Submitted:
25 March 2026
Posted:
26 March 2026
You are already at the latest version
Abstract
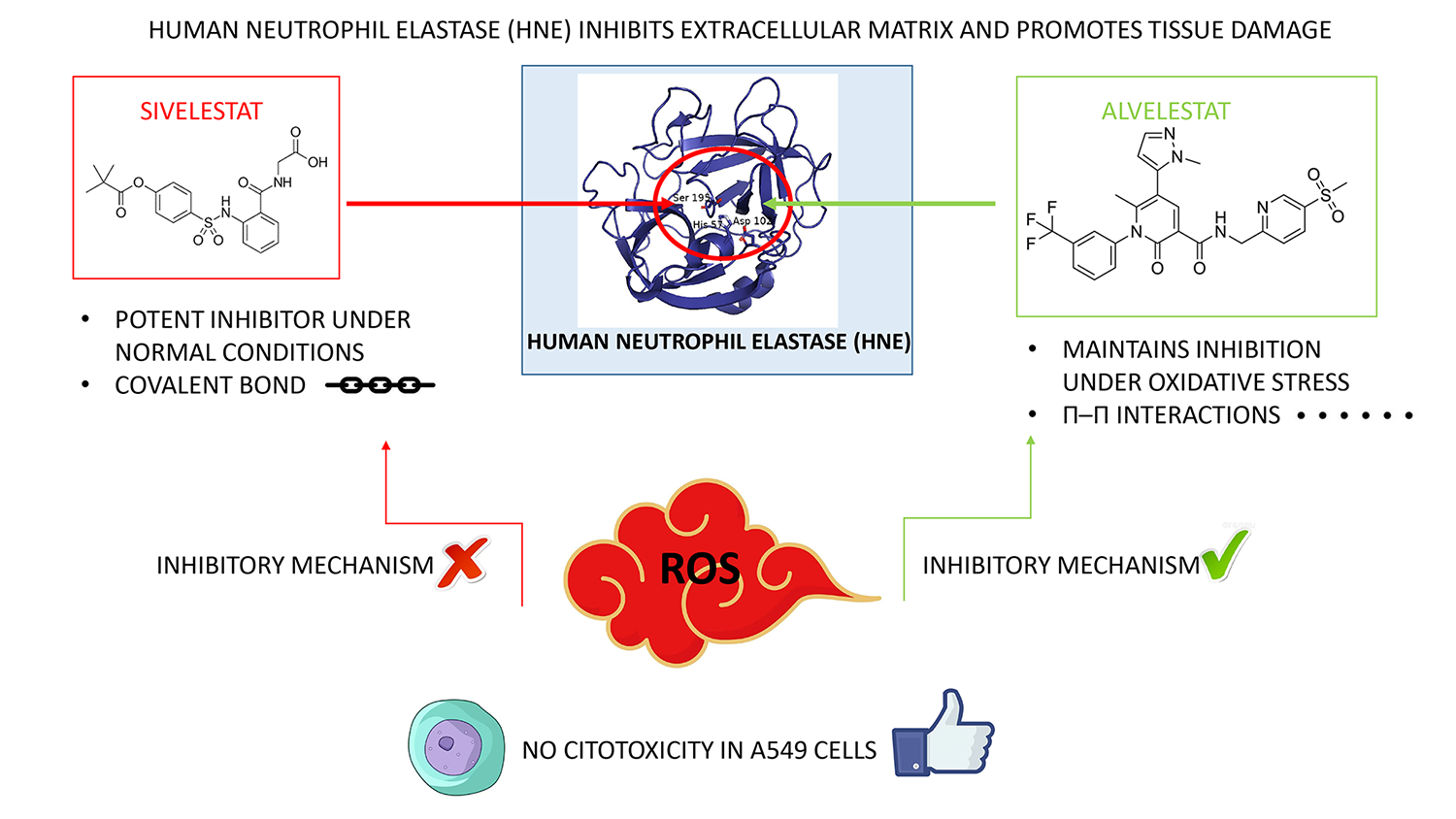
Human neutrophil elastase (HNE) is a key driver of inflammatory lung disorders, pro-moting extracellular matrix degradation and tissue damage. While inhibitors such as Sivelestat and Alvelestat are clinically relevant, their performance within the aggressive oxidative microenvironment of diseased lungs remains poorly understood. Here, we employed an integrated in vitro and in silico approach to investigate their behavior under physiological and oxidative conditions and to provide a molecular-level interpretation. Under physiological conditions, enzymatic assays and steady-state kinetics confirmed that both compounds act as competitive inhibitors, with Sivelestat displaying higher baseline potency. Under oxidative stress, however, Sivelestat exhibited a near-complete loss of activity, whereas Alvelestat retained its inhibitory efficacy. Molecular modeling and molecular dynamics simulations of native and oxidized HNE variants provided a structural rationale for this divergence. Alvelestat, as a non-covalent inhibitor, maintains stable binding despite increased flexibility of the active site induced by oxidative modifications. In contrast, Sivelestat, which acts through a reversible covalent mechanism, requires a precise pre-acylation geometry. Oxidation-induced remodeling of the S1 pocket disrupts the near-attack configuration required for covalent bond formation, thereby im-pairing inhibition. Overall, these findings indicate that oxidative stress selectively compromises covalent inhibition while preserving enzymatic activity, highlighting redox re-silience as a key parameter in the design of next-generation HNE inhibitors.
Keywords:
human neutrophil elastase (HNE)
; α1-antitrypsin (AAT)
; Alvelestat
; Sivelestat
; HNE inhibitors
; chronic lung diseases
1. Introduction
Human neutrophil elastase (HNE) is a potent serine protease stored within the azurophilic granules of neutrophils and released into the extracellular space during the inflammatory response [1,2,3]. While essential for the degradation of invading pathogens, its activity must be strictly regulated to prevent collateral damage to host tissues [4,5,6,7]. In healthy individuals, this regulation is primarily maintained by endogenous inhibitors such as alpha-1 antitrypsin (AAT) [8,9,10]. However, in severe inflammatory diseases, including chronic obstructive pulmonary disease (COPD), cystic fibrosis, acute respiratory distress syndrome (ARDS) and COVID-19 severe pneumonia, the protease–antiprotease balance collapses, leading to uncontrolled degradation of lung elastin and other extracellular matrix components [11,12,13]. A critical, yet often underestimated, driver of this pathology is the intense oxidative stress that characterizes the inflamed lung microenvironment [14,15]. Activated neutrophils generate a potent cocktail of reactive oxygen species (ROS) [15,16], including superoxide anions, hydrogen peroxide, and highly reactive oxidants such as singlet oxygen (1O₂) and hypochlorous acid (HOCl) [17,18,19,20]. This “oxidative storm” exerts a dual impact. First, it chemically inactivates AAT by oxidizing the critical methionine-358 residue [21,22]. Second, and more importantly from a drug design perspective, these reactive species can directly modify HNE itself, inducing post-translational modifications and conformational changes that may alter both its catalytic efficiency and its susceptibility to synthetic inhibitors [23].
Despite the clinical urgency, most drug discovery efforts targeting HNE inhibitors focus primarily on potency under optimized laboratory conditions, frequently neglecting the chemical complexity of the oxidative environment in which the enzyme operates [24,25,26]. This raises a fundamental question regarding the resilience of current therapeutics: to what extent does oxidative stress impair the binding and efficacy of different classes of inhibitors? In this context, Sivelestat, representing the class of covalent “suicide” inhibitors [27,28,29], and Alvelestat, a model for non-covalent inhibitors [30,31,32], provide an ideal comparative framework. These two compounds enable investigation of how distinct binding strategies, one relying on precise covalent anchoring and the other on conformational adaptability, respond to structural perturbations of the enzyme target.
