Submitted:
09 March 2026
Posted:
10 March 2026
You are already at the latest version
Abstract
Behcet disease (BD) is an inflammatory disorder with manifestation in mucosal tissues. Unlike autoimmune diseases that generate autoantibodies, BD is believed to be an autoinflammatory disease triggered by innate immune cells rather than adaptive cells. Hyperactivation of neutrophils causes vasculitis and thrombosis, and they migrate into cutaneous and ocular lesions. Dominance of M1 macrophages promotes the differentiation of Th1 cells. Moreover, the cross-reaction of bacterial heat shock proteins induces production of cytokines such as IL-4 and IFN-γ, in γδT cells, which alters the balance between Th1 and Th2 phenotypes. Nevertheless, natural killer (NK) cells play more critical roles in BD pathogenesis than other innate immune cells because not only their activity is precisely controlled by the interaction between ligands and receptors but NK1 shift also elicits Th1 dominance. The genetic factors associated with BD are HLA-B51 and major histocompatibility complex class I-related chain A (MICA), which stimulate NK receptors as ligands. Improperly processed peptides dysregulate their interaction with NK receptors, triggering the inflammatory response. NK1 and NK2 subsets represent cytokine production in relapse and remission periods; however, the cytotoxicity of NK cells in relapse is lower than that in remission periods. It still remains unclear how NK cells are activated recurrently and expand cytokine production. This review highlights the regulation of gene expression encoding NK receptors, tissue-resident NK cells, and adaptive NK cells to discuss their potential for relapsing. Splicing variants and readthrough genes encoding NK receptors easily alter cytokine production. Moreover, tissue-resident NK cells in mucosal tissues and adaptive NK cells that memorize the virus infection have the potential to trigger hyperactivation in relapse.
Keywords:
Behcet disease
; NK cells
; NK1/NK2
; IFN-γ
; splicing
; readthrough
; tissue-resident NK cells
; memory-like NK cells
1. Introduction
Behcet disease (BD) is a chronic and recurrent inflammatory disorder. Due to its high incidence in regions along the Silk Road, environmental factors and genetic association have been considered as a cause of the disease. Although no specific bacterium or virus that triggers the onset of BD has not been identified, some microorganisms that associate with abnormal immune responses have been listed [1]. For instance, the frequency of Streptococcus sanguinis in oral flora correlates with the activation of neutrophils [2]. Regarding genetic factors, a higher prevalence of human leukocyte antigen (HLA) -B51 in patients with BD than healthy controls (HCs) was reported in Japan from a serological analysis [3] followed by genotyping studies including disequilibrium with MICA [4,5,6,7]. Inflammatory diseases such as ankylosing spondylitis, colitis, and psoriasis are strongly associated with HLA class1 alleles and are known as MHC-I-opathies [8]. In this concept, although autoimmune diseases exhibit common pathogenesis, they are attributed to the contacts of HLA class 1 molecules with NK cells and CD8-positive T cells.
The primarily manifestations of BD are cutaneous lesions, oral aphthous ulcers, uveitis, genital ulcers, and vascular and neurological complications. BD affects both sexes; however, the mortality rate in men is higher than in women. Ocular, vascular, and neurological manifestations are more frequently observed in men, whereas oral and genital ulcers, skin lesions, and arthritis occur more frequently in women [9]. A genetic association study comparing men with women demonstrated that association with HLA-B and MICA in men is higher than in women [10].
Although autoimmune and autoinflammatory diseases with similar phenotypes in the same tissues have a complicated diagnosis, network modularity analyses have identified 10 diseases [11] in which BD is different from systemic lupus erythematosus (SLE) and rheumatoid arthritis. The International Diagnostic Criteria for Behcet’s Disease (ICBD) has established a guide for the accurate diagnosis and classification of BD [12]. Nonetheless, no single definitive laboratory test has been available till date. A diagnostic model for vascular BD based on an increase in the level of inflammatory, hematological, and thrombosis parameters in vascular BD compared with those in nonvascular BD was recently proposed [13].
Lack of evidences concerning autoantibodies in BD has directed interests in the pathogenesis of innate immune cells. Previous research has shown that serum isolated from patients with BD improved the adherence of neutrophils to human umbilical vein endothelial cells monolayer in vitro [14]. Another study showed that activated neutrophils migrate into cutaneous lesions in pathergy tests [15]. Moreover, they form neutrophil extracellular traps (NETs) to eliminate pathogens and activate other immune cells, releasing inflammatory cytokines. NETs damage endothelial cells and contribute to thrombosis via the production of reactive oxygen species [16]. In uveitis, the feedback loop between NETs and IL-17A maintains the hyperactivation and infiltration of neutrophils [17].
In general, M1 macrophages play a proinflammatory role and eliminate pathogens via the release of IL-12, TNF-α, IL-β, and IL-6, whereas M2 macrophages play an anti-inflammatory role for wound repair. In BD, macrophages are polarized due to a shift toward the proinflammatory M1 phenotype and impairment of the M2 phenotype [18]. Decrease in IL-10 secretion along with the dominance of proinflammatory cytokine production causes M1 polarization [5,19], which consequently induces differentiation of Th1 cells.
Despite a quite low frequency of cell numbers among the entire T cell population, a higher portion of γδT cells was detected in mucocutaneous lesions [20]. These cells respond to antigens directly unlike CD8-positive T cells that recognize antigens presented by HLA class 1 molecules. Bacterial infection induces cross-reaction of heat shock proteins in γδT cells, triggering proliferation [21]. Moreover, γδT cells produce IFN-γ and promote the differentiation of Th1 cells along with producing IL-4 for Th2 cells, thereby altering the balance between Th1 and Th2 phenotypes [22]. TNF-α secreted from γδT cells increases IL-8 secretion and recruits additional neutrophils to the lesions [23].
The role of natural killer (NK) cells in BD pathogenesis has not been explored in detail because of limited information regarding their migration. As HLA class 1 molecules control the function of NK cells through their receptors, their dysregulation could cause hyperactivation. Immature NK cells generally develop into cytokine-producing cells (CD56bright) and finally acquire cytolytic functions (CD56dim) [24], which comprise the majority of peripheral NK cells. CD56bright NK cells secret IFN-γ by recycling endosomes [25]. NK cells undergo a licensing process in which they correctly recognize self-HLA through killer cell immunoglobulin-like receptor (KIR) [26,27]. Then, they acquire cytotoxicity and eliminate only unnecessary cells that do not possess self-HLA without damaging own cells [28]. Perforin forms pores in the membranes that deliver granzyme into target cells, resulting in cell death. Secretory lysosomes store perforin and granzyme, but LAMP1 (CD107a) is expressed on the cell surface and it reduces the binding of perforin to the NK cell’s own membrane [29,30].
This review summarizes the role of NK cells in BD pathogenesis where cytokine production and cytotoxicity are dysregulated by the interaction between ligands and receptors due to genetic polymorphism is summarized. Next, the potential pathogenesis of BD with transcriptional control of NK receptors, tissue-resident NK cells, and adaptive NK cells is discussed. Altered cell surface expression of NK receptors could easily modify the responses to ligands. Furthermore, some tissue-resident NK cells in mucosal tissues and adaptive NK cells are focused as latent populations to be involved in relapse.
2. Search Methods
A literature review was performed using the following terms: “Behcet disease”, “NK cells” (retrieved 107 articles with the term “Behcet disease”), “KLRK1” (8 articles), “KLRC4” (16 articles), “splicing”, “KLRK1” (retrieved 24 articles with the term “splicing”), “memory”, “NK cells”, IL-12”, “IL-15”, “IL-18” (retrieved 53 articles with “memory”), and “KLRC2” (36 articles). Relevant original and review articles were collected through the PubMed and MEDLINE between January and February 2026.
3. NK Cells in BD
3.1. Numbers, Cytokine Production, and Cytotoxicity
Table 1 summarizes the studies on the population and capability of peripheral NK cells in BD. Conflicting data exists regarding NK subsets. Increases in the numbers of NK cells (CD16+CD56+) were detected in peripheral blood collected from BD patients compared with that in HCs [20,31]. Conversely, a decrease in the numbers of NK cells (CD3−CD56+) was found in pulmonary patients [32]. Another study demonstrated no significant differences in numbers of NK cells (CD16+CD56+) between HCs and patients with BD despite increases in the number of CD94+ cells [33]. The numbers of NK cells (CD3−CD56+) in patients with BD are comparable to that in HCs [34]. CD16 and CD56 discriminate cytolytic cells and cytokine-producing cells; however, no significant differences were found between HCs and patients with BD [35].
As BD is a recurrent disorder, NK subsets in relapse and remission were analyzed, which revealed that although patients showed an increase in the number of terminally differentiated NK cells (CD16+CD57+) during relapse compared with that during remission, the cytotoxicity of peripheral NK cells against K562 cells during relapse was lower than that during remission [36]. Moreover, NK cells (CD16+CD56+) exhibited decreased cytotoxicity during relapse [37]. Both of CD56bright and CD56dim subsets were depleted in peripheral blood collected from patients with BD [38]. Nevertheless, CD56bright NK cells in patients with BD produce higher levels of IFN-γ than HCs, whereas CD56dim NK cells in patients with BD release comparable levels of perforin and granzyme to those in HCs [38].
