Submitted:
02 March 2026
Posted:
02 March 2026
You are already at the latest version
Abstract
Rising atmospheric CO2 levels have increased the demand for robust, scalable adsorbents for practical CO2 capture and separation. Porous organic polymers (POPs) are attractive candidates because their pore architecture and binding site properties can be precisely tuned via building blocks and linkage formation. This review summarizes experimental and computational studies of azo-linked POPs and, more broadly, nitrogen-nitrogen (N−N) linked systems, emphasizing how synthetic routes, building blocks, and framework topology govern CO2 uptake. We highlight key synthetic strategies and representative systems, including porphyrin-azo networks, and discuss the relatively sparse experimental literature on alternative N−N linked POPs incorporating azoxy and azodioxy motifs. Emphasis is placed on reversible nitroso/azodioxide chemistry as a potential pathway to ordered porous organic materials. Computational studies provide a practical route to connect structure with adsorption behavior in largely amorphous or partially ordered networks. We review hierarchical workflows combining periodic DFT and electrostatic potential properties, grand canonical Monte Carlo (GCMC) simulations and binding-energy calculations to rationalize trends and identify favorable binding environments. Computational findings demonstrate that pore accessibility and stacking models can strongly influence predicted CO2 adsorption. This review provides guidelines for designing POPs with enhanced CO2 adsorption, offering an outlook and discussing challenges for future studies.
Keywords:
azo linkages
; azoxy linkages
; azodioxy linkages
; CO2 adsorption
; DFT calculations
; GCMC simulations
; porous organic polymers
1. Introduction
The continued rise in atmospheric carbon dioxide (CO2) levels has intensified efforts to develop new materials capable of efficient capture and separation under practical conditions [1,2,3,4]. Since pre-industrial times (~280 ppm), atmospheric CO2 has increased to over 427 ppm, intensifying interest in scalable adsorption-based approaches for mitigation [5,6]. A wide range of porous solids — including zeolites, activated carbons, metal–organic frameworks (MOFs), and porous organic polymers (POPs) — have been explored as CO2 adsorbents [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22]. Among them, POPs offer an attractive balance of chemical and thermal stability, low skeletal density, and structural tunability, enabling pore architecture and functionality to be adjusted directly through monomer design and linkage chemistry [23,24,25,26].
For POPs, achieving high CO2 uptake and selectivity generally requires more than high surface area — the local chemical environment and the presence of heteroatoms often dominate adsorption strength and CO2/N2 selectivities [27,28]. Because CO2 has a larger quadrupole moment than N2, incorporating CO2-philic polar or Lewis-basic motifs (e.g., imines, triazines, carbazoles, and nitrogen-nitrogen type functionalities) can strengthen framework–CO2 interactions and improve selectivity, sometimes even when porosity is only moderate [23,24,25,26,29,30,31,32,33,34,35].
Azo linkages (−N=N−) have drawn sustained attention because they can simultaneously increase CO2 affinity and, as proposed in studies, promote “N2-phobic” behavior that boosts CO2/N2 selectivity — an especially desirable feature for post-combustion capture [27,28]. Several robust synthetic approaches exist for building azo-linked POP networks, including catalyst-free heterocoupling of aromatic nitro and amino monomers, copper(I)-catalyzed oxidative homocoupling of aromatic amino monomers, and Zn- or NaBH4-mediated reductive homocoupling of aromatic nitro monomers (Figure 1a). These and other methods (e.g., diazo-coupling reactions) enable broad chemical diversity across connector geometries and pore topologies [27,36,37,38,39].
A complementary strategy to strengthen CO2 binding is the integration of porphyrin units, which provide rigid, π-rich platforms and basic pyrrolic environments that can serve as additional adsorption sites [40,41,42,43,44,45,46]. Combining porphyrins with azo-based linkages has therefore emerged as a promising design concept, with literature examples reporting high CO2 uptakes and selectivities in azo-bridged porphyrinic polymers [47,48,49]. Recent work from our group used an integrated experimental and computational approach to interpret adsorption trends in azo-linked porphyrin-based POPs, with particular focus on topology, stacking/accessibility, and local polarity [33,34].
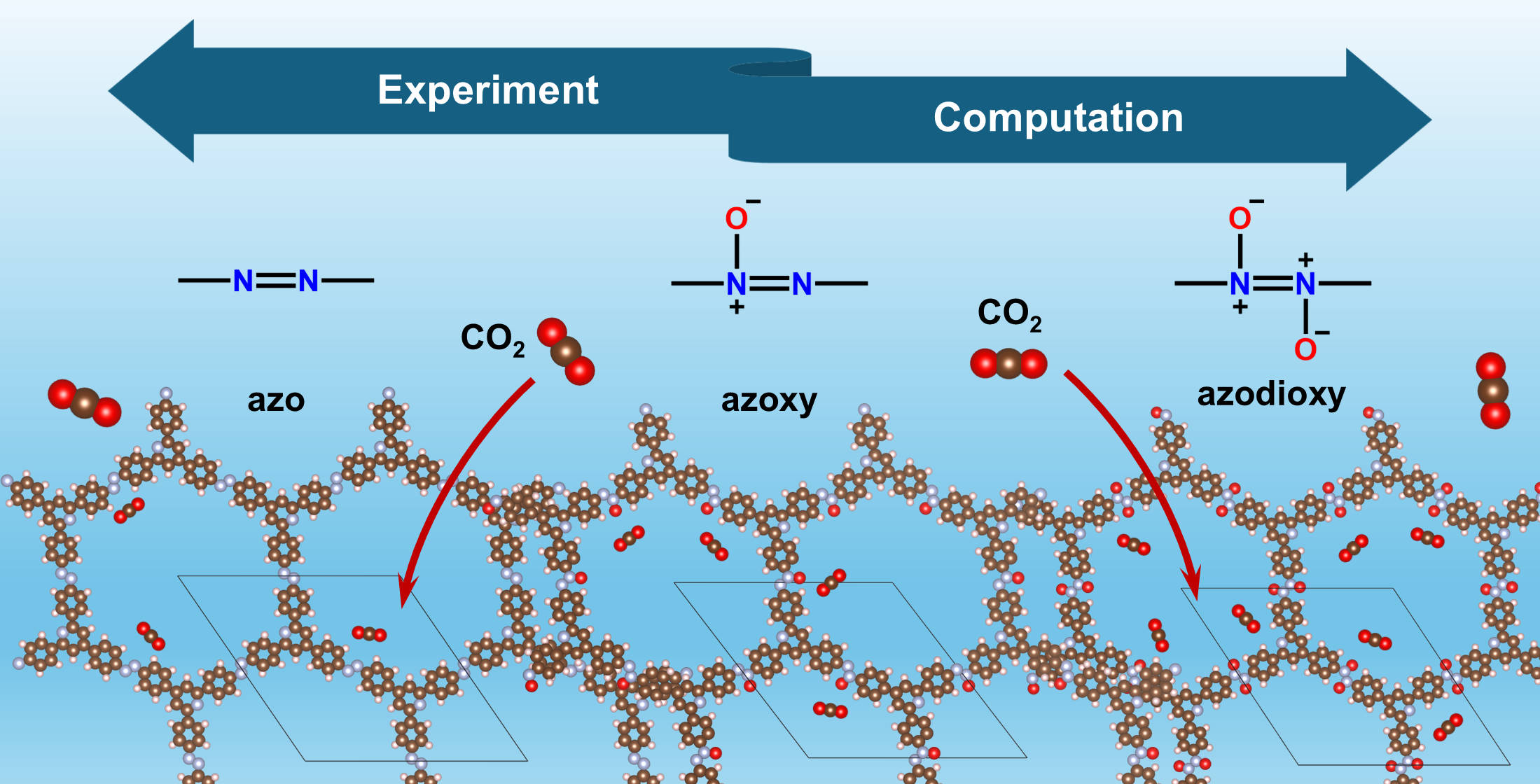
A potentially effective strategy to enhance CO2 binding affinity is to move beyond non-oxygenated azo bond (−N=N−) toward oxygenated N−N linkages, i.e. azoxy (−N=N(O)−) and azodioxy (−ON=NO−) groups, which are expected to increase linkage polarity and generate more negative electrostatic regions along the pore surfaces [50,51]. Unlike azo formation — which is typically irreversible and yields amorphous POPs — azodioxy linkages form via reversible dimerization/polymerization of aromatic C-nitroso species [52,53,54,55,56,57]. Such reversibility facilitates error correction during network formation [58], potentially leading to high structural order, as exemplified by the synthesis of monocrystalline diamondoid covalent organic networks from tetrahedral polynitroso monomers [59]. Azodioxy linkages have also been incorporated into porphyrin-based crystalline covalent organic framework (COF) prepared under ambient, homogeneous solution condition [60]. Importantly, azo/azoxy/azodioxy functionalities are chemically interrelated, offering a practical way to tune local polarity without fundamentally changing the underlying connector geometry [61]. Namely, while azodioxides are formed by the dimerization or polymerization of aromatic nitroso monomers, the condensation of nitroso and N-hydroxylamine species yields azoxy compounds, whereas the condensation of nitroso and amine monomers produces azo linkages.
In our computationally assisted CO2-adsorption studies on POP series built from trigonal units, periodic Density Functional Theory (DFT) and grand canonical Monte Carlo (GCMC) simulations indicated that replacing azo with azoxy, and especially azodioxy, linkages can enhance CO2 affinity and uptake, highlighting linkage oxidation level as a practical design variable [50,51]. In a subsequent study, we synthesized polynitroso precursors that self-associate in the solid state into E-azodioxy oligomers/polymers, demonstrating a practical nitroso-to-azodioxy route as an alternative pathway toward N−N linked porous networks [62].
A persistent challenge in POP research is that numerous materials are amorphous or only partially ordered, which limits direct structure determination and complicates quantitative structure–property relationships. Computational modelling therefore plays a central role in interpreting trends and guiding design: binding-energy calculations and electrostatic descriptors can rapidly rank functional motifs, while periodic models combined with GCMC simulations can connect pore geometry, stacking, and binding-site exposure to predicted isotherms, selectivities, and adsorption thermodynamics [33,34,50,51,62,63].
This review provides a comprehensive overview of experimental and computational approaches to azo-linked and, more broadly, N−N-linked (specifically azoxy and azodioxy) POPs. We focus on how synthetic routes, building blocks, and structural order govern CO2 adsorption, and how modelling choices — ranging from fragment vs. periodic representations to stacking treatments and descriptor selection — shape predicted CO2 adsorption trends. First, we discuss progress in the synthesis and CO2 capture performance of azo-linked POPs, highlighting the best-performing systems. We also present the relatively limited experimental studies on azodioxy and azoxy-linked POPs and their CO2 uptake characteristics. Subsequently, we review computational methodologies and theoretical studies concerning the structure and adsorption features of these materials, offering a direct comparison with experimental data. The final section summarizes the reported findings and outlines future directions for N−N linked POPs in advanced CO2 capture and catalytic conversion, with a brief perspective on emerging hybrid materials and machine learning guided design.
2. Synthesis and CO2 Capture Performance of Azo-Linked POPs
2.1. Synthesis of Azo-Linked POPs
Several synthetic routes toward the azo-linked POPs have been developed, incorporating diverse building blocks to fine-tune their functional (e.g., CO2 adsorption) properties (Figure 1). Patel et al. synthesized a series of POPs bearing azo linkages (azo-COPs) by metal catalyst-free heterocoupling of aromatic nitro and amino monomers under basic conditions (Figure 1a) [27,28]. The reaction proceeds in DMF at 150 °C for 24 h. The resulting azo-COPs exhibit various surface areas ranging from 11 to 730 m2 g−1. Another common synthesis approach to azo-linked POPs involves copper(I)-catalyzed oxidative homocoupling of aromatic amino monomers (Figure 1a). By using this method, Arab et al. synthesized highly porous azo-linked polymers (ALPs) with BET surface areas up to 1235 m2 g−1 [36,37]. They suggested that a stepwise increase in temperature and the use of mixed solvents (e.g., toluene and THF) are essential for obtaining ALPs with high surface areas. The reaction was carried out in three steps: at room temperature for 24 h, then at 60 °C for 12 h, and at 80 °C for a further 12 h. While polymerization at ambient temperature minimizes side reactions, increased catalytic activity of the catalyst at higher temperatures minimizes incomplete polymerization. Liu et al. synthesized azo-functionalized microporous organic polymers (Azo-MOPs) via oxidative polymerization of aromatic amines catalyzed by t-BuOCl/NaI [64]. The reaction proceeded rapidly (1 h) at room temperature, affording polymeric products with surface areas up to 706 m2 g−1 and high yields (> 95%). Azo-linked POPs are often synthesized by reductive homocoupling of aromatic nitro monomers. Lu et al. reported Zn-induced reductive homocoupling of four-folded tetragonal and tetrahedral nitro-containing monomers (Figure 1a) to synthesize azo-linked porous organic frameworks (POFs) [38]. The optimized reaction conditions included the use of a mixture of THF and DMF solvents, NaOH, a temperature of 65 °C, and a reaction time of 36 h, resulting in azo-POFs with moderate BET surface areas (up to 712 m2 g−1). Reductive homocoupling of four-folded monomers containing nitro groups was much more time-efficient when using NaBH4 instead of Zn. Namely, NaBH4-mediated reductive homocoupling on polynitro monomers produced azo-linked POPs (Figure 1a) of high BET surface area (up to 1478 m2 g−1) in only 30 min [39]. Nevertheless, this method was significantly more time-consuming, with a reaction time of 24 h, when employed for the synthesis of azo-linked POPs with trigonal triphenyltriazine and triphenylpyridine central units [63]. Recently, an optimized protocol for the rapid microwave-assisted synthesis of azo-linked POPs by NaBH4-mediated reductive homocoupling of three and four-folded aromatic nitro monomers was reported by our group [32]. The POPs were prepared in high yields with reduced reaction times when compared to their counterparts obtained by conventional heating, especially those bearing trigonal triphenyltriazine (15 min reaction time under microwave heating vs. 24 h under conventional heating) and triphenylpyridine units (5 min reaction time under microwave heating vs. 24 h under conventional heating). Notably, polymers obtained by microwave-assisted synthesis showed similar structural, thermal, and functional characteristics as their counterparts synthesized by conventional heating. In addition to metal-catalyzed oxidative homocoupling of aromatic amines, reductive homocoupling of nitroaromatic, and the environmentally friendly condensation of aromatic amino and nitro monomers, azo-linked POPs can also be synthesized via diazo-coupling reactions. Such approach was used for the synthesis of o-hydroxyazobenzene POPs (HAzo-POPs) [65], and triptycene-based azo-linked polymers (TAPs) [66]. Diazo-coupling reactions between the corresponding diazonium salts and polyhydroxybenzenes were performed under mild conditions in aqueous solution, yielding HAzo-POPs and TAPs with BET surface areas up to 593 and 772 m2 g−1, respectively.
