Submitted:
24 March 2026
Posted:
26 March 2026
You are already at the latest version
Abstract
Background/objective: Approximately 90% of breast cancer–related deaths result from recurrence and metastasis. Emerging evidence indicates that tumor recurrence, invasion, and metastatic spread are strongly influenced by both the tumor microenvironment (TME) and the metastatic niche. M2 macrophages promote immune suppression, inhibit inflammation, and facilitate epithelial-to-mesenchymal transition, invasion, angiogenesis, and tumor progression—effects that are particularly pronounced in triple-negative breast cancer (TNBC). The objectives of this study were to develop engineered exosomes to selectively deplete M2 macrophages to delay the growth of primary tumor and distal metastasis and enhance overall survival. Methods: Engineered exosomes were developed using our invented platform to selectively target and deplete alternatively activated CD206⁺ M2 macrophages in primary and metastatic TMEs via antibody-dependent cell-mediated cytotoxicity (ADCC). Engineered exosomes were characterized for size, zeta potential, and successful incorporation of targeting peptides and proteins. Whole-body and tumor-specific biodistribution were assessed. In vitro and in vivo experiments were conducted to evaluate targeting specificity. Toxicity and immunogenicity were examined in immune-competent animal models. Two treatment paradigms were employed. Results: Engineered exosomes containing M2-macrophage targeting peptides and Fc-mIgG2b were successfully made and there were no significant size differences between engineered and control exosomes. Both in vitro and in vivo studies confirmed the specificity of the engineered exosomes. Biodistribution studies showed no significant uptake to the resident macrophages in the lung and liver. No significant immune activation, based on cytokine profiling, or organ-specific toxicity was observed in immune-competent models. Flowcytometry studies using splenocytes showed significant depletion of M2-macrophages following treatments with engineered exosomes, however, there was no effect on the distribution of T-cells. M2-targeting therapeutic exosomes significantly delayed the growth of primary tumors and metastatic lesions. Conclusion: These findings support the potential of precision exosome-based strategies for enhancing therapeutic outcomes in breast cancer.
Keywords:
breast cancer
; exosomes
; tumor associated macrophages
; tumor microenvironment
; antibody dependent cellular cytotoxicity
1. Introduction
Approximately 90% of breast cancer–related deaths are attributable to recurrence and metastasis. Emerging evidence indicates that tumor recurrence, invasion, and metastatic spread are strongly influenced by both the tumor microenvironment (TME) and the metastatic niche. [1] The TME consists of a dynamic and complex network of cancer cells, stromal cells, immune cells (including myeloid cells, T cells, B cells), and endothelial cells. These components interact through the extracellular matrix, soluble mediators such as cytokines and chemokines, and environmental factors including hypoxia. [2,3,4,5]. Within this milieu, interactions among T cells, B cells, natural killer (NK) cells, macrophages, granulocytes (including neutrophils), and dendritic cells can either promote or inhibit tumor progression. [3,5,6,7] Macrophages exhibit remarkable plasticity and undergo functional reprogramming in response to environmental cues, broadly categorized into two polarization states: M1 (classically activated) and M2 (alternatively activated and immunosuppressive). [8,9] M1 macrophages typically express high levels of major histocompatibility complex class II (MHC II), CD68, and the co-stimulatory molecules CD80 and CD86, but lack CD206 expression. In contrast, M2 macrophages express high levels of MHC II, CD163, CD206 (also known as MRC1), and in mice arginase-1 (Arg-1), among other markers. Functionally, M2 macrophages secrete elevated levels of IL-10 and transforming growth factor-β (TGF-β), along with low levels of IL-12 and IL-23, reflecting a type-2 cytokine profile. They also produce chemokines such as CCL17, CCL22, and CCL24, which promote recruitment of regulatory T cells (Tregs), Th2 cells, eosinophils, and basophils, thereby establishing a highly immunosuppressive tumor microenvironment. [10,11] Clinical studies have demonstrated that tumors with high MRC1 expression are associated with poor prognosis, including reduced overall and disease-free survival. [12,13,14] CD206, or the mannose receptor (MR), is a 175 kDa type I transmembrane protein predominantly expressed on alternatively activated M2 macrophages and certain tissue-resident macrophages, particularly in the lungs, spleen, and liver. [15] Based on these observations, we hypothesize that selective targeting and depletion of M2 macrophages using a therapeutic strategy could reprogram the immunosuppressive TME, inhibit tumor growth and metastatic progression, and ultimately improve survival in breast cancer.
Exosomes are a subtype of extracellular vesicles (EVs), measuring approximately 30-150 nm in diameter, which are secreted by most cell types into the extracellular space. They modulate the microenvironment through cell-to-cell communication by fusing with the plasma membrane, undergoing endocytosis, and releasing their molecular cargo into recipient cells. [16,17,18,19,20,21] Regardless of their cellular origin, exosomes share common structural and molecular features, including tetraspanins (CD9, CD63, CD81), heat shock proteins (Hsp60, Hsp70, Hsp90), biogenesis-associated proteins (Alix, TSG101), membrane transport and fusion proteins (GTPases, annexins, Rab proteins), nucleic acids (mRNAs, miRNAs, long non-coding RNAs, and DNA), and lipids such as cholesterol and ceramide. [17,22,23] Due to their intrinsic biocompatibility, low toxicity and immunogenicity, ability to cross biological barriers including the blood–brain barrier (BBB) stability in circulation, and preferential accumulation at pathological sites, exosomes have emerged as promising therapeutic delivery vehicles. [24,25,26,27,28,29,30] Consequently, engineered exosomes have been developed to display biologically active proteins on their surface or encapsulate therapeutic agents for targeted delivery. [26,31,32,33,34] Recently, our laboratory has achieved several advances in exosome technology: (1) development of a proprietary platform (US Patent Application 17/083,124) for generating engineered exosomes that express specific cell-targeting peptides for in vivo detection; (2) application of these engineered exosomes as therapeutic probes to selectively deplete target cells; (3) optimization of scalable methods for rapid production of large quantities of uniform-sized exosomes from diverse cell sources; and (4) demonstration of differential biodistribution of exosomes in tumor-bearing models using clinically relevant single-photon emission computed tomography (SPECT) imaging. [35,36] We hypothesize that selectively targeting M2 macrophages within breast cancer TME using engineered therapeutic exosomes functionalized with an M2-specific targeting moiety and the Fc portion of mouse IgG2b (Fc-mIgG2b) to induce antibody-dependent cell-mediated cytotoxicity (ADCC) will reprogram the immunosuppressive microenvironment, inhibit tumor growth and metastasis, and improve survival outcomes.
In recent years, investigators have identified a peptide sequence, CSPGAK (6aa, targets both mouse and human cells) and its cyclic form CSPGAKVRC (9aa), that binds specifically to CD206+ M2 macrophages in tumors and sentinel lymph nodes in different tumor models. [37,38] The primary objective of this study was to evaluate the efficacy of non-tumorigenic HEK293 cell–derived engineered exosomes carrying these precision-targeting peptide and a therapeutic Fc-mIgG2b payload to selectively target and deplete alternatively activated CD206⁺ M2 macrophages within primary and metastatic TMEs in vivo via ADCC. We further aimed to determine whether modulation of the immunosuppressive TME using this strategy could reduce tumor burden and enhance survival.
2. Materials and Methods
All animals were kept under regular barrier conditions at room temperature with exposure to light for 12 hours and dark for 12 hours. Food and water were offered ad libitum. Syngeneic breast cancer cells were implanted orthotopically in the lower right breast in female animals with bodyweight between 20-25 g under isoflurane anesthesia. The depth of anesthesia was checked by pinching the skin or toe. The humane endpoint of the survival studies was the fulfilment of the criteria for euthanasia at the end of the survival studies (survival), which was by determining body weight (loss of more than 15% of baseline body weight), tumor size more than 2.0 cm in longitudinal diameter, ulceration, moribund, inability to drink/eat. All animals were checked 2-3 times a week. All animals were treated with soft chows, apples, and subcutaneous fluid when they started to show signs of weight loss and checked 2-3 times a week. The animals were humanely euthanized once the euthanasia criteria were achieved. All efforts were made to ameliorate the suffering of the animals. CO2 (displacement rate of 30-70% of the chamber volume with CO2 per minute) with a secondary method (bilateral thoracotomy or collection of major organs) was used to euthanize animals for tissue collection. Death was confirmed by established criteria of lack of breathing, lack of corneal reflex, lack of response to a firm toe pinch, and rigor mortis. All mice were randomized. Studies were conducted in multiple batches and each batch had animals for experimental and corresponding control groups. Personnel blinded to the various groups analyzed the data. The animals were of the same age and weight and had similar baseline behavior.
2.2. Cell lines, Peptides, and Proteins
Syngeneic, aggressive triple-negative breast cancer (TNBC) mouse cell lines 4T1 (for Balb/c mice) and AT3 (for C57BL/6 mice) were obtained from commercial sources and maintained in the laboratory in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), sodium pyruvate, non-essential amino acids, sodium glutamate, and antibiotics. Cells were harvested for implantation at 75–80% confluence. Approximately 2.5 × 10⁴ cells suspended in 20 µL of Matrigel were orthotopically implanted into the lower right mammary fat pad under anesthesia.
HEK293 cells were used to produce engineered exosomes. These cells were transduced with lentiviral vectors encoding CD206⁺ M2 macrophage–targeting peptides and Fc-mIgG2b, enabling surface expression of these constructs on the generated exosomes. Primary mouse bone marrow–derived macrophages (MBMDMs) were isolated from tumor-bearing mice, cultured in vitro, and polarized toward an M2 phenotype using GM-CSF, IL-13, and IL-4. Commercial mouse macrophage cell lines Raw 264.7 and J774A.1 were used for in vitro experiments and similarly polarized to the M2 phenotype with IL-13 and IL-4.
To evaluate the specificity of the precision peptide carrying engineered exosomes, targeting peptides (6-amino-acid and 9-amino-acid sequences for blocking studies) and a fusion protein consisting of the 9-amino-acid targeting peptide linked to Fc-mIgG2b were synthesized by a commercial vendor (GeneScript, NJ, USA).
2.3. Experimental Animal Model
For this study, syngeneic metastatic TNBC models were established in immunocompetent female Balb/c mice using 4T1 cells (aggressive TNBC) and in female C57BL/6 mice using AT3 cells (slow-growing TNBC). Mice were randomized prior to treatment allocation. Experiments were performed in multiple batches, with each batch including both experimental and corresponding control groups. Data analysis was conducted by personnel blinded to group assignments. Treatment was initiated on day 8 after tumor implantation, when most tumors measured approximately 3 mm in diameter.
