Submitted:
03 April 2026
Posted:
03 April 2026
You are already at the latest version
Abstract
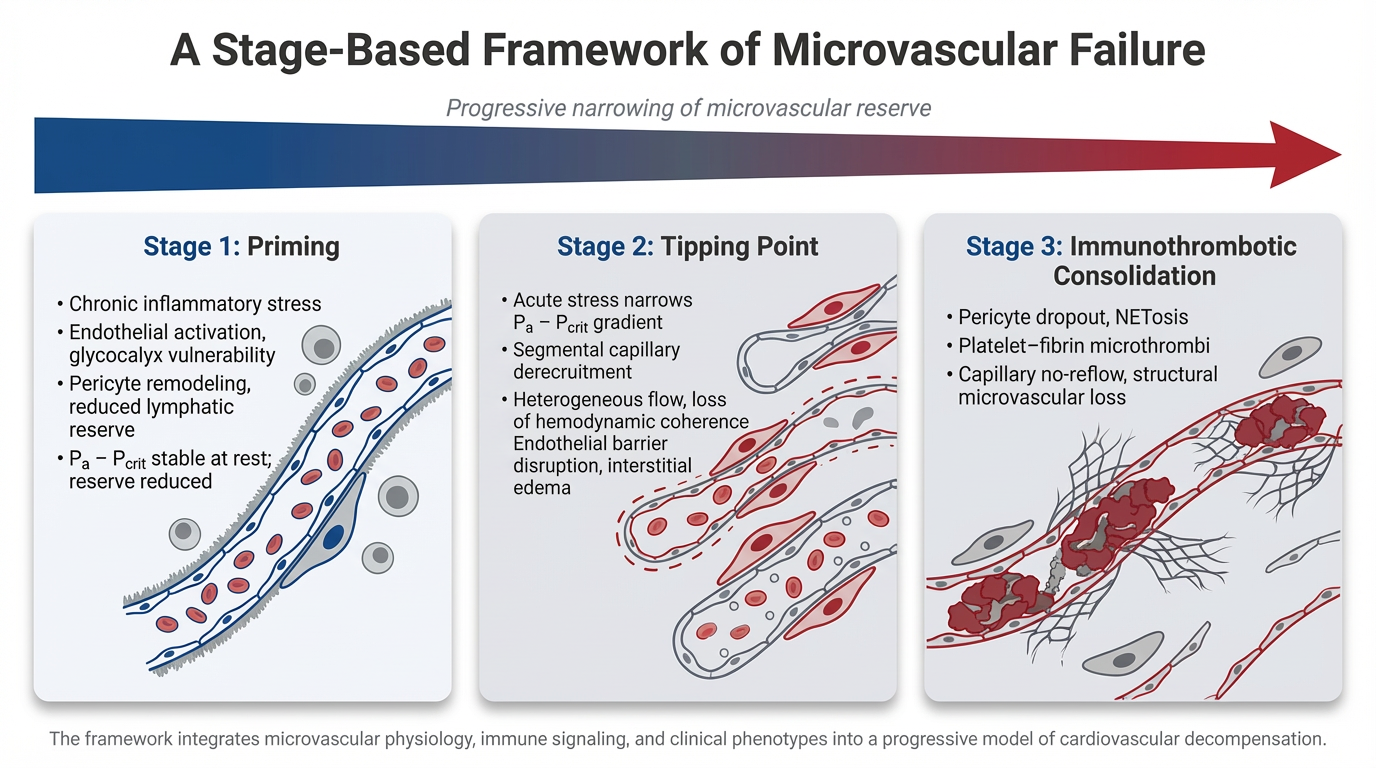
Cardiovascular disease is traditionally interpreted through macrocirculatory parameters such as cardiac output, vascular resistance, and epicardial coronary anatomy. However, clinical outcomes frequently diverge from predictions based solely on these indices, particularly in syndromes such as heart failure with preserved ejection fraction (HFpEF), cardiogenic shock, and sepsis-associated myocardial dysfunction. Increasing evidence suggests that the integrity of the microvascular–immune interface plays a central role in determining tissue perfusion and cardiovascular resilience. This review proposes a staged framework of cardiovascular decompensation centered on progressive failure of this interface.
In Stage 1, chronic cardiometabolic and inflammatory stress produces a primed but compensated microvascular state characterized by endothelial activation, glycocalyx vulnerability, pericyte remodeling, platelet sensitization, and reduced lymphatic reserve. Perfusion is preserved at rest, but vasodilatory reserve and microvascular stability are reduced, narrowing the effective perfusion window under physiologic stress.
In Stage 2, acute insults such as infection, ischemia, or neurohumoral activation precipitate threshold instability within the microcirculation. Perfusion becomes governed by the arterial pressure–critical closing pressure (Pa − Pcrit) relationship rather than traditional arterial–venous gradients. As this window narrows, segmental capillary derecruitment and heterogeneous flow emerge, producing loss of hemodynamic coherence in which systemic blood pressure and cardiac output may appear preserved despite impaired tissue perfusion.
In Stage 3, inflammatory amplification and immunothrombotic processes consolidate microvascular dysfunction. Pericyte contraction, endothelial injury, cytokine escalation, and neutrophil extracellular trap formation promote platelet–fibrin deposition and capillary obstruction, transforming reversible conductance failure into structural microvascular impairment.
This framework provides a unifying physiologic lens for diverse cardiovascular syndromes, including Type 2 myocardial infarction, HFpEF decompensation, and cardiogenic shock. It also suggests that therapeutic efficacy may depend less on macrocirculatory normalization alone and more on preserving microvascular integrity before immunothrombotic consolidation occurs. Although this model remains hypothesis-generating, it highlights the microvascular–immune interface as a central determinant of cardiovascular stability and a potential target for future precision hemodynamic and immunomodulatory strategies.
Keywords:
microcirculation
; glycocalyx
; immunothrombosis
; critical closing pressure
; hemodynamic coherence
; HFpEF
; cardiogenic shock
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



