Submitted:
01 March 2026
Posted:
03 March 2026
You are already at the latest version
Abstract
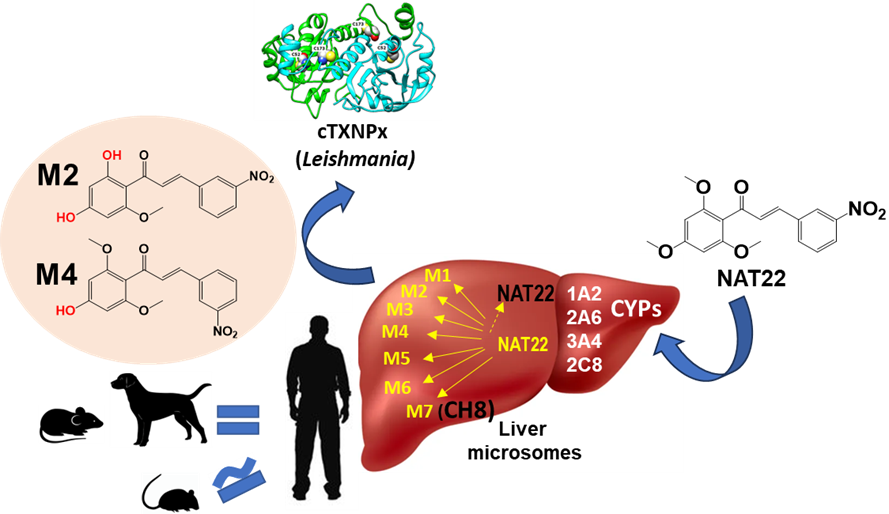
Background/Objectives: Human and canine leishmaniasis are neglected diseases with limited therapeutic options. The nitrochalcone NAT22, a high-affinity inhibitor of the essential parasite enzyme tryparedoxin peroxidase (cTXNPx), has emerged as a promising antileishmanial candidate. Interestingly, NAT22 demonstrated superior efficacy when administered orally rather than intralesionally, suggesting metabolism-driven enhancement of activity. Since in vivo studies with chalcones have been conducted exclusively in mice, this study aimed to evaluate whether mice are suitable models for oral chalcone therapies for human and canine leishmaniasis and to identify metabolites with potential antileishmanial activity. Methods: NAT22 hepatic metabolism was investigated using in silico prediction and in vitro liver microsomal assays from rats, mice, humans, and dogs. Metabolites were identified by LC-MS/MS and NMR, and docking studies were performed against cTXNPx. Results: In silico analysis predicted metabolism mainly by CYP1A2, CYP2A6, CYP2C8, and CYP3A4. Seven metabolites (M1–M7) were identified by LC-MS/MS and NMR in all species except mice, whose microsomes did not generate M6. Structural analyses indicated preservation of the α,β-enone system and nitro-substituted B ring in all metabolites. Docking studies showed that metabolites M2 and M4 displayed stronger predicted binding energies than NAT22. Conclusions: NAT22 undergoes hepatic phase I metabolism generating two metabolites with enhanced predicted interaction with cTXNPx. The similarity between human and canine metabolic profiles supports the translational relevance of oral NAT22 therapy in leishmaniasis, while metabolites M2 and M4 emerge as candidates for validation in local treatment of cutaneous leishmaniasis.
Keywords:
chalcone
; liver microsomes
; CYP
; cTXNPx
; leishmaniasis
1. Introduction
Chalcones are naturally occurring compounds characterized by the 1,3-diphenyl-2-propen-1-one scaffold and exhibit a broad spectrum of biological activities, including antimalarial, antibacterial, antiviral, and anti-inflammatory effects [1,2]. Among these properties, their antileishmanial potential has been extensively investigated through in silico, in vitro, and in vivo studies, highlighting chalcones as promising scaffolds for drug development against leishmaniases [3,4,5,6,7,8,9,10,11,12].
Leishmaniases are vector-borne neglected tropical diseases (NTDs) that manifest in a wide range of clinical forms, from cutaneous leishmaniasis—the most common and disfiguring presentation—to visceral leishmaniasis, which is frequently fatal in humans and dogs [13]. Current treatments remain suboptimal. Conventional therapies are prolonged and administered parenterally, often causing significant systemic toxicity. The only oral drug available, miltefosine, is limited by its teratogenic potential [14]. For cutaneous leishmaniasis, intralesional meglumine antimoniate reduces systemic toxicity but requires repeated administrations due to rapid systemic absorption associated with its high hydrophilicity, occasionally leading to systemic effects [15]. Therefore, the development of safe and effective drugs suitable for both oral and local administration remains an urgent unmet need.
Previously, we demonstrated the therapeutic efficacy of the natural chalcone 2’,6’-dihydroxy-4’-methoxychalcone (DMC, Figure 1A) [4] and its synthetic analogue 3-nitro-2’-hydroxy-4’,6’-dimethoxychalcone (CH8, Figure 1B) [5] in murine cutaneous leishmaniasis using intralesional administration to minimize systemic toxicity. More recently, the analogue NAT22, which retains the antileishmanial activity of CH8 while offering a more accessible synthetic route, has become the focus of further investigation (Figure 1C).
NAT22 antileishmanial mechanism involves inhibition of tryparedoxin peroxidase (cTXNPx), an essential parasite detoxification enzyme [16]. The local therapeutic performance of chalcones has been further enhanced through the use of microparticulate controlled-release implants, enabling single-dose intralesional injection with improved efficacy compared to the free compound [10,17,18,19]. Interestingly, NAT22 demonstrated superior efficacy when administered orally rather than intralesionally, suggesting that hepatic metabolism may generate metabolites with enhanced antileishmanial activity relative to the parent compound.
