Submitted:
03 April 2026
Posted:
03 April 2026
You are already at the latest version
Abstract
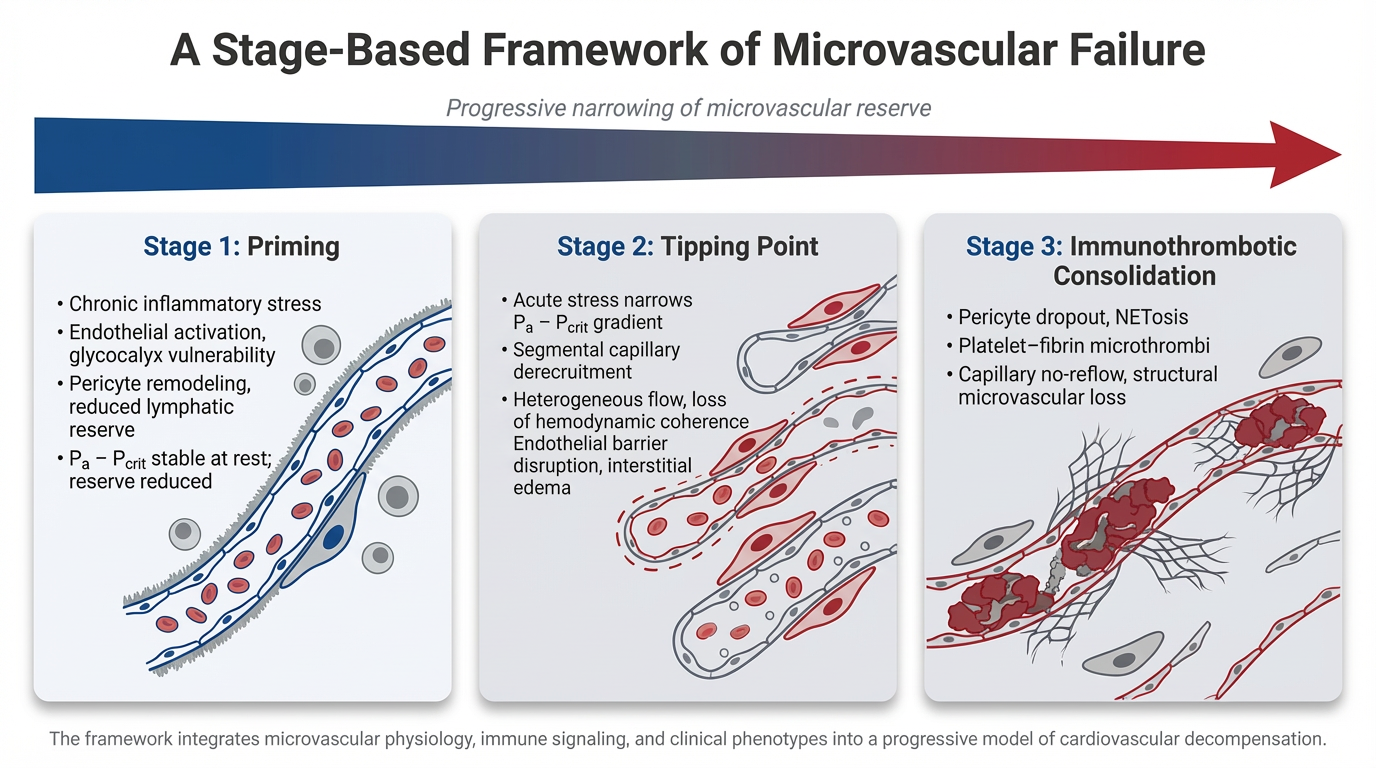
Cardiovascular disease is traditionally interpreted through macrocirculatory parameters such as cardiac output, vascular resistance, and epicardial coronary anatomy. However, clinical outcomes frequently diverge from predictions based solely on these indices, particularly in syndromes such as heart failure with preserved ejection fraction (HFpEF), cardiogenic shock, and sepsis-associated myocardial dysfunction. Increasing evidence suggests that the integrity of the microvascular–immune interface plays a central role in determining tissue perfusion and cardiovascular resilience. This review proposes a staged framework of cardiovascular decompensation centered on progressive failure of this interface.
In Stage 1, chronic cardiometabolic and inflammatory stress produces a primed but compensated microvascular state characterized by endothelial activation, glycocalyx vulnerability, pericyte remodeling, platelet sensitization, and reduced lymphatic reserve. Perfusion is preserved at rest, but vasodilatory reserve and microvascular stability are reduced, narrowing the effective perfusion window under physiologic stress.
In Stage 2, acute insults such as infection, ischemia, or neurohumoral activation precipitate threshold instability within the microcirculation. Perfusion becomes governed by the arterial pressure–critical closing pressure (Pa − Pcrit) relationship rather than traditional arterial–venous gradients. As this window narrows, segmental capillary derecruitment and heterogeneous flow emerge, producing loss of hemodynamic coherence in which systemic blood pressure and cardiac output may appear preserved despite impaired tissue perfusion.
In Stage 3, inflammatory amplification and immunothrombotic processes consolidate microvascular dysfunction. Pericyte contraction, endothelial injury, cytokine escalation, and neutrophil extracellular trap formation promote platelet–fibrin deposition and capillary obstruction, transforming reversible conductance failure into structural microvascular impairment.
This framework provides a unifying physiologic lens for diverse cardiovascular syndromes, including Type 2 myocardial infarction, HFpEF decompensation, and cardiogenic shock. It also suggests that therapeutic efficacy may depend less on macrocirculatory normalization alone and more on preserving microvascular integrity before immunothrombotic consolidation occurs. Although this model remains hypothesis-generating, it highlights the microvascular–immune interface as a central determinant of cardiovascular stability and a potential target for future precision hemodynamic and immunomodulatory strategies.
Keywords:
microcirculation
; glycocalyx
; immunothrombosis
; critical closing pressure
; hemodynamic coherence
; HFpEF
; cardiogenic shock
1. Introduction
Cardiovascular disease has traditionally been conceptualized through the lenses of hemodynamics, myocardial structure, lipidology, and epicardial coronary pathology. Yet across a broad spectrum of clinical syndromes, ranging from heart failure with preserved ejection fraction (HFpEF) and cardiogenic shock to sepsis-associated myocardial dysfunction, outcomes often diverge sharply from what macroscopic anatomy or resting hemodynamics would predict. This discrepancy has led to a growing recognition that cardiovascular performance is heavily influenced the integrity of the microvascular-immune interface which regulates tissue perfusion dynamics, capillary-venular pressure/flow coupling, and is highly sensitive to inflammatory tone[1,2].
Chronic cardiometabolic conditions such as obesity, hypertension, diabetes, and chronic kidney disease are understood to exert their deleterious cardiovascular effects in part through sustained immune activation and endothelial dysfunction[3]. At the level of the microcirculation, low-grade inflammation progressively alters endothelial barrier properties, disrupts mechano-transduction, and reshapes cellular crosstalk within the capillary niche. Over time, this inflammatory priming renders the cardiovascular system vulnerable to acute insults, such as infection, ischemia, or volume stress, precipitating disproportionate clinical decompensation[4,5].
Recent experimental and clinical evidence has further expanded this paradigm in discussing the active roles of non-myocyte, non-endothelial cells in cardiovascular disease. Pericytes, long regarded as passive structural supports, have emerged as dynamic regulators of capillary stability, endothelial phenotype, and immune responsiveness. Parallel insights into lymphatic dysfunction have reframed intravascular and tissue congestion as consequences not merely of elevated filling pressures, but of impaired microvascular–interstitial–lymphatic coupling[6].
Congestion itself is increasingly recognized as a dynamic continuum rather than a static elevation in filling pressures, with residual congestion frequently persisting at discharge and manifesting in distinct intravascular, tissue, or combined phenotypes, each associated with differential diuretic responsiveness and mortality risk. This further suggests the need to interpret congestion through a microvascular–immune lens rather than solely through pressure.
Together, these observations compel a shift away from reductionist models of cardiovascular disease toward an integrated framework in which immune regulation and microvascular integrity are central determinants of morbidity and mortality.
In this review, we propose that cardiovascular decompensation follows a staged progression of microvascular-immune failure. Outcomes are determined less by the initial insult than by whether interventions are applied before the transition from a reversible state of microvascular stress to irreversible immuno-thrombotic collapse.
2. Literature Review
2.1. Stage 1: The Primed Microvascular–Immune Unit
2.1.1. The Microvascular–Immune Unit in Cardiovascular Disease
The microcirculation is a highly regulated vascular–immune interface composed of arterioles, capillaries, and venules that coordinates oxygen delivery, endothelial signaling, and inflammatory surveillance. In cardiovascular disease, the resilience of this interface determines whether hemodynamic stress is accommodated or propagated into tissue injury. When destabilized, microvascular failure can produce clinical decompensation despite preserved macroscopic anatomy or resting hemodynamics[2].
Within this microcirculatory network resides the microvascular–immune unit: an integrated functional ensemble composed of the intraluminal endothelial layer and its superimposed glycocalyx, the abluminal pericyte sheath, circulating immune and platelet elements, and the adjacent interstitial–lymphatic network. Together, they regulate capillary conductance, barrier integrity, and immune–vascular signaling, thereby preserving continuity of flow across the macro-to-micro transition and within the microcirculation itself. The stability of this unit underlies tissue homeostasis under both basal conditions and physiological stress.