To generate a resection model for evaluating tumor recurrence and metastasis, primary tumors were surgically removed under anesthesia on day 11 post-implantation, following the protocol described in our previously published report. [39]
2.4. Biogenesis, Characterization and Determination of In Vitro Specificity of Engineered Exosomes
Supplemental Figure 1 (Figure S1) illustrates the vector design and a schematic representation of antibody-dependent cellular cytotoxicity (ADCC) mediated by engineered exosomes. Plasmids encoding CD206⁺ M2 macrophage–targeting peptides (6-amino-acid or 9-amino-acid sequences) and Fc-mIgG2b were generated by a commercial provider (VectorBuilder, USA). Lentiviral particles carrying these constructs were produced in-house using HEK-293TN cells and a standard packaging system (Addgene, USA).
HEK293 cells were transduced with the lentiviral vectors and selected using puromycin. Successful transduction was confirmed by fluorescence microscopy based on mCherry expression. Stably transduced (mCherry⁺) cells were then cultured in exosome-depleted media, and conditioned supernatants were collected at 48 and 72 hours following media replacement. Typically, 6 × 10⁶ cells were seeded in a T75 flask, yielding approximately 2 × 10¹⁰ exosomes over 3 days.
Exosomes were isolated and concentrated from collected supernatants using our previously published and optimized protocols. [35,36] Purified exosomes were aliquoted and stored at -20°C until further use. Genomic DNA extracted from transduced (mCherry⁺) cells was analyzed by polymerase chain reaction (PCR) to confirm the presence of inserted sequences encoding the M2 macrophage–targeting peptides and Fc-mIgG2b. Primers were obtained from Integrated DNA Technologies (IDT, IA, USA). The primer sequences were as follows:
- CD206 9aa peptide (CSPGAKVRC)
Forward: 5′-TGCTCTCCGGGGGCGAAA-3′
Reverse: 5′-CAGTCCTGGTGCTGGATGGG-3′
- Fc-mIgG2b
Forward: 5′-GGGCCCATTTCAACAATCAACC-3′
Reverse: 5′-CTGATGTCTCCAGGGTTGAAGCCC-3′
Each batch of engineered exosomes was characterized by nanoparticle tracking analysis (NTA) to determine particle size distribution and zeta potential. Western blot analysis was performed to confirm the presence of exosomal markers (e.g., CD9). Immuno-transmission electron microscopy (TEM) was used to assess exosome morphology, size, and surface expression of inserted IgG2b. For immune-TEM, a specific antibody against Fc-mIgG2b (Jackson ImmunoResearch Laboratories, Inc., PA, USA) was utilized.
Mouse embryonic fibroblasts (negative control) and MBMDMs were used to assess targeting specificity. After plating and polarization of MBMDMs to the M2 macrophage phenotype, DiI-labeled engineered exosomes were added and incubated for 1-2 hours. For blocking experiments, polarized cells were pre-incubated with CD206⁺ M2 macrophage-targeting peptides prior to the addition of engineered exosomes. Following incubation, cells were washed with PBS, fixed with paraformaldehyde, and counterstained with DAPI. Fluorescence images were captured from randomly selected fields and quantified using ImageJ (color threshold method).
To evaluate whether engineered exosomes remained on the cell surface, murine macrophage J774A.1 cells were polarized to the CD206⁺ M2 phenotype and incubated with DiI-labeled engineered exosomes. After washing, cells were cultured in exosome-free media and collected at 0.5, 1, 3, 6, and 24 hours. Cells were fixed, counterstained with DAPI, and examined by fluorescence microscopy to determine the localization of DiI-labeled exosomes on the cell surface.
2.5. Biodistribution of Control and Engineered Exosomes
CD206, also known as the mannose receptor (MR), is a 175-kDa type I transmembrane protein predominantly expressed by alternatively activated M2 macrophages and tissue-resident macrophages, particularly in the lungs, spleen, and liver. [15] Given this expression pattern, it was essential to determine whether CD206-targeting engineered exosomes preferentially accumulate in organs enriched with resident macrophages, such as the lungs and liver.
Control exosomes (derived from HEK293 cells) and engineered CD206-targeting exosomes were radiolabeled with iodine-131 (I-131) according to our previously published protocol. [35] A total of 10 female Balb/c mice were used. Radiolabeled control or engineered exosomes were administered intravenously, and animals underwent single-photon emission computed tomography (SPECT) imaging at 5 minutes, 3 hours, and 24 hours post-injection.
Whole-body SPECT images were acquired using a dedicated four-head NanoScan high-sensitivity microSPECT/CT 4R system (Mediso, Boston, MA, USA) equipped with high-resolution multi-pinhole collimators (100 pinholes total). The microSPECT system operates across an energy range of 20–600 keV and provides a spatial resolution of 275 µm. Imaging was performed using 60 projection views at 30–60 seconds per projection with a medium field of view. Attenuation correction was applied using concurrently acquired CT images, followed by image reconstruction with low-iteration, low-filtered back projection. Animals were maintained under anesthesia throughout the imaging procedure. Body temperature was maintained at 37°C, and respiration was continuously monitored.
After image reconstruction, radioactivity in the whole body, lungs, heart, and liver was quantified using ImageJ software (NIH). Organ-specific activity (lungs, heart, liver) was normalized to total whole-body activity and expressed as percentage of whole-body activity (%WB). The biodistribution profiles of control and engineered exosomes in the lungs, liver, and heart were then compared.
2.6. In vivo Specificity by SPECT Imaging
The in vivo specificity of CD206⁺ M2 macrophage–targeting peptides and engineered exosomes has been previously reported. [36] In the present study, we further evaluated the specificity of engineered exosomes and a CD206-targeting fusion protein and compared them with control exosomes (HEK293-derived) and anti-CD206 antibodies.
A total of 10 female Balb/c mice bearing metastatic breast tumors were included (n = 3 engineered exosomes, n = 3 fusion protein, n = 2 HEK293 control exosomes, n = 2 anti-CD206 antibody). A metastatic breast cancer model was established in Balb/c mice, and SPECT imaging was performed 5 weeks after tumor implantation to assess whole-body biodistribution as well as distribution toprimary tumors and metastatic lesions.
Control and engineered exosomes, the fusion protein, and anti-CD206 antibody were radiolabeled with iodine-131 (I-131) according to our previously published protocols. [35,40] Radiolabeled agents were administered intravenously, and SPECT/CT imaging was obtained 3 hours post-injection, as described in our reported studies. [35,36]
2.7. In vivo Depletion of M2 Macrophages and CD11b⁺ Myeloid Cells
The in vivo specificity and dose-escalation studies of CD206⁺ M2 macrophage–targeting peptides and engineered exosomes were previously reported. [36] In the present study, we investigated the specificity of engineered exosomes and fusion protein and compared them with that of control exosomes (HEK293 exosomes) and anti-CD206 antibodies for depleting M2 macrophages and T-cells. Three doses (3 days/week) of control exosomes (HEK exo) engineered exosomes (CD206 exo, 3x10[9] exosomes/dose), fusion protein (50 µg/day), and anti-mCD206 antibody (50 µg/day) were administered in the respective group of animals (Balb/c, n = 3 per group). The spleens were collected 24 hours after the last dose and a single cell suspension was made, and different immune cell populations, including M2 macrophages and CD11b⁺ myeloid cells, were quantified by flow cytometry.
2.8. Immunogenicity and Toxicity of HEK293 Cell–Derived Engineered Exosomes
To evaluate potential immunogenic and inflammatory responses following systemic administration of HEK293-derived engineered exosomes, non–tumor-bearing female Balb/c (n = 6) and C57BL/6 (n = 6) mice received six intravenous doses of either PBS (vehicle control) or engineered exosomes over a 6-week period. Twenty-four hours after the final dose, animals were euthanized, and blood plasma was collected for membrane array analysis to quantify pro-inflammatory, immunogenic, and immunosuppressive cytokines, and growth factors.
In a separate long-term toxicity study, groups of mice (n = 3) were euthanized 6 months after receiving six doses of HEK293-derived exosomes. Blood plasma was collected to assess the presence of mouse anti-human antibodies (MAHA). Major organs including liver, spleen, lungs, kidneys, and heart were harvested for histopathological evaluation using hematoxylin and eosin (H&E) staining to assess potential cellular or tissue-level toxicities.
2.9. In vivo Validation of NK Cell–Mediated ADCC
In our previous study, we demonstrated that splenocytes containing NK cells contribute to the depletion of polarized M2 macrophages in vitro. [36] In the present study, we investigated whether in vivo depletion of CD206⁺ M2 macrophages by engineered exosomes was dependent on NK cell–mediated ADCC.
An orthotopic AT3 breast cancer model was established in female C57BL/6 mice. Beginning on day 8 after tumor implantation, randomly assigned animals received four doses of one of the following: vehicle (PBS; n = 3), isotype IgG (n = 3), anti-Gr1 antibody (n = 3), or anti-NK antibody (PK136; n = 3). Subsequently, all groups were treated with four doses of engineered exosomes over a 2-week period to deplete CD206⁺ M2 macrophages.
Tumor volumes and optical images were obtained on days 8 (baseline), 15, and 22. The study rationale was that if depletion of M2 macrophages is mediated through NK cell-dependent ADCC, then NK cell-depleted mice (anti-NK–treated group) would fail to reduce M2 macrophage accumulation in the TME following administration of engineered exosomes, and tumor growth inhibition would not occur. In contrast, vehicle-, isotype IgG–, and anti-Gr1–treated animals without NK cell depletion would retain NK cell function, allowing engineered exosomes to reduce M2 macrophage levels in the TME and consequently suppress tumor growth.
2.10. Changes in the TME Milieu and Cytokine Levels
An orthotopic syngeneic AT3 breast cancer model was established in C57BL/6 female mice. Following tumor implantation, animals were treated with engineered exosomes (3 × 10⁹ exosomes per dose, twice weekly) administered either intratumorally (local delivery) or intravenously (IV). A control group received targeting exosomes administered IV that carried the targeting peptide alone (without mFc-IgG2b). Treatments continued for 2 or 4 weeks. At the respective endpoints, blood plasma, lungs (metastatic site), and primary tumors were harvested, and total protein was extracted. A custom-designed membrane array was used to quantify pro-inflammatory and immunosuppressive cytokines, chemo attractants, and growth factors, as previously described. [35,41]
In a separate experiment, an orthotopic syngeneic 4T1 breast cancer model was generated in Balb/c female mice. Animals were treated with either vehicle (PBS) or engineered exosomes (3 × 10⁹ exosomes per dose, twice weekly for 3 weeks). At the end of the study, single-cell suspensions were prepared from the primary tumor, lungs (potential metastatic site), and spleen. Both surface and intracellular markers were analyzed by flow cytometry to characterize myeloid, T-cell, and dendritic cell populations. Each group included three animals.
2.11. Effect of Engineered Exosomes on Tumor Growth, Recurrence, Metastasis, and Survival
Previous preclinical studies have demonstrated an increase in immunosuppressive cell populations within primary and metastatic breast tumors following chemotherapy and radiotherapy [42,43,44,45,46]. Clinical investigations have similarly reported elevated levels of immunosuppressive cells, including CD206⁺ M2 macrophages, in primary, metastatic, and therapy-resistant breast cancers [42,47,48]. Based on these findings, we hypothesized that depletion of CD206⁺ M2 macrophages within the tumor microenvironment (TME) would delay primary tumor growth and improve overall survival.