Preclinical evaluation of orally administered antileishmanial agents is traditionally conducted in mice due to their ability to reproduce key features of human leishmaniasis [20]. Mice are also widely employed in drug metabolism studies because their liver microsomal cytochrome P450 (CYP) enzyme profiles exhibit substantial homology with those of humans, supporting their predictive value in metabolic investigations. In contrast, rats present a more distinct metabolic profile compared to dogs and humans [21]. Within the liver, CYP450 enzymes account for approximately 75% of phase I drug metabolism reactions [22]. However, metabolic pathways may vary considerably among species, and extrapolation of oral drug data from experimental models to target hosts must therefore be undertaken cautiously [23].
Given that in vivo metabolism studies are complex, costly, and raise ethical concerns related to the three Rs principles [24], liver microsomes provide a cost-effective and ethically preferable platform for preclinical metabolic assessment of therapeutic candidates, including alkaloids [25], lignans [26], and flavonoids [27], more specifically chalcones [28]. Previous microsomal studies of oxygenated chalcones have demonstrated that unsubstituted or mono-hydroxylated aromatic rings are susceptible to CYP-mediated hydroxylation, while methoxy substituents frequently undergo oxidative dealkylation [28,29,30]. However, the metabolic behavior of nitrochalcones remains unexplored.
In this study, we compare the hepatic metabolic profile of NAT22 using liver microsomes from four relevant mammalian species. This approach aims to evaluate the suitability of mice as a translational model for oral chalcone studies and to assess the metabolic plausibility of oral NAT22 therapy in humans and dogs. Additionally, identification of metabolites with enhanced predicted activity may contribute to the rational design of improved candidates for single-dose local treatment using appropriate delivery systems.
2. Materials and Methods
2.1. Materials
The chalcones CH8 and NAT22 were synthesized to 97% and 98% purity, respectively, as previously described [5,10]. HPLC grade acetonitrile and methyl tert-butyl ether (MTBE) were purchased from Tedia® (Fairfield, OH, USA). Potassium phosphate monobasic, chloride acid, Tris (hydroxymethyl)aminomethane was obtained from Vetec (Brazil). Potassium hydroxide and formic acid were from Sigma-Aldrich (Saint Louis, USA). Human (pooled from 50 donors), dog, rat and mouse liver microsomes (20 mg of protein/mL) were purchased from Thermo Fisher Scientific/ Gibco (USA). Glucose-6-phosphate sodium salt and nicotinamide adenine dinucleotide phosphate sodium (NADP) were from Across OrganicTM (New Jersey, USA). Glucose-6-phosphate dehydrogenase was obtained from MP Biomedicals (USA).
2.2. In Silico Prediction
ADMET PredictorTM (version 9.5, Simulations Plus, Lancaster, CA, USA) was used to predict physicochemical and ADMET properties of NAT22 and its metabolites.
2.3. Liver Microsomal Assay
NAT22 (2.5 µg in 2.5 µL acetonitrile) was pre-incubated with 10 mg liver microsomal protein in 10 mL phosphate-buffered saline (PBS) at 37 °C in a Dubnoff NT232 metabolic bath. The reaction was initiated by adding 2.5 mL of a cofactor solution containing NADP⁺ (0.25 mM), glucose-6-phosphate (5 mM), and glucose-6-phosphate dehydrogenase (0.5 U/mL) prepared in Tris-HCl buffer (pH 7.4) during 90 min. Controls were NAT22 incubated in the absence of either cofactors or microsomal proteins. The reaction was terminated by adding 40 mL of methyl tert-butyl ether. Samples were centrifuged at 3500 rpm for 15 min at 4 °C, and 30 mL of supernatants were evaporated under nitrogen. The dried extracts were subjected to LC-MS/MS and NMR analyses, as described below.
2.4. LC-MS/MS Analysis
Dried microsomal extracts were reconstituted in acetonitrile:water (3:2, v/v) to a final concentration of 0.1 µg/µL, and 4 µL were injected into a reversed-phase LC-MS/MS system equipped with a Thermo Hypersil GOLD C18 column (50 × 2.1 mm, 1.9 µm particle size). Mobile phase A consisted of water (HPLC grade) containing 0.1% formic acid and 5 mM ammonium formate, and mobile phase B consisted of acetonitrile containing 0.1% formic acid. The analytes were eluted using a gradient starting with 15% mobile phase B that increased to 95% over 13 min and rebalanced to 15% (3 min). The flow rate was 0.35 mL/min and the column was kept at 40 ºC. Tandem mass spectra were acquired using positive ion electrospray with a Q Exactive Plus Orbitrap mass spectrometer (Thermo Scientific). Argon was used as the collision gas at a collision energy of 30 eV.
2.5. NMR Spectroscopy
The Nuclear Magnetic Resonance analyses were conducted on 600 and 800 MHz Bruker spectrometer at 25 °C at the National Center of Nuclear Magnetic Resonance Jiri Jonas (UFRJ, Brazil). The obtained dry microsomal extracts and standard chalcones (NAT22 and CH8) were suspended, in 180 and 600 µL, respectively, of chloroform-d6. 1H NMR spectra were acquired using 3 mm and 5 mm TXI probes, with average of 1024 scans. Topspin (Bruker Biospin, Rheinstetten, Germany) was used for data acquisition, and the MestRENova version 6.0 was used for processing the NMR spectra.