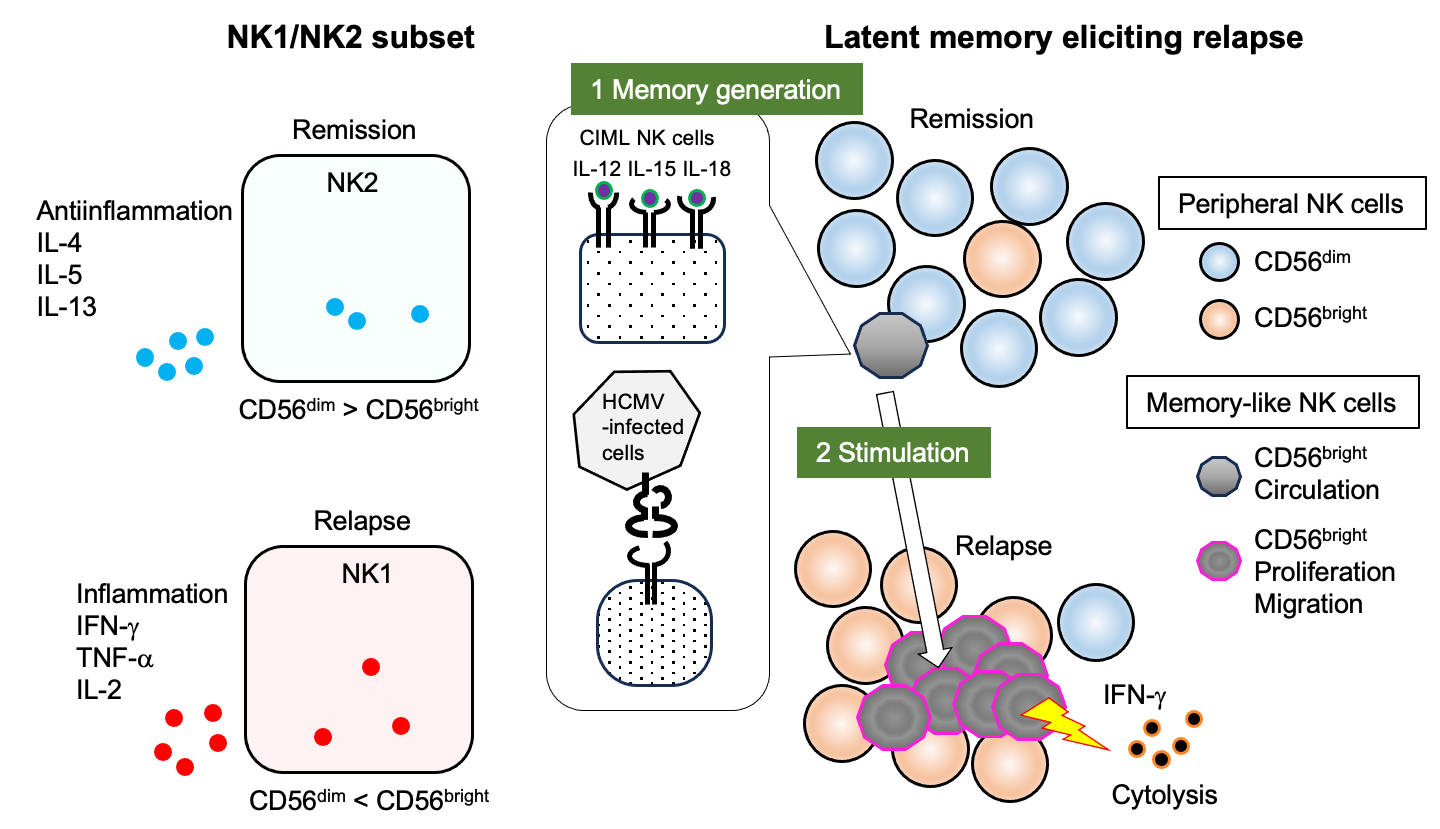
NK1 and NK2 subsets were originally investigated to determine whether they produce IFN-γ or not. IFN-γ non-secreting NK cells produce IL-4, IL-5, and IL-13 [39]. NK cells produce proinflammatory cytokines, such as IFN-γ, with their NK1 profile during relapse period and promoting Th1 inflammation. Conversely, NK cells produce IL-4 and IL-10 with their NK2 profile during the remission period, which suppress Th1 response. An increased NK1/NK2 ratio was detected in patients with uveitis [40] and mucocutaneous BD [35]. Remarkably, one study classified NK cells based on the data of scRNA-seq and cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) [41]. The NK1 subset includes cytotoxic cells (CD56dim), whereas NK2 subset represents cytokine-producing cells (CD56bright). As a distinct population from peripheral NK cells, NK3 cells include memory-like NK cells that express KLRC2.
Cytotoxicity against K562 cells is also estimated by means of LAMP1 expression; however, it does not conform with earlier studies (Table 2). The expression level of LAMP1 in NK cells (CD16+) isolated from BD cultured with K562 cells is comparable to HCs [35], whereas NK cells (CD3−CD56+) isolated from patients with BD exhibited higher expression of LAMP1 than those obtained from HCs [34]. The expression level of LAMP1 in NK cells (CD3−CD16+) during relapse period is higher than that during remission [42]. Therefore, studies concerning the number of NK cells and their capability indicate that the activities of NK cells are not proportionate to number of NK cells. Further functional assays are required to understand the dysregulation of NK cells.
3.2. NK Cell Receptors and Their Ligands
3.2.1. HLA-B and KIR
HLA-B51 is the highest genetic risk factor for BD. Furthermore, HLA-B15 and HLA-B27 are associated with HLA-B51-positive BD [7]. Patients with HLA-A02, HLA-A24, and HLA-B57 are at risk for developing BD, whereas HLA-A03, HLA-B35, and HLA-B58 are protective [43].
NK cells recognize their own cells through the interaction of HLA-B with KIR3DL1, by which normal cells are protected from cytolysis [44]. HLA-B variants are determined by specific residues at 77-83. Bw4 recognizes KIR3DL1, which possesses two immunoreceptor tyrosine-based inhibitory motifs (ITIMs) in the cytoplasmic region, and inhibits cytolytic signals, whereas Bw6 does not. Positions at 82 and 83 were identified as essential resides using HLA-B78 (HLA-B*1513), HLA-B75 (HLA-B*1502), and nonfunctional Bw6 motif [45,46].
The presence of neither inhibitory KIR3DL1 nor activating KIR3DS1 alleles increases susceptibility even if patients with BD carry Bw4 [47]. The frequency of KIR3DL1*004 encoding a misfolded protein [48] is lower in patients with BD than HCs in both HLA-B51-positive and -negative individuals [49]. The other component that influences the binding of HLA-B to KIR3DL1 is HLA class 1-bound peptides. The peptides bind to HLA-B51, but they exhibit weak affinity [50]. Insufficient licensing of NK cells may cause hyperactivation.
Next, HLA-B27 can be expressed as normal HLA-B27 with β-microglobulin (β2m) or as homodimers including free heavy chain forms (B272) [51]. Normal HLA-B27 binds to KIR3DL1; however, B272 binds to KIR3DL2 more strongly than other HLA class 1 molecules. Binding of B272 to KIR3DL2 expands Th17 proliferation and increases cytokine production in patients with ankylosing spondylitis [52]. A weak association between HLA-B27 and BD indicates that maintaining Th17 by IL-23 is more critical than B272.
Finally, KIR2DL4 possesses unique features because it contains a charged arginine in the transmembranes [53] and couples with an FcRγ adaptor [54]. KIR2DL4 induces IFN-γ production in NK cells, whereas CD16 and 2B4 induces cytolysis [55]. Although earlier studies found that HLA-G binds to KIR2DL4 and activates NK cells in the maternal vasculature during the initial period of pregnancy [56,57], KIR2DL4 was later identified as a novel susceptible gene in patients with severe uveitis [58].
3.2.2. MICA and KLRK1(NKG2D)
MICA is an MHC class 1 chain-related protein that does not require β2m to form a surface structure. KLRK1 homodimers bind monomeric MICA in solution [59]. MICA acts as a ligand of KLRK1 and causes cytolysis [60]. Pairs of KLRK1 and the adaptor protein HCST (DAP10) activate signaling molecules such as phosphatidylinositol 3-kinase (PI3K), GRB2, and VAV1 [61,62]. MICA gene has diverse polymorphisms, and its variance is associated with BD. In Slovak, Spanish, and other Eurasian populations, MICA*008 exhibited the highest frequency [63,64]. MICA exhibits strong linkage disequilibrium with HLA-B51 due to its proximity on the genome. MICA*009 is associated with HLA-B51 in Caucasian, Spanish, and Turkey patients [65,66,67]. MICA-TM A6 (MICA exon 5), which is a microsatellite polymorphism coding transmembrane region, detected in Greece patients [68] (Yabuki K et al., 1999) and MICA*006 found in Brazilian patients [69] (Marin ML et al., 2004) are associated with HLA-B51. The concentration of sMICA in serum patients with BD was comparable to that in HD despite its increases in patients with SLE [70].
