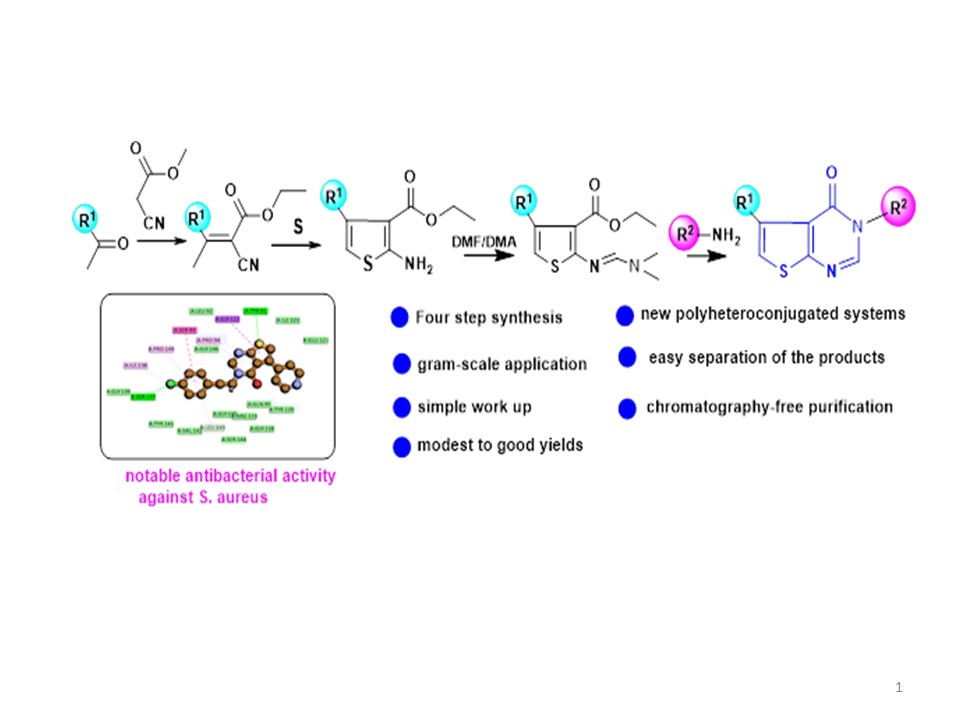
4. Materials and Methods
All chemicals were acquired from Sigma-Aldrich. They were used as received. The solvents were purified through standard procedures. 1H and 13C NMR spectra were recorded on Varian Mercury-300 MHz in DMSO-CCl4 mixture (1:3) or on Bruker AVANCE 400 MHz spectrometer in CDCl3. Chemical shifts (δ) in ppm are reported as quoted relative to the residual signals of CHCl3-d (7.25 for 1H NMR and 77.0 for 13C NMR) or DMSO-d6 (2.5 for 1H NMR and 39.5 for 13C NMR) as internal references. The coupling constants (J) are given in Hertz. Elemental analyses (C, H, and N) were performed using a Heraeus CHN–O–Rapid analyzer, and results agreed with calculated values. The purity of the isolated compounds and the reaction progress were monitored by TLC on “Silufol UV-254” plates using acetone–benzene (1 : 2) as eluent; spots were visualized by treatment with iodine vapor. The melting points were measured with an Electrothermal 9100 melting point apparatus.
Compounds
2a-2c were prepared according to the method [
28].
Compounds
3a-3c were prepared according to the Gewald’s method [
27].
General Procedure for the Syntheses of Compounds 2a-2c
A mixture of compounds 1a-1c (500.0 mmol), ethyl cyanoacetate (500.0 mmol), β-alanine (5.0 mmol), glacial acetic acid (24mL) and benzene (100mL) was refluxed with Dean-Stark trap for 18 hours. The mixture was allowed to cool to ambient temperature, benzene (30mL) was added, and then the mixture was washed with water (3 x 30mL). The combined aqueous washings were shaken with benzene (15mL), then the combined benzene solutions were dried (MgS04) and the solvent was removed in vacuum. The residual oil was distilled under reduced pressure to give compounds 2a-2c, which were used without further purification.
General Procedure for the Syntheses of Compounds 3a-3c
Method A. A mixture of compounds 2a-2c (50.0 mmol), sulphur (50.0 mmol), diethylamine (5mmol) and absolute ethanol (25mL) was refluxed for 7 h. The mixture was allowed to cool to ambient temperature and the solvent was removed under reduced pressure to give compounds 3a-3c, which were used without further purification.
Method B. A mixture of compounds 1b (500.0 mmol), ethyl cyanoacetate (500.0 mmol), β-alanine (5.0 mmol), sulphur (500.0 mmol), diethylamine (50mmol) and absolute ethanol (50mL) was refluxed with Dean-Stark trap for 25 h. The mixture was allowed to cool to ambient temperature and the solvent was removed under reduced pressure to give compound 3b.
General Procedure for the Syntheses of Compounds 4a-4c
A mixture of compounds 3a-3c (10 mmol) and DMF/DMA (12 mmol) in an anhydrous xylene (12 mL) was refluxed for 3 h. The mixture was allowed to cool to ambient temperature, then light petroleum (5 mL) was added into the mixture. The precipitate was filtered, washed with ether, dried.
Ethyl 2-(((dimethylamino)methylene)amino)-4-(naphthalen-2-yl)thiophene-3-carboxylate (4a). Yellow solid; yield 95%, m. p. 104-105oC. Found, %: C 68.54; H 6.08; N 8.29; S 9.38. C20H20N2O2S (352.45). Calculated, %: C 68.16; H 5.72; N 7.95; S 9.10. 1H NMR (300 MHz, DMSO-d6–CCl4, 1:3) δ 1.10 (t, J = 7.1Hz, 3H, OCH2CH3); 3.04 (br s, 3H, NCH3); 3.12 (br s, 3H, NCH3); 4.07 (q, J =7.1 Hz, 2H, OCH2CH3 ); 6.62 (s, 1H, Th-H); 7.39- 7.48 (m, 3H, NAP-H); 7.74 (s, 1H, N=CH); 7.78-8.01 (m, 4H, NAP-H).
