Ar-H), 7.46 (t, 3JHH = 6.5 Hz, 1H, Ar-H), 7.42 (t, 3JHH = 6.5 Hz, 1H, Ar-H), 7.30 (d, 3JHH = 8.0 Hz, 2H, Ts, Ts-H), 7.26 (d, 3JHH = 8.3 Hz, 1H, Np-H), 4.06 (t, 3JHH = 5.8 Hz, 2H, CH2), 2.74 (t, 3JHH = 5.8 Hz, 2H, CH2), 2.42 (s, CH3), 1.75–1.70 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 144.87, 139.24, 133.75, 133.34, 132.22, 130.01 (Ts, CH), 128.14 (CH), 128.06 (Ts, CH), 127.80 (CH), 127.58 (CH), 127.32 (CH), 126.63 (CH), 126.16 (CH), 125.42 (CH), 70.59 (CH2O), 35.40, 28.52, 27.13, 21.80 (CH3). HRMS calcd for C21H26NO3S [M + NH4]+: m/z = 372.1633, found 372.1638; error: 1.3 ppm.
4-(Pyridin-2-yl)butyl 4-methylbenzenesulfonate (X10). 4-(Pyridin-2-yl)butan-1-ol (5.0 g, 33.1 mmol), tosyl chloride (7.66 g, 40.2 mmol), and Et3N (12 mL, d = 0.726 g/mL, 86.1 mmol) were dissolved in ethyl acetate (100 mL). The reaction mixture was stirred at RT for 24 h. Reaction progress was monitored with HPLC. The next day, silica gel (60 mL) was added and the solvent removed under vacuum. The reaction mixture was then purified (Combi-Flash, silica gel, hexane and ethyl acetate) to give X10 as a thick colorless oil (2.09 g, yield 21%). 1H-NMR (CDCl3): δ 8.42 (d, 3JHH = 4.0 Hz, 1H, py-H), 8.36 (s, 1H, py-H), 7.76 (d, 3JHH = 12.0 Hz, 2H, Ts-H), 7.42 (d, 3JHH = 8.0 Hz, 1H, py-H), 7.32 (d, 3JHH = 12.0 Hz, 2H, Ts-H), 7.18 (dd, 3JHH = 8.0 Hz, 3JHH = 4.0 Hz, 1H, py-H), 4.02 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.55 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.42 (s, 3H, CH3), 1.66–1.64 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 149.99 (CH), 147.70 (CH), 144.96, 136.88, 135.85 (CH), 133.18, 130.00 (Ts, CH), 127.99 (Ts, CH), 123.46, 70.22 (CH2O), 32.34, 28.40, 27.00, 21.77 (CH3). HRMS calcd for C7H7O3S-: m/z = 171.0116, found 171.0119. Error (ppm): 1.8. HRMS calcd for C9H12N+: m/z = 134.0970, found 134.0967; error: –2.2 ppm.
4-(Pyridin-2-yl)but-3-yn-1-yl 4-methylbenzenesulfonate (X11). 4-(Pyridin-2-yl)but-3-yn-1-ol (1.99 g, 13.5 mmol), tosyl chloride (3.11 g, 16.3 mmol), and Et3N (6.0 mL, d = 0.726 g/mL, 43.0 mmol) were dissolved in DCM (100 mL). The reaction mixture was stirred at RT for 24 h. Reaction progress was monitored with HPLC (general method B). The next day, silica gel (60 mL) was added and the solvent removed under vacuum. The reaction mixture was then purified (Combi-Flash, silica gel, hexane/ethyl acetate) to give X11 as a clear waxy solid (0.88 g, yield 22%). 1H-NMR (CDCl3): δ 8.54 (d, 3JHH = 8.0 Hz, 1H, py-H), 7.82 (d, 3JHH = 8.0 Hz, 2H, Ts-H), 7.63 (dt, 3JHH = 8.0 Hz, 4JHH = 2.0 Hz, 1H, py-H), 7.34 (d, 3JHH = 8.0 Hz, 1H, py-H), 7.32 (d, 3JHH = 8.0 Hz, 2H, Ts-H), 7.21 (dd, 3JHH = 8.0 Hz, 3JHH = 4.0 Hz, 1H, py-H), 4.20 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.82 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.43 (s, 3H, CH3). 13C{1H}-NMR (CDCl3): δ 150.13 (py, CH), 145.21, 143.20, 136.33 (py, CH), 132.98, 130.13 (Ts, CH), 128.22 (Ts, CH), 127.20 (py, CH), 123.08 (py, CH), 84.36 (C2), 82.48 (C2), 67.49 (CH2O), 21.86 (CH3), 20.50. HRMS calcd for C16H16NO3S [M + H]+: m/z = 302.0851, found 302.0854; error: 1.0 ppm.
4-(6-Fluoropyridin-2-yl)but-3-yn-1-yl 4-methylbenzenesulfonate (X12). 4-(6-Fluoropyridin-2-yl)but-3-yn-1-ol (3, 2.65 g, 16.0 mmol), tosyl chloride (3.18 g, 16.7 mmol), and Et3N (10 mL, d = 0.726 g/mL, 71.7 mmol) were dissolved in DCM (40 mL). The reaction mixture was stirred at RT for 24 h. Reaction progress was monitored with HPLC (general method B). The next day silica gel (60 mL) was added and the solvent removed under vacuum. The reaction mixture was then purified (Combi-Flash, (silica gel, hexane/ethyl acetate) to give X12 as a clear waxy solid (0.68 g, yield 13%). 1H-NMR (CDCl3): δ 7.82 (d, 3JHH = 8.1 Hz, 2H, Ar-H), 7.73 (pseudo-q, 3JHH = 3JHF = 7.9 Hz, 1H, Ar-H), 7.34 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.23 (dd, 3JHH = 7.4 Hz, 5JHF = 1.9 Hz, 1H, Ar-H), 6.88 (dd, 3JHH = 8.3 Hz, 3JHF = 2.7 Hz, 1H, Ar-H), 4.19 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.81 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.44 (s, CH3). 13C{1H}-NMR (CDCl3): δ 163.1 (d, 1JCF = 250 Hz), 145.28, 141.38 (d, JCF = 8.1 Hz), 141.25 (d, JCF = 37.5 Hz), 132.93, 130.18 (Ts, CH), 128.22 (Ts, CH), 124.69 (d, JCF = 4.0 Hz), 109.66 (d, 1JCF = 36.6 Hz), 85.87 (C2), 81.23 (C2), 67.28 (CH2O), 21.86, 20.50 (CH3). HRMS calcd for C16H15NO3FS [M + H]+: m/z = 320.0757, found 320.0763; error: 1.9 ppm.
2-(Benzofuran-2-yl)ethyl 4-methylbenzenesulfonate (X13). Use of the method for X12 in the same molar proportions to 6 (910 mg, 5.61 mmol) gave X13 as a white crystalline solid (799 mg, yield 45%). Mp: 76–77 °C. 1H-NMR (CDCl3): δ 7.67 (d, 3JHH = 8.0 Hz, 2H, Ts, Ar-H), 7.46 (d, 3JHH = 7.3 Hz, 1H, BFu, Ar-H), 7.30 (d, 3JHH = 7.7 Hz, 1H, BFu, Ar-H,), 7.24‒7.18 (m, 2H, BFu Ar-H), 7.17 (d, 3JHH = 8.0 Hz, 2H, Ts, Ar-H), 6.42 (s, 1H, BFu, Ar-H), 4.36 (t, 3JHH = 6.3 Hz, 2H, CH2), 3.12 (t, 3JHH = 6.3 Hz, 2H, CH2), 2.37 (s, 3H, CH3). 13C{1H}-NMR (CDCl3): δ 154.90, 153.29, 144.98, 132.83, 129.92 (Ts, CH), 128.64, 127.99 (Ts, CH), 123.92 (BFu, CH), 122.87 (BFu, CH), 120.77 (BFu, CH), 111.02 (BFu, CH), 104.54 (BFu, CH), 67.55 (CH2O), 28.75 (CH2), 21.81 (CH3). HRMS calcd for C17H20NO4S [M + NH4]+: m/z = 334.1113, found 334.1114; error: 0.3 ppm.
2-(5-Bromobenzofuran-2-yl)ethyl 4-methylbenzenesulfonate (X14). Use of the method for X12 in the same molar proportions to 7 (790 mg, 3.28 mmol) gave X14 as a white crystalline solid (699 mg, yield 54%). Mp: 88–90 °C. 1H-NMR (CDCl3): δ 7.67 (d, 3JHH = 8.0 Hz, 2H, Ts, Ar-H), 7.58 (s, 1H, BFu, Ar-H), 7.31 (d, 3JHH = 8.7 Hz, 1H, BFu, Ar-H), 7.18 (d, 3JHH = 8.0 Hz, 3JHH = 8.7 Hz, 3H, BFu-Ts, Ar-H), 6.36 (s, 1H, BFu, Ar-H), 4.36 (t, 3JHH = 6.3 Hz, 2H, CH2), 3.11 (t, 3JHH = 6.3 Hz, 2H, CH2), 2.38 (s, 3H, CH3). 13C{1H}-NMR (CDCl3): δ 154.91, 153.65, 145.06, 132.84, 130.64, 129.93 (Ts, CH), 127.97 (Ts, CH), 126.82 (BFu, CH), 123.44 (BFu, CH), 115.93, 112.48 (BFu, CH), 104.10 (BFu, CH), 67.32 (CH2O), 28.76 (CH2), 21.83 (CH3). HRMS calcd for C17H19NO4SBr [M + NH4]+: m/z = 412.0218, found 412.0225; error: 1.7 ppm.
2-(5-Fluorobenzofuran-2-yl)ethyl 4-methylbenzenesulfonate (X15). Use of the method for X12 in the same molar proportions to 8 (610 mg, 3.39 mmol) gave X15 as a white crystalline solid (815 mg, yield 72%). Mp: 76–78 °C. 1H-NMR (CDCl3): δ 7.68 (d, 3JHH = 8.1 Hz, 2H, Ts, Ar-H), 7.23 (dd, 3JHF = 4.1 Hz, 3JHH = 8.9 Hz, 1H, BFu), 7.20 (d, 3JHH = 8.1 Hz, 2H, Ts, Ar-H), 7.11 (dd, 4JHF = 2.5 Hz, 3JHH = 9.0 Hz, 1H, BFu, Ar-H), 6.93 (td, 3JHF = 4JHH = 8.6 Hz, 4JHH = 2.5 Hz, 1H, BFu, Ar-H), 6.40 (s, 1H), 4.36 (t, 3JHH = 6.4 Hz, 2H, CH2), 3.11 (t, 3JHH = 6.4 Hz, 2H, CH2), 2.38 (s, 3H, CH3). 13C{1H}-NMR (CDCl3): δ 154.35 (d, 1JCF = 236.4 Hz), 155.32, 151.13, 145.04, 132.87, 129.94 (Ts, CH), 129.45 (d, JCF = 10.8 Hz, CH), 128.01 (Ts, CH), 111.64 (d, JCF = 2.7 Hz, CH), 111.46 (d, JCF = 13.8 Hz, CH), 106.35 (d, JCF = 25.0 Hz, CH), 104.81 (d, JCF = 4.0 Hz, CH), 67.37 (CH2O), 28.84 (CH2), 21.82 (CH3). HRMS calcd for C17H19NO4FS [M + NH4]+: m/z = 352.1019, found 352.1017; error:–0.6 ppm.