3.2.3. HLA-E and KLRD1/KLRC1 (CD94/NKG2A)
HLA-E is a nonclassical MHC class 1 molecule that loads nonapeptides processed from other classical HLA class 1 molecules. Heterodimers of KLRD1 with KLRC1 recognize HLA-E and inhibit cytolysis [71,72]. No single polymorphism of HLA-E was associated with BD; however, specific combinations of SNPs demonstrate susceptibility. Individuals with HLA-E*0101 show a decreased risk while individuals without HLA-E*0101, NKG2A c.-4258*G/*G, and c.338-90*G show an increased risk of developing BD [73,74].
Nonapeptides presented by HLA-E were classified into 10 groups based on sequence, and their effect on genetic association was investigated. The frequency of N2: VMAPRTLVL and N7: VTAPRTVLL was found to be higher in patients with BD than in controls, indicating that these nonapeptides confer inaccurate presentation and BD risk [75].
3.3. Expression of NK Receptors
In the past decade, there has been increasing research on the expression of cell surface markers. Genome editing has become available for knock-out experiments using human cells. For instance, knockout of CD56 does not affect cytotoxicity against cell lines [76]; however, loss of KLRC1 improves cytolytic function against myeloma and solid tumors [77,78].
The interaction of ligands with receptors is a trigger for an immediate response. KIR3DL1 is an inhibitory NK receptor. NKB1 antibody detects variant surface expression of KIR3DL1 among patients with BD [79]. Conversely, KLRK1 (NKG2D), NCR1 (NKp46), NCR2 (NKp44), and NCR3 (NKp30) activate NK receptors and induce cytolysis. No significant differences were detected in cell surface expression in NK cells (CD16+CD56dim) obtained from patients with BD and HCs [80]; however, increases in KLRK1 expression were detected in NK cells (CD3−CD56+, CD3−CD16+) obtained from patients with BD [34,42] (Table 2). An approach using genome editing to eliminate or improve the recognition of KLRK1 was proposed [81]. Exploring the role of NK receptors with variance in surface expression through knockout experiments might provide a link to the production of substantial IFN-γ in relapse.
3.4. Other Genetic Factors
ERAP1 trims precursor peptides in ER to the correct length for binding to HLA class 1 molecules [82]. GWAS demonstrated association at ERAP1 rs17482078 (R725Q) in patients with uveitis from a Turkish population [83]. ERAP1 rs17482078 fails to trim peptides due to decreased stability and enzymatic activity, suggesting that unprocessed longer peptides interfere with the binding of HLA class 1 molecules and KIR.
IL-23 maintains Th17 cells and induces the production of IL-17 in patients with uveitis [84]. The feedback loop between NETs and Th17 cells underlies the activation of neutrophils and infiltration [17]. Moreover, the expression of IL-12β2 mRNA in remission is lower than that in relapse. The shift to NK2 decreases IFN-γ production in NK cells [85]. However, IL23R/IL12Rβ2 rs924080 is a polymorphism in the noncoding region [5]. It remains unclear how these receptors mediate ligands signaling in BD pathogenesis.
4. Regulation of NK Receptor Genes
4.1. Genomic Organization and Gene Expression of the NK Complex
NKG2 receptors are expressed on NK cells and possess killer cell lectin-like domains. Liganded with MICA or HLA-E, they regulate cytokine production and cytotoxic function. They are located on human chromosome 12 and consist of KLRD1, KLRK1, and KLRC1-4 (Figure 1A). KLRD1 is a common counterpart for KLRC1, KLRC2, and KLRC3 to form heterodimers, whereas KLRK1 forms homodimers. KLRC1 with the ITIM motif in its cytoplasmic region works as an inhibitory receptor, whereas KLRC2 pairs with TYROBP (DAP12) to activate receptors [86,87]. KLRC3 pairs with TYROBP; however, they are retained in the endoplasmic reticulum rather than being expressed on the cell surface [88]. KLRC4 does not pair with KLRD1 but can pair with TYROBP, suggesting that the cytoplasmic distribution of KLRC4 suppresses the activating signal by depriving KLRC2 and KLRC3 of TYROBP instead of forming extracellular structures that are stimulated by ligands [89].
The amino acid sequences of KLRC4 are conserved between humans and other primates but not between rodents [88,90,91] (Figure 1A). Tree analysis of amino acid sequence clearly separates rodents Klrc1-3 from primates KLRC1-4 (Figure 1B). Judging from the similarity of KLRC1-4 amino acids and the proximate location on the genome, errors during DNA replication and recombination with transposable elements would have produced paralogs, and accumulation of mutations provides a functional difference.
4.2. Splicing of KLRK1 (NKG2D) and Its Function
Mice Klrk1 has short (NKG2D-S) and long (NKG2D-L) isoforms. NKG2D-S is only expressed only in activated NK cells pairing with HCST (DAP10) and TYROBP (DAP12), whereas NKG2D-L is found in both resting and activating NK cells pairing with HCST [92,93]. TYROBP includes the ITAM motif in its cytoplasmic region and activates SYK and ZAP70 [94].
In humans, NK cells possess full-length of KLRK1 and it does not interact with TYROBP but HCST [95,96]. In contrast, TYROBP mediates KIR2DS2 stimulation and activates downstream tyrosine kinases, such as SYK and ZAP70 (Lanier LL et al., 1998 nature, Wu J et al., 2000). KLRK1 activates PI3K, GRB2, and VAV1 and initiates tyrosine-phosphorylation events [61,62]. Truncated KLRK1 (KLRK1TR) is a human isoform that does not form an extracellular structure unlike mouse NKG2D-S. Splicing failure leaves intron four that provides a stop codon, generating KLRK1TR. KLRK1TR is bound to HCST, thereby interfering with the pairing of full length of KLRK1 (KLRK1Full) and HCST [98]. Splicing isoforms of KLRK1 are not conserved among species.
4.3. KLRK1-KLRC4 Readthrough Gene
Human KLRK1 and KLRC4 are located next to each other on chromosome 12. According to the NCBI reference sequence, they transcribe NM_007360.4 and NM_013431.2, respectively. In addition, they also form a readthrough gene, KLRC4–KLRK1 (NM_001199805.1), which entirely covers both of KLRC4 and KLRK1. Readthrough transcription is an event where transcription machinery fails to find the termination site in the transcription process under stress [99]. Therefore, KLRC4–KLRK1 could encode a hybrid protein. Alternatively, it is possible for KLRC4–KLRK1 can produce several variants using different promoters and start codons. However, only a translation product (NP_001186734.1), which is equivalent to KLRK1 translation (NP_031386.2), has been registered in the data base (Figure 2). KLRC4 protein is detectable using antibody because it has a shorter C-type lectin-like domain than KLRC1, KLRC2 and KLRC3. Nonetheless, it will be hard to identify each isoform using antibodies that recognize the structure unique to KLRC4 if KLRC4–KLRK1 uses multiple start codons and produce multiple isoforms. CAGE-seq analysis that covers isoforms more efficiently than conventional RNA-seq analysis will be useful to explore the transcription of KLRK1-KLRC4 thoroughly. Moreover, identification of transcription initiation sites could help predict translation.
Transcripts of these genes along with their translation are depicted.
4.4. Dysregulation of KLRC4 in BD
KLRC4 rs2617170 (N104S) was identified in patients with BD as a nonsynonymous variants (NSV) [83,100]. KLRC4 is less likely to form an extracellular structure [89]; however, it remains unclear how the lack of fucosylation affects the partnering of KLRC4 N104S with KLRD1. Gene Chip data obtained from the peripheral blood mononuclear cells (PBMCs) of patients with BD patients (mucocutaneous manifestations, ocular involvement, and large vein thrombosis) categorized genes into the following four groups: negative regulators of inflammation, neutrophil granule proteins, antigen processing and presentation proteins, and regulators of immune response. Regarding the role of KLRC4, it is a regulator of immune response [101]. Interestingly, the effect of polymorphisms on KLRC4 mRNA expression and cytokine production was analyzed, which revealed that the mRNA expression of KLRC4 rs2617170 from CC carriers was significantly higher than that in CT and TT individuals. Furthermore, PBMCs collected from patients with TT and stimulated with LPS secrete more IL-8 than PBMCs collected from CC and CT carriers [102].
4.5. Fucosylation of CD16a
FUT2 plays critical roles in the gut epithelium. FUT2 dysregulation impairs mucosal fucosylation. The weak barrier function increases gut inflammation. FUT2 variants are associated with inflammatory bowel disease (IBD) and Crohn’s disease [103]. In mice, chronic stress causes gut dysfunction and decreases of Fut2 expression and fucosylation on the intestine epithelium that tethers microbiota [104]. SNP array analysis found FUT2 variants rs601338 (W143X) and rs602662 (G258S) [105]. Fucose-deficient IgG1 bound more strongly to CD16a than fucosylated IgG1 and elevated antibody-dependent cell-mediated cytotoxicity (ADCC) [106].