Our recent study demonstrated how the choice of synthetic routes and building blocks critically influences the porosity of the resulting azo-linked polymer materials [63]. A series of benzene and triazine-based azo-linked polymers was synthesized by heterocoupling of aromatic nitro monomers and aromatic diamines of different lengths and rigidity, by oxidative homocoupling of aromatic amino monomers and by reductive homocoupling of aromatic nitro monomers. While all obtained polymers were isolated as amorphous materials with good thermal stability, they exhibited distinctly different BET surface areas. Specifically, triazine-based azo-linked polymers synthesized via NaBH4-mediated reductive homocoupling of the nitro monomer (AZO-T-P2) and copper(I)-catalyzed oxidative homocoupling of the amino monomer (AZO-T-P3) exhibited the highest BET surface areas (351 and 50.8 m2 g−1, respectively). Surprisingly, benzene and triazine-based azo linked polymers synthesized through the heterocoupling reactions of aromatic nitro monomers and various aromatic diamines exhibited very low BET surface areas. This stands in sharp contrast to azo-COPs prepared using three-dimensional (3D) building blocks, which typically display much higher porosity [27,28].
2.2. Azo-Linked POPs for CO2 Capture
The CO2 uptake capacity and selectivity of POPs are heavily influenced by their surface area, pore structure, and the presence of heteroatoms (e.g., nitrogen, oxygen, sulfur and phosphorus) [9,25]. Although a high surface area and abundant micropores are primary factors, the introduction of heteroatoms further improves the binding affinity towards CO2 via specific adsorbate-adsorbent interactions. The incorporation of nitrogen atoms has been proven particularly effective in enhancing the CO2 capture capacity and selectivity of POPs. Specifically, nitrogen atoms act as Lewis basic sites that can interact strongly with electron-deficient CO2 through dipole–quadrupole interactions. This selective interaction is facilitated by the substantially larger quadrupole moment of CO2 compared to other common gases, such as nitrogen (N2). Among various nitrogen-rich porous polymers, azo-linked POPs are particularly attractive due to their high affinity to CO2 and high CO2/N2 selectivity which increases with temperature, as reported by Patel [27]. This unusual behavior was attributed to the N2-phobicity of azo groups, originating from an entropic loss of N2 gas molecules upon binding, although the adsorption is enthalpically favorable[NO_PRINTED_FORM]. Patel et al. synthesized a family of 3D porous azo-COPs via metal catalyst-free direct coupling of aromatic nitro and amino monomers under basic conditions. To investigate the effects of building blocks on the CO2 and N2 gas sorption characteristics, a series of monomers with varying steric hindrance, rigidity, and π-surface area was utilized. Azo-COPs exhibited surface areas up to 729.6 m2 g−1 and CO2 uptake capacity up to 1.53 mmol g−1 at 298 K and 1 bar. Importantly, they showed excellent CO2/N2 selectivity, ranging from 95.6 to 165.2 at 298 K and 1 bar. The authors demonstrated that increasing the π-surface area resulted in an enhanced CO2-philic nature of the framework, reaching a remarkable CO2/N2 selectivity of 307.7 (323 K, 1 bar). The same group investigated the effect of four different polymerization routes on the CO2 sorption properties of chemically similar nanoporous azobenzene polymers (NABs) [67]. They revealed that the polymerization routes have a significant influence on the pore size distribution of the NABs and, subsequently, on the temperature dependence of their CO2/N2 selectivity. Specifically, it was suggested that pore-width maximum of 6–8 Å along with the narrow pore-size distribution and small particle size (20–30 nm) are critical for the CO2/N2 selectivity increase with rising temperatures. Given their high CO2/N2 selectivity, azo-COPs are promising candidates for post-combustion CO2 capture. Nevertheless, Arab et al. argued that the relatively low surface area of azo-COPs, which results in moderate CO2 uptake capacities, might limit their application in industrial CO2 capture and separation [36]. To overcome this limitation, they developed and optimized an alternative synthetic route to azo-linked porous organic polymers (ALPs) via the oxidative homocoupling of aromatic amine building blocks, utilizing a copper(I) bromide/pyridine system. This synthetic approach yielded ALPs with significantly higher surface areas (up to 1235 m2 g−1) and superior CO2 storage capacities (up to 5.37 mmol g−1 at 273 K and 1 bar) compared to previously reported azo-COPs. However, ALPs exhibited lower CO2/N2 selectivities (ranging from 26 to 56 at 298 K and 1 bar) compared to azo-COPs [36,37]. Moreover, while azo-COPs display enhanced CO2/N2 selectivity with increasing temperature, the selectivities of ALPs decrease or remain nearly constant as temperature increases. The authors have attributed this inconsistency to the differences in porosity parameters of ALPs and azo-COPs. It was suggested that the N2-phobicity in porous polymers is due to a relatively large mesopore portion which leads to a decrease in N2 uptake capacity and consequently enhanced CO2/N2 selectivity values at higher temperatures [68]. Comparison of N2 adsorption-desorption isotherms recorded at 77 K indicated that ALPs have a lower degree of mesoporosity than azo-COPs which show relatively large portion of mesopores. The role of porosity parameters on the N2-phobicity character of azo-linked polymers was further supported by the CO2/N2 selectivities of azo-POFs, which were synthesized via Zn-induced reductive homocoupling of aromatic nitro monomers [38]. In comparison to, e.g., ALP-1, which possesses a very high surface area (1235 m2 g−1) and a high CO2 uptake capacity (3.2 mmol g−1 at 298 K and 1 bar) but low CO2/N2 selectivity (28 at 298 K), azo-POFs exhibit much lower surface areas (up to 710 m2 g−1) and CO2 uptake capacities (up to 1.9 mmol g−1 at 298 K and 1 bar) but yield higher CO2/N2 selectivity values (ranging from 37 to 42 at 298 K). High CO2/N2 selectivities (from 91 to 103 at 298 K) were also reported for triptycene-based azo-linked polymers (TAPs) bearing phloroglucinol units [66]. TAPs display hierarchical porosity, characterized by the coexistence of micropores and mesopores, exhibiting moderate surface areas (up to 772 m2 g−1) and high CO2 uptakes (up to 150 mg g−1 at 273 K and 1 bar). Another azo-linked POP with an incorporated triptycene moiety (Azo-Trip) was synthesized via Zn-induced reductive homocoupling of the corresponding nitro monomer [69]. Azo-Trip exhibits a somewhat lower surface area (510.4 m2 g−1), CO2 uptake capacity (119.6 mg g−1 at 273 K and 1 bar), and CO2/N2 selectivity (38.6 at 273 K) relative to the best-performing TAPs.
In our recent studies, we investigated the structural and functional properties of azo-linked POPs featuring various trigonal (e.g., triphenylbenzene, triphenyltriazine and triphenylpyridine) and tetragonal (tetraphenylporphyrin) building blocks. The pyridine-based azo-linked polymer (TPP-azo), synthesized via the copper(I)-catalyzed oxidative homocoupling of 2,4,6-tris(4-aminophenyl)pyridine (TAPP), exhibited a significant BET surface area of 606 m2 g−1 and a CO2 uptake of 32 mg g−1 (at 303 K) [51]. In another study, a series of azo-linked porphyrin-based POPs (APPs), incorporating linear, bent, and trigonal linkers between the tetraphenylporphyrin units (APP-1 to APP-6), was synthesized via the direct heterocoupling of 5,10,15,20-tetrakis(4-nitrophenyl)-21H,23H-porphyrin (TNPPR) and various aromatic diamines and triamines [33]. Furthermore, oxidative homocoupling of 5,10,15,20-tetrakis(4-aminophenyl)-21H,23H-porphyrin (TAPPR) and reductive homocoupling of TNPPR and TNPPR-Zn were used for the synthesis of APP-7a, APP-7b and APP-8, respectively, with directly connected tetraphenylporphyrin units. A comparison of the BET surface areas of the synthesized APPs emphasized the influence of the synthetic method and building blocks on the porosity of the final polymer materials. Namely, APP-1 to APP-6 bearing linkers between the tetraphenylporphyrin moieties exhibit much higher BET surface areas (from 469 to 608 m2 g−1) compared to APP-7a, APP-7b and APP-8 containing tetraphenylporphyrin units directly connected through azo bonds (from 0.3 to 23 m2 g−1). The BET surface areas of APP-1 to APP-6 are comparable to the values previously reported for similar azo-linked porphyrin-based POPs synthesized by the same heterocoupling method [45,46,47,48,49]. In general, greater surface areas resulted in higher CO2 capture capacities at 306 K, particularly for APP-1 (41 mg g−1) and APP-2 (38 mg g−1), which feature a flexible methylene and ethylene bridge, respectively, and APP-5 (38 mg g−1), which contains a nitrogen-rich melamine linker. Nevertheless, the variations in CO2 uptake among the APPs with and without linkers between the tetraphenylporphyrin moieties are notably less pronounced than the differences in their BET surface areas. This suggests that the presence of CO2-philic azo groups and pyrrole rings contributes significantly to the CO2 binding affinity of the APPs. The CO2 capture capacities of the APPs are comparable to those measured for analogous azo-linked porphyrin-based POPs featuring linkers between the tetraphenylporphyrin units, such as Azo-CPPs (39.9–55.4 mg g−1 at 303 K) [48]. For Azo-CPPs, the calculated CO2/N2 selectivities ranged from 27.4 to 53.9 at 273 K, and from 31.2 to 107.8 at 303 K. These values are lower than those reported for Azo-COPs (63–124 at 273 K). Similarly to the Azo-COPs, the CO2/N2 selectivity of Azo-CPPs increases at higher temperatures, highlighting these azo-linked porphyrin-based polymers as potential candidates for post-combustion CO2 capture and sequestration technologies. Other reported porphyrin-based POPs containing azo linkages shown excellent CO2 uptake capacities (up to 3.98 mmol−1 at 273 K) and high CO2/N2 selectivity (up to 64.3 at 273 K) [45,46,47]. In our most recent work, we investigated the influence of heteroatom-containing linkers – specifically hydroxylated biphenyl (APP-BP-OH) and carbonyl-bearing anthraquinone (APP-AQ) – as well as sterically hindered linkers (methylated biphenyl and phenyl in APP-BP-Me and APP-Ph-Me, respectively) on the CO2 capture performance of APPs [34]. Although BET surface areas of these APPs vary widely from 167 to 673 m2 g−1, the variations in CO2 uptake capacity remain notably less pronounced. Specifically, APP-BP-OH (Figure 2) exhibited the highest CO2 uptake of 49 mg g−1 (at 303 K), while the remaining APPs yielded comparable values of 40 mg g−1 (APP-AQ) and 41 mg g−1 (APP-BP-Me and APP-Ph-Me). These results demonstrate that CO2 adsorption is governed by a complex interplay of factors, including the local chemical environment and linker topology, rather than surface area alone. Beyond the influence of the nitrogen-rich porphyrin and azo units, the presence of polar hydroxyl groups facilitates additional favorable interactions between CO2 and the framework. This is evident from a direct comparison of CO2 uptakes: APP-BP-OH achieves 49 mg g−1 (at 303 K), whereas its counterpart with an unsubstituted biphenyl linker, Azo-CPP-5,[NO_PRINTED_FORM] reaches 39.9 mg g−1 (at 303 K) [48]. Furthermore, the biphenyl linker itself appears to enhance CO2 capture capacity regardless of the surface area. Despite APP-BP-Me having a significantly lower BET surface area (167 m2 g−1) than APP-Ph-Me (673 m2 g−1), both materials exhibit identical CO2 uptakes of 41 mg g−1.