2-(5-Trifluoromethylbenzofuran-2-yl)ethyl 4-methylbenzenesulfonate (X16). Use of the method for X12 in the same molar proportions to 9 (1.07 g, 4.65 mmol) gave X16 as a white crystalline solid (697 mg, yield 39%). Mp: 94–95 °C. 1H-NMR (CD3OD): δ 7.74 (s, 1H, Ar-H), 7.53 (dd, 3JHH = 6.6 Hz, 4JHH = 1.6 Hz, 2H, Ar-H), 7.44 (AB, 3JHH = 8.7 Hz, 4JHH = 1.7 Hz, 1H, Ar-H), 7.40 (AB, 4JHH = 8.7 Hz, 1H, Ar-H), 7.09 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 6.52 (d, 4JHH = 0.7 Hz, 1H, Ar-H), 4.32 (t, 3JHH = 5.7 Hz, 2H, CH2), 3.07 (t, 3JHH = 5.7 Hz, 2H, CH2), 2.24 (s, 3H, Ar-CH3). 13C{1H}-NMR (CD3OD): δ 157.93, 157.68, 146.47, 134.19, 130.96 (Ts, CH), 130.44, 128.95 (Ts, CH), 126.37 (q, 1JCF = 271.0 Hz, 1C, CF), 126.51 (q, 2JCF = 32.0 Hz, 1C), 122.06 (q, 3JCF = 3.6 Hz, 1C, BFu, CH), 119.38 (q, 3JCF = 4.3 Hz, 1C, BFu CH), 112.58 (BFu, CH), 105.70 (BFu, CH), 69.02, (CH2O), 29.40 (CH 2), 21.66 (CH3). HRMS calcd for C18H19NO4F3S [M + NH4]+: m/z = 402.0987, found 402.0992; error: 1.2 ppm.
4-(Benzofuran-2-yl)butyl 4-methylbenzenesulfonate (X17). Use of the method for X12 in the same molar proportions to 10 (710 mg, 3.73 mmol) gave X17 as a clear waxy solid (977 mg, yield 76%). 1H-NMR (CDCl3): δ 7.78 (d, 3JHH = 8.0 Hz, 2H, Ts, Ar-H), 7.47 (d, 3JHH = 7.4 Hz, 1H, BFu, Ar-H), 7.39 (d, 3JHH = 7.6 Hz, 1H, BFu, Ar-H), 7.32 (d, 3JHH = 8.0 Hz, 2H, Ts, Ar-H), 7.21–7.17 (m, 2H, BFu, Ar-H), 6.34 (s, 1H, BFu, Ar-H), 4.07 (t, 3JHH = 5.7 Hz, 2H, CH2), 2.72 (t, 3JHH = 5.7 Hz, 2H, CH2), 2.43 (s, 3H, CH3), 1.76 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 158.49, 154.83, 144.95, 132.83, 130.05 (Ts, CH), 128.98, 128.08 (Ts, CH), 123.48 (BFu, CH), 122.69 (BFu, CH), 120.47 (BFu, CH), 111.93 (BFu, CH), 102.53 (BFu, CH), 70.29 (CH2O), 28.41 (CH2), 27.84 (CH2), 23.82 (CH2), 21.82 (CH3). HRMS calcd for C19H24NO4S [M + NH4]+: m/z = 362.1426, found 362.1419; error: –1.9 ppm.
4-(5-(Trifluoromethyl)benzofuran-2-yl)butyl 4-methylbenzenesulfonate (X18). Use of the method for X12 in the same molar proportions to 11 (425 mg, 1.65 mmol) gave X18 as a clear waxy solid (380 mg, yield 56%). 1H-NMR (CDCl3): δ 7.78 (d, 3JHH = 8.0 Hz, 2H, Ts-H), 7.76 (s, 1H, Ar-H), 7.49-7.45 (AB, 3JHH = 8.0 Hz, 2H, Ar-H), 7.32 (d, 3JHH = 8.0 Hz, 2H, Ts-H), 6.42 (s, 1H, Ar-H), 4.07 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.77 (t, 3JHH = 6.0 Hz, 2H, CH2), 2.43 (s, 3H, CH3), 1.83–1.61 (m, 4H, CH2CH2). 13C{1H}-NMR (CDCl3): δ 160.62, 156.22, 145.02 (Ts), 133.26 (Ts), 130.06 (Ts, CH), 129.07, 128.08 (Ts, CH), 125.44 (q, 2JCF = 32 Hz), 124.93 (q, 1JCF = 271 Hz, CF3), 120.72 (q, 2JCF = 3.0 Hz, CHCF), 110.13 (q, 2JCF = 4.0 Hz, CHCF), 70.14 (CH2O), 28.43, 27.87, 23.73, 21.82. HRMS calcd for C20H23NO4F3S [M + NH4]+: m/z = 430.1300, found 430.1305; error: 1.2 ppm.
7-Methoxy-3-(4-(phenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L1). 7-Methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (190 mg, 0.98 mmol), X1 (390 mg, 1.28 mmol), and K2CO3 (610 mg, 4.4 mmol) were suspended in acetonitrile (10 mL) and refluxed for 6 d. Reaction progress was monitored with HPLC (general method A). The reaction mixture was cooled to RT and passed through a 0.2 μm syringe filter. The reaction mixture was then purified with reversed phase HPLC (general method C). The solvent was then removed under vacuum and the product was redissolved in acetonitrile, passed through a 0.2 μm syringe filter, and then dried with Centrifan to give L1 as a white crystalline solid (0.23 g, yield 72%). Mp: 78–79 °C. 1H-NMR (CDCl3): δ 7.28 (t, 3JHH = 8.0 Hz, 2H, m-Ar-H), 7.18 (t, 3JHH = 8.0 Hz, 1H, p-Ar-H), 7.18 (d, 3JHH = 8.0 Hz, 2H, o-Ar-H), 7.10 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 6.64 (dd, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, 1H, Ar-H), 6.63 (brs, 1H, Ar-H), 4.59 (d, 3JHH = 4.0 Hz, 1H, CH-OH), 3.76 (s, 3H, OCH3), 3.26 (vt, 2JHH = 12 Hz, 1H, CH2), 3.18–3.14 (m, 1H, CH2), 3.02–2.97 (m, 1H, CH2), 2.68–2.59 (m, 5H, CH2), 2.53 (d, 2JHH = 12 Hz, 1H, CH2), 2.43 (vt, 2JHH = 12 Hz, 1H, CH2), 1.69–1.62 (m, 2H, CH2), 1.61–1.54 (m, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 159.09, 142.43, 141.25, 135.57, 129.86, 128.57 (CH), 128.52 (CH), 125.97 (CH), 116.74 (CH), 110.39 (CH), 72.42 (CHO), 60.88 (CN), 59.76 (CN), 56.19 (CN), 55.38 (OCH3), 36.52, 35.92, 29.28, 26.66. HRMS calcd for C21H28NO2 [M + H]+: m/z = 326.2120, found 326.2126; error: 1.8 ppm. HPLC (general method A): tR = 5.14 min, purity 99.62%.
7-Methoxy-3-(4-(4-iodophenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L2) and enantiomers. 7-Methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (420 mg, 2.17 mmol), X2 (1.13 g, 2.63 mmol), and Na2HPO4 (1.30 g, 9.2 mmol) were suspended in acetonitrile (8 mL) and heated at 75 oC for 5 d. Reaction progress was monitored with HPLC (general method A). Next, the reaction mixture was cooled to RT and passed through a 0.2 μm syringe filter and then purified with reversed phase HPLC (general method C). The solvent was then removed under vacuum. The residue was redissolved in ethanol, passed through a 0.2 µm syringe filter and then dried with Centrifan to give L2 as a white crystalline solid (0.714 g, yield 73%). Mp: 100–101 °C. 1H-NMR (CDCl3): δ 7.60 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.16 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 6.92 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 6.69 (dd, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, 1H, Ar-H), 6.65 (d, 4JHH = 4.0 Hz, 1H, Ar-H), 4.76 (brs, 1H, CHOH), 3.78 (s, 3H, OCH3), 3.39–3.32 (m, 2H, CH2), 3.16–3.12 (m, 1H, CH2), 2.75 (brs, 4H, CH2), 2.60–2.58 (m, 3H, CH2), 1.63 (m, 4H, CH2). 13C{1H}-NMR (CDCl3): δ 159.34, 141.60, 140.31, 137.64 (CH), 134.50, 130.67 (CH), 129.80, 116.62 (CH), 110.89 (CH), 91.16 (CAr-I), 71.47 (CHO), 60.33 (CN), 59.45 (CN), 55.79 (CN), 55.46 (OCH3), 42.47, 35.20, 28.81, 25.46, 11.26. HRMS calcd for C21H27INO2 [M + H]+: m/z = 452.1086, found 452.1092; error: 1.3 ppm. HPLC (general method A): tR = 7.31 min, purity 100%. Chiral HPLC analysis (general method E): (S)-L2 (tR = 6.97 min), 50.35%; (R)-L2 (tR = 10.96 min), 47.92%.
The enantiomers were resolved and purified with chiral HPLC (general method F). (S)-L2: chiral HPLC (general method E): tR = 6.89 min, purity 98.14%; [α]D²⁰ = –39.02° (c 1.0, CHCl3). [α]D²⁰ = +8.89° (c 1.0, EtOH). (R)-L2: chiral HPLC (general method E): tR = 11.24 min, HPLC purity 99.2%; [α]D²⁰ = +34.03° (c 1.0, CHCl3), [α]D²⁰ = –3.67° (c 1.0, EtOH).