In this study, we address this gap by investigating the performance of Alvelestat and Sivelestat under simulated oxidative conditions. By combining in vitro enzymatic assays with molecular dynamics (MD) simulations of oxidized HNE models, we demonstrate how oxidative perturbations in the active site differentially affect these two clinically relevant inhibitors. Our findings provide a structural rationale for the superior resilience of non-covalent, induced-fit inhibitors in oxidative environments, offering new insights for the rational development of next-generation therapies for chronic inflammatory lung diseases.
2. Results
2.1. Structural and Dynamic Characterization of Alvelestat and Sivelestat Binding to HNE by Molecular Modelling
To define the putative binding modes of Alvelestat and Sivelestat within the HNE catalytic site, a structure-based molecular modeling strategy combining induced-fit docking and molecular dynamics (MD) was employed. Because of the intrinsic conformational adaptability of the catalytic cleft [33,34], side-chain rearrangements upon ligand binding were explicitly considered during docking, and the most representative complexes were subsequently subjected to MD simulation to evaluate the stability and dynamics of the predicted binding modes.
Induced-fit docking of Alvelestat into the crystal structure of HNE (PDB ID: 2Z7F), revealed a binding mode spanning the S1–S4 subsites of the catalytic cleft. In the predicted pose, the m-(trifluoromethyl)phenyl moiety was deeply accommodated within the S1 pocket, forming hydrophobic contacts with Val216, Ala188, Phe192, and Ser195, whereas the central pyridone core anchored the ligand within the S2 sub-pocket via two hydrogen bonds with the backbone of Val216. The N-methylpyrazole moiety pointed toward a deeper S2 sub-pocket, consistent with an induced local opening of this region [33,35]. (Figure 1).
Cross-docking of Alvelestat into the 5ABW crystallographic structure of HNE in complex with a closely related Alvelestat analogue (WQQ), produced a binding orientation closely matching that of the cognate ligand within the cleft. A structured water molecule was observed bridging the sulfonyl group of Alvelestat with Arg217, acting as a key contributor to S4 anchoring and reproducing the interaction network observed in the crystallographic complex (Figure S1).
The stability of the docking-derived complex was evaluated by 100 ns MD simulation. During the simulation, the protein Cα backbone RMSD fluctuated around 2.1 ± 0.8 Å, while Alvelestat RMSD remained around 1.2 ± 0.5 Å relative to the initial docking pose (Figure S1, panel A left). Ligand RMSF analysis (Figure S1, panel C left) revealed limited fluctuation of the core scaffold, with RMSF value of approximately 0.5–0.6 Å for the pyridone and trifluoromethylphenyl rings. The hydrogen bonds with Val216 were maintained throughout the trajectory. A T-shaped π–π interaction with Phe215 and a water-mediated interaction involving Arg217 were also observed during the simulation, further supporting the stability of the binding mode.
Sivelestat is a reversible covalent inhibitor that forms an acyl–enzyme intermediate involving the catalytic Ser195. To investigate the geometry of the pre-covalent complex, induced-fit docking of Sivelestat was performed using the HNE crystal structure (PDB ID: 2Z7F). The predicted pre-covalent pose placed the pivaloyl ester group within the S1 pocket, forming hydrophobic contacts with Val216, Ala188, and Phe192. The para-substituted aromatic ring extended toward the S2 region, while the sulfonanilide moiety was oriented toward the S1′ subsite (Figure 2, panels A and B).
Importantly, in the predicted complex, the carbonyl oxygen of the pivaloyl group formed hydrogen bonds with the backbone NH groups of Gly193 and Ser195 in the oxyanion hole. The electrophilic carbon of the ester group was positioned close to the hydroxyl oxygen of Ser195, supporting a catalytically competent pre-reactive geometry. Covalent docking was subsequently performed to model the acyl-enzyme intermediate. (Figure 2, panel C) The resulting model predicted an acylated complex in which the pivaloyl group is anchored in the S1 sub-pocket while the carbonyl oxygen is stabilized in the oxyanion hole. Superposition of the predicted complex with crystallographic structures of elastase acyl-enzyme intermediates (PDB IDs: 8B53 and 8B1Y) showed a consistent positioning of the acyl group relative to the catalytic Ser195 (Figure 2, panel D).
The stability of the pre-covalent Sivelestat-HNE complex was evaluated by 100 ns of MD simulation. During the simulation, the Cα RMSD of the protein remained between approximately 1.1 and 1.3, indicating overall structural stability. After an initial increase corresponding to a rearrangement of the distal aryl-glycine moiety, the ligand RMSD converged to values of approximately 2.0–2.3 Å relative to the initial docking pose (Figure S2, panels A and B). Throughout the trajectory, the pivaloyl carbonyl oxygen remained engaged in hydrogen-bonding interactions with the oxyanion hole. The Bürgi–Dunitz angle defined by Ser195:Oγ–C(ester)–O(carbonyl) sampled values between 90° and 120° in approximately 28% of the simulation frames, consistent with geometries compatible with nucleophilic attack (Figure S2, panel C). Moreover, the Gly193:N–SIL:O distance averaged approximately 2.9 Å and the Ser195:N–SIL:O distance approximately 3.1 Å, further supporting the persistence of a pre-reactive configuration (Figure S2, panels D and F).
2.2. In Vitro Inhibition of HNE by Alvelestat and Sivelestat
To validate the in silico predictions, the inhibitory effects of Alvelestat and Sivelestat on standard HNE solutions were experimentally evaluated using the synthetic peptide substrate MeOSuc-Ala-Ala-Pro-Val-pNA. Preliminary kinetic characterization of native HNE yielded a Km of 0.30±0.05 mM and a kcat of 15 s−1, values that are in excellent agreement with established literature for this colorimetric assay [36], thereby confirming the reliability of the experimental setup. Subsequent inhibition assays, performed at a sub-saturating substrate concentration (0.15 mM), demonstrated that both compounds exert potent, concentration-dependent inhibition (Figure 3). Under these conditions, Sivelestat exhibited a higher inhibitory potency with an IC50 of 0.7±0.1 μM, while Alvelestat showed an IC50 of 2.7±0.5 μM.
To further characterize the nature of this inhibition, steady-state kinetic analyses were performed across a range of inhibitor concentrations. Global reciprocal (Lineweaver-Burk) plots (Figure 4, panels A and B) revealed a pattern characteristic of competitive inhibition for both ligands, as evidenced by the convergence of the regression lines at a common intercept on the y-axis. The calculated inhibition constants (Ki) were 7.0±0.9 μM for Sivelestat and 11.0±0.4 μM for Alvelestat. The consistency between Ki and IC₅₀ trends, despite the sub-saturating substrate concentration employed, further supports a competitive inhibition mechanism and indicates that both compounds directly target the catalytic site.
These experimental data corroborate the docking models, confirming that both inhibitors compete directly for the HNE active site, with Sivelestat showing a higher baseline affinity under standard, non-oxidative conditions.
2.3. Impact of Oxidative Stress on HNE Inhibition and Enzyme Stability
To evaluate the resilience of Alvelestat and Sivelestat in a biomimetic inflammatory environment, inhibition assays were conducted under oxidative stress induced by the addition of H2O2 (final concentration: 44 mM). As shown in Figure 5, panel A, Alvelestat maintained its inhibitory potency, with dose-response curves and IC50 values nearly identical to those obtained under native conditions. This highlights the robust efficacy of this non-covalent inhibitor even under strongly oxidizing conditions. In stark contrast, Sivelestat exhibited a complete loss of efficacy against the oxidized enzyme (Figure 5, panel B). While it remained a potent inhibitor of native HNE, it failed to achieve measurable inhibition of the oxidized form at any tested concentration, indicating that the oxidative environment selectively disrupts its inhibitory mechanism rather than the enzyme activity per se.
To verify whether oxidative treatment affected the intrinsic catalytic competence of HNE, steady-state kinetic parameters were determined after pre-incubation with H₂O₂. The oxidized enzyme displayed a Km of 0.32 ± 0.04 mM and a kcat of 16 s⁻¹, values essentially identical to those measured under native conditions. These results demonstrate that the overall catalytic efficiency of HNE is preserved under the applied oxidative conditions, thereby confirming that the observed loss of Sivelestat activity arises from alterations in inhibitor binding rather than from a loss of enzymatic function.
