Submitted:
19 March 2026
Posted:
20 March 2026
You are already at the latest version
Abstract
N-Alkyl urazoles are important heterocyclic compounds that serve as important precursors to potent N-alkyl 1,2,4-triazoline-3,5-dione electrophiles. Traditional methods for urazole synthesis have relied upon the use of toxic isocyanates. We have modified and optimized an overlooked and poorly-described literature method for the synthesis of urazoles that now avoids the use of isocyanates, limits the use of solvents, and provides urazoles without the need for purification steps. A variety of urazoles are afforded in good to high yields.
Keywords:
1
; 2
; 4-triazoline-3
; 5-dione
; urazole
; semicarbazide
; sustainable
1. Introduction
N-Alkyl urazoles (1, Scheme 1) are direct precursors, via oxidation, to the corresponding azo compounds N-alkyl 1,2,4-triazoline-3,5-diones (2, RTADs) [1]. RTADS are potent electrophilic reagents that readily engage in a variety of useful reactions including Diels-Alder cycloadditions with dienes, [2+2] cycloadditions with suitably-substituted alkenes, and ene reactions [1]. Such general organic reactivity has recently been directed towards practical applications such as “click”-type reactivity, polymer coupling, and surface modification [1,2].
The traditional method for the synthesis of N-alkyl urazoles was devised by Zinner in 1961 [3], and then refined and developed into an established Organic Synthesis procedure by Cookson in 1971 (Scheme 2) [4]. Unfortunately, however, this synthetic method relies upon access to appropriate alkyl isocyanates (3) as starting materials [5]. Due to the known dangers of working with isocyanates, the commercial availability of isocyanates has severely diminished, thereby hindering direct access to urazole compounds via the Cookson route. This has led to the development of alternative routes for the synthesis of urazoles that have been recently summarized by Du Prez [1].
Triazolinedione 2a is one of the most commonly used triazolinediones in the literature [6]. The lack of commercial availability of methylisocyanate, the Cookson precursor to N-methyl urazole (1a), is especially problematic because compound 2a is a convenient TAD derivative with which to work. Its particular usefulness derives from the unique chemical shift of the N-methyl signal in the 1H NMR spectrum (a singlet at ~3 ppm) that makes following reactions, and analyzing crude reaction mixtures, relatively straightforward [6]. Therefore, several years ago our group developed a method for the synthesis of urazole 1a via the in situ generation of methyl isocyanate from N-nitroso dimethylurea 5 (see Scheme 3), as well as via the reaction of methylamine with ethyl phenyl hydrazine-1,2-dicarboxylate 6 [6]. These reactions afforded semicarbazide 4a which could then be cyclized to the urazole 1a under standard Cookson conditions (see Scheme 2). Gratifyingly, we also found that derivatives of 4 other than just the N-methyl were also accessible from compound 6 (i.e., 1d-g) [6].
Recently, Sarlah reported a “scalable” synthesis of N-methyl urazole 1a that accessed semicarbazide intermediate 4 (R = Me) using carbonyl diimidazole (CDI) as the reactive carbonyl component (Scheme 4) [7]. While this method permits a multigram synthesis of N-methyl urazole, the process requires concentration of a reaction mixture under high vacuum followed by several recrystallization steps to afford the desired 4a. While still an excellent method for large scale synthesis of 1a, it is not particularly convenient for ordinary lab use, nor was it expanded to urazole substituents other than that of N-methyl. Finally, in related work Du Prez reported what was described as a “sustainable” synthetic route for urazoles [8]. This synthetic route eliminated the use of reaction solvents by substituting bulk heating of intermediate compounds (Scheme 5). The major limitation to the method was that the amines needed to be of relatively high boiling point (the smallest being butylamine) for the method to be viable.
We were recently fortunate to come across a previously described method for the synthesis of urazoles that seems to have been overlooked in the literature (or at least underappreciated). In a 1967 paper, Priehradny reported that heating neat 1,6-dialkylbiureas 7 to relatively high temperatures (~ 250 °C) resulted in spontaneous cyclization to form the corresponding N-alkyl urazoles (Scheme 6) [9]. Therefore, this synthesis can be considered to be a “sustainable” method akin to that described earlier by Du Prez (Scheme 5) in that solvent use is avoided in the cyclization step. The major downside to this method is that the synthesis of the required starting 1,6-dialkylbiureas 7 relied upon either the same toxic isocyanates as were used in the Cookson method, or the use of similarly toxic dialkyl azodicarboxylates via a two-step method (see Scheme 6). Additionally, there have also been some reports that the Priedhradny urazole synthesis method has been hard to reproduce [1,4,10]. The lack of reproducibility could be due to the lack of experimental detail provided in the original paper [9]. Therefore, for these reasons the use of Priehradny’s synthetic method for urazoles has been largely ignored.
