Submitted:
22 March 2026
Posted:
23 March 2026
You are already at the latest version
Abstract
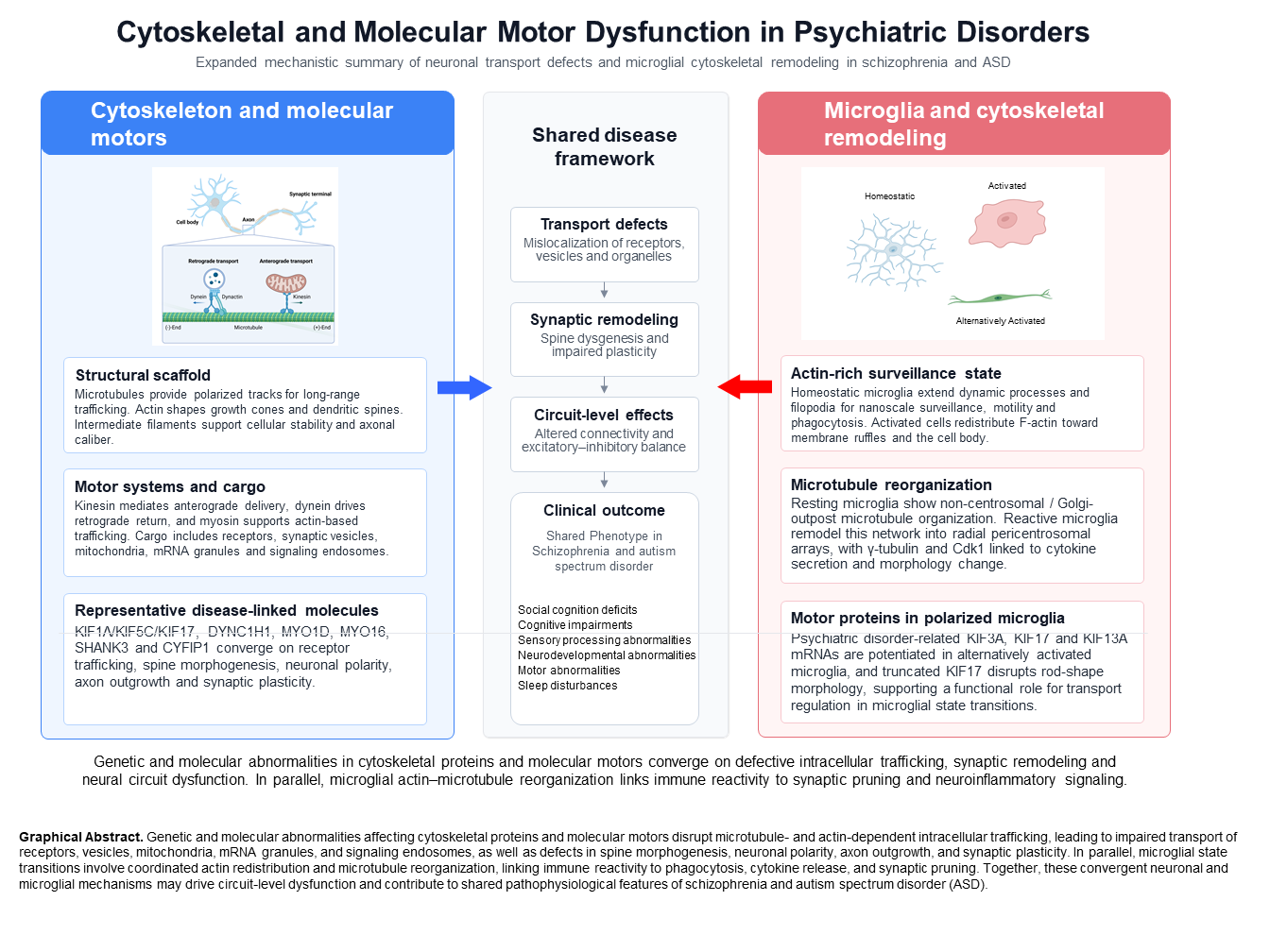
Elucidating the pathophysiological mechanisms of mental disorders remains a critical challenge in psychiatric research. Recent studies have highlighted the potential involvement of cytoskeletal and molecular motor abnormalities in the development of mental disorders such as schizophrenia and autism spectrum disorder (ASD). Although schizophrenia and ASD differ clinically, both disorders are increasingly regarded as neurodevelopmental conditions and share vulnerabilities in synapse formation and neural circuit maturation. This review synthesizes the latest findings on the relationship between cytoskeletal and molecular motor abnormalities and mental disorders. The cytoskeleton, composed of microtubules, actin filaments, and intermediate filaments, along with molecular motors such as kinesins, dyneins, and myosins, plays crucial roles in neurodevelopment, synapse formation, and neurotransmission. In schizophrenia, decreased expression of the microtubule-associated protein MAP2 and abnormalities in the DISC1 gene have been reported, potentially leading to dendritic morphological abnormalities and neurodevelopmental disorders. Additionally, abnormalities in molecular motors such as KIF17 and KIF1A have been implicated in schizophrenia pathophysiology. Myosin Id has been identified as a risk gene for ASD. Furthermore, abnormalities in actin-related proteins such as SHANK3 and CYFIP1 have been shown to cause synaptic dysfunction. These findings suggest that mental disorders arise from complex pathologies involving multiple cytoskeletal and molecular motor-related protein abnormalities. Future research should focus on elucidating the functions of individual proteins and adopting a comprehensive approach that includes glial cells. Advances in this field may deepen our understanding of the pathophysiological mechanisms of mental disorders and potentially lead to the development of novel therapeutic strategies.
Keywords:
cytoskeleton
; molecular motors
; schizophrenia
; autism spectrum disorder
; microglia
1. Introduction
Neurons possess a highly organized cytoskeletal system that enables them to maintain their unique morphology and specialized functions. This cytoskeletal network, composed primarily of microtubules, actin filaments, and intermediate filaments, plays essential roles in numerous cellular processes, including neuronal development, axonal and dendritic outgrowth, and synapse formation [1]. In addition, molecular motors—particularly members of the kinesin superfamily (KIFs)—travel along these cytoskeletal tracks to transport various intracellular cargos, thereby contributing critically to the maintenance of neuronal function [2,3].