3. Synthesis and CO2 Capture Performance of Azodioxy and Azoxy-Linked POPs
One of the drawbacks of azo-linked POPs is their amorphous nature, which stems from the irreversible formation of azo bonds. This structural disorder leads to several limitations in applications of these materials such as buried surface area where pores are either too narrow or physically blocked for CO2 access; a broad pore size distribution that permits the co-adsorption of N2 and CO2, thereby reducing CO2/N2 selectivity; and diminished reproducibility, as the synthesis is governed by kinetic factors rather than thermodynamic equilibrium. The formation of crystalline POPs, such as covalent organic frameworks (COFs), is guided by the principles of reticular chemistry. This process requires reversible bond cleavage and formation, which allows the system to correct structural errors and reach the most thermodynamically stable, ordered state. Aromatic C-nitroso compounds are well known for their ability to reversibly dimerize or polymerize via Z- or, more commonly, E-azodioxy linkages, presenting a promising avenue within dynamic covalent chemistry [52,53,70,71]. Indeed, Beaudoin et al. showed that the self-addition polymerization of suitably designed aromatic polynitroso monomers with tetrahedrally oriented nitroso groups resulted in the formation of large single crystals of diamondoid azodioxy networks (Figure 3a) [59]. These monocrystalline covalent organic networks typically crystallize as solvates, where the solvent molecules occupy the voids. Upon the removal of these guests, the resulting solids lose their crystallinity and do not exhibit permanent porosity. Spontaneous formation of azodioxy linkages was also employed for the synthesis of a porous, semiconductive porphyrin-based COF with a reported BET surface area of 447 m2 g−1, showing I2-doping enhanced photoconductivity [60]. To the best of our knowledge, these rare examples of azodioxy-linked POPs have not been investigated as CO2 adsorbents. To fill this gap, we recently synthesized novel aromatic polynitroso compounds with para-nitroso groups attached to the central triphenylbenzene, triphenylpyridine, triphenyltriazine, and triphenylamine cores, and investigated their polymerization into the corresponding azodioxy networks as well as their CO2 capture performance [62]. The synthesis of the pyridine and amine-based derivatives was performed via a standard procedure involving the reduction of aromatic nitro derivatives to N-arylhydroxylamines, followed by an oxidation step yielding 2,4,6-tris(4-nitrosophenyl)pyridine and the dinitroso derivative 4,4'-((4-nitrophenyl)methylene)bis(nitrosobenzene). Since attempts to synthesize benzene and triazine-based trinitroso compounds by a similar procedure proved unsuccessful, a novel synthetic strategy was implemented, involving the cyclotrimerization of 4-nitrosoacetophenone and 4-nitrosobenzonitrile, respectively (Figure 3b). Trinitroso derivatives containing triphenylbenzene and triphenyltriazine cores underwent self-polymerization to form E-azodioxy linked polymers (Figure 3b), whereas pyridine-based trinitroso and amine-based dinitroso compounds formed only E-oligomers in the solid state. The resulting materials exhibited modest BET surface areas (up to 48.1 m2 g−1) and relatively low CO2 uptake values (up to 6.2 mg g−1 at 303 K). For comparison, analogous POPs with azo linkages, such as triphenyltriazine-based AZO-T-P2 and triphenylpyridine-based TPP-azo, show significantly higher BET surface areas of 351 m2 g−1 [63] and 606 m2 g−1 [51], respectively, as well as CO2 uptake capacities of 22 mg g−1 (at 306 K) [32]and 32 mg g−1 (at 303 K) [51].
During the synthesis of 4-nitrosobenzonitrile, a starting material for 2,4,6-tris(4-nitrosophenyl)-1,3,5-triazine, a nitrile-substituted azoxybenzene derivative, 1,2-bis(4-cyanophenyl)diazene oxide, was formed as a by-product. This compound was subsequently subjected to a trifluoromethanesulfonic acid-catalyzed trimerization reaction under microwave conditions to yield an azoxy-linked triazine-based polymer [72]. The resulting polymer is an amorphous solid with good thermal stability and is characterized as a wide-bandgap semiconductor. However, it exhibits a rather low BET surface area of 10.2 m2 g−1 and, in contrast to its azo and azodioxy counterparts [32,62], shows no affinity for CO2 capture. To the best of our knowledge, there are no other reports on azoxy-linked POPs.
4. Computational Studies of N−N-Linked POPs
4.1. Computational Methodology for Studying Gas Adsorption in N−N-Linked POPs
Since azo-linked POPs are amorphous solids, structural information is typically limited to the expected connectivity (formation of −N=N− linkages), which was confirmed by IR and NMR spectroscopy. However, spectroscopic data alone provide little insight into how building units are arranged in 3D space. For computational modelling, certain assumption regarding the 3D organization and topology of the material are required, as adsorption properties strongly depend on the underlying framework geometry. Therefore, idealized periodic models are often constructed to represent the connectivity between the building units and the stacking arrangements of the layers. These models enable a systematic investigation of how linkage type and layer organization influence the adsorption properties of POPs. Although not ideal, such a computational approach can reproduce the experimentally observed trends reasonably well, supporting its use as a predictive tool when experimental findings are limited.
The computational methodology used throughout our studies follows a hierarchical strategy (Figure 4). When an experimentally resolved 3D structure was available — or when the material was closely analogous to previously characterized COFs, such as those constructed with imine linkages — the initial steps of the workflow could be omitted. Where no reliable periodic structure existed, or when the connectivity pattern was uncertain, the analysis began with the simplest level: isolated 0D molecular fragments (Figure 4a). These fragments were used to initially check binding preferences, electrostatic features of the building blocks, functional groups, and N−N linkages.
The next stage involved assembling the fragments into idealized 2D layers that captured the expected connectivity of the building blocks, such as azo bonds, but also azoxy and azodioxy linkages. Finally, these 2D layers were arranged into different plausible stacking modes to generate full 3D periodic models of the POPs (Figure 4b). This multilevel procedure allowed us to investigate the influence of connectivity, linkage type, and layer stacking even when direct structural information was unavailable, while maintaining the structural consistency of the investigated POPs.
Periodic DFT calculations are usually performed on idealized frameworks constructed from trigonal building blocks (e.g., triphenylbenzene, triphenylpyridine, triphenyltriazine and triphenylamine) [50,51,62,63], as well as tetragonal ones (e.g., tetraphenylporphyrin) [33,34], linked via nitrogen-nitrogen bonds. Several stacking arrangements (e.g., eclipsed AA, inclined AA′, serrated AA′, and staggered AB) were optimized using the PBE-D3 functional and pob-TZVP-rev2 basis sets in CRYSTAL23 [73]. Although these models represent perfectly ordered 3D architectures, they allow for a direct comparison of the relative stabilities and pore geometries of the corresponding POPs.
GCMC simulations in RASPA [74] are the final step that is performed on the optimized periodic structures to estimate CO2 and N2 adsorption capacities, as well as CO2/N2 selectivities, under experimentally relevant conditions (Figure 4c). In addition to adsorption isotherms, simulations generate 3D adsorption density plots that show the time-averaged spatial distribution of adsorbate molecules, providing direct insight into the preferred adsorption sites for gas molecules within the pores. Furthermore, GCMC simulations with a single adsorbate molecule in the framework enable the calculation of adsorption enthalpies, typically evaluated at the infinite-dilution, where adsorbate-adsorbate interactions are negligible.
In combination with ESP maps derived from periodic DFT, the simulations show that the most negative ESP regions of the framework correspond closely to the preferred CO2 adsorption sites. The combination of periodic DFT, ESP analysis, and GCMC simulations provides a robust protocol for interpreting the adsorption behavior of POPs. Although based on idealized structural models, computational predictions show good agreement with experimentally observed CO2 uptakes, demonstrating the utility of this hierarchical approach, particularly in systems where crystallographic information is lacking or limited.
4.2. Computational Studies of Azo-Linked Frameworks for CO2 Adsorption
Computational studies on azo-linked frameworks in the specific context of CO2 adsorption remain relatively scarce. Most published modelling has instead emphasized electronic structure and stacking stability or disorder, whereas adsorption thermodynamics and quantitative structure-property relationships (e.g., pore accessibility vs. binding-site polarity vs. stacking) are still comparatively less explored.
In our studies, computational analysis of azo-based frameworks primarily served to clarify how the intrinsic properties of the azo linkage, in combination with connector geometry and layer stacking (Figure 4a), influence CO2 adsorption (Table 1). Azo linkages in individual fragments showed the weakest interaction with CO2 among the three types of N−N linkages investigated (i.e., azo, azoxy, and azodioxy). Azo-linked benzene and triazine frameworks were modelled in their energetically preferred AA (eclipsed) and the less stable AB (staggered) configurations. For benzene-based azo systems, CO2 uptakes obtained from GCMC simulations increased from 19 mg g⁻¹ (AA) to 65 mg g⁻¹ (AB), demonstrating that interlayer displacement can expose additional binding regions and enhance CO2 uptake. In triazine-based azo systems, this effect was even more pronounced, and the more accessible triazine units in AB stacking increased the predicted CO2 uptake from 14 mg g⁻¹ (AA) to 92 mg g⁻¹ (AB) [50].
The study expanded the analysis to triphenylamine and triphenylpyridine azo frameworks and further introduced additional stacking modes, including inclined AA′ and serrated AA′ arrangements (Figure 5b) [51]. In the case of triphenylpyridine, the relative energies of these geometries lie within a relatively small energy window of about 7 kJ mol⁻¹, indicating the coexistence of differently stacked configurations. For triphenylpyridine, the CO2 uptake shows less pronounced variations compared to other investigated frameworks, ranging from 18 mg g⁻¹ for the AA and serrated stackings to 31 mg g⁻¹ in the AB mode, and reaching 37 mg g⁻¹ for the inclined configuration. For this compound, an excellent correlation was observed between the negative electrostatic potential values on the framework surface, particularly around the azo nitrogen atoms, and the simulated CO2 uptake. This supports the use of ESP as an informative qualitative descriptor for identifying the most favorable adsorption configurations in azo-linked systems (Figure 5c). These findings also provided a useful basis for subsequent comparison with azoxy and azodioxy analogues, where the introduction of oxygen substantially increases linkage polarity and influences CO2 binding affinity[51].
The computational study was extended to a series of azo-linked POPs constructed from trigonal benzene and triazine connectors combined with linear linkers of varying length, which expand the predicted hexagonal pores via insertion between the building blocks [63]. Idealized periodic models of azo-linked benzene and triazine-based frameworks were constructed and optimized using periodic DFT, assuming predominantly eclipsed AA stacking as a preferred reference structure. The calculated CO2 uptake capacities for the azo-linked systems typically fall within a range from 20 to 35 mg g−1, showing good qualitative agreement with experimentally measured values despite the amorphous nature of the real materials. A key outcome of the computational analysis was that variations in linker length and connector substitution had only a modest impact on CO2 uptake within the azo-linked series. Extending the distance between trigonal nodes or introducing additional phenyl spacers failed to yield a systematic increase in adsorption capacity[63].
Further computational analysis focused on frameworks constructed from tetragonal tetraphenylporphyrin building blocks, either directly connected through azo bonds or separated by linear, bent, or trigonal linkers [33]. In these systems, the porphyrin unit itself plays an active role in CO2 binding, serving as an additional adsorption site alongside the azo linkage. Periodic DFT and GCMC simulations revealed that such POPs can achieve moderate to relatively high CO2 uptakes, with adsorption performance strongly dependent on framework dimensionality, pore accessibility, and stacking arrangement. Systems featuring linker-bridged porphyrin units, including 2D layered and 3D pto-type networks, exhibited calculated CO2 capacities of up to 49 mg g−1, which is in good agreement with the experimentally measured values of approximately 38 mg g−1 for the most porous materials in the series. In contrast, frameworks composed of directly connected azo-linked porphyrin units exhibited significantly lower surface areas, yet maintained non-negligible CO2 uptakes (23 mg g−1), indicating that strong local interactions at the azo and porphyrin sites can partially compensate for the reduced porosity[33].
Most recently, the computational analysis was extended to a new set of azo-linked porphyrin POPs (APPs) in which the linker chemistry was tuned using heteroatom-containing (hydroxylated biphenyl and anthraquinone) versus sterically hindered (methylated biphenyl and methylated phenyl) diamines [34]. Although these polymers are amorphous, periodic DFT and GCMC simulations on ordered layered reference models reproduced the key qualitative trend seen experimentally — the hydroxylated derivative (APP-BP-OH) outperforms its methylated analogue in CO2 uptake despite a lower surface area. This confirms that adsorption is governed not only by porosity but also by the local chemical environment. Analysis of electrostatic potential maps and CO2 density distributions showed that adsorption is dominated by nitrogen-rich porphyrin/azo motifs, while hydroxyl and carbonyl groups introduce additional accessible binding regions that can further enhance uptake. Importantly, the study also explored alternative stacking arrangements and showed that small lateral shifts between layers can strongly increase pore accessibility and, consequently, the predicted CO2 uptake. For APP-BP-OH, the eclipsed AA model yields 25 mg g−1 (1750 m2 g−1), whereas a serrated AA′ offset significantly raises the calculated uptake to 213 mg g−1 (2716 m2 g−1), with the staggered AB model providing an intermediate 85 mg g−1 (1894 m2 g−1). In addition, uptake trends were complemented with zero-loading enthalpies (−13.6 to −14.9 kJ mol−1) and BSSE-corrected DFT interaction energies, showing stronger CO2 binding to hydroxyl than to carbonyl sites (−31 vs −27 kJ mol−1), consistent with the slightly higher simulated uptake of APP-BP-OH (25 mg g−1) compared with APP-AQ (23 mg g−1). Overall, these results provide a clear mechanistic link between linker functionalization, stacking accessibility, and CO2 adsorption in azo-linked porphyrin POPs[34].