Ethyl 2-(((dimethylamino)methylene)amino)-4-(pyridin-4-yl)thiophene-3-carboxylate (4b). Yellow solid; yield 87%, m. p. 97-98oC. Found, %: C 59.47, H 5.56, N 13.65, S 10.40. C15H17N3O2S (303.39). Calculated, %: C 59.39, H 5.65, N 13.85, S 10.57. 1H NMR (300 MHz, DMSO-d6–CCl4, 1:3) δ 1.10 (t, J =7.1 Hz, 3H, OCH2CH3); 3.04 (br s, 3H, NCH3); 3,12 (br s, 3H NCH3); 4.07 (q, 2H, J = 7.1 Hz, OCH2CH3); 6.69 (s,1H, Th-H); 7.19 (d, 2H, J = 5.0 Hz, Py-H-2,6); 7.74 (s, 1H, N=CH); 8.46 (d, 2H, J = 5.0 Hz, Py-H-3,5).
Ethyl 5’-(((dimethylamino)methylene)amino)-[2,3’-bithiophene]-4’-carboxylate (4c). Yellow solid; yield 94.9%, m. p. 85-86oC. Found, %: C 54.27, H 5.35, N 9.00, S 20.91. C14H16N2O2S2 (308.42). Calculated, %: C 54.52, H 5.23, N 9.08, S 20.79.1H NMR (300 MHz, DMSO-d6–CCl4, 1:3) δ 1.20 (t, J =7.1, 3H,OCH2CH3), 3.02 (br s, 3H, NCH3), 3.10 (br s, 3H, NCH3), 4.14 (q, 2H, J =7.1Hz, OCH2CH3), 6.62 (s, 1H, Th-H), 6.97–7.00 (m, 2H, Th-H-2,4), 7.23 (dd, J1 =4.7, J2 =1.6, 1H, Th-H-3), 7.72 (s, 1H, N=CH).
General Procedure for the Syntheses of Compounds 6a-6n
A mixture of dimethylaminomethyleneamino derivative 4a-4c (10 mmol) and the corresponding primary amine 5a-5l (35 mmol) in anhydrous xylene (7 mL) was refluxed for 30 h (extra 3 h after cessation of dimethyl amine isolation). The mixture was allowed to cool to ambient tem perature, then light petroleum (8 mL) was added into the mixture. The precipitate was filtered, washed with ether and dried.
3-(4-Fluorobenzyl)-5-(naphthalen-2-yl)thieno[2,3-d]pyrimidin-4(3H)-one(6a). Beige powder, yield 67%, m. p. 166-167oC. Found, %: 71.81; H, 4.30; N, 7.62; S, 8.64. C23H15FN2OS (386.44). Calculated, %: C 71.49; H, 3.91; N, 7.25; S, 8.30. 1H NMR (400 MHz, CDCl3) δ 5.15 (s, 2H, CH2); 6.98-7.05 (m, 2H, Ar-H-3,5), 7.26 (s, 1H, Th-H); 7.29-7.39 (m, 2H, Ar-H-2,6), 7.49-7.56 (m, 2H, NAP-H-3,4); 7.68-7.72 and 7.85-7.96 (m, 4H, NAP-H-5,6,7,8), 8.00 (s, 1H, NAP-H-1) 8.10 (s, 1H, N=CH). 13C NMR (125 MHz, CDCl3) δ 48.55 (CH2), 115.90 (C(3)-Ar), 116.12 (C(5)-Ar), 121.46 (C(1)-NAP), 121.62 (C(6)-NAP), 127.08 (C(7)-NAP), 127.74 (C(5)-Th), 127.90 (C(3)-NAP), 128.07(C(4)-NAP), 128.22 (C(8)-NAP), 130.01 (C(5)-NAP), 130.09(C(6)-Ar), 131.66(C(2)-Ar), 131.70 (C(3)-Th), 132.96 (C(1)-Ar), 133.12 (C-NAP), 133.15 (C-NAP), 139.83 (C(2)-NAP), 146.43 (C(4)-Th), 157.31 (C=N) 161.45 (C(2)-Th), 163.91 (C(4)-Ar), 165.03 (C=O).
3-(4-Chlorobenzyl)-5-(naphthalen-2-yl)thieno[2,3-d]pyrimidin-4(3H)-one (6b). Beige powder, yield 59%, m. p. 144-146oC. Found, %: C, 68.92; H, 3.99; N, 7.32; S, 8.27. C23H15ClN2OS (402.90). Calculated, %: C, 68.57; H, 3.75; N, 6.95; S, 7.96. 1H NMR (400 MHz, CDCl3) δ 5.15 (s, 2H, CH2); 7.26 (s, 1H, Th-H); 7.29-7.35 (m, 4H, Ar-H-2,3,5,6), 7.48-7.55 (m, 2H, NAP-H-3,4); 7.65-7.72 and 7.85-7.90 (m, 4H, NAP-H-5,6,7,8), 8.00 (s, 1H, NAP-H-1) 8.10 (s, 1H, N=CH). 13C NMR (125 MHz, CDCl3) δ 48.64 (CH2), 121.49 (C(1)-NAP), 121.69 (C(6,7)-NAP), 126.20 (C(5)-Th), 126.23 (C(3)-NAP), 127.11 (C(4)-NAP), 127.89 (C(5)-NAP), C(8)-NAP), 128.09 (C(3,5)-Ar), 128.24 (C(2,6)-Ar), 129.27 (C(3)-Th), 129.53 (C(4)-Ar), 132.98 (C-NAP), 133.13 (C-NAP), 134.33 (C(2)-NAP), 134.43 (C(1)-Ar), 139.88 (C(4)-Th), 146.40 (C=N), 157.30 (C(2)-Th), 165.06 (C=O).
3-(4-Methoxybenzyl)-5-(naphthalen-2-yl)thieno[2,3-d]pyrimidin-4(3H)-one (6c). Beige powder, yield 68%, m. p. 130-131oC. Found, %: C, 72.72; H, 4.89; N, 7.39; S, 8.41. C24H18N2O2S (398.48). Calculated, %: C, 72.34; H, 4.55; N, 7.03; S, 8.05. 1H NMR (400 MHz, CDCl3) δ 3.75 (s, 3H, OCH3); 5.10 (s, 2H, CH2); 6.85 (d, J = 7.9 Hz, 2H, Ar-H-3,5) 7.20 (s, 1H, Th-H); 7.28 (d, J = 7.9 Hz, 2H, Ar-H-2,6), 7.44-7.55 (m, 2H, NAP-H-3,4); 7.68-7.73 and 7.85-7.90 (m, 4H, NAP-H-5,6,7,8), 8.00 (s, 1H, NAP-H-1) 8.10 (s, 1H, N=CH). 13C NMR (125 MHz, CDCl3) δ 48.56 (CH2), 55.25 (OCH3), 114.34 (C(3,5)-Ar), 121.32 (C(1)-NAP), 126.04 (C(6)-NAP), 126.06 (C(7)-NAP), 126.94 (C(5)-Th), 127.64 (C(3)-NAP), 127.83 (C(4)-NAP), 127.90 (C(8)-NAP), 127.96 (C(5)-NAP), 128.14 (C(2,6)-Ar), 129.68 (C(3)-Th), 132.84 (C(1)-Ar), 133.03 (2C-NAP), 133.20 (C(2)-NAP), 139.69 (C(4)-Th), 146.52 (C=N), 157.29 (C(4)-Ar), 159.58 (C(2)-Th), 164.92 (C=O).