5. Distinct NK Subsets
5.1. Circulating and Tissue Resident NK Cells
It has been believed that circulating and resident NK cells are fundamentally separated during embryonic stages fundamentally. CD16−CD56bright is a major population in tissue resident NK cells, whereas peripheral NK cells predominantly include CD16+CD56dim predominantly [107]. Nonetheless, the frequency of CD16−CD56bright NK cells in patients with BD is higher than that in HCs [31]. Moreover, NK cells in the intestines decrease effector function and exhibit adaptation in their environment [107]. Despite these relevant observations, there has been limited evidence showing that tissue-resident NK cells are activated in the relapse period.
The other possibility is that some peripheral NK cells in patients with BD acquire tissue residency, although this assumption has not been supported. CD4+ T cells infiltrate predominantly; however, NK cells are infrequent in patients with uveitis [108]. Nevertheless, several studies have recently referred to the plasticity of peripheral NK cells. First, circulating NK cells acquire tissue residency when encountering acute infection [109]. Second, murine cytomegalovirus infection recruits NK cells to the salivary gland and forms a long-lived, tissue-resident, memory-like population (NKRM cells) [110]. NK cells appear to possess the ability to change their fate through virus infection and adapt to the tissue environment.
5.2. NK Cell Memory
Immediate immune response without prior sensitization is the capability of NK cells, although they can be sensitized. Some studies using anti-CD16a/CD30 bispecific antibodies, cancer cells, LPS, and cytokines have demonstrated the presence of memory-like NK cells [111]. Both mice and human NK cells cultured with IL-12, IL-15, and IL-18 produce substantial IFN-γ and persist for long time [112,113]. Moreover, these cytokine-induced memory-like (CIML) NK cells acquire cytotoxicity against myeloid leukemia [114]. Despite decreased expression of CD16 in CIML NK cells, they maintain the capacity to trigger antibody dependent cell-mediated cytotoxicity (ADCC) [115].
The other type of NK cells is memory/adaptive NK cells that have been infected by HCMV [116]. Human HCMV infection expands KLRC2-positive cells with increased cytokine production. These cells produce higher levels of IFN-γ in recipients with HCMV infection than in recipients without HCMV infection. KLRC2-positive NK cells maintain memory when they are transplanted to the recipient with antigen [117]. Furthermore, NK cells with a previous HMCV infection are FcRγ deficient and have distinct memory features. These cells persist for long term and produce IFN-γ against either HCMV or HSV-1 [118]. Memory/adaptive NK cells lack the expression of FcRγ and SYK due to epigenetic regulation and expand in an antibody-dependent manner [118,119].
Nevertheless, there are some ambiguous evidences when discussing the potential involvement of NK cell memory in BD. Firstly, HCMV IgG levels in patients with BD are lower than those in HCs [120]. which does not ensure that patients with BD have undergone the infection and generate memory. Secondly, periods of relapse alternate with periods of remission from weeks to years. NK cells in relapse exhibit lower cytotoxicity than those in remission [36,37], consistent with the dominance of CD56bright in relapse. Substantial IFN-γ production is common in both peripheral NK cells in BD and memory-like NK cells; however, there are some differences in population. Although peripheral NK cells in patients with BD are enriched CD56bright NK cells, CIML NK cells include both of CD56bright and CD56dim, and memory/adaptive NK cells predominantly have CD56dim. Seeking local environment where cytokine cocktails develop CIML NK cells or tracking cells infected with HCMV for identifying memory/adaptive NK cells among peripheral blood is difficult. Therefore, further studies using scRNA-seq data on CIML NK cells or memory/adaptive NK cells as reference will be effective in identifying original cells with memory that can be hyperactivated by specific stimulation and developing therapy to target those cells.
6. Conclusions
BD is a chronic autoinflammatory disorder characterized by unpredictable relapse. Innate immune cells contribute to its pathogenesis. The numbers of peripheral NK cells are variant among patients with BD and the cytotoxicity of peripheral NK cells tends to be lower during relapse than during remission. NK1/NK2 subsets clearly depict cytokine production in relapse and remission. Moreover, the strongest association with HLA-B51 indicates that the interaction with NK receptors is critical to BD pathogenesis. Polymorphisms and unprocessed peptides impair the accuracy of recognition and cause the hyperactivation of NK cells.
NK cells exhibit immediate response, including cytokine production and cytolysis, without prior sensitization, and their life is short. However, recent studies have demonstrated that NK cells can able to acquire memory through exposure to cytokine cocktails or HCMV infection and produce IFN-γ against specific stimulation. Although neither CIML NK cells nor memory/adaptive NK cells have been identified from patients with BD, hyperactivation in relapse is similar to the response of these adaptive NK cells. Alternatively, activation of tissue-resident NK cells and migration of peripheral NK cells to mucosal tissues are anticipated episodes. Moreover, transcriptional regulation of activating receptors, such as KLRK1 and KLRC4 could easily modify the responses to ligands and contribute to hyperactivation. Further studies focusing on adaptive NK cells among skewed innate NK cells in BD will clarify the mechanism underlying chronic and recurrent manifestations.
Author Contributions
Y.O. conceived and wrote the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- Moghoofei, M; Pajavand, H; Shahbazi, R; Rezaei, M; Taki, E. The Role of Viral and Bacterial Infections in the Etiology of Behçet’s Disease. J Clin Lab Anal. 2026, 40, e70133. [Google Scholar] [CrossRef] [PubMed]
- Isogai, E; Ohno, S; Kotake, S; Isogai, H; Tsurumizu, T; Fujii, N; Yokota, K; Syuto, B; Yamaguchi, M; Matsuda, H; et al. Chemiluminescence of neutrophils from patients with Behcet’s disease and its correlation with an increased proportion of uncommon serotypes of Streptococcus sanguis in the oral flora. Arch Oral Biol. 1990, 35, 43–48. [Google Scholar] [CrossRef]
- Ohno, S; Ohguchi, M; Hirose, S; Matsuda, H; Wakisaka, A; Aizawa, M. Close association of HLA-Bw51 with Behcet’s disease. Arch Ophthalmol. 1982, 100, 1455–1458. [Google Scholar] [CrossRef]
- Mizuki, N; Ohno, S; Tanaka, H; Sugimura, K; Seki, T; Mizuki, N; Kera, J; Inaba, G; Tsuji, K; Inoko, H. Association of HLA-B51 and lack of association of class II alleles with Behcet’s disease. Tissue Antigens. 1992, 40, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Remmers, EF; Cosan, F; Kirino, Y; Ombrello, MJ; Abaci, N; Satorius, C; Le, JM; Yang, B; Korman, BD; Cakiris, A; Aglar, O; Emrence, Z; Azakli, H; Ustek, D; Tugal-Tutkun, I; Akman-Demir, G; Chen, W; Amos, CI; Dizon, MB; Kose, AA; Azizlerli, G; Erer, B; Brand, OJ; Kaklamani, VG; Kaklamanis, P; Ben-Chetrit, E; Stanford, M; Fortune, F; Ghabra, M; Ollier, WE; Cho, YH; Bang, D; O’Shea, J; Wallace, GR; Gadina, M; Kastner, DL; Gül, A. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet’s disease. Nat Genet. 2010, 42, 698–702. [Google Scholar] [CrossRef]