2.6. Protein Modeling and Molecular Docking
Since crystal structure of cTXNPx protein from L. amazonensis is not available in the Protein Data Bank (PDB), homology modeling was employed to construct a model of cTXNPx. Through BLAST program [31], we identified a homologous protein template specifically, the crystallographic structure of the cTXNPx protein from L. major (Uniprot: ID code: Q4VKK8) [32]. The three-dimensional model of cTXNPx was constructed using MODELLER 10.2. Molecular docking of all chalcones into the binding site was performed using AutoDock Vina 1.2.0. The structural validation of the model was carried out using the PROCHECK program. All three-dimensional images of cTXNPx, as well as chalcone-protein complexes were generated using UCSF Chimera 1.16 program. The 2D diagrams representing the molecular interactions were generated using BIOVIA Discovery Studio Visualizer 21.1.0.20298 program.
3. Results
3.1. In Silico Prediction of Hepatic Metabolites of NAT22
To guide and complement the in vitro metabolism studies, potential phase I metabolites of NAT22 arising from first-pass hepatic metabolism were predicted using ADMET Predictor. The cytochrome P450 (CYP) isoforms evaluated included CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4. The predictions indicated that NAT22 is primarily metabolized by CYP1A2 and CYP2A6, generating 3-nitro-3’-hydroxy-2’,4’,6’-trimethoxychalcone and 3-nitro-2’-hydroxy-4’,6’-dimethoxychalcone (CH8). Additionally, CYP1A2, 2A6, 2C8, and 3A4 were predicted to produce 3-nitro-4’-hydroxy-2’,6’-dimethoxychalcone (Figure 2).
3.2. Identification of NAT22 Metabolites by LC-MS/MS and NMR
NAT22 was incubated with human, dog, rat and mouse liver microsomes and the resulting extracts were analyzed by LC-MS/MS. The total ion chromatogram and computer-reconstructed selected ion chromatograms are shown in Figure 3.
Seven metabolites (M1-M7) in addition to the parent NAT22 were detected. All seven compounds were formed in human, dog and rat microsomes, whereas six (M1–M5 and M7) were detected in mouse microsomes. Exact masses and molecular formulas are summarized in Table A1.
The untreated NAT22 exhibited Z and E isomers with m/z 344 ([M+H]⁺), eluting at 7.95 and 8.26 min, respectively. The CH8 standard (m/z 330, [M+H]⁺) showed Z and E isomers at 7.49 and 9.47 min. All detected metabolites predominantly corresponded to the E isomer. MS2 fragmentation of NAT22 generated characteristic ions at m/z 195 and 176, whereas CH8 produced ions at m/z 181 and 176. Similar acylium ion fragmentation patterns were observed for all metabolites, indicating preservation of the B ring and the α,β-enone system during metabolism (Figure 4).
Proposed fragmentation pathways the seven metabolites (M1-M7) are represented in Figure 4, whereas their structures are in Figure 7.
Metabolites M1, M3 and M6 (m/z 346, [M+H]⁺) generated major fragment ions at m/z 197 and 182, consistent with mono-demethylation and hydroxylation on ring A. These modifications are compatible with demethylation at ortho or para methoxy groups and hydroxylation at the unsubstituted meta position. Metabolite M2 (m/z 316, [M+H]⁺; tR = 6.08 min) showed a principal fragment at m/z 167, consistent with di-demethylation (28 Da mass loss).
Metabolites M4 and M7 (m/z 330, [M+H]⁺) yielded fragments at m/z 181 and 176, indicating mono-demethylation. M7 was confirmed as CH8 by comparison with the authentic standard. By exclusion, M4 was assigned as 3-nitro-4’-hydroxy-2’,6’-dimethoxychalcone. The shorter retention time observed for the ortho-demethylated metabolite (M7) relative to M4 is consistent with intramolecular hydrogen bonding between the ortho-hydroxyl group and the carbonyl, affecting chromatographic behavior. Based on retention time and fragmentation data, M6 (tR = 7.82 min) was inferred to involve ortho demethylation combined with hydroxylation.
Metabolite M5 (m/z 360, [M+H]⁺) produced fragment ions at m/z 211 and 196, indicating hydroxylation at the meta position.
3.3. NMR Analysis
To confirm LC-MS/MS-based structural assignments, 1H NMR analyses were performed on NAT22 and CH8, blank samples, control incubations (without microsomes), and NAT22 incubated with human liver microsomes (37 °C, 90 min).
Compared with the control, the metabolized sample showed decreased signal intensity corresponding to NAT22 (Figure 5A–G) and the appearance of new resonances. In the 3.7–3.9 ppm region, four singlets (3.76, 3.84, 3.85, and 3.87 ppm) were detected, corresponding to methoxy groups in the metabolites, alongside residual NAT22 signals (3.78 and 3.86 ppm) (Figure 5A and Figure 6A). Another singlet, not seen in the control could be observed at 6.17 ppm (Figure 5B). Integration of signals at 3.87 ppm (s, 6H) and a corresponding 2H singlet supported assignment to M4, the only metabolite retaining symmetry in the A ring (Figure 6A). In the aromatic region (7.06–8.35 ppm) (Figure 5C–E), signal overlap with microsomal background limited detailed interpretation. However, minimal changes in B-ring proton signals were observed, consistent with preferential oxidation occurring on ring A.
Downfield signals at 13.14, 13.85, and 14.10 ppm (Figure 5G) were consistent with strongly hydrogen-bonded phenolic OH groups, supporting the presence of ortho-hydroxylated metabolites (M3, M6, and M7), in agreement with LC-MS/MS data.