Analysis of CO2 density distributions and electrostatic potential maps showed that favorable adsorption occurs preferentially near the azo linkages and around the porphyrin rings, particularly when these sites are sterically accessible on both sides of the porphyrin plane, as in hypothetical AB stacking configurations. However, such arrangements were computationally found to be less stable than the eclipsed AA stacking. These findings are consistent with conclusions from trigonal azo-linked frameworks that in azo-based POPs, CO2 adsorption is governed not only by the overall surface area but also by the local electrostatic landscape and the accessibility of the nitrogen-rich motifs. Despite the idealized nature of the models and the lower experimental BET surface areas of the synthesized amorphous polymers, the calculations provide useful qualitative guidance. For example, the experimental CO2 uptake of 32 mg g−1 (at 303 K) for the pyridine-based azo-linked polymer (TPP-azo) closely matches the GCMC-predicted values for laterally displaced stacking models (31 mg g−1 for AB staggered and 37 mg g−1 for inclined AA’ configuration).
4.3. Computational Studies of Azo-Linked POPs Beyond CO2 Adsorption
An early example of computational study related to gas adsorption in a porous material containing azo bonds was reported for a framework obtained by the reaction of 4,4′-azodianiline with 1,3,5-triformylphloroglucinol, in which the azo linkage is indirectly introduced into the framework through the azodianiline moiety [75]. Adsorption of CO2, N2, CH4, and H2 was investigated experimentally. However, the computational analysis was limited to GCMC simulations of H2 adsorption only, performed on an idealized eclipsed structural model. Interpretations of CO2 selectivity relied primarily on experimental trends and qualitative arguments related to the CO2-philicity and N2-phobicity of azo groups [76]. The same framework served as a model system in a study that employed a dispersion-corrected periodic DFT to demonstrate that the widely assumed eclipsed AA stacking is not an energy minimum. Instead, calculations revealed several low-energy slipped configurations. Beyond structural implications, the study shows that stacking displacement can have a strong effect on electronic structure (e.g., band gap) [77]. This conclusion was further strengthened by a finite-temperature study based on machine-learning force fields trained on DFT data, which showed that eclipsed stacking is dynamically unstable under ambient conditions and that slipped configurations are entropically stabilized. Simulated PXRD patterns averaged over dynamic trajectories reproduced experimental diffraction features more accurately than static eclipsed models [78].
These findings are consistent with previous reports. Several computational and combined experimental-theoretical studies have systematically investigated interlayer stacking modes in 2D COFs and their influence on structural and electronic properties. Although performed on other systems, such as imine or boronate-based COFs, these works often demonstrate that fully eclipsed AA stacking is energetically less favorable, primarily due to interlayer π-π repuslion and electrostatic interactions between adjacent aromatic layers. Early molecular mechanics and DFT studies mapped the potential energy surfaces (PES) of dozens of COFs and showed that energy minima generally occur for horizontally displaced layers, with offsets of approximately 1–2 Å relative to the eclipsed geometry [79,80]. Subsequent diffraction-based analysis and simulations demonstrated that inclined (AA′) and zigzag (AB) stacking modes reproduce experimental PXRD patterns more accurately than eclipsed models, particularly for materials with limited crystallinity [81]. Experimental studies employing advanced techniques, such as solid-state NMR [82], further confirmed that stacking disorder and polymorphism [83] are intrinsic features of layered COFs, rather than defects or minor perturbations.
In parallel, a purely computational study used periodic DFT to propose hypothetical azo-linked COFs constructed from benzene, triphenylamine, triphenylbenzene, and triphenyltriazine connectors as conjugated semiconductors, and focused on electronic properties relevant for photocatalysis. In this work, the azo bond was treated as a backbone linkage enabling extended conjugation, but the calculations focused on band structures and band gaps from single-layer periodic 2D sheets. Although no adsorption simulations were performed, the study demonstrated that azo-linked frameworks exhibit electronic properties distinct from those of more commonly studied imine-linked analogues [84].
Finally, a practical synthetic route to true azo-linked COFs was established via imine to azo linkage exchange [85]. DFT was primarily employed to rationalize the electronic consequences of introducing the −N=N− linkage into the backbone, most notably bandgaps change consistent with experimental photophysical and photocatalytic trends. The computational study assumed idealized eclipsed AA stacking and focused on electronic structure, without directly addressing CO2 adsorption thermodynamics or stacking disorder.
4.4. Computational Study of Azodioxy and Azoxy-Linked Frameworks for CO2 Adsorption
One motivation for examining oxygenated N−N linkages was the hypothesis that azodioxy-based materials might exhibit a higher degree of structural order than their azo analogues. The idea was supported by the reported formation of crystalline 3D diamondoid networks from polynitroso monomers, enabling their characterization by single-crystal X-ray diffraction [59]. Computational methods proved to be a valuable tool for guiding synthetic procedures, as they are relatively inexpensive and can be applied to a wide range of systems. In the initial computational screening, six idealized 2D POPs were constructed from trigonal benzene and triazine-based connectors linked through azo, azoxy, or azodioxy bonds.
Binding-energy calculations on simple molecular fragments (B3LYP-D3(BJ)/def2-TZVP, BSSE-corrected) showed that introducing oxygen atoms into the N−N linkage systematically enhances the interaction with CO2 compared with azo units. Among the three linkage types, azodioxy fragments gave the most favorable CO2 binding energies of about −20.9 kJ mol−¹, followed by azoxy (–17.7 kJ mol⁻¹) and azo (–16.5 kJ mol⁻¹). This trend provided the computational indication that oxygenated N−N linkages could significantly improve the adsorption performance of POPs [50].
Periodic DFT (PBE-D3/pob-TZVP-rev2) was used to optimize energetically preferred AA (eclipsed) and the less stable AB (staggered) configurations of the corresponding 2D POPs, followed by GCMC simulations of CO2/N2 adsorption at 298 K and 1 bar. Azoxy and especially azodioxy linkages create highly negative ESP regions along the pore walls, which correlate with localized CO2 density near these linkages and significantly enhanced CO2 uptake compared with azo analogues. In benzene-based frameworks, both AA and AB azodioxy-linked systems exhibit the highest calculated CO2 capacity overall, reaching 30 mg g⁻¹ and 118 mg g⁻¹, respectively. In contrast, triazine-based analogues reveal a more balanced behavior: the more planar azo-linked triazine structure AB can outperform its azodioxy counterpart as layer distortion in the latter reduces the accessible adsorption sites. The study also highlighted the importance of slightly slipped stacking modes, such as AA′, where imperfect alignment of adjacent layers exposes additional high-affinity binding regions on both the linkages and the building blocks, further influencing the overall CO2 adsorption, from 31 mg g⁻¹ for AA to 118 mg g⁻¹ for AB. This observation is consistent with previously reported studies of boronate-based 2D COFs, where small lateral offsets between layers were energetically preferred and structurally more realistic than ideal AA packings [79,86].
This computational study was later extended to six additional POPs assembled from triphenylamine and triphenylpyridine building blocks linked through azo, azoxy, or azodioxy bonds. In this work, not only AA and AB but also inclined AA′ and serrated AA′ stacking modes were examined [51]. Modest lateral offsets between layers can enhance adsorption performance by exposing high-affinity regions on the framework that are partially shielded in case of perfectly eclipsed AA geometry. For triphenylamine based azodioxy systems, AB packings yielded the highest simulated CO2 uptakes of 177 mg g⁻¹. In contrast, triphenylpyridine-based frameworks exhibited small energy differences between AA and AA′, suggesting that multiple stacking arrangements may coexist and jointly influence their CO2 adsorption behavior.
A central outcome of the studies was the clear correlation between the most negative ESP regions on the framework, located either on the azo nitrogen atoms (Figure 5c) or, more likely, on azoxy/azodioxy oxygen atoms (Figure 5d), and both the magnitude of CO2 uptake and the spatial distribution of CO2 density within the pores. This supports the use of ESP as a simple and computationally relatively inexpensive descriptor for pre-screening of candidate linkages and connectors before undertaking full GCMC simulations and offering at least a qualitative indication of the expected CO2 adsorption capacity.
To validate the stacking models, we combined periodic DFT and PXRD simulations for azodioxy polymers derived from polynitroso compounds with triphenyl-substituted benzene, pyridine, triazine, and amine cores. Four stacking arrangements (AA, inclined AA′, serrated AA′, and AB) were optimized using PBE-D3/pob-TZVP-rev2 level of theory and employed to simulate PXRD patterns. Comparison with experimental data revealed that the PXRD of the triphenyltriazine-based azodioxy polymer is best matched by a serrated AA′ stacking, and not by the simple eclipsed AA geometry that is commonly assumed for 2D COFs (Figure 3b). These observations are consistent with earlier computational studies suggesting that slipped configurations[50,51] can be energetically competitive and structurally realistic [50,51]. Such layer offsets can enhance CO2 uptake by exposing additional adsorption sites, providing a possible explanation for the adsorption behavior found in amorphous or partially ordered frameworks.
4. Conclusion and Perspectives
Overall, this review highlights azo-linked POPs as a versatile platform for CO2 capture, where performance is governed by the interplay of building-block topology and functionalization, porosity, and the local binding-site environment. Across reported systems, enhanced CO₂ adsorption can be achieved, yet uptake does not scale uniformly with BET surface area — most notably in porphyrin-based networks, where nitrogen-rich motifs and linker polarity make substantial contributions to CO₂ affinity. Combined experimental and computational analysis further shows that pore accessibility and layer stacking can dominate adsorption trends — laterally displaced stackings often reproduce experimental uptakes better than ideal eclipsed models, particularly when several stacking arrangements are close in energy. Electrostatic descriptors (e.g., ESP) provide a practical route to locate high-affinity regions and rationalize CO2 density distributions.
The review also indicates, based on computational analysis, that oxygenated N−N linkages (azoxy/azodioxy) are a promising polarity-tuning handle that can strengthen CO2 binding in computational models, while reversible nitroso/azodioxide chemistry offers an alternative synthetic pathway toward more ordered N–N-linked networks. However, experimentally realized azoxy/azodioxy-linked POPs remain rare and, to date, have not been developed into high-capacity CO2 adsorbents, highlighting an opportunity for the design of new CO2 sorbents.
Although substantial progress has been made, the translation of POP adsorbents to industrial CO2 capture remains constrained by material stability under realistic operating conditions, production cost and scalability, and practical industrial implementation. Continued research should therefore prioritize improving durability and regenerability, lowering synthesis cost and complexity, and demonstrating performance under application-relevant conditions. Specifically, future work should focus on: (i) enhancing material stability, inlcuding thermal, chemical, and cycling durability under humidity and complex gas mixtures; (ii) reducing synthesis cost and time through greener, energy-efficient protocols such as solvent-free or green-solvent routes; and (iii) improving process practicality, including shaping, compatibility with existing capture infrastructure, and overall regeneration performance. In parallel, materials design should continue to exploit classical performance levers—pore volume, pore-size distribution and surface area — while also optimizing framework polarity and functional-site chemistry to boost CO2 affinity and selectivity.
Several directions highlighted in broader CO2-capture research are particularly relevant for POPs. One is the development of multifunctional porous materials that combine capture with post-capture conversion (e.g., incorporating catalytic sites for photo- or electro-driven utilization) without compromising adsorption performance. In this regard, azo-linked POPs with additional nitrogen-rich motifs (e.g., porphyrins, triazines, and pyridines) are especially attractive as heterogenous catalysts for CO2 conversion into high-value chemicals, such as cyclic carbonates. Furthermore, porphyrin-based POPs allow for the incorporation of metal ions to enhance their catalytic performance. Another is the move toward composite and hybrid architectures, where combining different adsorbents can improve robustness, moisture tolerance, and regeneration efficiency under application-specific conditions. Finally, computational chemistry and machine learning are playing an increasingly important role — simulations can screen functional motifs and pore environments, estimate adsorption and regeneration-relevant descriptors, and help rationalize stability and performance trends; expanding, well-curated experimental/computational datasets will further improve machine learning predictiveness and accelerate optimization processes. Overall, advancing POPs adsorbents will require tighter coupling of synthetic innovation, stability/regeneration testing, and computation-guided design, supported by cross-disciplinary collaboration and practical and economic considerations.