To investigate the molecular origin of the different inhibitory behavior of Sivelestat and Alvelestat under oxidative conditions, both the intrinsic stability of the inhibitors and the structural response of HNE to oxidative modifications were examined. The intrinsic stability of both inhibitors was evaluated under oxidizing conditions. Although only Sivelestat exhibited loss of activity, both inhibitors were tested to rigorously exclude differential chemical instability as a confounding factor and to strengthen the interpretation that the observed effect originates from enzyme modification rather than compound degradation. Both compounds were incubated for 30 min in the presence of 44 mM H₂O₂ and subsequently analyzed by LC-MS/MS. The mass spectra obtained for the oxidized samples were directly compared with those of the untreated inhibitors (controls). In both cases, the MS profiles of the H2O2-treated compounds were indistinguishable from those of the respective controls.
In fact, no detectable oxidation products, mass shifts, or degradation species were observed for either compound. The LC-MS/MS profiles of oxidized Sivelestat and Alvelestat remained unchanged compared to the untreated controls within the sensitivity limits of the assay, thereby excluding direct chemical degradation or modification of the inhibitors as a contributing factor to the observed loss of activity (Figure S3).
On the protein side, HNE contains eight cysteine residues arranged into four disulfide bridges, structural motifs that are known to be sensitive to oxidative stress. Cysteine thiols and disulfide bonds can undergo stepwise oxidation to sulfenic, and sulfinic and, under severe or prolonged oxidative conditions, may irreversibly evolve toward sulfonic acid (cysteic acid) derivatives [37,38]. Among the disulfide bridges present in HNE, the Cys42-Cys58 and Cys191-Cys220 pairs are located in proximity to the catalytic region and the S1 specificity pocket, respectively. In model OX1, the Cys42-Cys58 disulfide bridge was converted to the corresponding cysteic acid pair. In model OX2, the same modification was applied to the Cys191-Cys220 disulfide bridge. Both oxidized models were subjected to 20 ns MD simulations to evaluate the structural effects of these modifications on the catalytic site with particular attention to the S1 pocket (Figure S3, panel A).
In the OX1 model, oxidation of the Cys42-Cys58 bridge produced detectable changes in the geometry of the catalytic triad during the MD trajectory. The distance between Ser195:Oγ and His57:Nε2 shifted toward longer values compared with the wild-type enzyme, and the corresponding hydrogen bond was largely disrupted relative to the native state (Figure S3, panels B and C) [39]. In addition, Ser195:Oγ became engaged in a persistent hydrogen bond with Cys42-SO₃H, promoting a reorientation of the serine away from His57 throughout the simulation and indicating a perturbation of the catalytic machinery. In the OX2 model, oxidation of the Cys191-Cys220 disulfide bridge did not produce major alterations in the catalytic triad geometry during the MD trajectory (Figure S3, panel D). The Ser195:Oγ-His57:Nε2 distance remained within the hydrogen-bonding range for most of the simulation and showed fluctuations comparable to those observed in the wild-type enzyme. However, the RMSF profile of the protein Cα atoms indicated increased mobility in the region surrounding the Cys191-Cys220 bridge, which is located at the periphery of the S1 subsite (Figure S3, panel A), suggesting localized rather than global structural perturbations.
To evaluate the effect of this structural modification on inhibitor binding, Sivelestat was docked into the OX2 model, and the resulting complex was subjected to a 100 ns MD simulation (Figure S4, panel A center). During the first 10 ns of the trajectory, the ligand maintained a binding orientation similar to that observed in the wild-type enzyme. As the simulation progressed, the pivaloyl ester group gradually moved away from its deeply buried position within the S1 pocket, as reflected by the increased ligand RMSF, especially for the pivaloyl moiety (Figure S4, panel C). This displacement was accompanied by disruption of the hydrogen-bond interactions between the ester carbonyl oxygen and the oxyanion-hole NH groups of Gly193 and Ser195. During the trajectory, the ligand did not re-establish a stable geometry comparable to the initial pre-covalent complex, indicating a loss of the pre-reactive configuration required for covalent inhibition. Concomitantly, the S1 subsite exhibited increased local flexibility, with enhanced motion of the loop region containing the oxidized Cys191–Cys220 pair (Figure S4, panel B right), further supporting a mechanism in which localized structural perturbations impair the formation of a productive enzyme–inhibitor complex.
3.4. Cytotoxicity Profile in Human Alveolar Epithelial Cells
To evaluate the safety profile of Alvelestat and Sivelestat in a lung-relevant model, cytotoxicity assays were performed using the human alveolar basal epithelial cell line A549. Metabolic activity was quantified via the resazurin reduction assay across a broad concentration range (0.1 to 1000 µM). As shown in Figure 6, cell viability remained consistently close to 100% for both compounds throughout the tested range. These results indicate that neither Alvelestat nor Sivelestat induces detectable cytotoxic effects in A549 cells, even at concentrations far exceeding their IC50 values.
3. Discussion
Inhibition of human neutrophil elastase is a cornerstone in the management of inflammatory lung diseases, such as COPD, ARDS, cystic fibrosis and COVID-19 severe pneumonia. In these conditions, dysregulated HNE activity drives the uncontrolled degradation of the extracellular matrix, making the development of selective inhibitors a vital therapeutic strategy. Among these, Sivelestat and Alvelestat represent two of the most clinically relevant compounds. This study clarifies their binding modes and inhibitory mechanisms through an integrated in silico and in vitro approach, providing a mechanistic framework to plausibly explain their performance under both physiological and pathological conditions.
Docking and MD analysis indicate that Alvelestat engages HNE through a non-covalent, induced-fit binding mode stabilized by a distributed interaction network across the S1–S4 subsites. The m-trifluoromethylphenyl group is deeply accommodated within the S1 pocket, forming hydrophobic contacts with Val216, Ala188, Phe192, and Ser195, while the central pyridone core establishes persistent backbone hydrogen bonds with Val216 in S2. The N-methylpyrazole and sulfonyl moieties project toward solvent-exposed regions, where they interact with Arg217 via a structured water molecule, further consolidating S4 anchoring. MD simulations indicate that this interaction network remains stable throughout the trajectory, supporting the formation of a persistent and dynamically stable enzyme-inhibitor complex. Sivelestat, in contrast, behaves as a reversible covalent inhibitor and relies on a much more stringently organized pre-acylation geometry. Induced-fit docking into wild-type HNE positions the pivaloyl ester deeply in S1, forming hydrophobic contacts with Val216, Ala188, and Phe192, while the para-substituted aromatic ring engages π–π stacking with Phe192 in S2 and the sulfonanilide extends toward S1′. The pivaloyl carbonyl oxygen forms bifurcated hydrogen bonds with the backbone NH groups of Gly193 and Ser195, thereby aligning the electrophilic carbon with Ser195:Oγ at distances and angles compatible with nucleophilic attack. MD simulations further refine this mechanistic view: in the pre-acylation complex, the oxyanion-hole contacts are highly persistent and the Bürgi–Dunitz angle consistently samples a “reactive” window, supporting a stable polarization environment for the ester carbonyl and efficient nucleophilic attack.
