Submitted:
02 June 2026
Posted:
04 June 2026
You are already at the latest version
Abstract
Alzheimer’s disease (AD) affects more women than men, and risk rises precipitously during and after the menopausal transition. Estrogen deficiency has been the most prominent hypothesis to explain this sex difference, but increasing evidence also implicates follicle-stimulating hormone (FSH) as an independent contributor to neurodegenerative risk. This narrative review integrates the literature on reproductive aging, AD pathobiology, and sex differences in AD, with an emphasis on endocrine, metabolic, and inflammatory mechanisms relevant to their relationship. PubMed and Google Scholar were searched for peer-reviewed human studies, animal models, and mechanistic investigations published through early 2026, prioritizing primary research and systematic reviews on FSH signaling, ApoE biology, and AD pathophysiology. FSH rises in a graded fashion across the menopausal transition and has been associated with multiple pathways implicated in AD, including C/EBPβ–δ-secretase signaling, mitochondrial function, neuronal glucose metabolism, and autophagic-lysosomal clearance - though the causal directionality of many of these relationships remains to be established in humans. Dysfunction in these interrelated systems has been associated with Aβ accumulation, tau pathology, and chronic neuroinflammation. FSH also appears to influence apolipoprotein biology, particularly ApoE, through actions on lipid metabolism, protein lipidation, and clearance, with downstream effects on Aβ aggregation and inflammatory signaling that differ by ApoE isoform. In addition, reproductive aging is associated with changes in vascular integrity and blood-brain barrier function that may precede classical AD pathology. This review describes the mechanistic pathways through which chronically elevated FSH may contribute to AD risk in women and discusses the potential therapeutic implications of FSH modulation, while acknowledging that much of the current mechanistic evidence derives from preclinical models and requires validation in human populations.
Keywords:
Alzheimers
; reproductive aging
; neurobiology
; endocrinology
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common cause of dementia worldwide, and it presents an urgent public health concern. In the United States, approximately one in nine persons aged 65 years and older (10.8%) have AD, and each year an additional 1,275 per 100,000 persons develop the disease (Zhang et al., 2024). AD is a clinical diagnosis with primary features of amnestic cognitive impairment. Signs and symptoms may begin as early-onset depression, anxiety, social withdrawal, and sleep disturbance, which over time can develop into disabling and progressive memory loss, neuropsychiatric symptoms such as hallucinations and delusions, and behavioral dysregulation. In a subset of cases, AD may present with non-amnestic symptoms, including memory impairment with relative sparing of other domains, which may involve visuospatial processing, language, executive function, or motor dysfunction.
AD is broadly classified as familial and sporadic. Familial AD (FAD) represents about 1-5% of cases and is typically caused by autosomal dominant mutations in APP, PS1, and PS2. The onset of FAD symptoms usually occurs between 30 and 65 years of age, and disease progression is rapid (Liu et al., 2019). Sporadic or late-onset AD (SAD) makes up over 95% of AD cases and generally has a later age of onset, after 65 years old, and is a multifactorial consequence of polygenic risk, environmental exposures, and medical comorbidities (Fedele, 2023; Suresh et al., 2023).
Major risk factors for late-onset AD include older age, female sex, and APOE genotype (Riedel et al., 2016). In the United States, nearly two-thirds of individuals with AD are women, and the majority of cases occur after 65 years old (Alzheimer’s Disease and Dementia, 2024). The increased risk among women is likely due to a complex coordination of changes in endocrine and neurobiological systems that occurs with reproductive aging. Age-related declines in levels of circulating estrogen and progesterone, along with changes in gonadotropins and other hypothalamic signaling peptides (e.g., FSH, LH, GnRH), correspond with objective alterations in brain structure and function. Reductions in bioavailable estrogen have been associated with a higher risk of global cognitive decline and deficits in verbal memory (Laughlin et al., 2010; Zsido et al., 2019). Peri- and postmenopausal women also experience faster rates of cognitive decline compared to men, as well as greater loss of hippocampal volume (Mosconi et al., 2018). Moreover, vasomotor symptoms, such as night-time hot flashes, have been associated with impaired memory performance and higher levels of amyloid biomarkers (Maki et al., 2008; Thurston et al., 2024).
Critically, however, reproductive aging is also associated with an accumulation of other hormonal changes in addition to estrogen decline. Among these, the progressive elevation of FSH has emerged as another potentially important driver of age-related pathology. Chronically high levels of FSH have been associated with several chronic disorders, including AD, osteoporosis, cardiovascular dysfunction, and metabolic abnormalities (Lizneva et al., 2019). These findings suggest that FSH may play an active endocrine role in driving multiple systems of aging in addition to serving as a compensatory biomarker of ovarian insufficiency.
Prior reviews of sex differences in AD have focused predominantly on estrogen deficiency as the primary endocrine driver of female vulnerability (Lopez-Lee et al., 2024; Scheyer et al., 2018; Rocca et al., 2024), while reviews of FSH biology have largely addressed its reproductive and metabolic roles in peripheral tissues (Lizneva et al., 2019). To date, no review has systematically examined the convergence of FSH signaling, ApoE isoform biology, and the upstream cellular dysregulation - including mitochondrial dysfunction, autophagic impairment, neuroinflammation, and blood-brain barrier disruption - that may collectively link reproductive aging to AD pathogenesis. A specific and underexamined question in this context is whether FSH elevation constitutes an independent, mechanistically active contributor to neurodegeneration, rather than merely a correlate of declining ovarian function. The present review addresses this gap by synthesizing mechanistic evidence across these intersecting domains, with particular attention to how FSH and ApoE4 interactions may compound neurodegenerative risk in aging women.
In this review, the mechanistic pathways through which chronically elevated FSH may drive downstream pathological changes relevant to AD are discussed, along with the potential therapeutic implications of FSH modulation.
Methods
This narrative review of the peer-reviewed literature explores mechanistic links between reproductive aging, FSH signaling, ApoE biology, and AD pathogenesis. The narrative review format was deemed appropriate for the stated aim, which is to integrate mechanistic and translational evidence across intersecting biological fields, rather than to quantitatively summarize a well-defined set of primary studies.
Literature Search
PubMed and Google Scholar were queried for relevant peer-reviewed literature through early 2026. Search terms used included, in various combinations, “follicle-stimulating hormone”, “FSH”, “reproductive aging”, “menopause”, “Alzheimer’s disease”, “neurodegeneration”, “ApoE”, “apolipoprotein E”, “APOE4”, “C/EBPβ”, “delta-secretase”, “mitochondrial dysfunction”, “autophagy”, “neuroinflammation”, “blood-brain barrier”, “BBB”, “amyloid-beta”, and “tau”. Reference lists of the articles retrieved were manually screened.
Inclusion and Exclusion Criteria
Criteria for inclusion of studies in the review consisted of a focus on FSH signaling, ApoE biology, or reproductive aging as it relates to neurodegeneration or AD-relevant cellular pathways. Eligible studies for review were peer reviewed original research articles, systematic reviews and meta-analyses, and mechanistic studies in human, animal or cell based models. Only articles in English were included. Articles within the past 15 years were prioritized, in order to capture the current state of the field and shifting hypotheses where applicable. However, no formal date restriction was applied, and where appropriate core mechanistic or epidemiological claims were supplemented with seminal studies outside of this window.
Study Quality and Synthesis Approach
The studies included in this review are diverse. In particular, they range from epidemiological cohorts to clinical trials, to genetically modified mouse models and mechanistic cell studies. A risk-of-bias assessment was therefore not performed. Statements of the plausibility of individual mechanistic hypotheses are presented in the text and are qualified depending on the available data (confirmations in independent human cohorts, limited evidence from animal or cell models or hypotheses that are mechanistic extrapolations). Conflicting results are discussed when appropriate.
Overview of Canonical Cellular Alterations Underlying Neurodegeneration in Alzheimer’s Disease
Cholinergic Hypothesis
The cholinergic hypothesis is one of the first proposed models for AD pathogenesis. The basis of the hypothesis is the selective loss of cholinergic neurons that originate in the nucleus basalis of Meynert (NBM) that results in decreased choline acetyltransferase (ChAT) activity and subsequent decrease in acetylcholine synthesis, leading to cortical cholinergic dysfunction (Berry & Harrison, 2023). The cholinergic deficits in the cerebral cortex are accompanied by a significant increase in the deposition of senile plaques in these same regions (Serrano-Pozo et al., 2011). Overall, this hypothesis represents a model in which there is a clear link between basal forebrain cholinergic dysfunction and AD cognitive deficits (Hampel et al., 2019). The NBM, in addition to the medial septum and diagonal band of Broca (MS/DBB) project cholinergic innervation throughout the cerebral cortex and are some of the first regions to undergo neuronal degeneration (Schmitz et al., 2018).
ACh is synthesized in cholinergic neurons by the catalytic action of choline acetyltransferase (ChAT) on choline and acetyl-coenzyme A. ACh is loaded into synaptic vesicles by the vesicular acetylcholine transporter (VAChT) and is released from presynaptic terminals into the synaptic cleft upon presynaptic depolarization. ACh diffuses across the synaptic cleft and can bind to mAChRs and nAChRs on the postsynaptic cell (Paul et al., 2022). ACh signaling is terminated primarily by rapid enzymatic degradation of ACh by AChE to form choline. The choline is then taken back up by the presynaptic neuron (Berry & Harrison, 2023; Stanciu et al., 2019). Alterations in ChAT expression or activity cause a deficiency of ACh that can result in decreased cholinergic signaling. Cholinergic signaling deficits can influence a range of physiological processes that are modulated by cholinergic signaling, including cognitive function, attention, motor behaviors, and sleep–wake regulation (Walczak-Nowicka & Herbet, 2021).
Figure 1.
Cholinergic projections originate from the basal forebrain nuclei (medial septum, abbreviated as MS, vertical limb of diagonal band of Broca, abbreviated as vDB, nucleus basalis of Meynert, abbreviated as NBM, and substantia innominata, abbreviated as SI), which project widely to the hippocampus, thalamus, olfactory bulb and cortical areas. Simultaneously, cholinergic neurons of the pontomesencephalon (laterodorsal tegmental nucleus, abbreviated as LDT and pedunculopontine tegmental nucleus, abbreviated as PPT) project to the hindbrain, thalamus, hypothalamus and basal forebrain, which are thought to be important for the regulation of arousal and behavioural state. The NBM and MS-DBB are two of the first regions in the brain to be selectively affected by neurodegeneration in Alzheimer’s disease and the loss of cholinergic innervation to the hippocampus and cortex is responsible for the deficits in memory and attention. Arrow thickness and color are for illustrative purposes and should not be assumed to be quantitative differences in projection density. Adapted from Paul et al. (2015).
Figure 1.
Cholinergic projections originate from the basal forebrain nuclei (medial septum, abbreviated as MS, vertical limb of diagonal band of Broca, abbreviated as vDB, nucleus basalis of Meynert, abbreviated as NBM, and substantia innominata, abbreviated as SI), which project widely to the hippocampus, thalamus, olfactory bulb and cortical areas. Simultaneously, cholinergic neurons of the pontomesencephalon (laterodorsal tegmental nucleus, abbreviated as LDT and pedunculopontine tegmental nucleus, abbreviated as PPT) project to the hindbrain, thalamus, hypothalamus and basal forebrain, which are thought to be important for the regulation of arousal and behavioural state. The NBM and MS-DBB are two of the first regions in the brain to be selectively affected by neurodegeneration in Alzheimer’s disease and the loss of cholinergic innervation to the hippocampus and cortex is responsible for the deficits in memory and attention. Arrow thickness and color are for illustrative purposes and should not be assumed to be quantitative differences in projection density. Adapted from Paul et al. (2015).

Figure 2.
Acetylcholine release and synaptic signaling. Diagram of cholinergic neurotransmission including (1) Ca²⁺-dependent exocytotic release of ACh from presynaptic vesicles, (2) activation of postsynaptic mAChRs and nAChRs by ACh to cause depolarization, and (3) hydrolysis of ACh by acetylcholinesterase (AChE) followed by reuptake of the choline moiety into the presynaptic cell via the high-affinity choline transporter. Impairments in this cycle, most notably reduced ChAT activity and ACh production, are well-known aspects of Alzheimer’s disease and the rationale for cholinesterase inhibitor therapy. Adapted from Stanciu et al., (2019).
Figure 2.
Acetylcholine release and synaptic signaling. Diagram of cholinergic neurotransmission including (1) Ca²⁺-dependent exocytotic release of ACh from presynaptic vesicles, (2) activation of postsynaptic mAChRs and nAChRs by ACh to cause depolarization, and (3) hydrolysis of ACh by acetylcholinesterase (AChE) followed by reuptake of the choline moiety into the presynaptic cell via the high-affinity choline transporter. Impairments in this cycle, most notably reduced ChAT activity and ACh production, are well-known aspects of Alzheimer’s disease and the rationale for cholinesterase inhibitor therapy. Adapted from Stanciu et al., (2019).

Aβ
An important upstream transcriptional regulator of δ-secretase (i.e., asparagine endopeptidase, AEP; LGMN) is C/EBPβ. The presence of C/EBPβ thus provides a mechanistic link between inflammatory signaling and proteolytic cascades thought to drive AD (Luo et al., 2025). C/EBPβ activity has been shown to increase the expression of AEP, which can in turn cleave APP at N585 and tau at N368 to produce fragments with a high propensity to aggregate (Wu et al., 2020). APP is a type I transmembrane glycoprotein and substrate of sequential cleavage in the amyloidogenic pathway by β-secretase and γ-secretase (Castellani et al., 2025). The γ-secretase complex is a multiprotein intramembrane protease complex that has either PS1 or PS2 as its catalytic subunit. Cleavage by this complex produces Aβ) peptides of variable lengths, most notably Aβ40 and Aβ42. Aβ42 is more prone to β-sheet formation and fibrillogenesis, due to its hydrophobic C-terminal end, and is the main component of the senile plaques that make up the AD lesions (Lin et al., 2019).
Aβ has been a constant throughout AD models, and this hypothesis is further strengthened by genetic evidence from familial cases. Mutations in APP on chromosome 21 cause autosomal dominant AD, and trisomy of chromosome 21 (Down syndrome) always results in AD pathology if the individual lives to an advanced age (Wiseman et al., 2015). This is also the case for duplications of APP, whereas rare mutations associated with decreased Aβ production have also been linked to delayed cognitive decline in aging (Castellani et al., 2025). The only instances of Down syndrome without early pathology are the very rare cases without triplication of APP (Wiseman et al., 2015). The applicability of these findings to sporadic AD, however, is less clear given the global genetic imbalances associated with trisomy 21 (Castellani et al., 2025).
Mutations in PS1 and PS2 also support a role for amyloidogenic processing in familial disease. PS1 mutations, however, often exhibit unique features of pathology and may not be directly applicable to sporadic AD (Heilig et al., 2010). In both familial and sporadic cases, insoluble Aβ is elevated in comparison to non-pathological controls (van Helmond et al., 2010; Wang et al., 1999). However, if Aβ accumulation is alone responsible for the full clinical and pathological features of the disease, is still under debate (Castellani et al., 2025).
Nonetheless, Aβ deposition is a feature of the disease, and plaque load and the wider dysregulation of Aβ metabolism is tightly linked to disease progression. As noted throughout this review, however, Aβ deposition takes place in the context of a wider network of upstream regulatory changes. It is important to note, then, that while Aβ plays a central role in disease, the exact placement of Aβ and the specific role of Aβ deposition within the temporal and mechanistic hierarchy of AD are considered in greater detail elsewhere (Castellani et al., 2025; Riku et al., 2025).
Tau
Tau is highly expressed in axons and more weakly in dendrites, soma, and glia (Sinsky et al., 2021; Wei et al., 2022). Tau is highly enriched in numerous phosphorylation sites in the N-terminal region, C-terminal domain, and microtubule-binding repeats. Tau phosphorylation is regulated by coordinated activity of kinases and phosphatases to maintain physiological steady-state levels in cells (Noble et al., 2013).
Loss of regulation of tau phosphorylation leads to hyperphosphorylation, which weakens tau binding to microtubules and causes tau to detach (Bjørklund et al., 2019; Almansoub et al., 2019). Detached tau molecules are misfolded and redistributed from the axon to the somatodendritic compartment, where tau oligomerizes to form soluble oligomeric intermediates. These intermediates can eventually form paired helical filaments (PHFs) and finally neurofibrillary tangles (NFTs) that are hallmarks of tauopathies and that are found in neuronal cell bodies and dendrites (Lo et al., 2020; Robbins et al., 2021).
Tau pathology accumulates in synaptically connected networks and spreads through the brain in an early stage that impairs synaptic transmission and axonal transport in a manner that is detectable prior to formation of tangles. This progressive accumulation can be described by Braak staging (Therriault et al., 2022) and may underlie a network-based propagation mechanism that precedes synaptic loss, axonal degeneration, neuronal death, and ultimately cognitive decline (Lo et al., 2020; Wu et al., 2017; Yin et al., 2021). Importantly, tau pathology burden is more closely associated with memory and neurodegeneration than Aβ (Doering et al., 2024; Iaccarino et al., 2017; Lamontagne-Kam et al., 2023).
Tau is also subject to a number of other post-translational modifications including truncation, glycosylation, glycation, acetylation, methylation, and sumoylation. Many of these modifications affect tau structure, aggregation propensity, or trafficking (Alquézar et al., 2021). The vast majority of tau modifications take place on lysine residues and impact protein turnover. In particular, ubiquitination of lysine residues often marks tau for proteasomal degradation. Ubiquitination is in competition with acetylation or methylation modifications on the same lysine residues that block ubiquitin attachment and thus can prevent clearance (Luo et al., 2014; Kontaxi et al., 2017). Indeed, lysine 254 (K254) is preferentially methylated in fibrillar tau, indicating that site-specific methylation can block degradation and contribute to persistence of tau species (Thomas et al., 2012).
As will be discussed in more detail in the following sections, accumulation of tau and Aβ, as well as the disruptions in neurotransmission that result, are a downstream consequence of perturbations in upstream processes that are exacerbated by age-related and reproductive aging–associated changes.
The Role of APOE Variants in AD Pathobiology
APOE is one of the most consistent genetic modifiers of AD risk. Carriers of one ε4 allele have about 2–3-fold increased risk while ε4 homozygotes have ~10–12-fold increased risk (Ayton & Bush, 2021). ApoE protein is present in neuritic plaques, and the brains of ε4 carriers have significantly more Aβ accumulation when compared to non-carriers (Baek et al., 2020). Multiple mechanistic studies implicate APOE4 in amyloid pathology in a variety of ways including altered APP processing that may promote Aβ production, promotion of Aβ aggregation with both soluble and fibrillar Aβ, and disruption of Aβ clearance through several mechanisms such as disrupted glial uptake, enzymatic degradation, and decreased vascular efflux (Koutsodendris et al., 2022; Serrano-Pozo et al., 2021; Troutwine et al., 2022).
ApoE is the major cholesterol carrier in the CNS, where it is secreted by astrocytes, microglia, and neurons to redistribute lipids throughout the brain (Belaidi et al., 2025). ApoE is critical for neuronal function and brain health through mechanisms that are linked to maintenance of membrane integrity, lipid-dependent signaling, and clearance of neurotoxic substrates such as Aβ (Islam et al., 2025). Human ApoE is encoded by two alleles that code for 3 common isoforms ε2, ε3, and ε4. These ApoE isoforms differentially affect risk for AD. In summary, epidemiological studies have shown the lowest risk in ε2 carriers, near neutral risk in ε3 carriers, and highest risk in ε4 carriers, which is more severe in women (Stuchell-Brereton et al., 2023; Sun et al., 2023).
ApoE plays a role in Aβ clearance via vascular and cellular pathways. In the context of the blood-brain barrier, ApoE binds Aβ and forms Aβ-ApoE complexes that bind to lipoprotein receptors on the abluminal side of brain endothelial cells. These lipoprotein receptors include LDL receptor-related protein 1 (LRP1), the low-density lipoprotein receptor (LDLR), and very-low-density lipoprotein receptor (VLDLR) (Kanekiyo et al., 2013; Ruiz et al., 2005). ApoE2 and ApoE3 promote Aβ efflux primarily via LRP1-dependent mechanisms, which involves a fast rate of internalization and transcytosis into the circulation (Deane et al., 2008). ApoE4 shifts Aβ trafficking toward VLDLR-dependent pathways with a slower rate of clearance, leading to a higher propensity of brain retention and plaque formation.
Figure 3.
Isoform-dependent ApoE regulation of Aβ clearance kinetics and context-specific tau pathology. Isoform-dependent ApoE regulation of Aβ clearance kinetics and context-specific tau pathology. ApoE4 alters multi-compartment Aβ clearance kinetics - in part through lipoprotein receptors such as LRP1, LDLR, and HSPG - and results in impaired Aβ turnover, brain retention, and increased neuroinflammatory signaling. ApoE isoform relationships with tau pathology are context-specific and do not universally adhere to the canonical AD risk hierarchy: in primary tauopathy models (TauP301L and PSP) ApoE2 was associated with increased tau aggregation and hyperphosphorylation relative to ApoE3 or ApoE4 (ApoE2 > ApoE3 > ApoE4), while in the context of amyloid-β (as in AD) ApoE4 appeared to worsen tau-mediated neurodegeneration indirectly through mechanisms related to Aβ clearance and inflammation. Abbreviations: LRP1, low-density lipoprotein receptor-related protein 1; LDLR, low-density lipoprotein receptor; HSPG, heparan sulfate proteoglycan; BBB, blood-brain barrier; Aβ, amyloid-beta. Adapted from Liu et al., 2025.
Figure 3.
Isoform-dependent ApoE regulation of Aβ clearance kinetics and context-specific tau pathology. Isoform-dependent ApoE regulation of Aβ clearance kinetics and context-specific tau pathology. ApoE4 alters multi-compartment Aβ clearance kinetics - in part through lipoprotein receptors such as LRP1, LDLR, and HSPG - and results in impaired Aβ turnover, brain retention, and increased neuroinflammatory signaling. ApoE isoform relationships with tau pathology are context-specific and do not universally adhere to the canonical AD risk hierarchy: in primary tauopathy models (TauP301L and PSP) ApoE2 was associated with increased tau aggregation and hyperphosphorylation relative to ApoE3 or ApoE4 (ApoE2 > ApoE3 > ApoE4), while in the context of amyloid-β (as in AD) ApoE4 appeared to worsen tau-mediated neurodegeneration indirectly through mechanisms related to Aβ clearance and inflammation. Abbreviations: LRP1, low-density lipoprotein receptor-related protein 1; LDLR, low-density lipoprotein receptor; HSPG, heparan sulfate proteoglycan; BBB, blood-brain barrier; Aβ, amyloid-beta. Adapted from Liu et al., 2025.

In addition to vascular clearance, Aβ clearance occurs by cellular uptake from the interstitial fluid by glial cells. Aβ is then internalized in a complex with ApoE and transported to the lysosomes for degradation (Liu et al., 2025). Degradation of Aβ is then mediated by intralysosomal proteases such as neprilysin and the insulin-degrading enzyme (IDE). Clearance is also affected by the lipidation status of ApoE. Lipidated ApoE, which is regulated by ATP-binding cassette transporter A1 (ABCA1) among other factors, enhances cellular uptake and degradation of Aβ. In contrast, poorly lipidated ApoE is less efficient in Aβ clearance (Jiang et al., 2008).
The three common ApoE isoforms differ in two amino acid residues located at 112 and 158. These differences occur in the form of cysteine-to-arginine substitutions. ApoE2 has a cysteine residue at both sites, ApoE3 has cysteine at position 112 and arginine at position 158, and ApoE4 has arginine at both positions (Stuchell-Brereton et al., 2023). ApoE is known to have three functional domains. The N-terminal domain, which consists of residues 1-167, folds into a structurally conserved four-helix bundle domain. The hinge domain, which contains residues 168-205, links the helical bundle domain to the C-terminal domain. The C-terminal domain, which contains residues 206-299, has the most poorly understood structure and position (Frieden & Garai, 2013). Structural studies of the helical bundle of ApoE has been well resolved and reports largely similar tertiary structure in ApoE isoforms (Wilson et al., 1991; Grimaldi et al., 2024).