Chronic activation of innate immune pathways, via inflammasome signaling and persistent cytokine release, drives endothelial priming, lowering the activation threshold for barrier dysfunction and thrombo-inflammatory signaling during subsequent stress[7]. The endothelial glycocalyx is a mechanosensitive surface layer that regulates nitric oxide signaling, barrier permeability, and intercellular communication. Physiologic shear maintains its structure and vascular quiescence, whereas sustained inflammatory or disturbed flow states promote enzymatic shedding and surface destabilization. Even partial glycocalyx erosion enhances endothelial adhesiveness and permeability, lowering the threshold for leukocyte recruitment and thrombo-inflammatory activation. Glycocalyx integrity therefore functions as a critical determinant of microvascular resilience in cardiovascular disease.
Collectively, these processes do not produce immediate failure but reconfigure the microvascular–immune unit into a sensitized state. Persistent inflammatory and hemodynamic signaling remodel endothelial phenotype, alter pericyte behavior, and subtly shift microvascular tone, creating a circulation that remains functionally intact at rest yet increasingly reactive to secondary stress[8].
Persistent inflammatory signaling reprograms endothelial and pericyte phenotypes and establishes durable innate immune memory through epigenetic modification of hematopoietic progenitors, biasing systemic immunity toward a pro-inflammatory state[9]. As a result, normalization of macrocirculatory parameters alone may fail to restore microvascular homeostasis, explaining the frequent dissociation between stable hemodynamics and persistent tissue hypoperfusion observed in heart failure and shock[10]. The proposed structural and cellular features of this primed but compensated microvascular–immune state are illustrated in Figure 1.
2.1.2. Structural Components of Microvascular Vulnerability
Endothelial Surface: Baseline Vulnerability
Endothelial cells function as the principal immunologic and mechanical sensors of the circulation. Beyond serving as a passive barrier, they regulate leukocyte trafficking, platelet adhesion, nitric oxide bioavailability, and shear-dependent signaling[11].
These functions are critically modulated by the endothelial surface layer, whose glycocalyx governs mechanotransduction and limits inappropriate cellular adhesion. In addition, the glycocalyx contributes to maintenance of the intravascular oncotic gradient, thereby influencing transvascular fluid exchange[12]. Degradation of this surface layer through chronic inflammatory signaling or injury alters endothelial phenotype, increasing permeability, enhancing leukocyte adhesion, and promoting a procoagulant state[13].
Pericyte Remodeling: Chronic Sensitization
Pericytes constitute a second, often underappreciated pillar of the microvascular–immune unit. Positioned abluminally along capillaries, they regulate endothelial stability, capillary diameter, and flow distribution. Experimental models demonstrate that pericyte dysfunction precedes overt diastolic impairment and promotes endothelial activation, junctional disorganization, and microvascular rarefaction[14]. Under chronic inflammatory stress, pericytes undergo phenotypic reprogramming and adopt a pro-inflammatory profile that sensitizes adjacent endothelial cells to otherwise subthreshold stimuli. This shift amplifies local immune signaling and lowers the threshold for leukocyte recruitment within the microvascular niche[15],[16].
Lymphatic Reserve: Impaired Clearance Capacity
The lymphatic system forms a parallel vascular network responsible for interstitial fluid clearance and immune trafficking. By facilitating the removal of excess fluid, macromolecules, and immune cells, it serves as the principal regulator of tissue edema and inflammatory resolution. Within the heart, lymphatic drainage is tightly coupled to cardiac mechanics. Unlike peripheral collecting lymphatics, cardiac lymphatic vessels lack intrinsic smooth muscle and depend largely on myocardial contraction for propulsion. Consequently, lymphatic clearance is highly sensitive to changes in filling pressures, contractile function, and venous congestion. Even modest impairment of this clearance capacity promotes interstitial fluid accumulation, which can depress myocardial performance and impair microvascular oxygen delivery. In chronic cardiometabolic disease, lymphangiogenic signaling is often blunted, limiting adaptive remodeling and creating reduced clearance reserve even before overt congestion becomes clinically apparent[17].
Cardiometabolic disease and sustained inflammation blunt adaptive lymphangiogenic responses, limiting expansion and remodeling of the lymphatic network in the face of persistent capillary filtration. The result is relative clearance insufficiency: interstitial fluid, cytokines, and immune cells accumulate, sustaining a pro-inflammatory microenvironment that reinforces endothelial and pericyte activation[17].
The heart’s unique lymphatic anatomy, dependent on myocardial contraction for lymph propulsion, renders it particularly vulnerable in HFpEF, where diastolic dysfunction and elevated filling pressures simultaneously increase capillary filtration and impair drainage. This insufficiency not only contributes to congestion but sustains local inflammation by limiting immune cell egress and antigen clearance. [6,18]. This lymphatic insufficiency not only exacerbates volume-related symptoms but sustains local inflammation by limiting immune cell egress and antigen clearance.
Circulating Elements: Lowered Activation Threshold
Platelets function as rapid-response immune modulators at the microvascular interface. Beyond hemostasis, they express pattern-recognition receptors and respond to endothelial stress and inflammatory mediators. In the primed state, platelets exhibit lowered activation thresholds, promoting adhesion, microaggregate formation, and localized release of pro-inflammatory and pro-thrombotic mediators. This positions them as amplifiers of microvascular inflammation and lowers the threshold for thrombo-inflammatory escalation during acute stress[19]. Chronic priming similarly sensitizes neutrophils, predisposing to exaggerated inflammatory responses such as neutrophil extracellular trap formation under acute stress. These interactions further destabilize endothelial–pericyte crosstalk and prime the microvasculature for thrombo-inflammatory amplification[20].
2.1.3. Hemodynamic and Clinical Conditioning
Hemodynamic Vulnerability
Chronic priming does not initially produce flow cessation, but it narrows the safety margin sustaining microvascular conductance. Microvascular perfusion depends on the pressure gradient between arterioles and post-capillary venules. Persistent inflammatory tone, oxidative stress, and rising venous pressures subtly reduce this effective driving gradient while increasing forces opposing capillary patency. Although arterioles remain open at rest, the buffer protecting stable flow is progressively eroded. The circulation may appear hemodynamically adequate, yet it operates closer to the threshold at which regional perfusion becomes unstable.
Clinical Drivers
The biological vulnerability described above is not theoretical; it is continuously reinforced by the clinical milieu of contemporary cardiovascular disease. Chronic inflammatory states reshape endothelial behavior, microvascular architecture, and immune responsiveness long before overt decompensation becomes apparent.[21].
Cardiometabolic disorders, including obesity, insulin resistance, hypertension, diabetes, and chronic kidney disease, function as systemic inflammatory states rather than isolated metabolic derangements. Adipose dysfunction, oxidative stress, neurohormonal activation, and uremic toxins sustain low-grade innate immune activation, perpetuating endothelial stress even in clinically “stable” patients[5,22,23].
Heart failure with preserved ejection fraction exemplifies the clinical consequences of this chronic priming. Rather than arising from isolated myocardial injury, HFpEF reflects sustained endothelial inflammation and microvascular dysfunction driven by cardiometabolic comorbidities. Autopsy, imaging, and peripheral tissue studies consistently demonstrate capillary rarefaction, impaired vasodilator reserve, and systemic microvascular involvement, supporting the view of HFpEF as a multisystem microvascular disease rather than a purely myocardial disorder. Atherosclerosis similarly reflects cumulative endothelial conditioning over years, with plaque rupture representing a late manifestation of chronically dysregulated immune–vascular interactions[4,6].
Atherosclerosis similarly reflects the cumulative impact of chronic inflammatory conditioning. While plaque rupture and thrombosis define acute coronary syndromes, the vascular substrate upon which these events occur is shaped over years by endothelial activation, impaired shear sensing, and maladaptive immune–vascular interactions[24].
An increasingly important contributor to chronic inflammatory priming is gut dysbiosis. In cardiometabolic disease and chronic heart failure, venous congestion, intestinal edema, and impaired barrier integrity facilitate translocation of microbial products into the circulation. This low-grade endotoxemia does not typically produce overt sepsis, yet it sustains tonic activation of innate immune pathways and endothelial cells, reinforcing systemic inflammation even in the absence of acute infection.
Gut–Vascular Axis
This gut–vascular axis is particularly relevant in heart failure states, where elevated venous pressures precede hypoperfusion and promote intestinal barrier dysfunction early in the disease course.
When central venous congestion extends to the splanchnic circulation, vascular capacitance expands to accommodate increased effective vascular volume (EVV). The resultant EVV reduction triggers neurohumoral compensation through α-adrenergic venoconstriction and β2-adrenergic hepatic venodilation[25]. Upon reaching maximal splanchnic capacity, central venous and cardiac filling pressures rise sharply. The ensuing low-flow state from reduced cardiac output, splanchnic congestion, and vasoconstriction causes shunting of oxygenated blood from villous tips to bases, intensifying enterocyte hypoxic injury. Hypoxia-damaged enterocytes develop barrier dysfunction and increased permeability, allowing gram-negative bacterial endotoxins and lipopolysaccharides to enter circulation[26]. This triggers systemic vasodilation and proinflammatory cytokine release, causing cardiomyocyte dysfunction, which is amplified by impaired renal cytokine clearance with subsequent villous ischemia, producing acidosis which augments intestinal sodium and water reabsorption, perpetuating volume overload[27]. This process blunts mechanotransduction, attenuates nitric oxide–mediated vasodilation, and diminishes the ability of endothelial cells to appropriately sense and respond to laminar shear stress[11,28].
Concomitant visceral edema increases hydrostatic forces within the splanchnic bed, intensifying lymphatic filtration demands and challenging already reduced clearance reserve[27]. Experimental and clinical data suggest that this gut–vascular axis reinforces endothelial activation and immune dysregulation, thereby lowering the threshold for cardiovascular decompensation when secondary stressors supervene[10].