The enzymatic assays are fully consistent with these structural observations. The kinetic parameters obtained for native HNE are in close agreement with previously reported values [36], supporting the robustness and reliability of the experimental setup. Both inhibitors displayed potent, concentration-dependent inhibition, although Sivelestat exhibited higher baseline potency (IC₅₀ = 0.7 ± 0.1 μM) than Alvelestat (IC₅₀ = 2.7 ± 0.5 μM). This difference is consistent with their distinct inhibitory mechanisms: the higher apparent efficiency of Sivelestat likely reflects the formation of a covalent acyl–enzyme intermediate [25,29,40], whereas Alvelestat relies on an extensive network of non-covalent interactions to stabilize the enzyme-inhibitor complex. Steady-state kinetic analysis confirmed that both compounds behave as competitive inhibitors, with inhibition constants (Kᵢ = 7.0 ± 0.9 μM for Sivelestat and 11.0 ± 0.4 μM for Alvelestat) consistent with the observed IC₅₀ trends. However, their performance diverged markedly under oxidative conditions, revealing a clear difference in inhibitory resilience. While Alvelestat maintained its inhibitory capacity with only minor variations, Sivelestat exhibited a pronounced reduction in potency. Importantly, steady-state kinetic parameters of oxidized HNE remained essentially unchanged (Km = 0.32 ± 0.04 mM; kcat = 16 s⁻¹), demonstrating that the intrinsic catalytic competence of the enzyme is preserved under oxidative conditions. To exclude the possibility that this loss of potency originated from direct oxidation of the inhibitors, their intrinsic chemical stability was evaluated under the same oxidative conditions used in the enzymatic assays. LC–MS/MS analysis revealed no detectable degradation or oxidation products for either compound following incubation with H₂O₂, indicating that the observed effects are unlikely to arise from direct oxidative modification of the small molecules. These observations therefore point to oxidation-induced structural alterations of HNE as the most plausible origin of the altered inhibitory response.
Although the present data do not directly resolve the individual kinetic steps of covalent inhibition, the combined enzymatic and computational results consistently indicate that oxidative perturbations primarily affect the formation of a productive pre-acylation complex rather than completely preventing ligand binding. In this framework, the preservation of catalytic activity together with the marked loss of inhibition is most plausibly explained by a disruption of the geometric requirements necessary for covalent engagement.
A central point of discussion concerns the physiological relevance of the oxidative conditions employed. Although the H₂O₂ concentration used in this study exceeds the transient micromolar levels typically measured in bulk extracellular fluids and epithelial lining fluid of the lung, it is important to consider that within the confined microenvironment of the neutrophil oxidative burst, ROS are generated at high local fluxes and in combination with more reactive oxidants such as hypochlorous acid and singlet oxygen [17,18,19,20]. These oxidants possess a significantly higher oxidation potential and can induce irreversible oxidative modifications in susceptible amino acid residues beyond those typically produced by H2O2 alone. In particular, they are known to promote the terminal oxidation of cysteine residues to sulfonic acid [38,41,42], a modification that we explicitly reproduced in our computational models to simulate cumulative oxidative damage at the molecular level. Under these conditions, proteins located at the neutrophil–tissue interface may experience cumulative oxidative modifications over time. In vitro, such cumulative damage must necessarily be reproduced within a limited experimental timeframe. Therefore, the use of 44 mM H₂O₂ should be interpreted as an accelerated oxidative model designed to probe the structural robustness of the enzyme-inhibitor complex under sustained oxidative pressure, rather than as a direct quantitative representation of in vivo concentrations. Taken together, these results indicate that Sivelestat, as a covalent inhibitor that requires a precisely organized catalytic environment for acyl-enzyme formation, appears particularly sensitive to oxidative perturbations of the active site. In contrast, the retained inhibitory activity of Alvelestat suggests that a non-covalent, induced-fit binding mechanism can better tolerate moderate structural rearrangements of the catalytic pocket, thereby preserving inhibitory efficacy under oxidizing conditions.
To elucidate the potential molecular basis of this loss of potency, we investigated how disulfide bonds overoxidation impacts the structural integrity of HNE and how these modifications differentially impair Sivelestat binding relative to Alvelestat. By comparing the native enzyme with OX1 and OX2 variants where Cys42-Cys58 and Cys191-Cys220 were converted respectively into sulfonic acid residues, a significant divergence in stability emerged. MD simulations of OX1 revealed subtle but functionally relevant perturbations of the catalytic machinery. The Ser195:Oγ-His57:Nε2 distance shifted toward longer values and the corresponding hydrogen bond was largely lost, while Ser195:Oγ formed a persistent hydrogen bond with Cys42-SO₃H, reorienting the serine away from His57 throughout the simulation. This behavior is consistent with a partial decoupling of the Ser-His pair and a suboptimal orientation of Ser195 for nucleophile activation. Given that HNE retains measurable catalytic activity under oxidative conditions in the enzymatic assays, such destabilization of the triad makes extensive Cys42-Cys58 overoxidation an unlikely primary event in vivo.
By contrast, OX2 maintained the core architecture and competence of the catalytic triad and was comparable to that of the wild-type enzyme within thermal fluctuations, indicating that overoxidation at Cys191-Cys220 is structurally compatible with preservation of basal catalytic activity. At the same time, the loop bearing Cys191–Cys220 displayed increased mobility and the S1 pocket exhibited a modest widening, consistent with a localized “softening” of the S1 edge. Importantly, these features point to a decoupling between global catalytic competence and local structural plasticity, making the Cys191–Cys220 bridge a more plausible redox-sensitive hotspot, capable of altering the geometry and dynamics of the S1/oxyanion-hole region without fully compromising catalysis.
The functional consequences of Cys191–Cys220 overoxidation were probed by explicitly modeling inhibitor binding to OX2. Docking of Sivelestat into the OX2 model, followed by MD of the pre-acylation complex, revealed a progressive destabilization of the Michaelis-like geometry upon conversion of Cys191–Cys220 to cysteic acids. During the initial phase of the simulation, the ligand adopted a binding orientation similar to that seen in the wild-type complex. As the trajectory evolved, however, the pivaloyl ester gradually shifted out of its deeply buried S1 position, accompanied by disruption of the hydrogen-bond network between the carbonyl oxygen and the backbone NH groups of Gly193 and Ser195. The S1 subsite concomitantly exhibited increased local flexibility and modest expansion, and a stable near-attack geometry was not re-established over the remainder of the trajectory. Notably, this occurs without significant disruption of the catalytic triad, reinforcing the concept of a selective impairment of pre-reactive geometry rather than global enzymatic dysfunction. These observations, although derived from a modeled terminal oxidation state, support a mechanistic scenario in which localized remodeling of the S1/oxyanion-hole region selectively interferes with the stringent geometric requirements for covalent engagement by Sivelestat, while leaving the minimal catalytic competence of the enzyme largely intact.
In contrast, the resilience of Alvelestat likely lies in its non-covalent, induced-fit binding mechanism. Unlike covalent agents, Alvelestat appears to possess the conformational adaptability to rearrange its hydrophobic and hydrogen-bonding network (particularly with Val216 and Phe192) even when the active site is perturbed. By engaging a broader and more flexible binding surface rather than relying on a single covalent anchor point, Alvelestat could theoretically compensate for local oxidative damage in one sub-pocket by strengthening interactions in adjacent regions. This structural robustness provides a plausible rationale that while oxidative stress disrupts the rigid geometry necessary for acylation, the inherent flexibility of the induced-fit model may ensure sustained efficacy in the aggressive microenvironments of diseased lungs.
Another central question is whether the irreversible conversion of HNE cysteine residues into sulfonic acid groups represents a physiologically plausible event. Such terminal oxidation is well-documented in chronic inflammation; for instance, the trioxidation of serum albumin specifically at the conserved Cys34 residue is a recognized biomarker of cumulative oxidative stress in metabolic and systemic disorders [43,44]. In the lung environment, where COPD and ARDS patients exhibit a persistent “oxidative burst,” HNE is exposed to high concentrations of reactive oxidants such as hypochlorous acid and singlet oxygen. These species drive rapid and hierarchical oxidation pathways of protein thiols [15]. While the initial formation of cysteine-sulfenic acid often acts as a transient redox sensor [45], the intense ROS flux characteristic of diseased lungs can promote the progression beyond these reversible intermediates, ultimately leading. to stable and irreversible sulfonic acid forms, which significantly alter the protein’s biophysical properties and structural scaffolding [38,46].