The “domain interaction” model is a common explanation for differences in properties of the ApoE isoforms (Mahley et al., 2009). For ApoE4, the cysteine at position 112 is substituted with arginine. This substitution results in changes in electrostatic interactions in the helical bundle domain due to its positive charge. This results in a unique fold that can be propagated through the rest of the protein. Differences in the local charge environment as well as rearrangement of interactions with neighboring residues can cause global changes in the tertiary structure. These changes in protein conformation can cause changes in properties, including lipidation, receptor binding, susceptibility to proteases, and intracellular trafficking. These structural differences are also hypothesized to contribute to differences in autophagy and processing of Aβ and other substrates.
Figure 4.
Structural conformations of apolipoprotein E (apoE) isoforms. Schematic diagram of the different conformational properties of apoE2, apoE3, and apoE4, and the effect of amino acid substitutions at positions 112 and 158. The amino acid substitutions that change cysteine residues to positively charged arginine residues (apoE4 has arginine at both 112 and 158, whereas apoE2 has cysteine at both) changes the intramolecular interactions in the helical bundle domain, facilitating domain-domain (N-terminal and C-terminal) interactions. ApoE2 (cysteine at both positions) and apoE3 (cysteine at 112 and arginine at 158) show less intramolecular interaction and assume more flexible conformations. The panel on the right lists three other possible interactions that are unique to apoE4 as a result of these substitutions. These conformational differences affect the proteins’ binding to lipids and receptors, their sensitivity to proteolytic cleavage, and the isoform-specific mechanisms of the proteins in mediating risk for Alzheimer’s disease. Adapted from Butterfield and Mattson (2020).
Figure 4.
Structural conformations of apolipoprotein E (apoE) isoforms. Schematic diagram of the different conformational properties of apoE2, apoE3, and apoE4, and the effect of amino acid substitutions at positions 112 and 158. The amino acid substitutions that change cysteine residues to positively charged arginine residues (apoE4 has arginine at both 112 and 158, whereas apoE2 has cysteine at both) changes the intramolecular interactions in the helical bundle domain, facilitating domain-domain (N-terminal and C-terminal) interactions. ApoE2 (cysteine at both positions) and apoE3 (cysteine at 112 and arginine at 158) show less intramolecular interaction and assume more flexible conformations. The panel on the right lists three other possible interactions that are unique to apoE4 as a result of these substitutions. These conformational differences affect the proteins’ binding to lipids and receptors, their sensitivity to proteolytic cleavage, and the isoform-specific mechanisms of the proteins in mediating risk for Alzheimer’s disease. Adapted from Butterfield and Mattson (2020).

ApoE2 and ApoE3 possess cysteine residues at positions which provide thiol nucleophiles that can trap electrophilic lipid peroxidation products, which can limit their diffusion and secondary modification of proteins and membranes (Abeer et al., 2023). ApoE4 is deficient for cysteines at these critical positions, limiting its capacity for scavenging reactive aldehydes, and making ApoE4 more prone to oxidative protein modification (Abeer et al., 2023; Butterfield & Mattson, 2020). This is exacerbated by a deficit in glutathione-dependent redox buffering, as cysteine availability is the rate-limiting step for glutathione synthesis (Hara et al., 2017). Consistent with this, glutathione levels and GSH/GSSG ratios are lower in the brain of individuals with mild cognitive impairment and AD, indicating an imbalance in redox buffering capacity (Bermejo et al., 2008).
Not surprisingly, a large body of work shows that ApoE isoforms have distinct effects on Aβ pathology. ε4 carriers have higher brain Aβ deposition, an earlier age of amyloid positivity, and an accelerated rate of Aβ burden accrual over time (Liu et al., 2017). For decades, researchers tried to explain APOE-dependent AD risk almost exclusively in terms of differences in plaque load; however; this framework is limited, as plaque burden is highly variable and poorly correlated with synaptic dysfunction and cognitive decline (Xia et al., 2024). Instead, converging lines of evidence have led to the general consensus that soluble, early-stage Aβ assemblies - not mature plaques - are the species most intimately associated with neurotoxicity and cognitive impairment (Xia et al., 2024).
Accordingly, the role of fibrillization must be considered with this context in mind. On one hand, it is clear that fibril formation - by definition - ultimately contributes to plaque deposition. On the other hand, fibrillization can act as a negative feedback on acute neurotoxicity, sequestering highly diffusible, synaptotoxic oligomeric species into relatively inert fibrillar assemblies (Verma et al., 2015). Thus, the pathological relevance of fibril growth kinetics does not simply depend on whether elongation is increased or decreased, but how aggregation dynamics influence the abundance, persistence, and clearance of intermediate Aβ species. These intermediates, rather than mature fibrils, are now widely recognized as the primary drivers of synaptic dysfunction, neuroinflammation, and disease progression (Liu et al., 2025). In this light, it is important to recognize that while APOE2 and APOE3 more strongly inhibit fibril elongation than ApoE4, this is not associated with greater neurotoxicity. Rather, these isoforms promote more ordered aggregation pathways and facilitate clearance of early Aβ assemblies (Dasadhikari et al., 2025). In contrast, APOE4 preferentially stabilizes disordered, poorly cleared Aβ intermediates, prolonging their lifetime and exacerbating their synaptotoxic and pro-inflammatory effects (Xia et al. 2024).
Xia et al. (2024) used single-molecule imaging and ex vivo brain-derived aggregates to demonstrate that ApoE does not simply affect plaque deposition or mature fibril structure. Instead, ApoE transiently binds to Aβ during the earliest stages of aggregation, and the ApoE-Aβ interaction leads to the formation of highly populated yet short-lived APOE-Aβ co-aggregates which have a critical impact on aggregation pathways, toxicity, and clearance behavior (Verghese et al., 2013; Xia et al., 2024). While only 1-5% of Aβ particles are physically associated with APOE, these co-aggregates comprise approximately 20-50% of total Aβ mass, suggesting that these APOE-Aβ co-aggregates are disproportionately large and biologically consequential intermediates rather than rare byproducts.

Table 1.
Structural and functional hierarchy of amyloid-β aggregation states and their relevance to Alzheimer’s disease. This table summarizes the progressive aggregation states of amyloid-β (Aβ), spanning from soluble monomeric peptides to insoluble extracellular plaques, and highlights their distinct structural, biochemical, and pathological characteristics. Early-stage species, particularly soluble oligomers, are highly dynamic and exhibit the greatest neurotoxicity, with strong associations to synaptic dysfunction and cognitive decline. Intermediate assemblies such as protofibrils represent transitional forms with significant pathogenic potential. In contrast, mature fibrils and plaques are structurally stable end-products of aggregation that, while defining histopathological features of Alzheimer’s disease, show weaker correlations with clinical symptoms. Toxicity, stability, and AD relationship ratings represent qualitative characterizations based on the convergent literature rather than measured quantitative values. Aβ, amyloid-beta; AD, Alzheimer’s disease. Early-stage aggregates (oligomers) are consistently identified as the species most strongly associated with synaptic dysfunction and cognitive decline in human AD. Collectively, the table emphasizes that Aβ toxicity is not solely dependent on aggregate presence, but is critically influenced by aggregation state, solubility, and molecular dynamics.
Table 1.
Structural and functional hierarchy of amyloid-β aggregation states and their relevance to Alzheimer’s disease. This table summarizes the progressive aggregation states of amyloid-β (Aβ), spanning from soluble monomeric peptides to insoluble extracellular plaques, and highlights their distinct structural, biochemical, and pathological characteristics. Early-stage species, particularly soluble oligomers, are highly dynamic and exhibit the greatest neurotoxicity, with strong associations to synaptic dysfunction and cognitive decline. Intermediate assemblies such as protofibrils represent transitional forms with significant pathogenic potential. In contrast, mature fibrils and plaques are structurally stable end-products of aggregation that, while defining histopathological features of Alzheimer’s disease, show weaker correlations with clinical symptoms. Toxicity, stability, and AD relationship ratings represent qualitative characterizations based on the convergent literature rather than measured quantitative values. Aβ, amyloid-beta; AD, Alzheimer’s disease. Early-stage aggregates (oligomers) are consistently identified as the species most strongly associated with synaptic dysfunction and cognitive decline in human AD. Collectively, the table emphasizes that Aβ toxicity is not solely dependent on aggregate presence, but is critically influenced by aggregation state, solubility, and molecular dynamics.
| Term | Scale | Structure | Solubility | Stability | Primary Biological Relevance | Toxicity | Relationship to AD |
| Monomers | Molecular | Single Aβ peptides | Soluble | Unstable | Baseline physiological state | Low | Not directly pathogenic |
| Early-stage aggregates (oligomers) | Molecular-nanoscale | Small, flexible clusters (dimers, trimers, etc.) | Soluble | Transient | Synaptic signaling disruption, membrane interaction | Highest | Best correlate with cognitive decline |
| Protofibrils | Nanoscale | Elongated, partially ordered assemblies | Semi-soluble | Intermediate | Transitional species between oligomers and fibrils | High | Contribute to early pathology |
| Mature fibrils | Nanoscale–microscale | Highly ordered β-sheet polymers | Insoluble | Stable | Structural end-products of aggregation | Moderate–low | Build plaques but are not main toxic species |
| Plaques | Tissue-level | Macroscopic extracellular deposits of fibrils + cellular debris | Insoluble | Very stable | Histopathological hallmark | Variable | Poor correlation with symptoms |
In addition to ApoE isoform, formation and properties of these co-aggregates are heavily influenced by lipidation state (Nurmakova et al., 2026). In ApoE4 homozygotes, a larger fraction of Aβ is found in a co-aggregated form as compared to ApoE3 carriers, and a majority of these assemblies (~80%) incorporate non-lipidated ApoE (Xia et al., 2024). Rather than altering the final fibrillar end state, APOE likely modulates Aβ toxicity in a more upstream fashion by stabilizing or destabilizing toxic Aβ intermediates that mediate synapse dysfunction, membrane disruption, and neuroinflammatory signaling. Indeed, mature Aβ fibrils no longer contain ApoE and are functionally identical regardless of which APOE isoform was present during aggregation. These mechanisms are further supported by Xia et al. (2024), who show that lipidated ApoE-Aβ co-aggregates are more efficiently cleared by glial cells and provoke a comparatively mild inflammatory response. In contrast, non-lipidated ApoE4-Aβ co-aggregates are more poorly cleared and drive a robust pro-inflammatory glial phenotype. This long-lasting inflammatory milieu further impairs clearance of other Aβ species and therefore sets up a self-perpetuating cycle of progressive Aβ accumulation over time. This would mechanistically account for the increased Alzheimer’s risk due to ApoE4, which is less efficiently lipidated in vivo than ApoE2 or ApoE3. Poorer lipidation makes it more likely that ApoE4 will be incorporated into the composition of toxic, poorly cleared co-aggregates instead of protective, clearance-promoting aggregates. Indeed, consistent with this, genetic deletion of ABCA1-a master regulator of ApoE lipidation-worsens amyloid plaque deposition, while ABCA1 overexpression increases ApoE lipidation, decreases cerebral Aβ load, and improves cognitive performance in transgenic AD models (Wahrle et al., 2008; Tate et al., 2024).
Differential Hormonal Regulation of Apolipoprotein E
The functional units of the ovaries are the follicles, each consisting of an oocyte surrounded by specialized somatic cells, including granulosa and theca cells. These structures serve both endocrine and gametogenic roles, supporting steroid hormone and peptide synthesis as well as oocyte survival, growth, maturation, and eventual release for fertilization. Ovarian follicles are broadly classified into preantral and antral stages. Preantral follicles include primordial, intermediate, primary, and secondary follicles and collectively represent approximately 90-95% of the total follicular population in mammals (Smitz & Cortvrindt, 2002; van den Hurk & Zhao, 2005). As folliculogenesis progresses, follicles transition into the antral stage, where they can be further categorized based on size and developmental status into tertiary and preovulatory follicles. Being an abundantly expressed lipoprotein, not only is in the central nervous system, or by the liver, but also in mammalian ovarian tissue (Corraliza-Gomez et al., 2019; Oriá et al., 2020). Although ApoE is best known for its role in mediating cholesterol transport from peripheral tissues to the liver for metabolism, it also plays a critical role in reproductive physiology.
ApoE contributes to cholesterol trafficking within ovarian follicles, where it supports steroidogenesis by facilitating cholesterol availability to steroidogenic cells (Oriá et al., 2020). Cholesterol uptake by ovarian thecal and granulosa cells depends in part on ApoE, either through participation in lipoprotein-receptor complexes or via lipid endocytosis pathways (Getz et al., 2009). Structurally, ApoE is a key component of several lipoprotein particles, including VLDL remnants, a subclass of high-density lipoproteins (HDL) involved in reverse cholesterol transport, and chylomicrons that carry dietary lipids absorbed from the intestine (Kockx et al., 2018). Through these roles, ApoE serves as a molecular link between lipid metabolism, ovarian steroid hormone production, and systemic metabolic regulation.
ApoE production in ovarian cells is under direct hormonal control and shows a strong dependence on FSH signaling. Early in vitro studies demonstrated that exposure of rat granulosa cells to FSH (50 ng/mL) resulted in an approximately two-fold increase in ApoE secretion compared with cells maintained under FSH-free conditions (Driscoll et al., 1985). Mechanistically, this effect was mediated through activation of intracellular cyclic AMP (cAMP) signaling, as treatment with exogenous cAMP similarly enhanced ApoE release. These findings indicate that ApoE expression in granulosa cells is responsive to cAMP-dependent endocrine pathways downstream of FSH receptor activation. Consistent with these experimental observations, age-associated increases in circulating apolipoprotein E levels have also been reported in vivo in women, supporting a link between reproductive aging, increased gonadotropin signaling, and ApoE regulation (Von Wald et al., 2010).
Metabolic and Lipid Regulation of Apolipoprotein E
It is well established that both AD and reproductive aging are associated with extensive alterations in cellular metabolism and mitochondrial function (Muhammad, 2025; Spina et al., 2025; Wang et al., 2025; Xu et al., 2025). One of the most prominent consequences of impaired mitochondrial activity - particularly reduced oxidative phosphorylation (OXPHOS) - is the disruption of key components required for mitochondrial homeostasis, including proteins within the electron transport chain such as complex I. Beyond energetic deficits, emerging evidence suggests that mitochondrial dysfunction exerts broader regulatory effects on cellular signaling pathways, including those governing lipid metabolism and apolipoprotein expression. Notably, ApoE appears to be particularly sensitive to shifts in mitochondrial and metabolic state.
ApoE expression is highly responsive to cellular metabolic stress. Work by Wynne et al. (2023) demonstrates that disruption of mitochondrial electron transport - whether through genetic manipulation of mitochondrial solute carriers (e.g., SLC25A family members such as SLC25A1) or through pharmacologic and genetic inhibition of electron transport chain complexes I, III, or the copper-dependent complex IV - elicits robust upregulation of ApoEacross multiple cell types (Wynne et al. 2023) . This response is especially pronounced in human glial cells, which represent the primary source of ApoE within the central nervous system (Wynne et al., 2023; Wang et al., 2026).
Importantly, ApoE induction is not restricted to conditions of overt mitochondrial respiratory failure. Mutagenesis of the mitochondrial carnitine transporter SLC25A20 increases ApoE expression despite preserved oxidative phosphorylation and intact respiratory chain subunit abundance. Similarly, disruption of ATP citrate lyase (ACLY) - a cytosolic enzyme critical for acetyl-CoA production - also elevates ApoE levels (Wynne et al. 2023). Therefore, ApoE upregulation likely reflects an adaptive program linked to mitochondrial dysfunction, altered lipid and acetyl-CoA metabolism, and potentially mitochondria-initiated inflammatory signaling. While such responses may initially support membrane repair and metabolic homeostasis, they may become maladaptive in the aging brain, particularly in carriers of the ApoE4 isoform.
Under more canonical physiological conditions, ApoE expression is also regulated through sterol-sensitive transcriptional pathways. Reductions in neuronal cholesterol availability promote the formation of oxysterols, which activate the astrocyte-enriched liver X receptor (LXR) pathway (Wang et al., 2021). LXR functions as a ligand-activated nuclear receptor that induces the expression of genes central to cholesterol homeostasis, including ABCA1 and ApoE (Staurenghi et al., 2021). Together, these proteins facilitate the formation of HDL-like particles and mediate cholesterol transport within the brain (Lindner & Gavin, 2024). In parallel, peroxisome proliferator-activated receptor gamma (PPARγ) contributes to ApoE upregulation in astrocytes, linking APOE expression to broader metabolic programs involving glucose metabolism and insulin sensitivity (Tyagi et al., 2011; Qi et al., 2021).
Beyond sterol signaling, lipid availability itself further modulates ApoEexpression and processing. Oleic acid, a monounsaturated fatty acid that is esterified into triacylglycerols (TAGs) and stored in lipid droplets under conditions of excess, has been shown to alter APOE dynamics. Specifically, oleic acid exposure reduces intracellular levels of heavily glycosylated ApoEE while promoting its secretion (Huang et al., 2004). Chronic exposure enhances ApoE release from astrocytes, with a disproportionately greater increase observed for the ApoE4 isoform (Lindner et al., 2022). This hypersecretion is likely driven by impaired degradation of ApoE4 and increased interactions between ApoE4 and TAG-rich lipid droplets.
A Note on Other Apolipoprotein Variants
Hormonal perturbations associated with aging - particularly those influencing apoE dynamics - occur alongside broader age-related declines in multiple apolipoprotein species. Using data-independent acquisition (DIA) proteomics, McCoy et al. (2025) demonstrated that circulating apolipoproteins, including ApoA-I, ApoA-IV, ApoB-100, ApoC-I, ApoC-IV, ApoD, ApoE, and ApoM, decline significantly with age in postreproductive female mice.
Several of these apolipoproteins have established relevance to neurodegenerative processes. For example, ApoA-I can bind Aβ and has been implicated in modulating its aggregation and clearance; accordingly, circulating ApoA-I levels are significantly reduced in individuals with Alzheimer’s disease compared to healthy controls (Tong et al., 2022). Restoration of ApoA-I levels following ovarian tissue transplantation was associated with improved cognitive performance, reduced inflammatory signaling, and decreased gliosis in aged mice (McCoy et al., 2025).
Additional apolipoproteins exhibit similar patterns. ApoC-IV and ApoD both decline with age and are restored following transplantation of young ovarian tissue (McCoy et al., 2025). Notably, loss of ApoD function increases susceptibility to oxidative stress, elevates brain lipid peroxidation, and impairs locomotor and learning capacity, whereas ApoD overexpression confers protective effects, enhancing survival and limiting oxidative damage (Ganfornina et al., 2008).
Importantly, these restorative effects appear largely independent of follicular content. Both follicle-containing and follicle-depleted ovarian tissue transplants returned circulating apolipoprotein levels in aged mice to profiles resembling those of young animals, while also improving cognitive outcomes and attenuating gliosis (McCoy et al., 2025). In parallel, these interventions suppressed circulating FSH levels to those observed in young, reproductively active mice (McCoy et al., 2025; Uilenbroek et al., 1978). These effects occur alongside reductions in circulating FSH, supporting a model in which endocrine aging contributes to both altered lipid transport biology and neurodegenerative vulnerability. However, whether FSH directly regulates the synthesis of apolipoproteins beyond ApoE remains to be determined.
Stepwise Neurobiological Dysregulation Driven by Elevated FSH in Alzheimer’s Disease
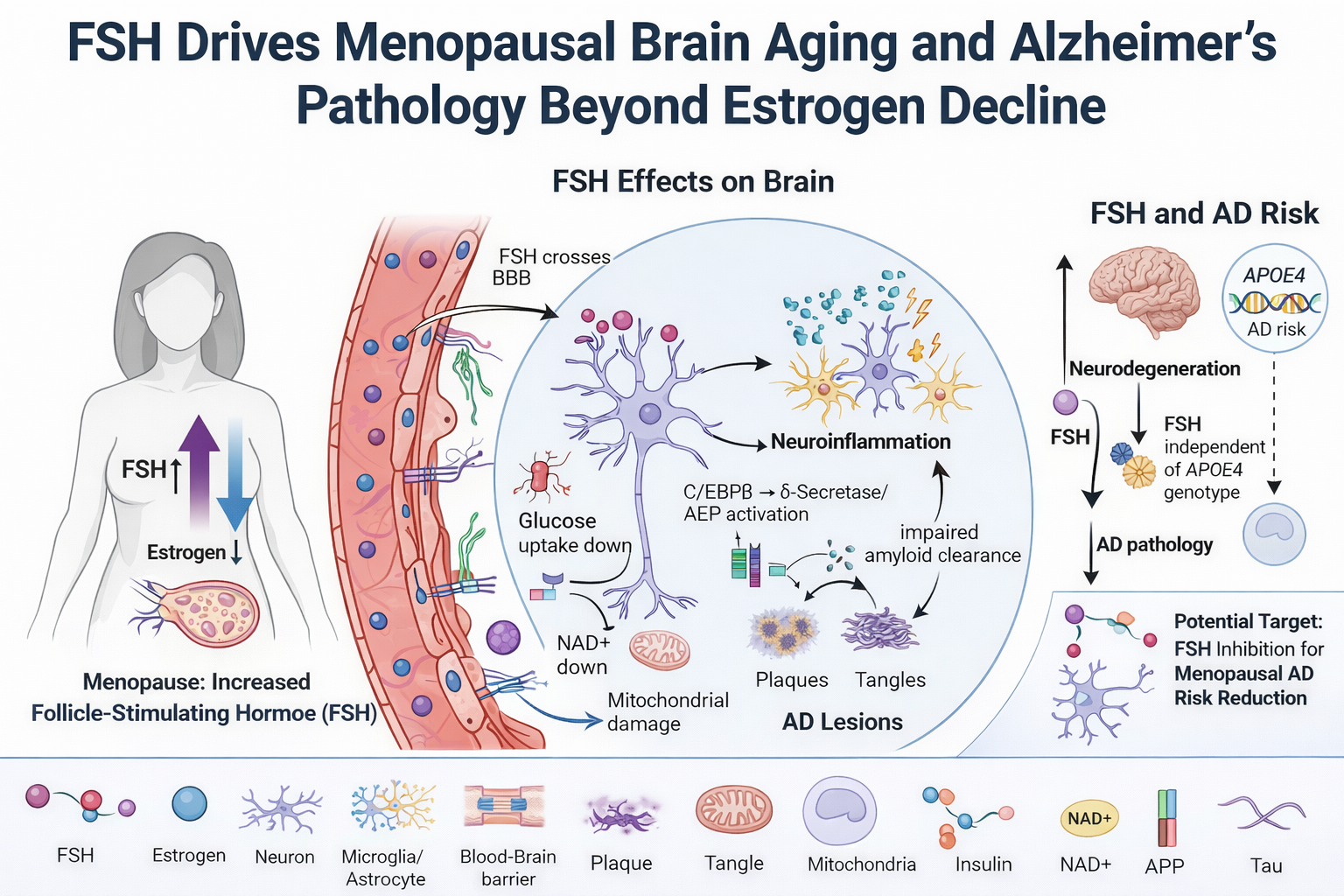
Elevated FSH Disrupts Oxidative Phosphorylation (OXPHOS) in Aging Neurons, Shifting Cellular Energetics Toward Dysfunction
Deficits in glucose metabolism and oxidative phosphorylation are recognized features of AD and are observed consistently in experimental and clinical studies. Cerebral hypometabolism in AD is regularly measured by fluorodeoxyglucose positron emission tomography (FDG-PET) as a diagnostic hallmark (Christodoulou et al., 2026). One major contributor to this phenotype is the C/EBPβ-δ-secretase signaling pathway, which is positively regulated by FSH (Xiong et al., 2023). C/EBPβ is a stress-responsive transcription factor that is induced during aging and in AD, and it inhibits neuronal glucose utilization by interfering with insulin signaling (Tsukada et al., 2011). C/EBPβ represses the activity of FOXO1 and inhibits Akt (protein kinase B), a central mediator of insulin-dependent glucose uptake. Inhibition of Akt, which is essential for glucose transport and subsequent metabolism, therefore creates a pseudo insulin-resistant state within neurons (Gabbouj et al., 2019). The inhibition of neuronal glucose uptake by C/EBPβ can therefore contribute to the impaired metabolic state observed by FDG-PET imaging in AD. This metabolic dysfunction of FSH is not only observed in the CNS, but also extends to other tissues that are dependent on FSH signaling, such as adipogenesis and cardiovascular system in both males and females, leading to systemic consequences (Kim et al., 2026).