Author Contributions
Conceptualization, I.K. and I.B.; methodology, I.K. and I.B.; writing—original draft preparation, I.K. and I.B.; writing—review and editing, I.K. and I.B.; visualization, I.K. and I.B.; project administration, I.B.; funding acquisition, I.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been fully supported by Croatian Science Foundation under project IP-2020-02-4467.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
The support of project CIuK co-financed by the Croatian Government and the European Union through the European Regional Development Fund—Competitiveness and Cohesion Operational Programme (Grant KK.01.1.1.02.0016) is acknowledged. The authors acknowledge the CERIC-ERIC Consortium for the access to experimental facilities.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ma, H.; Fu, H.; Tong, Y.; Umar, A.; Hung, Y.M.; Wang, X. Advances in CO2 Capture and Separation Materials: Emerging Trends, Challenges, and Prospects for Sustainable Applications. Carbon Capture Sci. Technol. 2025, 15, 100441. [Google Scholar] [CrossRef]
- Zhong, Z.; Wang, X.; Tan, B. Porous Organic Polymers for CO2 Capture and Catalytic Conversion. Chem. Eur. J. 2025, 31. [Google Scholar] [CrossRef]
- Yoro, K.O.; Daramola, M.O. CO2 Emission Sources, Greenhouse Gases, and the Global Warming Effect. In Advances in Carbon Capture; Elsevier, 2020; pp. 3–28. [Google Scholar]
- Nunes, L.J.R. The Rising Threat of Atmospheric CO2: A Review on the Causes, Impacts, and Mitigation Strategies. Environments 2023, 10, 66. [Google Scholar] [CrossRef]
- Carbon Dioxide Now More than 50% Higher than Pre-Industrial Levels. Available online: https://www.noaa.gov/news-release/carbon-dioxide-now-more-than-50-higher-than-pre-industrial-levels (accessed on 23 February 2026).
- Carbon Dioxide - Earth Indicator. Available online: https://science.nasa.gov/earth/explore/earth-indicators/carbon-dioxide/ (accessed on 23 February 2026).
- Zentou, H.; Hoque, B.; Abdalla, M.A.; Saber, A.F.; Abdelaziz, O.Y.; Aliyu, M.; Alkhedhair, A.M.; Alabduly, A.J.; Abdelnaby, M.M. Recent Advances and Challenges in Solid Sorbents for CO2 Capture. Carbon Capture Sci. Technol. 2025, 15, 100386. [Google Scholar] [CrossRef]
- Singh, G.; Lee, J.; Karakoti, A.; Bahadur, R.; Yi, J.; Zhao, D.; AlBahily, K.; Vinu, A. Emerging Trends in Porous Materials for CO2 Capture and Conversion. Chem. Soc. Rev. 2020, 49, 4360–4404. [Google Scholar] [CrossRef] [PubMed]
- Song, K.S.; Fritz, P.W.; Coskun, A. Porous Organic Polymers for CO2 Capture, Separation and Conversion. Chem. Soc. Rev. 2022, 51, 9831–9852. [Google Scholar] [CrossRef] [PubMed]
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.-H.; Long, J.R. Carbon Dioxide Capture in Metal–Organic Frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar] [CrossRef]
- Sai Bhargava Reddy, M.; Ponnamma, D.; Sadasivuni, K.K.; Kumar, B.; Abdullah, A.M. Carbon Dioxide Adsorption Based on Porous Materials. RSC Adv. 2021, 11, 12658–12681. [Google Scholar] [CrossRef]
- Kumar, S.; Srivastava, R.; Koh, J. Utilization of Zeolites as CO2 Capturing Agents: Advances and Future Perspectives. J. CO2 Util. 2020, 41, 101251. [Google Scholar] [CrossRef]
- Boer, D.G.; Langerak, J.; Pescarmona, P.P. Zeolites as Selective Adsorbents for CO2 Separation. ACS Appl. Energy Mater. 2023, 6, 2634–2656. [Google Scholar] [CrossRef]
- Li, Z.; Liu, P.; Ou, C.; Dong, X. Porous Metal–Organic Frameworks for Carbon Dioxide Adsorption and Separation at Low Pressure. ACS Sustain. Chem. Eng. 2020, 8, 15378–15404. [Google Scholar] [CrossRef]
- Ghanbari, T.; Abnisa, F.; Wan Daud, W.M.A. A Review on Production of Metal Organic Frameworks (MOF) for CO2 Adsorption. Sci. Total Environ. 2020, 707, 135090. [Google Scholar] [CrossRef] [PubMed]
- Trickett, C.A.; Helal, A.; Al-Maythalony, B.A.; Yamani, Z.H.; Cordova, K.E.; Yaghi, O.M. The Chemistry of Metal–Organic Frameworks for CO2 Capture, Regeneration and Conversion. Nat. Rev. Mater. 2017, 2, 17045. [Google Scholar] [CrossRef]
- Ding, M.; Flaig, R.W.; Jiang, H.-L.; Yaghi, O.M. Carbon Capture and Conversion Using Metal–Organic Frameworks and MOF-Based Materials. Chem. Soc. Rev. 2019, 48, 2783–2828. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xie, L.-H.; Li, J.-R.; Ma, Y.; Seminario, J.M.; Balbuena, P.B. CO2 Capture and Separations Using MOFs: Computational and Experimental Studies. Chem. Rev. 2017, 117, 9674–9754. [Google Scholar] [CrossRef]
- Bhanja, P.; Modak, A.; Bhaumik, A. Porous Organic Polymers for CO2 Storage and Conversion Reactions. ChemCatChem 2019, 11, 244–257. [Google Scholar] [CrossRef]
- Mohamed, M.G.; EL-Mahdy, Ahmed.F.M.; Kotp, M.G.; Kuo, S.-W. Advances in Porous Organic Polymers: Syntheses, Structures, and Diverse Applications. Mater. Adv. 2022, 3, 707–733. [Google Scholar] [CrossRef]
- Acevedo, S.; Giraldo, L.; Moreno-Piraján, J.C. Adsorption of CO2 on Activated Carbons Prepared by Chemical Activation with Cupric Nitrate. ACS Omega 2020, 5, 10423–10432. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Z.; Xue, C.; Chen, H.; Han, X.; Ren, Y. Amorphous Porous Organic Polymers Containing Main Group Elements. Commun. Chem. 2023, 6, 271. [Google Scholar] [CrossRef]
- Zhu, Y.; Long, H.; Zhang, W. Imine-Linked Porous Polymer Frameworks with High Small Gas (H2, CO2, CH4, C2H2) Uptake and CO2/N2 Selectivity. Chem. Mater. 2013, 25, 1630–1635. [Google Scholar] [CrossRef]
- Gu, C.; Liu, D.; Huang, W.; Liu, J.; Yang, R. Synthesis of Covalent Triazine-Based Frameworks with High CO2 Adsorption and Selectivity. Polym. Chem. 2015, 6, 7410–7417. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, M.; Yuan, D. Carbon Dioxide Capture in Amorphous Porous Organic Polymers. J. Mater. Chem. A 2017, 5, 1334–1347. [Google Scholar] [CrossRef]
- Nandi, S.; Werner-Zwanziger, U.; Vaidhyanathan, R. A Triazine–Resorcinol Based Porous Polymer with Polar Pores and Exceptional Surface Hydrophobicity Showing CO2 Uptake under Humid Conditions. J. Mater. Chem. A 2015, 3, 21116–21122. [Google Scholar] [CrossRef]
- Patel, H.A.; Hyun Je, S.; Park, J.; Chen, D.P.; Jung, Y.; Yavuz, C.T.; Coskun, A. Unprecedented High-Temperature CO2 Selectivity in N2-Phobic Nanoporous Covalent Organic Polymers. Nat. Commun. 2013, 4, 1357. [Google Scholar] [CrossRef]
- Patel, H.A.; Je, S.H.; Park, J.; Jung, Y.; Coskun, A.; Yavuz, C.T. Directing the Structural Features of N2 -Phobic Nanoporous Covalent Organic Polymers for CO2 Capture and Separation. Chem. Eur. J. 2014, 20, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Guo, J.; Huang, Q.; He, J.; Fu, Y.; Kuang, G.; Pan, C.; Yu, G. 1,3,5-Triazine-Based Microporous Polymers with Tunable Porosities for CO2 Capture and Fluorescent Sensing. Macromolecules 2017, 50, 8512–8520. [Google Scholar] [CrossRef]
- Zhu, X.; Mahurin, S.M.; An, S.-H.; Do-Thanh, C.-L.; Tian, C.; Li, Y.; Gill, L.W.; Hagaman, E.W.; Bian, Z.; Zhou, J.-H.; et al. Efficient CO2 Capture by a Task-Specific Porous Organic Polymer Bifunctionalized with Carbazole and Triazine Groups. Chem. Commun. 2014, 50, 7933. [Google Scholar] [CrossRef]
- Li, G.; Qin, L.; Yao, C.; Xu, Y. Controlled Synthesis of Conjugated Polycarbazole Polymers via Structure Tuning for Gas Storage and Separation Applications. Sci. Rep. 2017, 7, 15394. [Google Scholar] [CrossRef] [PubMed]
- Car, Ž.; Borovina, M.; Panić, B.; Biljan, I. Microwave-Assisted Reductive Homocoupling of Aromatic Nitro Monomers: Synthesis of Azo-Linked Porous Organic Polymers for CO2 Capture. RSC Adv. 2025, 15, 7332–7339. [Google Scholar] [CrossRef] [PubMed]
- Panić, B.; Frey, T.; Borovina, M.; Ištoković, P.; Kodrin, I.; Biljan, I. Synergistic Experimental and Computational Investigation of Azo-Linked Porphyrin-Based Porous Organic Polymers for CO2 Capture. RSC Adv. 2025, 15, 13774–13785. [Google Scholar] [CrossRef]
- Popović, M.; Frey, T.; Borovina, M.; Kodrin, I.; Biljan, I. Tuning Linkers in Azo-Linked Porphyrin-Based Porous Organic Polymers for Enhanced CO2 Capture. Langmuir 2026, 42, 4641–4652. [Google Scholar] [CrossRef]
- Rabbani, M.G.; El-Kaderi, H.M. Synthesis and Characterization of Porous Benzimidazole-Linked Polymers and Their Performance in Small Gas Storage and Selective Uptake. Chem. Mater. 2012, 24, 1511–1517. [Google Scholar] [CrossRef]
- Arab, P.; Rabbani, M.G.; Sekizkardes, A.K.; İslamoğlu, T.; El-Kaderi, H.M. Copper(I)-Catalyzed Synthesis of Nanoporous Azo-Linked Polymers: Impact of Textural Properties on Gas Storage and Selective Carbon Dioxide Capture. Chem. Mater. 2014, 26, 1385–1392. [Google Scholar] [CrossRef]
- Arab, P.; Parrish, E.; İslamoğlu, T.; El-Kaderi, H.M. Synthesis and Evaluation of Porous Azo-Linked Polymers for Carbon Dioxide Capture and Separation. J. Mater. Chem. A 2015, 3, 20586–20594. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, J. Facile Synthesis of Azo-Linked Porous Organic Frameworks via Reductive Homocoupling for Selective CO2 Capture. J. Mater. Chem. A 2014, 2, 13831–13834. [Google Scholar] [CrossRef]
- Zhou, J.-X.; Luo, X.-S.; Liu, X.; Qiao, Y.; Wang, P.; Mecerreyes, D.; Bogliotti, N.; Chen, S.-L.; Huang, M.-H. Azo-Linked Porous Organic Polymers: Robust and Time-Efficient Synthesis via NaBH4-Mediated Reductive Homocoupling on Polynitro Monomers and Adsorption Capacity towards Aniline in Water. J. Mater. Chem. A 2018, 6, 5608–5612. [Google Scholar] [CrossRef]
- Modak, A.; Nandi, M.; Mondal, J.; Bhaumik, A. Porphyrin Based Porous Organic Polymers: Novel Synthetic Strategy and Exceptionally High CO2 Adsorption Capacity. Chem. Commun. 2012, 48, 248–250. [Google Scholar] [CrossRef]
- Feng, L.; Wang, K.-Y.; Joseph, E.; Zhou, H.-C. Catalytic Porphyrin Framework Compounds. Trends Chem. 2020, 2, 555–568. [Google Scholar] [CrossRef]
- Ji, W.; Wang, T.-X.; Ding, X.; Lei, S.; Han, B.-H. Porphyrin- and Phthalocyanine-Based Porous Organic Polymers: From Synthesis to Application. Coord. Chem. Rev. 2021, 439, 213875. [Google Scholar] [CrossRef]
- Jin, L.; Lv, S.; Miao, Y.; Liu, D.; Song, F. Recent Development of Porous Porphyrin-based Nanomaterials for Photocatalysis. ChemCatChem 2021, 13, 140–152. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhu, D.; Chen, Y.; Yan, Q.; Liu, C.-Y.; Ling, K.; Liu, Y.; Lee, D.; Wu, X.; Senftle, T.P.; et al. Porphyrin-Based Donor–Acceptor COFs as Efficient and Reusable Photocatalysts for PET-RAFT Polymerization under Broad Spectrum Excitation. Chem. Sci. 2021, 12, 16092–16099. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Z.; Zhang, F.; Zhuang, X.; Zeng, Z.; Wei, J. New Nitrogen-Rich Azo-Bridged Porphyrin-Conjugated Microporous Networks for High Performance of Gas Capture and Storage. RSC Adv. 2016, 6, 30048–30055. [Google Scholar] [CrossRef]