Orthotopic AT3 tumor–bearing C57BL/6 mice were treated with either control or engineered exosomes (3 × 10⁹ exosomes per dose, twice weekly) for 3 weeks, beginning on day 8 post-implantation. Tumor growth and survival were monitored throughout the study. Tumor volume, body weight, and general health status were assessed at least twice weekly until predefined euthanasia criteria were met. Each group consisted of five animals.
In a post-surgical model, Balb/c female mice were orthotopically implanted in the lower right mammary fat pad with luciferase-expressing 4T1 breast cancer cells. On day 11 post-implantation, primary tumors were surgically resected. Twenty-four hours after surgery, all animals underwent bioluminescence imaging (BLI) to assess residual tumor burden. Animals with substantial residual disease (n = 3) were excluded from further analysis.
The remaining mice were treated intravenously with either control exosomes (n = 4) or engineered exosomes (n = 3) at a dose of 3 × 10⁹ exosomes per injection. Treatment was initiated 24 hours after tumor resection and administered weekly. Animals were monitored by serial BLI for the development of lung metastases or recurrence at the primary tumor site.
2.12. Statistical Analysis
All data were expressed in mean ± standard error of mean (SEM) unless otherwise stated. The effects of treatments (in vitro and in vivo) were analyzed using ANOVA across treatment groups, followed by pairwise comparisons with Tukey’s adjustment. Bonferroni corrections were applied to multiple related parameters. Survival studies were analyzed by Kaplan-Meier estimate. A p-value of <0.05 was considered significant difference.
3. Results
3.1. Biogenesis of Engineered Exosomes and Determination of Specificity
Figure 1 shows the representative images indicating successful generation of engineered exosomes carrying both M2-macrophage targeting peptides (9aa) and Fc-mIgG2b. Our selection process produced almost 100% mCherry positive cells. DNA collected from the transduced cells show the presence of sequences corresponding to the inserted peptides and Fc-mIgG2b. We have not observed any significant size differences between the control and engineered exosomes (146 ± 125nm vs 133 ± 72.3nm). Western blot study also showed the presence of CD9 markers in the collected exosomes. Immuno-TEM confirmed the size of the exosomes and the presence of Fc-mIgG2b on the surface of the exosomes. We also produced engineered exosomes using M2-macrophages targeting 6aa-peptides and compared with that of 9aa-peptides in respect of specificity using MBMDM. The in vitro studies using primary MBMDM polarized to M2-macrophages showed uptake of the engineered exosomes (carrying either 6aa or 9aa peptides), whereas MEFs showed no uptake of exosomes at all (Figure S2). When the polarized macrophages were pre-incubated with 6aa or 9aa peptides blocking peptides before adding corresponding engineered exosomes, the uptake of engineered exosomes carrying 9aa peptides showed significantly lower uptake by the cells indicating better specificity (Figure S2 and Figure 2). Based on the in vitro specificity and previously reported in vivo specificity of engineered exosomes carrying 9aa peptides, we have decided to perform subsequent studies using control, and therapeutic (carrying both targeting peptide plus mFc-IgG2b) engineered exosomes carrying 9aa peptides only.
To determine whether engineered exosomes remain on the surface of the polarized macrophages for extended time, DiI-labeled exosomes were added in the culture (glass chamber slides) of polarized M2-macrophages (J774A.1) cells and fluorescence microscopic images were taken at 0.5 to 24 hours after fixation of the cells. Figure 3 also showed that engineered exosomes remained on the cell surface for an extended period, indicating that they can act as a bridge to connect targeted cells with the effector cells for an extended period of time without being phagocytosed or internalized.
Figure 4 shows that engineered exosomes detected both primary tumors (white circles) and multiple metastatic foci in the lungs (white arrows) and other parts of the body (white arrows). Fusion protein showed similar results but fewer foci in the lungs, even though all animals showed multiple lung metastases. On the other hand, the anti-CD206 antibody failed to detect primary tumors or lung metastasis. Control (HEK) exosomes showed little uptake in the primary tumor and a few focal activities in the lungs. Randomly selected animals (from engineered and control exosomes administered) showed multiple lung metastases in 5 weeks following orthotopic tumor implantation in the breast. The primary tumor showed many CD206+ (green) cells that received control or engineered exosomes.
3.2. Engineered Exosomes Did not Show Increased Uptake to the Lungs And liver
We determined whole body biodistribution of I-131 tagged control and engineered exosomes using SPECT scanning. Semiquantitative analysis showed no significant differences between the control and engineered exosomes’ distribution of the lungs and liver between 5 minutes to 24 hours. The activity in the heart also showed no significant differences. Most of the activity from the lungs and livers cleared by 24 hrs. Figure 5 shows representative images from engineered exosomes administered animals and analysis of percent whole body activity of the lungs, liver, and heart.
3.3. Engineered Exosomes and Fusion Protein (Bispecific Protein) Depleted M2 Macrophages But Not T-Cells
Therapeutic exosomes and fusion protein (bispecific protein) depleted CD11b+ or CD206+ macrophages significantly compared to control exosomes, and antibody-treated animals (Figure 6). It is worth noting that the decrease in CD11b+ cells was related to the depletion of CD206+ macrophages, as macrophages are also CD11b+. However, there was no alteration of T-cells following the administration of therapeutic exosomes or fusion protein. Additionally, therapeutic exosomes did not deplete CD16+ or NKp46+ NK cells, and F4/80+/CD80+ M1 macrophages. Flowcytometric strategies are shown in Figures S3 and S4.
3.4. Immunogenic or Inflammatory Reaction Was Not Observed Following Administration Human HEK-293 Cells Derived Engineered Exosomes
Our studies have not detected mouse anti-human antibody (MAHA) over 6 months in immunocompetent animals after IV injections of exosomes (siz doses) derived from HEK293 cells (data not shown). We also found no changes in the expression of pro-inflammatory, immunogenic, and immunosuppressive cytokines, as well as chemo attractants in plasma compared to that of control animals (PBS treated) in different immunocompetent animal models (Figure 7).
3.5. NK Cells Were Involved in ADCC to Deplete Targeted M2-Macrophages
There was a significant (20-40 folds) increase (<0.05) in tumor volume (BLI photon intensity) in animals that were treated with anti-NK antibodies indicating that NK cell depletion prevented the killing of M2-macrophages through NK-cell mediated ADCC. However, there were no changes in tumor volume when Gr1+ cells were depleted (Figure 9). It is worth noting that the tumor volume measured by calipers also yielded similar results.
3.6. Changes in the TME Milieu and Cytokine Levels
Engineered exosomes administered locally or intravenously showed increased (>2 folds compared to control targeting exosomes) levels of inflammatory cytokines, different chemokines, and growth factors at the metastatic sites (lungs) following 2 weeks of treatment, which were not seen in plasma or primary tumors (Figure 10). However, following 4 weeks of therapy, most cytokines, chemokines, and growth factors were increased in plasma (Figure 11). It is to be noted that the primary tumor showed lower levels of cytokines, chemokines, and growth factors following both treatment schedules.
There were no significant differences observed in the population of granulocytes, monocytes and macrophages in the primary tumor or in the lungs. The number of cytotoxic T-cells (CD45+CD3+CD8+) was significantly higher in the primary and metastatic TME in therapeutic exosome-treated animals, and there was also an increased number of activated CD4+ T cells (CD45+CD3+CD25+CD69+) (Figure 12). We also observed an increased number of DCs in the lungs and spleen, although significant differences were not achieved (Figure 13). There were plenty of NK cells present in tumors, spleen, and peripheral blood.
3.7. Tumor Growth, Recurrence, Metastasis, and Survival
Engineered exosomes–treated animals showed lower tumor volumes throughout the study, although significant differences were not achieved. However, engineered exosomes–treated animals showed significantly longer survival compared to those of control exosomes–treated animals. (Figure 14).
In our previous studies, we have shown the timeline (6-7 weeks) of the development of distal metastasis in primary tumor resected models with 4T1 tumors. [39] Animals treated with control exosomes began to show local recurrence and lung metastasis starting on day 28 post-resection. On the other hand, none of the animals that received therapeutic exosomes to deplete CD206+ cells showed any recurrence or distal metastasis. It is noteworthy that a small leftover tumor (white arrow) in one of the therapeutic exosomes-treated animals disappeared following therapy on subsequent BLI (Figure 15). All animals treated with control exosomes died by 56 days post resection due to the development of recurrent and metastatic tumors. All animals in the engineered exosomes–treated group were alive when the study was terminated on day 63 post-resection.
4. Discussion
In this study we successfully generated immunosuppressive CD206+ M2-macrophages targeting engineered exosomes that carried CD206-targeting peptides and therapeutic payload (Fc-mIgG2b). We confirmed the presence of our inserted peptides or protein in the exosomes and showed their specificity in vitro and in vivo. Longterm administration of these exosomes did not show any sign of toxicity in the lungs, heart, liver, spleen, and kidneys. These engineered exosomes depleted targeted CD206+ M2 macrophages and delayed the growth of primary tumors and metastatic foci in mouse models of syngeneic breast cancers.
Recent reports indicated the importance of macrophages in breast cancer and macrophages have become a key player in breast cancer therapy resistance. [49,50] We have previously demonstrated the effect of engineered exosomes targeting CD206+ macrophages in the depletion of M2 Macrophages in animal models of breast cancer, showing delayed tumor growth and increased survival following the administration of therapeutic engineered exosome. [36] The anti-macrophage agents that are currently being used in preclinical or early clinical trials target both M1 and M2 populations, which cause unwanted depletion of M1 macrophages. M1 macrophages are important for antigen presentation and cytotoxic T-cell mediated tumor cell killing. [51,52] Antibody targeting TAM (both M1 and M2) is in its early stages of development. [53,54] Very recently, Jaynes, et al have shown the effectiveness of a small peptide (RP-182)-based therapeutics that activates the mannose receptor CD206 expressed on M2-macrophages to elicit endocytosis, phagosome-lysosome formation, autophagy, and reprogramming of M2-like TAM to an antitumor M1-phenotype. [55] However, it is unclear from the manuscript whether the investigators have used any delivery vehicle for intraperitoneal injections. It is known that many hydrophobic peptide-based therapeutics or imaging agents are not water-soluble and require a carrier (usually nanoparticles) for IV injection. [56] In this study, we used recombinant DNA technology to make engineered therapeutic exosomes, which are biocompatible, biodegradable, and water-soluble for IV or IP administration.