7-Methoxy-3-(4-(4-(methylthio)phenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L3), and its enantiomers. 7-Methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (200 mg, 1.03 mmol), X3 (450 mg, 1.28 mmol), and Na2HPO4 (620 mg, 4.4 mmol) were suspended in acetonitrile (2 mL) and heated at 75 oC for 2 d. Reaction progress was monitored with HPLC (general method A) . Next, the reaction mixture was cooled to RT and passed through a 0.2 μm syringe filter and then purified with reversed phase HPLC (general method C). The solvent was removed under vacuum, the product was redissolved in ethanol and Na2CO3 (1.0 g, 9.4 mmol) was added. The mixture was sonicated for 5 min and then stirred at RT for 1 min before finally being passed through a 0.2 µm syringe filter and then dried with a Centrifan to give L3 as a white crystalline solid (0.288 g, yield 75%). Mp: 89–91 °C. 1H-NMR (CDCl3): δ 7.18 (AB, 3JHH = 8.0 Hz, 2H, Ar-H), 7.09 (AB, 3JHH = 8.0 Hz, 1H, Ar-H), 7.08 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 6.63 (dd, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, 1H, Ar-H), 6.62 (brs, 1H, Ar-H), 4.56 (d, 3JHH = 4.0 Hz, 1H, CHOH), 3.75 (s, 3H, OCH3), 3.25 (vt, 2JHH = 12 Hz, 1H, CH2), 3.17–3.12 (m, 1H, CH2), 3.00–2.95 (m, 1H, CH2), 2.66–2.60 (m, 1H, CH2), 2.58 (t, 3JHH = 8.0 Hz, 4H, CH2), 2.50 (d, 2JHH = 12 Hz, 1H, CH2), 2.45 (s, 3H, SCH3), 2.39 (vt, 2JHH = 12 Hz, 1H, CH2), 1.62 (quint, 3JHH = 8.0 Hz, 2H, CH2), 1.54 (quint, 3JHH = 8.0 Hz, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 159.14, 141.36, 139.64, 135.69, 135.45, 129.94, 129.15 (CH), 127.39 (CH), 116.82 (CH), 110.40 (CH), 72.60 (CHO), 60.97 (CN), 59.79 (CN), 56.31 (CN), 55.42 (OCH3), 37.04, 35.40, 29.28, 26.77, 16.57 (SCH3). HRMS calcd for C22H30NO2S [M + H]+: m/z = 372.1997, found 372.1999; error: 0.51 ppm. HPLC (general method A): tR = 7.31 min, purity 100%. Chiral HPLC (general method E): (S)-L3: tR = 8.05 min, 48.03%; (R)-L3: tR = 13.22 min, 48.06%.
The enantiomers were resolved and purified with chiral HPLC (general method F). (S)-L3 chiral HPLC (general method E): tR = 8.10 min, 98.01%. [α]D²⁰ = –39.02° (c 1.0, CHCl3), [α]D²⁰ = +8.89° (c 1.0, EtOH). (R)-L3: chiral HPLC (general method E: tR = 13.27 min, 99.19%. [α]D²⁰ = +34.03° (c 1.0, CHCl3), [α]D²⁰ = –3.67° (c 1.0, EtOH).
7-Methoxy-3-(4-(4-trifluoromethylphenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L4). Use of the method for L3 in the same molar proportions to X4 (240 mg, 0.64 mmol) gave L4 as a white crystalline solid (0.232 g, yield 71%). Mp: 79–81 °C. 1H-NMR (CDCl3): δ 7.52 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.26 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.14 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 6.67 (dd, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, 1H, Ar-H), 6.63 (d, 4JHH = 4.0 Hz, 1H, Ar-H), 4.74 (brs, 1H, CHOH), 3.76 (s, 3H, OCH3), 3.35‒3.27 (m, 2H, CH2), 3.17–3.08 (m, 1H, CH2), 2.73-2.49 (m, 7H, CH2), 1.65 (brs, 4H, CH2). 13C{1H}-NMR (CD3OD): δ 160.26, 148.55, 141.78, 137.22, 130.23 (2C, CH),129.29 (q, 2JCF = 31.9 Hz, 1C, CCF), 127.67 (CH), 126.34 (q,3JCF = 3.9 Hz, 2C, CHCHCF), 126.08 (q, 1JCF = 271.0 Hz, 1C, CF), 116.57 (CH), 111.72 (CH), 72.37 (OCH), 63.71 (CN), 60.11 (CN), 56.53 (CN), 55.75 (OCH3), 36.58 (CH2), 30.31 (CH2), 27.24 (CH2). HRMS calcd for C22H27F3NO2 [M + H]+: m/z = 394.1994, found 394.1998; error: 1.0 ppm. HPLC (general method A): tR = 7.21 min, purity 100%.
7-Methoxy-3-(4-(4-methoxyphenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L5). 7-Methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (100 mg, 0.52 mmol), X5 (270 mg, 0.81 mmol), and Na2HPO4 (430 mg, 3.0 mmol) were suspended in acetonitrile (2 mL) and heated at 90oC for 2 d. Reaction progress was monitored with HPLC (general method A). The reaction mixture was cooled to RT, passed through a 0.2 μm syringe filter, and then purified with reversed phase HPLC (general method C). The solvent was then removed under vacuum. The recovered product was then redissolved in acetonitrile, passsed through a 0.2 μm syringe filter, and dried with Centrifan to give L5 as a white crystalline solid (0.21 g, yield 87%). Mp: 86–89 °C. 1H-NMR (CDCl3): δ 7.18 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 7.06 (3JHH = 8 Hz, 2H, Ar-H), 6.81 (3JHH = 8.0 Hz, 2H, Ar-H), 6.69 (dd, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, 1H, Ar-H), 6.62 (d, 4JHH = 4.0 Hz, 1H, Ar-H), 4.83 (d, 3JHH = 4.0 Hz, 1H, CHOH), 3.77 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.37 (vt, 2JHH = 12 Hz, 1H, CH2), 3.20–3.29 (m, 1H, CH2), 3.15 (m, 1H, CH2), 2.86–2.76 (m, 5H, CH2), 2.57 (vt, 2JHH = 12 Hz, 2H, CH2), 1.61 (m, 4H, CH2). 13C{1H}-NMR (CDCl3): δ 159.31, 158.02, 139.94, 134.24, 133.88, 129.56, 129.40 (2C, CH), 116.43 (CH), 114.00 (2C, CH), 111.01 (CH), 70.85 (CHO), 60.08 (CN), 59.39 (CN), 55.48 (CN), 55.41 (OCH3), 34.65, 34.33, 29.09, 24.92, 21.20. HRMS calcd for C22H30NO3 [M + H]+: m/z = 356.2226, found 356.2223; error: –0.8 ppm. HPLC (general method A): tR = 5.05 min, purity 99.52%.
7-Methoxy-3-(4-(4-methylphenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L6) and enantiomers. Use of the method for L5 in the same molar proportions with 7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (130 mg, 0.67 mmol), X6 (260 mg, 0.82 mmol), and Na2HPO4 (630 mg, 4.4 mmol) in acetonitrile (10 mL) gave L6 as a white crystalline solid (88 mg, 37%). Mp: 86–87 °C. 1H-NMR (CDCl3): δ 7.11 (d, 3JHH = 12 Hz, 1H, Ar-H), 7.10 (AB, 3JHH = 8 Hz, 2H, Ar-H), 7.07 (AB, 3JHH = 8.0 Hz, 2H, Ar-H), 6.66 (dd, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, 1H, Ar-H), 6.54 (d, 4JHH = 4.0 Hz, 1H, Ar-H), 4.60 (d, 3JHH = 4.0 Hz, 1H, CHOH), 3.78 (s, 3H, OCH3), 3.28 (vt, 2JHH = 12 Hz, 1H, CH2), 3.20–3.16 (m, 1H, CH2), 3.04–2.99 (m, 1H, CH2), 2.69–2.58 (m, 5H, CH2), 2.54 (d, 2JHH = 12 Hz, 1H, CH2), 2.43 (vt, 2JHH = 12 Hz, 1H, CH2), 2.32 (s, 3H, CH3), 1.68–1.61 (m, 2H, CH2), 1.59–1.53 (m, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 159.14, 141.31, 139.36, 135.63, 135.41, 129.91, 129.23 (CH), 128.46 (CH), 116.79 (CH), 110.43 (CH), 72.49 (CHO), 60.92 (CN), 59.83 (CN), 56.24 (CN), 55.42 (OCH3), 35.89, 35.48, 29.42, 26.70, 21.20 (CH3). HRMS calcd for C22H30NO2 [M + H]+: m/z = 340.2277, found 340.2271; error: –1.8 ppm. HPLC (general method A): tR = 6.27 min, purity 100%.
The enantiomers were resolved and purified with HPLC (general method F): Chiral HPLC (general method E): (S)-L6, tR = 5.76 min, 44.43%; (R)-L6, tR = 9.44 min, 51.46%.
(S)-L6: chiral HPLC (general method E): tR = 5.93min, 98.28%; [α]D²⁰ = –24.23° (c 1.0, CHCl3). (R)-L6: chiral HPLC (general method E): tR = 9.57 min, 99.28%. [α]D²⁰ = +28.23° (c 1.0, CHCl3).
7-Methoxy-3-(4-(4-bromophenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L7). Use of the method for L5 in the same molar proportions with 7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (210 mg, 1.1 mmol), X7 (400 mg, 1.04 mmol), and Na2HPO4 (700 mg, 4.9 mmol) in acetonitrile (2 mL) heated at 90 oC for 5 d gave L7 as a white crystalline solid (0.35 g, yield 62%). Mp: 93–96 °C. 1H-NMR (CDCl3): δ 7.40 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.11 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 7.05 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 6.66 (dd, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, 1H, Ar-H), 6.64 (d, 4JHH = 4.0 Hz, 1H, Ar-H), 4.62 (d, 3JHH = 4.0 Hz, 1H, CHOH), 3.77 (s, 3H, OCH3), 3.28 (vt, 2JHH = 12 Hz, 1H, CH2), 3.20–3.15 (m, 1H, CH2), 3.04–2.99 (m, 1H, CH2), 2.70–2.55 (m, 6H, CH2), 2.46 (vt, 2JHH = 12 Hz, 1H, CH2), 1.66–1.60 (m, 2H, CH2), 1.58–1.53 (m, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 159.16, 141.31, 141.13, 135.44, 131.59 (CH), 130.34 (CH), 129.87, 119.72 (CH), 116.76 (CH), 110.49 (CH), 72.36 (CHO), 60.85 (CN), 59.65 (CN), 56.19 (CN), 55.41 (OCH3), 36.64, 35.29, 29.08, 26.48. HRMS calcd for C21H27BrNO2 [M + H]+: m/z = 404.1225, found 404.1229; error: 1.0 ppm. HPLC (general method A): tR = 6.90 min, purity 96.64%.
7-Methoxy-3-(4-(4-fluorophenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L8). Use of the method for L5 in the same molar proportions with 7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (120 mg, 0.62 mmol), X8 (260 mg, 0.81 mmol), and Na2HPO4 (450 mg, 3.2 mmol) in acetonitrile (2 mL) gave L8 as a brown oil (0.23 g, yield 79%). 1H-NMR (CDCl3): δ 7.13 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 7.11 (d, 3JHH = 8.0 Hz, 2H, Ar-H), 6.96 (t, 3JHH = 3JHF = 8.0 Hz, 2H, Ar-H), 6.65 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 6.63 (brs, 1H, Ar-H), 4.62 (d, 3JHH = 4.0 Hz, 1H, CHOH), 3.77 (s, 3H, OCH3), 3.27 (vt, 2JHH = 12 Hz, 1H, CH2), 3.19‒3.15 (m, 1H, CH2), 3.03‒2.99 (m, 1H, CH2), 2.70–2.55 (m, 6H, CH2), 2.46 (vt, 2JHH = 12 Hz, 1H, CH2), 1.67–1.60 (m, 2H, CH2), 1.58‒1.53 (m, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 162.64 (C-F), 160.23 (C-F), 159.16, 141.13, 137.97, 137.94, 135.46, 129.89 (CH, JCF), 129.81 (CH, JCF), 116.75 (CH), 115.36 (CH, JCF), 115.15 (CH, JCF), 110.49 (CH), 72.34 (CHO), 60.86 (CN), 59.69 (CN), 56.17 (CN), 55.41 (OCH3), 36.65, 35.07, 29.37, 26.47. HRMS calcd for C21H27NO2F [M + H]+: m/z = 344.2026, found 344.2025; error: –0.3 ppm. HPLC (general method A): tR = 5.55 min, purity 98.74%.