Figure 7.
The seven metabolites that were generated following incubation of NAT22 with human, dog, rat and mouse liver microsomes. M6 was not produced by mice. Blue circles indicate the three metabolites that had been predicted in silico with ADMET Predictor software.
Figure 7.
The seven metabolites that were generated following incubation of NAT22 with human, dog, rat and mouse liver microsomes. M6 was not produced by mice. Blue circles indicate the three metabolites that had been predicted in silico with ADMET Predictor software.

3.4. Molecular Docking of NAT22 Metabolites
To evaluate their potential interaction with the parasite target, all metabolites were docked into a structural model of cTXNPx from Leishmania amazonensis. The model was based on the crystallographic structure of cTXNPx from L. major [33], with 88.4% sequence identity, and preservation of the catalytically essential Cys52 and Cys173 residues. The active site is located within a narrow cavity formed at the dimer interface, at the end of the helix that accommodates Cys-52 of one monomeric unit of the dimer and Cys-173 of the other unit [16,34].
All metabolites were predicted to interact with the active site (Figure 8). Metabolites M2 (-6.045 kcal/mol) and M4 (-5.970 kcal/mol) showed higher predicted affinity than NAT22 (-5.964 kcal/mol) and the other metabolites (Table A2). All chalcones formed a hydrogen bond between the nitro group and Gln289, as well as π-alkyl interactions between the B-ring aromatic system and Pro188. π-σ interactions with Thr248 were observed only for NAT22 and M1. Methoxy groups at the ortho position contributed to alkyl interactions with Pro186, Pro188, and Pro249, whereas para-methoxy groups interacted with Leu245. Notably, demethylation at ortho (M7), para (M4), or both positions (M2) altered the interaction profile toward increased van der Waals contributions, correlating with improved predicted binding energies.
4. Discussion
The nitrochalcone NAT22 has previously demonstrated potent antileishmanial activity in both in vitro and in vivo models [10,16,34]. However, most preclinical studies with chalcones, including those conducted in leishmaniasis, have relied on murine models. Given known interspecies differences in drug-metabolizing enzymes, extrapolation of metabolic and pharmacological data from mice to humans may not always be straightforward.
To address this limitation, we investigated the hepatic phase I metabolism of NAT22 using liver microsomes from different mammalian species. Our objectives were to characterize its metabolic profile, identify potentially active metabolites, and validate mice as suitable experimental model for therapeutic studies with chalcones. Phase I metabolism was prioritized because approximately 70-80% of drug metabolism involves oxidative, reductive, and hydrolytic reactions mediated by cytochrome P450 (CYP) enzyme family. Additionally, species-specific microsomes can serve as ethical substitutes for human volunteers and live animals, and provide reliable comparisons of metabolic profiles across species [35,36]. Overall, the metabolic profiles obtained in dog and rat microsomes were similar to those observed in human microsomes, since only mouse microsomes did not generate the metabolite M6. This finding suggests that murine metabolism of NAT22 may differ from the human profile to a greater extent than anticipated [21]. These interspecies differences highlight the need for caution when translating oral efficacy data obtained in mice to humans and dogs.
A good correlation was observed between in silico predictions and in vitro findings. Although only three metabolites (M7 = CH8, M5, and M4) were predicted using ADMET Predictor (Figure 2), seven phase I metabolites (M1–M7) were experimentally identified by LC-MS/MS and supported by NMR analysis. This discrepancy underscores the importance of complementing computational approaches with experimental validation. Among the seven metabolites identified, five are newly described, whereas two have been previously reported: CH8 (M7), which has documented antileishmanial activity similar to NAT22 [10], and M2, which has been associated with anticancer activity [37]. Importantly, the α,β-enone system and the nitro-substituted B ring were preserved in all metabolites, suggesting maintenance of structural features considered relevant for biological activity.
Docking studies indicated that all metabolites retained the ability to interact with the catalytic cavity of cTXNPx. Metabolites M2 and M4 showed slightly more favorable predicted binding energies compared with NAT22. Although the differences in calculated affinity were modest and require experimental confirmation, these findings suggest that phase I metabolism does not impair predicted target engagement and may subtly modulate binding interactions. The previously reported biological activity of CH8 [10], further supports the hypothesis that certain metabolites could contribute to the overall antileishmanial effect of NAT22. Nevertheless, given the pleiotropic nature of chalcones, additional molecular targets cannot be excluded.
Although further phase II metabolism in vivo is likely, the identification of phase I metabolites with preserved—or slightly enhanced—predicted affinity for cTXNPx is noteworthy.
5. Conclusions
This study provides the first cross-species comparative characterization of chalcone hepatic metabolism and demonstrates that phase I biotransformation of NAT22 generates metabolites with preserved or even enhanced predicted interaction with the validated parasite target cTXNPx. The identification of M2 and M4 as metabolites with stronger predicted binding affinity highlights metabolism as a potential contributor to the overall antileishmanial activity of NAT22 and supports their chemical synthesis for biological validation.
The strong concordance between human and canine metabolic profiles establishes a robust translational framework and reinforces the relevance of these species for the development of orally administered chalcone-based therapies. These findings support continued investigation of NAT22 as an oral therapeutic candidate while also encouraging complementary evaluation of localized delivery strategies for cutaneous leishmaniasis.
Conversely, the distinct metabolic profile observed in mice reveals meaningful interspecies differences and indicates that murine models may not reliably predict hepatic metabolism relevant to oral chalcone therapy. Importantly, this limitation may not be restricted to leishmaniasis but could extend to other disease contexts involving chalcone-based drug candidates, underscoring the need for comparative metabolism studies to strengthen translational drug development strategies.