- Bera, R.; Ansari, M.; Alam, A.; Das, N. Nanoporous Azo Polymers (NAPs) for Selective CO2 Uptake. J. CO2 Util. 2018, 28, 385–392. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, Y.; Liu, J.; Luo, Y.; Lyu, Y. Facile Synthesis of Porous Organic Polymers Bifunctionalized with Azo and Porphyrin Groups. RSC Adv. 2015, 5, 98508–98513. [Google Scholar] [CrossRef]
- Tao, L.; Niu, F.; Zhang, D.; Liu, J.; Wang, T.; Wang, Q. Azo-Bridged Covalent Porphyrinic Polymers (Azo-CPPs): Synthesis and CO2 Capture Properties. RSC Adv. 2015, 5, 96871–96878. [Google Scholar] [CrossRef]
- Youm, K.; Choi, Y.; Byun, H.; Kumar, S.; Cho, Y.; Hsan, N.; Koh, J. A Highly Efficient Porphyrin-Based Azo-Porous Organic Polymer for Selective CO2 Capture and Conversion. J. CO2 Util. 2024, 84, 102854. [Google Scholar] [CrossRef]
- Šutalo, P.; Pisačić, M.; Biljan, I.; Kodrin, I. Benzene and Triazine-Based Porous Organic Polymers with Azo, Azoxy and Azodioxy Linkages: A Computational Study. CrystEngComm 2022, 24, 4748–4763. [Google Scholar] [CrossRef]
- Frey, T.; Panić, B.; Šutalo, P.; Borovina, M.; Biljan, I.; Kodrin, I. Prediction of CO2 Adsorption Properties of Azo, Azoxy and Azodioxy-Linked Porous Organic Polymers Guided by Electrostatic Potential. CrystEngComm 2023, 25, 3870–3884. [Google Scholar] [CrossRef]
- Beaudoin, D.; Wuest, J.D. Dimerization of Aromatic C-Nitroso Compounds. Chem. Rev. 2016, 116, 258–286. [Google Scholar] [CrossRef]
- Vančik, H. Aromatic C-Nitroso Compounds; Springer Netherlands: Dordrecht, 2013; ISBN 978-94-007-6336-4. [Google Scholar]
- Rončević, I.; Bibulić, P.; Vančik, H.; Biljan, I. Solution Equilibria of Aromatic Dinitroso Compounds: A Combined NMR and DFT Study. Struct. Chem. 2018, 29, 1489–1497. [Google Scholar] [CrossRef]
- Bibulić, P.; Rončević, I.; Špadina, M.; Biljan, I.; Vančik, H. Isothermal and Isoconversional Modeling of Solid-State Nitroso Polymerization. J. Phys. Chem. A 2020, 124, 10726–10735. [Google Scholar] [CrossRef] [PubMed]
- Bibulić, P.; Rončević, I.; Varga, K.; Mihalić, Z.; Vančik, H. Structure and Topochemistry of Azodioxide Oligomers in Solid State. J. Mol. Struct. 2016, 1104, 85–90. [Google Scholar] [CrossRef]
- Gowenlock, B.G.; Richter-Addo, G.B. Dinitroso and Polynitroso Compounds. Chem. Soc. Rev. 2005, 34, 797. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.J.; Cantrill, S.J.; Cousins, G.R.L.; Sanders, J.K.M.; Stoddart, J.F. Dynamic Covalent Chemistry. Angew. Chem. Int. Ed. 2002, 41, 898–952. [Google Scholar] [CrossRef]
- Beaudoin, D.; Maris, T.; Wuest, J.D. Constructing Monocrystalline Covalent Organic Networks by Polymerization. Nat. Chem. 2013, 5, 830–834. [Google Scholar] [CrossRef]
- Nath, B.; Li, W.-H.; Huang, J.-H.; Wang, G.-E.; Fu, Z.; Yao, M.-S.; Xu, G. A New Azodioxy-Linked Porphyrin-Based Semiconductive Covalent Organic Framework with I2 Doping-Enhanced Photoconductivity. CrystEngComm 2016, 18, 4259–4263. [Google Scholar] [CrossRef]
- Huskić, I.; Halasz, I.; Friščić, T.; Vančik, H. Mechanosynthesis of Nitrosobenzenes: A Proof-of-Principle Study in Combining Solvent-Free Synthesis with Solvent-Free Separations. Green Chem. 2012, 14, 1597. [Google Scholar] [CrossRef]
- Cindro, N.; Car, Ž.; Petrović Peroković, V.; Borovina, M.; Panić, B.; Kodrin, I.; Biljan, I. Synthesis of Aromatic Polynitroso Compounds: Towards Functional Azodioxy-Linked Porous Polymers. Heliyon 2023, 9, e21781. [Google Scholar] [CrossRef]
- Panić, B.; Frey, T.; Borovina, M.; Konopka, K.; Sambolec, M.; Kodrin, I.; Biljan, I. Synthesis and Characterization of Benzene- and Triazine-Based Azo-Bridged Porous Organic Polymers. Polymers (Basel). 2023, 15, 229. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, H.; Yu, B.; Zhao, Y.; Ma, Z.; Ji, G.; Han, B.; Liu, Z. Azo-Functionalized Microporous Organic Polymers: Synthesis and Applications in CO2 Capture and Conversion. Chem. Commun. 2015, 51, 11576–11579. [Google Scholar] [CrossRef]
- Ji, G.; Yang, Z.; Zhang, H.; Zhao, Y.; Yu, B.; Ma, Z.; Liu, Z. Hierarchically Mesoporous o-Hydroxyazobenzene Polymers: Synthesis and Their Applications in CO2 Capture and Conversion. Angew. Chem. 2016, 128, 9837–9841. [Google Scholar] [CrossRef]
- Bera, R.; Ansari, M.; Alam, A.; Das, N. Triptycene, Phenolic-OH, and Azo-Functionalized Porous Organic Polymers: Efficient and Selective CO2 Capture. ACS Appl. Polym. Mater. 2019, 1, 959–968. [Google Scholar] [CrossRef]
- Buyukcakir, O.; Je, S.H.; Park, J.; Patel, H.A.; Jung, Y.; Yavuz, C.T.; Coskun, A. Systematic Investigation of the Effect of Polymerization Routes on the Gas-Sorption Properties of Nanoporous Azobenzene Polymers. Chem. Eur. J. 2015, 21, 15320–15327. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, H.J.; Lim, S.Y.; Kim, B.G.; Choi, J.W. Combined CO2 -Philicity and Ordered Mesoporosity for Highly Selective CO2 Capture at High Temperatures. J. Am. Chem. Soc. 2015, 137, 7210–7216. [Google Scholar] [CrossRef]
- Dang, Q.-Q.; Wang, X.-M.; Zhan, Y.-F.; Zhang, X.-M. An Azo-Linked Porous Triptycene Network as an Absorbent for CO2 and Iodine Uptake. Polym. Chem. 2016, 7, 643–647. [Google Scholar] [CrossRef]
- Varga, K.; Biljan, I.; Tomišić, V.; Mihalić, Z.; Vančik, H. Quantum Chemical Calculations of Monomer–Dimer Equilibria of Aromatic C -Nitroso Compounds. J. Phys. Chem. A 2018, 122, 2542–2549. [Google Scholar] [CrossRef]
- Biljan, I.; Vančik, H. Aromatic C-Nitroso Compounds: From Solid-State Reactivity to New Materials. ChemistrySelect 2025, 10. [Google Scholar] [CrossRef]
- Borovina, M.; Petrović Peroković, V.; Panić, B.; Biljan, I. Structural Characterization of 1,2-Bis(4-Cyanophenyl)Diazene Oxide and Its Cyclotrimerization to a Triazine-Linked Polymer. ACS Omega 2026. [Google Scholar] [CrossRef]
- Erba, A.; Desmarais, J.K.; Casassa, S.; Civalleri, B.; Donà, L.; Bush, I.J.; Searle, B.; Maschio, L.; Edith-Daga, L.; Cossard, A.; et al. CRYSTAL23: A Program for Computational Solid State Physics and Chemistry. J. Chem. Theory Comput. 2023, 19, 6891–6932. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.A.; Sharma, S.; Balestra, S.R.G.; Li, Z.; Calero, S.; Vlugt, T.J.H.; Snurr, R.Q.; Dubbeldam, D. RASPA3: A Monte Carlo Code for Computing Adsorption and Diffusion in Nanoporous Materials and Thermodynamics Properties of Fluids. J. Chem. Phys. 2024, 161. [Google Scholar] [CrossRef]
- Chandra, S.; Kundu, T.; Kandambeth, S.; BabaRao, R.; Marathe, Y.; Kunjir, S.M.; Banerjee, R. Phosphoric Acid Loaded Azo (−N═N−) Based Covalent Organic Framework for Proton Conduction. J. Am. Chem. Soc. 2014, 136, 6570–6573. [Google Scholar] [CrossRef]
- Ge, R.; Hao, D.; Shi, Q.; Dong, B.; Leng, W.; Wang, C.; Gao, Y. Target Synthesis of an Azo (N═N) Based Covalent Organic Framework with High CO 2 -over-N 2 Selectivity and Benign Gas Storage Capability. J. Chem. Eng. Data 2016, 61, 1904–1909. [Google Scholar] [CrossRef]
- Huang, J.; Golomb, M.J.; Kavanagh, S.R.; Tolborg, K.; Ganose, A.M.; Walsh, A. Band Gap Opening from Displacive Instabilities in Layered Covalent-Organic Frameworks. J. Mater. Chem. A 2022, 10, 13500–13507. [Google Scholar] [CrossRef]
- Huang, J.; Shin, S.-J.; Tolborg, K.; Ganose, A.M.; Krenzer, G.; Walsh, A. Room-Temperature Stacking Disorder in Layered Covalent-Organic Frameworks from Machine-Learning Force Fields. Mater. Horiz. 2023, 10, 2883–2891. [Google Scholar] [CrossRef]
- Koo, B.T.; Dichtel, W.R.; Clancy, P. A Classification Scheme for the Stacking of Two-Dimensional Boronate Ester-Linked Covalent Organic Frameworks. J. Mater. Chem. 2012, 22, 17460. [Google Scholar] [CrossRef]
- Lukose, B.; Kuc, A.; Heine, T. The Structure of Layered Covalent-Organic Frameworks. Chem. Eur. J. 2011, 17, 2388–2392. [Google Scholar] [CrossRef] [PubMed]
- Pütz, A.M.; Terban, M.W.; Bette, S.; Haase, F.; Dinnebier, R.E.; Lotsch, B. V. Total Scattering Reveals the Hidden Stacking Disorder in a 2D Covalent Organic Framework. Chem. Sci. 2020, 11, 12647–12654. [Google Scholar] [CrossRef]
- Kang, C.; Zhang, Z.; Usadi, A.K.; Calabro, D.C.; Baugh, L.S.; Yu, K.; Wang, Y.; Zhao, D. Aggregated Structures of Two-Dimensional Covalent Organic Frameworks. J. Am. Chem. Soc. 2022, 144, 3192–3199. [Google Scholar] [CrossRef]
- Emmerling, S.T.; Schuldt, R.; Bette, S.; Yao, L.; Dinnebier, R.E.; Kästner, J.; Lotsch, B. V. Interlayer Interactions as Design Tool for Large-Pore COFs. J. Am. Chem. Soc. 2021, 143, 15711–15722. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wang, L.; Xu, H.; Wu, X.; Yang, J. A Simple Molecular Design Strategy for Two-Dimensional Covalent Organic Framework Capable of Visible-Light-Driven Water Splitting. J. Am. Chem. Soc. 2020, 142, 4508–4516. [Google Scholar] [CrossRef]
- Zhou, Z.-B.; Tian, P.-J.; Yao, J.; Lu, Y.; Qi, Q.-Y.; Zhao, X. Toward Azo-Linked Covalent Organic Frameworks by Developing Linkage Chemistry via Linker Exchange. Nat. Commun. 2022, 13, 2180. [Google Scholar] [CrossRef] [PubMed]
- Spitler, E.L.; Koo, B.T.; Novotney, J.L.; Colson, J.W.; Uribe-Romo, F.J.; Gutierrez, G.D.; Clancy, P.; Dichtel, W.R. A 2D Covalent Organic Framework with 4.7-nm Pores and Insight into Its Interlayer Stacking. J. Am. Chem. Soc. 2011, 133, 19416–19421. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Synthesis and building blocks for azo-linked porous organic polymers (POPs). (a) Schematic representation of common synthetic approaches toward azo-linked POPs, including: a1) heterocoupling of aromatic nitro and amino monomers, and a2) oxidative homocoupling of aromatic amino monomers and reductive homocoupling of aromatic nitro monomers. (b) Representative building blocks successfully employed in the synthesis of azo-linked POPs.
Figure 1.
Synthesis and building blocks for azo-linked porous organic polymers (POPs). (a) Schematic representation of common synthetic approaches toward azo-linked POPs, including: a1) heterocoupling of aromatic nitro and amino monomers, and a2) oxidative homocoupling of aromatic amino monomers and reductive homocoupling of aromatic nitro monomers. (b) Representative building blocks successfully employed in the synthesis of azo-linked POPs.