3-(3,4-Dimethoxybenzyl)-5-(naphthalen-2-yl)thieno[2,3-d]pyrimidin-4(3H)-one (6d). Beige powder, yield 65%, m. p. 154-155oC. Found, %: C 70.41; H 5.05; N 6.89; S 7.80. C25H20N2O3S (428.05). Calculated, %: C 70.07; H 4.70; N 6.54; S 7.48. 1H NMR (400 MHz, CDCl3) δ 3.85 (s, 3H, 2OCH3); 5.10 (s, 2H, CH2); 6.78-6.90 (m, 3H, Ar-H-2,5,6), 7.20 (s, 1H, Th-H); 7.48-7.52 (m, 2H, NAP-H-3,4); 7.68-7.70 and 7.85-7.90 (m, 4H, NAP-H-5,6,7,8), 8.00 (s, 1H, NAP-H-1) 8.10 (s, 1H, N=CH). 13C NMR (125 MHz, CDCl3) δ 48.93 (CH2), 55.99 (OCH3), 56.08 (OCH3), 111.43 (C(2)-Ar), 111.69 (C(5)-Ar), 120.91(C(6)-Ar), 121.38 (C(1)-NAP), 121.46 (C(6)-NAP), 126.14 (C(7)-NAP), 126.16 (C(5)-Th), 127.02 (C(3)-NAP), 127.73 (C(4)-NAP), 127.94 (C(8)-NAP), 128.05 (C(5)-NAP), 128.20 (C(3)-Th), 128.32 (C-NAP), 132.92 (C-NAP), 133.10 (C(2)-NAP), 133.26 (C(1)-Ar), 139.77 (C(4)-Th), 146.53 (C=N), 149.26 (C(4)-Ar), 149.47(C(3)-Ar), 157.40 (C(2)-Th), 165.00 (C=O).
5-(Naphthalen-2-yl)-3-phenethylthieno[2,3-d]pyrimidin-4(3H)-one (6e). Beige powder, yield 77%, m. p. 118-119oC. Found, %: C 75.72; H 5.04; N, 7.69; S 8.71. C24H18N2OS (382.48). Calculated, %: C 75.37; H 4.74; N 7.32; S 8.38. 1H NMR (400 MHz, CDCl3) δ 3.05 (t, 2H, J = 7.2 Hz, CH2CH2Ph); 4.18 (t, 2H, J = 7.2 Hz, CH2CH2Ph); 7.10-7.18 (m, 2H, Ph), 7.25 (s, 1H, Th-H); 7.28-7.38 (m, 3H, Ph), 7.48-7.52 (m, 2H, NAP-H-3,4); 7.68-7.70 and 7.85-7.90 (m, 4H, NAP-H-5,6,7,8), 7.65 (s, 1H, NAP-H-1), 8.05 (s, 1H, N=CH). 13C NMR (125 MHz, CDCl3) δ 35.22 (CH2), 48.61 (CH2), 121.22 (C(1)-NAP), 126.08 (C(6)-NAP), 126.11 (C(7)-NAP), 126.99 (C(5)-Th), 127.01 (C(4)-NAP), 127.68 (C(3)-NAP), 127.90 (C(2,6)-Ar), 128.00 (C(8)-NAP), 128.18 (C(5)-NAP), 128.85 (C(3,5)-Ar), 128.91 (C(3)-Th), 132.89 (C-NAP), 133.07 (C-NAP), 133.17 (C(2)-NAP), 137.39 (C(1)-Ar), 139.58 (C(4)-Th), 146.60 (C=N), 157.28 (C(2)-Th), 165.16 (C=O).
3-(2-Chlorophenethyl)-5-(naphthalen-2-yl)thieno[2,3-d]pyrimidin-4(3H)-one (6f). Beige powder, yield 58%, m. p. 151-153oC. Found, %: C 69.49; H 4.40; Cl 8.87, N 7.02; S, 7.98. C24H17ClN2OS (416.92). Calculated, %: C 69.14; H 4.11; Cl 8.50, N 6.72; S, 7.69. 1H NMR (400 MHz, CDCl3) δ 3.22 (t, J =7.2 Hz, 2H, CH2CH2C6H4); 4.25 (t, J =7.2 Hz, 2H, CH2CH2C6H4), 7.1- 7.28 and 7.35-7.45 (m, 4H, Ar-H-3,4,5,6), 7.26 (s, 1H, Th-H); 7.48-7.55 (m, 2H, NAP-H-3,4); 7.65 (s, 1H, NAP-H-1), 7.62-7.72 and 7.85-7.96 (m, 4H, NAP-H-5,6,7,8), 8.02 (s, 1H, N=CH). 13C NMR (125 MHz, CDCl3) δ 33.12 (CH2), 46.56 (CH2), 121.26 (C(1)-NAP), 121.33 (C(6)-NAP), 126.15 (C(7)-NAP), 126.17 (C(6)-Ar), 127.07 (C(5)-Ar), 127.37 (C(4)-Ar), 127.74 (C(5)-Th), 127.93 (C(4)-NAP), 128.03 (C(3)-NAP), 128.24 (C(8)-NAP), 128.78 (C(5)-NAP), 129.86 (C(3)-Ar), 131.48 (C(3)-Th), 132.97 (C(2)-Ar), 133.15 (C-NAP), 133.21 (C-NAP), 134.15 (C(2)-NAP), 135.07 (C(4)-Th), 139.65 (C(1)-Ar), 146.59 (C=N), 157.44 (C(2)-Th), 165.23 (C=O).