Recent research has revealed that abnormalities in cytoskeletal structures and molecular motor proteins are deeply implicated in the pathophysiology of several psychiatric disorders, most notably schizophrenia and autism spectrum disorder (ASD) [4,5]. In this review, we summarize the fundamental functions of the cytoskeleton and molecular motors, and we discuss how disruptions in these systems contribute to psychiatric disease mechanisms, with a particular focus on schizophrenia and ASD.
Polygenic risk is enriched in genes involved in neurodevelopment and synaptic function, and increasing evidence implicates immune dysregulation and neuroinflammation (e.g., maternal immune activation and microglial activation) as modulators of circuit maturation [8,9]. These shared features provide a framework for examining how cytoskeletal regulation and intracellular transport shape synapse development, receptor trafficking, and network-level function. Schizophrenia is associated with cortical thinning, ventricular enlargement, and widespread functional dysconnectivity, particularly affecting prefrontal–temporal networks involved in cognitive control and working memory. In contrast, ASD has been associated with early brain overgrowth during childhood, alterations in cortical surface area development, and atypical functional connectivity within social brain networks and the default mode network [7].
Schizophrenia and ASD differ substantially in developmental trajectory and symptom profile: ASD presents in early childhood with persistent social-communication impairments and restricted/repetitive behaviors, whereas schizophrenia typically emerges in late adolescence with psychosis and cognitive disturbances. [4,5] Both disorders are highly heterogeneous, with diverse genetic architectures and environmental influences; cytoskeletal and motor dysfunction likely represents one of several converging biological pathways contributing to disease pathophysiology.
The neuronal cytoskeleton provides both structural support and dynamic scaffolding for morphogenesis and plasticity. Microtubules support axonal/dendritic growth and serve as tracks for long-range transport [12,13,14] and intermediate filaments contribute to cellular stability and axonal caliber [18,19]. Perturbations in these systems can therefore impact neuronal polarity, arborization, and synaptic architecture—cellular phenotypes repeatedly implicated in schizophrenia and ASD.
Molecular motors convert ATP hydrolysis into directed movement along cytoskeletal filaments. Kinesins primarily drive anterograde transport on microtubules, dynein mediates retrograde transport, and myosins regulate actin-based trafficking and spine dynamics [15,16,17]. By controlling the spatiotemporal delivery of receptors, vesicles, organelles, and mRNAs, these motors directly influence synaptogenesis, synaptic plasticity, and circuit maturation, providing mechanistic links between genetic/molecular perturbations and disease-relevant cellular phenotypes.
Increasing genetic and molecular evidence indicates that specific cytoskeletal and molecular motor proteins are directly linked to psychiatric disorders. For example, alterations in kinesin family members (such as KIF1A, KIF5C, and KIF17), dynein components (DYNC1H1), and actin-associated motors (MYO16 and MYO1D) have been reported in individuals with schizophrenia or ASD. These proteins regulate intracellular trafficking of synaptic vesicles, neurotransmitter receptors, and signaling complexes, suggesting that disruption of intracellular transport may contribute to synaptic dysfunction and abnormal circuit development [4,5,52].
Kinesin superfamily proteins (KIFs) mediate anterograde transport toward microtubule plusends, delivering cargos from the soma to axons and dendrites [20]. More than 45 KIF genes have been identified in humans, with individual KIFs transporting distinct cargo sets including synaptic vesicles, neurotransmitter receptors, mitochondria, and mRNA granules [11,20,21,22,23].
Among kinesins, KIF5A/B/C (kinesin-1 family) mediate long-range anterograde transport of mitochondria, membranous organelles, and receptor-containing vesicles, while KIF1A transports synaptic vesicle precursors and KIF17 delivers NMDA receptor subunits to dendrites [10,20].
Dynein, in contrast, mediates retrograde transport toward the minus-end of microtubules, moving cargos from axonal terminals back toward the cell body [24]. Its major roles include the retrograde trafficking of synaptic vesicles, neurotransmitter receptors, and various organelles; the transport of neurotrophic factor signaling endosomes; and the redistribution of cellular components during axon regeneration [25,26,27,28].
Myosins function primarily along actin filaments [29]. Numerous myosin family members are expressed in neurons, where they regulate the transport of diverse cargos and modulate actin architecture to influence synaptic activity. Myosins contribute to dendritic spine morphogenesis, local mobility of synaptic vesicles, growth cone motility, and the regulation of synaptic plasticity [30,31,32,33]. Myosin II, V, and VI to play essential roles in neuronal physiology.
Together, these molecular motor systems coordinate the highly dynamic intracellular transport processes required for neuronal development, synaptic function, and circuit maintenance. Dysfunction in any of these motor proteins can profoundly disrupt neuronal connectivity and is increasingly recognized as a key contributor to neurodevelopmental and psychiatric disorders [1,10].