In addition to inhibition of glucose utilization, C/EBPβ-δ-secretase signaling also impairs mitochondrial energetics through direct modulation of key regulators of mitochondrial function. Activation of this axis leads to degradation of nicotinamide phosphoribosyltransferase (NAMPT), resulting in depletion of nicotinamide adenine dinucleotide (NAD⁺) (Li et al., 2025), which is required for mitochondrial respiration and ATP production. Another important mechanism of relevance in aging is the upstream regulation of C/EBPβ. Physiologically, C/EBPβ is induced by gonadotropins, including follicle-stimulating hormone FSH and LH, to regulate genes involved in steroidogenesis and inflammatory responses, such as steroidogenic acute regulatory protein (StAR) and prostaglandin-endoperoxide synthase 2 (PTGS2/COX-2) (Sirois & Richards, 1993; Duffy et al., 2019). C/EBPβ induction during periovulation is produced by intermittent surges in LH, but prolonged increases in gonadotropins, such as chronic FSH elevation, can promote sustained activation of C/EBPβ (Zaidi et al., 2024). Prolonged induction and signaling by C/EBPβ can shift away from adaptive temporally restricted induction towards persistent inflammatory and proteolytic activity. Notably, both ApoE4 and FSH were found to additively activate the C/EBPβ-δ-secretase pathway, promoting proteolytic processing of APP and tau, which resulted in enhanced amyloid-β accumulation and neurofibrillary tangle formation (Xiong et al., 2022; Xiong et al., 2023; Zaidi et al., 2024).
The hypometabolic state observed in AD may not be an initiating factor, but rather preceded by a state of mitochondrial hyperactivity and neural hyperexcitability (Christodoulou et al., 2026; Naia et al., 2023). At this early stage of AD pathogenesis, C/EBPβ-dependent signaling transiently upregulates mitochondrial output - partly through increased TFAM promoter activity - which supports mitochondrial biogenesis and oxidative capacity during a period of increased demand (Sierra-Magro et al., 2023). At the same time, C/EBPβ-mediated repression of FOXO1 and REST results in decreased inhibitory neuronal programs, especially within GABAergic neuronal populations (Mattson M. P. 2020; Xia et al., 2022) . The combined effect results in loss of inhibitory tone and aberrant network excitation. This state of hyperexcitability is corroborated by early hypermetabolic changes detected in preclinical and prodromal phases of AD (Christodoulou et al., 2026; Naia et al., 2023).
Sustained activation of this program has been linked in preclinical models to a shift toward pathophysiology. Chronic C/EBPβ activation – as well as persistent Akt inhibition and mitochondrial stress – has been associated with a gradual buildup of mitochondrial dysfunction and declining metabolic efficiency (Shaerzadeh et al., 2014; Yu et al., 2014; Wang et al., 2025), though the extent to which this phenotype results from FSH-driven C/EBPβ activation in human aging is an open question. As mitochondrial quality control is eventually outstripped, the compensatory hypermetabolic state presumably gives way to the hypometabolism that is a well-known aspect of AD pathophysiology in humans (Naia et al., 2023; Christodoulou et al., 2026). The repercussions of this metabolic shift are self-reinforcing. The inability to sustain oxidative metabolism results in cytosolic acidification and lactate buildup, both of which also exacerbate lysosomal acidification and autophagic flux, two processes demonstrably altered in human AD brain (Chen et al., 2022; Hagihara & Miyakawa, 2024; Wang X et al., 2025). Lysosomal distention and autophagosome buildup, both of which are well-documented hallmarks of human AD, are downstream consequences that further contribute to the buildup of cellular debris and proteinaceous waste (Quick et al., 2023). These changes are also consistently associated with microglial activation and neuroinflammatory signaling in human disease, ultimately contributing to a self-perpetuating state in which metabolic dysfunction, impaired proteostasis, and inflammation all serve to drive neurodegeneration (Jung et al., 2025; Wu et al., 2025).
Figure 5.
Convergent drivers and consequences of mitochondrial perturbation in Alzheimer’s disease. Amyloid-β (Aβ) deposition, tau pathology, oxidative stress, and electron transport chain (ETC) dysfunction converge to perturb mitochondrial quality control. Excessive fission (via upregulation of dynamin-related protein 1 (DRP1), fission protein 1 (FIS1), and mitochondrial fission factor (MFF)) at the expense of fusion (optic atrophy protein 1 (OPA1); mitofusins (MFN1/2)), mitophagy (PTEN-induced kinase 1 (PINK1); Parkin; microtubule-associated protein light chain 3 (LC3)) and MDV formation underlies a shift in mitochondrial dynamics, turnover and trafficking that favors accumulation of fragmented, dysfunctional mitochondria. Adapted from Li et al., (2022).
Figure 5.
Convergent drivers and consequences of mitochondrial perturbation in Alzheimer’s disease. Amyloid-β (Aβ) deposition, tau pathology, oxidative stress, and electron transport chain (ETC) dysfunction converge to perturb mitochondrial quality control. Excessive fission (via upregulation of dynamin-related protein 1 (DRP1), fission protein 1 (FIS1), and mitochondrial fission factor (MFF)) at the expense of fusion (optic atrophy protein 1 (OPA1); mitofusins (MFN1/2)), mitophagy (PTEN-induced kinase 1 (PINK1); Parkin; microtubule-associated protein light chain 3 (LC3)) and MDV formation underlies a shift in mitochondrial dynamics, turnover and trafficking that favors accumulation of fragmented, dysfunctional mitochondria. Adapted from Li et al., (2022).

FSH-Mediated Enhancement of ApoE4 as a Barrier to Autophagy: Implications for Proteinopathy
Autophagy is another cellular process that may be influenced by FSH during folliculogenesis, but appears to have diametric opposite consequences in neurons. During folliculogenesis, FSH is considered to function as a general suppressor of autophagy. In a context-dependent manner, FSH signaling via the PI3K-AKT-mTOR pathway, FSHR activation of AKT-mTOR (growth factor) under basal conditions, contributes to cell growth and survival while inhibiting autophagy (Liu et al., 2023). However, in antral and pre-ovulatory stages, follicles experience rapid cellular proliferation and increased metabolic demand, leading to the establishment of localized hypoxic microenvironments. In these conditions, FSH signaling cross-talks with hypoxia-dependent pathways and causes dynamic regulation of mTOR activity. In particular, FSHR promotes transient activation of mTOR. However, as a consequence of hypoxia and HIF-1α stabilization, FSH-dependent mTOR activity is eventually attenuated relative to HIF-1α activity and the crosstalk between these two pathways promotes a shift towards autophagy induction in an FSHR-mTOR-HIF-1α–dependent manner (Zhou et al., 2017). This cellular transition allows follicular cells to maintain metabolic homeostasis despite facing the challenges of increased energetic and oxidative stress. Functionally, autophagy seems to play a crucial protective role in follicular survival. Autophagy inhibition and subsequent impairment of autophagic pathways is associated with the accumulation of intracellular waste, compromised cellular integrity and depletion of primordial follicle pool (Gawriluk et al., 2011; Wu et al., 2015; Song et al., 2015; Zhou et al., 2017). Taken together, stress-induced and upregulated autophagy during late stages of folliculogenesis seems to be an adaptive mechanism to preserve follicular viability under the conditions of increased metabolic demand.
Consequences of an upregulated FSH signaling may be highly context-dependent and might even go in the opposite direction in the CNS. Most likely this is due to tissue-specific FSHRs coupling to different intracellular signaling pathways. In typical endocrine cells such as ovarian cells FSHR predominantly couples to Gαs proteins and promotes cAMP synthesis. This then leads to protein kinase A (PKA) activation, which can further integrate with Akt-mTOR to promote cell survival (Casarini & Crépieux., 2019). In neurons, FSHR may predominantly couple to alternative G-protein subtypes. For example, Gαi-mediated signaling. Instead of triggering a cAMP-dependent anabolic program, such coupling would trigger Akt, ERK1/2, and SRPK2 activation, resulting in upregulation of the transcription factor C/EBPβ. After phosphorylation, SRPK2 translocates to the nucleus, which is thought to be important for δ-secretase (also known as AEP) activation. This secretase cleaves APP and tau and generates amyloid-β and tau aggregation-prone fragments (Li et al., 2024). Thus, as a result of FSH up-regulation protein metabolites are created that the cells are not able to clear. These include Aβ, tau, and ApoE (notably APOE4).
Accumulation of autophagic vesicles in dystrophic neurites is a well-described pathological hallmark of Alzheimer’s disease, which can be caused by impaired autophagosome-lysosome fusion and defective degradation of autophagic cargo (Nixon et al., 2005; Nixon & Yang, 2011; Zhang et al., 2022). Consistent with this hypothesis, animal models have demonstrated elevated levels of LC3-II, a marker of stalled or incomplete autophagosome turnover (Yang et al., 2011; Zhang et al., 2022). ApoE4 carriers have been found to have a pronounced accumulation of autophagic vesicles, suggesting that this isoform further exacerbates the impairment in autophagic flux (Sohn et al., 2021). Another mechanism has been proposed based on FSH-induced ApoE nuclear localization, which has been shown to negatively impact transcriptional control of autophagy by competing with TFEB for occupancy of coordinated lysosomal expression and regulation (CLEAR) elements of autophagy-related genes including SQSTM1, MAP1LC3B, and LAMP2 (Parcon et al., 2018). Suppression of TFEB-mediated transcription of lysosomal and autophagy-related genes has been specifically characterized for ApoE4, with no such effect demonstrated for ApoE2 and ApoE3. This isoform-specific mechanism provides a molecularly precise account of how ApoE4 may compound autophagic failure in the context of elevated FSH, and represents a compelling candidate pathway linking endocrine aging to the lysosomal dysfunction that is well established in human AD. Direct in vivo validation of this mechanism in the aging brain, and in the context of FSH-driven ApoE dynamics specifically, remains an important direction for future work.
Aberrant accumulation of autophagosomes has been shown to occur even before amyloid plaque deposition, suggesting that it is an early pathogenic event rather than a consequence of amyloidosis (Yu et al., 2005). Experimental induction or inhibition of macroautophagy in neuronal cells results in parallel changes in autophagic vesicle abundance and amyloid-β production (Yu et al., 2005). Defective PS1 function leads to mTORC1 activation, which in turn suppresses TFEB-dependent autophagy and lysosomal biogenesis in neuronal cells (Reddy et al., 2016), while PS2 mutations cause further inhibition of autophagosome - lysosome fusion (Fedeli et al., 2019). Notably, the efficiency of both autophagic and lysosomal pathways decreases with age even in individuals who do not carry presenilin mutations or have complete loss of function, which further increases vulnerability to disrupted protein homeostasis later in life (Beckman et al., 2020; Lim et al., 2024; Shen et al., 2007; Thakur & Ghosh, 2007).
FSH-Mediated Augmentation of Glial Activation in Alzheimer’s Disease
C/EBPβ serves as a central regulator of inflammatory signaling. Numerous pro-inflammatory genes contain consensus C/EBPβ binding sites, and both macrophages and glial cells exhibit marked upregulation of C/EBPβ in response to inflammatory stimuli (Wang et al., 2025). Consistent with this role, C/EBPβ-deficient brains display reduced expression of pro-inflammatory genes and attenuated neurotoxic effects of activated microglia (Straccia et al., 2011; (Yao et al., 2024). ). Moreover, loss of C/EBPβ activity confers neuroprotection in experimental models of ischemic and excitotoxic injury, underscoring its contribution to neuroinflammatory damage (Wu et al., 2023).
C/EBPβ plays a central role in regulating genes involved in inflammasome activation (Wang, J. et al., 2025). Inflammasomes - particularly the NLRP3 complex - are activated by a range of cellular stressors, including misfolded protein aggregates, oxidative stress, and mitochondrial dysfunction (Wu, D. et al., 2022; Baragetti et al., 2020; Hamilton & Anand, 2019; Halliday & Mallucci, 2015; Jones et al., 2021). Within this context, C/EBPβ directly regulates the transcription of key inflammasome components by binding to their promoter regions. Notably, C/EBPβ has been shown to upregulate expression of NLRP3, a core structural component of the inflammasome complex (Ma et al., 2018; Chen, J. et al., 2021). It similarly enhances expression of AIM2, a cytosolic DNA sensor that forms a caspase-1–activating inflammasome (Hornung et al., 2009; Zou et al., 2023). Through these actions, C/EBPβ contributes to the priming phase of inflammasome activation, increasing cellular sensitivity to downstream activating signals. In addition to its direct transcriptional effects, C/EBPβ functions synergistically with other inflammatory transcription factors, most notably NF-κB, to amplify cytokine production (McClure et al., 2017). NF-κB is a well-established regulator of inflammatory gene expression, and its cooperation with C/EBPβ enhances transcriptional output at shared target loci. For example, both factors bind to adjacent promoter regions of IL-1β and IL-18, driving coordinated upregulation of these cytokines (Ma et al., 2018).
Figure 6.
C/EBPβ-dependent transcriptional regulation of inflammasome activation and pro-inflammatory signaling. This figure illustrates the role of C/EBPβ as a central transcriptional regulator linking upstream inflammatory cues to inflammasome activation. In panel A, C/EBPβ cooperates with NF-κB to induce expression of key inflammasome components, including NLRP3 and AIM2, promoting assembly of ASC-containing complexes, activation of caspase-1, and subsequent maturation of IL-1β and IL-18. In panel B, distinct C/EBPβ isoforms differentially regulate inflammasome gene expression, thereby modulating downstream inflammatory responses through calcium-dependent signaling and cytokine production. Collectively, the figure highlights C/EBPβ as a key integrator of transcriptional control and innate immune activation, coordinating inflammasome pathways implicated in neuroinflammation and neurodegenerative disease. Adapted from Wang, J., et al. (2025).
Figure 6.
C/EBPβ-dependent transcriptional regulation of inflammasome activation and pro-inflammatory signaling. This figure illustrates the role of C/EBPβ as a central transcriptional regulator linking upstream inflammatory cues to inflammasome activation. In panel A, C/EBPβ cooperates with NF-κB to induce expression of key inflammasome components, including NLRP3 and AIM2, promoting assembly of ASC-containing complexes, activation of caspase-1, and subsequent maturation of IL-1β and IL-18. In panel B, distinct C/EBPβ isoforms differentially regulate inflammasome gene expression, thereby modulating downstream inflammatory responses through calcium-dependent signaling and cytokine production. Collectively, the figure highlights C/EBPβ as a key integrator of transcriptional control and innate immune activation, coordinating inflammasome pathways implicated in neuroinflammation and neurodegenerative disease. Adapted from Wang, J., et al. (2025).

Evidence that microglia directly express the FSH receptor is lacking at this time, precluding mechanistic interpretation of this step. However, as C/EBPβ is strongly implicated in microglial inflammatory activation, it is reasonable to hypothesize that increased FSH signaling may secondarily drive a pro-inflammatory microglial phenotype in an indirect, C/EBPβ-dependent fashion. (This should be explicitly tested.) As described above, FSH also appears to broadly drive neuroinflammatory and glial activation processes. To directly evaluate this, Xiong et al. (2023) generated an AD mouse model based on ovariectomy, a surgical procedure which permanently disturbs the endogenous balance of sex hormones but can be mimicked to maintain physiological levels of estrogens through exogenous replacement. FSH elevation in this context was associated with an accumulation of other disease-associated markers in the brain, including Aβ and hyperphosphorylated tau (Tau368), implicating a potential, estrogen-independent pathologic contribution by FSH. (Results of ovariectomy models should be interpreted cautiously when considering human menopause.
Figure 7.
FSH-FSHR signaling as a driver of amyloidogenic, tau-directed, and inflammatory pathology in Alzheimer’s disease. FSH-FSHR signaling as a driver of amyloidogenic, tau-directed, and inflammatory pathology in Alzheimer’s disease. FSH binding to the FSH receptor (FSHR) in cortical and hippocampal neurons activates Gαi-dependent extracellular signal-regulated kinase 1/2 (ERK1/2) and protein kinase B (AKT) signaling, leading to phosphorylation of CCAAT/enhancer-binding protein beta (C/EBPβ) and serine-arginine protein kinase 2 (SRPK2). Phosphorylated SRPK2 translocates to the nucleus where it contributes to upregulation of asparagine endopeptidase (AEP; also known as δ-secretase), which cleaves amyloid precursor protein (APP) and tau into aggregation-prone fragments, generating amyloid-beta (Aβ) and truncated tau (Tau368). These products further activate microglial and inflammasome signaling, amplifying cytokine production and sustaining neuroinflammation in a self-reinforcing cycle. Yellow circles labeled P denote phosphorylation events; the green wavy arrow indicates transcriptional activation at the C/EBPβ target gene promoter. Adapted from Li et al., (2024).
Figure 7.
FSH-FSHR signaling as a driver of amyloidogenic, tau-directed, and inflammatory pathology in Alzheimer’s disease. FSH-FSHR signaling as a driver of amyloidogenic, tau-directed, and inflammatory pathology in Alzheimer’s disease. FSH binding to the FSH receptor (FSHR) in cortical and hippocampal neurons activates Gαi-dependent extracellular signal-regulated kinase 1/2 (ERK1/2) and protein kinase B (AKT) signaling, leading to phosphorylation of CCAAT/enhancer-binding protein beta (C/EBPβ) and serine-arginine protein kinase 2 (SRPK2). Phosphorylated SRPK2 translocates to the nucleus where it contributes to upregulation of asparagine endopeptidase (AEP; also known as δ-secretase), which cleaves amyloid precursor protein (APP) and tau into aggregation-prone fragments, generating amyloid-beta (Aβ) and truncated tau (Tau368). These products further activate microglial and inflammasome signaling, amplifying cytokine production and sustaining neuroinflammation in a self-reinforcing cycle. Yellow circles labeled P denote phosphorylation events; the green wavy arrow indicates transcriptional activation at the C/EBPβ target gene promoter. Adapted from Li et al., (2024).

Importantly, Aβ and hyperphosphorylated tau act as reciprocal amplifiers of neuroinflammation. Aβ facilitates microglial recruitment and clustering within plaque-dense regions (Gao et al., 2023) and induces activation of the NLRP3 inflammasome, leading to the release of apoptosis-associated speck-like protein containing a CARD (ASC) aggregates. These ASC specks can bind Aβ, further promoting its aggregation and propagation (Venegas et al., 2017). In parallel, chronic activation of microglia and astrocytes enhances amyloidogenic APP processing and upregulates tau-directed kinases, including glycogen synthase kinase-3β (GSK-3β), thereby accelerating tau hyperphosphorylation, synaptic dysfunction, and neurodegenerative spread (Chen & Yu, 2023; Zhang et al., 2021). Further supporting a causal role, treatment with FSH-neutralizing antibodies has been shown to reduce Aβ accumulation and tau pathology in experimental models, reinforcing the contribution of FSH signaling to Alzheimer’s disease progression (Xiong et al., 2022).
FSH-Mediated Disruption of the Blood-Brain Barrier
The blood-brain barrier (BBB) is formed by specialized brain endothelial cells connected by tight junctions and supported by perivascular cell populations, including pericytes, astrocytes, microglia, and oligodendrocytes. Together, these components tightly regulate molecular exchange between the circulation and the central nervous system, restrict the entry of neurotoxic substances, and maintain neural homeostasis (Kim et al., 2024).
Circulating FSH levels are inversely associated with vascular compliance, such that higher FSH concentrations correlate with increased arterial stiffness and endothelial dysfunction (Ferretti et al., 2018; Laakkonen et al., 2021; Wang et al., 2026; Xue et al., 2025). Experimental and clinical studies further indicate that elevated FSH promotes vascular calcification and progressive loss of arterial elasticity, contributing to age-related vascular remodeling (Li et al., 2024; Roelofs et al., 2022). In parallel, FSH has been shown to disrupt endothelial junctional integrity by altering the expression of adherens junction proteins, including E-cadherin and V-cadherin, thereby increasing vascular permeability (Kolnes et al., 2020; Rocca et al., 2024).
Beyond its effects on peripheral vasculature and neural cells (Xue et al., 2025), emerging evidence suggests that FSH can also influence cerebrovascular function (Alkhalifa et al., 2023; Wilson et al., 2008). BBB dysfunction is now recognized as an early and consistent feature of AD, emerging prior to overt neurodegeneration and contributing to disease progression through increased permeability to inflammatory mediators, lipids, and circulating factors (Alkhalifa et al., 2023). Because BBB integrity depends on adherens and tight junction mechanisms shared across vascular beds, FSH-mediated disruption of endothelial junctional signaling observed in peripheral tissues has been proposed to extend to the BBB - though direct evidence in cerebrovascular tissue specifically remains limited, and this mechanistic link should be regarded as a plausible hypothesis requiring BBB-specific investigation.
In addition to adherens junction disruption, BBB impairment may also arise through FSH-associated alterations in gap junction signaling. In a mouse model, ovariectomy followed by treatment with a gonadotropin agonist (leuprolide acetate) resulted in increased expression of connexin-43 and enhanced Evans blue dye extravasation into the brain, indicating elevated BBB permeability (Wilson et al., 2008). Notably, BBB dysfunction is now recognized as an early feature of Alzheimer’s disease, emerging prior to overt neurodegeneration and clinical symptoms (Alkhalifa et al., 2023).
Astrocyte aging represents an additional layer of vulnerability. Aged astrocytes exhibit reduced capacity to support neuronal function and maintain homeostasis, and have been identified as a strong cellular predictor of Alzheimer’s disease risk (Bronzuoli et al., 2019; Cohen & Torres, 2019; Monterey et al., 2021). Given that astrocytic endfeet provide near-complete coverage of the cerebral vasculature (Mathiisen et al., 2010), age-related astrocytic dysfunction is likely to further compromise BBB stability. Consistent with this, aging itself is associated with progressive increases in BBB permeability, with older women demonstrating greater BBB vulnerability than younger individuals (Goodall et al., 2018; Knox et al., 2022).
Figure 8.
FSH-induced blood–brain barrier (BBB) dysfunction and cerebrovascular disease. Elevated FSH is associated with BBB breakdown, vascular stiffness, and decreased cerebral blood flow (CBF), mediated through inflammation - including lipopolysaccharide (LPS), tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β) - lipid dysregulation involving apolipoprotein E4 (ApoE-4) and low-density lipoprotein (LDL) via lipoprotein receptor-related protein 1 (LRP-1), and vascular structural alterations. The right panel illustrates a proposed two-hit model: a first hit comprising BBB disruption, CBF decline, vascular stiffness, and increased leakage of peripheral risk factors; and a second hit comprising neuroinflammation, lipid deposition, cerebral amyloid angiopathy (CAA), and intracranial atherosclerosis. Together these converging processes establish the cerebrovascular and neuroinflammatory conditions that promote Alzheimer’s disease-like pathology. Adapted from Xue, Y., et al. (2025).
Figure 8.