5-(Naphthalen-2-yl)-3-(thiophen-2-ylmethyl)thieno[2,3-d]pyrimidin-4(3H)-one (6g). Beige powder, yield 58%, m. p. 147-148oC. Found, %: C 67.36; H 3.77; N7.48; S 17.12. C21H14N2OS2 (374.48). Calculated, %: C 67.36; H 3.77; N7.48; S 17.12. . 1H NMR (400 MHz, CDCl3) δ 5.3 (s, 2H, CH2); 6.92 and 6.96 (dd, 1H, J1=5.1, J2=3.5Hz), 7.10 and 7.12 (dd, 1H, J1=3.5, J2=1.2Hz), 7.22 (s, 1H, Th-H); 7.27(dd, J1=5.1, J2=1.2Hz), 7.48-7.58 (m, 2H, NAP-H-3,4); 7.62-7.72 and 7.85-7.96 (m, 4H, NAP-H-5,6,7,8), 8.01 (s, 1H, NAP-H-1), 8.12 (s, 1H, N=CH). 13C NMR (125 MHz, CDCl3) δ 43.72 (CH2), 121.22 (C(5)-Th(2)), 121.47 (C(1)-NAP), 126.05 (C(6)-NAP), 126.08 (C(7)-NAP), 126.64 (C(3)-Th(2)), 126.99 (C(4)-Th(2)), 127.09 (C(5)-Th(1)), 127.64 (C(4)-NAP), 127.84 (C(3)-NAP), 127.96 (C(8)-NAP), 127.98 (C(5)-NAP), 128.14 (C(3)-Th), 132.85 (C-NAP), 133.02 (C-NAP), 133.07 (C(2)-NAP), 137.46 (C(4)-Th(1)), 139.69 (C(2)-Th(2)), 146.08 (C=N), 156.90 (C(2)-Th(1)), 164.94 (C=O).
3-(Furan-2-ylmethyl)-5-(naphthalen-2-yl)thieno[2,3-d]pyrimidin-4(3H)-one (6h). Beige powder, yield 44%, m. p. 120-121oC. Found, %: C 70.71; H 4.29; N 8.15; S 9.31. C21H14N2O2S (358.42). Calculated, %: C 70.37; H 3.94; N 7.82; S 8.94. 1H NMR (400 MHz, CDCl3) δ 5.15 (s, 2H, CH2); 6.31 and 6.32 (dd, 1H, J1=5.1, J2=3.5Hz, Fur-H-3), 6.44 and 6.46 (dd, 1H, J1=3.5, J2=1.2Hz, Fur-H-4), 7.22 (s, 1H, Th-H); 7.72 and 7.74 (dd, J1=5.1, J2=1.2Hz, Fur-H-5), 7.48-7.58 (m, 2H, NAP-H-3,4); 7.85-7.95 (m, 4H, NAP-H-5,6,7,8), 7.98 (s, 1H, NAP-H-1), 8.18 (s, 1H, N=CH). 13C NMR (125 MHz, CDCl3) δ 41.61 (CH2), 110.27 (C(3)-Fur), 110.27 (C(4)-Fur), 121.34 (C(1)-NAP), 121.42 (C(6)-NAP), 126.11 (C(7)-NAP), 127.14 (C(5)-Th), 127.03 (C(4)-NAP), 127.70 (C(3)-NAP), 127.89 (C(8)-NAP), 128.03 (C(5)-NAP), 128.20 (C(3)-Th), 132.92 (C-NAP), 133.09 (C-NAP), 133.16 (C(2)-NAP), 139.76 (C(4)-Th), 143.23 (C(5)-Fur), 146.3 (C(2)-Fur), 148.34 (C=N), 156.92 (C(2)-Th), 165.00 (C=O).
5-(Naphthalen-2-yl)-3-(pyridin-2-ylmethyl)thieno[2,3-d]pyrimidin-4(3H)-one (6i). Beige powder, yield 44%, m. p. 120-121oC. Found, %: C 71.91; H 4.42; N 11.71; S 9.01. C22H15N3OS (369.44). Calculated, %: C 71.52; H 4.09; N 11.37; S 8.68. 1H NMR (400 MHz, CDCl3) δ 5.24 (s, 2H, CH2); 7.16, 7.18 and 7.20 (ddd, 1H, J=7.5; 4.8; 1.0 Hz, Py-H-), 7.22 (s, 1H, Th-H); 7.36 and 7.38 (dd, J1=5.1, J2=1.2Hz, Py-H-5), 7.48-7.58 (m, 2H, NAP-H-3,4); 7.60, 7.62 and 7.64 (ddd, 1H, J=7.5; 4.8; 1.0 Hz, Py-H-4), 7.66 and 7.68 (dd, 1H, 1H, J1=5.1, J2=3.5Hz, Py-H-5), 7.83-7.91 (m, 3H, NAP-H-5,6,7,8), 7.98 (s, 1H, NAP-H-1), 8.38 (s, 1H, N=CH). 8.51 and 8.53 (dd, 1H, J1=5.1, J2=3.5Hz, Py-H-6). 13C NMR (125 MHz, CDCl3) δ 50.60 (CH2), 121.29 (C(5)-Py), 121.33 (C(3)-Py), 123.14 (C(6)-NAP), 123.20 (C(7)-NAP), 126.09 (C(1)-NAP), 126.11 (C(5)-Th), 127.02 (C(4)-NAP), 127.70 (C(3)-NAP), 127.91 (C(8)-NAP), 127.98 (C(5)-NAP), 128.20 (C(3)-Th), 132.91 (C-NAP), 133.10 (C-NAP), 133.24 (C(2)-NAP), 137.02 (C(4)-Py), 139.73 (C(4)-Th), 147.46 (C=N), 149.71 (C(6)-Py), 154.78 (C(2)-Py), 157.28 (C(2)-Th), 165.32 (C=O).
5-(Pyridin-4-yl)-3-(thiophen-2-ylmethyl)thieno[2,3-d]pyrimidin-4(3H)-one (6j). Beige crys tals, yield 81.5%, m. p. 228-229oC. Found, %: C 58.84, H 3.63, N 12.78, S 19.55. C16H11N3OS2 (325.40). Calculated, %: C 59.06, H 3.41, N 12.91, S 19.71. 1H NMR (300 MHz, DMSO) δ 5.38 (s, 2H, CH2), 6.92 (dd, 1H, J1=5.1, J2=3.5Hz), 7.22 (dd, 1H, J1=3.5, J2=1.2Hz), 7.25-7.28 (m, 2H), 7.47 (s, 1H, Th-H), 7.48-7.50 (m, 2H), 8.53-8.56 (m, 2H), 8.57 (s, 1H, N=CH).