It is necessary to precisely target immunosuppressive M2-macrophages in the TME. It is noted that CD206+ macrophages are seen in a limited number in the lungs, spleen, and liver as resident macrophages. [15] Dendritic cells may also express mannose receptor/s under inflammatory skin disease conditions. [57,58] A subpopulation of endothelial cells in the liver may express the CD206 marker. However, our studies showed no changes in the liver and other major organs on hematoxylin-eosin (H&E) staining following 6 weeks of treatments with therapeutic exosomes, and flow cytometry did not show any depletion of dendritic cells. SPECT study also did not show increased uptake of engineered exosomes in the lungs or liver compared to that of control exosomes, indicating that engineered exosomes will not have detrimental effects on the liver and lungs. Based on the above discussion and published reports of different agents targeting TAM in tumors, our strategies of using engineered exosomes derived from HEK293 cells in targeting immunosuppressive M2-macrophages in TME has great translational potential. Previous investigation, including ours, showed no immunogenic reaction following administration HEK293 cell-derived exosomes. Notably, HEK293-derived exosomes have shown no immune activation or suppression following long-term injections in animal models, [59] and in our ongoing studies, we have not seen any changes in the cytokine profiles and morphology of different organs following multiple injection of HEK293 cell-derived exosomes containing cell targeting moieties and Fc-mIgG2b. The engineered exosomes targeting CD206+ M2 macrophages we developed showed comparable specificity to that of targeting peptides[36] and these exosomes are stable in -20° or -80° for longer shelf life and can be produced continuously using stable transduced HEK-293 cells. Compared to synthetic nanoparticles like liposomes or metallic particles, exosomes offer advantages such as better biocompatibility, low toxicity, minimal immunogenicity, enhanced permeability (including through the BBB), and stability in biological fluids. Additionally, exosomes accumulate more specifically in target cells without being trapped in endosome-lysosome pathways. [24,25,26,27,28,29,60] As the exosome can cross blood brain barrier easily, investigators have used engineered exosomes targeting brain cancer and stroke. [61,62,63,64]
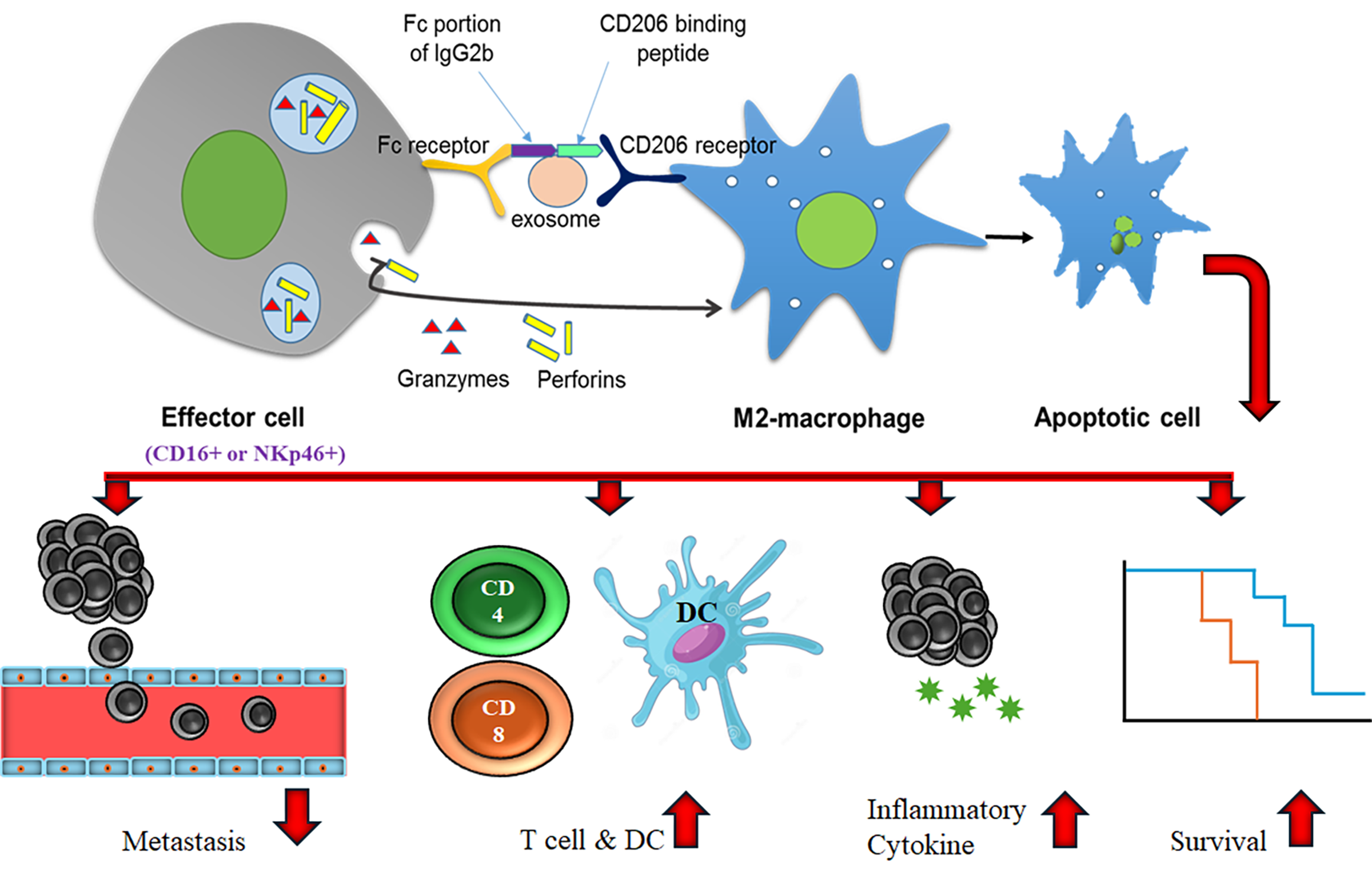
Antibody-dependent cell-mediated cytotoxicity (ADCC) is a non-phagocytic mechanism by which most NK cells (effector cells) can kill antibody-coated target cells in the absence of complement and without the major histocompatibility complex (MHC). [65] Targeted therapy utilizing monoclonal antibodies has revolutionized immunotherapies against several diseases including cancer, [66,67] and ADCC has become more applicable in a clinical context.[68] Antibodies serve as a bridge between Fc receptors on effector cells (such as natural killer (NK) cells) and the target antigen on the cells to be killed. Crosslinking of receptors in both effector and target cells is required for triggering the cytotoxic event. ADCC occurs through various pathways, including (a) release of cytotoxic granules; (b) TNF family death receptors signaling; and (c) release of pro-inflammatory cytokines, such as IFN-γ. [69] Here, we utilized the Fc gamma-receptor (FcγR) based platform to deplete M2 macrophages. We have identified the sequence of the Fc-mIgG2b that triggers FcγR mediated phagocytosis and cytotoxicity [70] and recently we have reported the utility of engineered exosomes as imaging and therapeutic probes. [36] This study has shown an abundant number of NK cells in the tumors, spleens and peripheral blood of tumor-bearing animals. Investigators have used either synthetic nanoparticles or fusion protein[71,72,73] to deliver Fc-IgG2b to initiate ADCC but because of the rigid body, synthetic nanoparticles rely mostly on the enhanced permeability and retention (EPR) effect, and reports are showing a lack of ADCC following tagging with gold nanoparticles.[74,75,76,77,78] Moreover, due to size-dependent manner, synthetic nanoparticles can be cleared by the kidneys or reticuloendothelial system, even with targeting moieties. [79,80,81] On the other hand, fusion protein-based ADCC did not show promise due to rapid clearance and non-specific bindings.[82,83,84,85] Antibody-mediated ADCC also depends on the antibody design with intact Fc-portion and specific attachment to the target cells [86] and most of these antibodies are monoclonal. [87]
Previous investigations proved the enhanced accumulation of systemically administered exosomes to the sites of lesions. [35,60,88] Due to their cellular origin, exosomes exhibit enhanced permeability, which is an advantage over synthetic nanoparticles. [89,90,91,92] Exosomes are also shown to utilize EPR effects. [92,93] Due to higher stability in biological fluids and enhanced permeability, exosomes are better for the targeted delivery of therapeutic payloads. [89,90,91,92,94] Our current studies showed active accumulation of engineered exosomes at the sites of primary and metastatic breast cancer foci and depletion of CD206+ macrophages in the spleens. The depletion of CD206+ cells was related to NK cell-mediated ADCC; additionally, there was increased activity of T-cells in the primary tumors and metastatic sites (lungs).
There is always a risk that depleting immunosuppressive cells may initiate hyperactive inflammation or immunogenic reactions. We have studied the effect of depletion of immunosuppressive CD206+ macrophages in normal and tumor bearing animals. We have not observed any changes in the levels of inflammation, immunogenicity, immunosuppressive cytokines, or other growth factors in the plasma in two different syngeneic immunocompetent animals (C57BL/6 and Balb/c) following six doses of therapeutic and control exosomes. However, we have noticed differential expressions of different cytokines in metastatic sites (lungs) and plasma at 2 and 4 weeks, respectively, following administration of therapeutic engineered exosomes either intravenous or intratumorally compared to that of targeting engineered exosomes (engineered exosomes without therapeutic payload). There were more inflammatory cytokines in the metastatic sites (lungs tissues) in 2 weeks following administration of engineered exosomes indicating active depletion of immunosuppressive macrophages and accumulation of cytotoxic T-cells (both flow cytometry and membrane array). These findings support the idea that depletion of immunosuppressive macrophages will change the TME to more immunogenic status and may open the window for immunotherapy using immune check point inhibitors. [95,96,97] However, the changes in the cytokine level at the metastatic site were not sustained until 4 weeks following therapy, but there was a global increase in cytokine and growth factor levels in the plasma following 4 weeks of therapy. These combined findings are difficult to explain but the global increase of cytokine and growth factor level in the plasma at 4 weeks following therapy could be an indication of the development of therapy resistance. Many investigators have already pointed out the development of therapy resistance following myeloid cells or macrophage targeted therapies in different tumors models due to the heterogeneity of macrophages and the dynamic nature of the TME that leads to treatment failure. [98] Therefore, a newer approach is needed to counteract the effect of immunosuppressive M2-macrophages.
5. Conclusions
In this study, we have used two different treatment models; 1) treatment was given to breast cancer bearing animals starting on day 8 following implantation and continued for 2 weeks (6 doses) to mimic neoadjuvant model, and 2) treatment was given in animals following resection of implanted tumors on day 11 following implantation to mimic post-surgical model. In both cases, therapeutic exosomes targeting M2-macrophages delayed the growth of primary tumors and metastatic foci, indicating the effectiveness of the developed engineered exosomes in altering TME and preventing the invasive and metastatic potential of the AT3 breast cancer cells. M2 macrophages are known to induce immune suppression and inhibit inflammation, epithelial-to-mesenchymal transition, invasion, angiogenesis, and subsequent tumor progression, particularly in TNBC. [42,99,100,101]
6. Potential Clinical Applications, Benefits, and Risks
Exosomes are natural, nano-sized vesicles (30–150 nm) involved in intercellular communication. They can be engineered to carry specific proteins for targeted therapy. We have generated engineered exosomes from FDA-approved HEK293 cells, which are widely used to produce therapeutic proteins and gene therapy vectors. Engineered exosomes developed in HEK293 cells can easily be translated to clinics provided optimum dose is determined by dose escalation studies, toxicity studies are done thoroughly, and usefulness of M2-macrophages targeting engineered exosomes are determined in conjunction with immunotherapy. Clinical trials using engineered exosomes (e.g., mesenchymal stem cell-derived exosomes carrying siRNA for pancreatic cancer) are already underway. Our approach holds significant promise for rapid clinical translation to modulate immunosuppressive TME and improve survival in patients with metastatic breast cancer.
