Submitted:
24 March 2026
Posted:
25 March 2026
You are already at the latest version
Abstract
Oleanolic acid (OA) is a hydrophobic pentacyclic triterpene widely distributed in the plant kingdom and characterized by broad biological activity, including antioxidant, anti-inflammatory, neuroprotective, renoprotective, and anticancer effects. Increasing evidence suggests, however, that many of these actions are better explained not by single molecular targets, but by OA-dependent modulation of an integrated organelle stress network involving mitochondria, the endoplasmic reticulum (ER), autophagy, mitophagy, and apoptosis. This review critically analyzes the available evidence on the effects of OA on the mitochondria–ER–autophagy–apoptosis axis, with particular emphasis on mechanisms governing the transition between cellular adaptation and cell death. The available literature indicates that, in non-cancer models, OA most commonly lowers reactive oxygen species (ROS), stabilizes mitochondrial function, attenuates the ER stress signature, and promotes adaptive autophagy and mitophagy. In contrast, in many cancer models, OA may enhance mitochondrial dysfunction, lower the threshold for mitochondrial apoptosis, and induce autophagy that can be either protective or cytotoxic depending on the biological context. Overall, the current evidence supports a model in which OA acts as a context-dependent modulator of the organelle stress threshold rather than as a uniformly cytoprotective or uniformly proapoptotic compound. At the same time, the literature remains heterogeneous with respect to models, doses, exposure times, and markers used, while poor aqueous solubility and limited bioavailability continue to constrain translation. Future studies should therefore integrate analyses of mitochondria, ER, mitochondria–ER contact sites (MERCS), autophagy, apoptosis, pharmacokinetics, formulation, and safety in order to define the true potential of OA as a modulator of biological stress.
Keywords:
oleanolic acid
; organelle stress
; mitochondria
; endoplasmic reticulum stress
; autophagy
; mitophagy
; apoptosis
; MERCS
; ROS
; unfolded protein response
1. Introduction
Oleanolic acid (OA) is a natural pentacyclic triterpene widely distributed in medicinal and edible plants. In recent years, OA has attracted growing attention because of its pleiotropic biological activities, including antioxidant, anti-inflammatory, hepatoprotective, renoprotective, neuroprotective, antidiabetic, and anticancer effects [1,2,3]. Importantly, many of these effects appear to arise not from a single molecular target, but from OA-dependent modulation of integrated cellular stress responses involving redox homeostasis, mitochondrial function, endoplasmic reticulum (ER) stress, autophagy, and apoptosis [1,2].
In eukaryotic cells, mitochondria and the ER represent two major hubs integrating the response to metabolic, toxic, and inflammatory injury. Mitochondrial dysfunction leads to excessive ROS production, loss of mitochondrial membrane potential, impairment of bioenergetics, and activation of the intrinsic apoptotic pathway [4,5,6,7]. In parallel, accumulation of misfolded proteins in the ER activates the unfolded protein response (UPR), which is initially adaptive but can switch to a pro-death program when stress is prolonged or severe [8,9,10,11].
However, mitochondrial stress and ER stress should not be viewed as isolated events. These organelles are functionally and structurally coupled through mitochondria–ER contact sites (MERCS), also referred to as mitochondria-associated membranes (MAMs), which regulate Ca2+ transfer, lipid metabolism, autophagy initiation, mitophagy, and apoptotic signaling [12,13,14]. In practice, this means that the cellular response to organelle stress is shaped by a coupled mitochondria–ER–autophagy–apoptosis axis rather than by independent pathways. This perspective is especially important for OA, because many studies report simultaneous effects on ROS, UPR markers, BCL-2 family proteins, caspases, and AMPK/mTOR or PI3K/AKT/mTOR signaling [12,13,14,15,16,17].
The currently available literature indicates that OA exerts highly context-dependent effects. In non-cancer models of toxic, metabolic, or degenerative injury, OA is often cytoprotective, attenuating oxidative stress, reducing ER stress, stabilizing mitochondrial function, and limiting apoptosis [18,19,20,21,22,23,24,25]. By contrast, in many cancer models, OA enhances autophagy, mitophagy, and/or mitochondrial apoptosis, suggesting that it may switch cells from adaptive survival toward programmed death [26,27,28,29,30,31,32,33,34]. This apparent duality is not necessarily contradictory; rather, it suggests that OA modulates critical stress-response nodes whose output depends on cell type, basal stress level, dose, and duration of exposure.
Despite the growing number of experimental studies, there is still no integrated review specifically addressing OA within the conceptual framework of organelle stress. Most existing reviews focus on the broad pharmacological activity of OA or on specific disease categories, whereas the relationship between OA, mitochondrial dysfunction, ER stress, autophagy, mitophagy, and apoptosis remains scattered across the literature [1,2,3,35]. Accordingly, the aim of this review is to critically discuss the available evidence on OA as a modulator of the integrated mitochondria–ER–autophagy–apoptosis network, with particular attention to the mechanisms governing the shift between cellular adaptation and cell death.
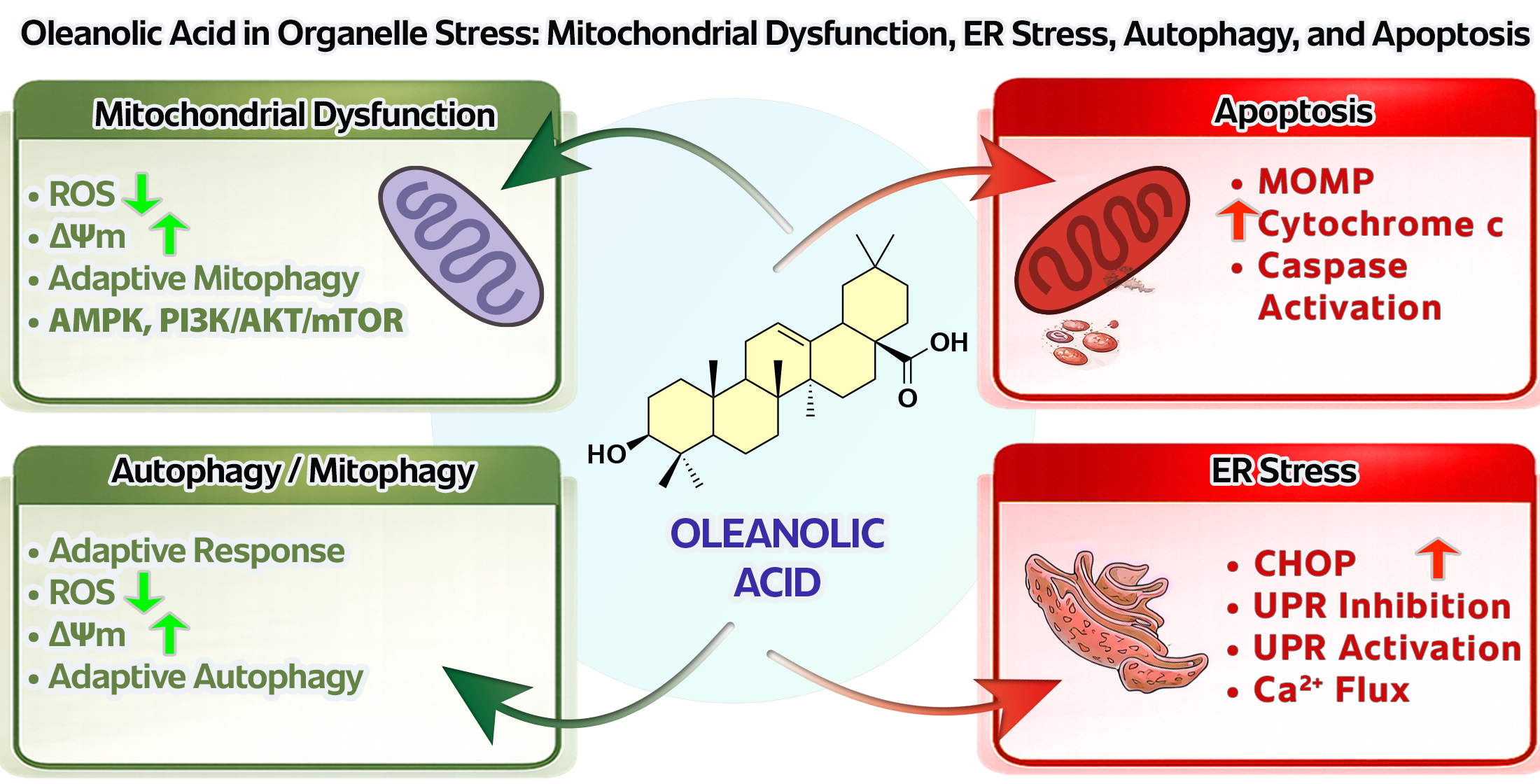
The following sections discuss the biological basis of organelle stress, the effects of OA on mitochondrial function, its role in ER stress and UPR regulation, the significance of autophagy and mitophagy in the OA response, and the relationship of these processes with apoptosis. Special emphasis is placed on whether OA acts primarily as a homeostasis-supporting modulator or as a compound capable of selectively overloading already stressed cellular networks, especially in cancer cells [1,2,12,13,14]. The conceptual framework adopted in this review is summarized in Figure 1.