In this paper we revitalize Priedhradny’s synthetic method by finding a general and highly convenient (i.e., free of purification steps) route for the synthesis of the 1,6-dialkylbiureas precursors (7) that eliminates the need for isocyanates and azodicarboxylates. We also refine the solvent-free cyclization of compounds 7 to the corresponding urazoles and provide important reaction details missing from the Priehradny paper that now allow for practical and reproducible results. We hope that by avoiding toxic reagents, optimizing reaction steps, and providing key details to the cyclization step that ensures reproducibility that this very convenient, but previously overlooked method, might enjoy greater use.
2. Materials and Methods
2.1. General Methods
All solvents and reagents were obtained commercially and used as received. Heating of 1,6-dialkylbisureas 7 were conducted in 5 mL conical vials left open to the atmosphere (in a suitable hood) using a metal hot plate whose temperature was closely controlled by a digitally-controlled unit. Chemical shifts (1H and 13C) are reported in units of parts per million downfield from TMS. IR spectra were collected as solids pressed against a ZnSe ATR crystal. High-resolution mass spectra (HRMS) were acquired via electron spray ionization on an LTQ-FTMS hybrid mass spectrometer.
2.2. Experimental Procedures
2.2.1 N,N-Diphenyl-1,2-hydrazinedicarboxamide (8). To a stirring mixture of 1.05 g (12.6 mmol) of hydrazine hydrate (60% by weight) and 2.76 g (2 equiv) of sodium carbonate in 125 mL of THF at 0 °C was added 4 g (25.5 mmol) of phenyl chloroformate dropwise via Pasteur pipette. The mixture was allowed to warm to room temperature and stirred overnight after which a white precipitate appeared. 50 mL of 0.5 M aq. HCl was added to the mixture and the THF removed via rotary evaporation to leave the white solid suspended in the aqueous layer. The mixture was filtered and the separated solid rinsed well with 100 mL of water. After air drying, the product was dried in the oven at 90 °C for at least two hours after which 84.7 g (96%) of 8 was isolated as a fluffy white solid, m.p. 153-154 °C: 1H NMR (DMSO-d6) δ 9.44-10.10 (multiple s, 2H, NH), 7.42 (t, J = 7.8 Hz, 4H), 7.25 (t, J = 7.8 Hz, 2H), 7.13 (d, J = 7.8 Hz, 4H); 13C NMR (DMSO-d6) δ 154.8, 150.5, 129.6, 125.6, 121.6. The spectra were consistent with those reported in the literature [11].
2.2.3 1,6-Dimethylbisurea (7a). To a stirring solution of 0.5 g (1.83 mmol) of compound 8 in 15 mL of CH3CN was added 5.50 mL (6 eq) of a 2 M solution of methyl amine in THF via syringe. A precipitate began to form some minutes later and stirring was continued overnight. The resulting white precipitate was isolated via vacuum filtration and washed with 10 mL of water. Air drying afforded 0.25 g (94% yield) of 7a as a white powder, m.p. 243-244 °C: IR (ATR) cm-1 3311.6, 1662.0, 1561.1, 1413.9, 1325.6,; 1H NMR (CF3CO2D) δ 11.82 (br s, 4H, NH), 2.99 (s, 6H); 13C NMR (CF3CO2D) δ 164.2, 28.1; HRMS (ESI) m/z [M+H]+ Calcd for C4H11N4O2 147.08765; Found 147.08695.
2.2.4 1,6-Diethylbisurea (7b). To a stirring solution of 0.5 g (1.83 mmol) of compound 8 in 15 mL of CH3CN was added 5.50 mL (6 eq) of a 2 M solution of ethyl amine in THF via syringe. A precipitate began to form some minutes later and stirring was continued overnight. The resulting white precipitate was isolated via vacuum filtration and washed with 10 mL of water. Air drying afforded 0.28 g (88% yield) of 7b as a white powder, m.p. 238-239 °C: IR (ATR) cm-1 3297.7, 1665.0, 1554.2, 1328.6,; 1H NMR (CF3CO2D) δ 11.73 (br s, 4H, NH), 3.47 (q, J = 7.7 Hz, 4H), 1.28 (t, J = 7.7 Hz, 6H); 13C NMR (CF3CO2D) δ 160.3, 34.9, 11.7; HRMS (ESI) m/z [M+H]+ Calcd for C6H15N4O2 175.11895; Found 175.11874.