FSH-induced blood–brain barrier (BBB) dysfunction and cerebrovascular disease. Elevated FSH is associated with BBB breakdown, vascular stiffness, and decreased cerebral blood flow (CBF), mediated through inflammation - including lipopolysaccharide (LPS), tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β) - lipid dysregulation involving apolipoprotein E4 (ApoE-4) and low-density lipoprotein (LDL) via lipoprotein receptor-related protein 1 (LRP-1), and vascular structural alterations. The right panel illustrates a proposed two-hit model: a first hit comprising BBB disruption, CBF decline, vascular stiffness, and increased leakage of peripheral risk factors; and a second hit comprising neuroinflammation, lipid deposition, cerebral amyloid angiopathy (CAA), and intracranial atherosclerosis. Together these converging processes establish the cerebrovascular and neuroinflammatory conditions that promote Alzheimer’s disease-like pathology. Adapted from Xue, Y., et al. (2025).

Integrative Mechanistic Framework: How Reproductive Aging in biological females Reconfigures Alzheimer’s Disease Risk
The biological features commonly associated with AD, as with many age-related disorders, are not unique to individuals who meet diagnostic criteria for the condition. Although amyloid-β accumulation, tau pathology, and α-synuclein aggregation are central to the pathobiology of neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and dementia with Lewy bodies, these same molecular alterations are also observed during normative aging (Sengupta & Kayed 2022). Indeed, postmortem studies have repeatedly identified amyloid plaques and tauopathy, of varying degrees, in cognitively normal for older adults and even middle aged carriers (Krishnadas et al., 2022; Morris et al., 2010 ; Pletnikova et al., 2015; Sengupta & Kayed 2022). Moreover, the proteins involved in these processes are produced constitutively throughout life, yet the majority of individuals never develop clinical neurodegenerative disease (Pearson et al., (1999). Thus, pathology does not arise simply from protein presence, but from failures in the regulatory systems that govern protein folding, trafficking, clearance, and cellular context, which are commonly aberrant among various neurodegenerative conditions outside of the one subject to this review (Jamerlan, A., & Hulme, J. (2026).
The earliest indicators of impending neurodegeneration - often preceding measurable cognitive impairment - include impaired proteostatic and autophagic clearance, altered cellular metabolic activity (Gazestani et al., 2023), chronic microglial activation (Li et al., 2018), increased blood-brain barrier permeability, and astrocytic dysfunction. Collectively, these upstream disturbances compromise cellular homeostasis, inflammatory regulation, and metabolic support well before overt neuronal loss becomes evident (Li et al., 2018). In this context, classical pathological hallmarks such as amyloid-β and tau accumulation, autophagosome buildup, and progressive neuronal degeneration are more accurately understood as downstream consequences of sustained regulatory failure rather than primary initiating events. With these points in mind, it is prudent to consider factors that pre-emptively perturb these aspects of the condition at an early stage so that the risk of disease progression may be mollified.
In the United States, the majority of AD cases occur in women, suggesting that female-specific biological factors contribute meaningfully to disease risk, progression, and severity. Consistent with this, women - particularly those carrying the ApoE4 allele - tend to exhibit earlier onset and more severe clinical manifestations of AD. Notably, approximately 40-60% of AD cases involve at least one ApoE4 allele, underscoring its substantial contribution to the overall disease burden (Pires & Rego, 2023). Beyond its role in AD, ApoE4 may also influence responses to other conditions associated with reproductive aging. For example, female APOE4 carriers appear to be at increased risk for treatment resistance in the context of menopausal symptoms, including vasomotor instability and sleep disturbances (Muhammad, 2025). These observations suggest that ApoE4 may interact with endocrine aging processes in ways that extend beyond classical neurodegenerative pathways.
Although this review focuses on mechanisms underlying increased AD risk in women, it is important to note that age-related increases in FSH are also observed in men, albeit to a lesser extent (Kim et al., 2024). This raises the possibility that FSH-related mechanisms may contribute to neurodegenerative processes across sexes, and warrants further investigation into the role of FSH in AD pathophysiology in male populations.
Endocrine and Metabolic Targets: Translational Opportunities in Alzheimer’s Disease
While various pharmacologic approaches are currently employed in the treatment of AD (cholinesterase inhibitors, NMDA receptor antagonists, and more recently anti-amyloid agents, etc) (Zhang et al., 2024), a thorough discussion of these approaches is outside of the scope of the present review. Instead, the present review focuses on the mechanistic link between reproductive aging and the pathophysiology of AD, with a focus on the endocrine transition, gonadotropin signaling, and the downstream consequences of this process (inflammation, metabolism, etc) and the impact on cellular homeostasis. Targeted intervention within this upstream, system-level process (FSH signaling, for example) could therefore be an important therapeutic approach, providing complementary strategies in the treatment of AD that build upon and enhance currently employed treatments.
Therapeutic Targeting of FSH in Alzheimer’s Disease
Given the emerging role of FSH in modulating AD risk and pathology, therapeutic targeting of FSH signaling represents a promising avenue for intervention. Preclinical studies demonstrate that ovariectomy induces AD-like pathology and cognitive deficits in female ApoE4 knock-in mice, effects that are significantly attenuated by treatment with FSH-neutralizing antibodies. These interventions reduce amyloid burden, mitigate hippocampal and cortical neurodegeneration, and improve cognitive performance (Xiong et al., 2022; Xiong et al., 2023). Subsequent work has reinforced these findings, showing that disruption of FSH signaling - either through FSHR knockout or antibody-mediated blockade - improves spatial learning and recognition memory in experimental models (Korkmaz et al., 2025; Pallapat et al., 2025). Despite these encouraging results, FSH-targeted therapies remain in the preclinical stage, although efforts toward translation into human trials are currently underway (Pallapat et al., 2025).
Hormone Therapy and Estrogen Receptor Modulation
The use of menopause hormone therapy (MHT) for the treatment of menopausal symptoms and its effects on cognition and dementia risk remains an area of ongoing clinical and public debate, largely due to conflicting evidence and concerns regarding potential adverse outcomes (Rocca et al., 2024). While experimental and laboratory studies often suggest neuroprotective or cognitive benefits of hormone therapy, these findings have not been consistently replicated in human populations (Ali et al., 2023; Andy et al., 2024).
Some clinical and epidemiological studies report that MHT may increase the risk of dementia and cognitive decline (Craig et al., 2005; Yuk et al., 2023), and it has been associated with increased Alzheimer’s-related tau pathology (Coughlan et al., 2023). However, the overall certainty of this evidence remains limited, as many systematic reviews fail to meet rigorous standards (e.g., GRADE) due to bias, heterogeneity, and inconsistency across studies (Hilton Boon et al., 2021). Early observational studies suggested that MHT might reduce dementia risk, particularly when initiated during midlife and used over extended durations (Henderson et al., 1994; Henderson et al., 2005; Kim et al., 2021; Whitmer et al., 2011; Zandi et al., 2002). However, these findings are highly susceptible to confounding - especially healthy user bias - and have not been consistently reproduced in randomized controlled trials (Rahman et al., 2019).
The Women’s Health Initiative Memory Study (WHIMS) reported that combined MHT (conjugated equine estrogens plus medroxyprogesterone acetate) approximately doubled dementia risk, while estrogen-only therapy increased the risk of mild cognitive impairment in women aged 65 years or older (Shumaker et al., 2003; Shumaker et al., 2004). Although these findings are often cited, the late age of therapy initiation limits their generalizability to typical clinical use, where treatment is often initiated earlier in the menopausal transition.
Subsequent observational studies and meta-analyses have yielded inconsistent results. Some large observational cohorts suggest that combined MHT may increase dementia risk, particularly with prolonged use (Pourhadi et al., 2023; Vinogradova et al., 2021), whereas others report a potential reduction in risk - especially with estrogen-only therapy initiated during midlife (Nerattini et al., 2023; Mosconi et al., 2025). However, more stringent umbrella reviews applying risk-of-bias assessments and GRADE criteria have found no reliable evidence supporting a protective “therapeutic window” for MHT and emphasize substantial uncertainty in the literature (Melville et al., 2025). Across randomized and observational evidence, findings remain heterogeneous and frequently limited by methodological constraints, including imprecision, study design variability, and cohort overlap. As such, current evidence does not support the use of MHT solely for dementia prevention, nor does it definitively establish a causal relationship between MHT and increased dementia risk (Livingston et al., 2024).
Notably, the analysis by Melville et al. (2025) did not account for potential modifying effects of genetic factors such as ApoE4 status, which may partially explain heterogeneity across studies. Evidence suggests that responses to estrogen-based therapies are genotype- and age-dependent. In younger postmenopausal women (mean age <65 years), estrogen treatment has been associated with beneficial effects on brain structure and amyloid dynamics, including reduced amyloid deposition and preservation of hippocampal and entorhinal cortex volumes - effects that appear most pronounced in ApoE3/4 carriers (Kantarci et al., 2016). However, outcomes in ε4 homozygotes remain uncertain (Saleh et al., 2023; Depypere et al., 2023), and these benefits are not consistently observed in women over the age of 65 (Yaffe et al., 2000; Burkhardt et al., 2004; Kang et al., 2004; Kang & Grodstein, 2012).
Experimental data further support genotype-specific differences in estrogen responsiveness. In female mouse models, acute estradiol administration into the dorsal hippocampus enhanced object recognition and spatial memory consolidation, along with increased dendritic spine density in the CA1 region, in animals carrying ApoE3 or a single ApoE4 allele (E3FAD and E3/4FAD), but not in ApoE4 homozygous mice (E4FAD) (Taxier et al., 2022a; Taxier et al., 2022b). Although the mechanisms underlying this reduced responsiveness remain unclear, ApoE4 homozygous females exhibit elevated ERα expression without corresponding changes in ERβ levels, suggesting a shift in receptor balance that may influence estrogen signaling outcomes (Taxier et al., 2022a,b).
Importantly, clinical practices such as sequential hormone replacement therapy - where estrogen is administered continuously with intermittent progestin in cycling women - may introduce additional risks. Chronic estrogen exposure in this context has been associated with adverse effects including hyperprolactinemia, hypertension, and metabolic disturbances ( Kalenga et al., 2022; MohanKumar et al., 2018), warranting careful consideration when evaluating endocrine interventions.
A limitation of estrogen-based therapies in postmenopausal individuals with AD is their association with increased risk of hormone-sensitive cancers, including breast and endometrial cancer, largely mediated through the proliferative effects of estrogen receptor alpha (ERα) signaling (Shete et al., 2023). In contrast, activation of ERβ does not promote cancer cell proliferation (Jia et al., 2015) and has been associated with beneficial effects on cognition (Fleischer et al., 2021). Experimental studies demonstrate that ERβ activation enhances memory formation in rodent models (Schwabe et al., 2024), and this receptor is highly expressed in brain regions critical for cognition and neuroendocrine regulation, including the hippocampus, medial prefrontal cortex, and hypothalamus (Almey et al., 2014; Shughrue et al., 1998; Milner et al., 2005).
Importantly, ERβ signaling has been linked to reductions in amyloid-β levels and tau phosphorylation within the hippocampus (Tian et al., 2013; Xiong et al., 2015). Consistent with a protective role, constitutive knockout of ERβ in mice results in increased Aβ42 deposition and ApoE accumulation in the hippocampus and cortex (Zhang et al., 2004). However, evidence from preclinical models indicates that ERβ-mediated benefits may be genotype-dependent, with cognitive and neuropathological improvements observed primarily in E3FAD and E3/4FAD mice, but not in ApoE4 homozygous models. Interestingly, a similar pattern of differential responsiveness has been reported in clinical contexts, where ERβ-targeted approaches appear to confer more consistent benefits in subsets of women experiencing menopausal symptoms (Muhammad, 2025). Together, these findings suggest that while ERβ signaling may offer a more favorable therapeutic profile than non-selective estrogen approaches, its efficacy is likely influenced by underlying genetic and physiological context. Further studies need to be conducted with human subjects to confirm or deny its clinical utility.
Targeting Metabolic Perturbations in Alzheimer’s Disease
Metabolic perturbations in brain cells represent some of the earliest detectable changes preceding the AD phenotype (Naia et al., 2023). These disturbances are further amplified by FSH-associated signaling dynamics, establishing a metabolic environment that predisposes to downstream dysfunction, including impaired autophagy, microglial activation, disrupted glucose utilization, and the accumulation of pathogenic protein species. In this context, metabolic dysfunction is not merely a consequence of disease progression but a key upstream driver of neurodegenerative vulnerability.
Importantly, metabolic alterations within the hypothalamus may also contribute to dysregulation of FSH signaling itself (Zhang et al., 2013). The menopausal transition is characterized by transient elevations in gonadotropin-releasing hormone GnRH, followed by subsequent declines in postmenopausal stages (Hall et al., 2000). Reduced GnRH signaling has been linked to activation of NF-κB/IKK-β-mediated inflammatory pathways (Zhang et al., 2013), suggesting that hypothalamic metabolic reprogramming may precede and facilitate inflammatory signaling cascades that further disrupt neuroendocrine regulation. These interactions point to a bidirectional relationship in which metabolic dysfunction and endocrine dysregulation reinforce one another across aging.
Among therapeutic strategies targeting metabolic dysfunction in AD, ketogenic diets (KD) have emerged as one of the most promising non-pharmacological approaches. Ketogenic interventions have a well-established role in neurological conditions such as epilepsy and are increasingly being explored as adjunctive therapies in disorders including bipolar disorder, glioblastoma, and neurodegenerative diseases. In AD, a randomized trial demonstrated that a 12-week KD intervention improved quality of life and daily functional performance, although interpretation is limited by small sample size and short duration (Phillips et al., 2021). Additional studies report improvements in cognitive function alongside favorable changes in metabolic and biomarker profiles (Neth et al., 2020), including increased cerebral ketone uptake, enhanced brain perfusion, and improved peripheral lipid and glucose metabolism.
A broader range of trials using varied ketogenic formulations has consistently demonstrated beneficial effects in individuals with mild cognitive impairment (MCI) and AD, with evidence suggesting that efficacy may be greatest during early to moderate stages of disease progression (Oliveira et al., 2023). Although the literature remains heterogeneous with respect to study design, intervention protocols, and outcome measures, the collective evidence supports the therapeutic potential of ketogenic strategies in AD (Grammatikopoulou et al., 2020). Additional pharmacologic and non-pharmacologic approaches targeting metabolic dysfunction in AD have also been explored, and readers are referred to Stefaniak et al. (2022) and Wang et al. (2023) for further discussion.
Limitations
Several limitations of the underlying evidence base warrant acknowledgment. First, a substantial proportion of the mechanistic evidence linking FSH to AD-relevant pathways derives from transgenic animal models and cell-based studies, and direct replication in human populations remains limited for many of the pathways discussed. Second, the majority of human epidemiological data examining FSH and cognitive outcomes is observational, precluding causal inference. Third, this review focuses primarily on mechanisms underlying increased AD risk in biological females undergoing reproductive aging; while age-related FSH elevation also occurs in men, the extent to which the mechanisms described here apply across sexes remains to be determined and warrants further investigation. Fourth, the heterogeneity, methodological inconsistency, and cohort overlap that characterize the menopause hormone therapy and dementia literature have been extensively documented and corroborated across independent systematic reviews and umbrella analyses, including those applying formal risk-of-bias criteria (Melville et al., 2025); this reflects the state of the field rather than a limitation of the present synthesis. Fifth, findings from the Women’s Health Initiative Memory Study regarding hormone therapy and dementia risk are subject to important generalizability constraints, as therapy was initiated late in the menopausal transition in women aged 65 and older — a context that differs meaningfully from clinical practice, where treatment typically begins earlier. Outcomes in APOE4 homozygotes within this cohort specifically remain uncertain, and conclusions drawn from this population should not be extended uncritically to younger postmenopausal women initiating therapy closer to the menopausal transition. These constraints are noted not to diminish the mechanistic framework presented, but to identify the specific gaps in human evidence that future work should prioritize.
Conclusions
Alzheimer’s disease emerges not solely from the accumulation of pathogenic proteins, but from progressive failures in the systems that regulate metabolism, proteostasis, inflammation, and vascular integrity. Reproductive aging represents a critical inflection point in female neurobiology, characterized by coordinated endocrine, immunological, and metabolic shifts that reshape vulnerability to neurodegeneration.
Among these changes, sustained elevations in FSH appear to exert broad and multifaceted effects that extend well beyond reproductive physiology. Evidence synthesized in this review suggests that FSH contributes to AD pathogenesis through convergent mechanisms involving dysregulated glucose metabolism, mitochondrial dysfunction, impaired autophagic clearance, altered apolipoprotein dynamics, and amplification of neuroinflammatory signaling. These effects are further compounded by interactions with genetic risk factors such as ApoE4, as well as age-associated declines in vascular and blood–brain barrier integrity. Importantly, many of the molecular alterations traditionally regarded as defining features of AD - such as amyloid-β accumulation and tau pathology - are more accurately understood as downstream consequences of earlier systemic dysregulation. This perspective shifts emphasis toward upstream drivers of disease susceptibility, particularly those emerging during the menopausal transition.
Although therapeutic strategies targeting FSH signaling remain in early stages of development, preclinical evidence suggests that modulation of this axis may attenuate multiple facets of AD pathology. Notably, such modulation need not rely solely on direct FSH blockade; selective targeting of estrogen receptor pathways, particularly ERβ, may offer an alternative means of mitigating FSH-associated effects while preserving beneficial estrogenic signaling. Future work should aim to clarify the extent to which FSH influences neurodegenerative outcomes in both men and women, as well as how progressive hypothalamic–pituitary alterations in neuroendocrine, metabolic, and immune regulation shape FSH dynamics across aging. Advancing this understanding may enable the development of more precise, biology-informed interventions that address upstream drivers of neurodegeneration rather than its late-stage manifestations.
Author Contributions
Manuscript drafting and revision, as well as figure development and acquisition, were all conducted by Yasin Ali Muhammad.
Funding
There was no funding.
Institutional Review Board Statement
Ethics declaration not applicable.
Conflicts of Interest
There are no competing interests.