3-(4-Chlorophenyl)-ethyl-5-pyridin-4-yl)thieno[2,3-d]pyrimidin-4(3H)-one (6k). Beige crystals, yield 41.36%, m. p.173-174oC. Found, %: C 61.81, H 3.80, Cl 9.48, N 11.27, S 8.61. C19H14ClN3OS (367.86). Calculated, %: C 62.04, H 3.84, Cl 9.64, N 11.42, S 8.72. 1H NMR (300 MHz, DMSO) δ 3.01 (t, 2H, J2 = 3.5Hz, CH2), 4.00 (t, 2H, J2 = 3.5Hz, CH2), 7.24 – 7.27 (m, 4H, C6H4), 7.44 (d, 2H, J= 3.5Hz, Py-H), 7.48 (s, 1H, Th-H), 8.24 (s, 1H, N=CH), 8.56 (d, 2H, J=3.5Hz, Py-H).
3-(4-Fluorobenzyl)-5-(pyridin-4-yl)thieno[2,3-d]pyrimidin-4(3H)-one (6l). Beige crystals, yield 87.0%, m. p. 177-178oC. Found, %: C 63.80, H 3.51, F 5.56, N 12.32, S 9.38. C18H12FN3OS (337.37). Calculated, %: C 64.08, H 3.59, F 5.63, N 12.45, S 9.50. 1H NMR (300 MHz, DMSO) δ 5.18 (s, 2H, CH2), 6.97–7.06 (m, 2H, C6H4), 7.43-7.52 (m, 2H, C6H4), 7.47 (s, 1H, Th-H), 7.52 (d, 2H, J=3.5Hz, Py-H), 8.53 (d, 2H, J=3.5Hz, Py-H), 8.60 (s, 1H, N=CH).
3-(2-Hydroxyethyl)-5-(thiophen-2-yl)thieno[2,3-d]pyrimidin-4(3H)-one (6m): Beige crystals, yield 39.9%, m. p. 134-135oC. Found, %: C 51.99, H 3.53, N 9.97, S 22.79. C12H10N2O2S2 (278.34). Calculated, %: C 51.78, H 3.62, N 10.06, S 23.04. 1H NMR (300 MHz, DMSO) 3.69 (gt, 2H, J1 = 5.3, J2=5.1Hz, CH2CH2OH); 4.06 (t, 2H, J=5.1Hz, CH2CH2OH); 4.70 (t, 1H, J=5.3Hz, OH); 7.03 (dd, 1H, J1 = 5.1, J2=3.6Hz, Th(2)-H-4); 7.31 (dd, 1H, J1=5.1, J2=1.0Hz, Th(2)-H-3); 7.34 (s, 1H, Th(1)-H); 7.60 (dd, 1H, J1=3.6, J2=1.0Hz, Th(2)-H-5); 8.20 (s, 1H, N=CH).
3-(3-Hydroxypropyl)-5-(thiophen-2-yl)thieno[2,3-d]pyrimidin-4(3H)-one (6n): Beige cry stals, yield 34.7%, m. p. 115-116oC. Found, %: C 53.28, H 4.01, N 9.51, S 21.73. C13H12N2O2S2 (292.37). Calculated, %: C 53.40, H 4.14, N 9.58, S 21.93. 1H NMR (300 MHz, DMSO) 1.89 (tt, 2H, J1 =6.8, J2=5.7Hz, CH2CH2CH2OH); 3.49 (t, 2H, J=5.7 Hz, CH2CH2CH2OH); 4.10 (t, 2H, J=6.8 Hz, CH2CH2CH2OH); 4.36 (br s, 1H, OH), 7.03 (dd, 1H, J1=5.1, J2=3.6Hz,); 7.30 (dd, 1H, J1=5.1, J2=1.0Hz); 7.34 (s, 1H, Th(1)-H); 7.59 (dd, 1H, J1=3.6, J2=1.0Hz); 8.30 (s, 1H, N=CH).
Molecular Docking Studies
To investigate the structural basis of the antimicrobial activity of thieno-pyrimidone derivatives 6e and 6k, molecular docking studies were carried out using AutoDock Vina within the PyRx 0.8 virtual screening environment [
29]. The molecular determinants underlying the antidiabetic potential of the synthesized compounds were investigated through computational docking studies performed against Staphylococcus aureus Aminoglycoside Phosphotransferase (APH(2″)-Ia, co-crystallized with Gentamicin C1 (51G) (PDB ID: 5IQC, 2.30 Å resolution) [
30] and Pseudomonas aeruginosa tRNA (Guanine37-N1)-Methyltransferase (TrmD) protein targets.The TrmD (PDB ID: 5ZHM, 2.65 Å resolution) is co-crystallized with a thienopyrimidinone derivative (9D3) [
31] were utilized for the docking simulations.
Aminoglycoside Phosphotransferase and TrmD were chosen as relevant targets to rationalize and validate the in vitro antimicrobial activity of the tested compounds. The binding affinities and interaction patterns of the compounds were analyzed and compared with those of the co-crystallized reference inhibitors, Gentamicin and 9D3, to gain insights into potential inhibitory mechanisms. These targets were specifically selected because the compounds demonstrated notable antibacterial activity, particularly against Staphylococcus aureus and P. aeruginosa strains. In line with the in vitro assays using Gentamicin as a comparator, S. aureus Aminoglycoside Phosphotransferase co-crystallized with Gentamicin was included for docking studies. Similarly, TrmD from P. aeruginosa co-crystallized with 9D3, structurally similar to the tested compounds due to its thienopyrimidone scaffold was selected to explore structure-based interactions potentially underlying the observed antibacterial effects.
Protein structures were preprocessed for molecular docking using the Dock Prep tool in UCSF Chimera. The preparation steps involved removal of co-crystallized ligands and water molecules, reconstruction of missing atoms or residues, and energy minimization to optimize geometry. Polar hydrogens were added, and Gasteiger charges were assigned to accurately represent electrostatics. The finalized protein models were then saved in PDBQT format for subsequent docking simulations.