7. Study Limitations
In this study, we have shown both in vitro and in vivo specificity of engineered exosomes in targeting CD206+ M2-macrophages in primary tumors and metastatic foci in mouse models of syngeneic breast cancers. The study also showed more activated T-cells in the metastatic sites following therapies with engineered exosomes. Although the sample size was limited, both survival and flowcytometry analysis showed the effectiveness of engineered exosomes in depleting M2-macrophages and delaying tumor growth significantly. Our studies were set to be proof of principle study and we have not determined the mechanisms by which engineered exosomes made bridges between NK cells and M2-macrophages in the TME or lungs and how ADCC was initiated. The mechanistic studies can be performed in our future investigations. Although the number of T-cells has increased in primary tumor and metastatic sites, we did not initiate immunotherapy. Addition of immunotherapy following engineered exosome treatments could be beneficial.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1. Strategies for making engineered exosomes and modes of action; Figure S2. Determination of specificity of 9aa peptide to target M2-macrophages; Figure S3. Flow cytometry panel for myeloid cells; Figure S4. Flow cytometry panel for T-cells.
Author Contributions
Mahrima Parvin: did the in vitro and in vivo experiments, compiled the data, and reviewed the manuscript, Ahmet Alptekin: did the in vitro and in vivo experiments, compiled the data, and reviewed the manuscript, Sawaiz Kashif: analyze the data, Fowzia A Selina: animals maintenance, collection of tissues, and reviewed the manuscript, Mst Anika Bushra: animals maintenance, collection of tissues, and reviewed the manuscript, Mohammad Syam: collection of tissue, flowcytometry, and reviewed the manuscript, Mohammad H Rashid: concept of the exosome design, Alicia Arnold: Reviewed the manuscript, advised on translational potential, Yutao Liu: helped characterization of the exosomes by NTA, and reviewed the manuscript, Santhakumar Manicassamy: advised the immunological experiments, interpretation of flow cytometry data, and reviewed the manuscript, Hasan Korkaya: creation of animal models and reviewed of the manuscript, Ali S Arbab: overall management of the studies, supplied all the materials, supports the lab personnel, analysis of flow data, imaging studies and interpretation, and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by Paceline (2023 Paceline), institutional pilot grants (IGPCT0016 and IGPP0048) and startup from Georgia Cancer Center.
Institutional Review Board Statement
All the experiments were performed according to the National Institutes of Health (NIH) guidelines and regulations. The Institutional Animal Care and Use Committee (IACUC) of Augusta University (protocol #2014–0625) approved all the experimental procedures. The study is reported in accordance with the ARRIVE guidelines.
Data Availability Statement
Most of the data is included in the manuscript. All the data will be available from the corresponding author on demand.
Acknowledgments
This study was supported by Paceline (2023 Paceline), institutional pilot grants (IGPCT0016 and IGPP0048) and startup from Georgia cancer center. We also acknowledge the help from small animal imaging, flowcytometry (RRID: SCR_025747), and histology cores. Dr. Brilynn Janckila of Augusta University Center for Writing Excellence proofread the English.
Conflicts of Interest
The authors declare no conflict of interest.
Declaration of generative AI and AI-assisted technologies in the writing process
No generative AI was used to write the manuscript or interpret the data. English was corrected using the online Grammarly program and proofread by Augusta University Center for Writing Excellence.
References
- Lahmar Q, Keirsse J, Laoui D, et. al. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim Biophys Acta 2016;1865:23-34. [CrossRef]
- Binnewies M, Roberts EW, Kersten K, et. al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine 2018;24:541-550. [CrossRef]
- Neophytou CM, Panagi M, Stylianopoulos T, Papageorgis P. The Role of Tumor Microenvironment in Cancer Metastasis: Molecular Mechanisms and Therapeutic Opportunities. Cancers (Basel) 2021;13. [CrossRef]
- Giraldo NA, Sanchez-Salas R, Peske JD, et. al. The clinical role of the TME in solid cancer. British Journal of Cancer 2019;120:45-53. [CrossRef]
- Khalaf K, Hana D, Chou JT-T, et. al. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Frontiers in Immunology 2021;12. [CrossRef]
- Elia I, Haigis MC. Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nature Metabolism 2021;3:21-32. [CrossRef]
- Danenberg E, Bardwell H, Zanotelli VRT, et. al. Breast tumor microenvironment structures are associated with genomic features and clinical outcome. Nature Genetics 2022;54:660-669. [CrossRef]
- Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012;122:787-795. [CrossRef]
- Mantovani A, Sozzani S, Locati M, et. al. Infiltration of tumours by macrophages and dendritic cells: tumour-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Novartis Found Symp 2004;256:137-145; discussion 146-138, 259-169.
- Germano G, Allavena P, Mantovani A. Cytokines as a key component of cancer-related inflammation. Cytokine 2008;43:374-379. [CrossRef]
- Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett 2008;267:204-215. [CrossRef]
- Wu Z, Huang C. Unveiling the Impact of MRC1 on Immune Infiltration and Patient's Prognosis: A Pan-Cancer Analysis Based on Single-Cell and Bulk Sequencing. Int J Gen Med 2024;17:2575-2592. [CrossRef]
- Debacker JM, Gondry O, Lahoutte T, Keyaerts M, Huvenne W. The Prognostic Value of CD206 in Solid Malignancies: A Systematic Review and Meta-Analysis. Cancers (Basel) 2021;13. [CrossRef]
- Ma R-Y, Black A, Qian B-Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends in Immunology 2022;43:546-563. [CrossRef]
- Gordon S, Plüddemann A. Tissue macrophages: heterogeneity and functions. BMC biology 2017;15:53-53. [CrossRef]
- van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nature Reviews Molecular Cell Biology 2018;19:213. [CrossRef]
- Li X, Corbett AL, Taatizadeh E, et. al. Challenges and opportunities in exosome research—Perspectives from biology, engineering, and cancer therapy. APL Bioengineering 2019;3:011503. [CrossRef]
- Valadi H, Ekstrom K, Bossios A, et. al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007;9:654-659. [CrossRef]
- Lorentzen E, Dziembowski A, Lindner D, Seraphin B, Conti E. RNA channelling by the archaeal exosome. EMBO Rep 2007;8:470-476. [CrossRef]
- Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci 2000;113 Pt 19:3365-3374.
- Zhang W, Peng P, Shen K. Role of Exosome Shuttle RNA in Cell-to-Cell Communication. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2016;38:480-483. [CrossRef]
- Colombo M, Raposo G, Théry C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annual Review of Cell and Developmental Biology 2014;30:255-289. [CrossRef]
- Kalluri R. The biology and function of exosomes in cancer. The Journal of Clinical Investigation 2016;126:1208-1215. [CrossRef]
- S ELA, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov 2013;12:347-357. [CrossRef]
- Lener T, Gimona M, Aigner L, et. al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J Extracell Vesicles 2015;4:30087. [CrossRef]
- Jiang XC, Gao JQ. Exosomes as novel bio-carriers for gene and drug delivery. Int J Pharm 2017;521:167-175. [CrossRef]
- Alvarez-Erviti L, Seow Y, Yin H, et. al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature Biotechnology 2011;29:341-345. [CrossRef]
- El Andaloussi S, Lakhal S, Mager I, Wood MJ. Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev 2013;65:391-397. [CrossRef]
- El-Andaloussi S, Lee Y, Lakhal-Littleton S, et. al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc 2012;7:2112-2126. [CrossRef]
- Mashouri L, Yousefi H, Aref AR, et. al. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Molecular Cancer 2019;18:75. [CrossRef]
- Bellavia D, Raimondi L, Costa V, et. al. Engineered exosomes: A new promise for the management of musculoskeletal diseases. Biochimica et Biophysica Acta (BBA) - General Subjects 2018;1862:1893-1901. [CrossRef]
- Sterzenbach U, Putz U, Low L-H, et. al. Engineered Exosomes as Vehicles for Biologically Active Proteins. Molecular Therapy 2017;25:1269-1278. [CrossRef]
- Luan X, Sansanaphongpricha K, Myers I, et. al. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacologica Sinica 2017;38:754. [CrossRef]
- Tian T, Zhang H-X, He C-P, et. al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials 2018;150:137-149. [CrossRef]
- Rashid MH, Borin TF, Ara R, et. al. Differential in vivo biodistribution of 131I-labeled exosomes from diverse cellular origins and its implication for theranostic application. Nanomedicine: Nanotechnology, Biology and Medicine 2019;21:102072. [CrossRef]
- Rashid MH, Borin TF, Ara R, et. al. Generation of Novel Diagnostic and Therapeutic Exosomes to Detect and Deplete Protumorigenic M2 Macrophages. Adv Ther (Weinh) 2020;3. [CrossRef]
- Scodeller P, Simón-Gracia L, Kopanchuk S, et. al. Precision Targeting of Tumor Macrophages with a CD206 Binding Peptide. Scientific Reports 2017;7:14655. [CrossRef]
- Asciutto EK, Kopanchuk S, Lepland A, et. al. Phage-Display-Derived Peptide Binds to Human CD206 and Modeling Reveals a New Binding Site on the Receptor. J Phys Chem B 2019;123:1973-1982. [CrossRef]
- Piranlioglu R, Lee E, Ouzounova M, et. al. Primary tumor-induced immunity eradicates disseminated tumor cells in syngeneic mouse model. Nature Communications 2019;10. ARTN 1430. [CrossRef]
- Cowell JK, Teng Y, Bendzunas NG, et. al. Suppression of Breast Cancer Metastasis Using Stapled Peptides Targeting the WASF Regulatory Complex. Cancer Growth Metastasis 2017;10:1179064417713197. [CrossRef]
- Rashid MH, Borin TF, Ara R, et. al. Critical immunosuppressive effect of MDSCderived exosomes in the tumor microenvironment. Oncol Rep 2021;45:1171-1181. [CrossRef]
- Qiu S-Q, Waaijer SJH, Zwager MC, et. al. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treatment Reviews 2018;70:178-189. [CrossRef]
- Yang C, He L, He P, et. al. Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Medical Oncology 2015;32:14. [CrossRef]
- Ouzounova M, Lee E, Piranlioglu R, et. al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nature Communications 2017;8. ARTN 14979 10.1038/ncomms14979.