References
- Abeer, M. I.; Abdulhasan, A.; Haguar, Z.; Narayanaswami, V. Isoform-specific modification of apolipoprotein E by 4-hydroxynonenal: protective role of apolipoprotein E3 against oxidative species. FEBS J. 2023, 290(11), 3006–3025. [Google Scholar] [CrossRef]
- Ali, N.; Sohail, R.; Jaffer, S. R.; Siddique, S.; Kaya, B.; Atowoju, I.; Imran, A.; Wright, W.; Pamulapati, S.; Choudhry, F.; Akbar, A.; Khawaja, U. A. The Role of Estrogen Therapy as a Protective Factor for Alzheimer’s Disease and Dementia in Postmenopausal Women: A Comprehensive Review of the Literature. Cureus 2023, 15(8), e43053. [Google Scholar] [CrossRef]
- Alkhalifa, A. E.; Al-Ghraiybah, N. F.; Odum, J.; Shunnarah, J. G.; Austin, N.; Kaddoumi, A. Blood-Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies. Int. J. Mol. Sci. 2023, 24(22), 16288. [Google Scholar] [CrossRef]
- Almansoub, H. A. M. M.; Tang, H.; Wu, Y.; Wang, D. Q.; Mahaman, Y. A. R.; Wei, N.; Almansob, Y. A. M.; He, W.; Liu, D. Tau Abnormalities and the Potential Therapy in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2019, 67(1), 13–33. [Google Scholar] [CrossRef]
- Almey, A.; Cannell, E.; Bertram, K.; Filardo, E.; Milner, T. A.; Brake, W. G. Medial prefrontal cortical estradiol rapidly alters memory system bias in female rats: ultrastructural analysis reveals membrane-associated estrogen receptors as potential mediators. Endocrinology 2014, 155(11), 4422–4432. [Google Scholar] [CrossRef]
- Alquezar, C.; Arya, S.; Kao, A. W. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2021, 11, 595532. [Google Scholar] [CrossRef]
- Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2024, 20(5), 3708–3821. [CrossRef]
- Andy, C.; Nerattini, M.; Jett, S.; Carlton, C.; Zarate, C.; Boneu, C.; Fauci, F.; Ajila, T.; Battista, M.; Pahlajani, S.; Christos, P.; Fink, M. E.; Williams, S.; Brinton, R. D.; Mosconi, L. Systematic review and meta-analysis of the effects of menopause hormone therapy on cognition. Front. Endocrinol. 2024, 15, 1350318. [Google Scholar] [CrossRef] [PubMed]
- Apátiga-Pérez, R.; Soto-Rojas, L. O.; Campa-Córdoba, B. B.; Luna-Viramontes, N. I.; Cuevas, E.; Villanueva-Fierro, I.; Ontiveros-Torres, M. A.; Bravo-Muñoz, M.; Flores-Rodríguez, P.; Garcés-Ramirez, L.; de la Cruz, F.; Montiel-Sosa, J. F.; Pacheco-Herrero, M.; Luna-Muñoz, J. Neurovascular dysfunction and vascular amyloid accumulation as early events in Alzheimer’s disease. Metab. Brain Dis. 2022, 37(1), 39–50. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Bush, A. I. β-amyloid: The known unknowns. Ageing Res. Rev. 2021, 65, 101212. [Google Scholar] [CrossRef] [PubMed]
- Baek, M. S.; Cho, H.; Lee, H. S.; Lee, J. H.; Ryu, Y. H.; Lyoo, C. H. Effect of APOE ε4 genotype on amyloid-β and tau accumulation in Alzheimer’s disease . Alzheimer’s Res. Ther. 2020, 12, 140. [Google Scholar] [CrossRef] [PubMed]
- Ball, A. B.; Jones, A. E.; Nguyễn, K. B.; Rios, A.; Marx, N.; Hsieh, W. Y.; Yang, K.; Desousa, B. R.; Kim, K. K. O.; Veliova, M.; Del Mundo, Z. M.; Shirihai, O. S.; Benincá, C.; Stiles, L.; Bensinger, S. J.; Divakaruni, A. S. Pro-inflammatory macrophage activation does not require inhibition of oxidative phosphorylation. EMBO Rep. 2025, 26(4), 982–1002. [Google Scholar] [CrossRef] [PubMed]
- Baragetti, A.; Catapano, A. L.; Magni, P. Multifactorial Activation of NLRP3 Inflammasome: Relevance for a Precision Approach to Atherosclerotic Cardiovascular Risk and Disease. Int. J. Mol. Sci. 2020, 21(12), 4459. [Google Scholar] [CrossRef]
- Bermejo, P.; Martín-Aragón, S.; Benedí, J.; Susín, C.; Felici, E.; Gil, P.; Villar, Á. M. Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from Mild Cognitive Impairment. Free Radic. Res. 2008, 42(2), 162–170. [Google Scholar] [CrossRef]
- Berry, A. S.; Harrison, T. M. New perspectives on the basal forebrain cholinergic system in Alzheimer’s disease. Neurosci. Biobehav. Rev. 2023, 150, 105192. [Google Scholar] [CrossRef]
- Beckman, M.; Knox, K.; Koneval, Z.; Smith, C.; Jayadev, S.; Barker-Haliski, M. Loss of presenilin 2 age-dependently alters susceptibility to acute seizures and kindling acquisition. Neurobiol. Dis. 2020, 136, 104719. [Google Scholar] [CrossRef]
- Bethlehem, R. A. I.; Seidlitz, J.; White, S. R.; Vogel, J. W.; Anderson, K. M.; Adamson, C.; Adler, S.; Alexopoulos, G. S.; Anagnostou, E.; Areces-Gonzalez, A.; Astle, D. E.; Auyeung, B.; Ayub, M.; Bae, J.; Ball, G.; Baron-Cohen, S.; Beare, R.; Bedford, S. A.; Benegal, V.; Beyer, F.; Alexander-Bloch, A. F. Brain charts for the human lifespan. Nature 2022, 604(7906), 525–533. [Google Scholar] [CrossRef]
- Bjørklund, G.; Aaseth, J.; Dadar, M.; Chirumbolo, S. Molecular Targets in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56(10), 7032–7044. [Google Scholar] [CrossRef]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M. T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol. Neurobiol. 2014, 50(2), 534–544. [Google Scholar] [CrossRef] [PubMed]
- Bronzuoli, M. R.; Facchinetti, R.; Valenza, M.; Cassano, T.; Steardo, L.; Scuderi, C. Astrocyte Function Is Affected by Aging and Not Alzheimer’s Disease: A Preliminary Investigation in Hippocampi of 3xTg-AD Mice. Front. Pharmacol. 2019, 10, 644. [Google Scholar] [CrossRef]
- Burkhardt, M. S.; Foster, J. K.; Laws, S. M.; Baker, L. D.; Craft, S.; Gandy, S. E.; Stuckey, B. G.; Clarnette, R.; Nolan, D.; Hewson-Bower, B.; Martins, R. N. Oestrogen replacement therapy may improve memory functioning in the absence of APOE epsilon4. J. Alzheimer’s Dis. JAD 2004, 6(3), 221–228. [Google Scholar] [CrossRef]
- Butterfield, D. A.; Mattson, M. P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef]
- Casarini, L.; Crépieux, P. Molecular Mechanisms of Action of FSH. Front. Endocrinol. 2019, 10, 305. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R. J.; Jamshidi, P.; Plascencia-Villa, G.; Perry, G. The Amyloid Cascade Hypothesis: A Conclusion in Search of Support. Am. J. Pathol. 2025, 195(11), 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fang, H. Q.; Liu, J. T.; Chang, S. Y.; Cheng, L. B.; Sun, M. X.; Feng, J. R.; Liu, Z. M.; Zhang, Y. H.; Rosen, C. J.; Liu, P. Atlas of Fshr expression from novel reporter mice. eLife 2025, 13, RP93413. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Q.; Wang, J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proceedings of the National Academy of Sciences of the United States of America 2011, 108(36), 14813–14818. [Google Scholar] [CrossRef]
- Chen, J.; Sun, J.; Hu, Y.; Wan, X.; Wang, Y.; Gao, M.; Liang, J.; Liu, T.; Sun, X. MicroRNA-191-5p ameliorates amyloid-β1-40 -mediated retinal pigment epithelium cell injury by suppressing the NLRP3 inflammasome pathway. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 2021, 35(4), e21184. [Google Scholar] [CrossRef]
- Chen, T. H.; Koh, K. Y.; Lin, K. M.; Chou, C. K. Mitochondrial Dysfunction as an Underlying Cause of Skeletal Muscle Disorders. Int. J. Mol. Sci. 2022, 23(21), 12926. [Google Scholar] [CrossRef]
- Chen, Y.; Jin, H.; Chen, J.; Li, J.; Găman, M. A.; Zou, Z. The multifaceted roles of apolipoprotein E4 in Alzheimer’s disease pathology and potential therapeutic strategies. Cell Death Discov. 2025, 11(1), 312. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y. J.; Lin, C. H.; Lane, H. Y. From Menopause to Neurodegeneration-Molecular Basis and Potential Therapy. Int. J. Mol. Sci. 2021, 22(16), 8654. [Google Scholar] [CrossRef]
- Chong, C. M.; Ke, M.; Tan, Y.; Huang, Z.; Zhang, K.; Ai, N.; Ge, W.; Qin, D.; Lu, J. H.; Su, H. Presenilin 1 deficiency suppresses autophagy in human neural stem cells through reducing γ-secretase-independent ERK/CREB signaling. Cell Death Dis. 2018, 9(9), 879. [Google Scholar] [CrossRef]
- Christodoulou, R. C.; Eller, D.; Papageorgiou, P. S.; Angelopoulou, E.; Vassiliou, E.; Papageorgiou, S. G. Metabolic Dysfunction in Alzheimer’s Disease: Brain Glucose Hypometabolism as an Early Precursor to Amyloid and Tau Pathology. J. Clin. Med. 2026, 15(5), 1884. [Google Scholar] [CrossRef]
- Cobley, J. N.; Fiorello, M. L.; Bailey, D. M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18(3), e12937. [Google Scholar] [CrossRef]
- Combs, B.; Mueller, R. L.; Morfini, G.; Brady, S. T.; Kanaan, N. M. Tau and Axonal Transport Misregulation in Tauopathies. Adv. Exp. Med. Biol. 2019, 1184, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Corraliza-Gomez, M.; Sanchez, D.; Ganfornina, M. D. Lipid-Binding Proteins in Brain Health and Disease. Front. Neurol. 2019, 10, 1152. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Canteli, M.; Iadecola, C. Alzheimer’s Disease and Vascular Aging: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75(8), 942–951. [Google Scholar] [CrossRef]
- Coughlan, G. T.; Betthauser, T. J.; Boyle, R.; Koscik, R. L.; Klinger, H. M.; Chibnik, L. B.; Jonaitis, E. M.; Yau, W. W.; Wenzel, A.; Christian, B. T.; Gleason, C. E.; Saelzler, U. G.; Properzi, M. J.; Schultz, A. P.; Hanseeuw, B. J.; Manson, J. E.; Rentz, D. M.; Johnson, K. A.; Sperling, R.; Johnson, S. C.; Buckley, R. F. Association of Age at Menopause and Hormone Therapy Use With Tau and β-Amyloid Positron Emission Tomography. JAMA Neurol. 2023, 80(5), 462–473. [Google Scholar] [CrossRef] [PubMed]
- Craig, M. C.; Maki, P. M.; Murphy, D. G. The Women’s Health Initiative Memory Study: findings and implications for treatment. Lancet. Neurol. 2005, 4(3), 190–194. [Google Scholar] [CrossRef]
- Dani, M.; Wood, M.; Mizoguchi, R.; Fan, Z.; Walker, Z.; Morgan, R.; Hinz, R.; Biju, M.; Kuruvilla, T.; Brooks, D. J.; Edison, P. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain A J. Neurol. 2018, 141(9), 2740–2754. [Google Scholar] [CrossRef]
- Dasadhikari, S.; Ghosh, S.; Pal, S.; Knowles, T. P. J.; Garai, K. A single fibril study reveals that ApoE inhibits the elongation of Aβ42 fibrils in an isoform-dependent manner. Commun. Chem. 2025, 8(1), 133. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M. B.; Holtzman, D. M.; Zlokovic, B. V. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118(12), 4002–4013. [Google Scholar] [CrossRef]
- Deming, Y.; Filipello, F.; Cignarella, F.; Cantoni, C.; Hsu, S.; Mikesell, R.; Li, Z.; Del-Aguila, J. L.; Dube, U.; Farias, F. G.; Bradley, J.; Budde, J.; Ibanez, L.; Fernandez, M. V.; Blennow, K.; Zetterberg, H.; Heslegrave, A.; Johansson, P. M.; Svensson, J.; Nellgård, B.; Cruchaga, C. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med. 2019, 11(505), eaau2291. [Google Scholar] [CrossRef]
- Depypere, H.; Vergallo, A.; Lemercier, P.; Lista, S.; Benedet, A.; Ashton, N.; Cavedo, E.; Zetterberg, H.; Blennow, K.; Vanmechelen, E.; Hampel, H.; Neurodegeneration Precision Medicine Initiative (NPMI). Menopause hormone therapy significantly alters pathophysiological biomarkers of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2023, 19(4), 1320–1330. [Google Scholar] [CrossRef]
- Descalzi, G.; Gao, V.; Steinman, M.Q.; Suzuki, A.; Alberini, C.M. Lactate from astrocytes fuels learning-induced mRNA translation in excitatory and inhibitory neurons. Commun. Biol. 2019, 2, 247. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Doering, S.; McKay, N. S.; Jana, N.; Dombrowski, K.; McCullough, A.; Millar, P. R.; Hobbs, D. A.; Agrawal, R.; Flores, S.; Llibre-Guerra, J. J.; Huey, E. D.; Ances, B. M.; Xiong, C.; Aschenbrenner, A. J.; Hassenstab, J.; Morris, J. C.; Gordon, B. A.; Benzinger, T. L. S. Domain-specific cognitive impairment is differentially affected by Alzheimer disease tau pathologic burden and spread. Imaging Neurosci. 2024, 2, imag-2-00405. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D. M.; Schreiber, J. R.; Schmit, V. M.; Getz, G. S. Regulation of apolipoprotein E synthesis in rat ovarian granulosa cells. J. Biol. Chem. 1985, 260(15), 9031–9038. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D. M.; Ko, C.; Jo, M.; Brannstrom, M.; Curry, T. E. Ovulation: Parallels With Inflammatory Processes. Endocr. Rev. 2019, 40(2), 369–416. [Google Scholar] [CrossRef]
- Erten-Lyons, D.; Dodge, H. H.; Woltjer, R.; Silbert, L. C.; Howieson, D. B.; Kramer, P.; Kaye, J. A. Neuropathologic basis of age-associated brain atrophy. JAMA Neurol. 2013, 70(5), 616–622. [Google Scholar] [CrossRef] [PubMed]
- Eskandari-Sedighi, G.; Crichton, M.; Zia, S.; Gomez-Cardona, E.; Cortez, L. M.; Patel, Z. H.; Takahashi-Yamashiro, K.; St Laurent, C. D.; Sidhu, G.; Sarkar, S.; Aghanya, V.; Sim, V. L.; Tan, Q.; Julien, O.; Plemel, J. R.; Macauley, M. S. Alzheimer’s disease associated isoforms of human CD33 distinctively modulate microglial cell responses in 5XFAD mice. Mol. Neurodegener. 2024, 19(1), 42. [Google Scholar] [CrossRef]
- Fedeli, C.; Filadi, R.; Rossi, A.; Mammucari, C.; Pizzo, P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca2+ homeostasis. Autophagy 2019, 15(12), 2044–2062. [Google Scholar] [CrossRef]
- Fedele, E. Anti-Amyloid Therapies for Alzheimer’s Disease and the Amyloid Cascade Hypothesis. Int. J. Mol. Sci. 2023, 24(19), 14499. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D. J. The machinery of macroautophagy. Cell Res. 2014, 24(1), 24–41. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M. T.; Iulita, M. F.; Cavedo, E.; Chiesa, P. A.; Schumacher Dimech, A.; Santuccione Chadha, A.; Baracchi, F.; Girouard, H.; Misoch, S.; Giacobini, E.; Depypere, H.; Hampel, H. & Women’s Brain Project and the Alzheimer Precision Medicine Initiative (2018). Sex differences in Alzheimer disease - the gateway to precision medicine. Nat. Rev. Neurol. 14(8), 457–469. [CrossRef]
- Fleischer, A. W.; Schalk, J. C.; Wetzel, E. A.; Hanson, A. M.; Sem, D. S.; Donaldson, W. A.; Frick, K. M. Long-term oral administration of a novel estrogen receptor beta agonist enhances memory and alleviates drug-induced vasodilation in young ovariectomized mice. Horm. Behav. 2021, 130, 104948. [Google Scholar] [CrossRef]
- Frieden, C.; Garai, K. Concerning the structure of apoE. Protein Sci. A Publ. Protein Soc. 2013, 22(12), 1820–1825. [Google Scholar] [CrossRef]
- Frieden, C.; Wang, H.; Ho, C. M. W. A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain-domain interactions. Proceedings of the National Academy of Sciences of the United States of America 2017, 114(24), 6292–6297. [Google Scholar] [CrossRef] [PubMed]
- Fruhwürth, S.; Zetterberg, H.; Paludan, S. R. Microglia and amyloid plaque formation in Alzheimer’s disease - Evidence, possible mechanisms, and future challenges. J. Neuroimmunol. 2024, 390, 578342. [Google Scholar] [CrossRef]
- Fung, Y. L.; Ng, K. E. T.; Vogrin, S. J.; Meade, C.; Ngo, M.; Collins, S. J.; Bowden, S. C. Comparative Utility of Manual versus Automated Segmentation of Hippocampus and Entorhinal Cortex Volumes in a Memory Clinic Sample. J. Alzheimer’s Dis. JAD 2019, 68(1), 159–171. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; Natunen, T. Altered Insulin Signaling in Alzheimer’s Disease Brain - Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef] [PubMed]
- Ganfornina, M. D.; Do Carmo, S.; Lora, J. M.; Torres-Schumann, S.; Vogel, M.; Allhorn, M.; González, C.; Bastiani, M. J.; Rassart, E.; Sanchez, D. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell 2008, 7(4), 506–515. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8(1), 359. [Google Scholar] [CrossRef]
- Gawriluk, T. R.; Hale, A. N.; Flaws, J. A.; Dillon, C. P.; Green, D. R.; Rucker, E. B., 3rd. Autophagy is a cell survival program for female germ cells in the murine ovary. Reproduction 2011, 141(6), 759–765. [Google Scholar] [CrossRef]
- Gazestani, V.; Kamath, T.; Nadaf, N. M.; Dougalis, A.; Burris, S. J.; Rooney, B.; Junkkari, A.; Vanderburg, C.; Pelkonen, A.; Gomez-Budia, M.; Välimäki, N. N.; Rauramaa, T.; Therrien, M.; Koivisto, A. M.; Tegtmeyer, M.; Herukka, S. K.; Abdulraouf, A.; Marsh, S. E.; Hiltunen, M.; Nehme, R.; Macosko, E. Z. Early Alzheimer’s disease pathology in human cortex involves transient cell states. Cell 2023, 186(20), 4438–4453.e23. [Google Scholar] [CrossRef]
- Getz, G. S.; Reardon, C. A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res. 2009, 50 Suppl(Suppl), S156–S161. [Google Scholar] [CrossRef]
- Gonos, E. S.; Kapetanou, M.; Sereikaite, J.; Bartosz, G.; Naparło, K.; Grzesik, M.; Sadowska-Bartosz, I. Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging 2018, 10(5), 868–901. [Google Scholar] [CrossRef] [PubMed]
- Goodall, E. F.; Wang, C.; Simpson, J. E.; Baker, D. J.; Drew, D. R.; Heath, P. R.; Saffrey, M. J.; Romero, I. A.; Wharton, S. B. Age-associated changes in the blood-brain barrier: comparative studies in human and mouse. Neuropathol. Appl. Neurobiol. 2018, 44(3), 328–340. [Google Scholar] [CrossRef] [PubMed]
- Grammatikopoulou, M. G.; Goulis, D. G.; Gkiouras, K.; Theodoridis, X.; Gkouskou, K. K.; Evangeliou, A.; Dardiotis, E.; Bogdanos, D. P. To Keto or Not to Keto? A Systematic Review of Randomized Controlled Trials Assessing the Effects of Ketogenic Therapy on Alzheimer Disease. Adv. Nutr. 2020, 11(6), 1583–1602. [Google Scholar] [CrossRef]
- Grimaldi, L.; Bovi, E.; Formisano, R.; Sancesario, G. ApoE: The Non-Protagonist Actor in Neurological Diseases. Genes 2024, 15(11), 1397. [Google Scholar] [CrossRef]
- Gyparaki, M. T.; Arab, A.; Sorokina, E. M.; Santiago-Ruiz, A. N.; Bohrer, C. H.; Xiao, J.; Lakadamyali, M. Tau forms oligomeric complexes on microtubules that are distinct from tau aggregates. Proceedings of the National Academy of Sciences of the United States of America 2021, 118(19), e2021461118. [Google Scholar] [CrossRef] [PubMed]
- Hagihara, H.; Miyakawa, T. Decreased Brain pH Correlated With Progression of Alzheimer Disease Neuropathology: A Systematic Review and Meta-Analyses of Postmortem Studies. Int. J. Neuropsychopharmacol. 2024, 27(10), pyae047. [Google Scholar] [CrossRef] [PubMed]
- Hall, J. E.; Lavoie, H. B.; Marsh, E. E.; Martin, K. A. Decrease in gonadotropin-releasing hormone (GnRH) pulse frequency with aging in postmenopausal women. J. Clin. Endocrinol. Metab. 2000, 85(5), 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Mallucci, G. R. Review: Modulating the unfolded protein response to prevent neurodegeneration and enhance memory. Neuropathol. Appl. Neurobiol. 2015, 41(4), 414–427. [Google Scholar] [CrossRef]
- Hamilton, C.; Anand, P. K. Right place, right time: localisation and assembly of the NLRP3 inflammasome. F1000Research 2019, 8, F1000 Faculty Rev–676. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M. M.; Cuello, A. C.; Khachaturian, A. S.; Vergallo, A.; Farlow, M. R.; Snyder, P. J.; Giacobini, E.; Khachaturian, Z. S. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J. Prev. Alzheimer’s Dis. 2019, 6(1), 2–15. [Google Scholar] [CrossRef]
- Hammond, T. R.; Marsh, S. E.; Stevens, B. Immune Signaling in Neurodegeneration. Immunity 2019, 50(4), 955–974. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; McKeehan, N.; Dacks, P. A.; Fillit, H. M. Evaluation of the Neuroprotective Potential of N-Acetylcysteine for Prevention and Treatment of Cognitive Aging and Dementia. J. Prev. Alzheimer’s Dis. 2017, 4(3), 201–206. [Google Scholar] [CrossRef]
- Harrison, T. M.; La Joie, R.; Maass, A.; Baker, S. L.; Swinnerton, K.; Fenton, L.; Mellinger, T. J.; Edwards, L.; Pham, J.; Miller, B. L.; Rabinovici, G. D.; Jagust, W. J. Longitudinal tau accumulation and atrophy in aging and alzheimer disease. Ann. Neurol. 2019, 85(2), 229–240. [Google Scholar] [CrossRef]
- Hatters, D. M.; Budamagunta, M. S.; Voss, J. C.; Weisgraber, K. H. Modulation of apolipoprotein E structure by domain interaction: differences in lipid-bound and lipid-free forms. J. Biol. Chem. 2005, 280(40), 34288–34295. [Google Scholar] [CrossRef]
- Heilig, E. A.; Xia, W.; Shen, J.; Kelleher, R. J., 3rd. A presenilin-1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of gamma-secretase activity. J. Biol. Chem. 2010, 285(29), 22350–22359. [Google Scholar] [CrossRef]
- Henderson, V. W.; Benke, K. S.; Green, R. C.; Cupples, L. A.; Farrer, L. A.; MIRAGE Study Group. Postmenopausal hormone therapy and Alzheimer’s disease risk: interaction with age. J. Neurol. Neurosurg. Psychiatry 2005, 76(1), 103–105. [Google Scholar] [CrossRef]
- Henderson, V. W.; Paganini-Hill, A.; Emanuel, C. K.; Dunn, M. E.; Buckwalter, J. G. Estrogen replacement therapy in older women. Comparisons between Alzheimer’s disease cases and nondemented control subjects. Arch. Neurol. 1994, 51(9), 896–900. [Google Scholar] [CrossRef]
- Heneka, M. T.; Kummer, M. P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T. C.; Gelpi, E.; Halle, A.; Korte, M.; Latz, E.; Golenbock, D. T. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493(7434), 674–678. [Google Scholar] [CrossRef]