2. Oleanolic Acid: Sources, Chemical Features, and Translational Limitations
Oleanolic acid is a pentacyclic oleanane-type triterpene widely distributed in the plant kingdom. It occurs both as a free acid and as the aglycone of triterpenoid saponins, and from a nutritional perspective it is a natural constituent of numerous plant-derived foods and medicinal herbs. OA is particularly abundant in olives and olive-derived products, but it has also been identified in apples, grapes, and other fruits and botanical materials [1,2,3].
Chemically, OA has the molecular formula C30H48O3 and belongs to hydrophobic pentacyclic triterpenes. Its structure consists of five fused rings, a hydroxyl group at C-3, a carboxyl group at C-28, and a double bond between C-12 and C-13. These features are not merely descriptive, because the biological activity of OA and its derivatives is strongly influenced by stereochemistry and by the possibility of structural modification at positions such as C-3 and C-28, which has been exploited in medicinal chemistry approaches aimed at improving bioactivity and pharmacokinetics [35,36]. The chemical structure of OA, including ring labeling and carbon atom numbering, is shown in Scheme 1.
The biomedical interest in OA stems from its broad spectrum of biological activities. Multiple reviews and experimental studies have described antioxidant, anti-inflammatory, hepatoprotective, cardioprotective, neuroprotective, antidiabetic, and anticancer properties [2,3]. For the purposes of this review, however, the key point is that many of these effects may be secondary to the ability of OA to modulate more fundamental processes controlling cellular homeostasis, such as redox balance, mitochondrial integrity, UPR signaling, and organelle quality control [1,2].
At the same time, the physicochemical properties of OA constitute one of the major obstacles to translation. OA is strongly hydrophobic, poorly soluble in water, and characterized by limited permeability, resulting in low oral bioavailability [1,2,37,38]. Consequently, much of the evidence on OA efficacy comes from in vitro models employing concentrations whose relevance to in vivo exposure is not always straightforward. This issue is particularly important when interpreting the literature on organelle stress, because mitochondrial, ER, and autophagic effects observed in cell culture may not directly reflect OA action in whole organisms.
To address these limitations, multiple formulation strategies have been explored, including cyclodextrin complexes, solid dispersions, nanosuspensions, self-emulsifying delivery systems, phospholipid complexes, liposomes, and other nanoformulations [37,38]. Such approaches improve solubility, stability, and systemic exposure, but translational challenges remain, including formulation robustness, scalability, and the lack of sufficient clinical data [37,38].
Human data are still limited. In the BIO-OLTRAD study, administration of a single 30 mg dose of OA formulated in functional olive oil to healthy volunteers produced measurable systemic exposure, with OA circulating in association with serum albumin and triglyceride-rich lipoproteins [37]. These findings suggest that OA bioavailability depends strongly on the lipid matrix and delivery vehicle rather than on dose alone. Therefore, OA should be considered not only a promising natural compound, but also a molecule whose biological action is tightly shaped by formulation, exposure, and experimental context [1,2,37,38].
3. Biology of Organelle Stress
Organelle stress can be defined as disruption of the homeostasis of specialized cellular compartments, especially mitochondria and the ER, resulting in activation of adaptive programs or progression toward cell death. Importantly, these processes are not isolated. Mitochondria and the ER are tightly coupled functionally, and their stress responses intersect with autophagy, mitophagy, and apoptosis. Accordingly, organelle stress is best viewed as a dynamic network rather than as a sum of independent pathways [6,12,15,17,39,40].
3.1. Mitochondrial Dysfunction
Mitochondria are central to ATP production, calcium homeostasis, intermediate metabolism, redox signaling, and programmed cell death. Mitochondrial dysfunction therefore encompasses not only reduced energy production, but also electron transport chain impairment, excessive ROS generation, loss of mitochondrial membrane potential, abnormal mitochondrial dynamics, mitochondrial DNA damage, and defective mitochondrial quality control [5,6,7,15]. Mitochondrial ROS should not be regarded solely as harmful by-products; they also serve important signaling functions. The problem arises when redox signaling becomes chronic oxidative stress and causes structural and functional damage [5,6].
A major determinant of mitochondrial stress biology is the integrity of the outer mitochondrial membrane. When stress exceeds the adaptive capacity of the cell, proapoptotic BCL-2 family proteins, primarily BAX and BAK, trigger mitochondrial outer membrane permeabilization (MOMP). MOMP enables cytochrome c release and caspase activation, thereby marking a critical step toward intrinsic apoptos. For this reason, mitochondrial membrane potential, ROS, the BCL-2/BAX ratio, cytochrome c release, and caspase-9/caspase-3 activation are widely used as core readouts of mitochondrial stress in OA studies [6,7].
Importantly, the mitochondrial response to stress is not binary. Mitochondria are highly dynamic organelles, and the balance among fission, fusion, selective degradation by mitophagy, and network remodeling determines whether the cell adapts or progresses toward irreversible injury [5,6,7,15]. This distinction is central for OA, because in some models OA stabilizes mitochondrial homeostasis, whereas in others it amplifies the processes leading to MOMP and apoptotic death.
3.2. Endoplasmic Reticulum Stress and the Unfolded Protein Response
The ER is responsible for the folding and maturation of secretory and membrane proteins, lipid synthesis, and calcium storage. When ER proteostasis is disrupted, ER stress develops and activates the unfolded protein response. In metazoan cells, the major UPR branches are controlled by three ER membrane sensors: IRE1, PERK, and ATF6. Initially, the UPR is cytoprotective because it decreases the burden of newly synthesized proteins, increases molecular chaperone expression, and promotes protein quality control. Under chronic or severe stress, however, the UPR can switch to a proapoptotic program [8,9,10,11,39,40].
Contemporary understanding of ER stress goes beyond the classic misfolded-protein paradigm. The UPR also senses membrane integrity, lipid bilayer stress, altered lipid metabolism, calcium imbalance, and signals originating from other organelles. This is highly relevant here because in many disease models OA-associated changes in GRP78/BiP, CHOP, PERK, IRE1α, or ATF6 occur together with mitochondrial alterations, implying regulation at the level of a broader stress network rather than of a single isolated pathway [8,9,10,11,39,40].
3.3. Mitochondria–ER Contact Sites
One of the most useful concepts in modern organelle stress biology is that of mitochondria–ER contact sites (MERCS), also known as mitochondria-associated membranes. These specialized interfaces are not merely structural bridges. They act as signaling hubs that regulate Ca2+ flux, lipid exchange, autophagy, mitophagy, and apoptosis [13,14]. Increasingly, they are regarded as crucial cell-fate decision platforms under stress.
MERCS are especially important in apoptosis because elements of the BCL-2 machinery localize there and influence not only mitochondrial membrane permeabilization but also calcium signaling and autophagy. Disturbances in MERCS dynamics and composition have been implicated in neurodegeneration, cancer, and inflammatory disorders. For OA, MERCS provide a powerful interpretive framework because they allow the simultaneous changes observed in ER stress, mitochondrial function, autophagy, and apoptosis to be understood as components of a single integrated system [6,7,13,14,17].
3.4. Autophagy and Mitophagy
Autophagy is a lysosomal degradative pathway that removes damaged proteins and organelles and recycles metabolic substrates under stress. In most settings it is adaptive and supports survival, but under certain conditions it may also participate in cell death [15,16,17]. Thus, increased autophagy does not automatically imply protection, and its suppression does not necessarily indicate greater injury.
Mitophagy is a selective form of autophagy targeting damaged mitochondria. It is essential for mitochondrial quality control because it limits the accumulation of mitochondria that generate excess ROS, lose membrane potential, or disturb calcium homeostasis [14,15,16]. Mitophagy may therefore be protective by restoring homeostasis, but excessive or dysregulated mitophagy may also contribute to loss of functional mitochondria and worsening cellular injury.
At the signaling level, autophagy and mitophagy are strongly regulated by the AMPK–mTOR axis. AMPK acts as an energy sensor that activates catabolic pathways, including autophagy, and contributes to mitochondrial quality control through ULK1 and other downstream effectors [15,17]. In OA studies this axis is repeatedly implicated, because many reports interpret OA action in terms of AMPK activation and/or mTOR inhibition, which may favor either adaptive autophagy or cell death depending on context.
3.5. Apoptosis and Crosstalk with Autophagy
Apoptosis remains the best-characterized form of regulated cell death, and mitochondria occupy a central role in its intrinsic pathway. MOMP, governed by the BCL-2 family network, leads to cytochrome c release, caspase activation, and a relatively orderly dismantling of the cell [6,7]. Modern studies also emphasize that mitochondrial apoptotic signaling can have non-lethal functions affecting aging, innate immunity, and tumorigenesis [7].
The relationship between autophagy and apoptosis is complex and bidirectional. Autophagy can delay apoptosis by removing damaged cellular material, but it can also facilitate the transition toward cell death under prolonged or overwhelming stress [16,17]. A key point of intersection is the Beclin 1 network, because Beclin 1 interacts with BCL-2/BCL-XL, whereas caspase-mediated cleavage of Beclin 1 favors apoptotic progression [17]. Therefore, in OA studies, concurrent changes in LC3, p62, Beclin 1, BCL-2/BAX, and caspases should be interpreted not as unrelated readouts, but as markers of dynamic crosstalk between adaptation and death.