These local perturbations are likely critical for covalent inhibitors like Sivelestat, which require a precise spatial alignment of their electrophilic warhead. within the catalytic machinery. Conceptually, the difference can be compared to how enzymes process natural substrates versus covalent inhibitors. A natural substrate can be likened to a slightly deformable object: even if moderately misshapen, it may still be accommodated and processed by the catalytic site. In contrast, Sivelestat behaves more like a precision lockpick, requiring an exact geometry fit within the keyhole of the active site to enable efficient acyl-enzyme formation. When oxidative stress subtly distorts the S1 pocket, this precise engagement may no longer occur. Alvelestat, however, maintains its inhibitory efficacy by potentially tolerating the increased flexibility of the binding site, highlighting the superior resilience of non-covalent, induced-fit inhibitors in aggressive inflammatory environments. Collectively, these findings reveal that oxidative stress likely exerts a dual impact on HNE: while the global structural integrity of the enzyme appears largely preserved, localized distortions within the catalytic pocket selectively compromise the molecular machinery required for covalent inhibition. These insights underscore the superior therapeutic resilience of non-covalent, induced-fit ligands, suggesting they may be better suited for the aggressive oxidative environments characteristic of chronic inflammatory diseases.
4. Materials and Methods
4.1. Reagents
Standard HNE was purchased from Abcam (Cambridge, UK). The synthetic non-peptide inhibitors Alvelestat and Sivelestat were obtained from Selleckchem (Munich, Germany) and Abcam (ab146184), respectively. The standard p-nitroaniline (p-NA) and the peptide substrate MeOSuc-Ala-Ala-Pro-Val-pNA used for the determination of HNE activity were obtained from Bachem (Bachem GmbH, Germany). Unless otherwise stated, all other analytical grade reagents were purchased from Sigma Aldrich (St. Louis, MO, USA). All buffers used were prepared using double-distilled water obtained with a Millipore (Bedford, MA, USA) Milli-Q purification system.
4.2. Molecular Modelling
Molecular docking was performed using Flare (Cresset, version 5). The X-ray crystal structure of HNE in complex with the SLPI (secretory leukocyte protease inhibitor) peptide (PDB ID: 2Z7F; resolution: 1.70 Å) and the reversible inhibitor WQQ (PDB ID: 5ABW; resolution: 1.85 Å) was retrieved from the RCSB Protein Data Bank. Chain A was retained for all calculations, as it provided complete and high-quality structural information. Missing atoms and side chains were rebuilt using Flare’s preparation tools. Protonation states of ionizable residues were assigned at pH 7.4, and hydrogen atoms were added. The protein was subjected to restrained energy minimization using the MMFF94 force field to relieve local steric clashes. The chemical structures of Alvelestat and Sivelestat were generated using the molecule editor of Flare and protonation and tautomeric states at physiological pH (7.4) were computed using Ligand Prep tool. The binding site was defined by centering the docking grid on the hydroxyl oxygen atom of the catalytic Ser195 residue and extended to include the S1-4 and S1’-2’ subsites (for docking against the structure of HNE in complex with SLPI, PDB ID: 2Z7F) and the cavity occupied by the co-crystallized ligand (for docking against the structure of HNE in complex with WQQ, PDB ID: 5ABW). Default parameters were used for ensemble docking. To assess the reliability of the docking workflow, the co-crystallized ligand WQQ was extracted from the 5ABW structure and re-docked into the prepared HNE binding site. The resulting pose reproduced the experimental binding conformation with a root-mean-square deviation (RMSD) of 0.95 Å, thereby validating the docking setup for subsequent ligand placement and scoring. For each ligand, ten poses were generated. Docked poses were visually inspected within Flare to examine key molecular interactions such as hydrogen bonding, electrostatic complementarity, and hydrophobic contacts.
4.3. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations were performed using GROMACS on an NVIDIA GeForce RTX 4060 GPU. The protein-ligand complex was solvated in a cubic box using the TIP3P water model, with a minimum distance of 1.0 nm between the complex and box edges. The system was neutralized by adding Na+ counterions. Energy minimization was performed using the steepest descent algorithm until the maximum force was less than 1000 kJ/mol/nm. The system was equilibrated in two phases. First, NVT (constant Number of particles, Volume, and Temperature) ensemble equilibration was conducted using Brownian dynamics at 10 K for 100 ps, followed by 12 ps with position restraints on solute heavy atoms. Second, NPT (constant Number of particles, Pressure, and Temperature) ensemble equilibration was performed without heavy atom restraints at 10 K for 12 ps and at 300 K for 24 ps. Pressure was maintained at 1 bar using the Parrinello-Rahman barostat, and temperature coupling was achieved using the modified Berendsen thermostat. Production MD simulations were conducted with a 2 fs time step under NPT conditions (1 bar, 300 K). 1000 frames per simulation were saved for analysis. Root mean square deviation (RMSD) and root mean square fluctuation (RMSF) analyses were performed using GROMACS analysis tools to assess the stability of the protein-ligand complex and identify regions of conformational flexibility.
To investigate the structural effects of oxidative stress on HNE, the initial structure was derived from the crystallographic model of HNE in complex with SILP (PDB ID: 2Z7F). The disulfide bonds between Cys191 and Cys220, and between Cys42 and Cys58 were manually cleaved, and each cysteine residue were modified to the sulfonic acid form. The protein was solvated in a cubic TIP3P water box and neutralized with appropriate counterions. The system was minimized as reported above, and production MD was carried out at 310 K in the NPT ensemble with periodic boundary conditions. Analyses were performed using GROMACS analysis tools and Python 3.x.
4.4. HNE Enzyme Activity, Steady State Kinetics and Inhibition Assays
HNE activity was assessed through a colorimetric assay based on the hydrolysis of the synthetic peptide substrate MeOSuc-Ala-Ala-Pro-Val-pNA [36]. To determine the steady-state kinetic parameters, 0.4 μg of HNE were added to an incubation buffer consisting of 50 mM Tris-HCl, pH 7.8 containing 500 mM NaCl. The reaction was initiated by adding different aliquots of the substrate (from a 120 mg/mL stock solution in DMSO) to achieve final concentrations ranging from 0.075 to 2.4 mM within a total reaction volume of 200 μL. The mixtures were incubated at 37 °C for 45 min, then quenched with 20 μL of 0.45 M trichloroacetic acid (TCA). After cooling on ice for 30 min, the samples were centrifuged at 11,500 rpm for 10 min to remove precipitates. The concentration of p-nitroaniline (p-NA) released into the supernatant was determined by measuring the absorbance at 410 nm. All assays were performed in triplicate, and the kinetic constants were determined by fitting the experimental data to the Michaelis-Menten equation using GraphPad Prism 8 software.
In parallel, HNE activity was investigated in the presence of Alvelestat and Sivelestat. Initially, IC50 values were determined by assaying the enzyme activity as described above, using a fixed substrate concentration of 0.15 mM and varying inhibitor concentrations ranging from 0.1 μM to 100 μM. IC50 values were estimated according to Equation 1, where A[I] and A [0] represent the enzymatic activity in the presence and absence of the inhibitor, respectively.
Subsequently, the inhibition constants (Ki) were determined by assaying HNE activity at varying concentrations of both substrate and inhibitors. Data were analyzed using an adapted equation for competitive inhibition [47] (Equation 2) with GraphPad Prism 8 software.
4.5. Inhibition Assays in the Presence of H2O2
HNE activity under oxidative conditions was determined using the colorimetric assay described above. Briefly, HNE was pre-incubated with 1 μL of 30% (w/v) H₂O₂ (final concentration 44 mM in a total reaction volume of 200 μL) for 60 min at 37 °C. These conditions were selected to promote cumulative oxidative modifications of redox-sensitive residues within a controlled experimental timeframe, thereby enabling comparison of inhibitor performance under sustained oxidative pressure.
The kinetic parameters of oxidized HNE were determined as described above. Subsequently, inhibition assays were performed by initiating the reaction with substrate (final concentration 0.15 mM) in the presence of increasing concentrations of inhibitor (0.1–100 μM). The reaction mixtures were then incubated at 37 °C for 45 min and then terminated by the addition of 20 μL of 0.45 M TCA. Samples were cooled on ice, centrifuged, and analyzed as described in Section 2.4.