- Hughes, T; Coit, P; Adler, A; Yilmaz, V; Aksu, K; Düzgün, N; Keser, G; Cefle, A; Yazici, A; Ergen, A; Alpsoy, E; Salvarani, C; Casali, B; Kötter, I; Gutierrez-Achury, J; Wijmenga, C; Direskeneli, H; Saruhan-Direskeneli, G; Sawalha, AH. Identification of multiple independent susceptibility loci in the HLA region in Behçet’s disease. Nat Genet. 2013, 45, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Ombrello, MJ; Kirino, Y; de Bakker, PI; Gül, A; Kastner, DL; Remmers, EF. Behçet disease-associated MHC class I residues implicate antigen binding and regulation of cell-mediated cytotoxicity. Proc Natl Acad Sci U S A. 2014, 111, 8867–8872. [Google Scholar] [CrossRef]
- McGonagle, D; Aydin, SZ; Gül, A; Mahr, A; Direskeneli, H. ‘MHC-I-opathy’-unified concept for spondyloarthritis and Behcet disease. Nat Rev Rheumatol. 2015, 11, 731–740. [Google Scholar] [CrossRef]
- Ucar-Comlekoglu, D; Fox, A; Sen, HN. Gender Differences in Behçet’s Disease Associated Uveitis. J Ophthalmol. 2014, 2014, 820710. [Google Scholar] [CrossRef] [PubMed]
- Jo, YG; Ortiz-Fernández, L; Coit, P; Yilmaz, V; Yentür, SP; Alibaz-Oner, F; Aksu, K; Erken, E; Düzgün, N; Keser, G; Cefle, A; Yazici, A; Ergen, A; Alpsoy, E; Salvarani, C; Kısacık, B; Kötter, I; Henes, J; Çınar, M; Schaefer, A; Nohutcu, RM; Takeuchi, F; Harihara, S; Kaburaki, T; Messedi, M; Song, YW; Kaşifoğlu, T; Martin, J; González Escribano, MF; Saruhan-Direskeneli, G; Direskeneli, H; Sawalha, AH. Sex-specific analysis in Behcet’s disease reveals higher genetic risk in male patients. J Autoimmun. 2022, 132, 102882. [Google Scholar] [CrossRef]
- Liu, A; Su, Y; Zhu, J; Li, YY. Disease association study of Autoimmune and autoinflammatory diseases by integrating multi-modal data and hierarchical ontologies. Front Immunol. 2025, 16, 1575490. [Google Scholar] [CrossRef]
- Davatchi, F; Assaad-Khalil, S; Calamia, KT; Crook, JE; Sadeghi-Abdollahi, B; Schirmer, M; Tzellos, T; Zouboulis, CC; Akhlagi, M; Al-Dalaan, A; Alekberova, ZS; Ali, AA; Altenburg, A; Arromdee, E; Baltaci, M; Bastos, M; Benamour, S; Ben Ghorbel, I; Boyvat, A; Carvalho, L; Chen, W; Ben-Chetrit, E; Chams-Davatchi, C; Correia, JA; Crespo, J; Dias, C; Dong, Y; Paix~ao-Duarte, F; Elmuntaser, K; Elonakov, AV; Gra~na Gil, J; Haghdoost, AA; Hayani, RM; Houman, H; Isayeva, AR; Jamshidi, AR; Kaklamanis, P; Kumar, A; Kyrgidis, A; Madanat, W; Nadji, A; Namba, K; Ohno, S; Olivieri, I; Vaz Patto, J; Pipitone, N; de Queiroz, MV; Ramos, F; Resende, C; Rosa, CM; Salvarani, C; Serra, MJ; Shahram, F; Shams, H; Sharquie, KE; Sliti-Khanfir, M; Tribolet de Abreu, T; Vasconcelos, C; Vedes, J; Wechsler, B; Cheng, YK; Zhang, Z; Ziaei, N. The International Criteria for Behcet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014, 28, 338–347. [Google Scholar]
- Zhan, H; Cheng, L; Chen, H; Liu, Y; Feng, X; Li, H; Li, Z; Li, Y. Evaluation of inflammatory-thrombosis panel as a diagnostic tool for vascular Behcet’s disease. Clin Rheumatol. 2025, 44, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Sahin, S; Akoğlu, T; Direskeneli, H; Sen, LS; Lawrence, R. Neutrophil adhesion to endothelial cells and factors affecting adhesion in patients with Behçet’s disease. Ann Rheum Dis. 1996, 55, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Ergun, T; Gürbüz, O; Harvell, J; Jorizzo, J; White, W. The histopathology of pathergy: a chronologic study of skin hyperreactivity in Behçet’s disease. Int J Dermatol 1998, 37, 929–933. [Google Scholar] [CrossRef]
- Le Joncour, A; Martos, R; Loyau, S; Lelay, N; Dossier, A; Cazes, A; Fouret, P; Domont, F; Papo, T; Jandrot-Perrus, M; Bouton, MC; Cacoub, P; Ajzenberg, N; Saadoun, D; Boulaftali, Y. Critical role of neutrophil extracellular traps (NETs) in patients with Behcet’s disease. Ann Rheum Dis. 2019, 78, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y; Ning, K; Huang, Z; Chen, B; Chen, J; Wen, Y; Bu, J; Hong, H; Chen, Q; Zhang, Z; Jia, R; Su, W. NETs-CD44-IL-17A Feedback Loop Drives Th17-Mediated Inflammation in Behcet’s Uveitis. Adv Sci (Weinh). 2025, 12, e2411524. [Google Scholar] [CrossRef]
- Hirahara, L; Takase-Minegishi, K; Kirino, Y; Iizuka-Iribe, Y; Soejima, Y; Yoshimi, R; Nakajima, H. The Roles of Monocytes and Macrophages in Behçet’s Disease With Focus on M1 and M2 Polarization. Front Immunol. 2022, 13, 852297. [Google Scholar] [CrossRef]
- Nakano, H; Kirino, Y; Takeno, M; Higashitani, K; Nagai, H; Yoshimi, R; Yamaguchi, Y; Kato, I; Aoki, I; Nakajima, H. GWAS-identified CCR1 and IL10 loci contribute to M1 macrophage-predominant inflammation in Behcet’s disease. Arthritis Res Ther. 2018, 20, 124. [Google Scholar] [CrossRef]
- Suzuki, Y; Hoshi, K; Matsuda, T; Mizushima, Y. Increased peripheral blood gamma delta+ T cells and natural killer cells in Behçet’s disease. J Rheumatol. 1992, 19, 588–592. [Google Scholar]
- Hasan, A; Fortune, F; Wilson, A; Warr, K; Shinnick, T; Mizushima, Y; van der Zee, R; Stanford, MR; Sanderson, J; Lehner, T. Role of gamma delta T cells in pathogenesis and diagnosis of Behcet’s disease. Lancet. 1996, 347, 789–794. [Google Scholar] [CrossRef]
- Raziuddin, S; al-Dalaan, A; Bahabri, S; Siraj, AK; al-Sedairy, S. Divergent cytokine production profile in Behçet’s disease. Altered Th1/Th2 cell cytokine pattern. J Rheumatol. 1998, 25, 329–333. [Google Scholar]
- Hasan, MS; Bergmeier, LA; Petrushkin, H; Fortune, F. Gamma Delta (γδ) T Cells and Their Involvement in Behçet’s Disease. J Immunol Res. 2015, 2015, 705831. [Google Scholar] [CrossRef]
- Poli, A; Michel, T; Thérésine, M; Andrès, E; Hentges, F; Zimmer, J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology. 2009, 126, 458–465. [Google Scholar] [CrossRef]
- Reefman, E; Kay, JG; Wood, SM; Offenhäuser, C; Brown, DL; Roy, S; Stanley, AC; Low, PC; Manderson, AP; Stow, JL. Cytokine secretion is distinct from secretion of cytotoxic granules in NK cells. J Immunol. 2010, 184, 4852–4862. [Google Scholar] [CrossRef]
- Anfossi, N; André, P; Guia, S; Falk, CS; Roetynck, S; Stewart, CA; Breso, V; Frassati, C; Reviron, D; Middleton, D; Romagné, F; Ugolini, S; Vivier, E. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006, 25, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, WM; Kim, S. Licensing of natural killer cells by self-major histocompatibility complex class I. Immunol Rev. 2006, 214, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Kärre, K. Natural killer cell recognition of missing self. Nat Immunol. 2008, 9, 477–480. [Google Scholar] [CrossRef]
- Fukuda, M. Lysosomal membrane glycoproteins. Structure, biosynthesis, and intracellular trafficking. J Biol Chem. 1991, 266, 21327–21330. [Google Scholar] [CrossRef] [PubMed]
- Alter, G; Malenfant, JM; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004, 294, 15–22. [Google Scholar] [CrossRef]
- Sakly, K; Lahmar, R; Nefzi, F; Hammami, S; Harzallah, O; Sakly, N; Sakly, W; Hassine, M; Mahjoub, S; Ghedira, I; Feki, S. Phenotypic abnormalities of peripheral blood mononuclear cells in patients with Behçet’s disease and association with HLA-B51 expression. Immunol Invest. 2014, 43, 463–478. [Google Scholar] [CrossRef]
- Hamzaoui, K; Berraies, A; Kaabachi, W; Ammar, J; Hamzaoui, A. Pulmonary manifestations in Behcet disease: impaired natural killer cells activity. Multidiscip Respir Med. 2013, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Saruhan-Direskeneli, G; Uyar, FA; Cefle, A; Onder, SC; Eksioglu-Demiralp, E; Kamali, S; Inanç, M; Ocal, L; Gül, A. Expression of KIR and C-type lectin receptors in Behcet’s disease. Rheumatology (Oxford). 2004, 43, 423–427. [Google Scholar] [CrossRef]