Ligand structures were initially sketched in 2D using ChemDraw and subsequently converted into 3D geometries for further refinement. Energy minimization was carried out using the MMFF94 force field with the conjugate gradient algorithm in PyRx’s Open Babel module, producing stable low-energy conformations. The optimized ligands were then exported in PDBQT format for docking studies with AutoDock.
Docking simulations in AutoDock Vina were performed by defining a focused grid around the active sites of the target proteins. For S. aureus Aminoglycoside Phosphotransferase, the grid center was set at X: 34.328, Y: -0.559, Z: 64.661, while for P. Aeruginosa TrmD, it was positioned at X: 45.373, Y: 113.745, Z: 16.516. A cubic grid box measuring 25 Å on each side with a spacing of 0.375 Å was employed to allow accurate sampling of ligand orientations within the binding pocket.
Following docking, the ligand–receptor complexes were examined to characterize binding modes and interaction patterns using ChimeraX [
32] and BIOVIA Discovery Studio 2021.
Molecular docking was carried out using the software’s default parameters, producing ten binding conformations per compound. The conformation exhibiting the lowest predicted binding energy was chosen for detailed analysis of ligand–receptor interactions.
The docking protocol was validated by re-docking 9D3 into its native co-crystallized binding site. The superimposition of the re-docked ligand onto the original crystal structure yielded an RMSD of 0.93 Å, demonstra ting that the docking setup reliably reproduces the experimentally observed binding conformation.
Figure 3. Binding affinity plot of 6e and 6k compounds with S.aureus Aminoglycoside Phosphotransferase and P.aeruginosa TrmD.
Figure 4.
2D molecular representation of interactions of compounds A)6e, B)6k, and C) Gentamicin with the active site residues of the S. aureus Aminoglycoside Phosphotransferase protein. Interactions were displayed as color coded dashed lines, green lines indicated the H–bonds.
Figure 4.
2D molecular representation of interactions of compounds A)6e, B)6k, and C) Gentamicin with the active site residues of the S. aureus Aminoglycoside Phosphotransferase protein. Interactions were displayed as color coded dashed lines, green lines indicated the H–bonds.
Table 2.
Binding energy and interaction summary with S.aureus Aminoglycoside Phosphotransferase.
Table 2.
Binding energy and interaction summary with S.aureus Aminoglycoside Phosphotransferase.
| Compounds |
Binding Energy (K.cal/mol) |
Interacting Amino acids |
Nature of interactions |
| 6e |
-7.3 |
TYR408, TYR212, GLU411, TYR448, VAL444, ASP374, GLU451, GLU415, SER413, GLU445, ARG475, ASN324, ASN373, ASP396 |
H-bond, π-sulfur, π-π stacked, π-π T shaped, π-alkyl, π-anion, π-donor hydrogen bond, van der waals |
| 6k |
-7.8 |
ASN378, TYR212, ASP374, GLU445, VAL444, TYR408, GLU451, TYR448, SER376, HIS379, ASP393, GLY211, GLU411 |
H-bond, π-anion, π-π stacked, π-π T shaped, halogen, alkyl, π-anion, π-donor hydrogen bond, van der waals |
| Gentamicin |
-8.2 |
ASP396, ASP374, SER376, GLU416, GLU415, GLU411, GLU445, TYR408, TYR448, GLU451, TYR212, ASN378 |
H-bond, π-sigma, π-alkyl, C-H bond, van der waals |
Figure 5.
2D molecular representation of interactions of compounds A)6e, and B)6k, and C)9D3 with the active site residues of the P. aeruginosa TrmD protein. Interactions were displayed as color coded dashed lines, green lines indicated the H–bonds.
Figure 5.
2D molecular representation of interactions of compounds A)6e, and B)6k, and C)9D3 with the active site residues of the P. aeruginosa TrmD protein. Interactions were displayed as color coded dashed lines, green lines indicated the H–bonds.
Table 3.
Binding energy and interaction summary with P.aeruginosa TrmD.
Table 3.
Binding energy and interaction summary with P.aeruginosa TrmD.
| Compounds |
Binding Energy (K.cal/mol) |
Interacting Amino acids |
Nature of interactions |
| 6e |
-7.1 |
TYR91, TYR120, SER93, PRO94, LEU143, PRO149, ILE123, GLY122, GLN95, GLY118, ARG119, SER144, GLY145, VAL142, GLY146, LEU92, TYR141, ILE138, SER137, TRP136, GLY139 |
H-bond, π-sulfur, π-π T shaped, Amide-π stacked, π-alkyl, van der waals |
| 6k |
-7.5 |
TYR91, SER137, SER93, GLY122, PRO94, PRO149, ILE138, LEU143, GLY139, TYR141, VAL142, SER144, GLY118, ARG119, GLY145, GLN95, TYR120, GLU121, ILE123, GLY146, LEU92 |
H-bond, π-sigma, π-sulfur, π-alkyl, alkyl, Amide-π stacked, C-H bond, van der waals |
| 9D3 |
-6.8 |
LEU143, TYR120, SER93, PRO149, ILE138, GLY139, PRO94, SER137, TRP136, LEU92, GLY146, GLY145, GLY118, GLY122, ARG119, GLU121, SER144, VAL142, TYR141 |
H-bond, π-π T shaped, Amide-π stacked, π-alkyl, C-H bond, van der waals, unfavourable acceptor-acceptor interactions |
Figure 6.
Molecular imension of the binding poses of compounds 6e (grey), 6k (orange) and Gentamicin(A) and 9D3 (B) in gold color in the actives of the S. aureus Aminoglycoside 9(A) and P. aeruginosa TrmD (B) proteins for comparative three imensional steric space visulazation with the reference compounds.
Figure 6.
Molecular imension of the binding poses of compounds 6e (grey), 6k (orange) and Gentamicin(A) and 9D3 (B) in gold color in the actives of the S. aureus Aminoglycoside 9(A) and P. aeruginosa TrmD (B) proteins for comparative three imensional steric space visulazation with the reference compounds.
Figure 7.
Protein ligand complexes representing the A) binding site of 6e and 6k (yellow) with Gentamicin (green) in stick model and B) their respective orientations and topology of the active site pocket of S. aureus Aminoglycoside Phosphotransferase.
Figure 7.
Protein ligand complexes representing the A) binding site of 6e and 6k (yellow) with Gentamicin (green) in stick model and B) their respective orientations and topology of the active site pocket of S. aureus Aminoglycoside Phosphotransferase.
Figure 8.