Psychiatric disorders such as schizophrenia and ASD impose a substantial societal burden, yet their molecular–cellular underpinnings remain incompletely understood. While synaptic and neurodevelopmental frameworks have been extensively discussed, the cytoskeleton and intracellular transport machinery—which directly govern neuronal morphogenesis, cargo trafficking, and synaptic organization—are often treated as background cell biology rather than as central disease mechanisms. The present review is a narrative review based on studies identified through PubMed and Google Scholar, using search terms including cytoskeleton, molecular motors, kinesin, dynein, myosin, schizophrenia, and autism spectrum disorder, primarily covering the period from 2000 to the present. Studies were prioritized based on the provision of (i) genetic evidence (e.g., GWAS, rare variants, copy-number variants), (ii) human brain evidence (e.g., postmortem expression or proteomics), and/or (iii) mechanistic support from cellular or animal models linking a specific cytoskeletal or motor protein alteration to a neuronal or glial phenotype relevant to disease. No formal systematic-review guidelines were applied; instead, the review focused on mechanistically informative and recent evidence, summarized in Table 1. The novelty of this review lies in its integration of foundational cytoskeletal biology with evidence from recent large-scale genomic studies, including a genome-wide association study of schizophrenia identifying 287 loci with convergence on synaptic and cytoskeletal gene networks [72], and a large-scale exome-sequencing study in ASD implicating more than 100 risk genes enriched in neurodevelopmental pathways [73]. These converging lines of genomic evidence substantially reinforce cytoskeletal and motor dysfunction as a shared pathological axis in both disorders. Furthermore, this review reflects the research interests of our laboratory, which has been investigating the roles of molecular motors and microtubule-associated proteins in neuronal function and psychiatric disease. Our group has demonstrated that Myosin Id accumulates in dendritic spines through its TH1 domain [53] and functions as a risk gene for ASD, and has shown that KIF17-mediated transport of NMDA receptor subunits is regulated in an activity-dependent manner [71]. In addition, our recent work has revealed that the expression of psychiatric disorder-related kinesin superfamily proteins is upregulated in alternatively activated microglia [77], suggesting a neuroimmune dimension to motor protein dysregulation. This review therefore integrates our laboratory’s findings with the broader field to provide a perspective on cytoskeletal and molecular motor dysfunction as a convergent and therapeutically relevant mechanism in schizophrenia and ASD.
2. Cytoskeletal and Molecular Motor Abnormalities in Schizophrenia
Table 1 summarizes representative cytoskeletal and molecular motor genes implicated in schizophrenia and ASD, together with reported variants, molecular functions, and proposed cellular consequences.
Schizophrenia is a psychiatric disorder characterized by hallucinations, delusions, cognitive impairments, and disturbances in emotion and behavior. Accumulating evidence indicates that abnormalities in cytoskeletal components and molecular motor proteins are present in the brains of individuals with schizophrenia and are closely linked to disease pathophysiology.
Among microtubule-related abnormalities, reduced expression of the microtubule-associated protein MAP2 has been reported in the prefrontal cortex and hippocampus of patients with schizophrenia [34,35]. Because MAP2 plays a critical role in the formation and maintenance of dendrites, decreased expression is thought to contribute to dendritic morphological abnormalities and impaired dendritic function. In addition, DISC1 (Disrupted in Schizophrenia 1), a susceptibility gene for schizophrenia, regulates microtubule stabilization and intracellular transport. Dysfunction of DISC1 is believed to impair neurodevelopment and synaptic plasticity [36,37]. Among the reported variants, the missense single nucleotide polymorphism rs821616 (p.Ser704Cys) in DISC1 is among the best-studied; this variant disrupts the interaction between DISC1 and its binding partners NDEL1 and LIS1, which are essential for microtubule stabilization and nuclear migration during cortical development. Additional SNPs, including rs1000731, have been associated with reduced hippocampal volume and impaired working memory in schizophrenia cohorts.
Abnormalities in actin-related proteins have also been identified in schizophrenia. Postmortem studies have reported altered expression of components of the WAVE complex—such as CYFIP1, NCKAP1, and WASF1 [38]—which regulates actin polymerization and contributes to dendritic spine formation. Disruption of this complex is expected to lead to synaptic dysfunction [39]. At the genetic level, 15q11.2 microdeletions—which encompass CYFIP1—represent one of the most frequent copynumber variants (CNVs) associated with schizophrenia (OR ≈ 2.8), causing ~50% reduction in CYFIP1 protein levels. This haploinsufficiency impairs assembly of the WAVE regulatory complex and Rac1-mediated actin polymerization, resulting in reduced density and abnormal morphology of dendritic spines [38,39]. Increased expression of calponin-3, an actin-binding protein, has also been observed in the prefrontal cortex of patients [40]. Elevated levels of calponin-3 may contribute to aberrant stabilization of the actin cytoskeleton [41].
Regarding intermediate filaments, increased expression of GFAP (glial fibrillary acidic protein) has been detected in the prefrontal cortex and hippocampus of individuals with schizophrenia [42]. As a major intermediate filament protein in astrocytes, elevated GFAP expression likely reflects neuroinflammatory or neuroprotective responses [43].
Genetic studies have suggested that variants affecting the KIF1A pathway may influence synaptic vesicle transport mechanisms relevant to psychiatric disorders. Because KIF1A mediates the anterograde transport of synaptic vesicle precursors, its dysfunction may reduce synaptic transmission efficiency and alter neuronal connectivity. Regarding KIF17, the missense single nucleotide polymorphism rs2722519, together with reduced KIF17 protein expression, has been reported in schizophrenia patients [44,45]. This variant impairs anterograde transport of NR2B-containing NMDA receptors to synapses, disrupting synaptic plasticity [6]. Genetic association studies have also identified single nucleotide polymorphisms within KIF1A, including variants in the motor and stalk domains [46,47], predicted to reduce microtubule-based processivity and diminish anterograde delivery of synaptic vesicle precursors.In addition, a mutation in KIF3B has been shown to reduce trafficking of the NMDA receptor subunit NR2A and to cause schizophrenia-like behavioral phenotypes in mice [48]. Genetic studies have suggested potential links between KIF1A variants and synaptic vesicle transport deficits relevant to schizophrenia [47], with disruption of KIF1A processivity proposed as a contributing mechanism.
Dynein-related abnormalities include rare variants in the DYNC1H1 (Dynein Cytoplasmic 1 Heavy Chain 1) gene identified in individuals with schizophrenia [49]. Because DYNC1H1 is a major component of the dynein complex, its dysfunction is expected to impair retrograde axonal transport [50]. Variants in MYO16 (Myosin XVI), a myosin family protein involved in neuronal migration and dendrite formation, have also been reported in schizophrenia [51]. Disruption of MYO16 function may impair neural circuit formation. Specifically, de novo missense variants such as p.His306Arg in DYNC1H1, documented in patients with neurodevelopmental disorders [49,50],rare de novo missense variants in DYNC1H1 [49], are predicted to destabilize the dynein motor domain, impairing retrograde trafficking of BDNF-containing signaling endosomes and compromising neurotrophin-dependent neuronal survival signaling. Variants in MYO16 (Myosin XVI) associated with schizophrenia include rare missense and copy-number variants identified in Han Chinese GWAS cohorts [51]. MYO16 is expressed in postmitotic neurons and regulates cytoskeletal dynamics during neuronal migration and dendrite morphogenesis; disruption of MYO16 function is therefore expected to contribute to circuit formation abnormalities observed in schizophrenia.