- Borin TF, Shankar A, Angara K, et. al. HET0016 decreases lung metastasis from breast cancer in immune-competent mouse model. PLoS One 2017;12:e0178830. [CrossRef]
- Olson OC, Kim H, Quail DF, Foley EA, Joyce JA. Tumor-Associated Macrophages Suppress the Cytotoxic Activity of Antimitotic Agents. Cell reports 2017;19:101-113. [CrossRef]
- Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer cell 2015;27:462-472. [CrossRef]
- Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. The Journal of Experimental Medicine 2015;212:435-445. [CrossRef]
- Sami A, Raza A. Reprogramming the tumor microenvironment – macrophages emerge as key players in breast cancer immunotherapy. Frontiers in Immunology 2024;Volume 15 - 2024. [CrossRef]
- Zhan C, Jin Y, Xu X, Shao J, Jin C. Antitumor therapy for breast cancer: Focus on tumor-associated macrophages and nanosized drug delivery systems. Cancer Med 2023;12:11049-11072. [CrossRef]
- Bendell JC, Tolcher AW, Jones SF, et. al. Abstract A252: A phase 1 study of ARRY-382, an oral inhibitor of colony-stimulating factor-1 receptor (CSF1R), in patients with advanced or metastatic cancers. Molecular Cancer Therapeutics 2013;12:A252-A252. [CrossRef]
- Ries Carola H, Cannarile Michael A, Hoves S, et. al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell 2014;25:846-859. [CrossRef]
- Im JH, Buzzelli JN, Jones K, et. al. FGF2 alters macrophage polarization, tumour immunity and growth and can be targeted during radiotherapy. Nat Commun 2020;11:4064. [CrossRef]
- Gomez-Roca CA, Italiano A, Le Tourneau C, et. al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann Oncol 2019;30:1381-1392. [CrossRef]
- Jaynes JM, Sable R, Ronzetti M, et. al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Science Translational Medicine 2020;12:eaax6337. [CrossRef]
- Pellico J, Lechuga-Vieco AV, Almarza E, et. al. In vivo imaging of lung inflammation with neutrophil-specific 68Ga nano-radiotracer. Scientific Reports 2017;7:13242. [CrossRef]
- Wollenberg A, Mommaas M, Oppel T, et. al. Expression and function of the mannose receptor CD206 on epidermal dendritic cells in inflammatory skin diseases. J Invest Dermatol 2002;118:327-334. [CrossRef]
- Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology 2018;154:3-20. [CrossRef]
- Zhu X, Badawi M, Pomeroy S, et. al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J Extracell Vesicles 2017;6:1324730. [CrossRef]
- Webb RL, Kaiser EE, Scoville SL, et. al. Human Neural Stem Cell Extracellular Vesicles Improve Tissue and Functional Recovery in the Murine Thromboembolic Stroke Model. Transl Stroke Res 2018;9:530-539. [CrossRef]
- Chavda VP, Luo G, Bezbaruah R, et. al. Unveiling the promise: Exosomes as game-changers in anti-infective therapy. Exploration 2024;4:20230139. [CrossRef]
- Yang J, Li Y, Jiang S, et. al. Engineered brain-targeting exosome for reprogramming immunosuppressive microenvironment of glioblastoma. Exploration 2025;5:20240039. [CrossRef]
- Alptekin A, Khan MB, Parvin M, et. al. Effects of low-intensity pulsed focal ultrasound-mediated delivery of endothelial progenitor-derived exosomes in tMCAo stroke. Frontiers in Neurology 2025;16. ARTN 1543133 10.3389/fneur.2025.1543133.
- Selina FA, Kelleher J, Bushra A, et. al. Engineered Exosomes Carrying Rabies Viral Glycoprotein (RVG) and Neuroglobin (Ngb) Improved Stroke Outcome in Mouse Model of tMCAo. Stroke 2026;57. ARTN A073 10.1161/str.57.suppl_1.A073.
- Graziano RF, Guyre PM. Antibody-dependent Cell-mediated Cytotoxicity (ADCC) eLS: Wiley Online Library 2006.
- Polansky M, Eisenstadt R, DeGrazia T, et. al. Rituximab therapy in patients with bullous pemphigoid: A retrospective study of 20 patients. Journal of the American Academy of Dermatology 2019;81:179-186. [CrossRef]
- Sneller MC. Rituximab and Wegener's granulomatosis: Are B cells a target in vasculitis treatment? Arthritis & Rheumatism 2005;52:1-5. [CrossRef]
- Aldeghaither DS, Zahavi DJ, Murray JC, et. al. A Mechanism of Resistance to Antibody-Targeted Immune Attack. Cancer Immunology Research 2018:canimm.0266.2018. [CrossRef]
- Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front Immunol 2015;6:368. [CrossRef]
- Qin H, Lerman B, Sakamaki I, et. al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med 2014;20:676-681. [CrossRef]
- Xu Y, Xiao Y, Luo C, et. al. Blocking PD-1/PD-L1 by an ADCC enhanced anti-B7-H3/PD-1 fusion protein engages immune activation and cytotoxicity. Int Immunopharmacol 2020;84:106584. [CrossRef]
- Ochoa MC, Minute L, López A, et. al. Enhancement of antibody-dependent cellular cytotoxicity of cetuximab by a chimeric protein encompassing interleukin-15. Oncoimmunology 2018;7:e1393597. [CrossRef]
- Sioud M, Westby P, Olsen JK, Mobergslien A. Generation of new peptide-Fc fusion proteins that mediate antibody-dependent cellular cytotoxicity against different types of cancer cells. Mol Ther Methods Clin Dev 2015;2:15043. [CrossRef]
- Rückert R, Herz U, Paus R, et. al. IL-15-IgG2b fusion protein accelerates and enhances a Th2 but not a Th1 immune response in vivo, while IL-2-IgG2b fusion protein inhibits both. European Journal of Immunology 1998;28:3312-3320. [CrossRef]
- Czajkowsky DM, Hu J, Shao Z, Pleass RJ. Fc-fusion proteins: new developments and future perspectives. EMBO molecular medicine 2012;4:1015-1028. [CrossRef]
- Ahmed M, Pan DW, Davis ME. Lack of in vivo antibody dependent cellular cytotoxicity with antibody containing gold nanoparticles. Bioconjugate chemistry 2015;26:812-816. [CrossRef]
- Leon PE, He W, Mullarkey CE, et. al. Optimal activation of Fc-mediated effector functions by influenza virus hemagglutinin antibodies requires two points of contact. Proceedings of the National Academy of Sciences 2016;113:E5944-E5951. [CrossRef]
- Sioud M, Westby P, Olsen JKE, Mobergslien A. Generation of new peptide-Fc fusion proteins that mediate antibody-dependent cellular cytotoxicity against different types of cancer cells. Molecular therapy Methods & clinical development 2015;2:15043-15043. [CrossRef]
- Han SG, Lee JS, Ahn K, et. al. Size-dependent clearance of gold nanoparticles from lungs of Sprague–Dawley rats after short-term inhalation exposure. Archives of Toxicology 2015;89:1083-1094. [CrossRef]
- Hoshyar N, Gray S, Han H, Bao G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine (London, England) 2016;11:673-692. [CrossRef]
- Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Molecular Pharmaceutics 2008;5:505-515. [CrossRef]
- Arora T, Padaki R, Liu L, et. al. Differences in binding and effector functions between classes of TNF antagonists. Cytokine 2009;45:124-131. [CrossRef]
- Gillies SD, Young D, Lo KM, Roberts S. Biological activity and in vivo clearance of antitumor antibody/cytokine fusion proteins. Bioconjug Chem 1993;4:230-235. [CrossRef]
- Wu B, Sun Y-N. Pharmacokinetics of Peptide–Fc Fusion Proteins. Journal of Pharmaceutical Sciences 2014;103:53-64. [CrossRef]
- Datta-Mannan A, Lu J, Witcher DR, et. al. The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies. mAbs 2015;7:1084-1093. [CrossRef]
- Zahavi D, AlDeghaither D, O’Connell A, Weiner LM. Enhancing antibody-dependent cell-mediated cytotoxicity: a strategy for improving antibody-based immunotherapy. Antibody Therapeutics 2018;1:7-12. [CrossRef]
- Gül N, van Egmond M. Antibody-Dependent Phagocytosis of Tumor Cells by Macrophages: A Potent Effector Mechanism of Monoclonal Antibody Therapy of Cancer. Cancer Research 2015;75:5008-5013. [CrossRef]
- Bunggulawa EJ, Wang W, Yin T, et. al. Recent advancements in the use of exosomes as drug delivery systems. Journal of nanobiotechnology 2018;16:81-81. [CrossRef]
- Srivastava A, Filant J, Moxley KM, et. al. Exosomes: a role for naturally occurring nanovesicles in cancer growth, diagnosis and treatment. Curr Gene Ther 2015;15:182-192.
- Haqqani AS, Delaney CE, Tremblay TL, et. al. Method for isolation and molecular characterization of extracellular microvesicles released from brain endothelial cells. Fluids Barriers CNS 2013;10:4. [CrossRef]
- Lakhal S, Wood MJ. Exosome nanotechnology: an emerging paradigm shift in drug delivery: exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. Bioessays 2011;33:737-741. [CrossRef]
- Sun D, Zhuang X, Zhang S, et. al. Exosomes are endogenous nanoparticles that can deliver biological information between cells. Advanced Drug Delivery Reviews 2013;65:342-347. [CrossRef]
- Kibria G, Ramos EK, Wan Y, Gius DR, Liu H. Exosomes as a Drug Delivery System in Cancer Therapy: Potential and Challenges. Molecular pharmaceutics 2018;15:3625-3633. [CrossRef]
- Boukouris S, Mathivanan S. Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteomics Clinical applications 2015;9:358-367. [CrossRef]
- Wang Y, Johnson KCC, Gatti-Mays ME, Li Z. Emerging strategies in targeting tumor-resident myeloid cells for cancer immunotherapy. Journal of Hematology & Oncology 2022;15:118. [CrossRef]
- Peranzoni E, Ingangi V, Masetto E, Pinton L, Marigo I. Myeloid Cells as Clinical Biomarkers for Immune Checkpoint Blockade. Front Immunol 2020;11:1590. [CrossRef]
- Shuptrine CW, Perez VM, Selitsky SR, Schreiber TH, Fromm G. Shining a LIGHT on myeloid cell targeted immunotherapy. European Journal of Cancer 2023;187:147-160. [CrossRef]
- Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Research 2021;81:1201-1208. [CrossRef]
- Williams CN, Hodges DB, Reynolds JM, Bhat A. Tumor-associated macrophages: CD206 and CD68 expression and patient outcomes in locally advanced breast cancer. Journal of Clinical Oncology 2018;36:e24130-e24130. [CrossRef]
- Dijkgraaf EM, Heusinkveld M, Tummers B, et. al. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res 2013;73:2480-2492. [CrossRef]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002;23:549-555.
Figure 1.