Conditioning for Failure
Pericytes are particularly vulnerable to chronic inflammatory stress. Sustained cytokine exposure and oxidative injury promote detachment and phenotypic shift, reducing capillary stability and contributing to microvascular rarefaction. Loss of pericyte support increases diffusion distances, impairs perfusion reserve, and sensitizes the microvascular bed to further inflammatory activation; findings consistently observed in HFpEF and chronic ischemic syndromes1[14,29].
The central insight emerging from these observations is that chronic inflammation does not directly precipitate thrombosis or acute cardiovascular collapse. Rather, it conditions the vascular bed for failure. Through progressive erosion of endothelial surface integrity, impairment of shear-dependent signaling, destabilization of pericyte–endothelial interactions, and reduction of lymphatic reserve, chronic inflammatory priming establishes a fragile microvascular equilibrium poised for destabilization.
In this sensitized state, otherwise tolerable insults, modest infection, transient ischemia, or volume shifts, can provoke disproportionate endothelial injury, immune amplification, and regional perfusion instability. Thrombosis and immunothrombotic consolidation thus represent downstream manifestations of a long-standing conditioning process rather than isolated acute events.
Recognizing chronic priming as a preparatory phase of cardiovascular disease reframes both risk stratification and therapeutic timing. Interventions directed solely at late-stage hemodynamic abnormalities may fail if the underlying microvascular substrate has already been compromised. Preserving microvascular–immune integrity before threshold destabilization occurs represents a critical and underutilized opportunity in cardiovascular care. Functionally, this chronic inflammatory priming may modestly elevate baseline vascular tone and reduce endothelial-dependent vasodilatory reserve, narrowing the effective Pa − Pcrit window under stress while preserving resting perfusion, as illustrated in Figure 1.
2.2. Stage 2: The Acute Tipping Point – Loss of Microvascular Control
2.2.1. Established Physiology: The Arterial Waterfall and Critical Closing Pressure
Under resting conditions, microvascular perfusion is classically described as being driven by the gradient between arteriolar inflow pressure and post-capillary venular pressure. However, small resistance vessels behave as collapsible conduits; their patency depends on the difference between intraluminal pressure and a critical closing pressure (Pcrit) generated by vascular smooth muscle tone and surrounding tissue forces. Consequently, once Pcrit exceeds venular pressure, the effective microvascular driving pressure is better represented by the difference between arterial pressure (Pa) and Pcrit (Pa – Pcrit). This “waterfall” phenomenon means that flow becomes governed by the gradient across the arteriolar segment rather than by venular back-pressure alone[30],[31,32].
In the healthy circulation, Pa is substantially higher than Pcrit, ensuring a robust driving gradient and stable capillary recruitment. Pcrit itself is dynamic, modulated by neurohumoral tone, local metabolic factors, and endothelial function, but it normally remains well below Pa across a range of physiological conditions[10].
2.2.2. Effects of Acute Stressors on Microvascular Parameters
Under physiologic conditions, capillary perfusion is sustained by the microcirculatory driving pressure, defined as the gradient between arteriolar inflow pressure and post-capillary venular pressure[33].
Acute cardiovascular insults: infection, ischemia, hemorrhage, or neurohumoral surges. can erode the Pa – Pcrit gradient from both sides. On the inflow side, systemic vasodilation or reduced cardiac output may lower Pa. On the outflow side, venous congestion, interstitial edema, and vasoconstrictor hyper-responsiveness can elevate Pcrit. Inflammatory mediators, catecholamines, and oxidative stress all contribute to a rise in effective closing pressure, while endothelial dysfunction impairs shear-dependent vasodilation that normally helps maintain microvascular patency[11,12,28].
Importantly, these changes can occur while macrocirculatory indices (e.g., mean arterial pressure, cardiac output) remain within normal ranges. The microcirculation therefore becomes vulnerable to instability before systemic hypotension is evident.
The progressive narrowing and collapse of the arterial waterfall are schematically depicted in Figure 2, which we hypothesize as the second stage of microvascular failure.
In this framework, microvascular failure represents the collapse of the arterial waterfall: perfusion becomes discontinuous, spatially heterogeneous, and increasingly dissociated from macrocirculatory indices.
2.2.3. Threshold Instability: A Framework Interpretation
Within the conceptual framework proposed here, the narrowing of the Pa – Pcrit gradient represents a transition from compensated microvascular reserve to threshold instability. As Pa declines or Pcrit rises, some capillary territories approach the point where Pa becomes only marginally greater than Pcrit. Flow in those territories becomes intermittent, and when Pa falls below Pcrit, complete segmental collapse occurs.
Crucially, collapse is not uniform. Patchy capillary derecruitment produces heterogeneous flow distribution—well-perfused areas adjacent to non-perfused capillaries. This spatial heterogeneity creates loss of hemodynamic coherence: systemic hemodynamics no longer reliably reflect tissue oxygenation. Mixed venous oxygen saturation may remain normal or even elevated because oxygen extraction is impaired by reduced perfused capillary surface area. Similarly, the venous–arterial carbon dioxide gap (ΔPCO₂) may widen as stagnant regions fail to clear CO₂[10,28,33].
This heterogeneity impairs oxygen extraction efficiency. Regions of low or absent flow coexist with hyperperfused areas, producing regional tissue hypoxia despite preserved systemic indices. Such dissociation between macro-hemodynamics and capillary-level perfusion constitutes loss of hemodynamic coherence[33].
As shown in Figure 2C, segmental dropout produces spatially heterogeneous perfusion and loss of hemodynamic coherence.
At this stage, tissue hypoxia may coexist with preserved or even elevated mixed venous oxygen saturation, reflecting impaired oxygen extraction rather than inadequate global delivery. Microvascular collapse thus precedes overt systemic hypotension and may remain occult unless specifically assessed.
2.2.4. Barrier Failure and Fluid Geometry
When the endothelial surface layer is also compromised, the revised Starling principle teaches us that capillaries behave as pressure-dependent filters; fluid resuscitation intended to raise Pa may instead drive excess filtration into the interstitium. The resulting interstitial edema further elevates extravascular compressive forces, raising Pcrit and perpetuating capillary collapse[12].
When this layer is compromised under inflammatory stress,capillaries behave as pressure-driven filters. Increases in hydrostatic pressure no longer meaningfully expand effective intravascular volume but instead disproportionately drive fluid into the interstitium. The resulting edema widens diffusion distances and mechanically compresses surrounding capillaries, compounding convection failure with diffusion impairment[11].Patients may therefore be simultaneously congested and hypoperfused.
2.2.5. Lymphatic Overload and Congestive Amplification
During acute endothelial barrier failure, the sudden increase in capillary filtration rapidly overwhelms lymphatic reserve, particularly in patients with preexisting lymphatic impairment, such as those with HFpEF or chronic venous congestion. The resulting interstitial fluid accumulation amplifies tissue edema, sustains local inflammation, and prolongs microvascular dysfunction. Thus, congestion in acute cardiovascular illness should be viewed not solely as a hemodynamic phenomenon, but as a failure of coordinated microvascular–interstitial–lymphatic coupling[6,18].
Interstitial fluid accumulation increases tissue hydrostatic pressure, mechanically compresses vulnerable capillaries, and sustains inflammatory signaling, thereby amplifying both convection and diffusion failure[33].
Clinically, two distinct congestion phenotypes warrant differentiation. Intravascular congestion reflects elevated venous pressures that narrow the arteriolar–venular driving gradient, while tissue congestion reflects impaired interstitial clearance and edema accumulation.
2.2.6. Clinical Recognition of Microvascular Instability
Microvascular recovery frequently lags behind macrocirculatory normalization. Sublingual microvideoscopy demonstrates persistent impairment in microvascular density and flow in severe heart failure and cardiogenic shock, particularly among non-survivors[30]. Device-based augmentation does not consistently restore microcirculation: intra-aortic balloon pump therapy has not reliably improved microvascular parameters[34], raising MAP with norepinephrine does not necessarily improve sublingual perfusion[35], and in VA-ECMO–supported shock, failure to restore microcirculatory indices within 24 hours predicts mortality despite acceptable macro-hemodynamics[36].
The resulting fluid load exceeds lymphatic transport capacity, producing lymphatic overload and sustained congestion, which in turn magnifies inflammation, edema, and cardiac dysfunction[37]. The Venous Excess Ultrasound (VExUS) score provides a non-invasive means of detecting this congestion, with higher grades correlating with adverse outcomes[38] but cannot directly assess capillary recruitment.
These findings support a bedside strategy that integrates global hemodynamics with perfusion surrogates, capillary refill time, mottling, lactate kinetics, Mixed venous oxygen saturation (SvO₂) patterns, and, where feasible, direct microcirculatory visualization[33].
SvO₂ reflects the balance between global oxygen delivery and consumption. In states of heterogeneous microvascular collapse, SvO₂ may remain normal or even elevated despite regional hypoxia, reflecting impaired oxygen extraction rather than adequate delivery. Capillary derecruitment reduces effective perfused surface area, limiting extraction capacity even when cardiac output appears sufficient.
Similarly, the venous–arterial carbon dioxide gap (ΔPCO₂) serves as a marker of impaired tissue CO₂ clearance. When capillary flow becomes discontinuous, regional stagnation impairs washout of CO₂, widening ΔPCO₂ even in the setting of preserved arterial pressure and cardiac output. In this geometry, ΔPCO₂ may identify microvascular flow limitation earlier than lactate elevation[39]. Peripheral perfusion markers such as capillary refill time and mottling provide additional real-time indicators of capillary instability. These variables reflect effective microvascular conductance rather than macrocirculatory adequacy.