2.2.5 1,6-Di-n-propylbisurea (7c). To a stirring solution of 0.5 g (1.83 mmol) of compound 8 in 20 mL of CH3CN was added 0.64 g (6 eq) of n-propyl amine dropwise via Pasteur pipette. The solution became cloudy after approximately 10 mins, and stirring was continued overnight. The resulting white precipitate was isolated via vacuum filtration and washed with 10 mL of water. Air drying afforded 0.32 g (87% yield) of 7c as a white powder, m.p. 243-244 °C: IR (ATR) cm-1 3290.0, 2961.8, 1658.5, 1560.2, 1335.3; 1H NMR (CF3CO2D) δ 11.72 (br s, 4H, NH), 3.39 (t, J = 7.3 Hz, 4H), 1.68 (h, J = 7.3 Hz, 4H), 0.99 (t, J = 7.3 Hz, 6H); 13C NMR (CF3CO2D) δ 164.1, 44.8, 24.1, 11.5; HRMS (ESI) m/z [M+H]+ Calcd for C8H19N4O2 203.15025; Found 203.15006.
2.2.6 1,6-Di-n-butylbisurea (7d). To a stirring solution of 0.5 g (1.83 mmol) of compound 8 in 20 mL of CH3CN was added 0.80 g (6 eq) of n-butyl amine dropwise via Pasteur pipette. The solution became cloudy after approximately 10 mins, and stirring was continued overnight. The resulting white precipitate was isolated via vacuum filtration and washed with 10 mL of water. Air drying afforded 0.38 g (90% yield) of 7d as a white powder, m.p. 246-247 °C: IR (ATR) cm-1 3295.0, 2956.6, 1659.6, 1555.3, 1377.0,; 1H NMR (CF3CO2D) δ 11.82 (br s, 4H, NH), 3.43 (t, J = 7.3 Hz, 4H), 1.63 (p, J = 7.3 Hz, 4H), 1.42 (h, J = 7.3 Hz, 4H), 0.98 (t, J = 7.3 Hz, 6H); 13C NMR (CF3CO2D) δ 163.5, 42.8, 32.9, 21.3, 14.0; HRMS (ESI) m/z [M+H]+ Calcd for C10H23N4O2 231.18155; Found 231.18157.
2.2.7 1,6-Di-tert-butylbisurea (7e). To a stirring solution of 0.5 g (1.83 mmol) of compound 8 in 20 mL of CH3CN was added 0.80 g (6 eq) of tert-butyl amine dropwise via Pasteur pipette. After stirring overnight, the solution was concentrated via rotary evaporation to afford a pale orange solid. The solid was taken up in 20 mL of CH2Cl2 and washed 2 x 20 mL 0.5 M aq. NaOH. The organic layer was then dried over Na2SO4, filtered, and concentrated to afford 0.24 g (57% yield) of 7e as a white solid, m.p. 189-190 °C: IR (ATR) cm-1 3314.7, 2968.1, 1654.9, 1558.4, 1363.3, 1217.6; 1H NMR (CDCl3) δ 6.88 (br s, 2H, NH), 5.62 (br s, 2H, NH), 1.34 (s, 9H); 13C NMR (CDCl3) δ 158.1, 50.6, 29.1; HRMS (ESI) m/z [M+H]+ Calcd for C10H23N4O2 231.18155; Found 231.18111.
2.2.8 1,6-Dicyclohexylbisurea (7g). To a stirring solution of 0.5 g (1.83 mmol) of compound 8 in 20 mL of CH3CN was added 1.09 g (6 eq) of cyclohexyl amine dropwise via Pasteur pipette. After stirring overnight, the resulting white precipitate was isolated via vacuum filtration and washed with 10 mL of water. Air drying afforded 0.45 g (77% yield) of 7g as a white powder, m.p. 203-204 °C: IR (ATR) cm-1 3675.5, 2972.0, 1646.4, 1531.5, 1393.9, 1066.0; 1H NMR (CF3CO2D) δ 11.69 (br s, 4H, NH), 3.77 (m, 2H), 1.97 (br s, 4H), 1.87 (br s, 4H), 1.72 (br d, J = 12.4 Hz, 2H), 1.35-1.50 (m, 8H), 1.26 (m, 2H); 13C NMR (CF3CO2D) δ 162.6, 53.6, 34.6, 26.6, 26.5; HRMS (ESI) m/z [M+H]+ Calcd for C14H27N4O2 283.21285; Found 283.21103.