Collectively, these cytoskeletal and molecular motor abnormalities contribute to a range of pathological processes observed in schizophrenia, including impaired synapse formation and plasticity, abnormal receptor trafficking, disrupted neurodevelopment, dysfunctional neural circuits, and cognitive deficits. These findings underscore the crucial role of intracellular transport and cytoskeletal regulation in the neurobiology of schizophrenia.
3. Cytoskeletal and Molecular Motor Abnormalities in Autism Spectrum Disorder
Autism spectrum disorder (ASD) is a neurodevelopmental condition characterized by impairments in social communication and interaction, together with restricted, repetitive patterns of behavior and interests. In recent years, multiple abnormalities in cytoskeletal components and molecular motor proteins have been reported in the brains of individuals with ASD.
Myosin ID (MYO1D) has been identified as a risk gene for ASD [52]. We previously showed that EGFP-tagged Myosin Id, when expressed in cultured neurons, accumulates in dendritic spines [53]. Furthermore, this localization critically depends on the TH1 (tail homology 1) domain: deletion of the TH1 domain disrupts to dendrites and markedly reduces its accumulation in dendritic spines. These findings strongly suggest that MYO1D interacts with actin filaments within dendritic spines and contributes to the regulation of excitatory synaptic transmission, and that disruption of this function may underlie aspects of ASD pathophysiology. The TH1 domain of MYO1D has also been reported to interact with the C-terminus of aspartoacylase, an N-acetylaspartate (NAA)–acylating enzyme [54]. NAA is present at high concentrations in the mammalian brain, and reduced NAA levels have been observed in children with ASD compared with typically developing controls [55]. These observations further support a functional link between MYO1D and ASD.
Myosin IXb (MYO9B) is known to regulate RhoA activity and to control dendritic morphology in cortical neurons [56]. Variants in MYO9B have been suggested to influence ASD risk [5]. Rare missense variants in MYO9B affecting its RhoGAP domain have been identified in ASD patients; these variants are predicted to impair RhoA inactivation, leading to dysregulation of actin dynamics and dendritic arborization [56]. In addition, cohort studies have reported an association between Myosin XVI and ASD [57]. Myosin XVI contributes to neuronal migration and dendritic development, and its dysfunction is thought to lead to defects in neural circuit formation.
Mutations in SHANK3 (SH3 and multiple ankyrin repeat domains 3) are recognized as major genetic risk factors for autism spectrum disorder (ASD). [58]. SHANK3 plays a key role in organizing the actin cytoskeleton in the postsynaptic density; its disruption leads to defective dendritic spine formation and synaptic dysfunction [59]. Duplications and deletions of CYFIP1 (Cytoplasmic FMR1-interacting protein 1) have also been identified as risk factors for ASD [60]. CYFIP1 is a component of the WAVE complex and regulates actin polymerization; its dysregulation is believed to cause dendritic spine abnormalities [61].
Specifically, heterozygous deletions of chromosome 22q13.3 encompassing SHANK3, as well as truncating and frameshift mutations have been identified in individuals with ASD and Phelan-McDermid syndrome [58]. These loss-of-function variants lead to near-complete absence of SHANK3 at the postsynaptic density, resulting in severely impaired actin cytoskeleton organization and dendritic spine morphogenesis.
Rare variants in TUBG1 (γ-tubulin) have been reported in individuals with ASD [62]. γ-Tubulin functions as a nucleation factor for microtubule assembly, and its disruption is expected to affect neuronal polarity and migration [63]. Mutations in ASPM (abnormal spindle-like microcephaly-associated protein) have likewise been implicated as risk factors for ASD [64]. ASPM plays an important role in the division and differentiation of neural stem cells, and its dysfunction is thought to lead to abnormal cortical development [65]. Biallelic loss-of-function mutations in ASPM—including frameshift and nonsense variants distributed throughout the IQ (isoleucine-glutamine) calmodulin-binding motif repeats—cause autosomal recessive primary microcephaly; heterozygous rare variants have been identified in ASD cohorts and are thought to impair the symmetric division of neural progenitors, reducing the final neuronal output of the developing cortex [64,65]. The de novo missense variant p.Tyr92Cys in TUBG1, reported in individuals with lissencephaly-spectrum cortical malformations and ASD-associated features [62,63,68], disrupts the γ-tubulin ring complex, impairing microtubule nucleation at the centrosome and Golgi apparatus and thereby compromising neuronal polarity and radial migration.
Rare variants in KIF1A have been identified in patients with ASD [66]. KIF1A is essential for the anterograde transport of synaptic vesicle precursors, and its dysfunction is predicted to reduce the efficiency of synaptic transmission [67]. Mutations in KIF5C have also been associated with ASD. KIF5C contributes to neuronal polarity and axonal elongation, and its disruption likely impairs neural circuit formation [68]. Among the reported KIF5C mutations, the de novo missense variant p.Glu237Lys, identified in patients with malformations of cortical development and intellectual disability [68], is located within the kinesin motor domain and disrupts ATP hydrolysis, severely impairing microtubule-based anterograde transport and thereby causing abnormal axon elongation and cortical wiring defects. Rare de novo missense variants including c.773C>T (p.Pro258Leu) and other motor domain mutations have been identified in ASD patients [66,67]. These variants impair the ATPase activity and processivity of KIF1A, leading to insufficient anterograde delivery of dense-core vesicles and synaptic vesicle precursors to distal axons and dendrites.