3.6. Why This Framework Matters for OA
From the perspective of this review, the key conclusion is that organelle stress is not a single event, but a multilayered response involving redox control, calcium handling, proteostasis, mitochondrial quality control, MERCS signaling, autophagy, and the apoptotic machinery [4,5,6,7,8,9,10,11,12,13,14,15,16,17,39,40]. This network model explains why OA may in some settings suppress ER stress, stabilize mitochondria, and reduce apoptosis, whereas in others it enhances autophagy, mitophagy, and mitochondrial apoptosis. The following sections therefore analyze OA within this network framework rather than in a strictly linear pathway model.
4. Oleanolic Acid and Mitochondrial Dysfunction
Mitochondria represent one of the major hubs of OA action because ROS production, membrane potential maintenance, mitochondrial quality control, mitophagy, and intrinsic apoptosis all converge at this level. The available evidence indicates that OA is not a unidirectional mitochondrial effector. Rather, in non-cancer injury models it frequently stabilizes mitochondrial function and limits cell death, whereas in many cancer models it promotes mitochondrial damage and switches cells toward apoptosis [6,7,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,41].
4.1. OA as a Stabilizer of Mitochondrial Function
In models of aging and neurodegeneration, OA shows clear mitochondria-protective effects. In aged mice, OA administration improved cardiac function, ultrastructure, and mitochondrial integrity, and these benefits were linked to regulation of mitophagy [21]. In Parkinson’s disease models based on MPP+ and MPTP, OA reduced mitochondrial dysfunction and ROS accumulation, increased DJ-1 levels, and promoted mitophagy, whereas DJ-1 knockdown weakened OA-mediated neuroprotection [22]. Earlier work using a 6-hydroxydopamine model showed that OA lowered intracellular ROS in PC12 cells and attenuated neurotoxicity and microglial activation in vivo [23]. Together, these findings suggest that in non-cancer cells OA can preserve a functional mitochondrial pool by reducing oxidative injury and improving mitochondrial quality control [21,22,23].
A similar pattern has been reported in metabolic and vascular models. In pancreatic beta cells, OA inhibited mitochondrial apoptosis, and this effect was linked to ERK–NRF2 signaling and enhanced glutathione synthesis [24]. In endothelial cells exposed to high glucose, OA reduced ROS, preserved mitochondrial membrane potential, and limited apoptosis in association with PPARδ activation [18]. A recent 2026 study extended this picture to mitochondrial dynamics, showing that in atherosclerosis OA activated AMPK, reduced p-DRP1/DRP1, MFF, and FIS1 signaling, alleviated mitochondrial swelling and cristae disruption, and increased ATP content [25]. These observations suggest that OA acts not only as an antioxidant modulator, but also as a regulator of mitochondrial fission and functional quality [18,24,25].
4.2. OA, Mitophagy, and Mitochondrial Quality Control
One of the most interesting aspects of OA action is its influence on mitophagy. In A549 lung cancer cells, OA induced autophagy followed by mitochondrial changes consistent with mitophagy; LC3-II accumulation became evident over time, and the observed pattern was interpreted as selective lysosomal removal of damaged mitochondria [26]. In cardiac aging, OA was similarly linked to mitophagy regulation and preservation of mitochondrial integrity [21], while in Parkinson’s disease its neuroprotective effect was associated with the JNK–Sp1–DJ-1 axis, suggesting a more specific signaling mechanism of mitophagy induction [22]. These findings indicate that OA may reprogram the cellular response to mitochondrial stress not only by suppressing ROS, but also by promoting turnover of damaged mitochondria [21,22,26].
Nevertheless, OA-induced mitophagy does not always imply cytoprotection. Depending on the biological setting, the same process may either support survival by purging damaged mitochondria or contribute to loss of mitochondrial mass and eventual cell death. In non-cancer models, OA-driven mitophagy usually supports homeostatic recovery, whereas in cancer cells it may form part of a more aggressive stress response coupled to apoptosis [15,16,21,22,26]. Therefore, increased mitophagy markers should not automatically be interpreted as beneficial without simultaneous assessment of viability, mitochondrial membrane potential, ROS, and apoptotic signaling.
4.3. OA as an Inducer of Mitochondrial Damage and Apoptosis in Cancer
In many cancer models, OA acts in the opposite direction and activates classical mitochondrial apoptosis. In gallbladder cancer cells, OA reduced cell viability, increased the apoptotic fraction, lowered the BCL-2/BAX ratio, enhanced cytochrome c release, and activated caspase-9, caspase-3, and PARP, while also inhibiting xenograft growth in vivo [27]. Similar results were reported in hepatocellular carcinoma, where OA induced mitochondrial dysfunction, increased the BAX/BCL-2 ratio, promoted cytochrome c release, and inhibited survival-related signaling [28]. In mechanistic work spanning several cancer cell lines, OA activated ROS/ASK1/p38 MAPK signaling, promoted BAX and BIM translocation to mitochondria, increased BCL-2 phosphorylation, and p38 inhibition rescued cells from OA-induced apoptosis [29]. Collectively, these data strongly support the view that in cancer cells OA can destabilize mitochondria and activate the MOMP–cytochrome c–caspase cascade [27,28,29].
This picture is complemented by studies showing that mitochondrial death after OA is often intertwined with autophagy. In AGS gastric cancer cells, OA induced both apoptosis and autophagy via the PI3K/AKT/mTOR pathway [32]. In colon cancer, OA also activated AMPK and inhibited mTOR, with autophagy and apoptosis being largely AMPK-dependent [33]. In KRAS-transformed cells, OA induced autophagy, and interruption of autophagy weakened its antiproliferative and anti-invasive effects, indicating that autophagy can participate in OA-mediated anticancer activity [30]. Thus, in oncology OA should not be viewed simply as an antioxidant, but rather as a regulator of cellular stress thresholds that may overload already bioenergetically unstable cancer cells [28,29,30,31,32,33].
Importantly, even within cancer cells, the response to OA is not fully uniform. ERK-dependent Nrf2 activation has been reported to counteract OA-induced ROS accumulation and reduce apoptosis; conversely, ERK or Nrf2 inhibition sensitized cancer cells to OA [31]. This suggests that even in tumors there is tension between OA-induced pro-death signaling and compensatory responses that partially protect mitochondria and survival. This helps explain why different models and doses may show different balances between autophagy and apoptosis, and different intensities of mitochondrial injury.
4.4. Mechanistic Synthesis
At present, the most plausible model is that OA acts as a context-dependent modulator of mitochondrial stress. In normal, aging, or metabolically injured cells, OA usually lowers ROS, stabilizes membrane potential, improves mitochondrial quality, and promotes adaptive mitophagy [18,21,22,23,24,25]. In cancer cells, OA more often amplifies stress signaling, lowers the threshold for MOMP, activates caspases, and couples apoptosis with autophagy [26,27,28,29,30,31,32,33]. This duality is best explained by the fact that OA does not simply “repair” mitochondria but acts on central decision nodes of the stress response, including redox control, mitochondrial dynamics, mitophagy, BCL-2 family signaling, and AMPK/mTOR.
5. Oleanolic Acid and Endoplasmic Reticulum Stress
Compared with the relatively extensive literature on OA and mitochondrial regulation, direct studies examining the regulation of ER stress by OA are fewer. This does not mean that ER involvement is marginal. On the contrary, the available data suggest that OA influences one of the central nodes of the cellular stress response, where proteostasis, calcium signaling, redox balance, and ER–mitochondria communication converge [8,9,10,11,12,13,14,19,20,39,40].
5.1. OA as an ER Stress-Limiting Factor in Metabolic Models
One of the clearest demonstrations of OA-dependent attenuation of ER stress comes from diabetic nephropathy. In a classic study, chronic OA administration in OLETF rats improved renal morphology, increased insulin secretion and antioxidant defenses, and reduced renal fibrosis together with ER stress-related abnormalities. This effect was partially reproduced in vitro: in mesangial cells, high glucose and ER stress inducers increased ROS and ER stress signaling, whereas OA attenuated these changes [19]. These data are important because they place OA at the intersection of oxidative stress and UPR regulation, in line with the current understanding of diabetic nephropathy as a complex stress-driven pathology [9,10,11,19,39,40].
Mechanistically, this model is highly informative because chronic hyperglycemia does not induce a “pure” ER stress response, but rather a mixed state involving glucotoxicity, ROS, inflammatory signaling, and fibrogenesis. In this context, OA appears to function less as a selective inhibitor of a single UPR branch and more as a modulator of the total stress burden that reduces activation of the PERK–eIF2α–ATF4 axis and its downstream proapoptotic consequences [9,10,11,19,39].
5.2. OA, TRAP1, and the ER–Mitochondria Axis in Ochratoxin A Nephrotoxicity
More direct mechanistic evidence comes from the study by Zhang et al. on ochratoxin A nephrotoxicity. In HK-2 cells, ochratoxin A suppressed TRAP1 and simultaneously activated mitochondrial apoptosis markers (CypD, BAX, cytochrome c, caspase-9, caspase-3) and ER stress markers (GRP78, p-PERK, p-eIF2α, ATF4, CHOP). OA pretreatment clearly attenuated TRAP1 suppression and alleviated this integrated injury program [20]. This is one of the most important studies for the present review because it frames OA not only as an antioxidant, but also as a regulator of ER–mitochondria coupling through a protein located close to the core of this crosstalk [13,14,20].