2.2.9 1,6-Dibenzylbisurea (7g). To a stirring solution of 0.5 g (1.83 mmol) of compound 8 in 20 mL of CH3CN was added 1.17 g (6 eq) of benzyl amine dropwise via Pasteur pipette. After stirring overnight, the resulting white precipitate was isolated via vacuum filtration and washed with 10 mL of water. Air drying afforded 0.42 g (76% yield) of 7g as a white powder, m.p. 245-246 °C: IR (ATR) cm-1 3292.2, 1660.3, 1552.1, 1298.6, 1219.0; 1H NMR (CF3CO2D) δ 11.68 (br s, 4H, NH), 7.26-7.34 (m, 10H), 4.53 (s, 4H); 13C NMR (CF3CO2D) δ 163.6, 138.1, 130.89, 130.3, 129.5, 46.6; HRMS (ESI) m/z [M+H]+ Calcd for C16H19N4O2 299.15025; Found 299.15007.
2.2.10 N-Methylurazole (1a). 100 mg (0.68 mmol) of finely powdered 7a was heated in a vial to 260 °C over approximately a 10 min period. Visible fuming began to appear at a temperature of ~ 240 °C. As the sample was held at 260 °C it melted to provide a colorless oil. The melt was held at this temperature with occasional swirling to allow liberated methyl amine to escape. After 10 mins, fuming had ceased, and the vial was removed from the heat with swirling to aid in crystallization of the urazole product with cooling. The product was scraped from the vial to afford 62 mg of 1a as a white crystalline product (80% yield), m.p. 237-238 °C (lit. 232-233 °C [12]): 1H NMR (DMSO-d6) δ 9.59 (br s, 2H, NH), 2.84 (s, 3H); 13C NMR (DMSO-d6) δ 155.3, 24.3. The spectra were consistent with those reported in the literature [13].
Repeating this process on a larger scale starting with 0.50 g of 7a afforded 0.39 g (85% yield) of 1a.
2.2.11 N-Ethylurazole (1b). 100 mg (0.68 mmol) of finely powdered 7b was heated in a vial to 260 °C over approximately a 10 min period. Visible fuming began to appear at a temperature of ~ 240 °C. As the sample was held at 260 °C it melted to provide a colorless oil. The melt was held at this temperature with occasional swirling to allow liberated ethyl amine to escape. After 10 mins, fuming had ceased, and the vial was removed from the heat with swirling to aid in crystallization of the urazole product with cooling. The product was scraped from the vial to afford 68 mg of 1b as a white crystalline product (80% yield), m.p. 193-194 °C (lit. 195-196 °C [12]): 1H NMR (DMSO-d6) δ 9.90 (br s, 2H, NH), 3.37 (q, J = 7.3 ppm, 2H), 1.09 (t, J = 7.3 ppm, 3H); 13C NMR (DMSO-d6) δ 154.9, 32.9, 13.3. The spectra were consistent with those reported in the literature [14].
2.2.12 N-Propylurazole (1c). 100 mg (0.68 mmol) of finely powdered 7c was heated in a vial to 260 °C over approximately a 10 min period. Visible fuming began to appear at a temperature of ~ 240 °C. As the sample was held at 260 °C it melted to provide a colorless oil. The melt was held at this temperature with occasional swirling to allow liberated propyl amine to escape. After 10 mins, fuming had ceased, and the temperature was dropped to 190 °C where it was held for 25 min, again with occasional swirling. After the heating period, the vial was removed from the heat with swirling to aid in crystallization of the urazole product with cooling. The product was scraped from the vial to afford 62 mg of 1c as a white crystalline product (89% yield), m.p. 166-167 °C (lit. 168-169 °C [12]): 1H NMR (DMSO-d6) δ 9.90 (br s, 2H, NH), 3.30 (t, J = 7.4 ppm, 2H), 1.52 (h, J = 7.4 ppm, 2H), 0.81 (t, J = 7.5 ppm, 3H); 13C NMR (DMSO-d6) δ 155.2, 39.51, 21.0, 11.0. The spectra were consistent with those reported in the literature [15].
2.2.13 N-Butylurazole (1d). 100 mg (0.68 mmol) of finely powdered 7d was heated in a vial to 260 °C over approximately a 10 min period. Visible fuming began to appear at a temperature of ~ 240 °C. As the sample was held at 260 °C it melted to provide a colorless oil. The melt was held at this temperature with occasional swirling to allow liberated butyl amine to escape. After 15 mins, fuming had ceased, and the temperature was dropped to 190 °C where it was held for 25 min, again with occasional swirling. After the heating period, the vial was removed from the heat with swirling to aid in crystallization of the urazole product with cooling. The product was scraped from the vial to afford 53 mg of 1d as a white crystalline product (78% yield), m.p. 170-171 °C (lit. 167-168 °C [12]): 1H NMR (DMSO-d6) δ 10.00 (br s, 2H, NH), 3.34 (t, J = 7.4 ppm, 2H), 1.50 (p, J = 7.4 ppm, 2H), 1.04 (h, J = 7.4 ppm, 2H ), 0.87 (t, J = 7.5 ppm, 3H); 13C NMR (DMSO-d6) δ 155.2, 37.6, 29.6, 19.4, 13.5. The spectra were consistent with those reported in the literature [13].