- Hickman, S. E.; Kingery, N. D.; Ohsumi, T. K.; Borowsky, M. L.; Wang, L. C.; Means, T. K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16(12), 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Hilton Boon, M.; Thomson, H.; Shaw, B.; Akl, E. A.; Lhachimi, S. K.; López-Alcalde, J.; Klugar, M.; Choi, L.; Saz-Parkinson, Z.; Mustafa, R. A.; Langendam, M. W.; Crane, O.; Morgan, R. L.; Rehfuess, E.; Johnston, B. C.; Chong, L. Y.; Guyatt, G. H.; Schünemann, H. J.; Katikireddi, S. V.; GRADE Working Group. Challenges in applying the GRADE approach in public health guidelines and systematic reviews: a concept article from the GRADE Public Health Group. J. Clin. Epidemiol. 2021, 135, 42–53. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D. R.; Latz, E.; Fitzgerald, K. A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458(7237), 514–518. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z. H.; Gu, D.; Mazzone, T. Oleic acid modulates the post-translational glycosylation of macrophage ApoE to increase its secretion. J. Biol. Chem. 2004, 279(28), 29195–29201. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, L.; Tammewar, G.; Ayakta, N.; Baker, S. L.; Bejanin, A.; Boxer, A. L.; Gorno-Tempini, M. L.; Janabi, M.; Kramer, J. H.; Lazaris, A.; Lockhart, S. N.; Miller, B. L.; Miller, Z. A.; O’Neil, J. P.; Ossenkoppele, R.; Rosen, H. J.; Schonhaut, D. R.; Jagust, W. J.; Rabinovici, G. D. Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer’s Disease. NeuroImage. Clin. 2017, 17, 452–464. [Google Scholar] [CrossRef]
- Ioannou, K.; Bucci, M.; Tzortzakakis, A.; Savitcheva, I.; Nordberg, A.; Chiotis, K. & Alzheimer’s Disease Neuroimaging Initiative Tau PET positivity predicts clinically relevant cognitive decline driven by Alzheimer’s disease compared to comorbid cases; proof of concept in the ADNI study. Mol. Psychiatry 2025, 30(2), 587–599. [Google Scholar] [CrossRef]
- Islam, S.; Noorani, A.; Sun, Y.; Michikawa, M.; Zou, K. Multi-functional role of apolipoprotein E in neurodegenerative diseases. Front. Aging Neurosci. 2025, 17, 1535280. [Google Scholar] [CrossRef]
- https. [CrossRef]
- Ismail, R.; Parbo, P.; Madsen, L. S.; Hansen, A. K.; Hansen, K. V.; Schaldemose, J. L.; Kjeldsen, P. L.; Stokholm, M. G.; Gottrup, H.; Eskildsen, S. F.; Brooks, D. J. The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer’s disease: a longitudinal PET study. J. Neuroinflammation 2020, 17(1), 151. [Google Scholar] [CrossRef]
- Jamerlan, A.; Hulme, J. From Evasion to Collapse: The Kinetic Cascade of TDP-43 and the Failure of Proteostasis. Int. J. Mol. Sci. 2026, 27(3), 1136. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J. Å. Estrogen receptor alpha and beta in health and disease. Best practice & research. Clin. Endocrinol. Metab. 2015, 29(4), 557–568. [Google Scholar] [CrossRef]
- Jiang, Q.; Lee, C. Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T. M.; Collins, J. L.; Richardson, J. C.; Smith, J. D.; Comery, T. A.; Riddell, D.; Holtzman, D. M.; Tontonoz, P.; Landreth, G. E. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58(5), 681–693. [Google Scholar] [CrossRef] [PubMed]
- Jones, G. H.; Vecera, C. M.; Pinjari, O. F.; Machado-Vieira, R. Inflammatory signaling mechanisms in bipolar disorder. J. Biomed. Sci. 2021, 28(1), 45. [Google Scholar] [CrossRef]
- Jung, E. S.; Choi, H.; Mook-Jung, I. Decoding microglial immunometabolism: a new frontier in Alzheimer’s disease research. Mol. Neurodegener. 2025, 20(1), 37. [Google Scholar] [CrossRef]
- Kalenga, C. Z.; Hay, J. L.; Boreskie, K. F.; Duhamel, T. A.; MacRae, J. M.; Metcalfe, A.; Nerenberg, K. A.; Robert, M.; Ahmed, S. B. The Association Between Route of Post-menopausal Estrogen Administration and Blood Pressure and Arterial Stiffness in Community-Dwelling Women. Front. Cardiovasc. Med. 2022, 9, 913609. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Cirrito, J. R.; Liu, C. C.; Shinohara, M.; Li, J.; Schuler, D. R.; Shinohara, M.; Holtzman, D. M.; Bu, G. Neuronal clearance of amyloid-β by endocytic receptor LRP1. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33(49), 19276–19283. [Google Scholar] [CrossRef] [PubMed]
- Kang, J. H.; Grodstein, F. Postmenopausal hormone therapy, timing of initiation, APOE and cognitive decline. Neurobiol. Aging 2012, 33(7), 1129–1137. [Google Scholar] [CrossRef]
- Kang, J. H.; Weuve, J.; Grodstein, F. Postmenopausal hormone therapy and risk of cognitive decline in community-dwelling aging women. Neurology 2004, 63(1), 101–107. [Google Scholar] [CrossRef] [PubMed]
- Kantarci, K.; Lowe, V. J.; Lesnick, T. G.; Tosakulwong, N.; Bailey, K. R.; Fields, J. A.; Shuster, L. T.; Zuk, S. M.; Senjem, M. L.; Mielke, M. M.; Gleason, C.; Jack, C. R.; Rocca, W. A.; Miller, V. M. Early Postmenopausal Transdermal 17β-Estradiol Therapy and Amyloid-β Deposition. J. Alzheimer’s Dis. JAD 2016, 53(2), 547–556. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A. M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19(6), 365–381. [Google Scholar] [CrossRef]
- Kermath, B. A.; Riha, P. D.; Woller, M. J.; Wolfe, A.; Gore, A. C. Hypothalamic molecular changes underlying natural reproductive senescence in the female rat. Endocrinology 2014, 155(9), 3597–3609. [Google Scholar] [CrossRef]
- Kerr, J. S.; Adriaanse, B. A.; Greig, N. H.; Mattson, M. P.; Cader, M. Z.; Bohr, V. A.; Fang, E. F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40(3), 151–166. [Google Scholar] [CrossRef]
- Kim, S.; Jung, U. J.; Kim, S. R. The Crucial Role of the Blood-Brain Barrier in Neurodegenerative Diseases: Mechanisms of Disruption and Therapeutic Implications. J. Clin. Med. 2025, 14(2), 386. [Google Scholar] [CrossRef]
- Kim, S. M.; Sultana, F.; Sims, S.; Gimenez-Roig, J.; Laurencin, V.; Pallapati, A.; Rojekar, S.; Frolinger, T.; Zhou, W.; Gumerova, A.; Macdonald, A.; Ryu, V.; Lizneva, D.; Korkmaz, F.; Yuen, T.; Zaidi, M. FSH, bone, belly and brain. J. Endocrinol. 2024, 262(1), e230377. [Google Scholar] [CrossRef]
- Kim, Y. J.; Soto, M.; Branigan, G. L.; Rodgers, K.; Brinton, R. D. Association between menopausal hormone therapy and risk of neurodegenerative diseases: Implications for precision hormone therapy. Alzheimer’s Dement. 2021, 7(1), e12174. [Google Scholar] [CrossRef] [PubMed]
- Knox, E. G.; Aburto, M. R.; Clarke, G.; Cryan, J. F.; O’Driscoll, C. M. The blood-brain barrier in aging and neurodegeneration. Mol. Psychiatry 2022, 27(6), 2659–2673. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, W. C.; Lyoo, C. H.; McGwier, M.; Snow, J.; Jenko, K. J.; Kimura, N.; Corona, W.; Morse, C. L.; Zoghbi, S. S.; Pike, V. W.; McMahon, F. J.; Turner, R. S.; Innis, R. B.; Biomarkers Consortium PET Radioligand Project Team. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain A J. Neurol. 2013, 136 Pt 7, 2228–2238. [Google Scholar] [CrossRef]
- Kim, S. M.; Sultana, F.; Sims, S.; Gimenez-Roig, J.; Laurencin, V.; Pallapati, A.; Rojekar, S.; Frolinger, T.; Zhou, W.; Gumerova, A.; Macdonald, A.; Ryu, V.; Lizneva, D.; Korkmaz, F.; Yuen, T.; Zaidi, M. FSH, bone, belly and brain. J. Endocrinol. 2024, 262(1), e230377. [Google Scholar] [CrossRef] [PubMed]
- Krishnadas, N.; Doré, V.; Groot, C.; Lamb, F.; Bourgeat, P.; Burnham, S. C.; Huang, K.; Goh, A. M. Y.; Masters, C. L.; Villemagne, V. L.; Rowe, C. C.; AIBL research group. Mesial temporal tau in amyloid-β-negative cognitively normal older persons. Alzheimer’s Res. Ther. 2022, 14(1), 51. [Google Scholar] [CrossRef]
- Kockx, M.; Traini, M.; Kritharides, L. Cell-specific production, secretion, and function of apolipoprotein E. J. Mol. Med. 2018, 96(5), 361–371. [Google Scholar] [CrossRef]
- Kolnes, A. J.; Øystese, K. A. B.; Olarescu, N. C.; Ringstad, G.; Berg-Johnsen, J.; Casar-Borota, O.; Bollerslev, J.; Jørgensen, A. P. FSH Levels Are Related to E-cadherin Expression and Subcellular Location in Nonfunctioning Pituitary Tumors. J. Clin. Endocrinol. Metab. 2020, 105(8), 2587–2594. [Google Scholar] [CrossRef]
- Kontaxi, C.; Piccardo, P.; Gill, A. C. Lysine-Directed Post-translational Modifications of Tau Protein in Alzheimer’s Disease and Related Tauopathies. Front. Mol. Biosci. 2017, 4, 56. [Google Scholar] [CrossRef]
- Koutsodendris, N.; Nelson, M. R.; Rao, A.; Huang, Y. Apolipoprotein E and Alzheimer’s disease: Findings, hypotheses, and potential mechanisms. Annu. Rev. Pathol. Mech. Dis. 17 2022, 73–99. [Google Scholar] [CrossRef]
- Korkmaz, F.; Sims, S.; Sen, F.; Sultana, F.; Laurencin, V.; Cullen, L.; Pallapati, A.; Liu, A.; Chen, R.; Rojekar, S.; Pevnev, G.; Cheliadinova, U.; Vasilyeva, D.; Burganova, G.; Macdonald, A.; Saxena, M.; Goosens, K.; Rosen, C. J.; Barak, O.; Lizneva, D.; Zaidi, M. Gene-dose-dependent reduction of Fshr expression improves spatial memory deficits in Alzheimer’s mice. Mol. Psychiatry 2025, 30(5), 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Laakkonen, E. K.; Karppinen, J. E.; Lehti, S.; Lee, E.; Pesonen, E.; Juppi, H. K.; Kujala, U. M.; Haapala, E. A.; Aukee, P.; Laukkanen, J. A.; Ihalainen, J. K. Associations of Sex Hormones and Hormonal Status With Arterial Stiffness in a Female Sample From Reproductive Years to Menopause. Front. Endocrinol. 2021, 12, 765916. [Google Scholar] [CrossRef] [PubMed]
- La Joie, R.; Visani, A. V.; Lesman-Segev, O. H.; Baker, S. L.; Edwards, L.; Iaccarino, L.; Soleimani-Meigooni, D. N.; Mellinger, T.; Janabi, M.; Miller, Z. A.; Perry, D. C.; Pham, J.; Strom, A.; Gorno-Tempini, M. L.; Rosen, H. J.; Miller, B. L.; Jagust, W. J.; Rabinovici, G. D. Association of APOE4 and Clinical Variability in Alzheimer Disease With the Pattern of Tau- and Amyloid-PET. Neurology 2021, 96(5), e650–e661. [Google Scholar] [CrossRef]
- Lamontagne-Kam, D.; Ulfat, A. K.; Hervé, V.; Vu, T. M.; Brouillette, J. Implication of tau propagation on neurodegeneration in Alzheimer’s disease. Front. Neurosci. 2023, 17, 1219299. [Google Scholar] [CrossRef]
- Laughlin, G. A.; Kritz-Silverstein, D.; Barrett-Connor, E. Endogenous oestrogens predict 4-year decline in verbal fluency in postmenopausal women: the Rancho Bernardo Study. Clin. Endocrinol. 2010, 72(1), 99–106. [Google Scholar] [CrossRef]
- Lee, J. H.; Yang, D. S.; Goulbourne, C. N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M. J.; Huo, C.; Peddy, J.; Pawlik, M.; Levy, E.; Rao, M.; Staufenbiel, M.; Nixon, R. A. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat. Neurosci. 2022, 25(6), 688–701. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Xie, Z.; Wang, M.; Nie, S.; Qian, Z.; Meng, X.; Liu, X.; Kang, S. S.; Ye, K. Neuronal C/EBPβ Shortens the Lifespan via Inactivating NAMPT. Adv. Sci. 2025, 12(21), e2414871. [Google Scholar] [CrossRef]
- 124; Li, C.; Ling, Y.; Kuang, H. Research progress on FSH-FSHR signaling in the pathogenesis of non-reproductive diseases. Front. Cell Dev. Biol. 2024, 12, 1506450. [Google Scholar] [CrossRef]
- Li, R.; Wang, X.; He, P. The most prevalent rare coding variants of TREM2 conferring risk of Alzheimer’s disease: A systematic review and meta-analysis. Exp. Ther. Med. 2021, 21(4), 347. [Google Scholar] [CrossRef]
- Li, J. W.; Zong, Y.; Cao, X. P.; Tan, L.; Tan, L. Microglial priming in Alzheimer’s disease. Ann. Transl. Med. 2018, 6(10), 176. [Google Scholar] [CrossRef]
- Li, X.; Fang, C.; Li, Y.; Xiong, X.; Xu, X.; Gu, L. Glycolytic reprogramming during microglial polarization in neurological diseases. Front Immunol. 2025, 16, 1648887. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lin, Y.; Im, H.; Diem, L. T.; Ham, S. Characterizing the structural and thermodynamic properties of Aβ42 and Aβ40. Biochem. Biophys. Res. Commun. 2019, 510(3), 442–448. [Google Scholar] [CrossRef]
- Li, Y.; Xia, X.; Wang, Y.; Zheng, J. C. Mitochondrial dysfunction in microglia: a novel perspective for pathogenesis of Alzheimer’s disease. J. Neuroinflammation 2022, 19(1), 248. [Google Scholar] [CrossRef] [PubMed]
- Lizneva, D.; Rahimova, A.; Kim, S. M.; Atabiekov, I.; Javaid, S.; Alamoush, B.; Taneja, C.; Khan, A.; Sun, L.; Azziz, R.; Yuen, T.; Zaidi, M. FSH Beyond Fertility. Front. Endocrinol. 2019, 10, 136. [Google Scholar] [CrossRef]
- Lindner, K.; Gavin, A. C. Isoform- and cell-state-specific APOE homeostasis and function. Neural Regen. Res. 2024, 19(11), 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Lim, S. H. Y.; Hansen, M.; Kumsta, C. Molecular Mechanisms of Autophagy Decline during Aging. Cells 2024, 13(16), 1364. [Google Scholar] [CrossRef] [PubMed]
- Lish, A. M.; Grogan, E. F. L.; Benoit, C. R.; Pearse, R. V., 2nd; Heuer, S. E.; Luquez, T.; Orme, G. A.; Galle, P. C.; Milinkeviciute, G.; Green, K. N.; Alexander, K. D.; Fancher, S. B.; Stern, A. M.; Fujita, M.; Bennett, D. A.; Seyfried, N. T.; De Jager, P. L.; Menon, V.; Young-Pearse, T. L. CLU alleviates Alzheimer’s disease-relevant processes by modulating astrocyte reactivity and microglia-dependent synaptic density. Neuron 2025, 113(12), 1925–1946.e11. [Google Scholar] [CrossRef]
- Liu, C. C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C. W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96(5), 1024–1032.e3. [Google Scholar] [CrossRef]
- Liu, C.; Liu, J.; Wang, Y. Y.; Xu, S. F.; Yu, L. M. APOE Lipoprotein Particles: Pathophysiology, Therapy, and the Crosstalk in Alzheimer’s Disease and Cardiovascular Disease. Mol. Neurobiol. 2025, 63(1), 325. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, X.; Chen, J.; Win, Y. Y.; Cai, J. Unnatural foldamers as inhibitors of Aβ aggregation via stabilizing the Aβ helix. Chem. Commun. 2025, 61(24), 4586–4594. [Google Scholar] [CrossRef]
- Liu, L.; Hao, M.; Zhang, J.; Chen, Z.; Zhou, J.; Wang, C.; Zhang, H.; Wang, J. FSHR-mTOR-HIF1 signaling alleviates mouse follicles from AMPK-induced atresia. Cell Rep. 2023, 42(10), 113158. [Google Scholar] [CrossRef]
- Liu, P. P.; Xie, Y.; Meng, X. Y.; Kang, J. S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Livingston, G.; Huntley, J.; Liu, K. Y.; Costafreda, S. G.; Selbæk, G.; Alladi, S.; Ames, D.; Banerjee, S.; Burns, A.; Brayne, C.; Fox, N. C.; Ferri, C. P.; Gitlin, L. N.; Howard, R.; Kales, H. C.; Kivimäki, M.; Larson, E. B.; Nakasujja, N.; Rockwood, K.; Samus, Q.; Mukadam, N. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet 2024, 404(10452), 572–628. [Google Scholar] [CrossRef]
- Lo, C. H.; Sachs, J. N. The role of wild-type tau in Alzheimer’s disease and related tauopathies. J. Life Sci. 2020, 2(4), 1–17. [Google Scholar] [CrossRef]
- Luo, H. B.; Xia, Y. Y.; Shu, X. J.; Liu, Z. C.; Feng, Y.; Liu, X. H.; Yu, G.; Yin, G.; Xiong, Y. S.; Zeng, K.; Jiang, J.; Ye, K.; Wang, X. C.; Wang, J. Z. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proceedings of the National Academy of Sciences of the United States of America 2014, 111(46), 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Liang, J.; Lei, X.; Sun, F.; Gong, M.; Liu, B.; Zhou, Z. C/EBPβ in Alzheimer’s disease: An integrative regulator of pathological mechanisms. Brain Res. Bull. 2025, 221, 111198. [Google Scholar] [CrossRef]
- Lopez-Lee, C.; Torres, E. R. S.; Carling, G.; Gan, L. Mechanisms of sex differences in Alzheimer’s disease. Neuron 2024, 112(8), 1208–1221. [Google Scholar] [CrossRef]
- Lutsenko, S.; Roy, S.; Tsvetkov, P. Mammalian copper homeostasis: physiological roles and molecular mechanisms. Physiol. Rev. 2025, 105(1), 441–491. [Google Scholar] [CrossRef]
- Mahley, R. W.; Weisgraber, K. H.; Huang, Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, 50 Suppl(Suppl), S183–S188. [Google Scholar] [CrossRef]
- Ma, J.; Liu, C.; Yang, Y.; Yu, J.; Yang, J.; Yu, S.; Zhang, J.; Huang, L. C/EBPβ Acts Upstream of NF-κB P65 Subunit in Ox-LDL-Induced IL-1β Production by Macrophages. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology 2018, 48(4), 1605–1615. [Google Scholar] [CrossRef]
- Mak, E.; Bethlehem, R. A. I.; Romero-Garcia, R.; Cervenka, S.; Rittman, T.; Gabel, S.; Surendranathan, A.; Bevan-Jones, R. W.; Passamonti, L.; Vázquez Rodríguez, P.; Su, L.; Arnold, R.; Williams, G. B.; Hong, Y. T.; Fryer, T. D.; Aigbirhio, F. I.; Rowe, J. B.; O’Brien, J. T. In vivo coupling of tau pathology and cortical thinning in Alzheimer’s disease. Alzheimer’s Dement. 2018, 10, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Maki, P. M.; Drogos, L. L.; Rubin, L. H.; Banuvar, S.; Shulman, L. P.; Geller, S. E. Objective hot flashes are negatively related to verbal memory performance in midlife women. Menopause 2008, 15(5), 848–856. [Google Scholar] [CrossRef]
- Malpetti, M.; Joie, R.; Rabinovici, G. D. Tau Beats Amyloid in Predicting Brain Atrophy in Alzheimer Disease: Implications for Prognosis and Clinical Trials. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2022, 63(6), 830–832. [Google Scholar] [CrossRef]
- Mathiisen, T. M.; Lehre, K. P.; Danbolt, N. C.; Ottersen, O. P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 2010, 58(9), 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M. P. Involvement of GABAergic interneuron dysfunction and neuronal network hyperexcitability in Alzheimer’s disease: Amelioration by metabolic switching. Int. Rev. Neurobiol. 2020, 154, 191–205. [Google Scholar] [CrossRef]
- Melville, M.; He, L.; Desai, R.; Nyamayaro, P.; Fox, C.; Kothari, K. U.; Condron, P.; Miao, M.; Hickey, M.; Spector, A. Menopause hormone therapy and risk of mild cognitive impairment or dementia: a systematic review and meta-analysis. Lancet. Healthy Longev. 2025, 6(12), 100803. [Google Scholar] [CrossRef] [PubMed]
- McClure, C.; McPeak, M. B.; Youssef, D.; Yao, Z. Q.; McCall, C. E.; El Gazzar, M. Stat3 and C/EBPβ synergize to induce miR-21 and miR-181b expression during sepsis. Immunol. Cell Biol. 2017, 95(1), 42–55. [Google Scholar] [CrossRef]
- McCoy, N. D.; Gawrys, S. P.; Mackintosh, S. G.; et al. Ovarian somatic tissue rejuvenates circulating apolipoproteins and promotes cognitive health in postreproductive female mice. In GeroScience; 2025. [Google Scholar] [CrossRef]
- Milkovic, L.; Zarkovic, N.; Marusic, Z.; Zarkovic, K.; Jaganjac, M. The 4-Hydroxynonenal-Protein Adducts and Their Biological Relevance: Are Some Proteins Preferred Targets? Antioxidants 2023, 12(4), 856. [Google Scholar] [CrossRef] [PubMed]
- Milner, T. A.; Ayoola, K.; Drake, C. T.; Herrick, S. P.; Tabori, N. E.; McEwen, B. S.; Warrier, S.; Alves, S. E. Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J. Comp. Neurol. 2005, 491(2), 81–95. [Google Scholar] [CrossRef] [PubMed]
- MohanKumar, S. M. J.; Balasubramanian, P.; Subramanian, M.; MohanKumar, P. S. Chronic estradiol exposure - harmful effects on behavior, cardiovascular and reproductive functions. Reproduction 2018, 156(5), R169–R186. [Google Scholar] [CrossRef]
- Monterey, M. D.; Wei, H.; Wu, X.; Wu, J. Q. The Many Faces of Astrocytes in Alzheimer’s Disease. Front. Neurol. 2021, 12, 619626. [Google Scholar] [CrossRef]
- Monsorno, K.; Ginggen, K.; Ivanov, A.; Buckinx, A.; Lalive, A. L.; Tchenio, A.; Benson, S.; Vendrell, M.; D’Alessandro, A.; Beule, D.; Pellerin, L.; Mameli, M.; Paolicelli, R. C. Loss of microglial MCT4 leads to defective synaptic pruning and anxiety-like behavior in mice. Nat. Commun. 2023, 14(1), 5749. [Google Scholar] [CrossRef]
- Morris, J. C.; Roe, C. M.; Xiong, C.; Fagan, A. M.; Goate, A. M.; Holtzman, D. M.; Mintun, M. A. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 2010, 67(1), 122–131. [Google Scholar] [CrossRef]
- Mosconi, L.; Andy, C.; Nerattini, M.; et al. Systematic Review and Meta-analysis of Menopause Hormone Therapy (MHT) and the Risk of Alzheimer’s Disease and All-cause Dementia: Effects of MHT Characteristics, Location, and APOE-4 Status. [CrossRef]
- Curr. Obstet. Gynecol. Rep. 2025, 14, 6. [CrossRef]
- Mosconi, L.; Rahman, A.; Diaz, I.; Wu, X.; Scheyer, O.; Hristov, H. W.; Vallabhajosula, S.; Isaacson, R. S.; de Leon, M. J.; Brinton, R. D. Increased Alzheimer’s risk during the menopause transition: A 3-year longitudinal brain imaging study. PLoS ONE 2018, 13(12), e0207885. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, Y. A. Reproductive aging in biological females: mechanisms and immediate consequences. Front. Endocrinol. 2025, 16, 1658592. [Google Scholar] [CrossRef] [PubMed]
- Naia, L.; Shimozawa, M.; Bereczki, E.; Li, X.; Liu, J.; Jiang, R.; Giraud, R.; Leal, N. S.; Pinho, C. M.; Berger, E.; Falk, V. L.; Dentoni, G.; Ankarcrona, M.; Nilsson, P. Mitochondrial hypermetabolism precedes impaired autophagy and synaptic disorganization in App knock-in Alzheimer mouse models. Mol. Psychiatry 2023, 28(9), 3966–3981. [Google Scholar] [CrossRef]
- Nerattini, M.; Jett, S.; Andy, C.; Carlton, C.; Zarate, C.; Boneu, C.; Battista, M.; Pahlajani, S.; Loeb-Zeitlin, S.; Havryulik, Y.; Williams, S.; Christos, P.; Fink, M.; Brinton, R. D.; Mosconi, L. Systematic review and meta-analysis of the effects of menopause hormone therapy on risk of Alzheimer’s disease and dementia. Front. Aging Neurosci. 2023, 15, 1260427. [Google Scholar] [CrossRef] [PubMed]
- Neth, B. J.; Mintz, A.; Whitlow, C.; Jung, Y.; Solingapuram Sai, K.; Register, T. C.; Kellar, D.; Lockhart, S. N.; Hoscheidt, S.; Maldjian, J.; Heslegrave, A. J.; Blennow, K.; Cunnane, S. C.; Castellano, C. A.; Zetterberg, H.; Craft, S. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: a pilot study. Neurobiol. Aging 2020, 86, 54–63. [Google Scholar] [CrossRef]