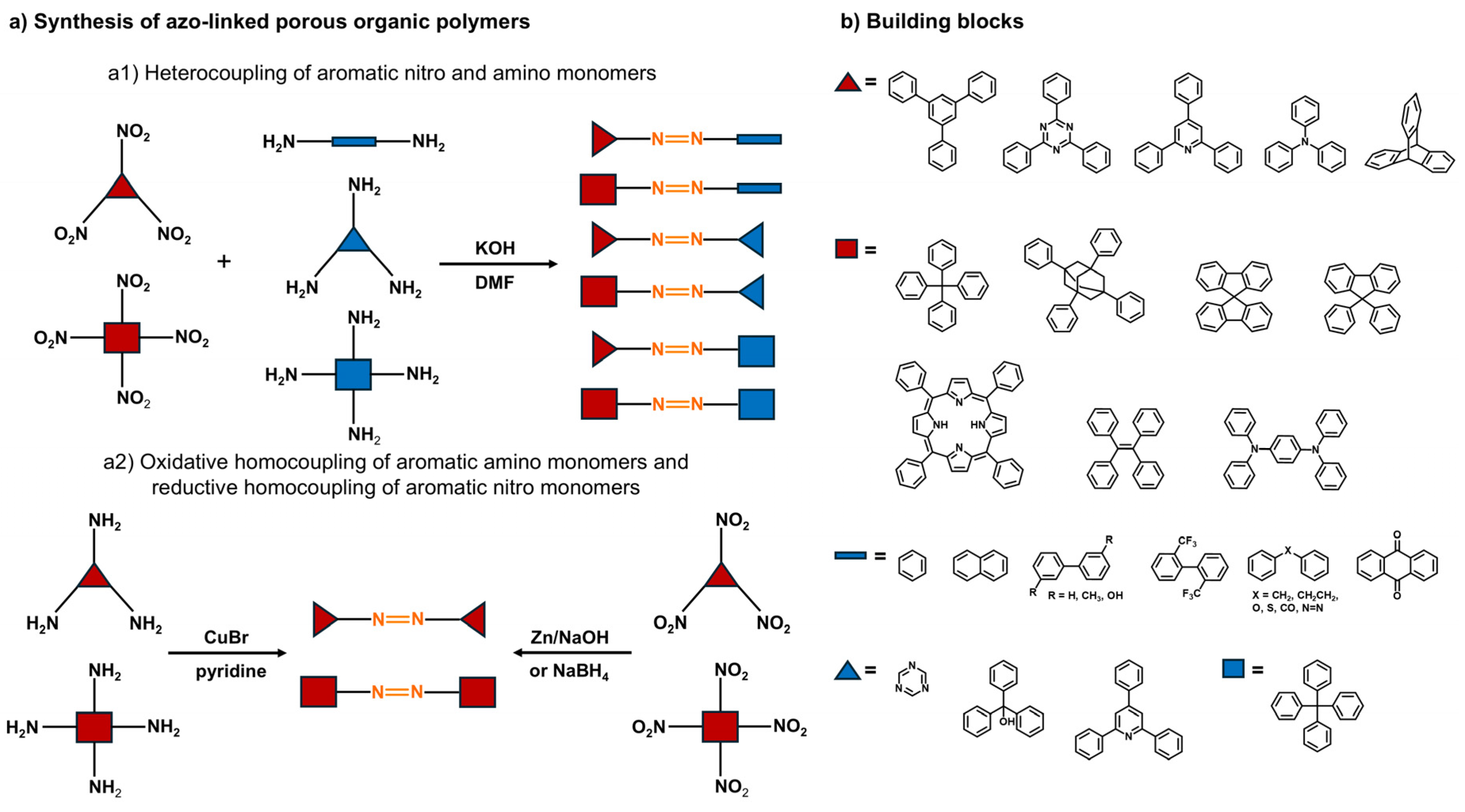
Figure 2.
Azo-linked porous organic polymer with hydroxylated biphenyl linker (APP-BP-OH). (a) Molecular structure with atom numbering. (b) 13C CP/MAS NMR spectrum. (c) PXRD pattern. (d) N2 adsorption-desorption isotherms measured at 77 K. Adsorption and desorption isotherms are depicted with filled and empty markers, respectively. (e) Pore size distribution. (f) Thermogravimetric CO2 adsorption and desorption profiles. (g) Scanning electron microscopy (SEM) image. Reprinted with permission of [34]. Copyright 2026, American Chemical Society.
Figure 2.
Azo-linked porous organic polymer with hydroxylated biphenyl linker (APP-BP-OH). (a) Molecular structure with atom numbering. (b) 13C CP/MAS NMR spectrum. (c) PXRD pattern. (d) N2 adsorption-desorption isotherms measured at 77 K. Adsorption and desorption isotherms are depicted with filled and empty markers, respectively. (e) Pore size distribution. (f) Thermogravimetric CO2 adsorption and desorption profiles. (g) Scanning electron microscopy (SEM) image. Reprinted with permission of [34]. Copyright 2026, American Chemical Society.
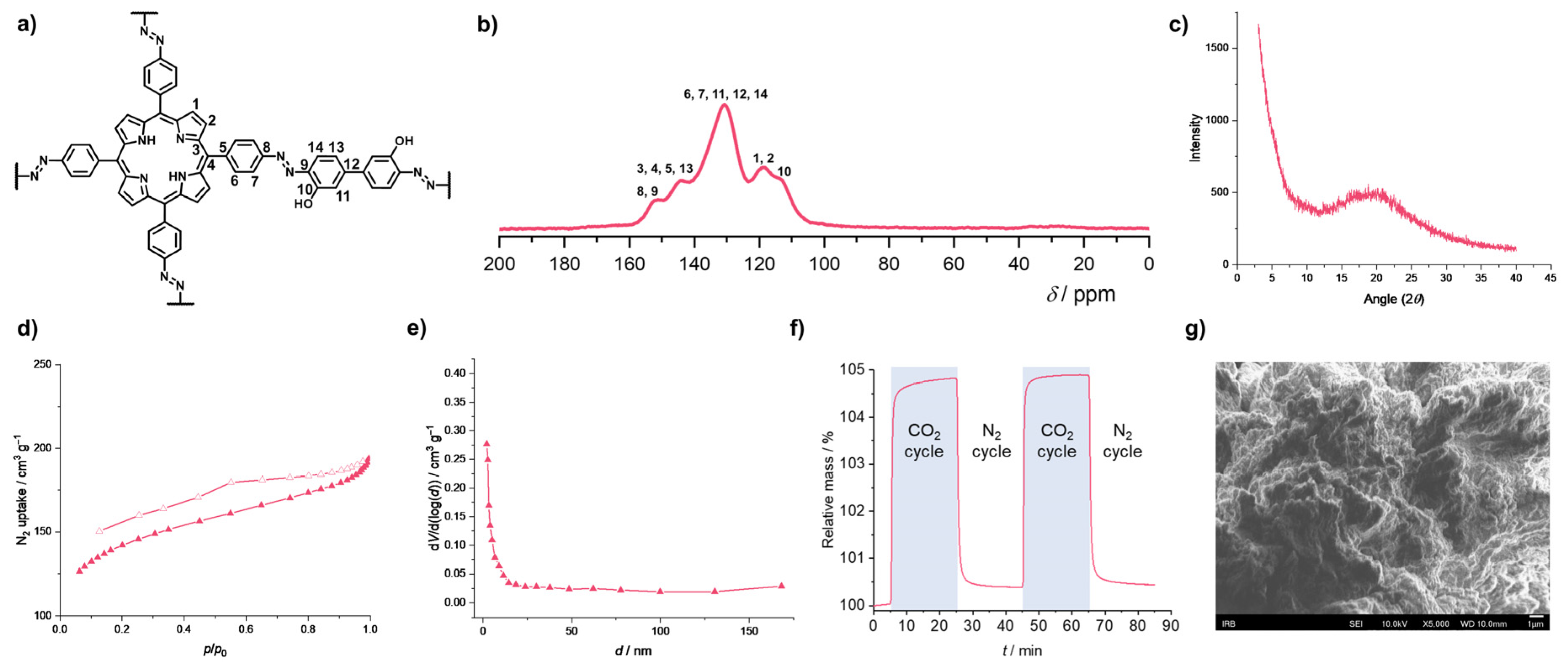
Figure 3.
Polymerization of aromatic polynitroso compounds into azodioxy networks. (a) Aromatic polynitroso compounds with tetrahedral symmetry. a1) Polymerization of tetrakis(4-nitrosophenylmethane) into covalent network NPN-1. a2) Representation of the structure of crystals of NPN-1 showing part of the diamondoid framework (ball-and-stick image at left), the degree of interpenetration (greyscale image in the middle) and the cross-sections of parallel channels viewed along the c-axis (space-filling image at right). Atoms of carbon are shown in grey, nitrogen in blue, oxygen in red and silicon in yellow. Guests are disordered and omitted for clarity. Reprinted with permission of [59]. Copyright 2016, American Chemical Society. (b) Aromatic polynitroso compounds with trigonal symmetry. b1) Synthesis and polymerization of 2,4,6-tris(4-nitrosophenyl)-1,3,5-triazine into azodioxy polymer. b2) Comparison of experimental and simulated (eclipsed AA, serrated AA’, staggered AB, and inclined AA’) PXRD patterns of azodioxy polymer. A peak at 2θ = 27.0° is marked by the grey dotted line.
Figure 3.
Polymerization of aromatic polynitroso compounds into azodioxy networks. (a) Aromatic polynitroso compounds with tetrahedral symmetry. a1) Polymerization of tetrakis(4-nitrosophenylmethane) into covalent network NPN-1. a2) Representation of the structure of crystals of NPN-1 showing part of the diamondoid framework (ball-and-stick image at left), the degree of interpenetration (greyscale image in the middle) and the cross-sections of parallel channels viewed along the c-axis (space-filling image at right). Atoms of carbon are shown in grey, nitrogen in blue, oxygen in red and silicon in yellow. Guests are disordered and omitted for clarity. Reprinted with permission of [59]. Copyright 2016, American Chemical Society. (b) Aromatic polynitroso compounds with trigonal symmetry. b1) Synthesis and polymerization of 2,4,6-tris(4-nitrosophenyl)-1,3,5-triazine into azodioxy polymer. b2) Comparison of experimental and simulated (eclipsed AA, serrated AA’, staggered AB, and inclined AA’) PXRD patterns of azodioxy polymer. A peak at 2θ = 27.0° is marked by the grey dotted line.
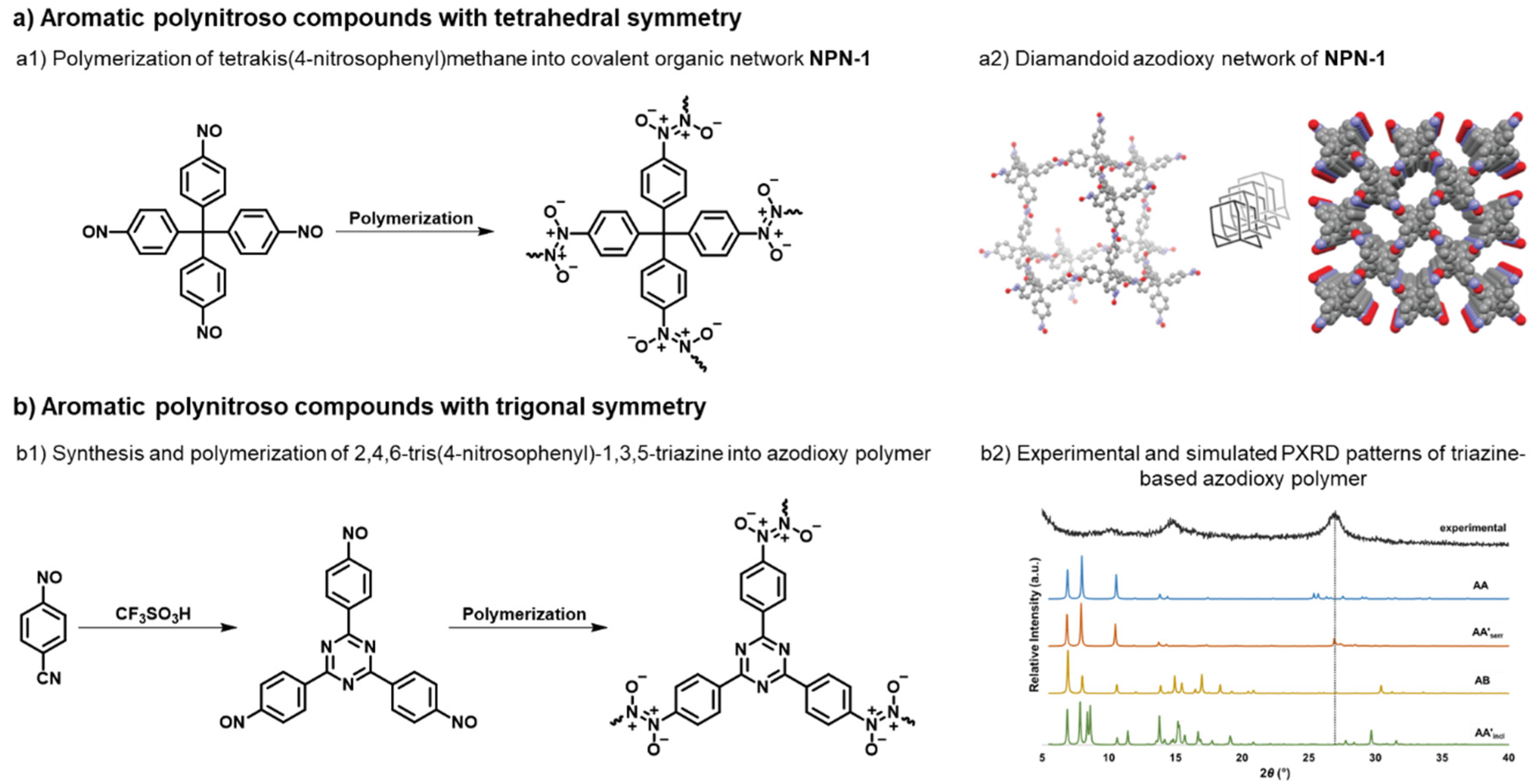
Figure 4.