Taken together, SvO₂, ΔPCO₂, peripheral perfusion assessment, and, when feasible, direct microcirculatory visualization provide a practical framework for detecting loss of hemodynamic coherence. Restoration of MAP alone should not be interpreted as restoration of perfusion if these indices suggest persistent instability of the Pa − Pcrit relationship.
2.3. Stage 3: Amplification and Consolidation – The Immunothrombotic Cascade
2.3.1. Pericyte Dysfunction as Amplifier
Following acute ischaemia-reperfusion, pericytes can undergo sustained contraction in response to oxidative stress and inflammatory mediators. Preclinical studies have demonstrated that this contraction narrows capillary lumens and contributes to flow obstruction even after epicardial patency is restored. Emerging evidence suggests that pericyte-mediated capillary constriction could contribute to the no-reflow phenomenon observed after ischaemia-reperfusion, though direct human data remain limited[40],[41],[42].
Stressed pericytes also adopt a pro-inflammatory signaling phenotype that reinforces endothelial activation and immune recruitment, creating a feed-forward loop that consolidates microvascular dysfunction at the moment perfusion reserve is most limited[43]. This feed-forward interaction intensifies microvascular dysfunction at precisely the moment when tissue perfusion is most vulnerable.
When inflammatory stress is sustained, pericyte dysfunction evolves from a potentially reversible response into a structural deficit. Progressive pericyte dropout promotes capillary rarefaction, reducing functional microvascular density and increasing diffusion distances for oxygen and metabolites[42]. Pericyte dysfunction may represent an early and underrecognized contributor HFpEF pathophysiology[6,15].
2.3.2. Pericytes in HFpEF and Coronary Microvascular Dysfunction
Pericyte dysfunction offers a unifying mechanism linking cardiometabolic inflammation to both HFpEF and coronary microvascular dysfunction. Critically, pericyte loss appears before diastolic dysfunction, capillary rarefaction, or fibrosis suggesting it initiates rather than follows microvascular disease. As pericytes fail, they destabilize endothelial barriers, widen capillaries, disorganize junctions, and adopt pro-inflammatory signaling that sensitizes the endothelium to immune stimuli, amplifying immune-vascular crosstalk at the capillary level[45].
This reframes HFpEF not as a disorder of myocardial stiffness alone, but as one of capillary dysregulation and immune amplification. Loss of pericyte support limits perfusion reserve, increases edema susceptibility, and magnifies the hemodynamic consequences of volume shifts or systemic inflammation collectively explaining exertional intolerance, congestion, and diuretic resistance in the setting of preserved systolic function. The same pericyte-driven pathology underlies coronary microvascular dysfunction, where capillary constriction, flow heterogeneity, and endothelial hypersensitivity produce stress-induced ischemia without obstructive disease. HFpEF and coronary microvascular dysfunction thus emerge as related expressions of a shared pericyte-centered failure of capillary integrity and immune containment[15].
2.3.3. Pericytes in Post-Myocardial Infarction Remodeling
Following myocardial infarction, pericytes undergo rapid and heterogeneous phenotypic transitions that shape infarct healing, neovascularization, and adverse remodeling. Lineage-tracing and single-cell studies indicate diversification into discrete programs across inflammatory, proliferative, and maturation phases. Early pericyte loss destabilizes capillaries and impairs perfusion, followed by proliferation and emergence of fibrogenic, migratory, and vascular-support states.
Activated subsets can adopt fibrogenic phenotypes, contributing to α-smooth muscle actin–expressing cells within the infarct while retaining pericyte markers, intermediate states characterized by extracellular matrix remodeling, integrin signaling, and TGF-β activation[43,45,46].
Other subsets localize to nascent vessels, supporting arteriolar maturation and modulating immune cell infiltration. Clinically, this heterogeneity helps explain divergent post-infarction trajectories: early disruption of pericyte–endothelial coupling promotes permeability and inflammatory infiltration, whereas maladaptive persistence of fibrogenic states stiffens the border zone and drives adverse remodeling.
2.3.4. Cytokine Escalation and Inflammatory Cell Death
Cytokine storm represents loss of immune proportionality, wherein inflammatory signaling becomes self-propagating and dissociated from pathogen burden or tissue repair needs[29].
In certain acute inflammatory states, immune signaling may become amplified beyond the requirements of host defense, producing a phenotype commonly described as “cytokine storm.” Rather than representing a discrete diagnosis, this state is better understood as a spectrum of dysregulated immune activation in which proinflammatory mediators such as interleukin-1β, interleukin-6, tumor necrosis factor, interferon-γ, and interleukin-18 rise disproportionately relative to the inciting trigger[29].
Within a primed cardiovascular substrate, even moderate cytokine elevations may exert disproportionate effects, impairing myocardial contractility and destabilizing endothelial signaling, thereby narrowing the Pa − Pcrit gradient. However, cytokine escalation alone does not uniformly precipitate collapse. Rather, its impact likely depends on preexisting microvascular–immune priming and threshold instability, functioning as a context-dependent amplifier of microvascular failure rather than a universal cause.
Recent work suggests that severe inflammatory states may be sustained by a self-amplifying interaction between cytokine signaling and inflammatory cell death pathways, including pyroptosis, apoptosis, necroptosis, and their integrated form, PANoptosis. In experimental systems, cytokine signaling can induce lytic cell death in immune and parenchymal cells, releasing damage-associated molecular patterns that further propagate inflammatory activation.
Within the cardiovascular microvasculature, such processes may exacerbate endothelial injury, impair barrier integrity, and intensify leukocyte–platelet interactions. Sustained cytokine exposure has been associated with reversible myocardial dysfunction and altered microvascular responsiveness, phenomena observed in sepsis-associated and stress-induced cardiomyopathy[29,47].
This framework helps explain why cytokine storm syndromes frequently produce stress-induced or sepsis-associated cardiomyopathy, characterized by acute biventricular dysfunction that is often disproportionate to ischemic burden and partially reversible with immune modulation rather than revascularization or inotropic escalation[21].
2.3.5. Septic Cardiomyopathy as Prototype
Sepsis-associated cardiomyopathy illustrates immune-driven cardiovascular dysfunction in its purest form. Myocardial depression arises from cytokine-mediated alterations in calcium handling, mitochondrial function, and microvascular perfusion[48].
Importantly, ventricular recovery often lags behind normalization of systemic hemodynamics, reinforcing that immune-mediated microvascular injury, not pump failure alone, governs trajectory. Similar patterns are observed in cardiogenic shock complicating acute-on-chronic heart failure, where restoration of macrocirculatory indices fails to reverse endothelial injury and microvascular collapse[49].
2.3.6. NETosis and Immunothrombosis
As inflammatory signaling intensifies, neutrophils may become central effectors of microvascular injury through the formation of neutrophil extracellular traps (NETs). NETosis represents a specialized form of inflammatory cell death in which neutrophils release chromatin scaffolds decorated with proteases and histones. While NET formation evolved as a host defense mechanism, excessive or dysregulated NETosis has been associated with endothelial injury, platelet activation, and activation of coagulation pathways[50].
Within a destabilized microvascular environment, NET deposition may contribute to capillary obstruction and reinforce thrombo-inflammatory signaling. Experimental and clinical studies have demonstrated NET involvement in microvascular thrombosis across inflammatory and ischemic conditions, though the degree to which NETs serve as primary drivers versus secondary amplifiers likely varies by phenotype[51].
In the context of prior threshold instability, NET-mediated platelet aggregation and fibrin deposition may consolidate previously reversible capillary derecruitment. Rather than initiating collapse independently, immunothrombotic processes may stabilize and propagate microvascular obstruction once mechanical and inflammatory perturbations are established.
Clinically, this amplification may manifest as rising D-dimer levels, laboratory evidence of coagulopathy, and persistent tissue hypoxia despite restoration of macrocirculatory targets. However, NET-driven immunothrombosis should be understood as one component of a broader immune–vascular interaction rather than a singular causal pathway.
Once immunothrombotic amplification becomes established, therapeutic reversibility may diminish, and conventional hemodynamic support alone may prove insufficient.
Microvascular obstruction, endothelial injury, and immune activation perpetuate one another in a self-sustaining cycle only partially responsive to conventional hemodynamic or antithrombotic therapy. Three reinforcing mechanisms converge:
(a) pericyte-driven inflammatory signaling that destabilizes capillary integrity;
(b) NET deposition within the microvasculature; and
(c) platelet–NET scaffold formation that consolidates thrombo-inflammatory niches.
The resulting no-reflow phenomenon, persistent tissue hypoperfusion despite macrocirculatory restoration, marks the physiological endpoint of this cascade and signals transition to consolidated microvascular failure. At this stage, thrombosis reflects systemic immune–vascular dysregulation rather than an isolated vascular event.
2.4. Clinical Cardiovascular Syndromes
We broadly conceptualize these syndromes as reflecting two dominant modes of microvascular failure: impaired arterial oxygen delivery and impaired venous–lymphatic clearance, a concept illustrated in Figure 3.
2.4.1. Type 2 Myocardial Infarction
Type 2 myocardial infarction (MI) illustrates how cardiovascular injury may arise from microvascular instability rather than acute atherothrombosis. Defined as myocardial injury due to oxygen supply–demand imbalance in the absence of plaque rupture, type 2 MI frequently occurs in the setting of systemic illness, infection, anemia, tachyarrhythmia, or hemodynamic stress. Importantly, contemporary data demonstrate that these patients carry substantial long-term cardiovascular risk, with survival rates comparable to or worse than those observed in type 1 MI[52].