4.6. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
All analyses were carried out on a liquid chromatography-tandem mass spectrometry (LC-MS/MS) system (Thermo Finnigan, San Jose, CA, USA) consisting of a thermostated column, a surveyor autosampler controlled at 25 °C, a quaternary gradient surveyor MS pump equipped with a diode array (DA) detector, and a linear trap quadrupole (LTQ) mass spectrometer with electrospray ionization (ESI) ion source controlled by Xcalibur software 1.4 (Thermo Fisher Scientific, Waltham, MA, USA). Analytes were separated by reverse phase high performance liquid chromatography (RP-HPLC) on a Jupiter (Phenomenex, Torrance, CA, USA) C18 column (150 × 2 mm, 4 µm, 90 Å particle size) using a linear gradient from 95% solvent A (0,1% aqueous formic acid, FA) to 60% solvent B (acetonitrile, ACN, containing 0.1% FA) in 60 min. The flow rate was 0.2 mL/min. Mass spectra were acquired in positive ion mode under constant instrumental conditions: source voltage, 5.0 kV; capillary voltage, 46 V; sheath gas flow, 40 arbitrary units; auxiliary gas flow, 10 arbitrary units; sweep gas flow, 1 arbitrary unit; capillary temperature, 200 °C; and tube lens voltage, −105 V. MS/MS spectra were generated by collision-induced dissociation (CID) in the linear ion trap, using an isolation width of 3 Th (m/z). The activation amplitude was set to 35% of the ejection RF amplitude, corresponding to 1.58 V. Data were processed using PEAKS Studio version 4.5 software (Bioinformatics Solutions Inc., Waterloo, Canada).
4.7. Determination of Silvelestat and Alvelestat Cytotoxicity on A549 Cell Monolayers
The cytotoxicity of Sivelestat and Alvelestat was evaluated using the human alveolar epithelial cell line A549. Cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and seeded into sterile 96-well plates at a density of 1×105 cells per well, allowing the formation of a confluent monolayer prior to treatment. After 24 h incubation at 37 °C (5% CO2), the culture medium was replaced with 200 μL of fresh medium containing ten-fold dilutions of the test compounds, ranging from 0.1 to 1000 µM.
After a 16 h exposure period, cell viability was assessed via the resazurin reduction assay [48]. Briefly, 30 μL of a 0.01% resazurin solution (Sigma-Aldrich) was added to each well and incubated for 4 h at 37 °C. The reduction of resazurin to resorufin by metabolically active cells was quantified using a ClarioSTAR microplate reader (BMG Labtech), with excitation and emission wavelengths set at 520 nm and 580 nm, respectively. All experiments were performed in triplicate, and cell viability was expressed as a percentage relative to untreated control wells.
5. Conclusions
This study provides a structural and mechanistic comparison of the inhibition of human neutrophil elastase by Sivelestat and Alvelestat under both physiological and oxidative conditions. While Sivelestat exhibits superior baseline potency under physiological conditions, its covalent inhibitory mechanism is selectively impaired under oxidative stress due to disruption of the precise geometric requirements for acyl–enzyme formation. The experimental data therefore indicate that oxidative stress selectively compromises the efficacy of the covalent inhibitor without significantly affecting the intrinsic catalytic competence of the enzyme. The computational analyses provide a plausible structural model to rationalize this observation. Oxidized HNE variants suggest that oxidative modifications affecting the region surrounding the Cys191-Cys220 disulfide bridge may increase the flexibility of the S1 subsite and perturb the geometry of the oxyanion-hole region. Such localized structural changes could interfere with the precise spatial arrangement required for formation of the acyl–enzyme intermediate with Sivelestat. In contrast, Alvelestat retains inhibitory efficacy following oxidative pre-treatment of HNE. This behavior is consistent with its non-covalent binding mode, which appears less sensitive to moderate structural perturbations of the catalytic pocket. Together, these results highlight the importance of considering redox-dependent structural perturbations of the target enzyme when evaluating inhibitor performance. In particular, they reveal a functional decoupling between preserved enzymatic activity and impaired covalent inhibition under oxidative stress. From a translational perspective, redox-resilient, conformationally adaptable inhibitors may therefore represent more robust therapeutic candidates for inflammatory lung diseases characterized by sustained ROS production, including COPD, cystic fibrosis, and severe viral pneumonia. More broadly, these observations support the inclusion of redox-resilience as an additional design parameter in the development of next-generation HNE inhibitors, emphasizing the need to evaluate inhibitor performance not only under ideal biochemical conditions but also within pathophysiologically relevant oxidative environments. Future studies should extend these findings to more complex disease models to further validate their translational relevance and guide the design of HNE inhibitors better suited to operate under sustained oxidative stress. Overall, these findings highlight that effective inhibition in inflammatory settings depends not only on binding affinity, but also on the ability of the inhibitor to withstand the dynamic and oxidative nature of the target microenvironment.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Molecular dynamics analysis of the Alvelestat (ALV) complex with HNE in the wild-type (wt) and oxidized (OX2) forms; Figure S2: 100 ns MD simulation of the wtHNE–Sivelestat pre-complex; Figure S3: Effect of disulfide bond overoxidation on HNE flexibility and catalytic geometry; Figure S4: Mass spectra of Alvelestat and Sivelestat under control and oxidative conditions..
Author Contributions
Conceptualization, M.D., P.I., and S.V.; methodology, M.D., P.L., L.R.C., P.I., and S.V.; formal analysis, M.D., P.L., L.R.C., G.P., P.I. and S.V.; investigation, M.D., P.L., L.R.C., and G.P.; resources, P.L., G.P., P.I., S.C., and S.V; writing—original draft preparation, M.D., P.L., L.R.C., P.I. and S.V.; writing—review and editing, M.D., P.L., L.R.C., G.P., P.I., M.A.G., M.G., T.R., and S.V.; visualization, P.L., L.R.C., G.P., and S.V.; supervision, P.I. and S.V.; funding acquisition, S.V. All authors have read and agreed to the published version of the manuscript.