7-Methoxy-3-(4-(naphthalen-2-yl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L9). Use of the method for L7 in the same molar proportions with7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (100 mg, 0.52 mmol), X9 (250 mg, 0.71 mmol), and Na2HPO4 (280 mg, 2.0 mmol) in acetonitrile (10 mL) gave L9 as a white crystalline solid (0.14 g, yield 74%). Mp: 86–88 °C. 1H-NMR (CDCl3): δ 7.81 (d, 3JHH = 8.0 Hz, 1H, Nap-H), 7.78 (d, 3JHH = 8.0 Hz, 1H, Nap-H), 7.77 (d, 3JHH = 8.0 Hz, 1H, Nap-H), 7.60 (s, 1H, Nap-H), 7.44 (p, 3JHH = 7.0 Hz, 2H, Nap-H), 7.31 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 7.14 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 6.68 (dd, 3JHH = 8.0 Hz, 4JHH = 4.0 Hz, 1H, Ar-H), 6.63 (d, 4JHH = 4.0 Hz, 1H, Ar-H), 4.72 (d, 3JHH = 4.0 Hz, 1H, CHOH), 3.76 (s, 3H, OCH3), 3.34 (vt, 2JHH = 12 Hz, 1H, CH2), 3.29–3.24 (m, 1H, CH2), 3.11–3.07 (m, 1H, CH2), 2.81–2.66 (m, 6H, CH2), 2.57 (vt, 2JHH = 12 Hz, 1H, CH2), 1.78–1.70 (m, 2H, CH2), 1.69‒1.62 (m, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 159.20, 139.69, 134.96, 133.78, 132.19, 129.79 (CH), 129.09, 128.12 (CH), 127.79 (CH), 127.61 (CH), 127.40 (CH), 126.68 (CH), 126.11 (CH), 125.34 (CH), 116.61 (CH), 110.67 (CH), 71.66 (CHO), 60.57 (CN), 59.62 (CN), 55.91 (CN), 55.40 (OCH3), 35.89, 35.69, 28.91, 25.89. HRMS calcd for C25H30NO2 [M + H]+: m/z = 376.2277, found 376.2272; error: –1.3 ppm. HPLC (general method A): tR = 5.69 min, purity 99.13%.
3-(4-(Pyridin-2-yl)butyl)-7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L10). Method 1. 7-Methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (15.3 mg, 0.52 mmol), X10 (39.7 mg, 0.66 mmol), and Na2HPO4 (73.3 mg, 2.0 mmol) were suspended in DMSO (1.0 mL). The reaction mixture was then heated in a microwave reactor (three conditions were utilized: i) 80 °C 10 min, 30 W, 250 psi; ii) 120 °C, 10 min, 50 W, 250 psi; iii) 150 °C, 10 min, 60 W, 250 psi). A cyclized by-product (Scheme 7) due to cyclization of X10 was the only product observed when analyzed with LC-MS.
Method 2. 7-Methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (150 mg, 0.52 mmol), X10 (300 mg, 0.66 mmol) and Na2HPO4 (510 mg, 2.0 mmol) were suspended in acetonitrile (10 ml) and heated at 90 oC for 22 d. Again, a by-product formed by cyclization of X10 (Scheme 7) was the only product observed when analyzed with LC-MS.
7-Methoxy-3-(4-(pyridin-2-yl)but-3-yn-1-yl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L11). Use of the method for L7 in the same molar proportions with X11 (240 mg, 0.80 mmol) gave L11 a brown oil (0.268 g, yield 77%). 1H-NMR (CD3OD): δ 8.34 (ddd, 3JHH = 5.0 Hz, 1 H, Ar-H), 7.68 (td, 3JHH = 7.8 Hz, 4JHH = 1.8 Hz, 1H, Ar-H), 7.37 (vt, 3JHH = 7.9 Hz, 1H, Ar-H), 7.23 (dd, 3JHH = 7.7 Hz ,1 H, Ar-H), 7.17 (d, 3JHH = 8.4 Hz, 1H, Ar-H), 6.62 (dd, 3JHH = 8.3 Hz, 4JHH = 2.7 Hz, 1H, Ar-H), 6.57 (d, 4JHH = 2.6 Hz, 1H, Ar-H), 4.67 (d, 3JHH = 7.6 Hz, 1H, CHOH), 3.66 (s, 3H, OCH3), 2.92 (m, 1H, CH2), 2.83 (m, 3H, CH2), 2.73 (m, 3H, CH2), 2.59 (m, 3H, CH2). 13C{1H}-NMR in (CD3OD): δ 160.35, 150.42 (CH), 144.54, 142.03, 138.66, 137.06, 128.66 (CH), 128.37 (CH), 124.42 (CH), 116.76 (CH), 111.69 (CH), 91.08 81. 69, 72.97 (CHO), 63.09 (CN), 58.77 (CN), 56.21 (CN), 55.76 (OCH3), 36.99 (CH2), 18.12 (CH2). HRMS calcd for C20H23N2O2 [M + H]+: m/z = 323.1760, found 323.1757; error: ‒0.9 ppm. HPLC (general method B): tR = 6.50 min, purity 100%.
3-(4-(6-Fluoropyridin-2-yl)but-3-yn-1-yl)-7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L12). 7-Methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (134 mg, 0.69 mmol) and X12 (274 mg, 0.86 mmol) were mixed and then heated in a microwave reactor (135 °C,10 min, 60 W, 250psi) to give a black solid upon cooling to RT. The solid was dissolved in DMF (3 mL), passed through a 0.2 μm syringe, and then purified with HPLC (general method C). The solvent was then removed under vacuum. The product was redissolved in acetonitrile, passed through a 0.2 μm syringe filter, and then dried with Centrifan to give L12 as a brown oil (29 mg, yield 12%). 1H-NMR (CDCl3) δ 7.70 (dd, 3JHH = 8.0 Hz, 3JHF = 16.0, 1H, Ar-H), 7.28 (dd, 3JHH = 7.1 Hz, 4JHH = 1.8 Hz, 1H, Ar-H), 7.11 (d, 3JHH = 7.9 Hz, 1H, Ar-H), 6.86 (dd, 3JHH = 8.3 Hz, 4JHH = 2.5 Hz, 1H, Ar-H), 6.67 (dd, 3JHH = 8.3 Hz, 4JHH = 2.6 Hz, 1H, Ar-H), 6.65 (s, 1H, Ar-H), 4.63 (d, 3JHH = 6.8 Hz, 1H, CH-OH), 3.78 (s, 3H, OCH3), 3.27 (m, 2H, CH2), 3.09 (m, 1H, CH2), 2.96 (t, 3JHH = 7.1 Hz, 2H, CH2), 2.68 (m, 4H, CH2), 2.58 (t, 3JHH = 12.0 Hz, 1H, CH2). 13C{1H}-NMR(CDCl3): δ 163.08 (d, 1JCF = 240 Hz, 1C, CF), 159.18, 141.62 (d, 3JCF = 15.1 Hz, 1C, CNCF), 141.37 (d, 3JCF = 8.2 Hz, 1C, CHCHCF), 141.19, 135.50, 130.06, 124.54 (d, 4JCF = 4.2 Hz, 1C, CHCHCHCF), 116.87 (CH), 110.50 (CH), 109.22 (d, 2JCF = 36.7 Hz, 1C, CHCF), 90.15, 80.64, 72.73 (CHO), 60.70 (CN), 58.07 (CN), 55.96 (CN), 55.42 (OCH3), 37.32 (CH2), 18.27 (CH2). HRMS calcd for C20H22N2O2F [M + H]+: m/z = 341.1665, found 323.1667. error: 0.6 ppm. HPLC (general method B): tR = 7.60 min, purity 98.69%.
3-(2-(Benzofuran-2-yl)ethyl)-7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L13). 7-Methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (100 mg, 0.52 mmol), X13 (184 mg, 0.582 mmol) and Na2HPO4 (250 mg, 1.76 mmol) were suspended in acetonitrile (10 mL) and heated at 90 oC for 22 d to give L13 as a brown oil (139 mg, yield 71%). 1H-NMR (CD3OD): δ 7.48 (dd, 3JHH = 7.1 Hz, 4JHH = 1.4, 1H, Ar-H), 7.39 (d, 3JHH = 8.1 Hz, 1H, Ar-H), 7.28 (d, 3JHH = 8.3 Hz, 1H, Ar-H), 7.20 (td, 3JHH = 7.3 Hz, 4JHH = 1.4 Hz, 1H, Ar-H), 7.16 (td, 3JHH = 7.4 Hz, 4JHH = 1.2 Hz, 1H, Ar-H), 6.73 (dd, 3JHH = 8.3 Hz, 4JHH = 2.6 Hz, 1H, Ar-H), 6.68 (d, 4JHH = 2.6 Hz, 1H, Ar-H), 6.53 (s, 1H, Ar-H), 4.79 (d, 3JHH = 8.0 Hz, 1H, CHOH), 3.77 (s, 3H, OCH3), 3.03 (t, 3JHH = 3.1 Hz, 4H, CH2), 2.97 (m, 2H, CH2), 2.86 (m, 2H, CH2), 2.78 (t, 3JHH = 11.7 Hz, 1H, CH2), 2.66 (t, 3JHH = 10.5 Hz, 1H, CH2). 13C{1H}-NMR (CD3OD): δ 160.31, 158.78, 156.28, 141.96, 137.06, 130.40, 128.27 (CH), 124.54 (CH), 123.72 (CH), 121.51 (CH), 116.75 (CH), 111.67 (CH), 103.90 (CH), 72.82 (CHO), 63.21 (CN), 58.41 (CN), 56.28 (CN) 55.75 (OCH3), 36.87 (CH2), 27.00 (CH2). HRMS calcd for C21H24NO3 [M + H]+: m/z = 338.1756, found 338.1751. error: ‒1.5 ppm. HPLC (general method A): tR = 4.59 min, purity 97.52%
3-(2-(5-Bromobenzofuran-2-yl)ethyl)-7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L14). Use of the method for L12 in the same molar proportions with X14 (142 mg, 0.359 mmol) gave L14 as a brown oil (76 mg, yield 51%). 1H-NMR (CDCl3): δ 7.61 (s, 1H, Ar-H), 7.33 (AB, 3JHH = 8.0 Hz, 1H, Ar-H), 7.27 (AB, 3JHH = 8.0 Hz, 1H, Ar-H), 7.21 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 7.13 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 6.72 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 6.65 (s, 1H, Ar-H), 6.46 (s, 1H, Ar-H), 4.92 (brs, 1H, CHOH), 3.78 (s, 3H, OCH3), 3.34 (brs, 2H, CH2), 3.21 (brs, 2H, CH2), 3.13 (q, 3JHH = 8.0 Hz, 2H, CH2), 2.75‒2.69 (m, 2H, CH2), 1.32 (t, 3JHH = 8.0 Hz, 2H, CH2). 13C{1H}-NMR (CDCl3): δ 159.30, 153.68, 130.84 (CH), 130.07, 129.12, 126.67 (CH), 126.13, 123.35 (CH), 116.77 (CH), 115.95 (CH), 112.46 (CH), 110.73 (CH), 103.10 (CBr), 72.03 (CHO), 60.52 (CN), 57.72 (CN), 56.00 (CN), 55.45 (OCH3), 45.90, 26.33. HRMS calcd for C21H23BrNO3 [M + H]+: m/z = 416.0861, found 416.0865; error: 1.0 ppm. HPLC (general method A): tR = 6.52 min, purity 100%.