Repeating this process on a larger scale starting with 0.95 g of 7d afforded 0.49 g (75% yield) of 1d.
2.2.14 N-Cyclohexylurazole (1f). 100 mg (0.68 mmol) of finely powdered 7f was heated in a vial to 260 °C over approximately a 10 min period. Visible fuming began to appear at a temperature of ~ 250 °C. As the sample was held at 260 °C it melted to provide a colorless oil. The melt was held at this temperature with occasional swirling to allow liberated butyl amine to escape. After 30 min, fuming had ceased, and the temperature was dropped to 190 °C where it was held for 25 min where it solidified. After the heating period, the vial was removed from the heat with swirling to aid in crystallization of the urazole product with cooling. The product was scraped from the vial to afford 55 mg of 1f as a white crystalline product (85% yield), m.p. 242-243 °C (lit. 239-241 °C [16]): 1H NMR (DMSO-d6) δ 9.95 (br s, 2H, NH), 3.68 (dt, J = 12.4, 3.9 Hz, 1H), 2.02 (dg, J = 12.4, 3.9 Hz, 1H), 1.76 (br d, 2H), 1.55-1.65 (m, 3H), 1.25 (qt, J = 12.4, 3.3 Hz, 1H), 1.12 (dt, J = 12.4, 3.0 Hz, 1H); 13C NMR (DMSO-d6) δ 155.3, 50.6, 29.5, 25.8, 25.3. The spectra were consistent with those reported in the literature [16].
2.2.15 N-Benzylurazole (1g). 100 mg (0.68 mmol) of finely powdered 7g was heated in a vial to 260 °C over approximately a 10 min period. Visible fuming began to appear at a temperature of ~ 250 °C. As the sample was held at 260 °C it melted to provide a colorless oil. The melt was held at this temperature with occasional swirling to allow liberated butyl amine to escape. After 1 hr, fuming had ceased, and the temperature dropped to 190 °C where it was held for 25 min, again with occasional swirling. After the heating period, the vial was removed from the heat with swirling to aid in crystallization of the urazole product with cooling. The product was scraped from the vial to afford 60 mg of 1g as a white crystalline product (94% yield), m.p. 181-182 °C (lit. 182-183 °C [12]): 1H NMR (DMSO-d6) δ 10.21 (br s, 2H, NH), 7.26-7.35 (m, 5H), 4.54 (s, 2H); 13C NMR (DMSO-d6) δ 154.8, 136.8, 128.6, 127.6, 127.5, 41.3. The spectra were consistent with those reported in the literature [13].
3. Results and Discussion
3.1. Improved Synthesis of 1,6-Dialkylbisureas
Based upon our earlier described success substituting phenol with amines from semicarbazide 6 (see Scheme 4) [6], we considered the possibility of forming the required 1,6-dialkylbisureas 7 via reaction of diphenyl hydrazine-1,2-dicarboxylate 8 (Scheme 7). Compound 8, itself, is easily synthesized in high yield and purity from the reaction of hydrazine hydrate with two equivalents of phenyl chloroformate in THF [see Materials and Methods section], and isolated via vacuum filtration of the crude reaction mixture without the need for any further purification.
Addition of amines to 8 successfully and cleanly afforded the desired bisureas 7. While 2 equivalents of amine at first glance seems adequate for the reaction, we soon realized that the progress of the reaction was quickly stifled by release of the acidic phenol which ties up unreacted amine. Therefore, at least 4 equivalents of amine are necessary to complete the reaction, but we observed that 6 equivalents of amine (i.e., 3 equivalents per carboxylate group) were optimal to afford the corresponding bisureas in good-to-high yield in reasonable time and in a reproducible manner without the need to heat the reaction. Given that the amines used are inexpensive, this did not prove to be a disadvantage. CH3CN proved to be the optimal solvent for the reaction although THF could be used with only slightly lower yields. Other than the di-tert-butyl derivative, the bisurea products are nearly insoluble in either solvent and can be isolated by simple filtration followed by washing with water (to remove phenol byproduct and excess amine) to afford compounds 7 as powdery white solids. The reaction yields are summarized in Table 1. The di-tert-butyl derivative 7e was soluble in both organic solvents, however, and necessitated removal of solvent for product isolation. The generally low solubility of the bisureas in typical organic solvents (even in DMSO) necessitated the use of deuterated trifluoroacetic acid as solvent for 1H and 13C NMR spectral analysis, in which they were all readily soluble. The NMR spectra of the isolated solids revealed essentially pure compounds free of any significant phenol contamination (see Supplementary Materials for the 1H and 13C NMR spectra).