The interpretation of TRAP1 extends beyond a single toxicological model. TRAP1 is a stress-associated chaperone linked to mitochondrial function, and its silencing in the same study further increased proapoptotic signaling. From the perspective of organelle stress biology, this suggests that OA can restrain the transition from an adaptive response to a terminal response in which UPR activation and mitochondrial stress reinforce one another and drive cell death [9,10,11,13,14,19,20,39,40].
5.3. Does OA Directly Regulate the UPR?
At present, it is difficult to conclude that OA is a direct and selective regulator of a specific UPR branch. What is better supported is the notion that OA reduces the overall burden of cellular stress and thereby secondarily dampens ER stress markers. This is supported by both the diabetic nephropathy model and the ochratoxin A model, where decreases in ROS, improved mitochondrial function, and reduced apoptosis co-occurred with attenuation of the ER stress signature [19,20]. In this sense, OA may act upstream of full UPR activation by limiting the signals that overload the ER, rather than by directly blocking only PERK, IRE1, or ATF6.
This interpretation also fits modern UPR biology, where ER stress is understood as a response not only to misfolded proteins but also to altered membrane composition, lipid metabolism, calcium homeostasis, and mitochondria-derived signals [9,10,11,39,40]. Since OA strongly affects redox signaling, NRF2, lipid metabolism, and mitochondria, it is not surprising that ER stress markers are also modulated.
5.4. The Liver as an Example of Protection and Adaptive Overload
In the hepatic literature, OA is usually described as hepatoprotective, particularly in models of cholestasis, oxidative stress, and disturbed bile acid metabolism. In a model of ANIT-induced cholestatic liver injury, OA reduced liver damage, decreased bile acid accumulation, enhanced expression of bile acid-related transporters and regulators, and promoted Nrf2 activation [42]. Although this study did not focus directly on the UPR, it fits the logic of ER stress biology because cholestasis and membrane-lipid disturbance are strongly connected to ER overload [9,10,11,39,40,42].
At the same time, hepatic studies also show that OA is not unconditionally protective. Repeated oral administration of OA in mice was reported to cause cholestatic liver injury [43], and a metabolomic study of OA-induced hepatotoxicity demonstrated disturbances in bile acid, amino acid, and energy metabolism [44]. These findings are highly relevant for the present review because they indicate that in the liver, too, OA may shift from adaptive support to homeostatic overload, depending on dose and duration of exposure.
5.5. Critical Synthesis
Taken together, the current evidence suggests that OA should not be regarded as a classical “ER stress inhibitor,” but rather as a context-dependent modulator of the ER–mitochondria axis. In metabolic and toxic models, OA generally attenuates UPR markers and the CHOP-dependent proapoptotic component, especially when this occurs together with ROS reduction and mitochondrial stabilization [19,20]. In hepatic models OA may be protective in certain settings [42], but at excessive or prolonged exposure it may itself contribute to injury and fibrosis [43,44]. This pattern strongly supports the central thesis of this review: OA does not act linearly, but shifts the balance between adaptation and decompensation, with ER stress being one of the major sites where this balance becomes evident.
6. Oleanolic Acid and Autophagy/Mitophagy
Autophagy and mitophagy most clearly illustrate the context dependence of OA action. Many studies show autophagy induction through inhibition of PI3K/AKT/mTOR or activation of AMPK, accompanied by changes in LC3, p62, and apoptosis-related proteins. Yet the biological meaning of this response is far from uniform. OA-induced autophagy has been interpreted as protective, adaptive, mitochondria-quality preserving, or cytotoxic depending on the model [15,16,17,18,21,22,26,27,28,29,30,31,32,33,41,43].
6.1. OA as an Inducer of Adaptive Autophagy in Non-Cancer Models
Compelling examples of protective autophagy come from degenerative and aging-related models. In osteoarthritis, OA alleviated pain and cartilage degeneration in a rat model, while in chondrocyte-like cells it suppressed the IL-1β-induced inflammatory response. Importantly, autophagy activation was demonstrated by multiple complementary methods and linked to inhibition of PI3K/AKT/mTOR signaling [41]. In this case, autophagy clearly functioned as a homeostatic response rather than as an indicator of irreversible damage.
Similarly, in cardiac aging OA improved cardiac function, tissue structure, mitochondrial integrity, and mitophagy-related parameters, with the authors implicating FUNDC1-dependent mitophagy [21]. In Parkinson’s disease models, OA reduced mitochondrial dysfunction and ROS accumulation, increased DJ-1, and promoted mitophagy through the JNK–Sp1–DJ-1 axis [22]. Collectively, these data indicate that in non-cancer settings OA most often engages autophagy and mitophagy as adaptive programs supporting organelle quality and stress resolution [21,22,41].
6.2. OA and Protective Autophagy in Cancer Cells
In cancer, the picture is more complex because autophagy induced by OA can paradoxically reduce its anticancer efficacy. A notable study showed that OA induced protective autophagy through JNK activation and mTOR inhibition, and that autophagy blockade enhanced apoptosis [34]. Likewise, in A549 cells, OA induced complete autophagic flux together with mitochondrial fragmentation and mitophagy, while inhibition of autophagy aggravated mitochondrial depolarization and decreased cell survival [26]. In these settings, autophagy/mitophagy served as a pro-survival mechanism allowing cells to cope with OA-induced stress.
These results are highly important for interpretation because they show that increased LC3-II or the presence of autophagosomes does not automatically indicate anticancer efficacy. In at least part of the cancer literature, OA-induced autophagy acts as a compensatory response that protects cells from death.
6.3. OA and Autophagy Participating in Cytotoxicity
By contrast, another group of studies indicates that OA-induced autophagy may also participate in cytotoxicity. In AGS gastric cancer cells, OA reduced viability and induced both apoptosis and autophagy via PI3K/AKT/mTOR signaling [32]. In colon cancer, OA induced autophagy and apoptosis largely through AMPK activation and mTOR inhibition [36]. In such models, autophagy appears not to rescue the cell but to form part of the OA-driven anticancer program.
These divergent findings do not support a simple conclusion that OA induces either “good” or “bad” autophagy. Rather, OA seems to shift the cellular set point between adaptation and collapse depending on cell type, basal stress burden, dose, exposure time, and the dominant signaling background. In highly stressed cells, especially many cancer cells, the same autophagic response may either transiently buffer stress or amplify the progression toward apoptosis.
6.4. Mitophagy as a Specific OA Target Node
Mitophagy deserves special emphasis in the OA literature because it most directly links mitochondrial stress, autophagy, and survival/death decisions. In A549 cells, OA induced mitochondrial fragmentation and colocalization of mitochondria with LC3, consistent with activation of mitophagy [26]. In the aging heart, OA increased mitophagy-related markers associated with FUNDC1 and improved mitochondrial integrity [21]. In Parkinson’s disease, OA promoted DJ-1-dependent sequestration of damaged mitochondria into autophagosomes and protected dopaminergic neurons [22]. Together, these studies suggest that one of the most important actions of OA may be selective turnover of damaged mitochondria rather than indiscriminate global activation of autophagy.
Still, the same mitophagic process may support either survival or death depending on context. This is methodologically important: without simultaneous assessment of membrane potential, ROS, viability, and caspase activation, it is easy to overinterpret the increase in LC3-II or mitophagy markers as inherently beneficial.
6.5. PI3K/AKT/mTOR and AMPK–mTOR as the Main Interpretive Axes
At the signaling level, most key studies converge on two axes: PI3K/AKT/mTOR and AMPK–mTOR. In chondrocytes, OA activated autophagy by suppressing PI3K/AKT/mTOR [41]. In AGS cells, the same pathway was associated with concurrent autophagy and apoptosis [32]. In colon cancer, AMPK–mTOR predominated [33]. More broadly, molecular reviews of OA in cancer have repeatedly identified AMPK activation and mTOR suppression as central recurring themes [2,3,15,32,33,41].
At the same time, one should distinguish studies reporting changes in autophagy-associated markers from those demonstrating functional autophagic flux. This distinction is essential for a critical review because not every decrease in mTOR activity or increase in Beclin 1 proves that a complete and biologically meaningful autophagic program has occurred.
6.6. Critical Synthesis
The available data support a model in which OA acts as a context-dependent regulator of organelle quality rather than as a simple “autophagy inducer.” In non-cancer, aging, and neurodegenerative settings, OA generally promotes adaptive autophagy/mitophagy and supports homeostasis [21,22,41]. In cancer, OA may trigger either protective autophagy that reduces treatment efficacy [26,34] or autophagy coupled to apoptosis that contributes to cytotoxicity [32,33]. This is why autophagy is central to the present review: it best illustrates that OA does not act linearly but shifts cells between adaptation and death through the integrated mitochondria–ER–mTOR/AMPK–apoptosis axis.
Representative non-cancer models of OA action within the organelle stress framework are summarized in Table 1.
7. Oleanolic Acid and Apoptosis
Apoptosis is one of the most frequently reported consequences of OA exposure, but the literature clearly shows that this effect is not unidirectional. Depending on the biological model, OA may either suppress apoptosis in normal or reversibly injured cells or induce apoptosis in cancer cells. This duality fits with current understanding of intrinsic apoptosis, in which BAX/BAK, MOMP, cytochrome c, and caspases interact closely with autophagy and ER stress [6,7,17].