3-(2-(5-Fluorobenzofuran-2-yl)ethyl)-7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L15). Use of the method for L12 in the same molar proportions with X15 (214 mg, 0.640 mmol) gave L16 as a brown wax (134 mg, yield 59%). 1H-NMR (CD3OD): δ 7.38 (dd, 3JHH = 3.9 Hz, 1H, Ar-H), 7.26 (d, 3JHH = 8.3 Hz, 1H, Ar-H), 7.20 (dd, 3JHH = 8.8, 4JHH = 2.4 Hz, 1H, Ar-H), 6.96 (td, 3JHH = 9.3 Hz, 4JHH = 2.9 Hz, 1H, Ar-H), 6.74 (dd, 3JHH = 8.4 Hz, 4JHH = 2.4 Hz, 1H, Ar-H), 6.71 (d, 4JHH = 2.4 Hz, 1H, Ar-H), 6.58 (s, 1H, Ar-H), 4.82 (t, 3JHH = 4.6 Hz CH-OH), 3.77 (s, 3H, O-CH3), 3.13 (br, 5H, CH2), 2.95 (br, 5H, CH2). 13C{1H}-NMR (CD3OD): δ 160.78 (d, 1J CF = 235 Hz, 1C, CF), 160.66, 159.93, 152.60, 141.64, 136.24, 131.29 (d, 3JCF = 11.0 H, 1C, CHCHCF), 129.17, 116.94 (CH), 112.53 (d, 3JCF = 9.9 Hz, 1C, CHCHCF), 112.20 (CH), 111.95 (CH), 107.06 (d, 2J CF = 25.2 Hz, 1C, CHCF), 104.71 (d, 4JCF = 3.7 Hz, 1C, CHCHCHCF), 72.34 (CHO), 62.21 (CN), 58.09 (CN), 56.62 (CN), 55.80 (OCH3), 35.75 (CH2), 26.43 (CH2). HRMS calcd for C21H23NO3F [M + H]+: m/z = 356.1662, found 356.1658; error: –1.1 ppm. HPLC (general method A): tR = 5.38 min, purity 99.57%.
3-(2-(5-Trifluoromethylbenzofuran-2-yl)ethyl)-7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L16). Use of the method for L11 in the same molar proportions with X16 (178 mg, 0.463 mmol) gave L16 as a thick brown oil (120 mg, yield 64%). 1H-NMR (CD3OD): δ 7.85 (s, 1H, Ar-H), 7.58 (AB, 3JHH = 8.6 Hz, 1H, Ar-H), 7.52 (AB, 3JHH = 8.6 Hz, 4JHH = 1.5 Hz, 1H, Ar-H), 7.28 (d, 3JHH = 8.4 Hz, 1H, Ar-H), 6.73 (dd, 3JHH = 8.4 Hz, 4JHH = 2.6 Hz, 1H, Ar-H), 6.68 (m, 3JHH = 4.0 Hz, 2H, Ar-H), 4.78 (d, 3JHH = 7.6 Hz, 1H, CH-OH), 3.77 (s, 3H, OCH3), 3.06 (m, 4H, CH2), 2.99 (m, 1H, CH2), 2.95 (m, 1H, CH2), 2.83 (m, 2H, CH2), 2.77 (m, 1H, CH2), 2.67 (m, 1H, CH2). 13C{1H}-NMR (CD3OD): δ 161.42, 160.34, 157.71 (d, 4JCF = 1.67 Hz, 1C, CHCHCHCF), 141.96, 130.75, 128.28 (CH), 126.41 (q, 1JCF = 270.0 Hz, 1C, CF), 126.41 (q, 2JCF = 31.4 Hz, 1C, CHCF), 121.64 (q, 3JCF = 3.6 Hz, 1C, CHCHCF), 119.15 (q, 3JCF = 4.3 Hz, 1C, CHCHCF), 116.74 (CH), 112.35 (CH), 111.68 (CH), 104.31 (CH), 72.89 (CHO), 63.23 (CN), 58.14 (CN), 56.28 (CN), 55.75 (OCH3), 36.87 (CH2), 27.03 (CH2). HRMS calcd for C22H23NO3F3 [M + H]+: m/z = 406.1630, found 406.1625; error: ‒1.2 ppm. HPLC (general method A): tR = 6.81 min, purity 96.43%
3-(4-(Benzofuran-2-yl)butyl)-7-methoxy-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L17). Use of the method for L12 in same molar proportions with X17 (257 mg, 0.746 mmol) gave L17 as a white crystalline solid (194 mg, yield 71%). Mp: 90–92 °C. 1H-NMR (CD3OD): δ 7.47 (dd, 3JHH = 7.0 Hz, 4JHH = 1.9 Hz, 1H, Ar-H), 7.37 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 7.29 (d, 3JHH = 8.5 Hz, 1H, Ar-H), 7.18 (td, 3J HH = 7.2 Hz, 4JHH = 1.5 Hz, 1H, Ar-H), 7.14 (td, 3JHH = 7.3 Hz, 4JHH = 1.3 Hz, 1H, Ar-H), 6.72 (dd, 3JHH = 8.4 Hz, 4JHH = 2.6 Hz, 1H, Ar-H), 6.66 (d, 4JHH = 2.6 Hz, 1H, Ar-H), 6.47 (d, 4JHH = 0.6 Hz, 1H, Ar-H), 4.79 (d, 3JHH = 8.1 Hz, 1H, CH-OH), 3.75 (s, 3H, OCH3), 2.89 (m, 6H, CH2), 2.60 (m, 3H, CH2), 2.44 (br, 1H, CH2), 1.79 (quint, 2H, CH2), 1.65 (quint, 2H, CH2). 13C{1H}-NMR (CD3OD): δ 160.59, 160.24, 156.28, 141.79, 137.22, 130.50 (CH), 127.68 (CH), 124.38, 123.64, 121.41 (CH), 116.57 (CH), 111.71 (CH), 111.62 (CH), 103.26 (CH), 72.37 (CHO), 63.70 (CN), 60.01 (CN), 56.52 (CN), 55.75 (OCH3), 36.51 (CH2), 29.18 (CH2), 27.20 (CH2), 26.89 (CH2). HRMS: calcd for C23H28NO3 [M + H]+: m/z = 366.2069, found 366.2069; error: 0.0 ppm. HPLC (general method A): tR = 6.34 min, purity 99.00%.
7-Methoxy-3-(4-(5-(trifluoromethyl)benzofuran-2-yl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L18). Use of the method for L12 in same molar proportions with X18 (197 mg, 0.478 mmol) gave L18 as a thick brown oil (141 mg, yield 68%). 1H-NMR (CD3OD): δ 7.83 (s, 1H, Ar-H), 7.56 (AB, 3JHH = 8.6 Hz, 1H, Ar-H), 7.51 (AB, 3JHH = 8.6 Hz, 4JHH = 1.3 Hz, 1 H, Ar-H), 7.29 (d, 3JHH = 8.4 Hz, 1 H, Ar-H), 6.72 (dd, 3JHH = 8.4 Hz, 4JHH = 2.6 Hz, 1 H, Ar-H), 6.67 (d, 4JHH = 2.5 Hz, 1 H, Ar-H), 6.63 (d, 4JHH = 0.8 Hz, 1 H, Ar-H), 4.79 (d, 3JHH = 7.8 Hz, 1H, CH-OH), 3.76 (s, 3H, OCH3), 2.90 (m, 6H, CH2), 2.61 (br, 3H, CH2), 2.51 (br, 1H, CH2), 1.82 (quint, 3JHH = 7.8 Hz, 2H, CH2), 1.67 (quint, 3JHH = 7.8 Hz, 2H, CH2). 13C{1H}-NMR (CD3OD): δ 163.13, 160.28, 157.71, 144.77, 137.15, 130.83, 127.78 (CH), 126.44 (q, 1JCF = 270.8 Hz, 1C, CF), 126.35 (q, 2JCF = 31.6 Hz, 1C, CHCF), 121.52 (q, 3JCF = 3.7 Hz, 1C, CHCHCF), 119.06 (q, 3JCF = 4.1 Hz, 1C, CHCHCF), 116.59 (CH), 112.30 (CH), 111.73 (CH), 103.64 (CH), 72.39 (CHO), 63.60 (CN), 59.94 (CN), 56.56 (CN), 55.75 (OCH3), 36.46 (CH2), 29.14 (CH2), 27.15 (CH2), 26.70 (CH2). HRMS calcd for C23H28NO3 [M + H]+: m/z = 434.1943, found 434.1948; error: 1.2 ppm. HPLC (general method A): tR = 8.06 min, purity 98.37%.