Author Contributions
Conceptualization, MFM, TB, PGS and BR-B; X.X. and Y.Y.; methodology, BR-B, OAS-F, PGS and APCV; software, AMTS, BAAV,VMA and OAS-F; validation, ARRB, OAS-F X.X., Y.Y. and Z.Z.; formal analysis, APCV, investigation, ARRB, resources, BR-B, PGS and APCV; data curation, ARRB; writing—original draft preparation, ARRB, writing—review and editing, BR-B, OAS-F, PGS; visualization, MFM and TB; supervision, BR-B and PGS; project administration, BR-B, funding acquisition, PGS and BR-B.
Funding
This research was funded by FAPERJ grant number E-26/010.000982/2019, the Royal Society grant number IC160044, and The APC was funded by CNPq grant number 315438/2021-5.
Institutional Review Board Statement
The animal study protocol was approved by the Federal University of Rio de Janeiro Institutional Animal Care and Use Committee with protocol number 030/17.
Informed Consent Statement
Not applicable.
Data Availability Statement
The authors declare that all the data supporting the findings of this study are available within the paper and its Supplemental Data.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
- NAT22 — 3-nitro-2’-hydroxy-4’,6’-dimethoxychalcone
- cTXNPx — Tryparedoxin peroxidase
- CH8 — 3-nitro-2’-hydroxy-4’,6’-dimethoxychalcone
- DMC — 2’,6’-dihydroxy-4’-methoxychalcone
- CYP — Cytochrome P450
- LC-MS/MS — Liquid Chromatography coupled to Tandem Mass Spectrometry
- NMR — Nuclear Magnetic Resonance
- PBS — Phosphate-Buffered Saline
- MTBE — Methyl tert-butyl ether
- HPLC — High Performance Liquid Chromatography
- NADP⁺ — Nicotinamide Adenine Dinucleotide Phosphate
- Tris-HCl — Tris(hydroxymethyl)aminomethane hydrochloride buffer
- ADMET — Absorption, Distribution, Metabolism, Excretion, and Toxicity
- BLAST — Basic Local Alignment Search Tool
- PDB — Protein Data Bank
- TXI probe — Probe type for NMR spectroscopy (1H NMR)
Appendix A

Table A1.
Exact masses of NAT22 and its metabolites generated by microsomes from different mammals were obtained after LC-ESI-MS/MS.
Table A1.
Exact masses of NAT22 and its metabolites generated by microsomes from different mammals were obtained after LC-ESI-MS/MS.
| Analyte | Accurate mass (m/z) |
Human | Dog | Rat | Mouse | Molecular Formula | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Exact mass (m/z) |
Error (ppm) |
Exact mass (m/z) |
Error (ppm) |
Exact mass (m/z) |
Error (ppm) |
Exact mass (m/z) |
Error (ppm) |
|||
| NAT22 (Z) | 344,11286 | 344,11246 | 1,16 | 344,11209 | 2,24 | 344,11209 | 2,24 | 344,11215 | 2,06 | C18H17NO6 |
| NAT22 (E) | 344,11230 | 1,63 | 344,11206 | 2,32 | 344,11191 | 2,76 | 344,11218 | 1,98 | ||
| M1 (E) | 346,09213 | 346,09186 | 2,92 | 346,09149 | 1,85 | 346,09186 | 0,78 | 346,09171 | 1,21 | C17H15NO7 |
| M2 (E) | 316,08156 | 316,08112 | 1,39 | 316,08099 | 1,80 | 316,08081 | 2,37 | 316,08145 | 0,35 | C16H13NO6 |
| M3 (E) | 346,09213 | 346,09189 | 0,69 | 346,09143 | 2,02 | 346,09140 | 2,11 | 346,09164 | 1,42 | C17H15NO7 |
| M4 (Z) | 330,09721 | 330,09702 | 0,58 | 330,09653 | 2,06 | 330,09631 | 2,73 | 330,09665 | 1,70 | C17H15NO6 |
| M4 (E) | 330,09680 | 1,24 | 330,09641 | 2,42 | 330,09637 | 2,54 | 330,09650 | 2,15 | ||
| M5 (E) | 360,10778 | 360,10745 | 0,92 | 360,10690 | 2,44 | 360,10687 | 2,53 | 360,10709 | 1,92 | C18H17NO7 |
| M6 (E) | 346,09213 | 346,09180 | 0,95 | 346,09140 | 2,11 | 346,09131 | 2,37 | - | - | C17H15NO7 |
| M7 (Z) | 330,09721 | 330,09702 | 0,58 | 330,09647 | 2,24 | 330,09647 | 2,24 | 330,09653 | 2,06 | C17H15NO6 |
| M7 (E) | 330,09708 | 0,39 | 330,09644 | 2,33 | 330,09659 | 1,88 | 330,09677 | 1,33 | ||
Table A2.
Docking energy (ordered by best interaction energy) and types of intermolecular interactions observed between the ligands and the cTXNPx enzyme.
Table A2.