7.1. OA and Mitochondrial Apoptosis
In cancer models, OA most often activates the classical intrinsic apoptotic pathway. In gallbladder cancer, OA reduced viability, increased the apoptotic fraction, lowered the BCL-2/BAX ratio, enhanced cytochrome c release, and activated caspase-9, caspase-3, and PARP [27]. Similar results were obtained in hepatocellular carcinoma, where OA increased the BAX/BCL-2 ratio and promoted mitochondrial apoptosis [28]. Additional mechanistic studies linked OA-induced apoptosis to ROS/ASK1/p38 MAPK signaling and translocation of BAX and BIM to mitochondria [29]. These data strongly support the conclusion that in cancer cells OA often acts at the level of the core mitochondrial death module.
7.2. OA as an Antiapoptotic Agent in Non-Cancer Models
In non-cancer models, the picture is often reversed. In pancreatic beta cells, OA reduced mitochondrial apoptosis through ERK–NRF2 signaling and increased glutathione synthesis [24]. In diabetic nephropathy, OA attenuated oxidative stress and ER stress while improving renal injury [19]. In ochratoxin A nephrotoxicity, OA lowered expression of CypD, BAX, cytochrome c, caspase-9, and caspase-3, together with ER stress markers such as GRP78, p-PERK, p-eIF2α, ATF4, and CHOP [20]. Thus, in non-tumor tissues OA more often stabilizes mitochondria and the ER than activates terminal apoptosis.
This apparent contradiction is mechanistically coherent. In normal or reversibly injured cells, OA typically lowers ROS, improves redox balance, and suppresses secondary activation of the apoptotic machinery. In cancer cells, which already operate close to a bioenergetic stress threshold, the same compound may tip the balance toward MOMP and caspase activation [6,7,19,20,24].
7.3. ER Stress-Associated Apoptosis
An important aspect of OA action is its effect on ER stress-associated apoptosis. Modern UPR biology holds that sustained or intense ER stress drives the shift from adaptive signaling to a proapoptotic state involving CHOP, ATF4, PERK/eIF2α, calcium signaling, and mitochondrial engagement [8,9,10,11,39,40]. In the ochratoxin A model, OA attenuated both the UPR signature and mitochondrial apoptotic markers, suggesting that OA can interfere earlier in the cascade by reducing the transition from adaptive UPR to terminal, pro-death UPR [20].
This distinction is important. If OA reduces CHOP-dependent UPR and simultaneously stabilizes mitochondria, its action can be understood as interruption of a positive feedback loop between the ER and mitochondria. Such a mechanism is fully consistent with current knowledge on MERCS as integration sites for calcium, redox, and proapoptotic signals [13,14].
7.4. Autophagy–Apoptosis Crosstalk After OA
One of the most difficult interpretive issues is the relationship between autophagy and apoptosis after OA treatment. In AGS gastric cancer cells, OA induced both autophagy and apoptosis via PI3K/AKT/mTOR [32]. In colon cancer, a similar pattern was seen through AMPK–mTOR signaling [33]. In these cases, autophagy appears to cooperate with cytotoxicity. In other cancer models, however, OA-induced autophagy was protective, and its inhibition increased apoptosis [26,30,31]. These discrepancies fit well with the broader biology of Beclin 1 and BCL-2 network interactions, where the same molecular interface can promote either adaptation or death depending on stress intensity and cellular background [17].
From a methodological perspective, OA studies should therefore not treat autophagy and apoptosis as alternative endpoints, but as coupled processes. LC3-II, Beclin 1, or p62 alone are insufficient to infer whether autophagy is beneficial or harmful; similarly, caspase-3 activation alone does not define whether a cell has irreversibly crossed the death threshold. Reliable interpretation requires parallel assessment of autophagic flux, membrane potential, ROS, BAX/BCL-2 balance, cytochrome c release, and caspase activation.
7.5. What Determines the Direction of OA Action?
At present, the most plausible determinants of whether OA acts pro- or antiapoptotically include cell type, basal stress load, mitochondrial state, activity of the PI3K/AKT/mTOR and AMPK pathways, the strength of antioxidant responses, and dose and duration of exposure [15,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,41,45]. In normal or metabolically injured cells, OA more often reinforces compensatory responses and limits apoptosis. In cancer, it more often overloads the stress network and facilitates MOMP, caspase activation, and cell death.
7.6. Critical Synthesis
Taken together, apoptosis appears to be one of the main downstream effectors of OA action, but not necessarily the primary event in every model. OA may suppress apoptosis by reducing ROS, attenuating ER stress, and stabilizing mitochondria, or induce it by increasing stress burden, activating BAX/BIM, lowering BCL-2, releasing cytochrome c, and activating caspases [19,20,22,23,24,27,28,29,30,31,32,33]. The final outcome depends on the starting condition of the cell and on whether autophagy/mitophagy still functions adaptively or can no longer compensate for damage.
Representative cancer models linking mitochondrial stress, autophagy, and apoptosis are summarized in Table 2.
8. Integrated Model of OA Action: The Mitochondria–ER–Autophagy–Apoptosis Axis
Based on the evidence reviewed above, OA should be interpreted not as a regulator of a single pathway, but as a context-dependent modulator of an integrated organelle stress network. In this model, OA influences redox homeostasis, mitochondrial function, UPR signaling, autophagy/mitophagy, and the apoptotic machinery in a coordinated manner [4,5,6,7,8,9,10,11,12,13,14,15,16,17,39,40]. This framework is fully consistent with current understanding of MERCS as signaling platforms integrating Ca2+ transfer, lipid metabolism, autophagy, and apoptosis [13,14].
Under non-cancer injury conditions such as aging, neurodegeneration, nephrotoxicity, or metabolic stress, OA most often shifts the system toward adaptation and restoration of homeostasis. Functionally, this usually means lower ROS, improved mitochondrial quality, mitophagic removal of damaged organelles, and reduced transition from adaptive UPR to CHOP-driven proapoptotic signaling [18,19,20,21,22,23,24,25]. In Parkinson’s disease models, OA reduced mitochondrial dysfunction and promoted DJ-1-dependent mitophagy [22], while in ochratoxin A nephrotoxicity it attenuated both mitochondrial and ER-linked apoptotic signaling through a TRAP1-related mechanism [20].
In cancer cells, the situation is different. Many tumors already operate under elevated basal stress, with disturbed bioenergetics, deregulated PI3K/AKT/mTOR signaling, and enhanced dependence on compensatory survival systems. In this context, OA may push the stress network beyond its tolerance threshold. The resulting phenotype may include increased autophagy, mitophagy, and mitochondrial apoptosis, but the biological interpretation is not uniform: in some models autophagy is protective, whereas in others it contributes to cytotoxicity [26,27,28,29,30,31,32,33,41]. OA should therefore be understood as a regulator of stress thresholds rather than as a universal antioxidant or a universal cell-death inducer.
The integrated model can be summarized as follows: OA → redox/Ca2+/UPR–mitochondria crosstalk → autophagy/mitophagy → survival or apoptosis. During the adaptive phase, OA lowers ROS, stabilizes mitochondria, unloads the ER, and promotes repair-oriented autophagy/mitophagy. During decompensation, especially in cancer cells or under excessive stress, OA may facilitate MOMP, cytochrome c release, caspase activation, and cell death. MERCS provide the most plausible integration level for this model because calcium signaling, BCL-2 family signaling, autophagy, and stress-related inflammatory cues intersect there. At present, however, this remains mainly a mechanistic synthesis based on convergent evidence, rather than a fully validated direct target model.
9. Critical Appraisal of the Literature
Despite the rapidly increasing number of publications, the OA literature in organelle stress remains highly heterogeneous. Biological models differ widely, as do OA doses, exposure times, solvents, and marker panels. Some studies emphasize ROS and membrane potential, others focus on GRP78/CHOP, and still others on LC3-II, p62, or caspases. As a result, direct comparison among studies is difficult. This is especially true in cancer research, where cell lines differ substantially in metabolic state and genetic background, which may explain why OA-induced autophagy is reported as protective in some cases and cytotoxic in others [26,27,28,29,30,31,32,33,41].
A second important limitation is that much of the literature is phenotypic and marker-based rather than fully causal. Many studies document changes in mitochondrial, ER, autophagic, or apoptotic markers, but far fewer establish which process is primary and which is secondary. This is particularly relevant for autophagy, where increased LC3-II, decreased p62, or autophagosome formation do not necessarily prove complete autophagic flux, let alone define whether autophagy is adaptive or harmful [15,16,26,32,33,41].
A third limitation is the lack of direct studies examining integrative nodes of ER–mitochondria communication such as MERCS or direct OA targets within stress-sensing machinery. The available evidence strongly suggests that MERCS provide an excellent interpretive framework for OA, especially considering the data on TRAP1, DJ-1-dependent mitophagy, BCL-2 family signaling, and UPR–mitochondria coupling [13,14,20,22]. Nevertheless, for OA itself this remains more of an integrative hypothesis than a widely tested experimental field.
A fourth major issue concerns translation. OA has poor aqueous solubility, limited permeability, and low bioavailability, which are longstanding barriers to its pharmacological and nutraceutical application [1,2,37,38].Formulation research has produced improved delivery strategies, but even so, clinical translation remains limited. Human pharmacokinetic data are still scarce, and currently available evidence is insufficient to directly map most cellular observations onto expected clinical effects [37,38].