3.2. Thermal Cyclization of 1,6-Dialkylbisureas 7 to N-Alkyl Urazoles 1.
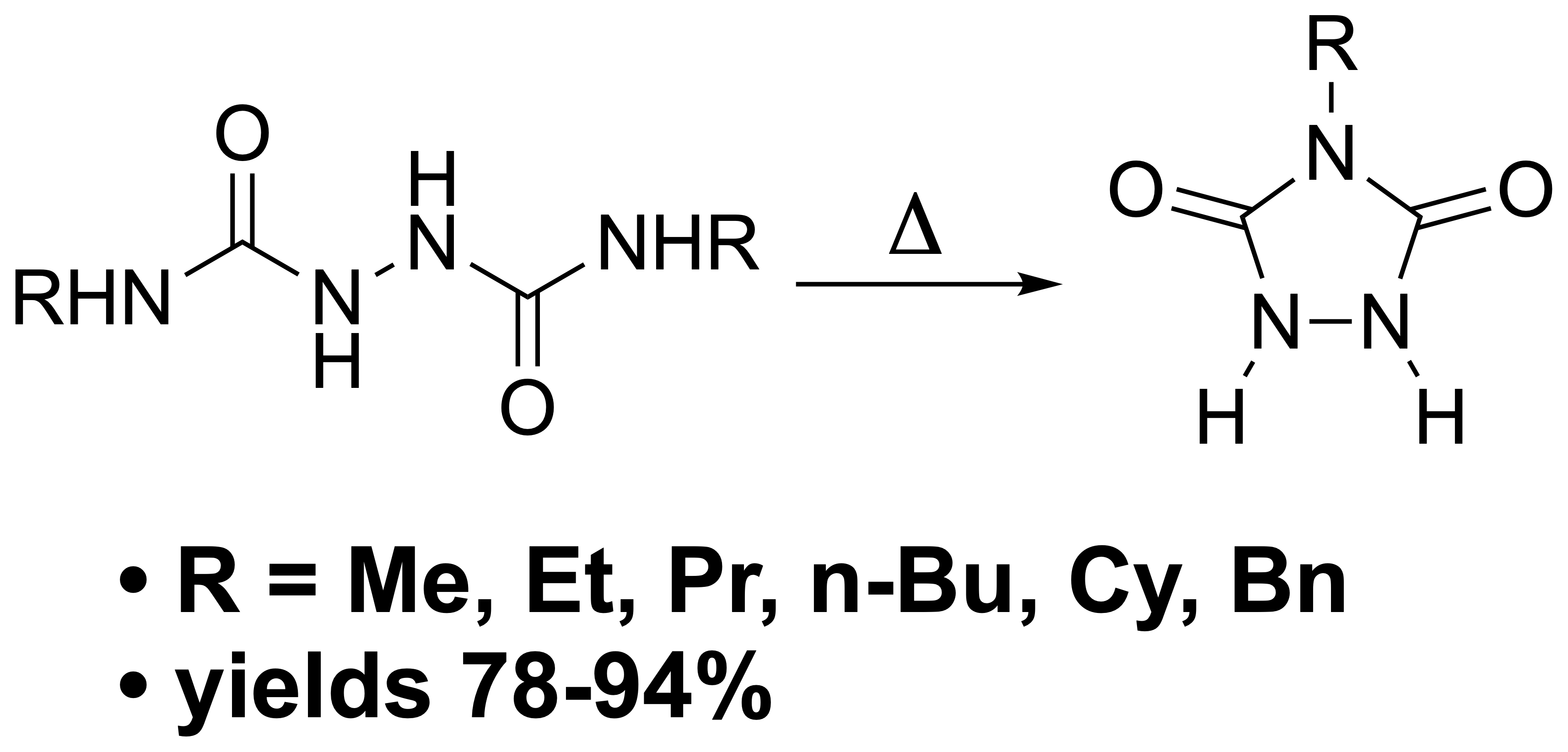
The bisureas 7 all had high melting points (> 180 °C) with most in the range of 230 – 250 °C. Conversion to the urazoles (100 mg samples) occurred readily upon heating neat samples of 7 [9]. At temperatures of ~230-250 °C, amine was released and could be observed to escape via either visible fuming and/or condensation of the amine near the top of the reaction vial. A reasonable mechanism for the conversion is provided in Scheme 8. All of the powders were heated to a final pot temperature of 260 °C to induce the cyclization reaction.