- Bonacini, M; Soriano, A; Zerbini, A; Calò, E; Cimino, L; Muratore, F; Fontana, L; Braglia, L; Parmeggiani, M; Salvarani, C; Croci, S. Higher Frequencies of Lymphocytes Expressing the Natural Killer Group 2D Receptor in Patients With Behcet Disease. Front Immunol. 2018, 9, 2157. [Google Scholar] [CrossRef]
- Cosan, F; Aktas Cetin, E; Akdeniz, N; Emrence, Z; Cefle, A; Deniz, G. Natural Killer Cell Subsets and Their Functional Activity in Behcet’s Disease. Immunol Invest. 2017, 46, 419–432. [Google Scholar] [CrossRef]
- Kaneko, F; Takahashi, Y; Muramatsu, R; Adachi, K; Miura, Y; Nakane, A; Minagawa, T. Natural killer cell numbers and function in peripheral lymphoid cells in Behcet’s disease. Br J Dermatol. 1985, 113, 313–318. [Google Scholar] [CrossRef]
- Onder, M; Bozkurt, M; Gürer, MA; Gülekon, A; Sezgin, P; Imir, T. Natural cellular cytotoxicity in Behcet’s disease. J Dermatol. 1994, 21, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Hasan, MS; Ryan, PL; Bergmeier, LA; Fortune, F. Circulating NK cells and their subsets in Behçet’s disease. Clin Exp Immunol. 2017, 188, 311–322. [Google Scholar] [CrossRef]
- Deniz, G; Akdis, M; Aktas, E; Blaser, K; Akdis, CA. Human NK1 and NK2 subsets determined by purification of IFN-gamma-secreting and IFN-gamma-nonsecreting NK cells. Eur J Immunol. 2002, 32, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Kucuksezer, UC; Aktas-Cetin, E; Bilgic-Gazioglu, S; Tugal-Tutkun, I; Gül, A; Deniz, G. Natural killer cells dominate a Th-1 polarized response in Behcet’s disease patients with uveitis. Clin Exp Rheumatol. 2015, 33, S24–S29. [Google Scholar]
- Rebuffet, L; Melsen, JE; Escalière, B; Basurto-Lozada, D; Bhandoola, A; Björkström, NK; Bryceson, YT; Castriconi, R; Cichocki, F; Colonna, M; Davis, DM; Diefenbach, A; Ding, Y; Haniffa, M; Horowitz, A; Lanier, LL; Malmberg, KJ; Miller, JS; Moretta, L; Narni-Mancinelli, E; O’Neill, LAJ; Romagnani, C; Ryan, DG; Sivori, S; Sun, D; Vagne, C; Vivier, E. High-dimensional single-cell analysis of human natural killer cell heterogeneity. Nat Immunol. 2024, 25, 1474–1488. [Google Scholar] [CrossRef]
- Sallakci, N; Tahrali, I; Kucuksezer, UC; Cetin, EA; Gul, A; Deniz, G. Effect of different cytokines in combination with IL-15 on the expression of activating receptors in NK cells of patients with Behcet’s disease. Immunol Res. 2022, 70, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Montes-Cano, MA; Conde-Jaldón, M; García-Lozano, JR; Ortiz-Fernández, L; Ortego-Centeno, N; Castillo-Palma, MJ; Espinosa, G; Graña-Gil, G; González-Gay, MA; Barnosi-Marín, AC; Solans, R; Fanlo, P; Camps, T; Castañeda, S; Sánchez-Bursón, J; Núñez-Roldán, A; Martín, J; González-Escribano, MF. HLA and non-HLA genes in Behçet’s disease: a multicentric study in the Spanish population. Arthritis Res Ther. 2013, 15, R145. [Google Scholar] [CrossRef] [PubMed]
- Litwin, V; Gumperz, J; Parham, P; Phillips, JH; Lanier, LL. NKB1: a natural killer cell receptor involved in the recognition of polymorphic HLA-B molecules. J Exp Med. 1994, 180, 537–543. [Google Scholar] [CrossRef]
- Gumperz, JE; Litwin, V; Phillips, JH; Lanier, LL; Parham, P. The Bw4 public epitope of HLA-B molecules confers reactivity with natural killer cell clones that express NKB1, a putative HLA receptor. J Exp Med. 1995, 181, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Sanjanwala, B; Draghi, M; Norman, PJ; Guethlein, LA; Parham, P. Polymorphic sites away from the Bw4 epitope that affect interaction of Bw4+ HLA-B with KIR3DL1. J Immunol. 2008, 181, 6293–6300. [Google Scholar] [CrossRef]
- Erer, B; Takeuchi, M; Ustek, D; Tugal-Tutkun, I; Seyahi, E; Özyazgan, Y; Duymaz-Tozkir, J; Gül, A; Kastner, DL; Remmers, EF; Ombrello, MJ. Evaluation of KIR3DL1/KIR3DS1 polymorphism in Behçet’s disease. Genes Immun. 2016, 17, 396–399. [Google Scholar] [CrossRef]
- Pando, MJ; Gardiner, CM; Gleimer, M; McQueen, KL; Parham, P. The protein made from a common allele of KIR3DL1 (3DL1*004) is poorly expressed at cell surfaces due to substitution at positions 86 in Ig domain 0 and 182 in Ig domain 1. J Immunol. 2003, 171, 6640–6649. [Google Scholar] [CrossRef]
- Castaño-Núñez, Á; Montes-Cano, MA; García-Lozano, JR; Ortego-Centeno, N; García-Hernández, FJ; Espinosa, G; Graña-Gil, G; Sánchez-Bursón, J; Juliá, MR; Solans, R; Blanco, R; Barnosi-Marín, AC; Gómez de la Torre, R; Fanlo, P; Rodríguez-Carballeira, M; Rodríguez-Rodríguez, L; Camps, T; Castañeda, S; Alegre-Sancho, JJ; Martín, J; González-Escribano, MF. Association of Functional Polymorphisms of KIR3DL1/DS1 With Behçet’s Disease. Front Immunol. 2019, 10, 2755. [Google Scholar] [CrossRef]
- Gebreselassie, D; Spiegel, H; Vukmanovic, S. Sampling of major histocompatibility complex class I-associated peptidome suggests relatively looser global association of HLA-B*5101 with peptides. Hum Immunol. 2006, 67, 894–906. [Google Scholar] [CrossRef]
- Kollnberger, S; Bird, L; Sun, MY; Retiere, C; Braud, VM; McMichael, A; Bowness, P. Cell-surface expression and immune receptor recognition of HLA-B27 homodimers. Arthritis Rheum. 2002, 46, 2972–2982. [Google Scholar] [CrossRef]
- Wong-Baeza, I; Ridley, A; Shaw, J; Hatano, H; Rysnik, O; McHugh, K; Piper, C; Brackenbridge, S; Fernandes, R; Chan, A; Bowness, P; Kollnberger, S. KIR3DL2 binds to HLA-B27 dimers and free H chains more strongly than other HLA class I and promotes the expansion of T cells in ankylosing spondylitis. J Immunol. 2013, 190, 3216–3224. [Google Scholar] [CrossRef]
- Faure, M; Long, EO. KIR2DL4 (CD158d), an NK cell-activating receptor with inhibitory potential. J Immunol. 2002, 168, 6208–6214. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi-Maki, A; Catina, TL; Campbell, KS. Cutting edge: KIR2DL4 transduces signals into human NK cells through association with the Fc receptor gamma protein. J Immunol. 2005, 174, 3859–3863. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S; Fu, J; Long, EO. Cutting edge: induction of IFN-gamma production but not cytotoxicity by the killer cell Ig-like receptor KIR2DL4 (CD158d) in resting NK cells. J Immunol. 2001, 167, 1877–1881. [Google Scholar] [CrossRef]
- Rajagopalan, S; Long, EO. A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J Exp Med. 1999, 189, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S; Long, EO. KIR2DL4 (CD158d): An activation receptor for HLA-G. Front Immunol. 2012, 3, 258. [Google Scholar] [CrossRef]
- Kim, SJ; Lee, S; Park, C; Seo, JS; Kim, JI; Yu, HG. Targeted resequencing of candidate genes reveals novel variants associated with severe Behçet’s uveitis. Exp Mol Med. 2013, 45, e49. [Google Scholar] [CrossRef]
- Steinle, A; Li, P; Morris, DL; Groh, V; Lanier, LL; Strong, RK; Spies, T. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics. 2001, 53, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S; Groh, V; Wu, J; Steinle, A; Phillips, JH; Lanier, LL; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Billadeau, DD; Upshaw, JL; Schoon, RA; Dick, CJ; Leibson, PJ. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, JL; Arneson, LN; Schoon, RA; Dick, CJ; Billadeau, DD; Leibson, PJ. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat Immunol. 2006, 7, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Saá, I; Cambra, A; Pallarés, L; Espinosa, G; Juan, A; Pujalte, F; Matamoros, N; Milà, J; Julià, MR. Allelic diversity and affinity variants of MICA are imbalanced in Spanish patients with Behçet’s disease. Scand J Immunol. 2006, 64, 77–82. [Google Scholar] [CrossRef]
- Durmanová, V; Tirpakova, J; Stuchlikova, M; Shawkatova, I; Kuba, D; Sapak, M; Buc, M. Characterization of MICA gene polymorphism of HLA complex in the Slovak population. Ann Hum Biol. 2011, 38, 570–576. [Google Scholar] [CrossRef]