Funding
The authors wish to thank the Italian Ministry of Research and University for funding this paper in the framework of the project PRIN 2022F5N25M.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Acknowledgments
The authors are deeply grateful to Dr. Monica Campagnoli (Department of Molecular Medicine, University of Pavia, Italy) for her tireless input in the paper drafting.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| AAT | α1-antitrypsin deficiency |
| ACN | Acetonitrile |
| ARDS | Acute respiratory distress syndrome |
| CID | Collision-induced dissociation |
| COPD | Chronic obstructive pulmonary disease |
| DA | Diode array |
| DMEM | Dulbecco’s modified eagle medium |
| DMSO | Dimethyl sulfoxide |
| ESI | Electrospray ionization |
| FBS | Fetal bovine serum |
| HNE | Human neutrophil elastase |
| IC50 | Half maximal inhibitory concentration |
| Ki | Inhibitory constant |
| LTQ | Linear trap quadrupole |
| LC-MS/MS | Liquid chromatography tandem mass spectrometry |
| MD | Molecular dynamics |
| ROS | Reactive oxygen species |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| RP-HPLC | Reverse-phase high performance liquid chromatography |
| SLPI | Secretory leukocyte protease inhibitor |
| TCA | Trichloroacetic acid |
| TIP3P | Transferable Intermolecular Potential 3-Point |
References
- Weissmann, G.; Smolen, J.E.; Korchak, H.M. Release of inflammatory mediators from stimulated neutrophils. N. Engl. J. Med. 1980, 303, 27–34. [Google Scholar] [CrossRef]
- Owen, C.A.; Campbell, M.A.; Sannes, P.L.; Boukedes, S.S.; Campbell, E.J. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J. Cell. Biol. 1995, 131, 775–789. [Google Scholar] [CrossRef]
- Korkmaz, B.; Moreau, T.; Gauthier, F. Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie 2008, 90, 227–242. [Google Scholar] [CrossRef]
- Weiss, S.J. Tissue destruction by neutrophils. N. Engl. J. Med. 1989, 320, 365–376. [Google Scholar] [CrossRef]
- Smallman, L.A.; Hill, S.L.; Stockley, R.A. Reduction of ciliary beat frequency in vitro by sputum from patients with bronchiectasis: a serine proteinase effect. Thorax 1984, 39, 663–667. [Google Scholar] [CrossRef]
- Amitani, R.; Wilson, R.; Rutman, A.; Read, R.; Ward, C.; Burnett, D.; Stockley, R.A.; Cole, P.J. Effects of human neutrophil elastase and Pseudomonas aeruginosa proteinases on human respiratory epithelium. Am. J. Respir. Cell Mol. Biol. 1991, 4, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Demkow, U.; van Overveld, F.J. Role of elastases in the pathogenesis of chronic obstructive pulmonary disease: implications for treatment. Eur. J. Med. Res. 2010, 15, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Stockley, R.A. Neutrophils and protease-antiprotease imbalance. Am. J. Respir. Crit. Care Med. 1999, 160, S49–S52. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The discovery of α1-antitrypsin and its role in health and disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef]
- De Serres, F.; Blanco, I. Role of alpha-1 antitrypsin in human health and disease. J. Intern. Med. 2014, 276, 311–335. [Google Scholar] [CrossRef]
- Jackson, P.L.; Xu, X.; Wilson, L.; Weathington, N.M.; Clancy, J.P.; Blalock, J.E.; Gaggar, A. Human neutrophil elastase-mediated cleavage sites of MMP-9 and TIMP-1: implications to cystic fibrosis proteolytic dysfunction. Mol. Med. 2010, 16, 159–166. [Google Scholar] [CrossRef]
- Sandhaus, R.A.; Turino, G. Neutrophil elastase-mediated lung disease. COPD 2013, 10, 60–63. [Google Scholar] [CrossRef]
- Voynow, J.A.; Shinbashi, M. Neutrophil elastase and chronic lung disease. Biomolecules 2021, 11, 1065. [Google Scholar] [CrossRef]
- Rahman, I.; Adcock, I.M. Oxidative stress and redox regulation in lung inflammation. Eur. Respir. J. 2006, 28, 219–242. [Google Scholar] [CrossRef]
- Boukhenouna, S.; Wilson, M.A.; Bahmed, K.; Kosmider, B. Reactive oxygen species in chronic obstructive pulmonary disease. Oxid. Med. Cell. Longev. 2018, 2018, 5730395. [Google Scholar] [CrossRef]
- Park, W.H. The mitochondrial nexus: Dysfunction, inhibition, and therapeutic frontiers in lung disease. Respir. Med. 2025, 250, 108506. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J. Myeloperoxidase: friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef] [PubMed]
- Nishinaka, Y.; Arai, T.; Adachi, S.; Takaori-Kondo, A.; Yamashita, K. Singlet oxygen is essential for neutrophil extracellular trap formation. Biochem. Biophys. Res. Commun. 2011, 413, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.J.; Cooper, P.R.; Ling, M.R.; Wright, H.J.; Huissoon, A.; Chapple, I.L. Hypochlorous acid regulates neutrophil extracellular trap release in humans. Clin. Exp. Immunol. 2012, 167, 261–268. [Google Scholar] [CrossRef]
- Lin, W.; Chen, H.; Chen, X.; Guo, C. The roles of neutrophil-derived myeloperoxidase (MPO) in diseases: the new progress. Antioxidants (Basel) 2024, 13, 132. [Google Scholar] [CrossRef]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef]
- Siddiqui, T.; Zia, M.K.; Ali, S.S.; Rehman, A.A.; Ahsan, H.; Khan, F.H. Reactive oxygen species and anti-proteinases. Arch. Physiol. Biochem. 2016, 122, 1–7. [Google Scholar] [CrossRef]
- Vogt, W. Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radic. Biol. Med. 1995, 18, 93–105. [Google Scholar] [CrossRef]
- Marinaccio, L.; Stefanucci, A.; Scioli, G.; Della Valle, A.; Zengin, G.; Cichelli, A.; Mollica, A. Peptide human neutrophil elastase inhibitors from natural sources: an overview. Int. J. Mol. Sci. 2022, 23, 2924. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, M.P.; Schepetkin, I.A.; Quinn, M.T.; Cantini, N.; Crocetti, L.; Guerrini, G.; Iacovone, A.; Paoli, P.; Rossi, P.; Bartolucci, G. Synthesis, biological evaluation, and molecular modelling studies of potent human neutrophil elastase (HNE) inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 1108–1124. [Google Scholar] [CrossRef] [PubMed]
- Ocampo-Gallego, J.S.; Pedroza-Escobar, D.; Caicedo-Ortega, A.R.; Berumen-Murra, M.T.; Novelo-Aguirre, A.L.; de Sotelo-León, R.D.; Delgadillo-Guzmán, D. Human neutrophil elastase inhibitors: Classification, biological-synthetic sources and their relevance in related diseases. Fundam. Clin. Pharmacol. 2024, 38, 13–32. [Google Scholar] [CrossRef]
- Iwata, K.; Doi, A.; Ohji, G.; Oka, H.; Oba, Y.; Takimoto, K.; Igarashi, W.; Gremillion, D.H.; Shimada, T. Effect of neutrophil elastase inhibitor (Sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): a systematic review and meta-analysis. Intern. Med. 2010, 49, 2423–2432. [Google Scholar] [CrossRef] [PubMed]
- Sahebnasagh, A.; Saghafi, F.; Safdari, M.; Khatami, S.; Sadremomtaz, A.; Talaei, Z.; Rezai Ghaleno, H.; Bagheri, M.; Habtemariam, S.; Avan, R. Neutrophil elastase inhibitor (Sivelestat) may be a promising therapeutic option for management of acute lung injury/acute respiratory distress syndrome or disseminated intravascular coagulation in COVID-19. J. Clin. Pharm. Ther. 2020, 45, 1515–1519. [Google Scholar] [CrossRef]
- Crocetti, L.; Giovannoni, M.P.; Cantini, N.; Guerrini, G.; Vergelli, C.; Schepetkin, I.A.; Khlebnikov, A.I.; Quinn, M.T. Novel sulfonamide analogs of Sivelestat as potent human neutrophil elastase inhibitors. Front. Chem. 2020, 8, 795. [Google Scholar] [CrossRef] [PubMed]
- Vogelmeier, C.; Aquino, T.O.; O’Brien, C.D.; Perrett, J.; Gunawardena, K.A. A randomised, placebo-controlled, dose-finding study of AZD9668, an oral inhibitor of neutrophil elastase, in patients with chronic obstructive pulmonary disease treated with tiotropium. COPD 2012, 9, 111–120. [Google Scholar] [CrossRef]
- Stockley, R.; De Soyza, A.; Gunawardena, K.; Perrett, J.; Forsman-Semb, K.; Entwistle, N.; Snell, N. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir. Med. 2013, 107, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Nordenmark, L.H.; Taylor, R.; Jorup, C. Feasibility of computed tomography in a multicenter COPD trial: a study of the effect of AZD9668 on structural airway changes. Adv. Ther. 2015, 32, 548–566. [Google Scholar] [CrossRef]