Methyl 3-((4-(4-(1-hydroxy-7-methoxy-1,2,4,5-tetrahydro-3H-benzo[d]azepin-3-yl)butyl)phenyl)thio)propanoate (L19) and enantiomers. L2 (0.2067 g, 0.458 mmol), methyl 3-mercaptopropanoate (62 µL, d = 1.085 g/mL, 0.560 mmol), N,N,1,1,1-pentamethylstannanamine (90 µL, d =1.274 g/mL, 0.552 mmol), and Pd(PPh3)4 (120 mg, 0.104 mmol) were dissolved in DMSO (4.0 mL). The reaction mixture was then heated in a microwave reactor (110 °C, 10 min, 50 W, 250 psi). LC-MS analysis showed consumption of the starting material L2, and this was confirmed with HPLC (general method A). The reaction mixture was purified with reversed phase HPLC (general method D). The product fractions were collected and dried under vacuum. The product was then redissolved in ethanol, passed through a 0.2 μm syringe filter, and then dried with Centrifan to give L19 a brown waxy material (174.3 mg, yield 86%). 1H-NMR (CD3OD): δ 7.30 (d, 3JHH = 6.7 Hz, 2H, Ar-H), 7.30 (t, 3JHH = 4.5 Hz, 1H, Ar-H), 7.16 (d, 3JHH = 8.2 Hz, 2H, Ar-H), 6.73 (dd, 3JHH = 8.3 Hz, 4JHH = 2.6 Hz, 1H, Ar-H), 6.67 (d, 4JHH = 2.5 Hz, 1H. Ar-H), 4.79 (d, 3JHH = 8.0 Hz, 1H, CHOH), 3.76 (s, 3H, OCH3), 3.65 (s, 3H, OCH3), 3.11 (t, 3JHH = 7.1 Hz, 2H, CH2), 2.88 (m, 4H, CH2), 2.61 (m, 8H, CH2), 1.63 (m, 4H, CH2). 13C{1H}-NMR (CD3OD): δ 174.10 (C=O), 160.31, 142.71, 141.71, 138.65, 137.04, 133.59 (CH), 132.03 (CH), 130.43 (CH), 127.88 (CH), 116.61 (CH), 111.76 (CH), 72.24 (OCH), 63.45 (CN), 60.18 (CN), 56.55 (CN), 55.77 (OCH3), 52.36 (OCH3), 36.29 (CH2), 35.34 (CH2), 30.92 (CH2), 30.62 (CH2), 30.44 (CH2), 27.04 (CH2). HRMS calcd for C25H34NO4S [M + H]+: m/z = 444.2209, found 444.2203. error: ‒1.4 ppm. HPLC (general method A): tR = 6.46 min, purity 97.56%. Chiral HPLC (general method E): (S)-L19, tR = 13.91 min, 50.24%; (R)-L19, tR = 21.48 min, 48.23%.
The enantiomers were resolved and purified with HPLC (general method F). (S)-L19: tR = 14.04 min, 97.24%. [α]D²⁰ = +2.47° (c 1.0, CHCl3). (R)-L19: tR = 20.25 min, 97.71%. [α]D²⁰ = –1.81° (c 1.0, CHCl3).
7-Methoxy-3-(4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (L20). Method 1. L2 (105.7 mg, 0.234 mmol), 4,4,4’,4’,5,5,5’,5’-octamethyl-2,2’-bi(1,3,2-dioxaborolane) (86.9 mg, 0.342 mmol), KOAc (83.7 mg, 0.853 mmol), and DPPF-PdCl2·CH2Cl2 (34.8 mg, 0.0426 mmol) were dissolved in DMSO (3.0 mL). The reaction mixture was then heated in a microwave reactor (80 °C, 60 min, 60 W, 250 psi). LC-MS analysis showed consumption of the starting material L2 and this was confirmed with HPLC (general method A). The reaction mixture was purified with reversed phase HPLC (general method D). A saturated NaCl solution was immediately added to each product fraction as they eluted, followed by a saturated NaHCO3 solution to prevent decomposition. The aqueous phase was then extracted with acetonitrile and dried (MgSO4), filtered, and then dried under vacuum. The product was redissolved in ethanol, passed through a 0.2 μm syringe filter, and then dried with Centrifan to give L20 purple wax (39.0 mg, yield 37%). 1H-NMR (CD3CN): δ 7.63 (d, 3JHH = 7.7 Hz, 2H, Ar-H), 7.23 (d, 3JHH = 7.6 Hz, 2H, Ar-H), 7.17 (d, 3JHH = 8.0 Hz, 1H, Ar-H), 6.68 (m, 2H, Ar-H), 4.61 (d, 3JHH = 7.3 Hz, 1H, CHOH), 3.74 (s, 3H, OCH3), 2.85 (m, 1H, CH2), 2.73 (m, 2H, CH2), 2.65 (m, 2H, CH2), 2.58 (m, 2H, CH2), 1.64 (q, 3JHH = 7.6 Hz, 2H, CH2), 1.54 (m, 2H, CH2), 1.31 (s, 12H, CH3). 13C{1H}-NMR (CD3CN): δ 159.27, 146.85, 136.67, 135.12, 128.70 (CH), 128.57 (CH), 117.88 (CH), 116.35 (CH), 110.76 (CH), 84.23 (COC3), 72.02 (COH), 62.06 (CN), 59.21 (CN), 55.98 (CN), 55.35 (OCH3), 36.29 (CH2), 35.94 (CH2), 29.27 (CH2), 26.68 (CH2), 24.77 (CH3). HRMS calcd for C27H39BNO4 [M+H]+: m/z = 452.2972, found 452.2976; error: 0.9 ppm. HPLC (general method A): tR = 7.64 min, purity 76.67%.
Method 2. Compound L7 (18.7 mg, 0.0462 mmol), 4,4,4’,4’,5,5,5’,5’-octamethyl-2,2’-bi(1,3,2-dioxaborolane) (57 mg, 0.224 mmol), KOAc (7.9 mg, 0.0805 mmol), and DPPF-PdCl2·CH2Cl2 (13.2 mg, 0.0162 mmol) were dissolved in DMSO (0.5 mL). The reaction mixture was then heated in a microwave reactor (110 °C, 30 min, 50 W, 250 psi). LC-MS analysis showed consumption of the starting material L7and this was confirmed with HPLC (general method A). The reaction mixture was purified with reversed phase HPLC (general method D) and the collected fractions were dried under vacuum. The product was redissolved in ethanol, passed through a 0.2 μm syringe filter, and then dried with Centrifan to give L20 as a purple wax (11.2 mg, yield 54%).
Infrared/Vibrational Circular Dichroism. The enantiomers of L3 were subjected to a computational infrared (IR) and vibrational circular dichroism (VCD) study to allow comparison of computed spectra with those determined experimentally, and to define the absolute configurations of the enantiomers (see Supplementary Information). The first eluting enantiomer in chiral HPLC analysis (general method E) was found to be the S-enantiomer.
Verification of absolute configuration of enantiomers of L6, L2, L19, and L20 by chemical transformations originating from (S)-L3.
Synthesis of (S)-L19 from (S)-L2. Compound (S)-L2 (46.8 mg, 103.7 μmol), methyl 3-((trimethylstannyl)thio)propanoate (26.7 μL, 124.4 μmol), triethylamine (14.5 μL, 103.7 μmol), and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (35.5 mg, 48.52 μmol) were dissolved in acetonitrile (1 mL). The reaction mixture was then heated in a microwave reactor (90 °C. 20 min, 120 W, 250 psi) and then purified with normal phase HPLC (method G) to give compound (S)-L19 as a brown oil (38.8 mg, 84.4%). HPLC (general method E): tR = 14.04 min, purity 97.24%.
Synthesis of (S)-L3 from (S)-L19. Compound (S)-L19 (27.4 mg, 61.77 μmol) in DCM (1 mL) was mixed with methyl iodide (3.86 μL, 61.77 μmol) and 1 M tetra-n-butylammonium hydroxide (TBAOH;129.7 μL, 129.7 μmol) and stirred at rt for 20 min. The reaction mixture was then diluted with DCM (5 mL) and washed thrice with water (10 mL). The solvent was removed by rotary evaporator, redissolved in DCM (1 mL) and purified with HPLC (general method F) to give compound (S)-L3 as a colorless oil (17.3 mg, 75.4%). HPLC (general method E): tR = 8.10 min, purity 98.01%.
Synthesis of (S)-L20 from (S)-L2. Compound (S)-L2 (138 mg, 306 μmol), bis(pinacolato)diborane (200 mg, 788 μmol), potassium acetate (51.2 mg, 521 μmol), and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (62.8 mg, 85.8 μmol) were dissolved in acetonitrile (3 mL). The reaction mixture was then heated in a microwave reactor (80 °C, 20 min, 150 W, 250 psi). and passed through a 0.2 μm syringe filter and purified with normal phase HPLC in three equal portions (method G). Fractions containing the product were immediately neutralized with saturated aqueous sodium bicarbonate (1 mL), extracted with ethyl acetate, and dried to give (S)-L20 (43 mg, 31%) as a purple wax. HPLC (general method E): tR = 6.0 min, purity 93.5%.
Synthesis of (S)-L6 from (S)-L20. Compound (S)-L20 (20.0mg, 44.3 μmol) was dissolved in methanol (0.4 mL). Cesium fluoride (199.4 μmol) in methanol (1 M; 199.4 μL), methyl iodide (3.04 μL, 48.8 μmol) and tetrakis(triphenylphosphine)palladium(0) (5.2 mg, 4.50 μmol) were dissolved in acetonitrile (0.6 mL). The reaction mixture was then heated in a microwave reactor (90 °C, 20 min, 80 W, 250 psi) and then passed through a 0.2 μm syringe filter and purified by reverse phased HPLC (method G) to give (S)-L6 as a brown oil (12.6mg, 83.9%). HPLC (general method E): tR = 5.93 min, purity 98.28%
Methylation of L19 to synthesize L3 under conditions mimicking a11C-labeling reaction. A stock solution of L19 in DMF (1.25 mg/mL) was prepared and 0.4 mL of this solution was transferred to a V-vial. A stock solution of iodomethane in DMF (1% v/v; 1 mL) was prepared and 7.0 µL of this solution was added to the L19 solution and stirred for 2 min. TBAOH in methanol (1 M; 5.0 µL) was then added and this was stirred at RT for 5 min. LC-MS showed complete conversion of L19 into L3. The solution was then dried with Centrifan and the reaction mixture redissolved in ethanol (0.1 mL) for chiral HPLC (general method E) and LC-MS analysis.
Methylation of L20 to L6 under conditions mimicking a11C-labeling reaction. Pd2(dba)3 and P(o,p-tol)3 were mixed in the weight ratio of 1: 1.33. A portion of this mixture (0.6 mg; 0.562 µmol Pd) was taken and placed in a vial. A solution of L20 in methanol (1.25 mg/mL; 0.40 mL) was added, followed by a solution of iodomethane in methanol (10% w/w; 7 µL). A stock solution (20 µL) of cesium fluoride in methanol (1 M) was then added to the reaction mixture, which was heated at 80 oC for 90 min. LC-MS showed consumption of the starting material (L20) and product formation (L6). The reaction mixture was then purified with reversed phase HPLC (general method C) and the product dried with Centrifan. The purified product was analyzed with chiral HPLC (general method E) and LC-MS.