From a physiologic standpoint, the microcirculation accounts for the majority of total coronary vascular resistance. Accordingly, invasive assessment of microvascular function using indices such as the index of microcirculatory resistance (IMR) and coronary flow reserve (CFR) now carries a Class 1B recommendation in the 2024 ESC Guidelines for evaluation of patients with ischemia and non-obstructed coronary arteries[53].
In the inflamed or infected patient, a cascade of interconnected pathologies converges to critically lower the threshold for ischemia. Inflammatory activation, endothelial dysfunction, pericyte-mediated flow heterogeneity, and impaired oxygen extraction conspire to reduce myocardial oxygen delivery. Troponin release in this context often reflects reversible cardiomyocyte stress and microvascular ischemia, not extensive necrosis. This mechanistic distinction has profound therapeutic implications: it explains why aggressive antithrombotic strategies extrapolated from type 1 MI have not improved outcomes, and why correction of the triggering stressor alone is frequently insufficient.
Within the framework proposed here, type 2 MI may reflect failure of the coronary Pa − Pcrit gradient under systemic stress. Inflammatory activation, endothelial dysfunction, altered microvascular tone, and impaired oxygen extraction can narrow effective perfusion thresholds even in the absence of obstructive disease. Troponin elevation in this context may represent reversible microvascular ischemia and cardiomyocyte stress rather than large-territory necrosis.
Emerging experimental data support a role for immune–vascular crosstalk in this process. In a 2025 murine model, deletion of the co-stimulatory molecules CD80/86 attenuated ischemia–reperfusion injury by reducing endothelial E-selectin expression and limiting leukocyte infiltration into myocardial tissue[54].
While translational extrapolation must be cautious, these findings suggest that immune signaling may modulate microvascular injury severity independent of epicardial obstruction. Importantly, infection-related and inflammatory triggers of type 2 MI are associated with worse prognosis. In large registry data, adjusted cardiovascular event risk remains significantly elevated compared to patients without myocardial injury, even after accounting for comorbidities[52].
2.4.2. HFpEF Decompensation
Heart failure with preserved ejection fraction exemplifies chronic immune–microvascular and lymphatic dysfunction rather than isolated ventricular systolic impairment. Persistent diastolic dysfunction elevates filling pressures, produces systemic venous congestion, and impairs lymphatic clearance, establishing low-grade interstitial edema and immune activation even during clinical quiescence. This chronic congestion activates endothelial mechanoreceptors, promotes leukocyte adhesion in post-capillary venules, and impairs lymphatic clearance of inflammatory mediators, creating a self-perpetuating cycle of microvascular injury. Over time, these insults damage endothelial surfaces, promote capillary rarefaction, and reduce myocardial perfusion reserve, rendering the cardiovascular system vulnerable to additional stress[55].
Acute stressors, infection, surgery, hypertensive crises, atrial arrhythmias, trigger rapid decompensation in this primed substrate. The result is disproportionate congestion, diuretic resistance, and end-organ dysfunction, all unfolding despite a left ventricular ejection fraction that remains, by definition, preserved. Although the mechanistic literature on lymphatic overload and venous congestion has largely focused on acute right ventricular failure, the underlying physiology directly illuminates HFpEF pathobiology. In both conditions, chronically elevated right-sided and systemic venous pressures impair lymphatic drainage, producing persistent interstitial congestion with downstream cardiohepatic, cardiorenal, and cardiointestinal manifestations[56]. Organ dysfunction in this context reflects failed venous and lymphatic clearance, not merely inadequate forward flow.
These findings confirm HFpEF decompensation stems from congestive microvascular failure rather than simple volume overload, explaining why diuretic escalation or preload reduction alone often fails to restore effective tissue perfusion despite "adequate" macrocirculatory parameters[57].
“Just as Type 2 MI reveals failure of the arterial microcirculation to deliver oxygen, HFpEF decompensation reveals failure of the venous microcirculation and lymphatics to clear congestion.”
2.4.3. Targeted Anti-Inflammatory Therapy (CANTOS)
The CANTOS trial provides essential proof-of-principle that targeted immunomodulation can modify cardiovascular risk. Among 10,061 patients with prior myocardial infarction and residual inflammatory activity (hsCRP ≥2 mg/L), IL-1β inhibition with canakinumab reduced recurrent cardiovascular events independent of lipid lowering, establishing inflammation as a causal mechanism in atherothrombosis rather than a passive biomarker[58].
Equally instructive are the trial's constraints. CANTOS enrolled stable, chronically inflamed patients targeting the smoldering inflammatory activity that perpetuates microvascular dysfunction and plaque vulnerability. It was not designed for, and benefit would not be expected in, acute cytokine storm or established shock, where immunothrombotic consolidation may already be entrenched.
The mechanistic basis for this window of opportunity is well-defined. IL-1β inhibition downregulates endothelial adhesion molecules, reduces leukocyte trafficking, restores nitric oxide bioavailability, and attenuates platelet activation, collectively preserving microvascular integrity before irreversible barrier disruption occurs. Once capillary plugging, interstitial edema, and endothelial injury are established, immunomodulation is unlikely to reverse the injury and may impair reparative mechanisms.
CANTOS therefore validates both the promise and the boundaries of immune-targeted therapy: the immune–microvascular axis is therapeutically tractable, but intervention must be precisely timed. Delayed or indiscriminate immunosuppression risks futility, or harm.
2.5. Therapeutic and Research Implications
2.5.1. Preserving the Microvascular–Immune Unit
The following discussion translates the preceding mechanistic framework into potential therapeutic and investigative directions. Because direct clinical evidence for stage-specific, microvascular-targeted interventions is currently limited, these considerations are intended to highlight plausible strategies and to outline priorities for future research rather than to provide clinical recommendations.
If glycocalyx degradation and pericyte-mediated microvascular collapse represent the initiating structural events in decompensated heart failure, then therapeutic strategy must shift from pressure-centric resuscitation to preservation of the microvascular–immune unit. The failure in acute decompensation is not solely inadequate cardiac power generation, but loss of effective perfused capillary surface area and impaired lymphatic clearance. This section reframes current therapies and future directions through that lens. The stage-dependent therapeutic window proposed in this framework is summarized in Figure 4.
2.5.2. Glycocalyx Stabilization and Endothelial Preservation
The endothelial glycocalyx functions as a shear-sensitive mechanotransducer and the primary regulator of subglycocalyx oncotic gradients. Under the Revised Starling paradigm, once this layer is degraded, the capillary converts from a regulated exchange surface into a pressure-driven filtration membrane, amplifying edema formation. Restoration or preservation of the glycocalyx is therefore foundational to microvascular integrity. While direct clinical measurement of glycocalyx recovery remains limited, several established cardiovascular therapies exert endothelial-modulating effects that may indirectly support microvascular resilience.
Sodium–glucose cotransporter 2 (SGLT2) inhibitors reduce oxidative stress and inflammatory signaling and improve clinical outcomes in heart failure populations[59,60]. Although their primary benefits are multifactorial, it is plausible that improved endothelial signaling and interstitial fluid regulation contribute to raising the threshold for microvascular collapse.
Mineralocorticoid receptor antagonists (MRAs), including finerenone, attenuate aldosterone-mediated oxidative stress and vascular inflammation[61].
These effects may favorably influence microvascular stiffness and endothelial tone, though direct causal links to glycocalyx stabilization in humans remain to be established. RAAS inhibitors and statins confer robust mortality benefits that likely extend beyond blood pressure and lipid reduction. Both classes attenuate oxidative stress, dampen inflammatory signaling, and improve endothelial nitric oxide bioavailability mechanisms that may stabilize the endothelial surface layer.
Statins suppress matrix metalloproteinases, particularly MMP-9, a key mediator of syndecan shedding and glycocalyx degradation. Through metalloproteinase inhibition and reduction of redox-driven proteolysis, statins may indirectly preserve glycocalyx integrity and microvascular barrier function, although this remains a mechanistically plausible but unproven hypothesis in humans[62]. Supporting this concept, rosuvastatin partially restored systemic glycocalyx volume in patients with familial hypercholesterolemia despite complete LDL normalisation, suggesting that endothelial structural recovery could contribute to its pleiotropic benefits[63,64].
Similarly, RAAS inhibition reduces angiotensin II-mediated NADPH oxidase activation and downstream protease signaling, potentially limiting enzymatic shedding of glycocalyx components. Whether part of the clinical benefit of these established cardiovascular therapies reflects preservation of microvascular conductance or glycocalyx integrity represents an important area for translational investigation rather than a settled mechanism.
2.5.3. Acute Barrier Support and Fluid Strategy
In acute inflammatory states, endothelial surface disruption alters fluid responsiveness. Under such conditions, fluid loading may increase arterial pressure while simultaneously promoting interstitial edema and mechanical capillary compression. These observations suggest that resuscitation strategies might benefit from individualized consideration of microvascular stability rather than reliance on hydrostatic augmentation alone.
Albumin has theoretical advantages in this context. Beyond oncotic support, albumin transports sphingosine-1-phosphate and exhibits antioxidative properties that may support endothelial stability. However, current clinical evidence does not justify routine albumin administration solely for glycocalyx preservation, and phenotype-specific investigation is required[65,66].
Similarly, anticoagulation strategies may modulate inflammatory–thrombotic interactions at the microvascular level. While heparin and antithrombin exhibit anti-inflammatory properties, their net effects on endothelial surface integrity and immunothrombosis are complex and context-dependent. Prospective evaluation is needed before such approaches can be integrated into microvascular-targeted protocols.