Finally, safety must be considered critically. Although OA is often described as hepatoprotective, the literature also shows that repeated or prolonged administration can cause cholestatic liver injury and metabolic disturbances [43,44]. This is highly relevant for the present topic because it indicates that shifting organelle stress networks by OA may be beneficial only within a specific therapeutic window. The major translational limitations od OA are summarized below in Table 3.
Overall, the major strength of the current literature lies in its rich mechanistic insight at the cellular level, whereas its major weaknesses are insufficient standardization, incomplete causal validation, and limited translational depth. Future OA studies should integrate standardized dosing and formulation, simultaneous assessment of mitochondria, ER, MERCS, autophagy, and apoptosis, functional autophagic flux measurements, and better pharmacokinetic and safety studies.

Table 3.
Translational limitations of oleanolic acid.
| Limitation | Description | Consequence for interpretation | Possible solution / research direction |
Ref. |
|---|---|---|---|---|
| Poor aqueous solubility. | OA is strongly hydrophobic and poorly water-soluble. | Limits absorption and complicates extrapolation from in vitro to in vivo exposure. | Nanoformulations, cyclodextrin complexes, solid dispersions, lipid-based delivery systems. | [1,2,37,38] |
| Low oral bioavailability. | Systemic exposure after oral dosing is limited and formulation-dependent. | Many cellular concentrations may not be clinically achievable. | Pharmacokinetic-guided dose selection; optimized oral formulations. | [1,37,38] |
| Strong formulation dependence. | OA behavior changes with matrix and carrier system. | Biological effects may vary between studies using different vehicles. | Standardization of formulations across experimental studies. | [1,2,37,38] |
| Heavy reliance on in vitro data. | Many mechanistic findings come from cell culture models. | May overestimate efficacy or miss tissue-level toxicity and metabolism. | More in vivo validation and better PK/PD integration. | [1,2,3] |
| Heterogeneous doses and exposure times. | Studies use widely different concentrations and treatment schedules. | Direct comparison across models is difficult. | Standardized reporting and dose–response studies. | [1,2,3] |
| Incomplete organelle-level integration. | Many studies assess only one layer: ROS, ER stress, autophagy, or apoptosis. | Makes mechanistic interpretation fragmentary. | Simultaneous analysis of mitochondria, ER, MERCS, autophagic flux, and apoptosis. | [4,9,10,11,12,13,14,39,40] |
| Limited direct MERCS evidence. | MERCS are mechanistically plausible but rarely measured directly in OA studies. | Integrated model remains partly inferential. | Direct imaging and functional MERCS assays in OA-treated models. | [13,14] |
| Limited human data. | Human PK data remain scarce. | Translation to clinical or nutraceutical use is still uncertain. | More controlled human PK and safety studies. | [37] |
| Dose-dependent hepatotoxicity risk. | OA can be hepatoprotective in some settings but hepatotoxic in others. | Therapeutic window may be narrow or context dependent. | Long-term safety studies; dose-window definition. | [42,43,44] |
| Overinterpretation of autophagy markers. | LC3-II or p62 changes alone do not prove beneficial or harmful autophagy. | Risk of incorrect mechanistic conclusions. | Assess full autophagic flux together with viability, ROS, ΔΨm, and caspases. | [16,17,26,32,33,34] |
Note: The translational challenges are driven mainly by poor solubility, low and formulation-dependent bioavailability, limited human pharmacokinetic data, and a safety paradox in which OA may be protective in some liver-injury settings but hepatotoxic under repeated or prolonged exposure.
10. Conclusions and Future Perspectives
The currently available evidence supports the view that OA is a promising but clearly context-dependent modulator of organelle stress. In non-cancer models, OA most often lowers the cellular stress burden, stabilizes mitochondria, attenuates ER stress signatures, and promotes adaptive autophagy/mitophagy. In many cancer models, the same compound may shift the system toward decompensation by lowering the threshold for mitochondrial apoptosis and coupling this response to autophagy. The most coherent mechanistic picture emerges when OA is interpreted as a regulator of the integrated mitochondria–ER–autophagy–apoptosis axis rather than as a compound acting purely through antioxidant or purely through proapoptotic mechanisms [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,41].
The most important future direction is a move from marker-based descriptions toward integrative studies that simultaneously examine UPR signaling, MERCS, mitochondrial dynamics, mitophagy, apoptosis, and OA pharmacokinetics. Particularly valuable would be studies that directly test whether OA modulates MERCS components or stress sensors, and studies defining the range between protective and overload-inducing exposure. Equally important are formulation advances and better human translational studies, because OA currently remains a highly promising molecule for stress biology, but not yet one with fully defined translational utility [37,38].
Author Contributions
Conceptualization, A.G. and B.B.-C.; methodology, A.G. writing—original draft preparation, A.G.; writing—review and editing, A.G.; visualization, A.G.; supervision, A.G and B.B.-C.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
All authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 6-OHDA | 6-Hydroxydopamine |
| AGS | Human gastric adenocarcinoma cell line |
| AKT | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| ANIT | Alpha-naphthyl isothiocyanate |
| A549 | Human lung adenocarcinoma cell line |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| ATF4 | Activating transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| ATP | Adenosine triphosphate |
| BAK | BCL-2 antagonist/killer 1 |
| BAX | BCL-2-associated X protein |
| BCL-2 | B-cell lymphoma 2 |
| BCL-XL | B-cell lymphoma-extra large |
| BiP | Binding immunoglobulin protein |
| BIM | BCL-2-interacting mediator of cell death |
| Ca2+ | Calcium ion |
| CHOP | C/EBP homologous protein |
| CypD | Cyclophilin D |
| DJ-1 | DJ-1 protein |
| DRP1 | Dynamin-related protein 1 |
| eIF2α | Eukaryotic initiation factor 2 alpha |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| FIS1 | Mitochondrial fission 1 protein |
| FUNDC1 | FUN14 domain-containing protein 1 |
| GRP78 | Glucose-regulated protein 78 |
| HCC | Hepatocellular carcinoma |
| HK-2 | Human kidney-2 proximal tubular epithelial cell line |
| IGF-1 | Insulin-like growth factor 1 |
| IL-1β | Interleukin 1 beta |
| IRE1 | Inositol-requiring enzyme 1 |
| JNK | c-Jun N-terminal kinase |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| MAMs | Mitochondria-associated membranes |
| MAPK | Mitogen-activated protein kinase |
| MERCS | Mitochondria–ER contact sites |
| MFF | Mitochondrial fission factor |
| MOMP | Mitochondrial outer membrane permeabilization |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MPP+ | 1-Methyl-4-phenylpyridinium |
| mTOR | Mechanistic target of rapamycin |
| NRF2 / Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OA | Oleanolic acid |
| OLETF | Otsuka Long-Evans Tokushima Fatty |
| PARP | Poly(ADP-ribose) polymerase |
| PC12 | Rat pheochromocytoma cell line |
| PERK | Protein kinase RNA-like ER kinase |
| PI3K | Phosphoinositide 3-kinase |
| PK/PD | Pharmacokinetic/pharmacodynamic |
| PPARδ | Peroxisome proliferator-activated receptor delta |
| ROS | Reactive oxygen species |
| TRAP1 | Tumor necrosis factor receptor-associated protein 1 |
| ULK1 | Unc-51-like kinase 1 |
| UPR | Unfolded protein response |
References
- Castellano, J.M.; Ramos-Romero, S.; Perona, J.S. Oleanolic Acid: Extraction, Characterization and Biological Activity. Nutrients 2022, 14, 623. [CrossRef]
- Jannus, F.; Sainz, J.; Reyes-Zurita, F.J. Principal Bioactive Properties of Oleanolic Acid, Its Derivatives, and Analogues. Molecules 2024, 29, 3291. [CrossRef]
- Günther, A.; Bednarczyk-Cwynar, B. Oleanolic Acid: A Promising Antioxidant—Sources, Mechanisms of Action, Therapeutic Potential, and Enhancement of Bioactivity. Antioxidants 2025, 14, 598. [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial Dysfunction: Mechanisms and Advances in Therapy. Sig Transduct Target Ther 2024, 9, 124. [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS Production by Mitochondria: Function or Dysfunction? Oncogene 2024, 43, 295–303. [CrossRef]