Biogenesis of engineered exosomes and its characteristics: Lentivector containing plasmids to express CD206 targeting peptides and Fc-mIgG2b on the exosomes was used to transduce HEK293. (A) mCherry positive cells are indicative of transduced HEK293. Yellow bar is 300µm. (B) PCR was performed to determine the expression of inserted sequences in the DNA of transduced cells. Transduced HEK293 cells expressed the sequences of both CD206 targeting peptide (TP, 338bp) and Fc-mIgG2b (500bp). (C) Collected exosomes from different transduced cells showed the classical marker of exosome (CD9). CD9 is expressed in control HEK and engineered exosomes (Eng Exo). Nanoparticle Tracking Analysis showed typical (D) Zeta potential and (E) size distribution of collected engineered exosomes. (F) Immnu-transmission electron microscopy showed the expression of IgG2b (black dots) on the surface of the exosomes (white oval shaped structures). The TEM can also confirm size of the exosomes. White bar is 100nm. HEK = Human embryonic kidney, Immuno-TEM = Immnu-transmission electron microscopy PCR = polymer chain reaction, TEM = transmission electron microscopy, WB = western blotting.
Figure 1.
Biogenesis of engineered exosomes and its characteristics: Lentivector containing plasmids to express CD206 targeting peptides and Fc-mIgG2b on the exosomes was used to transduce HEK293. (A) mCherry positive cells are indicative of transduced HEK293. Yellow bar is 300µm. (B) PCR was performed to determine the expression of inserted sequences in the DNA of transduced cells. Transduced HEK293 cells expressed the sequences of both CD206 targeting peptide (TP, 338bp) and Fc-mIgG2b (500bp). (C) Collected exosomes from different transduced cells showed the classical marker of exosome (CD9). CD9 is expressed in control HEK and engineered exosomes (Eng Exo). Nanoparticle Tracking Analysis showed typical (D) Zeta potential and (E) size distribution of collected engineered exosomes. (F) Immnu-transmission electron microscopy showed the expression of IgG2b (black dots) on the surface of the exosomes (white oval shaped structures). The TEM can also confirm size of the exosomes. White bar is 100nm. HEK = Human embryonic kidney, Immuno-TEM = Immnu-transmission electron microscopy PCR = polymer chain reaction, TEM = transmission electron microscopy, WB = western blotting.

Figure 2.
Specificity of engineered exosomes: Mouse embryonic fibroblast (MEF) was used as negative control and polarized mouse bone marrow-derived macrophage (MBMDM) was used as positive control. Uptake of engineered exosomes (DiI-labeled, red) was investigated with or without blocking peptides. (A) MEF did not show any uptake of DiI-labeled exosomes. (B) Polarized MBMDM (green) showed plenty exosomes (red) uptake without blocking. (C) Polarized MBMDM (green) showed very low exosomes (red) uptake with blocking. (D & E) color threshold masked images from without or with blocking of CD206. (F) Quantitative analysis shows significantly (*p<0.01) lower uptake following blocking of CD206 indicating the specificity of engineered exosomes. n=3 per group. MEF = mouse embryonic fibroblast, MBMDM = mouse bone marrow derived macrophages.
Figure 2.
Specificity of engineered exosomes: Mouse embryonic fibroblast (MEF) was used as negative control and polarized mouse bone marrow-derived macrophage (MBMDM) was used as positive control. Uptake of engineered exosomes (DiI-labeled, red) was investigated with or without blocking peptides. (A) MEF did not show any uptake of DiI-labeled exosomes. (B) Polarized MBMDM (green) showed plenty exosomes (red) uptake without blocking. (C) Polarized MBMDM (green) showed very low exosomes (red) uptake with blocking. (D & E) color threshold masked images from without or with blocking of CD206. (F) Quantitative analysis shows significantly (*p<0.01) lower uptake following blocking of CD206 indicating the specificity of engineered exosomes. n=3 per group. MEF = mouse embryonic fibroblast, MBMDM = mouse bone marrow derived macrophages.

Figure 3.
Surface attachment of DiI-labeled engineered exosomes (red) carrying precision peptide and mIgG2b from 0.5 hours to 24 hours. Murine macrophages (J774A.1) labeled with CFSE dye (green) were polarized to M2 macrophage phenotype using IL-13 and IL-4 for 24 hours and then exosomes were incubated with the cells. Cells were collected and fixed at different time points and stained with DAPI. The microscopic fluorescent images showed engineered exosomes (red) remained on the surface up to 24 hours, indicating that exosomes can bridge with NK cell for extended time. n = 2 separate culture wells. CFSE = Carboxyfluorescein succinimidyl ester, hr = hours, IL = interleukin, NK = natural killer.
Figure 3.
Surface attachment of DiI-labeled engineered exosomes (red) carrying precision peptide and mIgG2b from 0.5 hours to 24 hours. Murine macrophages (J774A.1) labeled with CFSE dye (green) were polarized to M2 macrophage phenotype using IL-13 and IL-4 for 24 hours and then exosomes were incubated with the cells. Cells were collected and fixed at different time points and stained with DAPI. The microscopic fluorescent images showed engineered exosomes (red) remained on the surface up to 24 hours, indicating that exosomes can bridge with NK cell for extended time. n = 2 separate culture wells. CFSE = Carboxyfluorescein succinimidyl ester, hr = hours, IL = interleukin, NK = natural killer.

Figure 4.
Differential biodistribution exosomes and protein: I-131 labeled control exosomes (HEK exo), anti-mouse CD206 antibody (CD206 Ab), fusion protein (fusion P) containing targeting peptide and Fc-mIgG2b, and engineered exosomes (Engi. Exo) carrying targeting peptide were administered IV in 4T1 breast cancer bearing animals and SPECT images were obtained 3 hrs. after administration. (A) SPECT images showed the distribution of the agents in the primary tumors (white circles) and in the lungs (yellow rectangle areas). White arrows show possible metastatic foci. Fusion protein and engineered exosomes accumulated in primary as well as metastatic tumors. (B) Randomly selected lung H&E stains showed multiple metastatic foci (black arrows). (C) Presence of numerous CD206+ cells is seen in the tumors (green cells). n =2-3 animals per group. Ab = antibody, Engi = engineered, Exo = exosomes, HEK = human embryonic kidney cells, HE = Hematoxylin and Eosin, P = protein, SPECT = single photon emission computed tomography.
Figure 4.
Differential biodistribution exosomes and protein: I-131 labeled control exosomes (HEK exo), anti-mouse CD206 antibody (CD206 Ab), fusion protein (fusion P) containing targeting peptide and Fc-mIgG2b, and engineered exosomes (Engi. Exo) carrying targeting peptide were administered IV in 4T1 breast cancer bearing animals and SPECT images were obtained 3 hrs. after administration. (A) SPECT images showed the distribution of the agents in the primary tumors (white circles) and in the lungs (yellow rectangle areas). White arrows show possible metastatic foci. Fusion protein and engineered exosomes accumulated in primary as well as metastatic tumors. (B) Randomly selected lung H&E stains showed multiple metastatic foci (black arrows). (C) Presence of numerous CD206+ cells is seen in the tumors (green cells). n =2-3 animals per group. Ab = antibody, Engi = engineered, Exo = exosomes, HEK = human embryonic kidney cells, HE = Hematoxylin and Eosin, P = protein, SPECT = single photon emission computed tomography.

Figure 5.
Whole body and organ specific biodistribution of administered HEK and engineered exosomes: I-131 tagged control and engineered exosomes were IV administered in separate groups of animals. Whole-body (WB) single photon emission computed tomography (SPECT) images were obtained in 5 minutes, 3 hrs., and 24 hrs. WB as well as organ specific (Lu-=lung, H=heart, Li=Liver) activities were determined and expressed as percent WB activity. (A) Representative SPECT images showing the distribution of administered exosomes. (B & C) Semiquantitative analysis showed no retention of activity in the lungs or liver (for both HEK and engineered exosomes) indicating minimum uptake to the resident macrophages in the lungs and liver. Activities in the stomach (ST), thyroid (Th) and Bladder (BL) are due to free I-131. n = 5 per group. BL = bladder, H = heart, hrs = hours, Lu =lungs, Li = liver, ns = not significant, ST stomach, Th = thyroid, WB = whole body.
Figure 5.
Whole body and organ specific biodistribution of administered HEK and engineered exosomes: I-131 tagged control and engineered exosomes were IV administered in separate groups of animals. Whole-body (WB) single photon emission computed tomography (SPECT) images were obtained in 5 minutes, 3 hrs., and 24 hrs. WB as well as organ specific (Lu-=lung, H=heart, Li=Liver) activities were determined and expressed as percent WB activity. (A) Representative SPECT images showing the distribution of administered exosomes. (B & C) Semiquantitative analysis showed no retention of activity in the lungs or liver (for both HEK and engineered exosomes) indicating minimum uptake to the resident macrophages in the lungs and liver. Activities in the stomach (ST), thyroid (Th) and Bladder (BL) are due to free I-131. n = 5 per group. BL = bladder, H = heart, hrs = hours, Lu =lungs, Li = liver, ns = not significant, ST stomach, Th = thyroid, WB = whole body.

Figure 6.
The effect of engineered exosomes (CD206 exo) vs fusion protein vs antibody: Groups of animals were treated separately with control and engineered exosomes (CD206-exo), fusion protein, and anti CD206 antibody. Animals were euthanized at the end of the treatments and spleens were collected and single cell suspension were made for flowcytometry using myeloid and T-cell specific multi-color flow panels. Flow-cytometric analysis of splenic cells (collected from treated animals) showed significantly higher depletion of CD11b+ or CD206+ cells by CD206 exosomes and fusion protein compared to the control exosomes and antibody-treated group only. There was no significant change in T-cell and B-cell populations. Quantitative data are expressed in mean ± SEM. *p<0.05 compared to all groups. n=3 per group. Exo = exosomes.
Figure 6.
The effect of engineered exosomes (CD206 exo) vs fusion protein vs antibody: Groups of animals were treated separately with control and engineered exosomes (CD206-exo), fusion protein, and anti CD206 antibody. Animals were euthanized at the end of the treatments and spleens were collected and single cell suspension were made for flowcytometry using myeloid and T-cell specific multi-color flow panels. Flow-cytometric analysis of splenic cells (collected from treated animals) showed significantly higher depletion of CD11b+ or CD206+ cells by CD206 exosomes and fusion protein compared to the control exosomes and antibody-treated group only. There was no significant change in T-cell and B-cell populations. Quantitative data are expressed in mean ± SEM. *p<0.05 compared to all groups. n=3 per group. Exo = exosomes.

Figure 7.
Heat maps of cytokines and chemokines profiles: Both Balb/c and C57BL/6 (B6) animals were treated with 6 doses of either PBS or engineered exosomes (CD206 exo) over six weeks. 24 hours after the last dose, animals were euthanized, and plasma was subjected to membrane array (53 targets) analysis to determine inflammatory, immunogenic, immunosuppressive, invasive, and growth factors. There were no significant differences among the groups. n=4 per group. B6 = C57BL/6, Exo =exosomes, PBS = phosphate buffered saline.
Figure 7.