Protein ligand complexes representing the A) binding site of 6e and 6k (yellow) with 9D3 (green) in stick model and B) their respective orientations and topology of the active site pocket of P. aeruginosa TrmD.
Figure 8.
Protein ligand complexes representing the A) binding site of 6e and 6k (yellow) with 9D3 (green) in stick model and B) their respective orientations and topology of the active site pocket of P. aeruginosa TrmD.
Molecular interaction profile of compounds with Aminoglycoside Phosphotransferase
Docking-based binding affinity results against
S. aureus Aminoglycoside phosphotransferase showed that compound
6k (–7.8 kcal/mol) binds more strongly than compound
6e (–7.3 kcal/mol) and displays a binding affinity close to that of gentamicin (–8.2 kcal/mol). These results correspond well with the relative
in-vitro antibacterial activities of the compounds (
Table 2).
Gentamicin C1 comprises of three structural moieties; garosamine, 2-deoxystreptamine, and purpurosamine rich in hydroxyl and amine functionalities. These functional groups enable an extensive hydrogen-bonding network with key residues, particularly involving the –NH₂ groups, which form multiple hydrogen bonds with ASP396, ASP374, SER376, GLU416, GLU415, GLU411, and GLU445. In addition, TYR408 engages in a π–σ interaction with the 2-deoxystreptamine ring. The methylamine substituent exhibits a π–alkyl interaction with TYR448, while GLU451 contributes a C–H bond interaction with the garosamine ring, collectively stabilizing the ligand within the binding pocket.
Compound 6e, bearing naphthyl and phenethyl substituents on the thienopyrimidinone scaffold, exhibits two key hydrogen-bond interactions with the active-site residues TYR212 and TYR408. The residues GLU411, GLU451, and ASP374 participate in π–anion interactions with the phenyl and thieno rings. Furthermore, VAL444 and TYR408 engage in π–alkyl, π–π T-shaped, and π-donor hydrogen-bond interactions with the naphthyl moiety, while TYR448 forms a π–π stacking interaction with the pyrimidinone core. Collectively, these interactions indicate that both the substituent aromatic rings and the heterocyclic backbone contribute to a diverse and complementary interaction profile that stabilizes compound 6e within the active site. In addition, nearby residues SER413, GLU415, ARG475, GLU445, ASN324, ASN373, and ASP396 contribute van der Waals interactions, contributing to the further reinforced ligand binding.
Compound 6k, featuring pyridyl and 4-chlorphenethyl substituents on the core scaffold, exhibited a well-defined interaction profile within the active site. A hydrogen-bond interaction was observed between the sulfur atom of the thieno ring and ASN378, while the pyridine nitrogen and 4-oxo group of the pyrimidone ring formed hydrogen bonds with TYR212. In addition, ASP374 engaged in a π–anion interaction with the thieno-pyrimidone scaffold. The TYR212 residue further contributed a π–π stacking interaction with the pyridyl ring, whereas TYR408 participated in a π-donor hydrogen-bond interaction with the chloro-substituted phenyl ring. The chloro substituent established halogen bonding interactions with GLU445 and alkyl contacts with VAL444. Additionally, surrounding residues including GLU411, TYR448, GLU451, SER376, HIS379, GLY211, and ASP393 formed stabilizing van der Waals interactions, collectively reinforcing the binding stability of compound 6k within the active site.
Gentamicin C1 exhibited a predominantly hydrogen-bond–driven interaction network with aromatic contributions limited to weak π–σ or π–alkyl contacts. In contrast, compound
6e showed strong aromatic and hydrophobic π-interactions, arising mainly from its bulky naphthyl and phenethyl substituents. Compound
6k had a balanced profile, combining hydrogen bonding, π-interactions, and halogen contacts, offering a more diversified interaction pattern than 6e including halogen bonding (GLU445) and additional hydrogen bonds contacts through its pyridyl nitrogen. This broader interaction network distinguishes compound
6k from
6e, resulting in improved relative binding affinity and enhanced adaptability within the binding pocket. Gentamicin,
6e, and
6k exhibit distinct binding modes arising from their diverse interaction profiles, as illustrated in
Figure 6A, with a summary of the corresponding interactions provided in
Table 2.
Molecular interaction profile of compounds with TrmD
Binding affinity outcomes against Pseudomonas aeruginosa TrmD indicated that compound
6k (–7.5 kcal/mol) exhibited a stronger predicted binding affinity than compound
6e (–7.1 kcal/mol) and 9D3 (–6.8 kcal/mol) (
Table 3). Although compound
6k showed a better predicted binding affinity toward TrmD than
6e and 9D3, the
in vitro activity favoured
6e, suggesting that binding affinity alone does not fully account for antibacterial efficacy. This highlights the contribution of non-binding factors, such as physicochemical properties and bacterial cell barriers, in determining overall biological performance.
Compound 9D3 shares a scaffold similar to the evaluated compounds but features a predominant substitution on the thieno ring, consisting of a carboxamide linker connected to a methylphenyl ring bearing a diethylamino substituent. The interaction profile of 9D3 includes a hydrogen-bond interaction with LEU143. The phenyl ring forms a π–π T-shaped interaction with TYR120, while an amide–π stacking interaction is observed between SER93 and the thieno-pyrimidinone core. Additionally, PRO94, ILE138, and PRO149 participate in π–alkyl interactions, and GLY139 contributes a C–H bond interaction. An unfavourable acceptor–acceptor interaction is observed with TYR141 involving the backbone scaffold, whereas TYR120 also engages in alkyl interactions with the ethyl moiety of the diethylamine substituent. Furthermore, nearby binding-site residues SER137, TRP136, LEU92, GLY146, GLY118, GLY145, VAL142, ARG119, GLU121, and SER144 contribute stabilizing van der Waals interactions.
Although
6e and
6k share a similar core scaffold with 9D3, substitutions on both the thieno and pyrimidinone rings give rise to a slightly distinct interaction profile and binding orientation, as illustrated in
Figure 4B.