The urazole products 1 all have melting points lower than 260 °C. Therefore, as the cyclization reaction took place, a melt was formed. The melt was occasionally swirled to aid in removal of liberated amine. For bisureas 7a and 7b, once fuming stopped, the mixture could be cooled immediately to afford crystalline urazoles 1a and 1b. However, for bisureas derived from medium sized amines (i.e., 7c,d), after fuming ceased, the sample temperature was dropped to 190 °C with continued heating for 25 min to allow time for the amine to completely escape. Cooling of the melt then afforded crystalline products. Longer heating times were generally required for bisureas derived from higher boiling amines (e.g., benzyl and cyclohexyl). Larger scale reactions were also conducted starting with bisureas 7a (0.5 g) and 7d (1 g) to afford the corresponding urazole products with comparable yields (see Materials and Methods section).
The identities of the known urazole products were confirmed by 1H and 13C NMR spectroscopy (spectra are provided in the Supporting Information) and melting point comparison with literature data. Specific heating protocols for each of the bisureas are provided in the Materials and Methods section. The yields for the urazole products are provided in Table 2 (column 2) which are generally good to high.
Unfortunately, the tert-butyl derivative 7e failed to provide the corresponding urazole cleanly despite many efforts to optimize the heating protocol. In addition to formation of urazole 1e as a major product, other unknown products were also formed. The reluctance of 1e to cleanly cyclize can likely be traced to the sterically bulky tert-butyl group that inhibits initial nucleophilic attack of the nitrogen atom on the neighboring carbonyl group in the cyclization process (see Scheme 8).
4. Conclusion
The literature synthesis of urazoles via thermal cyclization of 1,6-dialkylbisureas 7 was a largely forgotten synthetic method [1,9]. The reason for its neglect is presumably due to the poor experimental procedures that were provided at the time which led many to the conclusion that the results were difficult to reproduce. Furthermore, the dependence of the method on toxic reagents such as isocyanates for the synthesis of the bisureas made it unattractive. We have now provided a high yielding and convenient method for the synthesis of the bisurea precursors that not only eliminates the use of isocyanates, but also allows for their isolation via a simple filtration process. Finally, experimental details for the thermal conversion of the bisureas to the desired urazoles have been provided that allow for good-to-high yields of the urazoles in a reproduceable manner.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, 1H and 13C NMR spectra and IR spectra for compounds 7a-g, and 1H and 13C NMR spectra for compounds 8 and 1a-d, 1f-g.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, G.W.B; methodology, C.B.D, A.B.J, O.N.S, A.J.T., B.L.Z, G.B.; validation, C.B.D, A.B.J, O.N.S, A.J.T., B.L.Z, G.B.; formal analysis, G.W.B.; investigation, C.B.D, A.B.J, O.N.S, A.J.T., B.L.Z, G.B.; data curation, C.B.D, A.B.J, O.N.S, A.J.T., B.L.Z, G.B.; writing—original draft preparation, G.W.B.; writing—review and editing, C.B.D, A.B.J, O.N.S, A.J.T., B.L.Z, G.B.; project administration, G.W.B.; funding acquisition, G.W.B. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- De Bruycker, K.; Billiet, S.; Houck, H.A.; Chattopadhyay, S.; Winne, J.M. Triazolinediones as Highly Enabling Synthetic Tools. Chem. Rev. 2016, 116, 3919–3974. [Google Scholar] [CrossRef] [PubMed]
- Laure, W.; De Bruycker, K.; Espeel, P.; Fournier, D.; Woisel, P.; Du Prez, F.E.; Lyskawa, J. Ultrafast Tailoring of Carbon Surfaces via Electrochemically Attached Triazolinediones. Langmuir 2018, 34, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Zinner, G.; Deucker, W. Zur Synthese Von Urazolen. Arch. Pharm. 1961, 294, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Cookson, R.C.; Gupte, S.S.; Stevens, I.D.R.; Watts, C.T. 4-Phenyl-1,2,4-triazoline-3,5-dione. Org. Synth. 1971, 51, 121–127. [Google Scholar] [CrossRef]