Taken together, these cytoskeletal and molecular motor abnormalities appear to contribute to ASD pathophysiology through several interrelated mechanisms. First, disturbances in actin-regulating proteins such as SHANK3 and CYFIP1 result in defective dendritic spine formation and synaptic dysfunction, thereby disrupting the normal development and function of neural circuits. Second, abnormalities in kinesins are expected to impair axonal transport and neuronal migration, leading to structural and functional abnormalities in neural networks. Third, defects in proteins such as MYO9B that regulate neuronal migration and axon guidance may interfere with the precise positioning of neurons and the accurate extension of axons, thereby contributing to macroscopic brain abnormalities. Fourth, dysfunction of ASPM is predicted to alter the division and differentiation of neural stem cells and to disrupt the proper formation of cortical lamination. Fifth, impaired synaptic vesicle transport due to abnormalities in KIF1A and KIF5C may reduce the efficiency of neurotransmission and compromise information-processing capacity. The convergence of genetic, molecular, and cellular findings suggests that cytoskeletal and molecular motor abnormalities may represent a mechanistic bridge between diverse ASD risk genes and the synaptic and connectivity phenotypes frequently observed in this disorder.
These findings together suggest that ASD is not caused by a single molecular defect but rather arises from a complex interplay among multiple abnormalities in cytoskeletal and molecular motor–related proteins, which collectively perturb neurodevelopment, synaptic function, and network-level information processing.
4. Conclusions
Accumulating evidence indicates that abnormalities in cytoskeletal organization and molecular motor function—particularly within the kinesin (KIF) and myosin families—are deeply involved in the pathophysiology of psychiatric disorders such as schizophrenia and ASD. These abnormalities affect multiple aspects of brain function, including neurodevelopment, synapse formation, neurotransmission, and neuronal plasticity, and are thought to contribute to the characteristic symptoms and cognitive impairments observed in these conditions.
Recent findings on the localization and function of Myosin Id in dendritic spines provide important clues for understanding the disease mechanisms of ASD. In our laboratory, we plan to further dissect the roles of Myosin Id and to clarify in greater detail how its dysfunction contributes to ASD onset and progression. In parallel, elucidating the mechanisms by which KIFs transport neurotransmitter receptors and synaptic vesicles has provided new perspectives on the pathogenesis of psychiatric disorders [71]. A more precise understanding of the functions of individual KIFs and their links to specific psychiatric phenotypes is expected to facilitate the development of novel, mechanism-based therapeutic strategies.
Recent large-scale human genetics studies published in the 2020s further reinforce the relevance of synaptic and neurodevelopmental pathways that intersect with cytoskeletal regulation and intracellular transport. For schizophrenia, a large two-stage GWAS reported associations at 287 genomic loci and highlighted convergence on synaptic organization and neuronal differentiation [72]. For ASD, large-scale exome sequencing has identified more than 100 risk genes and emphasized both developmental and functional mechanisms in excitatory and inhibitory neuronal populations [73]. In parallel, recent work has expanded the microglia-focused literature in psychiatric and neurodevelopmental disorders, including evidence linking microglial activation to interneuron metabolic disruptions relevant to schizophrenia [74], reviews integrating microglia–neuron interactions in schizophrenia [75], and functional genomics studies in human iPSC-derived microglia implicating ASD risk genes in endocytosis and synaptic pruning [76].
Growing evidence indicates that microglial cytoskeletal dysfunction is directly implicated in the pathophysiology of both schizophrenia and ASD, rather than representing a generic neuroinflammatory response. In schizophrenia, activated microglia have been shown to cause persistent metabolic disruptions in developing cortical interneurons, including cytoskeletal dysregulation that may underlie the GABAergic circuit abnormalities characteristic of the disorder [74]. A comprehensive review of microglia–neuron interactions in schizophrenia further highlights synaptic pruning dysregulation and altered cytoskeletal dynamics as key contributors to aberrant connectivity [75]. In ASD, a CRISPRi-based screen of ASD risk genes in microglia revealed that ADNP—a microtubule end-binding protein—regulates microglial endocytosis and synaptic pruning, establishing a direct mechanistic link between microglial cytoskeletal function and ASD-associated synaptic pathology [76]. Importantly, the morphological changes accompanying microglial activation involve a shift from acentrosomal to centrosomal microtubule arrays [69] and polarized microtubule remodeling that drives cytokine release [70], providing a mechanistic basis for how cytoskeletal dysfunction in microglia could amplify neuroinflammatory responses relevant to both schizophrenia and ASD. Dysregulated synaptic pruning and altered neuroimmune signaling driven by abnormal microglial cytoskeletal remodeling have been proposed as convergent contributors to aberrant circuit development in both disorders [9,56,77]. Furthermore, upregulation of psychiatric disorder-associated kinesin family members (including KIF3A, KIF17, and KIF13A) in alternatively activated primary cultured microglia [77] suggests that dysregulation of microtubule-based motor transport is not confined to neurons but extends to glial cells, providing a novel mechanistic dimension to the cytoskeletal dysfunction framework in psychiatric disease. Together, these findings highlight the characterization of microglial cytoskeletal and motor protein abnormalities in schizophrenia and ASD as an important priority for future investigation, with potential implications for biomarker development and therapeutic targeting.
Key future directions include:
- Clarifying the relationships between glia-specific cytoskeletal and molecular motor gene variants and psychiatric disorders;
- Developing novel therapeutic strategies that directly target glial cytoskeletal and molecular motor pathways;
- Elucidating the roles of cytoskeletal and motor systems in neuron–glia interactions; and
- Defining how morphological changes in glial cells relate to functional alterations in the context of psychiatric illnesses.
Through these lines of investigation, we anticipate further advances in our understanding of the pathophysiological mechanisms underlying psychiatric disorders, which in turn should contribute to the development of new therapeutic and preventive strategies. Approaches that specifically target microglial cytoskeletal and molecular motor dynamics may exert their effects for patients with treatment-resistant conditions. Research on cytoskeletal and molecular motor systems lies at the interface of neuroscience and psychiatry and is expected to become increasingly important in the coming years. Ultimately, progress in this field may help improve the quality of life of individuals suffering from psychiatric disorders.