Docking energy (ordered by best interaction energy) and types of intermolecular interactions observed between the ligands and the cTXNPx enzyme.
| Ligands | Docking energy (kcal/mol) | H-bond | Pi-sigma | Pi-alkyl | Alkyl | Van der Waals |
|---|---|---|---|---|---|---|
| M2 | -6,045 | Gln-289 | - | Pro-188 | - | Pro-186, Glu-187, Ser-191, Val-192, Phe-196, Phe-247, Leu-245, Pro-249, Lys-293, Gly-294 |
| M4 | -5,970 | Gln-289 | - | Pro-188 | Pro-249 | Pro-186, Glu-187, Ser-191, Val-192, Phe-196, Phe-247, Leu-245, Thr-248, Lys-293, Gly-294 |
| NAT22 | -5,964 | Gln-289 | Thr-248 | Pro-188 | Leu-245, Pro-249 | Pro-186, Glu-187, Ser-191, Val-192, Phe-196, Phe-247, Lys-293, Gly-294 |
| M7 | -5,927 | Gln-289 | - | Pro-188 | Leu-245 | Pro-186, Glu-187, Ser-191, Val-192, Phe-196, Phe-247, Thr-248, Lys-293, Pro-249, Gly-294 |
| M3 | -5,886 | Gln-289 | - | Pro-188 | Pro-188, Pro-249 | Glu-187, Ser-191, Val-192, Phe-196, Leu-245, Phe-247, Thr-248, Lys-293, Gly-294 |
| M1 | -5,851 | Gln-289 | Thr-248 | Pro-188 | - | Pro-186, Glu-187, Ser-191, Val-192, Phe-196, Phe-247, Pro-249, Lys-293, Gly-294 |
| M6 | -5,841 | Gln-289 | - | Pro-188 | Leu-245 | Pro-186, Glu-187, Ser-191, Val-192, Phe-196, Phe-247, Thr-248, Lys-293, Pro-249, Gly-294 |
| M5 | -5,831 | Gln-289 | - | Pro-188 | Pro-186, Leu-245, Pro-249 | Glu-187, Ser-191, Val-197, Phe-196, Phe-247, Thr-248, Lys-293, Gly-294 |
References
- Adhikari, S.; Nath, P.; Deb, V. K.; Das, N.; Banerjee, A.; Pathak, S.; Duttaroy, A. K. Pharmacological potential of natural chalcones: a recent studies and future perspective. Front Pharmacol. 2025, 16, 1570385. [Google Scholar] [CrossRef]
- Nematollahi, M. H.; Mehrabani, M.; Hozhabri, Y.; Mirtajaddini, M.; Iravani, S. Antiviral and antimicrobial applications of chalcones and their derivatives: from nature to greener synthesis. Heliyon 2023, 9, e20428. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Christensen, S. B.; Blom, J.; Lemmich, E.; Nadelmann, L.; Fich, K.; Theander, A.; Kharazmi, T. G. Licochalcone A, a novel antiparasitic agent with potent activity against human pathogenic protozoan species of Leishmania. Antimicrob Agents and Chemother. 1993, 37, 2550–2556. [Google Scholar] [CrossRef] [PubMed]
- Torres-Santos, E. C.; Moreira, D. L.; Kaplan, M. A. C.; Meirelles, M. N.; Rossi-Bergmann, B. Selective effect of 2',6'-dihydroxy-4'-methoxychalcone isolated from Piper aduncum on Leishmania amazonensis. Antimicrob Agents Chemother. 1999, 43, 1234–1241. [Google Scholar] [CrossRef]
- Boeck, P.; Falcão, C. A. B.; Leal, P. C.; Yunes, R. A.; Cechinel-Filho, V.; Torres-Santos, E. C.; Rossi-Bergmann, B. Synthesis of chalcone analogues with increased antileishmanial activity. Bioorg Med Chem. 2006, 14, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Andrighetti-Fröhner, C. R.; de Oliveira, K. N.; Gaspar-Silva, D.; Pacheco, L. K.; Joussef, A. C.; Steindel, M.; Simões, C. M. O.; de Souza, A. M. T.; Magalhães, U. O.; Afonso, I. F.; Rodrigues, C. R.; Nunes, R. J.; Castro, H. C. Synthesis, biological evaluation and SAR of sulfonamide 4-methoxychalcone derivatives with potential antileishmanial activity. Eur J Med Chem 2009, 44, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Aponte, J. C.; Castillo, D.; Estevez, Y.; Gonzalez, G.; Arevalo, J.; Hammond, G. B.; Sauvain, M. In vitro and in vivo anti-Leishmania activity of polysubstituted synthetic chalcones. ACS Med Chem Lett. 2010, 20, 100–103. [Google Scholar] [CrossRef]
- Gupta, S.; Shivahare, R.; Korthikunta, V.; Singh, R.; Gupta, S.; Tadigoppula, N. Synthesis and biological evaluation of chalcones as potential antileishmanial agents. Eur J Med Chem. 2014, 81, 359–366. [Google Scholar] [CrossRef] [PubMed]
- De Mello, M. V. P.; Abrahim-Vieira, B. D. A.; Domingos, T. F. S.; de Jesus, J. B.; de Sousa, A. C. C.; Rodrigues, C. R.; Souza, A. M. T. D. A comprehensive review of chalcone derivatives as antileishmanial agents. Eur J Med Chem 2018, 150, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Batista, A. J.; Arruda-Costa, N.; Escrivani, D. O.; Reynaud, F.; Steel, P. G.; Rossi-Bergmann, B. Single-dose treatment for cutaneous leishmaniasis with an easily synthesized chalcone entrapped in polymeric microparticles. Parasitol 2020, 147, 1032–1037. [Google Scholar] [CrossRef]
- Garcia, A. R.; Oliveira, D. M. P.; Jesus, J. B.; Souza, A. M. T.; Sodero, A. C. R.; Vermelho, A. B.; Leal, I. C. R.; Souza, R. O. M. A.; Miranda, L. S. M.; Pinheiro, A. S.; Rodrigues, I. A. Identification of chalcone derivatives as inhibitors of Leishmania infantum arginase and promising antileishmanial agents. Front Chem 2021, 8, 624678. [Google Scholar] [CrossRef] [PubMed]
- De Santiago-Silva, K. M.; Bortoleti, B. T. da S.; Oliveira, L. do N.; Maia, F. L. de A.; Castro, J. C.; Costa, I. C.; Lazarin, D. B.; Wardell, J. L.; Wardell, S. M. S. V.; Albuquerque, M. G.; Lima, C. H. da S.; Pavanelli, W. R.; Bispo, M. de L. F.; Gonçalves, R. S. B. Antileishmanial Activity of 4,8-Dimethoxynaphthalenyl chalcones on Leishmania amazonensis. Antibiotics 2022, 11, 1402. [Google Scholar] [CrossRef] [PubMed]
- Alvar, J.; Velez, I. D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, L. R.; Silva, S. N.; Saliba, M. F.; Carvalho, J. de P.; Cota, G. PloS One 2024, 19, e0315710.