In Vitro Binding Assays
PDSP binding assay. Selected compounds early in the project were assayed by the National Institutes of Health (NIH) Psychoactive Drug Screening Program (PDSP) for Ki at the GluN2B subtype (https://pdspdb.unc.edu/pdspweb/). A suspension of transient transfected mouse fibroblast cell membrane homogenates was prepared as reported at a concentration of ~500,000 cells/mL. This suspension was sonicated, and aliquots (100 μL) were added to each of four tubes (total 48 in a rack). A solution of [3H]ifenprodil (2.22 TBq/mmol, 0.01 kBq/μL; ARC, https://arcincusa.com/) in PBS (100 μL) was added to each tube. Ifenprodil or other displacer was dissolved in DMSO to give a 1-mM stock solution, which was further diluted with DMSO to give solutions ranging in known concentration from 10-5 to 10-10 M. Then 10 μL of each solution was added to a separate tube. The content of each tube was then diluted to 1.0 mL with PBS, vortexed, and incubated at 37 °C for 2 h. After separation of tube contents with a cell harvester, filter paper (GF/B; Whatman; Derwood, MD), pretreated with 0.5% polyethyleneimine solution, was washed with PBS (3 mL × 3). Each filter was then placed in a 7-mL plastic vial. Scintillation fluid (4 mL) was added to each vial. The scintillation vials were incubated overnight and then counted for radioactivity. The data were analyzed with Prism 7 version 7.03 (GraphPad Software; San Diego, CA) with ‘One site competition’ curve-fitting. Ki values were calculated according to the Cheng-Prusoff equation(57): Ki = IC50/(1 + [L]/KD) where [L] is the concentration (0.4 nM) and KD the equilibrium dissociation constant (7.6 nM) of the reference radioligand. The latter was determined with ‘Scatchard analysis’ of homologous displacement from multiple runs with self-displacement from membrane homogenates.
In-house binding assay. The assay procedure was the same as that described above but with a different protein source. The rat brain was homogenized in PBS (1×) at 1: 24, and the protein concentration measured. The protein concentration used for the binding assay ranged from 0.8 to 3.5 mg/mL. A solution of [3H]ifenprodil (2.22 TBq/mmol, 0.01 kBq/μL) or [3H]NR2B-Me ([3H]L6) (2.96 TBq/mmol, 37 kBq/μL; American Radiochemicals; St Louis, MO) in PBS (100 μL) was added to each tube (10 nM).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, NMR spectra; HPLC general methods; analytical and preparative HPLC chromatograms, including chiral HPLC chromatograms; ligand physicochemical properties used in CNS PET MPO scoring.
Author Contributions
LC, SC, LM, and AW, chemistry and analysis; LC, pharmacology; LC, study concept; LC and VWP, project supervision and manuscript drafting; all authors contributed to analysis of data and manuscript writing and reviewed the final manuscript.
Funding
This study was supported by the Intramural Research Programs of the National Institutes Health (NIH) (project: ZIAMH002793 to VWP). The contributions of the NIH authors were made as part of their official duties as NIH federal employees and comply with agency policy requirements. They are considered Works of the United States Government. However, the findings and conclusions presented in this paper are those of the authors and do not necessarily reflect the views of the NIH or the U.S. Department of Health and Human Services.
Acknowledgments
We thank Dr. John Lloyd (NIDDK Advanced Mass Spectrometry Facility) for high-resolution mass spectrometry measurements, Dr. Peter Herscovitch (NIH Clinical PET Center) for radioisotope production, and Prof. Bryan L. Roth (NIMH Psychoactive Drug Screening Program) for ligand assays at the University of North Carolina at Chapel Hill (Contract # 75N95023C00021; Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA; https://pdsp.unc.edu/ims/investigator/web/).
Abbreviations
CNS, central nervous system; DCM, dichloromethane; DMF, N,N’-dimethylformamide; DMSO, dimethyl sulfoxide; MPO, multi parameter optimization; MW, microwave; NIMH, National Institute of Mental Health; NMDA, N-methyl-D-aspartate; PDSP, psychoactive drug screening program; RT, room temperature; THF; TFA, trifluoroacetic acid; VCD/IR, vibrational circular dichroism/infrared; USP, Unites States Pharmacopeia.
References
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; et al. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Li, X.H.; Miao, H.H.; Zhuo, M. NMDA Receptor Dependent Long-term Potentiation in Chronic Pain. Neurochem Res. 2018, 3, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Serajee, F.; Huq, A.M. Clinical Benefit of NMDA Receptor Antagonists in a Patient With ATP1A2 Gene Mutation. Pediatrics 2018, 141, S390–S394. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sheng, M. NMDA receptors in nervous system diseases. Neuropharmacology 2013, 74, 69–75. [Google Scholar] [CrossRef]
- Song, X.; Jensen, M.O.; Jogini, V.; et al. Mechanism of NMDA receptor channel block by MK-801 and memantine. Nature 2018, 556, 515–519. [Google Scholar] [CrossRef]
- Folch, J.; Busquets, O.; Ettcheto, M.; et al. Memantine for the Treatment of Dementia: A Review on its Current and Future Applications. J. Alzheimers Dis. 2018, 62, 1223–1240. [Google Scholar] [CrossRef]
- Zhuo, M. Ionotropic glutamate receptors contribute to pain transmission and chronic pain. Neuropharmacology 2017, 112, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Chazot, P.L. The NMDA receptor NR2B subunit: a valid therapeutic target for multiple CNS pathologies. Curr. Med. Chem. 2004, 11, 389–396. [Google Scholar] [CrossRef]
- Monaco, S.A.; Gulchina, Y.; Gao, W.J. NR2B subunit in the prefrontal cortex: A double-edged sword for working memory function and psychiatric disorders. Neurosci. Biobehav Rev. 2015, 56, 127–138. [Google Scholar] [CrossRef]
- Politis, M.; Piccini, P. Positron emission tomography imaging in neurological disorders. J. Neurol. 2012, 259, 1769–1780. [Google Scholar] [CrossRef]
- Hooker, J.M.; Carson, R.E. Human Positron Emission Tomography Neuroimaging. Annu Rev. Biomed. Eng. 2019, 21, 551–581. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. Overview of clinically available radiotracers for imaging in neurodegenerative disorders. In Molecular Imaging of Neurodegenerative Disorders; Cross, D.J., Mosci, K., Minoshima, S., Eds.; Switzerland AG Springer Nature, 2023; pp. 35–55. [Google Scholar]
- Ferrando, R.; Hernandes, D.; Ghini, B.G.; Coutinho, A.M. PET Imaging in Psychiatric Disorders. Semin Nucl. Med. 2025, 55, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Naganawa, M.; Gallezot, J.D.; Rossano, S.; Carson, R.E. Quantitative PET Imaging in Drug Development: Estimation of Target Occupancy. Bull. Math. Biol. 2019, 81, 3508–3541. [Google Scholar] [CrossRef]
- Ghosh, K.K.; Padmanabhan, P.; Yang, C.T.; et al. Positron emission tomographic imaging in drug discovery. Drug. Discov. Today 2022, 27, 280–291. [Google Scholar] [CrossRef]
- Pike, V.W. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef]
- Zhang, L.; Villalobos, A.; Beck, E.M.; et al. Design and Selection Parameters to Accelerate the Discovery of Novel Central Nervous System Positron Emission Tomography (PET) Ligands and Their Application in the Development of a Novel Phosphodiesterase 2A PET Ligand. J. Med. Chem. 2013, 56, 4568–4579. [Google Scholar] [CrossRef]
- Pike, V.W. Considerations in the Development of Reversibly Binding PET Radioligands for Brain Imaging. Curr. Med. Chem. 2016, 23, 1818–1869. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Chen, Z.; Josephson, L.; Li, Z.; Liang, S.H. Positron Emission Tomography (PET) Ligand Development for Ionotropic Glutamate Receptors: Challenges and Opportunities for Radiotracer Targeting N-methyl-D-aspartate (NMDA), alpha-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA) and Kainate Receptors. J. Med. Chem. 2018, 62, 403–419. [Google Scholar]
- Kassenbrock, A.; Vasdev, N.; Liang, S.H. Selected PET Radioligands for Ion Channel Linked Neuroreceptor Imaging: Focus on GABA, NMDA and nACh Receptors. Curr. Top. Med. Chem. 2016, 16, 1830–1842. [Google Scholar] [CrossRef]
- Erlandsson, K.; Galovic, M. Recent Progress in NMDA Glutamate Receptor Imaging. In Molecular Imaging for Brain Diseases.; Sone, D., Ed.; Humana: New York, 2025; Volume 222, pp. 1–20. [Google Scholar]
- Kim, J.H.; Marton, J.; Ametamey, S.M.; Cumming, P. A Review of Molecular Imaging of Glutamate Receptors. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Rischka, L.; Vraka, C.; Pichler, V.; et al. First-in-Humans Brain PET Imaging of the GluN2B-Containing N-methyl-d-aspartate Receptor with (R)-11C-Me-NB1. J. Nucl. Med. 2022, 63, 936–941. [Google Scholar] [CrossRef]
- Cai, L.; Liow, J.S.; Morse, C.L.; et al. Evaluation of 11C-NR2B-SMe and its Enantiomers as PET Radioligands for Imaging the NR2B Subunit within the NMDA Receptor Complex in Rats. J. Nucl. Med. 2020, 61, 00–000. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Liow, J.S.; Morse, C.L.; et al. Candidate 3-benzazepine-1-ol type GluN2B receptor radioligands (11C-NR2B-Me enantiomers) have high binding in cerebellum but not to sigma1 receptors. EJNMMI Res. 2023, 13, 28. [Google Scholar] [CrossRef]
- Smart, K.; Zheng, M.Q.; Ahmed, H.; et al. Comparison of three novel radiotracers for GluN2B-containing NMDA receptors in non-human primates: (R)-[11C]NR2B-Me, (R)-[18F]of-Me-NB1, and (S)-[18F]of-NB1. J. Cereb. Blood Flow. Metab. 2022, 42, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Kramer, S.D.; Betzel, T.; Mu, L.; et al. Evaluation of 11C-Me-NB1 as a Potential PET Radioligand for Measuring GluN2B-Containing NMDA Receptors, Drug Occupancy, and Receptor Cross Talk. J. Nucl. Med. 2018, 59, 698–703. [Google Scholar] [CrossRef]
- Haider, A.; Iten, I.; Ahmed, H.; et al. Identification and Preclinical Evaluation of a Radiofluorinated Benzazepine Derivative for Imaging the GluN2B Subunit of the Ionotropic NMDA Receptor. J. Nucl. Med. 2019, 60, 259–266. [Google Scholar] [CrossRef]