- von Nussbaum, F.; Li, V.M. Neutrophil elastase inhibitors for the treatment of (cardio)pulmonary diseases: Into clinical testing with pre-adaptive pharmacophores. Bioorg. Med. Chem. Lett. 2015, 25, 4370–4381. [Google Scholar] [CrossRef] [PubMed]
- Hansen, G.; Gielen-Haertwig, H.; Reinemer, P.; Schomburg, D.; Harrenga, A.; Niefind, K. Unexpected active-site flexibility in the structure of human neutrophil elastase in complex with a new dihydropyrimidone inhibitor. J. Mol. Biol. 2011, 409, 681–691. [Google Scholar] [CrossRef]
- Sjö, P. Neutrophil elastase inhibitors: recent advances in the development of mechanism-based and nonelectrophilic inhibitors. Future Med. Chem. 2012, 4, 651–660. [Google Scholar] [CrossRef]
- Viglio, S.; Zanaboni, G.; Luisetti, M.; Cetta, G.; Guglielminetti, M.; Iadarola, P. Micellar electrokinetic chromatography: a convenient alternative to colorimetric and high performance liquid chromatographic detection to monitor protease activity. Electrophoresis 1998, 19, 2083–208. [Google Scholar] [CrossRef]
- Griffiths, S.W.; King, J.; Cooney, C.L. The reactivity and oxidation pathway of cysteine 232 in recombinant human alpha 1-antitrypsin. J Biol Chem 2002, 277, 25486–25492. [Google Scholar] [CrossRef]
- Garrido Ruiz, D.; Sandoval-Perez, A.; Rangarajan, A.V.; Gunderson, E.L.; Jacobson, M.P. Cysteine oxidation in proteins: structure, biophysics, and simulation. Biochemistry 2022, 61, 2165–2176. [Google Scholar] [CrossRef]
- Du, S.; Kretsch, R.C.; Parres-Gold, J.; Pieri, E.; Cruzeiro, V.W.D.; Zhu, M.; Pinney, M.M.; Yabukarski, F.; Schwans, J.P.; Martínez, T.J.; et al. Conformational ensembles reveal the origins of serine protease catalysis. Science 2025, 387, eado5068. [Google Scholar] [CrossRef] [PubMed]
- Viglio, S.; Bak, E.G.; Schouten, I.G.M.; Iadarola, P.; Stolk, J. Protease-specific biomarkers to analyse protease inhibitors for emphysema associated with alpha 1-antitrypsin deficiency. An overview of current approaches. Int. J. Mol. Sci. 2021, 22, 1065. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef]
- Peskin, A.V.; Winterbourn, C.C. Kinetics of the reactions of hypochlorous acid and amino acid chloramines with thiols, methionine, and ascorbate. Free Radic. Biol. Med. 2001, 30, 572–579. [Google Scholar] [CrossRef]
- Oettl, K.; Stauber, R.E. Physiological and pathological changes in the redox state of human serum albumin critically influence its binding properties. Br. J. Pharmacol. 2007, 151, 580–590. [Google Scholar] [CrossRef]
- Paramasivan, S.; Adav, S.S.; Ngan, S.C.; Dalan, R.; Leow, M.K.; Ho, H.H.; Sze, S.K. Serum albumin cysteine trioxidation is a potential oxidative stress biomarker of type 2 diabetes mellitus. Sci. Rep. 2020, 10, 6475. [Google Scholar] [CrossRef] [PubMed]
- Sharar, M.; Saied, E.M.; Rodriguez, M.C.; Arenz, C.; Montes-Bayón, M.; Linscheid, M.W. Elemental labelling and mass spectrometry for the specific detection of sulfenic acid groups in model peptides: a proof of concept. Anal. Bioanal. Chem. 2017, 409, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Höhn, A. Protein oxidation - Formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol. 2021, 42, 101901. [Google Scholar] [CrossRef]
- Copeland, R.A. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis; John Wiley & Sons Inc.: New York, NY, USA, 2000. [Google Scholar]
- Gonzalez, R.J.; Tarloff, J.B. Evaluation of hepatic subcellular fractions for Alamar blue and MTT reductase activity. Toxicol. In Vitro 2001, 15, 257–259. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Predicted binding mode of Alvelestat (yellow sticks) within the wt HNE binding site (PDB ID: 2Ζ7F, gray surface/cartoon). Key residues are shown as gray lines; H-bonds are indicated by yellow dashed lines. Atoms are colored coded: oxygen in red, nitrogen in blue, sulphur in yellow.
Figure 1.
Predicted binding mode of Alvelestat (yellow sticks) within the wt HNE binding site (PDB ID: 2Ζ7F, gray surface/cartoon). Key residues are shown as gray lines; H-bonds are indicated by yellow dashed lines. Atoms are colored coded: oxygen in red, nitrogen in blue, sulphur in yellow.

Figure 2.
Induced-fit and covalent docking results for Sivelestat. A, B) Most representative predicted pre-acylation complex of Sivelestat (red sticks) within the HNE binding site from MD simulation (PDB ID: 2Z7F, gray surface/cartoon with key residues shown as gray lines). C) Predicted acylated HNE complex with the pivaloyl moiety of Sivelestat from covalent docking. D) Superposition of the predicted acylated complex with crystal structures of porcine pancreatic elastase acylated at Ser195 with an m-toluoylcarbonyl group (PDB ID: 8B53, yellow) and a cyclopropylcarbonyl group (PDB ID: 8B1Y, pale green). H-bonds and π−π stackings are indicated by yellow and blue dashed lines, respectively. Atoms are colored coded: oxygen in red, nitrogen in blue, sulphur in yellow.
Figure 2.
Induced-fit and covalent docking results for Sivelestat. A, B) Most representative predicted pre-acylation complex of Sivelestat (red sticks) within the HNE binding site from MD simulation (PDB ID: 2Z7F, gray surface/cartoon with key residues shown as gray lines). C) Predicted acylated HNE complex with the pivaloyl moiety of Sivelestat from covalent docking. D) Superposition of the predicted acylated complex with crystal structures of porcine pancreatic elastase acylated at Ser195 with an m-toluoylcarbonyl group (PDB ID: 8B53, yellow) and a cyclopropylcarbonyl group (PDB ID: 8B1Y, pale green). H-bonds and π−π stackings are indicated by yellow and blue dashed lines, respectively. Atoms are colored coded: oxygen in red, nitrogen in blue, sulphur in yellow.

Figure 3.
IC50 determination of Alvelestat (black circle) and Sivelestat (red square) against HNE activity. Values represent the mean ± standard deviation obtained from three separate experiments.
Figure 3.
IC50 determination of Alvelestat (black circle) and Sivelestat (red square) against HNE activity. Values represent the mean ± standard deviation obtained from three separate experiments.

Figure 4.
Global reciprocal plot of data from HNE steady-state kinetics analysis, in the presence of different concentrations (from 0.1 μM to 100 μM ) of Alvelestat (panel A) or Sivelestat (Panel B). Values represent the mean ± SD obtained from three separate experiments.
Figure 4.
Global reciprocal plot of data from HNE steady-state kinetics analysis, in the presence of different concentrations (from 0.1 μM to 100 μM ) of Alvelestat (panel A) or Sivelestat (Panel B). Values represent the mean ± SD obtained from three separate experiments.

Figure 5.
IC50 determination of Alvelestat (panel A) and Sivelestat (panel B) under non-oxidizing (red circles) and oxidizing (black squares) conditions. Values represent the mean ± SD obtained from three separate experiments.
Figure 5.
IC50 determination of Alvelestat (panel A) and Sivelestat (panel B) under non-oxidizing (red circles) and oxidizing (black squares) conditions. Values represent the mean ± SD obtained from three separate experiments.

Figure 6.
Cytotoxicity evaluation of Alvelestat and Sivelestat in A549 cells. Cell viability was assessed after incubation with increasing concentrations of compounds (up to 1000 µM) using the resazurin assay. Resorufin fluorescence was measured at λexc=520 nm and λem=580 nm. Data are expressed as a percentage of the untreated control (100%) and represent the mean of three independent experiments.
Figure 6.
Cytotoxicity evaluation of Alvelestat and Sivelestat in A549 cells. Cell viability was assessed after incubation with increasing concentrations of compounds (up to 1000 µM) using the resazurin assay. Resorufin fluorescence was measured at λexc=520 nm and λem=580 nm. Data are expressed as a percentage of the untreated control (100%) and represent the mean of three independent experiments.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