2.5.4. Mechanical Circulatory Support and Microvascular Coherence
In SCAI stage E shock requiring Microaxial flow pumps, or Extracorporeal Support with ECMO, nonphysiologic shear stress, complement activation, hemolysis-derived free hemoglobin, and plasmin generation intensify ROS production and metalloproteinase activation. The result may be microvascular glycocalyx disruption and hemodynamic incoherence despite restoration of macrocirculatory parameters. In this context, controlled anticoagulation, albumin-supported barrier stabilization, and TEG-guided plasma replacement are mechanistically aligned with preservation of microvascular integrity and coherence, rather than pressure normalization alone[67].
While mechanistically plausible, this approach requires prospective evaluation before clinical adoption.
Prospective evaluation of glycocalyx integrity as a therapeutic target, using circulating biomarkers (syndecan-1, heparan sulfate) or emerging imaging techniques, represents the logical next step. Trials comparing glycocalyx-preserving strategies against pressure-centric resuscitation in acute decompensation could transform the management of cardiogenic shock and advanced heart failure.
2.5.5. Future Directions
Future investigation should prioritize validation of biomarkers and physiologic tools capable of detecting microvascular instability prior to structural consolidation. Circulating markers of endothelial injury and glycocalyx degradation, including syndecan-1 and heparan sulfate, alongside NET-associated proteins reflecting immunothrombotic activation, may delineate the transition from functional microvascular stress to inflammatory amplification. Advances in bedside microcirculatory assessment, including sublingual videomicroscopy, perfusion indices, and congestion scoring, may further enable real-time identification of hemodynamic incoherence in clinical settings.
Prospective trials are needed to evaluate whether stage-guided strategies, or perhaps targeting microvascular preservation early and immunothrombotic amplification, can alter the trajectory of cardiovascular decompensation. Integration of continuous physiologic monitoring with biomarker-based phenotyping may ultimately support precision approaches to circulatory support and immunomodulation across syndromes including HFpEF decompensation, cardiogenic shock, and Type 2 myocardial infarction.
3. Conclusions
Within this framework, immunothrombosis and NET-associated endothelial injury represent convergence points between inflammatory signaling and circulatory instability. Thrombosis at this stage reflects immune–vascular dysregulation that compromises capillary conductance despite restored macrocirculatory indices, suggesting that escalation of anticoagulation or pressure targets alone is insufficient when microvascular integrity remains unaddressed.
Pericytes and the endothelial surface layer emerge as critical integrators linking inflammatory tone to flow regulation, barrier stability, and structural remodeling. This reframes congestion in HFpEF not solely as volume excess, but as a failure of coordinated microvascular–interstitial–lymphatic coupling, and offers a physiologic basis through which SGLT2 inhibitors and mineralocorticoid receptor antagonists may confer benefit as modulators of endothelial signaling and microvascular resilience, particularly when applied early in the appropriate phenotypic context.
These observations remain mechanistically informed rather than prescriptive. Whether stage-guided preservation of the microvascular–immune unit alters clinical trajectory requires prospective validation. Nevertheless, across Type 2 MI, HFpEF decompensation, and cardiogenic shock, outcomes may ultimately be determined less by epicardial anatomy or isolated hemodynamic targets than by the timing, intensity, and reversibility of microvascular injury.
This framework positions the microvascular–immune interface not as a secondary consideration, but as a primary determinant of cardiovascular recovery, and offers a physiologically grounded foundation for future investigation into precision hemodynamic and immunomodulatory strategies.
Author Contributions
J.P. conceived the study, developed the conceptual framework, performed the literature synthesis, and drafted the manuscript. B.L. contributed to conceptual development of the graphical figures and provided input on the role of neutrophil extracellular traps and immunothrombosis. M.B. contributed to discussions and interpretation of the gut–vascular axis and its relevance to cardiovascular disease. A.G. contributed to the interpretation of HFpEF and venous–lymphatic mechanisms of microvascular dysfunction. All authors contributed to critical revisions of the manuscript, approved the final version, and agree to be accountable for the content of the work.
Funding
This research received no external funding.
Ethics Approval and Consent to Participate
Not applicable.
Data Availability Statement
This manuscript constitutes a narrative review and does not include primary research data. All referenced data are publicly available through the cited sources
Acknowledgments
The authors would like to acknowledge Dr. Carlos Jerjes-Sanchez Díaz for introducing the conceptual principles of immunothrombosis that influenced this work. The authors also thank Dr. Lakshmikanth, Dr. David London, and Dr. Nandyala for their mentorship and discussions on microvascular and congestive hemodynamics, which contributed to the development of the physiologic framework presented in this review.
Conflicts of Interest
The authors declare no conflict of interest.
Declaration of AI and AI-assisted Technologies in the Writing Process
During the preparation of this work the authors used ChatGpt-5.2 & Grammarly in order to check spell and grammar. Images designed in BioRender were enhanced with FigureLabsAI. After using these tools, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Abbreviations
HFpEF, heart failure with preserved ejection fraction; NO, nitric oxide; Pa, arterial pressure; Pcrit, critical closing pressure; Pmsf, mean systemic filling pressure; SvO₂, mixed venous oxygen saturation; ΔPCO₂, venous–arterial carbon dioxide difference; VExUS, venous excess ultrasound score; NETs, neutrophil extracellular traps; MI, myocardial infarction; IMR, index of microcirculatory resistance; CFR, coronary flow reserve; RAAS, renin–angiotensin–aldosterone system; SNS, sympathetic nervous system; SGLT2, sodium–glucose cotransporter 2; MRA, mineralocorticoid receptor antagonist; MMP, matrix metalloproteinase; ROS, reactive oxygen species; ECMO, extracorporeal membrane oxygenation; SCAI, Society for Cardiovascular Angiography and Interventions.
References
- Ince, C. [No Title Found]. Crit Care 2005, 9, S13. [CrossRef]
- Ince, C. Hemodynamic Coherence and the Rationale for Monitoring the Microcirculation. Crit Care 2015, 19, S8. [CrossRef]
- Młynarska, E.; Bojdo, K.; Frankenstein, H.; Krawiranda, K.; Kustosik, N.; Lisińska, W.; Rysz, J.; Franczyk, B. Endothelial Dysfunction as the Common Pathway Linking Obesity, Hypertension and Atherosclerosis. IJMS 2025, 26, 10096. [CrossRef]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure With Preserved Ejection Fraction. Journal of the American College of Cardiology 2013, 62, 263–271. [CrossRef]
- Libby, P.; Loscalzo, J.; Ridker, P.M.; Farkouh, M.E.; Hsue, P.Y.; Fuster, V.; Hasan, A.A.; Amar, S. Inflammation, Immunity, and Infection in Atherothrombosis. Journal of the American College of Cardiology 2018, 72, 2071–2081. [CrossRef]
- Rossitto, G.; Mary, S.; McAllister, C.; Neves, K.B.; Haddow, L.; Rocchiccioli, J.P.; Lang, N.N.; Murphy, C.L.; Touyz, R.M.; Petrie, M.C.; et al. Reduced Lymphatic Reserve in Heart Failure With Preserved Ejection Fraction. Journal of the American College of Cardiology 2020, 76, 2817–2829. [CrossRef]
- Han, J.-H. Immuno-Metabolic Diseases and Therapeutics: Molecular Mechanisms via Inflammasome Signaling. Cell Commun Signal 2025, 23, 373. [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The Immune System in Cardiac Homeostasis and Disease. Nat Rev Immunol 2018, 18, 733–744. [CrossRef]
- Isath, A.; Mehra, M.R. Cardio-Systemic Stress and Trained Innate Immune Memory in Heart Failure. Transplantation 2025, 109, 565–568. [CrossRef]
- Isath, A.; Desai, A.S.; Mehra, M.R. Beyond the Pump. JACC: Heart Failure 2026, 14, 102711. [CrossRef]
- Woodcock, T.E.; Woodcock, T.M. Revised Starling Equation and the Glycocalyx Model of Transvascular Fluid Exchange: An Improved Paradigm for Prescribing Intravenous Fluid Therapy. British Journal of Anaesthesia 2012, 108, 384–394. [CrossRef]
- Levick, J.R. Revision of the Starling Principle: New Views of Tissue Fluid Balance. The Journal of Physiology 2004, 557, 704–704. [CrossRef]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The Glycocalyx: A Novel Diagnostic and Therapeutic Target in Sepsis. Crit Care 2019, 23, 16. [CrossRef]
- Lee, L.L.; Chintalgattu, V. Pericytes in the Heart. In Pericyte Biology in Different Organs; Birbrair, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, 2019; Vol. 1122, pp. 187–210 ISBN 978-3-030-11092-5.