Heat maps of cytokines and chemokines profiles: Both Balb/c and C57BL/6 (B6) animals were treated with 6 doses of either PBS or engineered exosomes (CD206 exo) over six weeks. 24 hours after the last dose, animals were euthanized, and plasma was subjected to membrane array (53 targets) analysis to determine inflammatory, immunogenic, immunosuppressive, invasive, and growth factors. There were no significant differences among the groups. n=4 per group. B6 = C57BL/6, Exo =exosomes, PBS = phosphate buffered saline.

Figure 8.
Hematoxylin and eosin (H&E) staining of different organs: Both Balb/c and C57BL/6 (B6) animals were treated with 6 doses of either PBS or engineered exosomes (CD206 exo) over 6 weeks. 6 months after the last dose, animals were euthanized, and different organs were collected, fixed and sections were made for H&E staining to determine the effect of engineered exosomes at the cellular level due to presence of resident macrophages in the organs. There were no changes observed following treatments with engineered exosomes. The images were obtained using 40x optics and 9 images were stitched together. Yellow bar = 300µm. B6 = C57BL/6, PBS = phosphate buffered saline.
Figure 8.
Hematoxylin and eosin (H&E) staining of different organs: Both Balb/c and C57BL/6 (B6) animals were treated with 6 doses of either PBS or engineered exosomes (CD206 exo) over 6 weeks. 6 months after the last dose, animals were euthanized, and different organs were collected, fixed and sections were made for H&E staining to determine the effect of engineered exosomes at the cellular level due to presence of resident macrophages in the organs. There were no changes observed following treatments with engineered exosomes. The images were obtained using 40x optics and 9 images were stitched together. Yellow bar = 300µm. B6 = C57BL/6, PBS = phosphate buffered saline.

Figure 9.
Involvement of NK cells: Tumor volume as well as optical images were obtained on days 8 (baseline), 15, and 22 following specific cell depletion and engineered exosome therapies. The rationale behind the investigation was that in absence of NK cells tumor growth would not decrease even after administration of engineered exosomes. There were significant (20-40 folds) increased (# = p<0.05) BLI (bioluminescent imaging) photon intensity in tumors that were treated with anti-NK antibodies. However, there were no significant changes in photon intensity compared to that of vehicle or isotype IgG when Gr1+ cells were depleted. It is to note that tumor volume measured by calipers also showed similar results. n=3-4 per group. NK = natural killer cell.
Figure 9.
Involvement of NK cells: Tumor volume as well as optical images were obtained on days 8 (baseline), 15, and 22 following specific cell depletion and engineered exosome therapies. The rationale behind the investigation was that in absence of NK cells tumor growth would not decrease even after administration of engineered exosomes. There were significant (20-40 folds) increased (# = p<0.05) BLI (bioluminescent imaging) photon intensity in tumors that were treated with anti-NK antibodies. However, there were no significant changes in photon intensity compared to that of vehicle or isotype IgG when Gr1+ cells were depleted. It is to note that tumor volume measured by calipers also showed similar results. n=3-4 per group. NK = natural killer cell.

Figure 10.
Heat maps of cytokines and chemokines profiles following 2 weeks of treatments: Tumor-bearing animals were treated with engineered exosomes (either therapeutic (carrying both CD206 targeting peptides and Fc-mIgG2b) or targeting (carrying only CD206 targeting peptides)). Therapeutic exosomes were administered either IV or intratumor routes. Plasma and tissues (extracted proteins) from primary tumors and metastatic sites were subjected to membrane array (53 targets) analysis. These results are from pooled samples. Values were normalized to corresponding Targeting exosomes. n=3 per group. There were increased cytokines and chemokines in the lungs. Targeting -Exo = targeting exosomes, Thera-Exo = therapeutic exosomes.
Figure 10.
Heat maps of cytokines and chemokines profiles following 2 weeks of treatments: Tumor-bearing animals were treated with engineered exosomes (either therapeutic (carrying both CD206 targeting peptides and Fc-mIgG2b) or targeting (carrying only CD206 targeting peptides)). Therapeutic exosomes were administered either IV or intratumor routes. Plasma and tissues (extracted proteins) from primary tumors and metastatic sites were subjected to membrane array (53 targets) analysis. These results are from pooled samples. Values were normalized to corresponding Targeting exosomes. n=3 per group. There were increased cytokines and chemokines in the lungs. Targeting -Exo = targeting exosomes, Thera-Exo = therapeutic exosomes.

Figure 11.
Heat maps of cytokines and chemokines profiles following 4 weeks of treatments. Tumor-bearing animals were treated with engineered exosomes (either therapeutic (carrying both CD206 targeting peptides and Fc-mIgG2b) or targeting (carrying only CD206 targeting peptides)). Therapeutic exosomes were administered either IV or intratumor routes. Plasma and tissues (extracted proteins) from primary tumors and metastatic sites were subjected to membrane array (53 targets) analysis. These results are from pooled samples. Values were normalized to corresponding targeting exosomes. There were increased cytokines and chemokines in the plasma. n=3 per group. Targeting -Exo = targeting exosomes, Thera-Exo = therapeutic exosomes.
Figure 11.
Heat maps of cytokines and chemokines profiles following 4 weeks of treatments. Tumor-bearing animals were treated with engineered exosomes (either therapeutic (carrying both CD206 targeting peptides and Fc-mIgG2b) or targeting (carrying only CD206 targeting peptides)). Therapeutic exosomes were administered either IV or intratumor routes. Plasma and tissues (extracted proteins) from primary tumors and metastatic sites were subjected to membrane array (53 targets) analysis. These results are from pooled samples. Values were normalized to corresponding targeting exosomes. There were increased cytokines and chemokines in the plasma. n=3 per group. Targeting -Exo = targeting exosomes, Thera-Exo = therapeutic exosomes.

Figure 12.
Distribution of myeloid and T-cell population in the primary tumor and metastatic sites (lungs): Tumor (4T1) bearing animals were treated with vehicle (PBS) or engineered exosomes (CD206-exo). At the end of the treatments, animals were euthanized, primary tumors and lungs were collected for single cell suspension. Multi-color panel flow cytometry was conducted to determine the myeloid and T-cell populations. (A) No significant differences were observed in the population of granulocytes and M1-macrophages both in primary tumors and metastatic sites. (B) There were significantly increased number of different T-cell populations in the primary and metastatic site of 4T1 TNBC following treatment with PBS and engineered therapeutic exosomes targeting CD206+ cells (CD206). Total 100,000 counts were acquired from each sample for flow analysis. * = p<0.05. n=3.per group.
Figure 12.
Distribution of myeloid and T-cell population in the primary tumor and metastatic sites (lungs): Tumor (4T1) bearing animals were treated with vehicle (PBS) or engineered exosomes (CD206-exo). At the end of the treatments, animals were euthanized, primary tumors and lungs were collected for single cell suspension. Multi-color panel flow cytometry was conducted to determine the myeloid and T-cell populations. (A) No significant differences were observed in the population of granulocytes and M1-macrophages both in primary tumors and metastatic sites. (B) There were significantly increased number of different T-cell populations in the primary and metastatic site of 4T1 TNBC following treatment with PBS and engineered therapeutic exosomes targeting CD206+ cells (CD206). Total 100,000 counts were acquired from each sample for flow analysis. * = p<0.05. n=3.per group.

Figure 13.
Presence of dendritic and NK cells: Tumor bearing animals were treated with vehicle (PBS) and engineered exosomes (CD206-exo) and at the end of the treatment different organs were collected, single cell suspension was made, and flow cytometry was performed. (A) Number of dendritic cells was increased in the metastatic foci and in the spleen following treatment with therapeutic engineered exosomes. (B) There were plenty of NK cells present in the primary tumors, spleen and peripheral blood. Total 100,000 counts were acquired from each sample for flow analysis. * = p<0.05 compared to PBS-treated animals. n=3 per group. PBS = phosphate buffered saline, PBMC = peripheral blood mononuclear cells.
Figure 13.
Presence of dendritic and NK cells: Tumor bearing animals were treated with vehicle (PBS) and engineered exosomes (CD206-exo) and at the end of the treatment different organs were collected, single cell suspension was made, and flow cytometry was performed. (A) Number of dendritic cells was increased in the metastatic foci and in the spleen following treatment with therapeutic engineered exosomes. (B) There were plenty of NK cells present in the primary tumors, spleen and peripheral blood. Total 100,000 counts were acquired from each sample for flow analysis. * = p<0.05 compared to PBS-treated animals. n=3 per group. PBS = phosphate buffered saline, PBMC = peripheral blood mononuclear cells.

Figure 14.
Primary tumor growth and survival: Animals bearing AT3 (in B6) and 4T1 (in Balb/c) syngeneic TNBC were treated with control and engineered exosomes. 6 doses of therapeutic engineered exosomes were IV administered starting on day 8 of tumor implantation. Treatment of tumor-bearing animals with therapeutic engineered exosomes prevented tumor growth and improved survival by depleting CD206+ M2-macrophages. Data are expressed in mean±95%CI. n=5 per group. Exo = exosomes.
Figure 14.
Primary tumor growth and survival: Animals bearing AT3 (in B6) and 4T1 (in Balb/c) syngeneic TNBC were treated with control and engineered exosomes. 6 doses of therapeutic engineered exosomes were IV administered starting on day 8 of tumor implantation. Treatment of tumor-bearing animals with therapeutic engineered exosomes prevented tumor growth and improved survival by depleting CD206+ M2-macrophages. Data are expressed in mean±95%CI. n=5 per group. Exo = exosomes.

Figure 15.
Resected model of breast cancer and metastasis: All primary tumors were resected 11 days after tumor implantation and the animals were followed up to 63 days after resection. The figure shows representative animals from control (HEK) and therapeutic engineered exosomes treated groups with resected tumors. Animals treated with control exosomes began showing local recurrence and lung metastasis (black arrows show local recurrence and multiple distal metastasis) starting on day 28 post resection. On the other hand, none of the animals that received therapeutic engineered exosomes to deplete CD206+ cells show any recurrence or distal metastasis. It is to note that small leftover tumor (black arrow) in one of the therapeutic exosomes treated animals disappeared following therapy on subsequent bioluminescent imaging. n=5 per group HEK = human embryonic kidney cells, Pre-Sur= pre surgery, POD = post-operative day.
Figure 15.
Resected model of breast cancer and metastasis: All primary tumors were resected 11 days after tumor implantation and the animals were followed up to 63 days after resection. The figure shows representative animals from control (HEK) and therapeutic engineered exosomes treated groups with resected tumors. Animals treated with control exosomes began showing local recurrence and lung metastasis (black arrows show local recurrence and multiple distal metastasis) starting on day 28 post resection. On the other hand, none of the animals that received therapeutic engineered exosomes to deplete CD206+ cells show any recurrence or distal metastasis. It is to note that small leftover tumor (black arrow) in one of the therapeutic exosomes treated animals disappeared following therapy on subsequent bioluminescent imaging. n=5 per group HEK = human embryonic kidney cells, Pre-Sur= pre surgery, POD = post-operative day.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