- Rischka, L.; Vraka, C.; Pichler, V.; et al. First-in-human brain PET imaging of the GluN2B-containing N-methyl-D-aspartate receptor with (R)-11C-Me-NB1. J. Nucl. Med. 2021. [Google Scholar]
- Ahmed, H.; Haider, A.; Varisco, J.; et al. Structure-Affinity Relationships of 2,3,4,5-Tetrahydro-1H-3-benzazepine and 6,7,8,9-Tetrahydro-5H-benzo[7]annulen-7-amine Analogues and the Discovery of a Radiofluorinated 2,3,4,5-Tetrahydro-1H-3-benzazepine Congener for Imaging GluN2B Subunit-Containing N-Methyl-d-aspartate Receptors. J. Med. Chem. 2019, 62, 9450–9470. [Google Scholar]
- Zheng, M.Q.; Ahmed, H.; Holden, D.; et al. Comparative study of the two enantiomers of [18F]PF-NB1 for imaging the NMDA GluN2B receptors. J. Nucl. Med. 2023, 64. [Google Scholar]
- Ahmed, H.; Wallimann, R.; Gisler, L.; et al. Characterization of (R)- and (S)-[18F]OF-NB1 in Rodents as Positron Emission Tomography Probes for Imaging GluN2B Subunit-Containing N-Methyl-d-Aspartate Receptors. ACS Chem. Neurosci. 2023, 14, 4323–4334. [Google Scholar] [CrossRef]
- Korff, M.; Chaudhary, A.; Li, Y.; et al. Synthesis and Biological Evaluation of Enantiomerically Pure (R)- and (S)-[18F]OF-NB1 for Imaging the GluN2B Subunit-Containing NMDA receptors. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Korff, M.; Chaudhary, A.; Li, Y.; et al. Synthesis and Biological Evaluation of Enantiomerically Pure (R)- and (S)-[18F]OF-NB1 for Imaging the GluN2B Subunit-Containing NMDA Receptors. J. Med. Chem. 2023, 66, 16018–16031. [Google Scholar] [CrossRef]
- Korff, M.; Steigerwald, R.; Bechthold, E.; et al. Chemical, pharmacodynamic and pharmacokinetic characterization of the GluN2B receptor antagonist 3-(4-phenylbutyl)-2,3,4,5-tetrahydro-1H-3-benzazepine-1,7-diol - starting point for PET tracer development. Biol. Chem. 2023, 404, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Gisler, L.; Elghazawy, N.H.; et al. Development and Validation of [3H]OF-NB1 for Preclinical Assessment of GluN1/2B Candidate Drugs; Pharmaceuticals (Basel), 2022; p. 15. [Google Scholar]
- Zheng, M.; Ahmed, H.; Smart, K.; et al. Characterization in nonhuman primates of (R)-[18F]OF-Me-NB1 and (S)-[18F]OF-Me-NB1 for imaging the GluN2B subunits of the NMDA receptor. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 2153–2162. [Google Scholar] [CrossRef] [PubMed]
- Burger, P.B.; Yuan, H.; Karakas, E.; et al. Mapping the binding of GluN2B-selective N-methyl-D-aspartate receptor negative allosteric modulators. Mol. Pharmacol. 2012, 82, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Bechthold, E.; Schreiber, J.A.; Lehmkuhl, K.; et al. Ifenprodil Stereoisomers: Synthesis, Absolute Configuration, and Correlation with Biological Activity. J. Med. Chem. 2021, 64, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Borgel, F.; Szermerski, M.; Schreiber, J.A.; et al. Synthesis and Pharmacological Evaluation of Enantiomerically Pure GluN2B Selective NMDA Receptor Antagonists. ChemMedChem 2018, 13, 1580–1587. [Google Scholar] [CrossRef]
- Karakas, E.; Furukawa, H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 2014, 344, 992–997. [Google Scholar] [CrossRef]
- Karakas, E.; Simorowski, N.; Furukawa, H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature 2011, 475, 249–253. [Google Scholar] [CrossRef]
- Tewes, B.; Frehland, B.; Schepmann, D.; Schmidtke, K.U.; Winckler, T.; Wunsch, B. Conformationally constrained NR2B selective NMDA receptor antagonists derived from ifenprodil: Synthesis and biological evaluation of tetrahydro-3-benzazepine-1,7-diols. Bioorg Med. Chem. 2010, 18, 8005–8015. [Google Scholar] [CrossRef]
- Tewes, B.; Frehland, B.; Schepmann, D.; et al. Enantiomerically Pure 2-Methyltetrahydro-3-benzazepin-1-ols Selectively Blocking GluN2B Subunit Containing N-Methyl-D-aspartate Receptors. J. Med. Chem. 2015, 58, 6293–6305. [Google Scholar] [CrossRef]
- Schreiber, J.A.; Schepmann, D.; Frehland, B.; et al. A common mechanism allows selective targeting of GluN2B subunit-containing N-methyl-D-aspartate receptors. Commun. Biol. 2019, 2, 420. [Google Scholar] [CrossRef] [PubMed]
- Tewes, B.; Frehland, B.; Schepmann, D.; Schmidtke, K.U.; Winckler, T.; Wunsch, B. Design, Synthesis, and Biological Evaluation of 3-Benzazepin-1-ols as NR2B-Selective NMDA Receptor Antagonists. ChemMedChem 2010, 5, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Haider, A.; Herde, A.M.; Kramer, S.D.; et al. Preclinical Evaluation of Benzazepine-Based PET Radioligands (R)- and (S)-11C-Me-NB1 Reveals Distinct Enantiomeric Binding Patterns and a Tightrope Walk Between GluN2B- and sigma1-Receptor-Targeted PET Imaging. J. Nucl. Med. 2019, 60, 1167–1173. [Google Scholar] [CrossRef]
- Wang, L.; Jacobson, O.; Avdic, D.; et al. Ortho-Stabilized F-18-Azido Click Agents and their Application in PET Imaging with Single-Stranded DNA Aptamers. Angew. Chem.-Int. Ed. 2015, 54, 12777–12781. [Google Scholar] [CrossRef]
- Pingali, H.; Jain, M.; Shah, S.; et al. Discovery of a highly orally bioavailable c-5-[6-(4-Methanesulfonyloxyphenyl)hexyl]-2-methyl-1,3-dioxane-r-2-carboxylic acid as a potent hypoglycemic and hypolipidemic agent. Bioorg Med. Chem. Lett. 2008, 18, 5586–5590. [Google Scholar] [CrossRef] [PubMed]
- Sooriyaarachchi, S.; Chofor, R.; Risseeuw, M.D.P.; et al. Targeting an Aromatic Hotspot in Plasmodium falciparum 1-Deoxy-d-xylulose-5-phosphate Reductoisomerase with -Arylpropyl Analogues of Fosmidomycin. Chemmedchem 2016, 11, 2024–2036. [Google Scholar] [CrossRef]
- Gu, Z.S.; Zhou, A.N.; Xiao, Y.; Zhang, Q.W.; Li, J.Q. Synthesis and antidepressant-like activity of novel aralkyl piperazine derivatives targeting SSRI/5-HT1A/5-HT7. Eur. J. Med. Chem. 2018, 144, 701–715. [Google Scholar] [CrossRef]
- Banerjee, T.S.; Paul, S.; Sinha, S.; Das, S. Synthesis of iboga-like isoquinuclidines: Dual opioid receptors agonists having antinociceptive properties. Bioorg Med. Chem. 2014, 22, 6062–6070. [Google Scholar] [CrossRef]
- Cowart, M.; Faghih, R.; Curtis, M.P.; et al. 4-(2-[2-(2(R)-methylpyrrolidin-1-yl)ethyl]benzofuran-5-yl)benzonitrile and related 2-aminoethylbenzofuran H3 receptor antagonists potently enhance cognition and attention. J. Med. Chem. 2005, 48, 38–55. [Google Scholar] [CrossRef]
- Huang, H.M.; McDouall, J.J.W.; Procter, D.J. SmI2-catalysed cyclization cascades by radical relay. Nat. Catal. 2019, 2, 211–218. [Google Scholar] [CrossRef]
- Larsen, P.; Ulin, J.; Dahlstrom, K.; Jensen, M. Synthesis of [11C]iodomethane by iodination of [11C]methane. Appl. Radiat. Isot. 1997, 48, 153–157. [Google Scholar] [CrossRef]
- Lu, S.Y.; Giamis, A.M.; Pike, V.W. Synthesis of [18F]Fallypride in a Micro-Reactor: Rapid Optimization and Multiple-Production in Small Doses for Micro-PET Studies. Curr. Radiopharm. 2009, 2, 49–55. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between Inhibition Constant (K1) and Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic-Reaction. Biochem Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
Chart 1.
Radioligands that have been evaluated for PET imaging of brain GluN2B in animals and humans.
Chart 1.
Radioligands that have been evaluated for PET imaging of brain GluN2B in animals and humans.

Chart 2.

Scheme 1.
Strategy for ligand synthesis.

Scheme 2.
Synthesis of alcohols 1 and 2.

Scheme 3.
Synthesis of alcohols 3 and 4.

Scheme 4.
Synthesis of alcohol 5.

Scheme 5.
Synthesis of alcohols 6 to 11.

Scheme 6.
Syntheses of tosylates X1–X18.

Scheme 7.
Syntheses of ligands L1–L9 and L11–L18.

Scheme 8.
Attempted synthesis of L10.

Scheme 9.
Synthesis of L12.

Scheme 10.
Synthesis of L20.

Scheme 11.
Determination of the absolute configuration of enantiomers of, L3, L6, L19, and L20, through relation to the VCD/IR experimentally determined absolute configuration of (S)-L3 and chemical reactions that do not alter stereochemistry.
Scheme 11.
Determination of the absolute configuration of enantiomers of, L3, L6, L19, and L20, through relation to the VCD/IR experimentally determined absolute configuration of (S)-L3 and chemical reactions that do not alter stereochemistry.


Table 1.
CNS MPO scores, CNS PET MPO scores, clogD values, and Ki values for the binding of ligands L1–L9 and L11–L19 to GluN2B.
Table 1.
CNS MPO scores, CNS PET MPO scores, clogD values, and Ki values for the binding of ligands L1–L9 and L11–L19 to GluN2B.
 |
a Calculated according to literature methods.(17) b Assays performed by the Psychoactive Drug Screening Program with cells overexpressing GluN2B (see Experimental). c From in-house assay, as described in the Experimental. d Ro 25-6981 gave Ki values between 10 and 26 nM in the in-house assay, as described in the Experimental using rat brain tissue. First reported in Cai et al. (25) f Ro 25-6981 had a Ki of 13 ± 4 nM in the same assay session.
Table 2.
Inhibition of binding at σ1 and σ2 receptors by various GluN2B ligands at 10 mM concentration.
Table 2.
Inhibition of binding at σ1 and σ2 receptors by various GluN2B ligands at 10 mM concentration.
| Ligand | Inhibition at cellular σ1 receptora (%) |
σ1 receptor Ki (nM) |
Inhibition at cellular σ2 receptorb (%) |
|---|---|---|---|
|
L1 (WMS 1405) |
89 ± 11 | 88 ± 9 | |
| L3 | 84c | 90c | |
| (R)-L3 | 88d | ||
| (S)-L3 | 24d | ||
| L5 | 94 ± 12 | 91 ± 10 | |
| L6 | 90 ± 8 | 91 ± 12c | |
| (–)-L6 | 42e | ||
| (+)-L6 | 130e | ||
| L7 | 84 ± 9 | 90 ± 11 | |
| L8 | 58 ± 7 | 77 ± 8 | |
| L9 | 84 ± 9 | 94 ± 12 | |
| L11 | 66 ± 8 | 74 ± 9 | |
| L12 | 41 ± 6 | 56 ± 4 | |
| a Reference radioligand [3H]Ifenprodil. b Reference radioligand [3H]DTG. The assays were performed by the PDSP. c Data first reported in reference 24. d Data first reported in reference 24. e Data first reported in reference 25. | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