- Dadsena, S.; King, L.E.; García-Sáez, A.J. Apoptosis Regulation at the Mitochondria Membrane Level. Biochimica et Biophysica Acta (BBA) - Biomembranes 2021, 1863, 183716. [CrossRef]
- Glover, H.L.; Schreiner, A.; Dewson, G.; Tait, S.W.G. Mitochondria and Cell Death. Nat Cell Biol 2024, 26, 1434–1446. [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat Rev Mol Cell Biol 2020, 21, 421–438. [CrossRef]
- Acosta-Alvear, D.; Harnoss, J.M.; Walter, P.; Ashkenazi, A. Homeostasis Control in Health and Disease by the Unfolded Protein Response. Nat Rev Mol Cell Biol 2025, 26, 193–212. [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Molecular Cell 2018, 69, 169–181. [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, Autophagy, and Mitochondria Crosstalk Underlies the ER Stress Response. Trends in Biochemical Sciences 2015, 40, 141–148. [CrossRef]
- Rühmkorf, A.; Harbauer, A.B. Role of Mitochondria–ER Contact Sites in Mitophagy. Biomolecules 2023, 13, 1198. [CrossRef]
- Larrañaga-SanMiguel, A.; Bengoa-Vergniory, N.; Flores-Romero, H. Crosstalk between Mitochondria–ER Contact Sites and the Apoptotic Machinery as a Novel Health Meter. Trends in Cell Biology 2025, 35, 33–45. [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat Rev Mol Cell Biol 2018, 19, 121–135. [CrossRef]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a Decisive Process for Cell Death. Exp Mol Med 2020, 52, 921–930. [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 Network Regulates Autophagy and Apoptosis. Cell Death Differ 2011, 18, 571–580. [CrossRef]
- Zhang, Z.; Jiang, M.; Xie, X.; Yang, H.; Wang, X.; Xiao, L.; Wang, N. Oleanolic Acid Ameliorates High Glucose-Induced Endothelial Dysfunction via PPARδ Activation. Sci Rep 2017, 7, 40237. [CrossRef]
- Lee, E.S.; Kim, H.M.; Kang, J.S.; Lee, E.Y.; Yadav, D.; Kwon, M.-H.; Kim, Y.M.; Kim, H.S.; Chung, C.H. Oleanolic Acid and N -Acetylcysteine Ameliorate Diabetic Nephropathy through Reduction of Oxidative Stress and Endoplasmic Reticulum Stress in a Type 2 Diabetic Rat Model. Nephrol. Dial. Transplant. 2016, 31, 391–400. [CrossRef]
- Zhang, Q.; Chen, W.; Zhang, B.; Li, C.; Zhang, X.; Wang, Q.; Wang, Y.; Zhou, Q.; Li, X.; Shen, X.L. Central Role of TRAP1 in the Ameliorative Effect of Oleanolic Acid on the Mitochondrial-Mediated and Endoplasmic Reticulum Stress-Excitated Apoptosis Induced by Ochratoxin A. Toxicology 2021, 450, 152681. [CrossRef]
- Gong, Y.; Luo, Y.; Liu, S.; Ma, J.; Liu, F.; Fang, Y.; Cao, F.; Wang, L.; Pei, Z.; Ren, J. Pentacyclic Triterpene Oleanolic Acid Protects against Cardiac Aging through Regulation of Mitophagy and Mitochondrial Integrity. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2022, 1868, 166402. [CrossRef]
- Yang, H.-B.; Lee, C.-H.; Nhung, N.T.; Hung, S.-Y. Oleanolic Acid Activates the JNK-Sp1-DJ-1 Axis to Promote Mitophagy-Mediated Neuroprotection in Dopaminergic Neurons for Parkinson’s Disease Treatment. Arch. Pharm. Res. 2025, 48, 528–548. [CrossRef]
- Msibi, Z.N.P.; Mabandla, M.V. Oleanolic Acid Mitigates 6-Hydroxydopamine Neurotoxicity by Attenuating Intracellular ROS in PC12 Cells and Striatal Microglial Activation in Rat Brains. Front. Physiol. 2019, 10, 1059. [CrossRef]
- Wang, X.; Chen, H.L.; Liu, J.Z.; Liao, N.; Yu, W.H.; Zhang, X.D.; Zhang, T.; Li, W.L.; Hai, C.X. Protective Effect of Oleanolic Acid against Beta Cell Dysfunction and Mitochondrial Apoptosis: Crucial Role of ERK-NRF2 Signaling Pathway. J Biol Regul Homeost Agents 2013, 27, 55–67.
- Xie, J.-Z.; Cui, W.-J.; Zhong, W.-T.; Ning, L. Oleanolic Acid Inhibits Mitochondrial Fission by Improving Mitochondrial Dysfunction and Reducing Atherosclerosis via Regulating AMPK/Drp1 Pathway: An in Vivo and in Vitro Study. Asian Pacific Journal of Tropical Biomedicine 2026, 16, 24–39. [CrossRef]
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Baltierra-Uribe, S.L.; Castillo-Cruz, J.; Campillo-Navarro, M.; Hernández-Pérez, A.D.; Luna-Angulo, A.B.; Chacón-Salinas, R.; Coral-Vázquez, R.M.; Estrada-García, I.; et al. Ursolic and Oleanolic Acids Induce Mitophagy in A549 Human Lung Cancer Cells. Molecules 2019, 24, 3444. [CrossRef]
- Li, H.; Wang, X.; Xiang, S.; Hu, Y.; Jiang, L.; Shu, Y.; Li, M.; Wu, X.; Zhang, F.; Ye, Y.; et al. Oleanolic Acid Induces Mitochondrial-Dependent Apoptosis and G0/G1 Phase Arrest in Gallbladder Cancer Cells. DDDT 2015, 3017. [CrossRef]
- Wang, X.; Bai, H.; Zhang, X.; Liu, J.; Cao, P.; Liao, N.; Zhang, W.; Wang, Z.; Hai, C. Inhibitory Effect of Oleanolic Acid on Hepatocellular Carcinoma via ERK–P53-Mediated Cell Cycle Arrest and Mitochondrial-Dependent Apoptosis. Carcinogenesis 2013, 34, 1323–1330. [CrossRef]
- Liu, J.; Wu, N.; Ma, L.-N.; Zhong, J.-T.; Liu, G.; Zheng, L.-H.; Lin, X.-K. P38 MAPK Signaling Mediates Mitochondrial Apoptosis in Cancer Cells Induced by Oleanolic Acid. Asian Pacific Journal of Cancer Prevention 2014, 15, 4519–4525. [CrossRef]
- Liu, J.; Zheng, L.; Ma, L.; Wang, B.; Zhao, Y.; Wu, N.; Liu, G.; Lin, X. Oleanolic Acid Inhibits Proliferation and Invasiveness of Kras-Transformed Cells via Autophagy. The Journal of Nutritional Biochemistry 2014, 25, 1154–1160. [CrossRef]
- Liu, J.; Ma, L.; Chen, X.; Wang, J.; Yu, T.; Gong, Y.; Ma, A.; Zheng, L.; Liang, H. ERK Inhibition Sensitizes Cancer Cells to Oleanolic Acid-Induced Apoptosis through ERK/Nrf2/ROS Pathway. Tumor Biol. 2016, 37, 8181–8187. [CrossRef]
- Lee, J.-H.; Yoo, E.-S.; Han, S.-H.; Jung, G.-H.; Han, E.-J.; Jung, S.-H.; Seok Kim, B.; Cho, S.-D.; Nam, J.-S.; Choi, C.; et al. Oleanolic Acid Induces Apoptosis and Autophagy via the PI3K/AKT/mTOR Pathway in AGS Human Gastric Cancer Cells. Journal of Functional Foods 2021, 87, 104854. [CrossRef]
- Hu, C.; Cao, Y.; Li, P.; Tang, X.; Yang, M.; Gu, S.; Xiong, K.; Li, T.; Xiao, T. Oleanolic Acid Induces Autophagy and Apoptosis via the AMPK-mTOR Signaling Pathway in Colon Cancer. Journal of Oncology 2021, 2021, 1–17. [CrossRef]
- Liu, J.; Zheng, L.; Zhong, J.; Wu, N.; Liu, G.; Lin, X. Oleanolic Acid Induces Protective Autophagy in Cancer Cells through the JNK and mTOR Pathways. Oncology Reports 2014, 32, 567–572. [CrossRef]
- Yang, Y.-H.; Dai, S.-Y.; Deng, F.-H.; Peng, L.-H.; Li, C.; Pei, Y.-H. Recent Advances in Medicinal Chemistry of Oleanolic Acid Derivatives. Phytochemistry 2022, 203, 113397. [CrossRef]
- Günther, A.; Kulawik, M.; Sip, S.; Zalewski, P.; Jarmołowska-Jurczyszyn, D.; Stawicki, P.; Bednarczyk-Cwynar, B. Targeting Oxidative Stress in Carcinogenesis: Oleanolic Acid and Its Molecular Pathways. Antioxidants 2026, 15, 67. [CrossRef]
- García-González, A.; Espinosa-Cabello, J.M.; Cerrillo, I.; Montero-Romero, E.; Rivas-Melo, J.J.; Romero-Báez, A.; Jiménez-Andreu, M.D.; Ruíz-Trillo, C.A.; Rodríguez-Rodríguez, A.; Martínez-Ortega, A.J.; et al. Bioavailability and Systemic Transport of Oleanolic Acid in Humans, Formulated as a Functional Olive Oil. Food Funct. 2023, 14, 9681–9694. [CrossRef]
- Das, I.; Rajagopal, N.; Reddy, Y.; Kampa Sundara, B. Oleanolic Acid in Nanomedicine: Emerging Formulation Strategies and Applications. J Pharm Innov 2025, 20, 266. [CrossRef]
- Chipurupalli, S.; Samavedam, U.; Robinson, N. Crosstalk Between ER Stress, Autophagy and Inflammation. Front. Med. 2021, 8, 758311. [CrossRef]
- Kettel, P.; Karagöz, G.E. Endoplasmic Reticulum: Monitoring and Maintaining Protein and Membrane Homeostasis in the Endoplasmic Reticulum by the Unfolded Protein Response. The International Journal of Biochemistry & Cell Biology 2024, 172, 106598. [CrossRef]
- Yu, Y.; Ma, T.; Lv, L.; Jia, L.; Ruan, H.; Chen, H.; Zhang, J.; Gao, L. Oleanolic Acid Targets the Regulation of PI3K/AKT/mTOR Pathway and Activates Autophagy in Chondrocytes to Improve Osteoarthritis in Rats. Journal of Functional Foods 2022, 94, 105144. [CrossRef]
- Liu, J.; Liu, J.; Meng, C.; Huang, C.; Liu, F.; Xia, C. Oleanolic Acid Alleviates ANIT-Induced Cholestatic Liver Injury by Activating Fxr and Nrf2 Pathways to Ameliorate Disordered Bile Acids Homeostasis. Phytomedicine 2022, 102, 154173. [CrossRef]
- Lu, Y.-F.; Wan, X.-L.; Xu, Y.; Liu, J. Repeated Oral Administration of Oleanolic Acid Produces Cholestatic Liver Injury in Mice. Molecules 2013, 18, 3060–3071. [CrossRef]
- Feng, H.; Wu, Y.-Q.; Xu, Y.-S.; Wang, K.-X.; Qin, X.-M.; Lu, Y.-F. LC-MS-Based Metabolomic Study of Oleanolic Acid-Induced Hepatotoxicity in Mice. Front. Pharmacol. 2020, 11, 747. [CrossRef]
- Xu, Y.; Wei, J.; Wang, W.; Mao, Z.; Wang, D.; Zhang, T.; Zhang, P. Oleanolic Acid Slows Down Aging Through IGF-1 Affecting the PI3K/AKT/mTOR Signaling Pathway. Molecules 2025, 30, 740. [CrossRef]
Figure 1.