Compound 6e formed a hydrogen-bond interaction with TYR91 involving the sulfur atom of the thieno ring. Notably, its hydrophobic stacking interactions showed similarities to those observed for 9D3, engaging the same key residues TYR120 and SER93 in a comparable manner. Specifically, TYR120 exhibited a π–π T-shaped interaction with the naphthyl moiety, while SER93 participated in an amide–π stacking interaction with the phenyl ring. In addition, PRO94 displayed π–alkyl interactions with both the scaffold and the phenyl ring, whereas PRO149 engaged in π–alkyl interactions exclusively with the phenyl ring; both proline residues exhibited interaction patterns similar to those observed in 9D3. LEU143, which participates in a π–alkyl interaction in 6e, formed a hydrogen bond in the 9D3 complex. Furthermore, multiple van der Waals interactions were observed with surrounding binding-site residues, including ILE123, GLY122, GLN95, GLY118, ARG119, SER144, GLY145, VAL142, TYR141, GLY139, ILE138, SER137, TRP136, GLY146, and LEU92, collectively contributing to enhanced ligand stabilization within the active site
Compound 6k exhibited a hydrogen-bond interaction pattern similar to that of 6e, involving TYR91 through the thieno ring. In addition, an extra hydrogen-bond interaction was observed with SER137, mediated by the chloro substituent on the phenyl ring. A distinct π–σ interaction with GLY122, involving the thieno ring, was also identified. Furthermore, a triad of alkyl and π–alkyl interactions involving PRO94, ILE138, and PRO149 was observed; notably, this same set of residues displayed comparable interactions in the 9D3 complex. LEU143 engaged in both a π–alkyl interaction with the phenyl ring and a C–H bond interaction with the ethyl linker. Additionally, multiple surrounding binding-site residues, including LEU92, ILE123, GLU121, GLY146, GLY139, TYR141, VAL142, GLY145, ARG119, GLN95, TYR120, GLY118, and SER144, contributed stabilizing van der Waals interactions, collectively supporting the binding of compound 6k within the active site.
For both
6e and
6k, TYR91 contributes to ligand stabilization through concurrent hydrogen-bonding and π–sulfur interactions with the thieno moiety. A summary of these interaction details is presented in
Table 3.
Compounds 9D3,
6e and
6k share a common thieno-pyrimidinone scaffold and display overlapping binding modes within the same active-site pocket, engaging a conserved set of residues and adopting closely aligned binding orientations (
Figure 6B). Across all three protein-ligand complexes, TYR120 and SER93 play central roles in aromatic stacking interactions, while PRO94, ILE138, and PRO149 consistently contribute π–alkyl or alkyl interactions, reflecting a shared hydrophobic interaction framework. LEU143 is involved in ligand stabilization in all cases, although the nature of the interaction varies (hydrogen bonding in 9D3 versus π–alkyl or C–H interactions in
6e and
6k). Overall, the similarities highlight a conserved recognition framework, with differences arising mainly from substituent-driven modulation of interaction types.
Figure 4 and
Figure 5 provide a comprehensive visualization of the distinct interaction profiles of compounds
6e and
6k within the active sites of
S. aureus aminoglycoside phosphotransferase and
P. aeruginosa TrmD, respectively.
Figure 7 and
Figure 8 further depict the corresponding binding sites and pocket conformations of the evaluated compounds, offering deeper insight into their molecular recognition patterns and binding behavior
Determination of ADMET profile, Lipinski rule, and pharmacokinetics
ADMET profiling was performed to evaluate the pharmacokinetic behavior of the selected compounds. Absorption, distribution, metabolism, and excretion properties were predicted using the SwissADME platform (
http://www.swissadme.ch/) [
33], offering insights into their drug-likeness and key physicochemical features. Toxicological profile was assessed via the pkCSM webserver (
https://biosig.lab.uq.edu.au/pkcsm/prediction) [
34]. These computational assessments provided preliminary insights into the safety and pharmaceutical characteristics of the compounds.
In silico ADME assessment revealed that compounds
6e and
6k share largely overlapping pharmacokinetic characteristics. Both candidates are predicted to exhibit efficient high gastrointestinal (GI) absorption, absence of BBB permeation suggesting minimal CNS exposure and an identical bioavailability score (0.55). Compliance with Lipinski’s criteria was observed for both molecules; however, compound
6e had a violation due to elevated lipophilicity (MLOGP > 4.15). Structural differences between the two compounds are manifested in their polarity-related descriptors, with
6k possessing an additional hydrogen bond acceptor and a correspondingly increased topological polar surface area (TPSA) (76.02 Ų vs. 63.13 Ų). These features translate into a more favorable solubility profile for 6k, which is predicted to have moderate aqueous solubility, whereas
6e is associated with limited solubility. Predicted CYP450 inhibition is similar for
6e and
6k across the major isoforms (CYP1A2, CYP2C19, CYP2C9, and CYP3A4). A summary of these details is provided in
Table 4 for reference.
The comparative analysis highlights 6k as pharmacokinetically more balanced than 6e. The lower molecular weight and reduced lipophilicity (LogP 3.98 vs. 5.19) of 6k are advantageous, as excessive lipophilicity in 6e may contribute to poor aqueous solubility, increased nonspecific binding, and higher metabolic liabilities. Compound 6k combines higher polarity with good permeability, resulting in improved solubility and maintained higher GI absorption.
Notably,
6e is predicted to be a P-gp substrate, which may limit its effective intracellular accumulation due to efflux mechanisms, whereas
6k lacks this liability, potentially offering superior systemic exposure. Despite comparable CYP450 inhibition profiles, the metabolic liability of drug–drug interactions is greater for
6e owing to its less favorable lipophilicity and solubility.
Figure 9 presents these pharmacokinetic features using the BOILED-Egg model, highlighting GI absorption, BBB permeability, and P-gp interactions.
Overall, within the scope of the exploratory in silico ADME evaluation, compound 6k exhibits a more favorable drug-like profile, characterized by improved solubility, balanced lipophilicity, higher predicted GI absorption, and lower efflux-associated liabilities. Collectively, these attributes support the prioritization of 6k as a promising lead for further optimization toward enhanced pharmaceutical performance and therapeutic potential.
Predicted safety endpoints suggest that both
6e and
6k are associated with hERG II inhibition and potential hepatotoxic effects, pointing to possible risks involving cardiac repolarization and liver function. Genotoxicity predictions distinguish the two compounds, as
6e is non-mutagenic, whereas
6k is predicted to be AMES-positive that raises concerns regarding potential mutagenicity (
Table 5). Thus, although
6k may present advantages in pharma co kinetic and drug-like properties, its predicted genotoxic risk introduces a key safety challenge that must be addressed during lead optimization with structural modifications to mitigate this liability. No significant risks of skin sensitization were observed with both the compounds. These findings highlight the importance of incorporating toxicity-mitigation strategies to improve overall safety.