- Hughes, EH; Collins, RW; Kondeatis, E; Wallace, GR; Graham, EM; Vaughan, RW; Stanford, MR. Associations of major histocompatibility complex class I chain-related molecule polymorphisms with Behcet’s disease in Caucasian patients. Tissue Antigens. 2005, 66, 195–199. [Google Scholar] [CrossRef]
- Muñoz-Saá, I; Cambra, A; Pallarés, L; Espinosa, G; Juan, A; Pujalte, F; Matamoros, N; Milà, J; Julià, MR. Allelic diversity and affinity variants of MICA are imbalanced in Spanish patients with Behçet’s disease. Scand J Immunol. 2006, 64, 77–82. [Google Scholar] [CrossRef]
- Mizuki, N; Meguro, A; Tohnai, I; Gül, A; Ohno, S; Mizuki, N. Association of Major Histocompatibility Complex Class I Chain-Related Gene A and HLA-B Alleles with Behçet’s Disease in Turkey. Jpn J Ophthalmol. 2007, 51, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Yabuki, K; Mizuki, N; Ota, M; Katsuyama, Y; Palimeris, G; Stavropoulos, C; Koumantaki, Y; Spyropoulou, M; Giziaki, E; Kaklamani, V; Kaklamani, E; Inoko, H; Ohno, S. Association of MICA gene and HLA-B*5101 with Behçet’s disease in Greece. Invest Ophthalmol Vis Sci. 1999, 40, 1921–1926. [Google Scholar]
- Marin, ML; Savioli, CR; Yamamoto, JH; Kalil, J; Goldberg, AC. MICA polymorphism in a sample of the São Paulo population, Brazil. Eur J Immunogenet. 2004, 31, 63–71. [Google Scholar] [CrossRef]
- Hervier, B; Ribon, M; Tarantino, N; Mussard, J; Breckler, M; Vieillard, V; Amoura, Z; Steinle, A; Klein, R; Kötter, I; Decker, P. Increased Concentrations of Circulating Soluble MHC Class I-Related Chain A (sMICA) and sMICB and Modulation of Plasma Membrane MICA Expression: Potential Mechanisms and Correlation With Natural Killer Cell Activity in Systemic Lupus Erythematosus. Front Immunol. 2021, 12, 633658. [Google Scholar] [CrossRef]
- Lee, N; Llano, M; Carretero, M; Ishitani, A; Navarro, F; López-Botet, M; Geraghty, DE. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A. 1998, 95, 5199–5204. [Google Scholar] [CrossRef]
- Borrego, F; Ulbrecht, M; Weiss, EH; Coligan, JE; Brooks, AG. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med. 1998, 187, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Seo, J; Park, JS; Nam, JH; Bang, D; Sohn, S; Lee, ES; Park, KS. Association of CD94/NKG2A, CD94/NKG2C, and its ligand HLA-E polymorphisms with Behcet’s disease. Tissue Antigens. 2007, 70, 307–313. [Google Scholar] [CrossRef]
- Park, KS; Park, JS; Nam, JH; Bang, D; Sohn, S; Lee, ES. HLA-E*0101 and HLA-G*010101 reduce the risk of Behcet’s disease. Tissue Antigens. 2007, 69, 139–144. [Google Scholar] [CrossRef]
- Castaño-Núñez, ÁL; Montes-Cano, MA; García-Lozano, JR; Ortego-Centeno, N; García-Hernández, FJ; Espinosa, G; Graña-Gil, G; Sánchez-Bursón, J; Juliá, MR; Solans, R; Blanco, R; Barnosi-Marín, AC; Gómez de la Torre, R; Fanlo, P; Rodríguez-Carballeira, M; Rodríguez-Rodríguez, L; Camps, T; Castañeda, S; Alegre-Sancho, JJ; Martín, J; González-Escribano, MF. The complex HLA-E-nonapeptide in Behçet disease. Front Immunol. 2023, 14, 1080047. [Google Scholar] [CrossRef]
- Picard, LK; Claus, M; Fasbender, F; Watzl, C. Human NK cells responses are enhanced by CD56 engagement. Eur J Immunol. 2022, 52, 1441–1451. [Google Scholar] [CrossRef]
- Bexte, T; Alzubi, J; Reindl, LM; Wendel, P; Schubert, R; Salzmann-Manrique, E; von Metzler, I; Cathomen, T; Ullrich, E. CRISPR-Cas9 based gene editing of the immune checkpoint NKG2A enhances NK cell mediated cytotoxicity against multiple myeloma. Oncoimmunology 2022, 11, 2081415. [Google Scholar] [CrossRef] [PubMed]
- Mac Donald, A; Guipouy, D; Lemieux, W; Harvey, M; Bordeleau, LJ; Guay, D; Roméro, H; Li, Y; Dion, R; Béland, K; Haddad, E. KLRC1 knockout overcomes HLA-E-mediated inhibition and improves NK cell antitumor activity against solid tumors. Front Immunol. 2023, 14, 1231916. [Google Scholar] [CrossRef]
- Takeno, M; Shimoyama, Y; Kashiwakura, J; Nagafuchi, H; Sakane, T; Suzuki, N. Abnormal killer inhibitory receptor expression on natural killer cells in patients with Behcet’s disease. Rheumatol Int. 2004, 24, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Gelmez, MY; Cinar, S; Cetin, EA; Ozcit-Gürel, G; Babuna-Kobaner, G; Erdugan, M; Gul, A; Akdag-Kose, A; Deniz, G. Inflammatory status might direct ILC and NK cells to IL-17 expressing ILC3 and NK subsets in Behcet’s disease. Immunol Lett. 2021, 235, 1–8. [Google Scholar] [CrossRef]
- Alves, E; McLeish, E; Blancafort, P; Coudert, JD; Gaudieri, S. Manipulating the NKG2D Receptor-Ligand Axis Using CRISPR: Novel Technologies for Improved Host Immunity. Front Immunol. 2021, 12, 712722. [Google Scholar] [CrossRef]
- Saric, T; Chang, SC; Hattori, A; York, IA; Markant, S; Rock, KL; Tsujimoto, M; Goldberg, AL. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol. 2002, 3, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Kirino, Y; Bertsias, G; Ishigatsubo, Y; Mizuki, N; Tugal-Tutkun, I; Seyahi, E; Ozyazgan, Y; Sacli, FS; Erer, B; Inoko, H; Emrence, Z; Cakar, A; Abaci, N; Ustek, D; Satorius, C; Ueda, A; Takeno, M; Kim, Y; Wood, GM; Ombrello, MJ; Meguro, A; Gül, A; Remmers, EF; Kastner, DL. Genome-wide association analysis identifies new susceptibility loci for Behçet’s disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. 2013, 45, 202–207. [Google Scholar] [CrossRef]
- Chi, W; Zhu, X; Yang, P; Liu, X; Lin, X; Zhou, H; Huang, X; Kijlstra, A. Upregulated IL-23 and IL-17 in Behcet patients with active uveitis. Invest Ophthalmol Vis Sci. 2008, 49, 3058–3064. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y; Takahashi, H; Satoh, T; Okazaki, Y; Mizuki, N; Takahashi, K; Ikezawa, Z; Kuwana, M. Natural killer cells control a T-helper 1 response in patients with Behcet’s disease. Arthritis Res Ther. 2010, 12, R80. [Google Scholar] [CrossRef]
- Le Dréan, E; Vély, F; Olcese, L; Cambiaggi, A; Guia, S; Krystal, G; Gervois, N; Moretta, A; Jotereau, F; Vivier, E. Inhibition of antigen-induced T cell response and antibody-induced NK cell cytotoxicity by NKG2A: association of NKG2A with SHP-1 and SHP-2 protein-tyrosine phosphatases. Eur J Immunol. 1998, 28, 264–276. [Google Scholar] [CrossRef]
- Lanier, LL; Corliss, B; Wu, J; Phillips, JH. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity. 1998, 8, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Orbelyan, GA; Tang, F; Sally, B; Solus, J; Meresse, B; Ciszewski, C; Grenier, JC; Barreiro, LB; Lanier, LL; Jabri, B. Human NKG2E is expressed and forms an intracytoplasmic complex with CD94 and DAP12. J Immunol. 2014, 193, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Kim, DK; Kabat, J; Borrego, F; Sanni, TB; You, CH; Coligan, JE. Human NKG2F is expressed and can associate with DAP12. Mol Immunol. 2004, 41, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Shum, BP; Flodin, LR; Muir, DG; Rajalingam, R; Khakoo, SI; Cleland, S; Guethlein, LA; Uhrberg, M; Parham, P. Conservation and variation in human and common chimpanzee CD94 and NKG2 genes. J Immunol. 2002, 168, 240–252. [Google Scholar] [CrossRef]
- Averdam, A; Kuhl, H; Sontag, M; Becker, T; Hughes, AL; Reinhardt, R; Walter, L. Genomics and diversity of the common marmoset monkey NK complex. J Immunol. 2007, 178, 7151–7161. [Google Scholar] [CrossRef]
- Gilfillan, S; Ho, EL; Cella, M; Yokoyama, WM; Colonna, M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat Immunol. 2002, 3, 1150–1155. [Google Scholar] [CrossRef]