- Simmonds, S.J.; Grootaert, M.O.J.; Cuijpers, I.; Carai, P.; Geuens, N.; Herwig, M.; Baatsen, P.; Hamdani, N.; Luttun, A.; Heymans, S.; et al. Pericyte Loss Initiates Microvascular Dysfunction in the Development of Diastolic Dysfunction. European Heart Journal Open 2023, 4, oead129. [CrossRef]
- Wu, Y.; Fu, J.; Huang, Y.; Duan, R.; Zhang, W.; Wang, C.; Wang, S.; Hu, X.; Zhao, H.; Wang, L.; et al. Biology and Function of Pericytes in the Vascular Microcirculation. Anim Models and Exp Med 2023, 6, 337–345. [CrossRef]
- Cooper, S.T.E.; Lokman, A.B.; Riley, P.R. Role of the Lymphatics in Cardiac Disease. ATVB 2024, 44, 1181–1190. [CrossRef]
- Fudim, M.; Salah, H.M.; Sathananthan, J.; Bernier, M.; Pabon-Ramos, W.; Schwartz, R.S.; Rodés-Cabau, J.; Côté, F.; Khalifa, A.; Virani, S.A.; et al. Lymphatic Dysregulation in Patients With Heart Failure. Journal of the American College of Cardiology 2021, 78, 66–76. [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circulation Research 2018, 122, 337–351. [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat Rev Immunol 2013, 13, 34–45. [CrossRef]
- Mann, D.L. Innate Immunity and the Failing Heart: The Cytokine Hypothesis Revisited. Circulation Research 2015, 116, 1254–1268. [CrossRef]
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [CrossRef]
- Hotamisligil, G.S. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 2017, 47, 406–420. [CrossRef]
- Libby, P. Mechanisms of Acute Coronary Syndromes and Their Implications for Therapy. New England Journal of Medicine 2013, 368, 2004–2013. [CrossRef]
- Braunwald, E. Heart Failure. JACC: Heart Failure 2013, 1, 1–20. [CrossRef]
- Sandek, A.; Swidsinski, A.; Schroedl, W.; Watson, A.; Valentova, M.; Herrmann, R.; Scherbakov, N.; Cramer, L.; Rauchhaus, M.; Grosse-Herrenthey, A.; et al. Intestinal Blood Flow in Patients With Chronic Heart Failure. Journal of the American College of Cardiology 2014, 64, 1092–1102. [CrossRef]
- Dodi, A.E.; Jacobs, M. Acute Right Ventricular Failure in the Medical ICU. Lung 2025, 203, 107. [CrossRef]
- Michel, C.C.; Curry, F.E. Microvascular Permeability. Physiological Reviews 1999, 79, 703–761. [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N Engl J Med 2020, 383, 2255–2273. [CrossRef]
- De Backer, D.; Orbegozo Cortes, D.; Donadello, K.; Vincent, J.-L. Pathophysiology of Microcirculatory Dysfunction and the Pathogenesis of Septic Shock. Virulence 2014, 5, 73–79. [CrossRef]
- Castro, R.; Retamal, J.; Hernández, G.; Kattan, E.; Pinsky, M.R. Critical Closing Pressure in the Circulation: Understanding the Vascular Waterfall Phenomenon. Journal of Critical Care 2026, 93, 155485. [CrossRef]
- Miller, A.; Rola, P.; Spiegel, R.; Haycock, K. Arteriolar Collapse and Haemodynamic Incoherence in Shock: Rethinking Critical Closing Pressure. JPM 2026, 16, 78. [CrossRef]
- Schemmelmann, M.; Kelm, M.; Jung, C. The Microcirculation in Cardiogenic Shock. European Heart Journal: Acute Cardiovascular Care 2024, 13, 802–809. [CrossRef]
- Jung, C.; Fuernau, G.; De Waha, S.; Eitel, I.; Desch, S.; Schuler, G.; Figulla, H.R.; Thiele, H. Intraaortic Balloon Counterpulsation and Microcirculation in Cardiogenic Shock Complicating Myocardial Infarction: An IABP-SHOCK II Substudy. Clin Res Cardiol 2015, 104, 679–687. [CrossRef]
- Dubin, A.; Pozo, M.O.; Casabella, C.A.; Pálizas, F.; Murias, G.; Moseinco, M.C.; Kanoore Edul, V.S.; Pálizas, F.; Estenssoro, E.; Ince, C. Increasing Arterial Blood Pressure with Norepinephrine Does Not Improve Microcirculatory Blood Flow: A Prospective Study. Crit Care 2009, 13, R92. [CrossRef]
- Chommeloux, J.; Montero, S.; Franchineau, G.; Bréchot, N.; Hékimian, G.; Lebreton, G.; Le Guennec, L.; Bourcier, S.; Nieszkowska, A.; Leprince, P.; et al. Microcirculation Evolution in Patients on Venoarterial Extracorporeal Membrane Oxygenation for Refractory Cardiogenic Shock. Critical Care Medicine 2020, 48, e9–e17. [CrossRef]
- Cooper, S.T.E.; Lokman, A.B.; Riley, P.R. Role of the Lymphatics in Cardiac Disease. ATVB 2024, 44, 1181–1190. [CrossRef]
- Garapati, H.N.; Dandamudi, S.; Kalagara, H. Venous Excess Ultrasound Score (VExUS). Best Practice & Research Clinical Anaesthesiology 2025, 39, 351–359. [CrossRef]
- Hørsdal, O.K. Can Utilization of the Venous-to-Arterial Carbon Dioxide Difference Improve Patient Outcomes in Cardiogenic Shock? A Narrative Review. Am Heart J Plus 2025, 50, 100504. [CrossRef]
- Bourguignon, C.; Grouthier, V.; Chapouly, C.; Couffinhal, T.; Renault, M.-A. Cardiovascular Risk Factors Have an Impact on the Biology of Pericytes. Archives of Cardiovascular Diseases 2025, 118, S180. [CrossRef]
- Hamilton, N.B. Pericyte-Mediated Regulation of Capillary Diameter: A Component of Neurovascular Coupling in Health and Disease. Front. Neuroenerg. 2010, 2. [CrossRef]
- Schrimpf, C.; Teebken, O.E.; Wilhelmi, M.; Duffield, J.S. The Role of Pericyte Detachment in Vascular Rarefaction. J Vasc Res 2014, 51, 247–258. [CrossRef]
- Moro, M.; Balestrero, F.C.; Grolla, A.A. Pericytes: Jack-of-All-Trades in Cancer-Related Inflammation. Front. Pharmacol. 2024, 15, 1426033. [CrossRef]
- Brandt, M.M.; Cheng, C.; Merkus, D.; Duncker, D.J.; Sorop, O. Mechanobiology of Microvascular Function and Structure in Health and Disease: Focus on the Coronary Circulation. Front. Physiol. 2021, 12, 771960. [CrossRef]
- Van Splunder, H.; Villacampa, P.; Martínez-Romero, A.; Graupera, M. Pericytes in the Disease Spotlight. Trends in Cell Biology 2024, 34, 58–71. [CrossRef]
- Moore-Morris, T.; Evans, S.M. Cardiac Pericyte Diversity in Infarct Remodeling: Not Just Vascular Support Cells? Circulation 2023, 148, 899–902. [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149-168.e17. [CrossRef]
- Boissier, F.; Aissaoui, N. Septic Cardiomyopathy: Diagnosis and Management. Journal of Intensive Medicine 2022, 2, 8–16. [CrossRef]
- Chommeloux, J.; Montero, S.; Franchineau, G.; Bréchot, N.; Hékimian, G.; Lebreton, G.; Le Guennec, L.; Bourcier, S.; Nieszkowska, A.; Leprince, P.; et al. Microcirculation Evolution in Patients on Venoarterial Extracorporeal Membrane Oxygenation for Refractory Cardiogenic Shock. Critical Care Medicine 2020, 48, e9–e17. [CrossRef]
- Bautista-Becerril, B.; Campi-Caballero, R.; Sevilla-Fuentes, S.; Hernández-Regino, L.M.; Hanono, A.; Flores-Bustamante, A.; González-Flores, J.; García-Ávila, C.A.; Aquino-Gálvez, A.; Castillejos-López, M.; et al. Immunothrombosis in COVID-19: Implications of Neutrophil Extracellular Traps. Biomolecules 2021, 11, 694. [CrossRef]
- Vazquez-Garza, E.; Jerjes-Sanchez, C.; Navarrete, A.; Joya-Harrison, J.; Rodriguez, D. Venous Thromboembolism: Thrombosis, Inflammation, and Immunothrombosis for Clinicians. J Thromb Thrombolysis 2017, 44, 377–385. [CrossRef]
- Chapman, A.R.; Taggart, C.; Boeddinghaus, J.; Mills, N.L.; Fox, K.A.A. Type 2 Myocardial Infarction: Challenges in Diagnosis and Treatment. European Heart Journal 2025, 46, 504–517. [CrossRef]
- Smilowitz, N.R.; Toleva, O.; Chieffo, A.; Perera, D.; Berry, C. Coronary Microvascular Disease in Contemporary Clinical Practice. Circ: Cardiovascular Interventions 2023, 16. [CrossRef]
- Liu, L.; Wang, X.-X.; Wang, S.-X.; Yang, H.; Xiao, X.; Li, N.; Chai, H.-J.; Wang, H.-X. Attenuation of Myocardial Ischemia–Reperfusion Injury in Mice through CD80/86 Deficiency: Improved Microvascular Obstruction via Reduced Macrophage and T Lymphocyte Infiltration. Basic Res Cardiol 2025, 120, 873–888. [CrossRef]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure With Preserved Ejection Fraction. Journal of the American College of Cardiology 2013, 62, 263–271. [CrossRef]
- Itkin, M.; Rockson, S.G.; Burkhoff, D. Pathophysiology of the Lymphatic System in Patients With Heart Failure. Journal of the American College of Cardiology 2021, 78, 278–290. [CrossRef]
- Landi, I.; Guerritore, L.; Iannaccone, A.; Ricotti, A.; Rola, P.; Garrone, M. Assessment of Venous Congestion with Venous Excess Ultrasound Score in the Prognosis of Acute Heart Failure in the Emergency Department: A Prospective Study. European Heart Journal Open 2024, 4, oeae050. [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 2017, 377, 1119–1131. [CrossRef]
- McLean, P.; Bennett, J.; “Trey” Woods, E.; Chandrasekhar, S.; Newman, N.; Mohammad, Y.; Khawaja, M.; Rizwan, A.; Siddiqui, R.; Birnbaum, Y.; et al. SGLT2 Inhibitors across Various Patient Populations in the Era of Precision Medicine: The Multidisciplinary Team Approach. npj Metab Health Dis 2025, 3, 29. [CrossRef]
- Durante, W.; Behnammanesh, G.; Peyton, K.J. Effects of Sodium-Glucose Co-Transporter 2 Inhibitors on Vascular Cell Function and Arterial Remodeling. IJMS 2021, 22, 8786. [CrossRef]
- Roman-Pepine, D.; Serban, A.M.; Capras, R.-D.; Cismaru, C.M.; Filip, A.G. A Comprehensive Review: Unraveling the Role of Inflammation in the Etiology of Heart Failure. Heart Fail Rev 2025, 30, 931–954. [CrossRef]
- Jacquet, C.; Gustafsson, R.; Patel, A.K.; Hansson, M.; Rankin, G.; Bano, F.; Byström, J.W.; Blomberg, A.; Rasmuson, J.; Satchell, S.; et al. Matrix Metalloproteinase-9 Mediates Endothelial Glycocalyx Degradation and Correlates with Severity of Hemorrhagic Fever with Renal Syndrome 2025.