- Esfandiarpour, I.; Farajzadeh, S.; Rahnama, Z.; Fathabadi, E. A.; Heshmatkhah, A. Adverse effects of intralesional meglumine antimoniate and its influence on clinical laboratory parameters in the treatment of cutaneous leishmaniasis. Int J Dermatol 2012, 51, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Escrivani, D. O.; Charlton, R. L.; Borsodi, M. P. G.; Zingali, R. B.; Souza, A. M. T.; Abrahim-Vieira, B.; Freitag-Pohl, S.; Pohl, E.; Denny, P. W.; Rossi-Bergmann, B.; Steel, P. G. Chalcones identify cTXNPx as a potential antileishmanial drug target. PLoS Negl Trop Dis 2021, 15, e0009951. [Google Scholar] [CrossRef]
- Sousa-Batista, A. J.; Escrivani-Oliveira, D.; Falcão, C. A. B.; Da S. Philipon, C. I. M.; Rossi-Bergmann, B. Broad spectrum and safety of oral treatment with a promising nitrosylated chalcone in murine leishmaniasis. Antimicrob Agents Chemother. 2018a, 62, e00792. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Batista, A. J.; Pacienza-Lima, W.; Arruda-Costa, N.; Falcão, C. A. B.; Ré, M. I.; Rossi-Bergmann, B. Depot subcutaneous injection with chalcone CH8-loaded poly (Lactic-Co-Glycolic Acid) microspheres as a single-dose treatment of cutaneous leishmaniasis. Antimicrob Agents Chemother 2018b, 62, e01822-17. [Google Scholar] [CrossRef]
- Sousa-Batista, A. J.; Arruda-Costa, N.; Rossi-Bergmann, B.; Ré, M. I. Improved drug loading via spray drying of a chalcone implant for local treatment of cutaneous leishmaniasis. Drug Dev Ind Pharm 2018c, 44, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Caridha, D.; Vesely, B.; van Bocxlaer, K.; Arana, B.; Mowbray, C. E.; Rafati, S.; Uliana, S.; Reguera, R.; Kreishman-Deitrick, M.; Sciotti, R.; Buffet, P.; Croft, S. L. Route map for the discovery and pre-clinical development of new drugs and treatments for cutaneous leishmaniasis. Int J Parasitol Drugs Drug Resist 2019, 11, 106–117. [Google Scholar] [CrossRef]
- Turpeinen, M.; Ghiciuc, C.; Opritoui, M.; Tursas, L.; Pelkonen, O.; Pasanen, M. Predictive value of animal models for human cytochrome P450 (CYP)-mediated metabolism: a comparative study in vitro. Xenobiotica 2007, 37, 1367–1377. [Google Scholar] [CrossRef]
- Guengerich, F. P.; Waterman, M. R.; Egli, M. Recent structural insights into cytochrome P450 function. Trends Pharmacol Sci 2016, 37, 625–640. [Google Scholar] [CrossRef]
- Esteves, F.; Rueff, J.; Karanendonk, M. The central role of cytochrome P450 in xenobiotic metabolism—A brief review on a fascinating enzyme family. J Xenobiot. 2021, 11, 94–114. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Roy, S.; Ghosh, D.; Nandi, S. K. Role of animal models in biomedical research: a review. Lab Anim Res 2022, 38, 18. [Google Scholar] [CrossRef] [PubMed]
- Marques, L. M. M.; Da Silva-Júnior, E. A.; Gouvea, D. R.; Vessecchi, R.; Pupo, M. T.; Lopes, N. P.; Kato, M. J.; De Oliveira, A. R. M. In vitro metabolism of the alkaloid piplartine by rat liver microsomes. J Pharm Biomed Anal 2014, 95, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.; Habenschus, M. D.; Moreira, F. L.; Ferreira, L. de S.; Lopes, N. P.; De Oliveira, A. R. M. In vitro metabolism of the lignan (-)-grandisin, an anticancer drug candidate, by human liver microsomes. Drug Test Anal 2015, 7, 780–786. [Google Scholar] [CrossRef]
- Nielsen, S. E.; Breinholt, V.; Justesen, U.; Cornett, C.; Dragsted, L. O. In vitro biotransformation of flavonoids by rat liver microsomes. Xenobiot 1998, 28, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Zenger, K.; Agnolet, S.; Schneider, B.; Kraus, B. Biotransformation of flavokawains A, B, and C, chalcones from Kava (Piper methysticum), by human liver microsomes. J Agric Food Chem 2015, 63, 6376–6385. [Google Scholar] [CrossRef]
- Guo, J.; Liu, D.; Nikolic, D.; Zhu, D.; Pezzuto, J. M.; van Breemen, R. B. In vitro metabolism of isoliquiritigenin by human liver microsomes. Drug Metab Dispos. 2008, 36, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Wang, P.; Duan, S.; Wan, X.; Xing, H.; Yang, J.; Zhang, X.; Yao, Z.; Yao, X. Potential determinants for metabolic fates and inhibitory effects of isobavachalcone involving in human cytochrome P450, UDP-glucuronosyltransferase enzymes, and efflux transporters. J Pharma Sci. 2021, 110, 2285–2294. [Google Scholar] [CrossRef]