- Diefenbach, A; Tomasello, E; Lucas, M; Jamieson, AM; Hsia, JK; Vivier, E; Raulet, DH. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol. 2002, 3, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Zompi, S; Hamerman, JA; Ogasawara, K; Schweighoffer, E; Tybulewicz, VL; Di Santo, JP; Lanier, LL; Colucci, F. NKG2D triggers cytotoxicity in mouse NK cells lacking DAP12 or Syk family kinases. Nat Immunol. 2003, 4, 565–572. [Google Scholar] [CrossRef]
- Wu, J; Cherwinski, H; Spies, T; Phillips, JH; Lanier, LL. DAP10 and DAP12 form distinct, but functionally cooperative, receptor complexes in natural killer cells. J Exp Med. 2000, 192, 1059–1068. [Google Scholar] [CrossRef]
- André, P; Castriconi, R; Espéli, M; Anfossi, N; Juarez, T; Hue, S; Conway, H; Romagné, F; Dondero, A; Nanni, M; Caillat-Zucman, S; Raulet, DH; Bottino, C; Vivier, E; Moretta, A; Paul, P. Comparative analysis of human NK cell activation induced by NKG2D and natural cytotoxicity receptors. Eur J Immunol. 2004, 34, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Lanier, LL; Corliss, BC; Wu, J; Leong, C; Phillips, JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998, 391, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Karimi, MA; Aguilar, O; Zou, B; Bachmann, MH; Carlyle, JR; Baldwin, CL; Kambayashi, T. A truncated human NKG2D splice isoform negatively regulates NKG2D-mediated function. J Immunol. 2014, 193, 2764–2771. [Google Scholar] [CrossRef]
- Caldas, P; Luz, M; Baseggio, S; Andrade, R; Sobral, D; Grosso, AR. Transcription readthrough is prevalent in healthy human tissues and associated with inherent genomic features. Commun Biol. 2024, 7, 100. [Google Scholar] [CrossRef]
- Padula, MC; Leccese, P; Lascaro, N; Padula, AA; Carbone, T; Martelli, G; D’Angelo, S. A First Step for the Molecular Characterization of Neurological Involvement of Behcet Syndrome: an Italian Pivotal Study. J Mol Neurosci. 2021, 71, 1284–1289. [Google Scholar] [CrossRef]
- Oğuz, AK; Yılmaz, ST; Oygür, ÇŞ; Çandar, T; Sayın, I; Kılıçoğlu, SS; Ergün, İ; Ateş, A; Özdağ, H; Akar, N. Behçet’s: A Disease or a Syndrome? Answer from an Expression Profiling Study. PLoS One. 2016, 11, e0149052. [Google Scholar] [CrossRef]
- Yang, Y; Tan, H; Deng, B; Yu, H; Su, G; Hu, J; Cao, Q; Yuan, G; Kijlstra, A; Yang, P. Genetic polymorphisms of C-type lectin receptors in Behcet’s disease in a Chinese Han population. Sci Rep. 2017, 7, 5348. [Google Scholar] [CrossRef] [PubMed]
- Parmar, AS; Alakulppi, N; Paavola-Sakki, P; Kurppa, K; Halme, L; Färkkilä, M; Turunen, U; Lappalainen, M; Kontula, K; Kaukinen, K; Mäki, M; Lindfors, K; Partanen, J; Sistonen, P; Mättö, J; Wacklin, P; Saavalainen, P; Einarsdottir, E. Association study of FUT2 (rs601338) with celiac disease and inflammatory bowel disease in the Finnish population. Tissue Antigens 2012, 80, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Omata, Y; Aoki, R; Aoki-Yoshida, A; Hiemori, K; Toyoda, A; Tateno, H; Suzuki, C; Takayama, Y. Reduced fucosylation in the distal intestinal epithelium of mice subjected to chronic social defeat stress. Sci Rep. 2018, 8, 13199. [Google Scholar] [CrossRef] [PubMed]
- Xavier, JM; Shahram, F; Sousa, I; Davatchi, F; Matos, M; Abdollahi, BS; Sobral, J; Nadji, A; Oliveira, M; Ghaderibarim, F; Shafiee, NM; Oliveira, SA. FUT2: filling the gap between genes and environment in Behçet’s disease? Ann Rheum Dis. 2015, 74, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Shields, RL; Lai, J; Keck, R; O’Connell, LY; Hong, K; Meng, YG; Weikert, SH; Presta, LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef]
- Dogra, P; Rancan, C; Ma, W; Toth, M; Senda, T; Carpenter, DJ; Kubota, M; Matsumoto, R; Thapa, P; Szabo, PA; Li Poon, MM; Li, J; Arakawa-Hoyt, J; Shen, Y; Fong, L; Lanier, LL; Farber, DL. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell. 2020, 180, 749–763.e13. [Google Scholar] [CrossRef]
- Charteris, DG; Barton, K; McCartney, AC; Lightman, SL. CD4+ lymphocyte involvement in ocular Behcet’s disease. Autoimmunity. 1992, 12, 201–206. [Google Scholar] [CrossRef]
- Torcellan, T; Friedrich, C; Doucet-Ladevèze, R; Ossner, T; Solé, VV; Riedmann, S; Ugur, M; Imdahl, F; Rosshart, SP; Arnold, SJ; Gomez de Agüero, M; Gagliani, N; Flavell, RA; Backes, S; Kastenmüller, W; Gasteiger, G. Circulating NK cells establish tissue residency upon acute infection of skin and mediate accelerated effector responses to secondary infection. Immunity 2024, 57, 124–140.e7. [Google Scholar] [CrossRef]
- Schuster, IS; Sng, XYX; Lau, CM; Powell, DR; Weizman, OE; Fleming, P; Neate, GEG; Voigt, V; Sheppard, S; Maraskovsky, AI; Daly, S; Koyama, M; Hill, GR; Turner, SJ; O’Sullivan, TE; Sun, JC; Andoniou, CE; Degli-Esposti, MA. Infection induces tissue-resident memory NK cells that safeguard tissue health. Immunity. 2023, 56, 531–546.e6. [Google Scholar] [CrossRef]
- Terrén, I; Orrantia, A; Astarloa-Pando, G; Amarilla-Irusta, A; Zenarruzabeitia, O; Borrego, F. Cytokine-Induced Memory-Like NK Cells: From the Basics to Clinical Applications. Front Immunol. 2022, 13, 884648. [Google Scholar] [CrossRef]
- Cooper, MA; Elliott, JM; Keyel, PA; Yang, L; Carrero, JA; Yokoyama, WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A. 2009, 106, 1915–1919. [Google Scholar] [CrossRef]
- Romee, R; Schneider, SE; Leong, JW; Chase, JM; Keppel, CR; Sullivan, RP; Cooper, MA; Fehniger, TA. Cytokine activation induces human memory-like NK cells. Blood 2012, 120, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Romee, R; Rosario, M; Berrien-Elliott, MM; Wagner, JA; Jewell, BA; Schappe, T; Leong, JW; Abdel-Latif, S; Schneider, SE; Willey, S; Neal, CC; Yu, L; Oh, ST; Lee, YS; Mulder, A; Claas, F; Cooper, MA; Fehniger, TA. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [PubMed]
- Ewen, EM; Pahl, JHW; Miller, M; Watzl, C; Cerwenka, A. KIR downregulation by IL-12/15/18 unleashes human NK cells from KIR/HLA-I inhibition and enhances killing of tumor cells. Eur J Immunol. 2018, 48, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, S; Sun, JC. Virus-specific NK cell memory. J Exp Med. 2021, 218, e20201731. [Google Scholar] [CrossRef] [PubMed]
- Foley, B; Cooley, S; Verneris, MR; Curtsinger, J; Luo, X; Waller, EK; Anasetti, C; Weisdorf, D; Miller, JS. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol. 2012, 189, 5082–5088. [Google Scholar] [CrossRef]
- Zhang, T; Scott, JM; Hwang, I; Kim, S. Cutting edge: antibody-dependent memory-like NK cells distinguished by FcRγ deficiency. J Immunol. 2013, 190, 1402–1406. [Google Scholar] [CrossRef]
- Lee, J; Zhang, T; Hwang, I; Kim, A; Nitschke, L; Kim, M; Scott, JM; Kamimura, Y; Lanier, LL; Kim, S. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity. 2015, 42, 431–442. [Google Scholar] [CrossRef]
- Lee, EB; Kwon, YJ; Shin, KC; Song, YW; Park, CG; Hwang, ES; Cha, CY. Decreased serum level of antibody against human cytomegalovirus in patients with Behçet’s disease. Rheumatol Int. 2005, 25, 33–36. [Google Scholar] [CrossRef]
Figure 1.
NKC and phylogenetic relationship between primate and rodent NKC genes. (A) Genomic organization of humans and mice is shown. (B) Phylogenetic relationship of amino acid sequences from humans, chimpanzees, mice, and rats is analyzed. Sequences were obtained from NCBI.
Figure 1.
NKC and phylogenetic relationship between primate and rodent NKC genes. (A) Genomic organization of humans and mice is shown. (B) Phylogenetic relationship of amino acid sequences from humans, chimpanzees, mice, and rats is analyzed. Sequences were obtained from NCBI.

Figure 2.
Gene structure of human KLRK1, KLRC4, and KLRC4-KLRK1.


Table 1.
NK subsets in BD pathogenesis.

Table 2.
The expression of marker genes.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