- Nixon, R. A.; Wegiel, J.; Kumar, A.; Yu, W. H.; Peterhoff, C.; Cataldo, A.; Cuervo, A. M. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64(2), 113–122. [Google Scholar] [CrossRef]
- Nixon, R. A.; Yang, D. S. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol. Dis. 2011, 43(1), 38–45. [Google Scholar] [CrossRef]
- Noble, W.; Hanger, D. P.; Miller, C. C.; Lovestone, S. The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 2013, 4, 83. [Google Scholar] [CrossRef]
- Nordengen, K.; Kirsebom, B. E.; Henjum, K.; Selnes, P.; Gísladóttir, B.; Wettergreen, M.; Torsetnes, S. B.; Grøntvedt, G. R.; Waterloo, K. K.; Aarsland, D.; Nilsson, L. N. G.; Fladby, T. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflammation 2019, 16(1), 46. [Google Scholar] [CrossRef]
- Nurmakova, K.; Levine, Z. Apolipoprotein E lipidation state modulates amyloid beta interaction and subsequent uptake by human astrocytes [Conference abstract]. Biophys. J. 2026, 125139a. [Google Scholar] [CrossRef]
- Oliveira, T. P. D.; Morais, A. L. B.; Dos Reis, P. L. B.; Palotás, A.; Vieira, L. B. A Potential Role for the Ketogenic Diet in Alzheimer’s Disease Treatment: Exploring Pre-Clinical and Clinical Evidence. Metabolites 2023, 14(1), 25. [Google Scholar] [CrossRef]
- Okello, A.; Edison, P.; Archer, H. A.; Turkheimer, F. E.; Kennedy, J.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.; Rossor, M.; Brooks, D. J. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology 2009, 72(1), 56–62. [Google Scholar] [CrossRef]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. BioEssays News Rev. Mol. Cell. Dev. Biol. 2018, 40(6), e1800008. [Google Scholar] [CrossRef] [PubMed]
- Oriá, R. B.; de Almeida, J. Z.; Moreira, C. N.; Guerrant, R. L.; Figueiredo, J. R. Apolipoprotein E Effects on Mammalian Ovarian Steroidogenesis and Human Fertility. Trends Endocrinol. Metab. TEM 2020, 31(11), 872–883. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Lyoo, C. H.; Sudre, C. H.; van Westen, D.; Cho, H.; Ryu, Y. H.; Choi, J. Y.; Smith, R.; Strandberg, O.; Palmqvist, S.; Westman, E.; Tsai, R.; Kramer, J.; Boxer, A. L.; Gorno-Tempini, M. L.; La Joie, R.; Miller, B. L.; Rabinovici, G. D.; Hansson, O. Distinct tau PET patterns in atrophy-defined subtypes of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2020, 16(2), 335–344. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Smith, R.; Mattsson-Carlgren, N.; Groot, C.; Leuzy, A.; Strandberg, O.; Palmqvist, S.; Olsson, T.; Jögi, J.; Stormrud, E.; Cho, H.; Ryu, Y. H.; Choi, J. Y.; Boxer, A. L.; Gorno-Tempini, M. L.; Miller, B. L.; Soleimani-Meigooni, D.; Iaccarino, L.; La Joie, R.; Baker, S.; Hansson, O. Accuracy of Tau Positron Emission Tomography as a Prognostic Marker in Preclinical and Prodromal Alzheimer Disease: A Head-to-Head Comparison Against Amyloid Positron Emission Tomography and Magnetic Resonance Imaging. JAMA Neurol. 2021, 78(8), 961–971. [Google Scholar] [CrossRef]
- Parbo, P.; Ismail, R.; Hansen, K. V.; Amidi, A.; Mårup, F. H.; Gottrup, H.; Brændgaard, H.; Eriksson, B. O.; Eskildsen, S. F.; Lund, T. E.; Tietze, A.; Edison, P.; Pavese, N.; Stokholm, M. G.; Borghammer, P.; Hinz, R.; Aanerud, J.; Brooks, D. J. Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer’s disease. Brain A J. Neurol. 2017, 140(7), 2002–2011. [Google Scholar] [CrossRef]
- Parcon, P. A.; Balasubramaniam, M.; Ayyadevara, S.; Jones, R. A.; Liu, L.; Shmookler Reis, R. J.; Barger, S. W.; Mrak, R. E.; Griffin, W. S. T. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14(2), 230–242. [Google Scholar] [CrossRef]
- Pallapati, A. R.; Korkmaz, F.; Rojekar, S.; Sims, S.; Misra, A.; Gimenez-Roig, J.; Gangadhar, A.; Laurencin, V.; Gumerova, A.; Cheliadinova, U.; Sultana, F.; Vasilyeva, D.; Cullen, L.; Schuermann, J.; Munitz, J.; Kannangara, H.; Parte, S.; Pevnev, G.; Burganova, G.; Tumoglu, Z.; Zaidi, M. Efficacy and safety of a therapeutic humanized FSH-blocking antibody in obesity and Alzheimer’s disease models. J. Clin. Investig. 2025, 135(17), e182702. [Google Scholar] [CrossRef]
- Palmer, J. E.; Wilson, N.; Son, S. M.; Obrocki, P.; Wrobel, L.; Rob, M.; Takla, M.; Korolchuk, V. I.; Rubinsztein, D. C. Autophagy, aging, and age-related neurodegeneration. Neuron 2025, 113(1), 29–48. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Jeon, W. K.; Bizon, J. L.; Han, J. S. Interaction of basal forebrain cholinergic neurons with the glucocorticoid system in stress regulation and cognitive impairment. Front. Aging Neurosci. 2015, 7, 43. [Google Scholar] [CrossRef]
- Paul, S. M.; Yohn, S. E.; Popiolek, M.; Miller, A. C.; Felder, C. C. Muscarinic Acetylcholine Receptor Agonists as Novel Treatments for Schizophrenia. Am. J. Psychiatry 2022, 179(9), 611–627. [Google Scholar] [CrossRef]
- Pearson, H. A.; Peers, C. Physiological roles for amyloid beta peptides. J. Physiol. 2006, 575 Pt 1, 5–10. [Google Scholar] [CrossRef]
- Phillips, M. C. L.; Deprez, L. M.; Mortimer, G. M. N.; Murtagh, D. K. J.; McCoy, S.; Mylchreest, R.; Gilbertson, L. J.; Clark, K. M.; Simpson, P. V.; McManus, E. J.; Oh, J. E.; Yadavaraj, S.; King, V. M.; Pillai, A.; Romero-Ferrando, B.; Brinkhuis, M.; Copeland, B. M.; Samad, S.; Liao, S.; Schepel, J. A. C. Randomized crossover trial of a modified ketogenic diet in Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13(1), 51. [Google Scholar] [CrossRef]
- Piccarducci, R.; Giacomelli, C.; Bertilacchi, M. S.; Benito-Martinez, A.; Di Giorgi, N.; Daniele, S.; Signore, G.; Rocchiccioli, S.; Vilar, M.; Marchetti, L.; Martini, C. Apolipoprotein E ε4 triggers neurotoxicity via cholesterol accumulation, acetylcholine dyshomeostasis, and PKCε mislocalization in cholinergic neuronal cells. Biochim. Et. Biophys. Acta. Mol. Basis Dis. 2023, 1869(7), 166793. [Google Scholar] [CrossRef]
- Pires, M.; Rego, A. C. Apoe4 and Alzheimer’s Disease Pathogenesis-Mitochondrial Deregulation and Targeted Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24(1), 778. [Google Scholar] [CrossRef]
- Planche, V.; Manjon, J. V.; Mansencal, B.; Lanuza, E.; Tourdias, T.; Catheline, G.; Coupé, P. Structural progression of Alzheimer’s disease over decades: the MRI staging scheme. Brain Commun. 2022, 4(3), fcac109. [Google Scholar] [CrossRef] [PubMed]
- Pletnikova, O.; Rudow, G. L.; Hyde, T. M.; Kleinman, J. E.; Ali, S. Z.; Bharadwaj, R.; Gangadeen, S.; Crain, B. J.; Fowler, D. R.; Rubio, A. I.; Troncoso, J. C. Alzheimer Lesions in the Autopsied Brains of People 30 to 50 Years of Age. Cognitive and behavioral neurology: official journal of the Society for Behavioral and Cognitive Neurology 2015, 28(3), 144–152. [Google Scholar] [CrossRef]
- Pourhadi, N.; Mørch, L. S.; Holm, E. A.; Torp-Pedersen, C.; Meaidi, A. Menopausal hormone therapy and dementia: nationwide, nested case-control study. BMJ (Clinical research ed.) 2023, 381, e072770. [Google Scholar] [CrossRef] [PubMed]
- Price, M. S.; Rastegari, E.; Gupta, R.; Vo, K.; Moore, T. I.; Venkatachalam, K. Intracellular lactate dynamics in Drosophila neurons. iScience 2025, 28(10), 113462. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R. D.; Yin, F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021, 34(1), 108572. [Google Scholar] [CrossRef] [PubMed]
- Quick, J. D.; Silva, C.; Wong, J. H.; Lim, K. L.; Reynolds, R.; Barron, A. M.; Zeng, J.; Lo, C. H. Lysosomal acidification dysfunction in microglia: an emerging pathogenic mechanism of neuroinflammation and neurodegeneration. J. Neuroinflammation 2023, 20(1), 185. [Google Scholar] [CrossRef]
- Rahman, A.; Jackson, H.; Hristov, H.; Isaacson, R. S.; Saif, N.; Shetty, T.; Etingin, O.; Henchcliffe, C.; Brinton, R. D.; Mosconi, L. Sex and Gender Driven Modifiers of Alzheimer’s: The Role for Estrogenic Control Across Age, Race, Medical, and Lifestyle Risks. Front. Aging Neurosci. 2019, 11, 315. [Google Scholar] [CrossRef]
- Rauchmann, B. S.; Brendel, M.; Franzmeier, N.; Trappmann, L.; Zaganjori, M.; Ersoezlue, E.; Morenas-Rodriguez, E.; Guersel, S.; Burow, L.; Kurz, C.; Haeckert, J.; Tatò, M.; Utecht, J.; Papazov, B.; Pogarell, O.; Janowitz, D.; Buerger, K.; Ewers, M.; Palleis, C.; Weidinger, E.; Perneczky, R. Microglial Activation and Connectivity in Alzheimer Disease and Aging. Ann. Neurol. 2022, 92(5), 768–781. [Google Scholar] [CrossRef]
- Reddy, K.; Cusack, C. L.; Nnah, I. C.; Khayati, K.; Saqcena, C.; Huynh, T. B.; Noggle, S. A.; Ballabio, A.; Dobrowolski, R. Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 2016, 14(9), 2166–2179. [Google Scholar] [CrossRef]
- Riedel, B. C.; Thompson, P. M.; Brinton, R. D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef]
- Riku, Y.; Brion, J. P.; Ando, K.; Uchihara, T.; Iwasaki, Y. The Determinant of Tau Spreading in Alzheimer’s Disease: Dependent on Senile Plaque, Neural Circuits, or Spatial Proximity? Int. J. Mol. Sci. 2025, 26(24), 12088. [Google Scholar] [CrossRef]
- Robbins, M.; Clayton, E.; Kaminski Schierle, G. S. Synaptic tau: A pathological or physiological phenomenon? Acta Neuropathol. Commun. 2021, 9(1), 149. [Google Scholar] [CrossRef] [PubMed]
- Rocca, M. S.; Pannella, M.; Bayraktar, E.; Marino, S.; Bortolozzi, M.; Di Nisio, A.; Foresta, C.; Ferlin, A. Extragonadal function of follicle-stimulating hormone: Evidence for a role in endothelial physiology and dysfunction. Mol. Cell. Endocrinol. 2024, 594, 112378. [Google Scholar] [CrossRef]
- Rocca, W. A.; Kantarci, K.; Faubion, S. S. Risks and benefits of hormone therapy after menopause for cognitive decline and dementia: A conceptual review. Maturitas 2024, 184, 108003. [Google Scholar] [CrossRef] [PubMed]
- Roda, A. R.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17(8), 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Roe, J. M.; Vidal-Piñeiro, D.; Sørensen, Ø.; Grydeland, H.; Leonardsen, E. H.; Iakunchykova, O.; Pan, M.; Mowinckel, A.; Strømstad, M.; Nawijn, L.; Milaneschi, Y.; Andersson, M.; Pudas, S.; Bråthen, A. C. S.; Kransberg, J.; Falch, E. S.; Øverbye, K.; Kievit, R. A.; Ebmeier, K. P.; Lindenberger, U.; Wang, Y. Brain change trajectories in healthy adults correlate with Alzheimer’s related genetic variation and memory decline across life. Nat. Commun. 2024, 15(1), 10651. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, E. J.; Dengel, D. R.; Wang, Q.; Hodges, J. S.; Steinberger, J.; Baker, K. S. The Role of Follicle-stimulating Hormone in Vascular Dysfunction Observed in Hematopoietic Cell Transplant Recipients. J. Pediatr. Hematol. 2022, 44(3), e695–e700. [Google Scholar] [CrossRef]
- Rosu, G. C.; Catalin, B.; Balseanu, T. A.; Laurentiu, M.; Claudiu, M.; Kumar-Singh, S.; Daniel, P. Inhibition of Aquaporin 4 Decreases Amyloid Aβ40 Drainage Around Cerebral Vessels. Mol. Neurobiol. 2020, 57(11), 4720–4734. [Google Scholar] [CrossRef]
- Ruiz, J.; Kouiavskaia, D.; Migliorini, M.; Robinson, S.; Saenko, E. L.; Gorlatova, N.; Li, D.; Lawrence, D.; Hyman, B. T.; Weisgraber, K. H.; Strickland, D. K. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J. Lipid Res. 2005, 46(8), 1721–1731. [Google Scholar] [CrossRef]
- Rutherford, S.; Fraza, C.; Dinga, R.; Kia, S. M.; Wolfers, T.; Zabihi, M.; Berthet, P.; Worker, A.; Verdi, S.; Andrews, D.; Han, L. K.; Bayer, J. M.; Dazzan, P.; McGuire, P.; Mocking, R. T.; Schene, A.; Sripada, C.; Tso, I. F.; Duval, E. R.; Chang, S. E.; Marquand, A. F. Charting brain growth and aging at high spatial precision. eLife 2022, 11, e72904. [Google Scholar] [CrossRef]
- Saleh, R. N. M.; Hornberger, M.; Ritchie, C. W.; Minihane, A. M. Hormone replacement therapy is associated with improved cognition and larger brain volumes in at-risk APOE4 women: results from the European Prevention of Alzheimer’s Disease (EPAD) cohort. Alzheimer’s Res. Ther. 2023, 15(1), 10. [Google Scholar] [CrossRef]
- Scheyer, O.; Rahman, A.; Hristov, H.; Berkowitz, C.; Isaacson, R. S.; Diaz Brinton, R.; Mosconi, L. Female Sex and Alzheimer’s Risk: The Menopause Connection. J. Prev. Alzheimer’s Dis. 2018, 5(4), 225–230. [Google Scholar] [CrossRef]
- Schmitz, T. W.; Mur, M.; Aghourian, M.; Bedard, M. A.; Spreng, R. N.; Alzheimer’s Disease Neuroimaging Initiative. Longitudinal Alzheimer’s Degeneration Reflects the Spatial Topography of Cholinergic Basal Forebrain Projections. Cell Rep. 2018, 24(1), 38–46. [Google Scholar] [CrossRef]
- Schwabe, M. R.; Fleischer, A. W.; Kuehn, R. K.; Chaudhury, S.; York, J. M.; Sem, D. S.; Donaldson, W. A.; LaDu, M. J.; Frick, K. M. The novel estrogen receptor beta agonist EGX358 and APOE genotype influence memory, vasomotor, and anxiety outcomes in an Alzheimer’s mouse model. Front. Aging Neurosci. 2024, 16, 1477045. [Google Scholar] [CrossRef]
- Sengupta, U.; Kayed, R. Amyloid β, Tau, and α-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog. Neurobiol. 2022, 214, 102270. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M. P.; Masliah, E.; Hyman, B. T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1(1), a006189. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Das, S.; Hyman, B. T. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet. Neurol. 2021, 20(1), 68–80. [Google Scholar] [CrossRef]
- Sierra-Magro, A.; Bartolome, F.; Lozano-Muñoz, D.; Alarcón-Gil, J.; Gine, E.; Sanz-SanCristobal, M.; Alonso-Gil, S.; Cortes-Canteli, M.; Carro, E.; Pérez-Castillo, A.; Morales-García, J. A. C/EBPβ Regulates TFAM Expression, Mitochondrial Function and Autophagy in Cellular Models of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24(2), 1459. [Google Scholar] [CrossRef]
- Shaerzadeh, F.; Motamedi, F.; Khodagholi, F. Inhibition of akt phosphorylation diminishes mitochondrial biogenesis regulators, tricarboxylic acid cycle activity and exacerbates recognition memory deficit in rat model of Alzheimer’s disease. Cell. Mol. Neurobiol. 2014, 34(8), 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S. B.; Nicholson, L. F. Effects of chronic low dose rotenone treatment on human microglial cells. Mol. Neurodegener. 2009, 4, 55. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Kelleher, R. J., 3rd. The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proceedings of the National Academy of Sciences of the United States of America 2007, 104(2), 403–409. [Google Scholar] [CrossRef] [PubMed]
- Shete, N.; Calabrese, J.; Tonetti, D. A. Revisiting Estrogen for the Treatment of Endocrine-Resistant Breast Cancer: Novel Therapeutic Approaches. Cancers 2023, 15(14), 3647. [Google Scholar] [CrossRef]
- Short, R. A.; Bowen, R. L.; O’Brien, P. C.; Graff-Radford, N. R. Elevated gonadotropin levels in patients with Alzheimer disease. Mayo Clin. Proc. 2001, 76(9), 906–909. [Google Scholar] [CrossRef]
- Shughrue, P. J.; Lane, M. V.; Scrimo, P. J.; Merchenthaler, I. Comparative distribution of estrogen receptor-alpha (ER-alpha) and beta (ER-beta) mRNA in the rat pituitary, gonad, and reproductive tract. Steroids 1998, 63(10), 498–504. [Google Scholar] [CrossRef]
- Shumaker, S. A.; Legault, C.; Rapp, S. R.; Thal, L.; Wallace, R. B.; Ockene, J. K.; Hendrix, S. L.; Jones, B. N., 3rd; Assaf, A. R.; Jackson, R. D.; Kotchen, J. M.; Wassertheil-Smoller, S.; Wactawski-Wende, J.; WHIMS Investigators. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003, 289(20), 2651–2662. [Google Scholar] [CrossRef]
- Shumaker, S. A.; Legault, C.; Kuller, L.; Rapp, S. R.; Thal, L.; Lane, D. S.; Fillit, H.; Stefanick, M. L.; Hendrix, S. L.; Lewis, C. E.; Masaki, K.; Coker, L. H.; Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004, 291(24), 2947–2958. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.; Silva, J.; Ferreira, R.; Trigo, D. Glymphatic system, AQP4, and their implications in Alzheimer’s disease. Neurol. Res. Pract. 2021, 3(1), 5. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Lieberburg, I. Cellular mechanisms of beta-amyloid production and secretion. Proceedings of the National Academy of Sciences of the United States of America 1999, 96(20), 11049–11053. [Google Scholar] [CrossRef]
- Sirois, J.; Richards, J. S. Transcriptional regulation of the rat prostaglandin endoperoxide synthase 2 gene in granulosa cells. Evidence for the role of a cis-acting C/EBP beta promoter element. J. Biol. Chem. 1993, 268(29), 21931–21938. [Google Scholar] [CrossRef]
- Singleton, E.; Hansson, O.; Pijnenburg, Y. A. L.; La Joie, R.; Mantyh, W. G.; Tideman, P.; Stomrud, E.; Leuzy, A.; Johansson, M.; Strandberg, O.; Smith, R.; Berendrecht, E.; Miller, B. L.; Iaccarino, L.; Edwards, L.; Strom, A.; Wolters, E. E.; Coomans, E.; Visser, D.; Golla, S. S. V.; Ossenkoppele, R. Heterogeneous distribution of tau pathology in the behavioural variant of Alzheimer’s disease. In Journal of neurology, neurosurgery, and psychiatry; Advance online publication, 2021; Volume 92, 8, pp. 872–880. [Google Scholar] [CrossRef]
- Sinsky, J.; Pichlerova, K.; Hanes, J. Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2021, 22(17), 9207. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B. S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54(10), 8071–8089. [Google Scholar] [CrossRef]
- Smitz, J. E.; Cortvrindt, R. G. The earliest stages of folliculogenesis in vitro. Reproduction 2002, 123(2), 185–202. [Google Scholar] [CrossRef]
- Sohn, H. Y.; Kim, S. I.; Park, J. Y.; Park, S. H.; Koh, Y. H.; Kim, J.; Jo, C. ApoE4 attenuates autophagy via FoxO3a repression in the brain. Sci. Rep. 2021, 11(1), 17604. [Google Scholar] [CrossRef] [PubMed]
- Spina, E.; Ferrari, R. R.; Pellegrini, E.; Colombo, M.; Poloni, T. E.; Guaita, A.; Davin, A. Mitochondrial Alterations, Oxidative Stress, and Therapeutic Implications in Alzheimer’s Disease: A Narrative Review. Cells 2025, 14(3), 229. [Google Scholar] [CrossRef]
- Stanciu, G. D.; Luca, A.; Rusu, R. N.; Bild, V.; Beschea Chiriac, S. I.; Solcan, C.; Bild, W.; Ababei, D. C. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules 2019, 10(1), 40. [Google Scholar] [CrossRef] [PubMed]
- Staurenghi, E.; Giannelli, S.; Testa, G.; Sottero, B.; Leonarduzzi, G.; Gamba, P. Cholesterol Dysmetabolism in Alzheimer’s Disease: A Starring Role for Astrocytes? Antioxidants 2021, 10(12), 1890. [Google Scholar] [CrossRef] [PubMed]
- Stefaniak, O.; Dobrzyńska, M.; Drzymała-Czyż, S.; Przysławski, J. Diet in the Prevention of Alzheimer’s Disease: Current Knowledge and Future Research Requirements. Nutrients 2022, 14(21), 4564. [Google Scholar] [CrossRef]
- Stuchell-Brereton, M. D.; Zimmerman, M. I.; Miller, J. J.; Mallimadugula, U. L.; Incicco, J. J.; Roy, D.; Smith, L. G.; Cubuk, J.; Baban, B.; DeKoster, G. T.; Frieden, C.; Bowman, G. R.; Soranno, A. Apolipoprotein E4 has extensive conformational heterogeneity in lipid-free and lipid-bound forms. Proceedings of the National Academy of Sciences of the United States of America 2023, 120(7), e2215371120. [Google Scholar] [CrossRef] [PubMed]
- St-Onge, F.; Chapleau, M.; Breitner, J. C.; Villeneuve, S.; Binette, A. P. Tau accumulation and its spatial progression across the Alzheimer’s disease spectrum. medRxiv Prepr. Serv. Health Sci. 2023, 23290880. [Google Scholar] [CrossRef]
- Stouffer, K. M.; Chen, C.; Kulason, S.; Xu, E.; Witter, M. P.; Ceritoglu, C.; Albert, M. S.; Mori, S.; Troncoso, J.; Tward, D. J.; Miller, M. I.; Alzheimer’s Disease Neuroimaging Initiative. Early amygdala and ERC atrophy linked to 3D reconstruction of rostral neurofibrillary tau tangle pathology in Alzheimer’s disease. NeuroImage. Clin. 2023, 38, 103374. [Google Scholar] [CrossRef]
- Straccia, M.; Gresa-Arribas, N.; Dentesano, G.; Ejarque-Ortiz, A.; Tusell, J. M.; Serratosa, J.; Solà, C.; Saura, J. Pro-inflammatory gene expression and neurotoxic effects of activated microglia are attenuated by absence of CCAAT/enhancer binding protein β. J. Neuroinflammation 2011, 8, 156. [Google Scholar] [CrossRef]
- Suresh, S.; Singh S, A.; Rushendran, R.; Vellapandian, C.; Prajapati, B. Alzheimer’s disease: the role of extrinsic factors in its development, an investigation of the environmental enigma. Front. Neurol. 2023, 14, 1303111. [Google Scholar] [CrossRef]
- Song, Y. J.; Li, S. R.; Li, X. W.; Chen, X.; Wei, Z. X.; Liu, Q. S.; Cheng, Y. The Effect of Estrogen Replacement Therapy on Alzheimer’s Disease and Parkinson’s Disease in Postmenopausal Women: A Meta-Analysis. Front. Neurosci. 2020, 14, 157. [Google Scholar] [CrossRef]
- Sung, Y. F.; Tsai, C. T.; Kuo, C. Y.; Lee, J. T.; Chou, C. H.; Chen, Y. C.; Chou, Y. C.; Sun, C. A. Use of Hormone Replacement Therapy and Risk of Dementia: A Nationwide Cohort Study. Neurology 2022, 99(17), e1835–e1842. [Google Scholar] [CrossRef]
- Sun, Y. Y.; Wang, Z.; Huang, H. C. Roles of ApoE4 on the Pathogenesis in Alzheimer’s Disease and the Potential Therapeutic Approaches. Cell. Mol. Neurobiol. 2023, 43(7), 3115–3136. [Google Scholar] [CrossRef] [PubMed]
- Song, Z. H.; Yu, H. Y.; Wang, P.; Mao, G. K.; Liu, W. X.; Li, M. N.; Wang, H. N.; Shang, Y. L.; Liu, C.; Xu, Z. L.; Sun, Q. Y.; Li, W. Germ cell-specific Atg7 knockout results in primary ovarian insufficiency in female mice. Cell Death Dis. 2015, 6(1), e1589. [Google Scholar] [CrossRef]