Oleanolic Acid as a Context-Dependent Modulator of Organelle Stress: Mitochondria–Endoplasmic Reticulum–Autophagy–Apoptosis Axis.
Figure 1.
Oleanolic Acid as a Context-Dependent Modulator of Organelle Stress: Mitochondria–Endoplasmic Reticulum–Autophagy–Apoptosis Axis.

Scheme 1.
Chemical structure of oleanolic acid (OA). (A) Parent structure of OA; (B) structure with A–E ring labeling; (C) structure with carbon atom numberings.
Scheme 1.
Chemical structure of oleanolic acid (OA). (A) Parent structure of OA; (B) structure with A–E ring labeling; (C) structure with carbon atom numberings.

Table 1.
Effects of oleanolic acid in non-cancer models of organelle stress.
| Model | Cell / tissue / organ |
Stressor / condition |
Main mitochondrial findings | Main ER stress findings | Autophagy / mitophagy findings | Apoptosis findings |
Main pathway / mechanistic axis | Outcome | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Type 2 diabetic nephropathy (OLETF rats; mesangial cells). | Kidney / mesangial cells. | Hyperglycemia / diabetic injury. | Indirect improvement through reduced oxidative burden. | Decreased ER stress markers; attenuation of stress response under high glucose. | Not primary endpoint. | Reduced injury-associated cell stress and damage. | Oxidative stress–ER stress coupling. | Protective. | [19] |
| Ochratoxin A nephrotoxicity (HK-2 cells). | Renal tubular epithelial cells. | Ochratoxin A. | Reduced CypD, Bax, cytochrome c, caspase-9, caspase-3; preservation of mitochondrial function. | Reduced GRP78, p-PERK, p-eIF2α, ATF4, CHOP. | Not primary endpoint. | Reduced mitochondrial- and ER stress-mediated apoptosis. | TRAP1-centered ER–mitochondria protection. | Protective. | [20] |
| Cardiac aging. | Heart / myocardium. | Aging. | Improved mitochondrial integrity and ultrastructure. | Not a primary endpoint. | Increased mitophagy; improved mitochondrial quality control. | Reduced injury-associated cell death signaling. | Mitophagy / FUNDC1-related regulation. | Protective. | [21] |
| Parkinson’s disease model (MPP+/MPTP). | Dopaminergic neurons. | Neurotoxic mitochondrial stress. | Reduced mitochondrial dysfunction and ROS. | Not directly assessed. | Increased mitophagy. | Reduced neuronal death. | JNK–Sp1–DJ-1 axis. | Protective. | [22] |
| 6-OHDA neurotoxicity. | PC12 cells / rat brain. | 6-Hydroxydopamine. | Reduced intracellular ROS; improved mitochondrial stress status indirectly. | Not directly assessed. | Not primary endpoint. | Reduced neurotoxicity. | ROS attenuation. | Protective. | [23] |
| β-cell dysfunction. | Pancreatic β-cells. | β-cell stress / dysfunction. | Reduced mitochondrial apoptosis; improved mitochondrial protection. | Not directly assessed. | Not primary endpoint. | Reduced apoptotic signaling. | ERK–NRF2 / glutathione axis. | Protective. | [24] |
| High glucose-induced endothelial dysfunction. | Endothelial cells. | High glucose. | Preserved mitochondrial membrane potential; reduced ROS. | Not directly assessed. | Not primary endpoint. | Reduced apoptosis. | PPARδ activation. | Protective. | [18] |
| Osteoarthritis. | Chondrocytes / cartilage. | IL-1β / OA model. | Indirect protection through reduced cellular stress. | Not directly assessed. | Activated autophagy. | Reduced degenerative response. | PI3K/AKT/mTOR inhibition. | Protective. | [41] |
| Cellular aging model. | Aging-related cell model. | Aging. | Indirect mitochondrial benefit through metabolic stress reduction. | Not directly assessed. | Suggestive pro-autophagic milieu via mTOR suppression. | Not primary endpoint. | IGF-1–PI3K/ AKT/mTOR modulation. | Protective/ anti-aging. | [45] |
Note: In several non-cancer studies, ER stress or autophagy was not measured directly, but mitochondrial and apoptotic findings strongly support an organelle stress interpretation. That pattern is especially clear in diabetic nephropathy, OTA nephrotoxicity, cardiac aging, Parkinson’s disease models, and osteoarthritis.
Table 2.
Effects of oleanolic acid in cancer models: mitochondrial stress, autophagy, and apoptosis.
Table 2.
Effects of oleanolic acid in cancer models: mitochondrial stress, autophagy, and apoptosis.
| Cancer type | Cell line / model |
OA dose / exposure |
Main mitochondrial effects | Role of autophagy |
Main apoptosis findings | Main signaling pathway | Overall outcome |
Ref. |
|---|---|---|---|---|---|---|---|---|
| Lung cancer. | A549 | Time-dependent exposure. | Mitochondrial fragmentation, swelling, mitophagy-associated changes. | Mainly protective mitophagy / protective autophagy. | Blocking autophagy aggravated mitochondrial depolarization and reduced survival. | Mitophagy-related stress response. | Stress adaptation / partial survival support. | [26] |
| Gallbladder cancer. | Gallbladder cancercells; xenograft model. | Dose dependent. |
Loss of mitochondrial integrity; cytochrome c release. | Not primary focus. | Increased Bax, decreased Bcl-2, activation of caspase-9, caspase-3, PARP. | Mitochondrial apoptosis. | Anticancer / proapoptotic. | [27] |
| Hepatocellular carcinoma. | HCC cells. | Dose dependent. |
Mitochondrial-dependent apoptosis. | Not primary focus. | Increased Bax/Bcl-2 ratio; cytochrome c release; apoptosis. | ERK-p53-mediated cell cycle arrest with mitochondrial apoptosis. |
Anticancer / proapoptotic. | [28] |
| Multiple cancer cell models. | Various. | Dose dependent. |
ROS increase; Bax/Bim translocation to mitochondria. | Not primary focus. | p38-mediated mitochondrial apoptosis. | ROS/ASK1/p38 MAPK. | Anticancer / proapoptotic. | [29] |
| KRAS-transformed cells. | KRAS-transformed cell model. | Dose dependent. |
Indirect mitochondrial stress participation. | Autophagy contributed to OA effect. | Antiproliferative / anti-invasive effect linked to autophagy. | Autophagy-associated signaling. |
Anticancer; autophagy assisted. |
[30] |
| Cancer cells with ERK/Nrf2 compensation. | Various cancer cell models. | Dose dependent. |
ROS-sensitive mitochondrial response. | Not central. | OA-induced apoptosis increased when ERK/Nrf2 defense was blocked. | ERK/Nrf2/ROS pathway. | Sensitization to OA-induced apoptosis. | [31] |
| Gastric cancer. | AGS | Dose dependent. |
Mitochondrial stress accompanies apoptosis. | Autophagy accompanied cytotoxic response. | Increased apoptosis with autophagy induction. | PI3K/AKT/mTOR pathway. |
Anticancer / apoptosis + autophagy. | [32] |
| Colon cancer. | Colon cancer model. | Dose dependent. |
Mitochondrial stress associated with apoptosis. | Autophagy coupled to cytotoxicity. | Increased apoptosis. | AMPK–mTOR pathway. | Anticancer / apoptosis + autophagy. |
[33] |
Note: The cancer literature does not support a single uniform model. In some systems OA induces protective autophagy, whereas in others autophagy appears to participate in cytotoxicity and apoptosis. This contrast is well illustrated by A549, AGS, colon cancer, KRAS-transformed cells, gallbladder cancer, and hepatocellular carcinoma studies.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