- Altschul, S. F.; Gish, W.; Miller, W.; Myers, E. W.; Lipman, D. J. Basic local alignment search tool. J Mol Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 2021, 49, D480–D489. [Google Scholar]
- Brindisi, M.; Brogi, S.; Relitti, N.; Vallone, V.; Butini, S.; Gemma, S.; Novellino, E.; Colotti, G.; Angiulli, G.; Di Chiaro, F.; Fiorillo, A.; Ilari, A.; Campiani, G. Structure-based discovery of the first non-covalent inhibitors of Leishmania major tryparedoxin peroxidase by high throughput docking. Sci Rep. 2015, 5, 9705. [Google Scholar] [CrossRef]
- De Oliveira, N. S.; De Souza, L. G.; De Almeida, V. M.; Barreto, A. R. R.; Carvalho-Gondim, F.; Schaeffer, E.; Santos-Filho, O. A; Rossi-Bergmann, B.; Da Silva, A. J. M. Synthesis and evaluation of hybrid sulfonamide-chalcones with potential antileishmanial activity. Arch Pharm (Weinheim) 2024, 357, e2300440. [Google Scholar] [CrossRef] [PubMed]
- Almazroo, O. A.; Miah, M. K.; Venkataramanan, R. Drug Metabolism in the liver. Clin Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Issa, N. T.; Wathieu, H.; Ojo, A.; Byers, S. W.; Dakshanamurthy, S. Drug metabolism in preclinical drug development: A survey of the discovery process, toxicology, and computational tools. Curr Drug Metab. 2017, 18, 556–565. [Google Scholar] [CrossRef]
- Zhang, B.; Duan, D.; Ge, C.; Yao, J.; Liu, Y.; Li, X.; Fang, J. Synthesis of xanthohumol analogues and discovery of potent thioredoxin reductase inhibitor as potential anticancer agent. J Med Chem. 2015, 58, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structures of some antileishmanial chalcones.

Figure 2.
In silico prediction of NAT22 products following phase I metabolism by human cytochrome P450 (CYP) enzymes.
Figure 2.
In silico prediction of NAT22 products following phase I metabolism by human cytochrome P450 (CYP) enzymes.

Figure 3.
Positive ion electrospray high resolution LC-MS/MS total ion chromatogram (TIC) and computer-reconstructed mass chromatograms showing chalcone NAT22 and its phase I metabolites (M1-M7) produced after in vitro incubation with (A) human; (B) dog; (C) rat and (D) mouse liver microsomes.
Figure 3.
Positive ion electrospray high resolution LC-MS/MS total ion chromatogram (TIC) and computer-reconstructed mass chromatograms showing chalcone NAT22 and its phase I metabolites (M1-M7) produced after in vitro incubation with (A) human; (B) dog; (C) rat and (D) mouse liver microsomes.

Figure 4.
Representative positive ion ESI-MS/MS mass spectra and proposed of fragmentation of NAT22 metabolites M1-M7.
Figure 4.
Representative positive ion ESI-MS/MS mass spectra and proposed of fragmentation of NAT22 metabolites M1-M7.

Figure 5.
Overlay of 1H NMR spectra of NAT22 prior to (Unmetabolized) and following in vitro incubation with human microsomes (Metabolized). The blue values above and below the peaks refer, respectively, to the chemical shifts and integrals of NAT22 hydrogens in the control Unmetabolized sample. The peaks indicated by arrows correspond to the hydrogen signals of the metabolites.
Figure 5.
Overlay of 1H NMR spectra of NAT22 prior to (Unmetabolized) and following in vitro incubation with human microsomes (Metabolized). The blue values above and below the peaks refer, respectively, to the chemical shifts and integrals of NAT22 hydrogens in the control Unmetabolized sample. The peaks indicated by arrows correspond to the hydrogen signals of the metabolites.

Figure 6.
Amplification of methoxy (A) and aromatic (B) hydrogens in 1H NMR spectrum from Figure 5.
Figure 6.
Amplification of methoxy (A) and aromatic (B) hydrogens in 1H NMR spectrum from Figure 5.

Figure 8.
Docking of NAT22 and representative metabolites M2 and M4 between the cysteines (Cys-52 and Cys-173) of each monomeric unit of the cTXNPx enzyme. The 3D (left) and 2D (right) docking images obtained for each chalcone show the structural orientation and types of intermolecular interactions observed between the ligands and amino acids of the cTXNPx enzyme.
Figure 8.
Docking of NAT22 and representative metabolites M2 and M4 between the cysteines (Cys-52 and Cys-173) of each monomeric unit of the cTXNPx enzyme. The 3D (left) and 2D (right) docking images obtained for each chalcone show the structural orientation and types of intermolecular interactions observed between the ligands and amino acids of the cTXNPx enzyme.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