- Sweeney, M. D.; Montagne, A.; Sagare, A. P.; Nation, D. A.; Schneider, L. S.; Chui, H. C.; Harrington, M. G.; Pa, J.; Law, M.; Wang, D. J. J.; Jacobs, R. E.; Doubal, F. N.; Ramirez, J.; Black, S. E.; Nedergaard, M.; Benveniste, H.; Dichgans, M.; Iadecola, C.; Love, S.; Bath, P. M.; Zlokovic, B. V. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2019, 15(1), 158–167. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.; Wijeratne, H. R. S.; Kim, B.; Philtjens, S.; You, Y.; Lee, D. H.; Gutierrez, D. A.; Sharify, D.; Wells, M.; Perez-Cardelo, M.; Doud, E. H.; Fernandez-Hernando, C.; Lasagna-Reeves, C.; Mosley, A. L.; Kim, J. Deletion of miR-33, a regulator of the ABCA1-APOE pathway, ameliorates neuropathological phenotypes in APP/PS1 mice. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2024, 20(11), 7805–7818. [Google Scholar] [CrossRef]
- Taxier, L. R.; Philippi, S. M.; Fleischer, A. W.; York, J. M.; LaDu, M. J.; Frick, K. M. APOE4 homozygote females are resistant to the beneficial effects of 17β-estradiol on memory and CA1 dendritic spine density in the EFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2022a, 118, 13–24. [Google Scholar] [CrossRef]
- Taxier, L. R.; Philippi, S. M.; York, J. M.; LaDu, M. J.; Frick, K. M. The detrimental effects of APOE4 on risk for Alzheimer’s disease may result from altered dendritic spine density, synaptic proteins, and estrogen receptor alpha. Neurobiol. Aging 2022b, 112, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Tang, Y.; Cai, D. Hypothalamic inflammation and GnRH in aging development. Cell Cycle 2013, 12(17), 2711–2712. [Google Scholar] [CrossRef]
- Thakur, M. K.; Ghosh, S. Age and sex dependent alteration in presenilin expression in mouse cerebral cortex. Cell. Mol. Neurobiol. 2007, 27(8), 1059–1067. [Google Scholar] [CrossRef]
- Therriault, J.; Pascoal, T. A.; Lussier, F. Z.; Tissot, C.; Chamoun, M.; Bezgin, G.; Servaes, S.; Benedet, A. L.; Ashton, N. J.; Karikari, T. K.; Lantero-Rodriguez, J.; Kunach, P.; Wang, Y. T.; Fernandez-Arias, J.; Massarweh, G.; Vitali, P.; Soucy, J. P.; Saha-Chaudhuri, P.; Blennow, K.; Zetterberg, H.; Rosa-Neto, P. Biomarker modeling of Alzheimer’s disease using PET-based Braak staging. Nat. Aging 2022, 2(6), 526–535. [Google Scholar] [CrossRef]
- Thomas, S. N.; Funk, K. E.; Wan, Y.; Liao, Z.; Davies, P.; Kuret, J.; Yang, A. J. Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol. 2012, 123(1), 105–117. [Google Scholar] [CrossRef]
- Thurston, R. C.; Maki, P.; Chang, Y.; Wu, M.; Aizenstein, H. J.; Derby, C. A.; Karikari, T. K. Menopausal vasomotor symptoms and plasma Alzheimer disease biomarkers. Am. J. Obstet. Gynecol. 2024, 230(3), 342.e1–342.e8. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Fan, J.; Zhao, Y.; Bi, S.; Si, L.; Liu, Q. Estrogen receptor beta treats Alzheimer’s disease. Neural Regen. Res. 2013, 8(5), 420–426. [Google Scholar] [CrossRef] [PubMed]
- Troutwine, B. R.; Hamid, L.; Lysaker, C. R.; Strope, T. A.; Wilkins, H. M. Apolipoprotein E and Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12(2), 496–510. [Google Scholar] [CrossRef]
- Tong, J. H.; Gong, S. Q.; Zhang, Y. S.; Dong, J. R.; Zhong, X.; Wei, M. J.; Liu, M. Y. Association of Circulating Apolipoprotein AI Levels in Patients With Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2022, 14, 899175. [Google Scholar] [CrossRef]
- Tsukada, J.; Yoshida, Y.; Kominato, Y.; Auron, P. E. The CCAAT/enhancer (C/EBP) family of basic-leucine zipper (bZIP) transcription factors is a multifaceted highly-regulated system for gene regulation. Cytokine 2011, 54(1), 6–19. [Google Scholar] [CrossRef]
- Twarowski, B.; Herbet, M. Inflammatory Processes in Alzheimer’s Disease-Pathomechanism, Diagnosis and Treatment: A Review. Int. J. Mol. Sci. 2023, 24(7), 6518. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A. S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2(4), 236–240. [Google Scholar] [CrossRef] [PubMed]
- Uilenbroek, J. T.; Tiller, R.; de Jong, F. H.; Vels, F. Specific suppression of follicle-stimulating hormone secretion in gonadectomized male and female rats with intrasplenic ovarian transplants. J. Endocrinol. 1978, 78(3), 399–406. [Google Scholar] [CrossRef]
- van Helmond, Z.; Miners, J. S.; Kehoe, P. G.; Love, S. Higher soluble amyloid beta concentration in frontal cortex of young adults than in normal elderly or Alzheimer’s disease. Brain Pathol. 2010, 20(4), 787–793. [Google Scholar] [CrossRef]
- van den Hurk, R.; Zhao, J. Formation of mammalian oocytes and their growth, differentiation and maturation within ovarian follicles. Theriogenology 2005, 63(6), 1717–1751. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B. S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M. P.; Griep, A.; Gelpi, E.; Beilharz, M.; Riedel, D.; Golenbock, D. T.; Geyer, M.; Walter, J.; Latz, E.; Heneka, M. T. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552(7685), 355–361. [Google Scholar] [CrossRef]
- Verghese, P. B.; Castellano, J. M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D. M. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proceedings of the National Academy of Sciences of the United States of America 2013, 110(19), E1807–E1816. [Google Scholar] [CrossRef]
- Verma, M.; Vats, A.; Taneja, V. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18(2), 138–145. [Google Scholar] [CrossRef]
- Vogel, J. W.; Young, A. L.; Oxtoby, N. P.; Smith, R.; Ossenkoppele, R.; Strandberg, O. T.; La Joie, R.; Aksman, L. M.; Grothe, M. J.; Iturria-Medina, Y.; Alzheimer’s Disease Neuroimaging Initiative; Pontecorvo, M. J.; Devous, M. D.; Rabinovici, G. D.; Alexander, D. C.; Lyoo, C. H.; Evans, A. C.; Hansson, O. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 27(5), 871–881. [Google Scholar] [CrossRef]
- Voloboueva, L.A.; Emery, J.F.; Sun, X.; Giffard, R.G. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett. 2013, 587(6), 756–62. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Von Wald, T.; Monisova, Y.; Hacker, M. R.; Yoo, S. W.; Penzias, A. S.; Reindollar, R. R.; Usheva, A. Age-related variations in follicular apolipoproteins may influence human oocyte maturation and fertility potential. Fertil. Steril. 2010, 93(7), 2354–2361. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, Y.; Dening, T.; Hippisley-Cox, J.; Taylor, L.; Moore, M.; Coupland, C.; et al. Use of menopausal hormone therapy and risk of dementia: nested case-control studies using QResearch and CPRD databases. BMJ 2021, 374, n2182. [Google Scholar] [CrossRef] [PubMed]
- Wahrle, S. E.; Jiang, H.; Parsadanian, M.; Kim, J.; Li, A.; Knoten, A.; Jain, S.; Hirsch-Reinshagen, V.; Wellington, C. L.; Bales, K. R.; Paul, S. M.; Holtzman, D. M. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Investig. 2008, 118(2), 671–682. [Google Scholar] [CrossRef] [PubMed]
- Walczak-Nowicka, Ł. J.; Herbet, M. Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in their Pathogenesis. Int. J. Mol. Sci. 2021, 22(17), 9290. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Xia, Y. C/EBPβ as a master regulator of inflammasome signaling in neurodegenerative diseases: mechanisms and therapeutic implications. Front. Immunol. 2025, 16, 1656165. [Google Scholar] [CrossRef]
- Wang, S.-M.; Jeong, C.; Um, Y.H.; Kang, D.W.; Kim, S.; Lee, S.; Lee, C.U.; Aizenstein, H.J.; Baek, K.-H.; Lim, H.K. Follicle-stimulating hormone linked to cognitive decline and amyloid burden in postmenopausal women. Front. Aging Neurosci. 2026, 17, 1697255. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, G.; Schindler, S. E.; Christensen, J.; McKay, N. S.; Liu, J.; Wang, S.; Sun, Z.; Hassenstab, J.; Su, Y.; Flores, S.; Hornbeck, R.; Cash, L.; Cruchaga, C.; Fagan, A. M.; Tu, Z.; Morris, J. C.; Mintun, M. A.; Wang, Y.; Benzinger, T. L. S. Baseline Microglial Activation Correlates With Brain Amyloidosis and Longitudinal Cognitive Decline in Alzheimer Disease. Neurology (R) Neuroimmunol. Neuroinflammation 2022, 9(3), e1152. [Google Scholar] [CrossRef]
- Wang, S.; Liao, Z.; Zhang, Q.; Han, X.; Liu, C.; Wang, J. Mitochondrial dysfunction in Alzheimer’s disease: a key frontier for future targeted therapies. Front. Immunol. 2025, 15, 1484373. [Google Scholar] [CrossRef]
- Wang, X.; Shao, X.; Yu, L.; Sun, J.; Yin, X. S.; Chen, Z.; Xu, Y.; Wang, N.; Zhang, D.; Qiu, W.; Liu, F.; Ma, C. Changes in the pH value of the human brain in Alzheimer’s disease pathology correlated with CD68-positive microglia: a community-based autopsy study in Beijing, China. Mol. Brain 2025, 18(1), 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, J.; Qian, S.; Zheng, Y.; Yu, X.; Jiang, T.; Ai, R.; Hou, J.; Ma, E.; Cai, J.; He, H.; Wang, X.; Xie, C. Amyloid-β but not tau accumulation is strongly associated with longitudinal cognitive decline. CNS Neurosci. Ther. 2024, 30(7), e14860. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, H.; Liu, X.; Guo, X. Hypoglycemic medicines in the treatment of Alzheimer’s disease: Pathophysiological links between AD and glucose metabolism. Front. Pharmacol. 2023, 14, 1138499. [Google Scholar] [CrossRef]
- Wang, J.; Dickson, D. W.; Trojanowski, J. Q.; Lee, V. M. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp. Neurol. 1999, 158(2), 328–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yutuc, E.; Griffiths, W. J. Neuro-oxysterols and neuro-sterols as ligands to nuclear receptors, GPCRs, ligand-gated ion channels and other protein receptors. Br. J. Pharmacol. 2021, 178(16), 3176–3193. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Iqbal, M.; Zheng, J.; Wines-Samuelson, M.; Shen, J. Partial loss of presenilin impairs age-dependent neuronal survival in the cerebral cortex. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34(48), 15912–15922. [Google Scholar] [CrossRef]
- Whitmer, R. A.; Quesenberry, C. P.; Zhou, J.; Yaffe, K. Timing of hormone therapy and dementia: the critical window theory revisited. Ann. Neurol. 2011, 69(1), 163–169. [Google Scholar] [CrossRef]
- Weber, D.; Davies, M. J.; Grune, T. Determination of protein carbonyls in plasma, cell extracts, tissue homogenates, isolated proteins: Focus on sample preparation and derivatization conditions. Redox Biol. 2015, 5, 367–380. [Google Scholar] [CrossRef]
- Wei, L.; Yang, X.; Wang, J.; Wang, Z.; Wang, Q.; Ding, Y.; Yu, A. H3K18 lactylation of senescent microglia potentiates brain aging and Alzheimer’s disease through the NFκB signaling pathway. J. Neuroinflammation 2023, 20(1), 208. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wei, Y.; Liu, M.; Wang, D. The propagation mechanisms of extracellular tau in Alzheimer’s disease. J. Neurol. 2022, 269(3), 1164–1181. [Google Scholar] [CrossRef]
- Wilson, A. C.; Clemente, L.; Liu, T.; Bowen, R. L.; Meethal, S. V.; Atwood, C. S. Reproductive hormones regulate the selective permeability of the blood-brain barrier. Biochim. Et. Biophys. Acta 2008, 1782(6), 401–407. [Google Scholar] [CrossRef]
- Wilson, C.; Wardell, M. R.; Weisgraber, K. H.; Mahley, R. W.; Agard, D. A. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 1991, 252(5014), 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Oh, H.; Mormino, E. C.; Markley, C.; Landau, S. M.; Jagust, W. J. The effect of amyloid β on cognitive decline is modulated by neural integrity in cognitively normal elderly. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2013, 9(6), 687–698.e1. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, F. K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V. L.; Fisher, E. M.; Strydom, A. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16(9), 564–574. [Google Scholar] [CrossRef] [PubMed]
- Wood Alexander, M.; Honer, W. G.; Saloner, R.; Galea, L. A. M.; Bennett, D. A.; Rabin, J. S.; Casaletto, K. B. The interplay between age at menopause and synaptic integrity on Alzheimer’s disease risk in women. Sci. Adv. 2025, 11(10), eadt0757. [Google Scholar] [CrossRef]
- Wu, D. M.; Liu, J. P.; Liu, J.; Ge, W. H.; Wu, S. Z.; Zeng, C. J.; Liang, J.; Liu, K.; Lin, Q.; Hong, X. W.; Sun, Y. E.; Lu, J. Immune pathway activation in neurons triggers neural damage after stroke. Cell Rep. 2023, 42(11), 113368. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhang, Y.; Zhao, C.; Li, Q.; Zhang, J.; Han, J.; Xu, Z.; Li, J.; Ma, Y.; Wang, P.; Xu, H. Disruption of C/EBPβ-Clec7a axis exacerbates neuroinflammatory injury via NLRP3 inflammasome-mediated pyroptosis in experimental neuropathic pain. J. Transl. Med. 2022, 20(1), 583. [Google Scholar] [CrossRef]
- Wu, J.; Carlock, C.; Zhou, C.; Nakae, S.; Hicks, J.; Adams, H. P.; Lou, Y. IL-33 is required for disposal of unnecessary cells during ovarian atresia through regulation of autophagy and macrophage migration. J. Immunol. 2015, 194(5), 2140–2147. [Google Scholar] [CrossRef]
- Wu, Q. L.; Yang, X.; Luo, J. X.; Liu, L.; Zhou, Y.; Lu, M. H. Microglia energy metabolism: A new perspective on Alzheimer’s disease treatment. J. Neurol. Sci. 2025, 475, 123585. [Google Scholar] [CrossRef]
- Wu, X. L.; Piña-Crespo, J.; Zhang, Y. W.; Chen, X. C.; Xu, H. X. Tau-mediated Neurodegeneration and Potential Implications in Diagnosis and Treatment of Alzheimer’s Disease. Chin. Med. J. 2017, 130(24), 2978–2990. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, Z. H.; Liu, X.; Zhang, Z.; Gu, X.; Yu, S. P.; Keene, C. D.; Cheng, L.; Ye, K. Traumatic brain injury triggers APP and Tau cleavage by delta-secretase, mediating Alzheimer’s disease pathology. Prog. Neurobiol. 2020, 185, 101730. [Google Scholar] [CrossRef]
- Wynne, M. E.; Ogunbona, O.; Lane, A. R.; Gokhale, A.; Zlatic, S. A.; Xu, C.; Wen, Z.; Duong, D. M.; Rayaprolu, S.; Ivanova, A.; Ortlund, E. A.; Dammer, E. B.; Seyfried, N. T.; Roberts, B. R.; Crocker, A.; Shanbhag, V.; Petris, M.; Senoo, N.; Kandasamy, S.; Claypool, S. M.; Faundez, V. APOE expression and secretion are modulated by mitochondrial dysfunction. eLife 2023, 12, e85779. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Qadota, H.; Wang, Z. H.; Liu, P.; Liu, X.; Ye, K. X.; Matheny, C. J.; Berglund, K.; Yu, S. P.; Drake, D.; Bennett, D. A.; Wang, X. C.; Yankner, B. A.; Benian, G. M.; Ye, K. Neuronal C/EBPβ/AEP pathway shortens life span via selective GABAnergic neuronal degeneration by FOXO repression. Sci. Adv. 2022, 8(13), eabj8658. [Google Scholar] [CrossRef]
- Xia, Z.; Prescott, E. E.; Urbanek, A.; Wareing, H. E.; King, M. C.; Olerinyova, A.; Dakin, H.; Leah, T.; Barnes, K. A.; Matuszyk, M. M.; Dimou, E.; Hidari, E.; Zhang, Y. P.; Lam, J. Y. L.; Danial, J. S. H.; Strickland, M. R.; Jiang, H.; Thornton, P.; Crowther, D. C.; Ohtonen, S.; De, S. Co-aggregation with Apolipoprotein E modulates the function of Amyloid-β in Alzheimer’s disease. Nat. Commun. 2024, 15(1), 4695. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Kang, S. S.; Wang, Z.; Liu, X.; Kuo, T. C.; Korkmaz, F.; Padilla, A.; Miyashita, S.; Chan, P.; Zhang, Z.; Katsel, P.; Burgess, J.; Gumerova, A.; Ievleva, K.; Sant, D.; Yu, S. P.; Muradova, V.; Frolinger, T.; Lizneva, D.; Iqbal, J.; Ye, K. FSH blockade improves cognition in mice with Alzheimer’s disease. Nature 2022, 603(7901), 470–476. [Google Scholar] [CrossRef]
- Xiong, J.; Kang, S. S.; Wang, M.; Wang, Z.; Xia, Y.; Liao, J.; Liu, X.; Yu, S. P.; Zhang, Z.; Ryu, V.; Yuen, T.; Zaidi, M.; Ye, K. FSH and ApoE4 contribute to Alzheimer’s disease-like pathogenesis via C/EBPβ/δ-secretase in female mice. Nat. Commun. 2023, 14(1), 6577. [Google Scholar] [CrossRef]
- Xiong, Y. S.; Liu, F. F.; Liu, D.; Huang, H. Z.; Wei, N.; Tan, L.; Chen, J. G.; Man, H. Y.; Gong, C. X.; Lu, Y.; Wang, J. Z.; Zhu, L. Q. Opposite effects of two estrogen receptors on tau phosphorylation through disparate effects on the miR-218/PTPA pathway. Aging Cell 2015, 14(5), 867–877. [Google Scholar] [CrossRef]
- Xu, X.; Pang, Y.; Fan, X. Mitochondria in oxidative stress, inflammation and aging: from mechanisms to therapeutic advances. Signal Transduct. Target. Ther. 2025, 10(1), 190. [Google Scholar] [CrossRef]
- Xue, Y.; Zuo, S.; Wang, F.; Qi, X. From hormones to neurodegeneration: how FSH drives Alzheimer’s disease. Front. Aging Neurosci. 2025, 17, 1578439. [Google Scholar] [CrossRef]
- https. [CrossRef]
- Yaffe, K.; Haan, M.; Byers, A.; Tangen, C.; Kuller, L. Estrogen use, APOE, and cognitive decline: evidence of gene-environment interaction. Neurology 2000, 54(10), 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Yang, D. S.; Stavrides, P.; Mohan, P. S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S. D.; Wesson, D. W.; Bandyopadhyay, U.; Jiang, Y.; Pawlik, M.; Peterhoff, C. M.; Yang, A. J.; Wilson, D. A.; St George-Hyslop, P.; Westaway, D.; Mathews, P. M.; Levy, E.; Cuervo, A. M.; Nixon, R. A. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 2011, 7(7), 788–789. [Google Scholar] [CrossRef]
- Yao, Q.; Long, C.; Yi, P.; Zhang, G.; Wan, W.; Rao, X.; Ying, J.; Liang, W.; Hua, F. C/EBPβ: A transcription factor associated with the irreversible progression of Alzheimer’s disease. CNS Neurosci. Ther. 2024, 30(4), e14721. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Mo, N.; Tong, L.; Dong, J.; Fan, Z.; Jia, M.; Yue, J.; Wang, Y. Microglia lactylation in relation to central nervous system diseases. Neural Regen. Res. 2025, 20(1), 29–40. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, Z.; Klionsky, D. J. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 2010, 12(9), 814–822. [Google Scholar] [CrossRef]
- Yin, X.; Zhao, C.; Qiu, Y.; Zhou, Z.; Bao, J.; Qian, W. Dendritic/Post-synaptic Tau and Early Pathology of Alzheimer’s Disease. Front. Mol. Neurosci. 2021, 14, 671779. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S. M.; Iwata, N.; Saido, T. C.; Maeda, J.; Suhara, T.; Trojanowski, J. Q.; Lee, V. M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53(3), 337–351. [Google Scholar] [CrossRef]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H. M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proceedings of the National Academy of Sciences of the United States of America 2014, 111(43), 15514–15519. [Google Scholar] [CrossRef]
- Yuk, J. S.; Lee, J. S.; Park, J. H. Menopausal hormone therapy and risk of dementia: health insurance database in South Korea-based retrospective cohort study. Front. Aging Neurosci. 2023, 15, 1213481. [Google Scholar] [CrossRef]
- Zaidi, M.; Ye, K. A pituitary hormone has a key role in Alzheimer’s disease. In Nature; Advance online publication, 2022. [Google Scholar] [CrossRef]
- Zandi, P. P.; Carlson, M. C.; Plassman, B. L.; Welsh-Bohmer, K. A.; Mayer, L. S.; Steffens, D. C.; Breitner, J. C.; Cache County Memory Study Investigators. Hormone replacement therapy and incidence of Alzheimer disease in older women: the Cache County Study. JAMA 2002, 288(17), 2123–2129. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Li, K.; Luo, X.; Wang, S.; Xu, X.; Jiaerken, Y.; Liu, X.; Hong, L.; Hong, H.; Li, Z.; Fu, Y.; Zhang, T.; Chen, Y.; Liu, Z.; Huang, P.; Zhang, M.; for behalf of Alzheimer’s Disease Neuroimaging Initiative (ADNI). The association of enlarged perivascular space with microglia-related inflammation and Alzheimer’s pathology in cognitively normal elderly. Neurobiol. Dis. 2022, 170, 105755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic programming of systemic aging involving IKK-β, NF-κB and GnRH. Nature 2013, 497(7448), 211–216. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17(9), 2181–2192. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent advances in Alzheimer’s disease: Mechanisms, clinical trials and new drug development strategies. Signal Transduct. Target. Ther. 2024, 9(1), 211. [Google Scholar] [CrossRef]
- Zhang, X. W.; Zhu, X. X.; Tang, D. S.; Lu, J. H. Targeting autophagy in Alzheimer’s disease: Animal models and mechanisms. Zool. Res. 2023, 44(6), 1132–1145. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, C.; Sun, J.; Shen, H. M.; Wang, J.; Yang, C. Impairment of the autophagy-lysosomal pathway in Alzheimer’s diseases: Pathogenic mechanisms and therapeutic potential. Acta Pharm. Sin. B 2022, 12(3), 1019–1040. [Google Scholar] [CrossRef]
- Zhou, J.; Yao, W.; Li, C.; Wu, W.; Li, Q.; Liu, H. Administration of follicle-stimulating hormone induces autophagy via upregulation of HIF-1α in mouse granulosa cells. Cell Death Dis. 2017, 8(8), e3001. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Chen, M.; Wang, X.; Yu, J.; Li, X.; Xie, Y.; Liu, J.; Liu, M.; Xu, L.; Zhang, Q.; Tian, X.; Zhang, F.; Guo, B. C/EBPβ isoform-specific regulation of podocyte pyroptosis in lupus nephritis-induced renal injury. J. Pathol. 2023, 261(3), 269–285. [Google Scholar] [CrossRef] [PubMed]
- Zsido, R. G.; Heinrich, M.; Slavich, G. M.; Beyer, F.; Kharabian Masouleh, S.; Kratzsch, J.; Raschpichler, M.; Mueller, K.; Scharrer, U.; Löffler, M.; Schroeter, M. L.; Stumvoll, M.; Villringer, A.; Witte, A. V.; Sacher, J. Association of Estradiol and Visceral Fat With Structural Brain Networks and Memory Performance in Adults. JAMA Netw. Open 2019, 2(6), e196126. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