Author Contributions
Conceptualization, T.S.; writing—original draft preparation, K.N. and T.S.; writing—review and editing, T.S., A.K., S.S., S.K., and K.I.; supervision, T.S. All authors have read and agreed to the published version of the manuscript.
Funding
The work conducted in our laboratory has been supported by Grants-in-Aid for Scientific Research (KAKENHI; 16H06276, 19K08065, 22K07611, 19H05201), as well as by the Advanced Pharmaceutical Research Promotion Foundation, the Naito Memorial Foundation for the Promotion of Science, the Takeda Science Foundation, the Kawano Pediatric Medical Research Foundation, the Daido Life Welfare Foundation, the Life Science Foundation of Japan, the Nakatomi Health Science Foundation, the Pharmacological Research Foundation, the Pharmaceutical Research Encouragement Foundation, the Mishima Kaiun Memorial Foundation, the Collaborative Research Program of the National Institute for Basic Biology, and the Open Facility Initiative of the University of Tsukuba.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
We would also like to express our sincere gratitude to all members of the Laboratory of Anatomy and Neuroscience for their valuable advice and support in preparing this review. The authors acknowledge the use of AI language tools (Claude, Anthropic) for assistance with English language editing and manuscript preparation in accordance with the MDPI AI use policy.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Fletcher, DA; Mullins, RD. Cell mechanics and the cytoskeleton. Nature 2010, 463(7280), 485–92. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N; Noda, Y; Tanaka, Y; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009, 10(10), 682–96. [Google Scholar] [CrossRef] [PubMed]
- Franker, MAM; Hoogenraad, CC. Microtubule-based transport - basic mechanisms, traffic rules and role in neurological pathogenesis. J Cell Sci. 2013, 126 Pt 11, 2319–29. [Google Scholar] [CrossRef] [PubMed]
- Marchisella, F. Abnormalities of the microtubule system and impaired neurite outgrowth in psychiatric disorders. In Neural Plast; 2016. [Google Scholar]
- De Rubeis, S; He, X; Goldberg, AP; Poultney, CS; Samocha, K; et al.; The DDD Study Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515(7526), 209–15. [Google Scholar] [CrossRef]
- Moghaddam, B; Javitt, D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012, 37(1), 4–15. [Google Scholar] [CrossRef]
- Padmanabhan, A; Lynch, CJ; Schaer, M; Menon, V. The default mode network in autism. Biol Psychiatry Cogn Neurosci Neuroimaging 2017, 2(6), 476–86. [Google Scholar] [CrossRef]
- Estes, ML; McAllister, AK. Maternal immune activation: Implications for neuropsychiatric disorders. Science 2016, 353(6301), 772–7. [Google Scholar] [CrossRef]
- Müller, N. Neuroinflammation in schizophrenia and autism spectrum disorders: Recent advances. In Prog Neuropsychopharmacol Biol Psychiatry; 2018. [Google Scholar]
- Hirokawa, N; Takemura, R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005, 6(3), 201–14. [Google Scholar] [CrossRef]
- Miki, H; Setou, M; Kaneshiro, K; Hirokawa, N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci U S A 2001, 98(13), 7004–11. [Google Scholar] [CrossRef]
- Conde, C; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci 2009, 10(5), 319–32. [Google Scholar] [CrossRef]
- Takei, Y; Teng, J; Harada, A; Hirokawa, N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000, 150(5), 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Kapitein, LC; Hoogenraad, CC. Building the neuronal microtubule cytoskeleton. Neuron 2015, 87(3), 492–506. [Google Scholar] [CrossRef] [PubMed]
- Dent, EW; Gupton, SL; Gertler, FB. The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb Perspect Biol. 2011, 3(3), a001800–a001800. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, LA; Goda, Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 2008, 9(5), 344–56. [Google Scholar] [CrossRef]
- Okamoto, K-I; Nagai, T; Miyawaki, A; Hayashi, Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004, 7(10), 1104–12. [Google Scholar] [CrossRef]
- Perrot, R; Eyer, J. Neuronal intermediate filaments and neurodegenerative disorders. Brain Res Bull. 2009, 80(4–5), 282–95. [Google Scholar] [CrossRef]
- Lendahl, U; Zimmerman, LB; McKay, RD. CNS stem cells express a new class of intermediate filament protein. Cell 1990, 60(4), 585–95. [Google Scholar] [CrossRef]
- Hirokawa, N; Niwa, S; Tanaka, Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68(4), 610–38. [Google Scholar] [CrossRef]
- Mandal, A; Drerup, CM. Axonal transport and mitochondrial function in neurons. Front Cell Neurosci 2019, 13, 373. [Google Scholar] [CrossRef]
- Kanai, Y; Dohmae, N; Hirokawa, N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 2004, 43(4), 513–25. [Google Scholar] [CrossRef]
- Cosker, KE; Segal, RA. Neuronal signaling through endocytosis. Cold Spring Harb Perspect Biol. 2014, 6(2), a020669–a020669. [Google Scholar] [CrossRef]
- Roberts, AJ; Kon, T; Knight, PJ; Sutoh, K; Burgess, SA. Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol. 2013, 14(11), 713–26. [Google Scholar] [CrossRef]
- Caviston, JP; Holzbaur, ELF. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 2006, 16(10), 530–7. [Google Scholar] [CrossRef] [PubMed]