- Prasad, A.M.; Perli, G.; Ximenis, M.; Tejero, A.; Mugica, A.; Fonseca, L.P.; Sangroniz, A.; Vidal, F.; Sardon, H. Lab Safety Alert: A Real Case of Isocyanate Exposure. Polym. Chem. 2025, 16, 2905–2909. [Google Scholar] [CrossRef]
- Breton, G.W.; Turlington, M. Alternative Synthetic Routes to N-Methyl-1,2,4-triazoline-3,5-dione (MeTAD) and Other Triazolinedione Derivatives. Tetrahedron Lett. 2014, 55, 4661–4663. [Google Scholar] [CrossRef]
- Siddiqi, Z.R.; Ungarean, C.N.; Bingham, T.W.; Sarlah, D. Development of a Scalable and Sublimation-Free Route to MTAD. Org. Process Res. Dev. 2020, 24, 2953–2959. [Google Scholar] [CrossRef] [PubMed]
- Vlaminck, L.; Van de Voorde, B.; Dr Prez, F.E. Sustainable Synthesis Routes towards Urazole Compounds. Green Chem. 2017, 19, 5659–5664. [Google Scholar] [CrossRef]
- Furdik, M.; Mikulasek, S.; Livar, M.; Priehradny, S. Synthesis of Azodicarboxylic Acid Imide, its N-Substituted Derivatives, and Their Use as Dienophiles in Diels-Alder Reactions. Chem. Zvesti 1967, 21, 427–442. [Google Scholar]
- Chandrasekhar, B.; Kumar, G.B.; Mallela, S.; Bhirud, S.B. A Reliable Multi-Kilogram Preparation of 4-Phenylurazole. Org. Prep. Proced. Int. 2004, 36, 469–472. [Google Scholar] [CrossRef]
- Banik, S.; Kumar, P.; Ghule, V.D.; Khanna, S.; Allimuthu, D.; Dharavath, S. Facile Synthesis of Nitroamino-1,3,4-oxadiazole with Azo Linkage; A New Family of High Performance and Biosafe Energetic Materials. J. Mater. Chem. A. 2022, 10, 22803–22811. [Google Scholar] [CrossRef]
- Tsuji, T. Researches on Chemotherapeutic Drugs against Viruses. XVII. Synthesis and Antiviral Properties of Urazoles and Related Compounds. Pharm. Bull. 1954, 2, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A.; Narangoda, C.J.; Johnson, T.L.; Barata, R.D.; Belue, F.; Solomon, E.E.; Bragg, A.A.; Whitehead, D.C. Telescoped Oxidation and Cycloaddition of Urazoles to Access Diazacyclobutenes. J. Org. Chem. 2022, 87, 7494–7500. [Google Scholar] [CrossRef] [PubMed]
- Fantazier, R.M.; Herweh, J.E. 1,1'-Azobisformamide. I. Photochemical Decomposition in Solution. J. Am. Chem. Soc. 1974, 96, 1187–1192. [Google Scholar] [CrossRef]
- Adibi, H.; Abiri, R.; Mallakpour, S.; Zolfigol, M.A.; Majnooni, M.B. Evaluation of In Vitro Antimicrobial and Antioxidant Activities of 4-Substituted1,2,4-Triazoline-3,5-Dione Derivatives. J. Rep. Pharm. Sci. 2012, 1, 87–93. [Google Scholar]
- Zinner, G.; Bohlke, B. For Knowledge of 1,2-Malonylurazoles. Arch. Pharm. Ber. Dtsch. Pharm. Ges. 1966, 299, 43–55. [Google Scholar] [CrossRef]
- Mallakpour, S.E.; Rostamizadeh, H. Synthesis of New Polyureas Derived from 4-Cyclohexylurea. J. Appl. Polym. Sci. 2001, 80, 1335–1341. [Google Scholar] [CrossRef]
Scheme 1.
The oxidation of N-alkyl urazoles 1 to the corresponding triazolinediones 2.

Scheme 2.
Zinner and Cookson method for the synthesis of urazoles.

Scheme 3.
Two other reported methods for the synthesis of semicarbazide 4a (R = Me), the precursor to urazole 1a.
Scheme 3.
Two other reported methods for the synthesis of semicarbazide 4a (R = Me), the precursor to urazole 1a.

Scheme 4.
Synthesis of semicarbazide 4a via the use of CDI.

Scheme 5.
Du Prez’s method for the synthesis of urazoles.

Scheme 6.
Priehradny’s synthesis of urazoles (1) via thermal cyclization of 1,6-dialkylbiureas 7.

Scheme 7.
Synthesis of 1,6-dialkylbisureas 7.

Scheme 8.
Proposed mechanism for formation of urazoles 1 from bisalkylureas 7.


Table 1.
Synthesis of 1,6-dialkylbisureas 7.
| R | Yield (%) |
| methyl | 96 |
| ethyl | 88 |
| n-propyl | 87 |
| n-butyl | 90 |
| tert-butyl | 57 |
| cyclohexyl | 77 |
| benzyl | 76 |
Table 2.
Yields from thermal cyclization of 1,6-dialkylbisureas 7 to afford N-alkyl urazoles 1.
| R = | % Yield |
| methyl | 80 |
| ethyl | 92 |
| n-propyl | 89 |
| n-butyl | 78 |
| cyclohexyl | 85 |
| benzyl | 94 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