- Meuwese, M.C.; Mooij, H.L.; Nieuwdorp, M.; Van Lith, B.; Marck, R.; Vink, H.; Kastelein, J.J.P.; Stroes, E.S.G. Partial Recovery of the Endothelial Glycocalyx upon Rosuvastatin Therapy in Patients with Heterozygous Familial Hypercholesterolemia. Journal of Lipid Research 2009, 50, 148–153. [CrossRef]
- Banerjee, S.; Mwangi, J.G.; Stanley, T.K.; Mitra, R.; Ebong, E.E. Regeneration and Assessment of the Endothelial Glycocalyx To Address Cardiovascular Disease. Ind. Eng. Chem. Res. 2021, 60, 17328–17347. [CrossRef]
- Sawashita, Y.; Kazuma, S.; Tokinaga, Y.; Kikuchi, K.; Hirata, N.; Masuda, Y.; Yamakage, M. Albumin Protects the Ultrastructure of the Endothelial Glycocalyx of Coronary Arteries in Myocardial Ischemia-Reperfusion Injury in Vivo. Biochemical and Biophysical Research Communications 2023, 666, 29–35. [CrossRef]
- Aldecoa, C.; Llau, J.V.; Nuvials, X.; Artigas, A. Role of Albumin in the Preservation of Endothelial Glycocalyx Integrity and the Microcirculation: A Review. Ann. Intensive Care 2020, 10, 85. [CrossRef]
- Levy, J.H.; Alexander, P.M.A.; Wolberg, A.S.; McCarty, O.J.T.; Pusateri, A.E.; Bartz, R.R.; Bergmeier, W.; Cohen, M.J.; Connors, J.M.; Morrissey, J.H.; et al. ECMO-Induced Coagulopathy: Strategic Initiatives for Research and Clinical Practice (a Workshop Report of the NHLBI). Blood Vessels, Thrombosis & Hemostasis 2025, 2, 100064. [CrossRef]
Figure 1.
The Primed Microvascular–Immune Unit. Panel A illustrates the homeostatic microvascular–immune interface, characterized by intact endothelial signaling, preserved glycocalyx structure, stable pericyte attachment, minimal leukocyte–platelet interaction, and functional lymphatic drainage. Under these conditions, the effective microvascular driving pressure (Pa − Pcrit) is stable, and perfusion is preserved with adequate microvascular reserve. Panel B depicts chronic inflammatory priming. Subtle glycocalyx thinning, endothelial activation, altered pericyte phenotype, platelet sensitization, and reduced lymphatic clearance coexist with preserved baseline perfusion. Although capillary conductance remains intact at rest, baseline vascular tone is modestly elevated (Pcrit increased but still < Pa), reducing reserve and lowering the threshold for subsequent instability. Abbreviations: Pa, arterial pressure; Pcrit, critical closing pressure.
Figure 1.
The Primed Microvascular–Immune Unit. Panel A illustrates the homeostatic microvascular–immune interface, characterized by intact endothelial signaling, preserved glycocalyx structure, stable pericyte attachment, minimal leukocyte–platelet interaction, and functional lymphatic drainage. Under these conditions, the effective microvascular driving pressure (Pa − Pcrit) is stable, and perfusion is preserved with adequate microvascular reserve. Panel B depicts chronic inflammatory priming. Subtle glycocalyx thinning, endothelial activation, altered pericyte phenotype, platelet sensitization, and reduced lymphatic clearance coexist with preserved baseline perfusion. Although capillary conductance remains intact at rest, baseline vascular tone is modestly elevated (Pcrit increased but still < Pa), reducing reserve and lowering the threshold for subsequent instability. Abbreviations: Pa, arterial pressure; Pcrit, critical closing pressure.

Figure 2.
Threshold Instability and Collapse of the Arterial Waterfall. Panel A represents the physiologic state in which arterial pressure (Pa) exceeds critical closing pressure (Pcrit), and Pcrit remains above mean systemic filling pressure (Pmsf), preserving a functional arterial waterfall. Microvascular driving pressure (Pa − Pcrit) is adequate, and hemodynamic coherence is intact. Panel B illustrates threshold narrowing. Reductions in Pa and/or elevations in effective closing pressure progressively narrow the Pa − Pcrit gradient, reducing microvascular reserve while perfusion remains partially preserved. Panel C depicts threshold collapse and amplification. As Pa approaches Pcrit, segmental capillary dropout occurs. Increased interstitial pressure and effective closing thresholds further narrow the perfusion window. Microvascular conductance becomes heterogeneous, resulting in loss of hemodynamic coherence, wherein macrocirculatory normalization may coexist with misleading SvO₂ values and widening venous–arterial CO₂ gradients (ΔPCO₂). Abbreviations: Pa, arterial pressure; Pcrit, critical closing pressure; Pmsf, mean systemic filling pressure; SvO₂, mixed venous oxygen saturation.
Figure 2.
Threshold Instability and Collapse of the Arterial Waterfall. Panel A represents the physiologic state in which arterial pressure (Pa) exceeds critical closing pressure (Pcrit), and Pcrit remains above mean systemic filling pressure (Pmsf), preserving a functional arterial waterfall. Microvascular driving pressure (Pa − Pcrit) is adequate, and hemodynamic coherence is intact. Panel B illustrates threshold narrowing. Reductions in Pa and/or elevations in effective closing pressure progressively narrow the Pa − Pcrit gradient, reducing microvascular reserve while perfusion remains partially preserved. Panel C depicts threshold collapse and amplification. As Pa approaches Pcrit, segmental capillary dropout occurs. Increased interstitial pressure and effective closing thresholds further narrow the perfusion window. Microvascular conductance becomes heterogeneous, resulting in loss of hemodynamic coherence, wherein macrocirculatory normalization may coexist with misleading SvO₂ values and widening venous–arterial CO₂ gradients (ΔPCO₂). Abbreviations: Pa, arterial pressure; Pcrit, critical closing pressure; Pmsf, mean systemic filling pressure; SvO₂, mixed venous oxygen saturation.

Figure 3.
Arterial supply versus venous–lymphatic clearance failure in cardiovascular syndromes. (A) Arterial microvascular failure (Type 2 MI). Narrowing of the arterial perfusion gradient (Pa − Pcrit) reduces myocardial oxygen delivery, producing capillary underperfusion and supply–demand mismatch despite patent epicardial vessels. (B) Venous–lymphatic microvascular failure (HFpEF). Elevated filling pressures and venous hypertension cause interstitial edema and lymphatic congestion, increasing diffusion distance and impairing metabolite clearance despite preserved arterial inflow.
Figure 3.
Arterial supply versus venous–lymphatic clearance failure in cardiovascular syndromes. (A) Arterial microvascular failure (Type 2 MI). Narrowing of the arterial perfusion gradient (Pa − Pcrit) reduces myocardial oxygen delivery, producing capillary underperfusion and supply–demand mismatch despite patent epicardial vessels. (B) Venous–lymphatic microvascular failure (HFpEF). Elevated filling pressures and venous hypertension cause interstitial edema and lymphatic congestion, increasing diffusion distance and impairing metabolite clearance despite preserved arterial inflow.

Figure 4.
Therapeutic Windows Across the Immune–Microvascular Continuum. This schematic illustrates a hypothesis-generating framework in which the timing of intervention relative to the evolution of microvascular-immune dysfunction may influence therapeutic efficacy. The interventions shown are mechanistically plausible examples based on preclinical and indirect clinical evidence; however, stage-specific benefit remains unproven. No direct comparative trials have validated this model. The gradient arrow represents the authors’ conceptual synthesis, not established clinical guidance. Future research should test whether stage-guided strategies improve outcomes.
Figure 4.
Therapeutic Windows Across the Immune–Microvascular Continuum. This schematic illustrates a hypothesis-generating framework in which the timing of intervention relative to the evolution of microvascular-immune dysfunction may influence therapeutic efficacy. The interventions shown are mechanistically plausible examples based on preclinical and indirect clinical evidence; however, stage-specific benefit remains unproven. No direct comparative trials have validated this model. The gradient arrow represents the authors’ conceptual synthesis, not established clinical guidance. Future research should test whether stage-guided strategies improve outcomes.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