- Vallee, RB; Tsai, J-W. The cellular roles of the lissencephaly gene LIS1, and what they tell us about brain development. Genes Dev. 2006, 20(11), 1384–93. [Google Scholar] [CrossRef] [PubMed]
- Heerssen, HM; Pazyra, MF; Segal, RA. Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat Neurosci. 2004, 7(6), 596–604. [Google Scholar] [CrossRef] [PubMed]
- Tojima, T; Itofusa, R; Kamiguchi, H. Steering neuronal growth cones by shifting the imbalance between exocytosis and endocytosis. J Neurosci. 2014, 34(21), 7165–78. [Google Scholar] [CrossRef]
- Hartman, MA; Spudich, JA. The myosin superfamily at a glance. J Cell Sci. 2012, 125 Pt 7, 1627–32. [Google Scholar] [CrossRef]
- Rex, CS; Gavin, CF; Rubio, MD; Kramar, EA; Chen, LY; Jia, Y; et al. Myosin IIb regulates actin dynamics during synaptic plasticity and memory formation. Neuron 2010, 67(4), 603–17. [Google Scholar] [CrossRef]
- Kneussel, M; Wagner, W. Myosin motors at neuronal synapses: drivers of membrane transport and actin dynamics. Nat Rev Neurosci. 2013, 14(4), 233–47. [Google Scholar] [CrossRef]
- Wang, Z; Edwards, JG; Riley, N; Provance, DW, Jr.; Karcher, R; Li, X-D; et al. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell 2008, 135(3), 535–48. [Google Scholar] [CrossRef]
- Huber, KM; Gallagher, SM; Warren, ST; Bear, MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 2002, 99(11), 7746–50. [Google Scholar] [CrossRef] [PubMed]
- Jones, LB; Johnson, N; Byne, W. Alterations in MAP2 immunocytochemistry in areas 9 and 32 of schizophrenic prefrontal cortex. Psychiatry Res Neuroimaging 2002, 114(3), 137–48. [Google Scholar] [CrossRef] [PubMed]
- DeGiosio, R; Kelly, RM; DeDionisio, AM; Newman, JT; Fish, KN; Sampson, AR; et al. MAP2 immunoreactivity deficit is conserved across the cerebral cortex within individuals with schizophrenia. NPJ Schizophr 2019, 5(1), 13. [Google Scholar] [CrossRef] [PubMed]
- Brandon, NJ; Sawa, A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci. 2011, 12(12), 707–22. [Google Scholar] [CrossRef]
- Lipska, BK; Peters, T; Hyde, TM; Halim, N; Horowitz, C; Mitkus, S; et al. Expression of DISC1 binding partners is reduced in schizophrenia and associated with DISC1 SNPs. Hum Mol Genet. 2006, 15(8), 1245–58. [Google Scholar] [CrossRef]
- Kähler, AK; Djurovic, S; Rimol, LM; Brown, AA; Athanasiu, L; Jönsson, EG; et al. Candidate gene analysis of the human natural killer-1 carbohydrate pathway and perineuronal nets in schizophrenia: B3GAT2 is associated with disease risk and cortical surface area. Biol Psychiatry 2011, 69(1), 90–6. [Google Scholar] [CrossRef]
- Soderling, SH; Guire, ES; Kaech, S; White, J; Zhang, F; Schutz, K; et al. A WAVE-1 and WRP signaling complex regulates spine density, synaptic plasticity, and memory. J Neurosci. 2007, 27(2), 355–65. [Google Scholar] [CrossRef]
- Föcking, M; Dicker, P; English, JA; Schubert, KO; Dunn, MJ; Cotter, DR. Common proteomic changes in the hippocampus in schizophrenia and bipolar disorder and particular evidence for involvement of cornu ammonis regions 2 and 3. Arch Gen Psychiatry 2011, 68(5), 477–88. [Google Scholar] [CrossRef]
- Rami, G; Caillard, O; Medina, I; Pellegrino, C; Fattoum, A; Ben-Ari, Y; et al. Change in the shape and density of dendritic spines caused by overexpression of acidic calponin in cultured hippocampal neurons. Hippocampus 2006, 16(2), 183–97. [Google Scholar] [CrossRef]
- Toro, CT; Hallak, JEC; Dunham, JS; Deakin, JFW. Glial fibrillary acidic protein and glutamine synthetase in subregions of prefrontal cortex in schizophrenia and mood disorder. Neurosci Lett. 2006, 404(3), 276–81. [Google Scholar] [CrossRef]
- Sofroniew, MV; Vinters, HV. Astrocytes: biology and pathology. Acta Neuropathol 2010, 119(1), 7–35. [Google Scholar] [CrossRef] [PubMed]
- Ratta-Apha, W; Mouri, K; Boku, S; Ishiguro, H; Okazaki, S; Otsuka, I; et al. A decrease in protein level and a missense polymorphism of KIF17 are associated with schizophrenia. Psychiatry Res. 2015, 230(2), 424–9. [Google Scholar] [CrossRef] [PubMed]
- Yin, X; Feng, X; Takei, Y; Hirokawa, N. Regulation of NMDA receptor transport: a KIF17-cargo binding/releasing underlies synaptic plasticity and memory in vivo. J Neurosci. 2012, 32(16), 5486–99. [Google Scholar] [CrossRef] [PubMed]
- Rivero, O; Sich, S; Popp, S; Schmitt, A; Franke, B; Lesch, K-P. Impact of the ADHD-susceptibility gene CDH13 on development and function of brain networks. Eur Neuropsychopharmacol 2013, 23(6), 492–507. [Google Scholar] [CrossRef]
- Okada, Y; Yamazaki, H; Sekine-Aizawa, Y; Hirokawa, N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995, 81(5), 769–80. [Google Scholar] [CrossRef]
- Alsabban, AH; Morikawa, M; Tanaka, Y; Takei, Y; Hirokawa, N. Kinesin Kif3b mutation reduces NMDAR subunit NR2A trafficking and causes schizophrenia-like phenotypes in mice. EMBO J 2020, 39(1), e101090. [Google Scholar] [CrossRef]
- Vissers, LELM; de Ligt, J; Gilissen, C; Janssen, I; Steehouwer, M; de Vries, P; et al. A de novo paradigm for mental retardation. Nat Genet. 2010, 42(12), 1109–12. [Google Scholar] [CrossRef]
- Schiavo, G; Greensmith, L; Hafezparast, M; Fisher, EMC. Cytoplasmic dynein heavy chain: the servant of many masters. Trends Neurosci. 2013, 36(11), 641–51. [Google Scholar] [CrossRef]
- Yue, W; Yu, X; Zhang, D. Progress in genome-wide association studies of schizophrenia in Han Chinese populations. NPJ Schizophr 2017, 3(1), 24. [Google Scholar] [CrossRef]
- Stone, JL; Merriman, B; Cantor, RM; Geschwind, DH; Nelson, SF. High density SNP association study of a major autism linkage region on chromosome 17. Hum Mol Genet. 2007, 16(6), 704–15. [Google Scholar] [CrossRef]
- Koshida, R; Tome, S; Takei, Y. Myosin Id localizes in dendritic spines through the tail homology 1 domain. Exp Cell Res. 2018, 367(1), 65–72. [Google Scholar] [CrossRef]
- Benesh, AE; Fleming, JT; Chiang, C; Carter, BD; Tyska, MJ. Expression and localization of myosin-1d in the developing nervous system. Brain Res. 2012, 1440, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Moffett, JR; Ross, B; Arun, P; Madhavarao, CN; Namboodiri, AMA. N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol 2007, 81(2), 89–131. [Google Scholar] [CrossRef] [PubMed]
- Long, H; Zhu, X; Yang, P; Gao, Q; Chen, Y; Ma, L. Myo9b and RICS modulate dendritic morphology of cortical neurons. Cereb Cortex 2013, 23(1), 71–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, K; Zhang, H; Ma, D; Bucan, M; Glessner, JT; Abrahams, BS; et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 2009, 459(7246), 528–33. [Google Scholar] [CrossRef]
- Durand, CM; Betancur, C; Boeckers, TM; Bockmann, J; Chaste, P; Fauchereau, F; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007, 39(1), 25–7. [Google Scholar] [CrossRef]
- Naisbitt, S; Kim, E; Tu, JC; Xiao, B; Sala, C; Valtschanoff, J; et al. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 1999, 23(3), 569–82. [Google Scholar] [CrossRef]
- Nishimura, Y; Martin, CL; Vazquez-Lopez, A; Spence, SJ; Alvarez-Retuerto, AI; Sigman, M; et al. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007, 16(14), 1682–98. [Google Scholar] [CrossRef]
- De Rubeis, S; Pasciuto, E; Li, KW; Fernández, E; Di Marino, D; Buzzi, A; et al. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 2013, 79(6), 1169–82. [Google Scholar] [CrossRef]
- Poirier, K; Saillour, Y; Bahi-Buisson, N; Jaglin, XH; Fallet-Bianco, C; Nabbout, R; et al. Mutations in the neuronal ß-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet. 2010, 19(22), 4462–73. [Google Scholar] [CrossRef]
- Tovey, CA; Conduit, PT. Microtubule nucleation by γ-tubulin complexes and beyond. Essays Biochem. 2018, 62(6), 765–80. [Google Scholar] [CrossRef]
- Bond, J; Roberts, E; Mochida, GH; Hampshire, DJ; Scott, S; Askham, JM; et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002, 32(2), 316–20. [Google Scholar] [CrossRef]
- Fish, JL; Kosodo, Y; Enard, W; Pääbo, S; Huttner, WB. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci U S A 2006, 103(27), 10438–43. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, FF; Gauthier, J; Araki, Y; Lin, D-T; Yoshizawa, Y; Higashi, K; et al. Excess of DE Novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet. 2011, 88(4), 516. [Google Scholar] [CrossRef]
- Yonekawa, Y; Harada, A; Okada, Y; Funakoshi, T; Kanai, Y; Takei, Y; et al. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol. 1998, 141(2), 431–41. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K; Lebrun, N; Broix, L; Tian, G; Saillour, Y; Boscheron, C; et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. 2013, 45(6), 639–47. [Google Scholar] [CrossRef]
- Rosito, M; Sanchini, C; Gosti, G; Moreno, M; De Panfilis, S; Giubettini, M; et al. Microglia reactivity entails microtubule remodeling from acentrosomal to centrosomal arrays. Cell Rep. 2023, 42(2), 112104. [Google Scholar] [CrossRef]
- Adrian, M; Weber, M; Tsai, M-C; Glock, C; Kahn, OI; Phu, L; et al. Polarized microtubule remodeling transforms the morphology of reactive microglia and drives cytokine release. Nat Commun. 2023, 14(1), 6322. [Google Scholar] [CrossRef]
- Iwata, S; Morikawa, M; Takei, Y; Hirokawa, N. An activity-dependent local transport regulation via degradation and synthesis of KIF17 underlying cognitive flexibility. Sci Adv 2020, 6(51), eabc8355. [Google Scholar] [CrossRef]
- Trubetskoy, V; Pardiñas, AF; Qi, T; Panagiotaropoulou, G; Awasthi, S; Bigdeli, TB; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef]
- Satterstrom, FK; Kosmicki, JA; Wang, J; Breen, MS; De Rubeis, S; An, JY; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020, 180(3), 568–584.e23. [Google Scholar] [CrossRef]
- Park, GH; Noh, H; Shao, Z; Ni, P; Qin, Y; Liu, D; et al. Activated microglia cause metabolic disruptions in developmental cortical interneurons that persist in interneurons from individuals with schizophrenia. Nature Neuroscience 2020, 23, 1352–1364. [Google Scholar] [CrossRef]
- Hartmann, S-M; Heider, J; Wüst, R; Fallgatter, AJ; Volkmer, H. Microglia-neuron interactions in schizophrenia. Frontiers in Cellular Neuroscience 2024, 18, 1345349. [Google Scholar] [CrossRef]
- Teter, OM; McQuade, A; Hagan, V; Liang, W; Dräger, NM; Sattler, SM; et al. CRISPRi-based screen of autism spectrum disorder risk genes in microglia uncovers roles of ADNP in microglia endocytosis and synaptic pruning. Molecular Psychiatry 2025, 30, 4176–4193. [Google Scholar] [CrossRef]
- Iwata, S; Hyugaji, M; Soga, Y; Morikawa, M; Sasaki, T; Takei, Y. Gene expression of psychiatric disorder-related kinesin superfamily proteins (Kifs) is potentiated in alternatively activated primary cultured microglia. BMC Res Notes 2025, 18(44), 1–8. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