Hierarchical computational workflow for the investigation of porous organic polymers (POPs) with azo, azoxy, and azodioxy linkages. (a) Molecular fragment-based calculations performed on isolated molecular building blocks, including the evaluation of interaction energies with gas molecules (a1) and electrostatic potential (ESP) maps used to identify the most favorable gas binding sites (a2). (b) Periodic DFT calculations of the extended framework, including geometry optimizations of 3D periodic structures (b1) and ESP maps providing insight into the local chemical environment (b2). (c) GCMC simulations of gas adsorption in the periodic frameworks, resulting in adsorption isotherms (c1), selectivities (c2), and density distribution of adsorbate inside the pores (c3). Depending on the investigated system, computational studies of molecular fragments can be performed independently, particularly when the 3D periodic structure is unknown or not experimentally resolved but can be predicted from the synthetic protocol. Reprinted with permission of [50]. Copyright 2022, Royal Society of Chemistry.
Figure 4.
Hierarchical computational workflow for the investigation of porous organic polymers (POPs) with azo, azoxy, and azodioxy linkages. (a) Molecular fragment-based calculations performed on isolated molecular building blocks, including the evaluation of interaction energies with gas molecules (a1) and electrostatic potential (ESP) maps used to identify the most favorable gas binding sites (a2). (b) Periodic DFT calculations of the extended framework, including geometry optimizations of 3D periodic structures (b1) and ESP maps providing insight into the local chemical environment (b2). (c) GCMC simulations of gas adsorption in the periodic frameworks, resulting in adsorption isotherms (c1), selectivities (c2), and density distribution of adsorbate inside the pores (c3). Depending on the investigated system, computational studies of molecular fragments can be performed independently, particularly when the 3D periodic structure is unknown or not experimentally resolved but can be predicted from the synthetic protocol. Reprinted with permission of [50]. Copyright 2022, Royal Society of Chemistry.
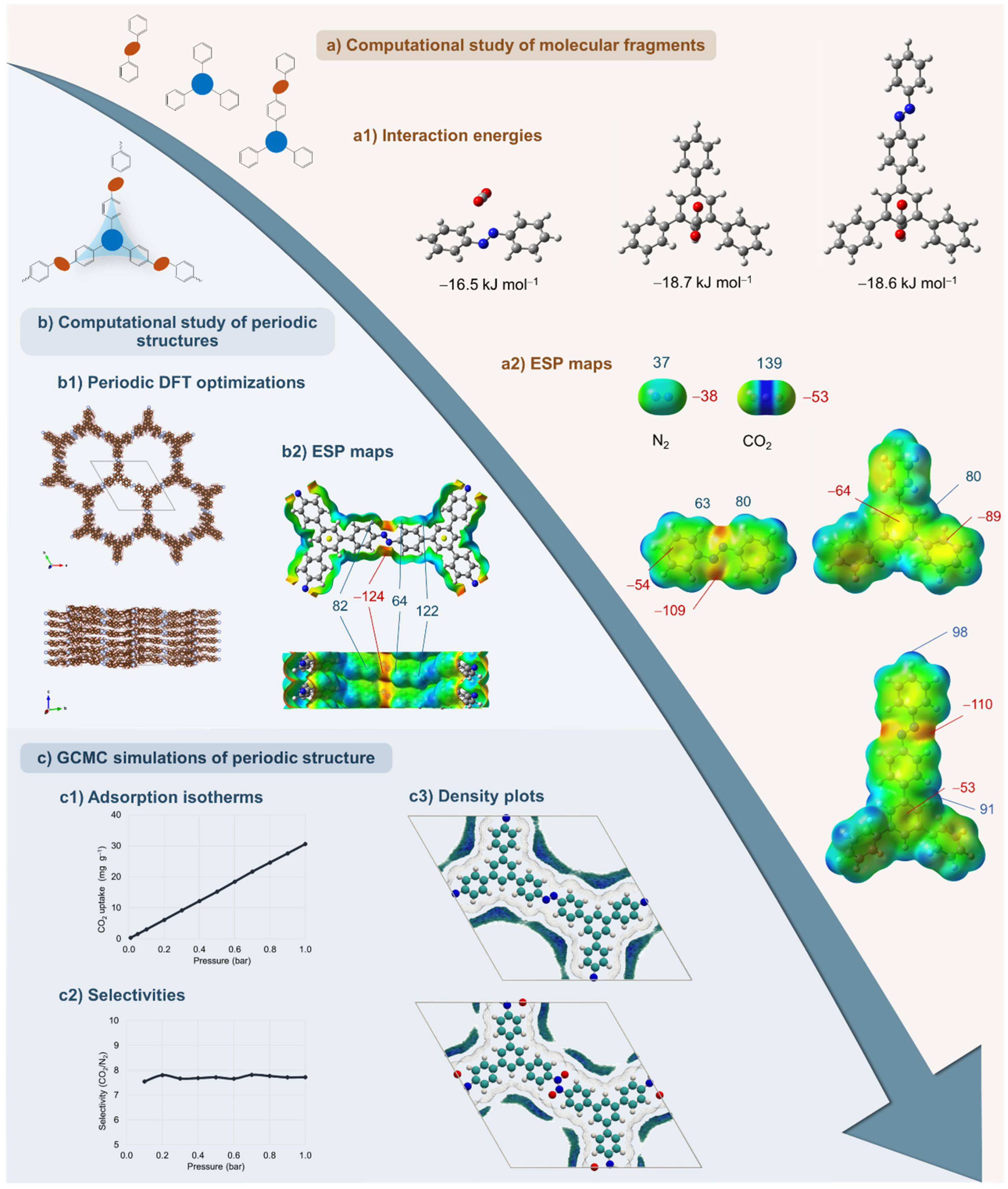
Figure 5.
Effect of interlayer stacking on CO2 adsorption in layered azo- and azodioxy-linked porous organic polymers. (a) Schematic representation of four idealized stacking modes between adjacent 2D layers: inclined, eclipsed, serrated, and staggered. (b) Optimized periodic structures highlighting how the stacking mode changes pore shape and size, and the local chemical environment in the azo-linked polymers derived from 2,4,6-tris(4-substituted phenyl)pyridine (TPP). (c) Structure-property correlation between ESP values at azo nitrogen atoms and calculated CO2 uptakes for the four stacking modes of the azo-linked pyridine-based connectors (TPP). (d) Structure-property correlation between ESP values at azodioxy oxygen atoms and calculated CO2 uptakes for the four stacking modes of the azodioxy-linked amine-based connectors (TPA). Reprinted with permission of [51]. Copyright 2023, Royal Society of Chemistry.
Figure 5.
Effect of interlayer stacking on CO2 adsorption in layered azo- and azodioxy-linked porous organic polymers. (a) Schematic representation of four idealized stacking modes between adjacent 2D layers: inclined, eclipsed, serrated, and staggered. (b) Optimized periodic structures highlighting how the stacking mode changes pore shape and size, and the local chemical environment in the azo-linked polymers derived from 2,4,6-tris(4-substituted phenyl)pyridine (TPP). (c) Structure-property correlation between ESP values at azo nitrogen atoms and calculated CO2 uptakes for the four stacking modes of the azo-linked pyridine-based connectors (TPP). (d) Structure-property correlation between ESP values at azodioxy oxygen atoms and calculated CO2 uptakes for the four stacking modes of the azodioxy-linked amine-based connectors (TPA). Reprinted with permission of [51]. Copyright 2023, Royal Society of Chemistry.
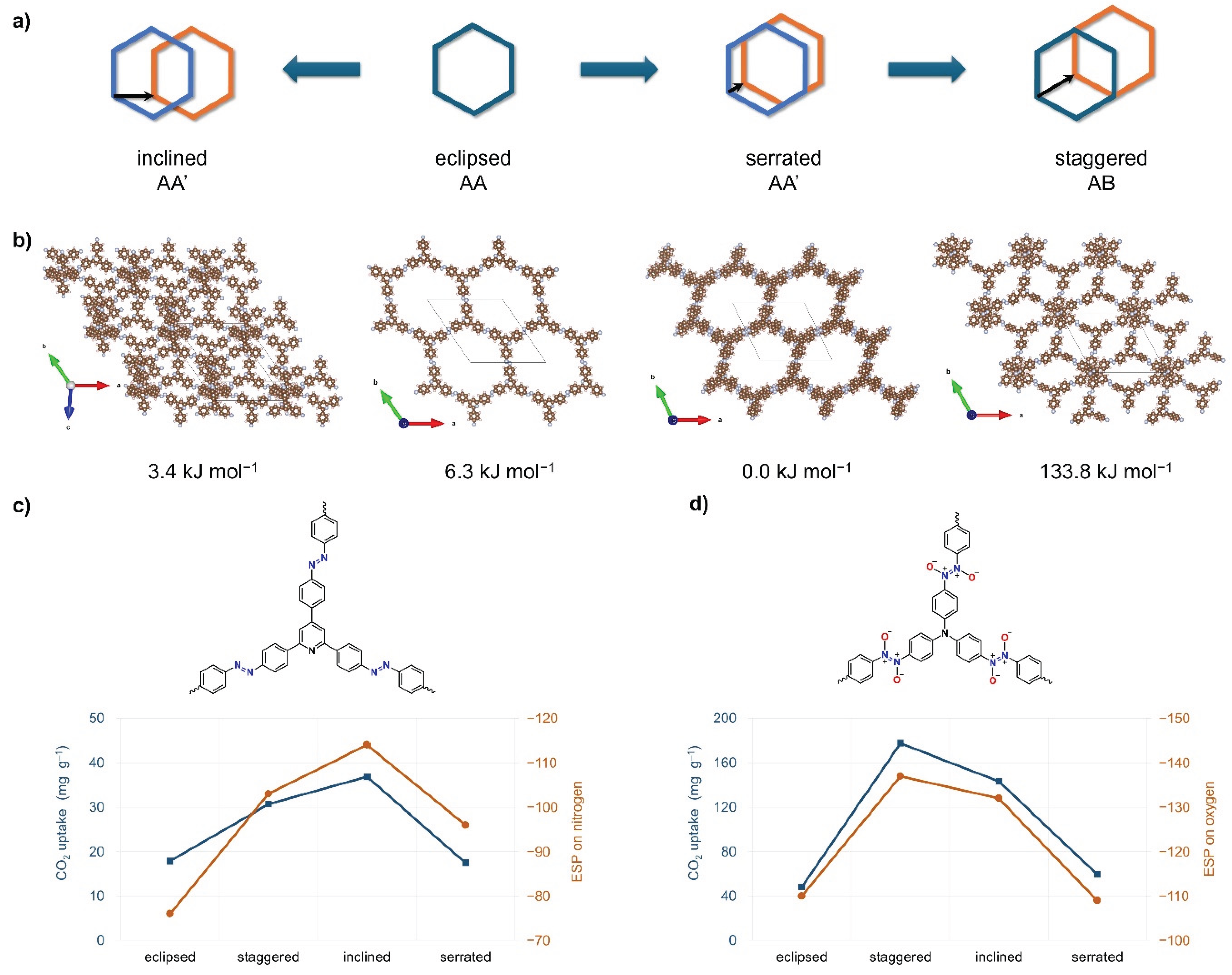

Table 1.
Calculated average surface areas and CO2 uptakes (at 298 K and 1 bar).
| Compound |
Average surface area (m2 g−1) |
CO2 uptake (mg g−1) |
| TPB-azo (AA)a | 1957 | 19 |
| TPB-azo (AB)a | 2462 | 65 |
| TPB-azoxy (AA)a | 1830 | 28 |
| TPB-azoxy (AB)a | 2284 | 73 |
| TPB-azodioxy (AA)a | 1691 | 31 |
| TPB-azodioxy (AB)a | 1949 | 118 |
| TPT-azo (AA)a | 1828 | 14 |
| TPT-azo (AB)a | 2411 | 92 |
| TPT-azoxy (AA)a | 1704 | 19 |
| TPT-azoxy (AB)a | 2077 | 73 |
| TPT-azodioxy (AA)a | 1596 | 21 |
| TPT-azodioxy (AB)a | 1635 | 44 |
| TPA-azo (AA)b | 1858 | 23 |
| TPA-azo (AB)b | 2327 | 153 |
| TPA-azo (AA’incl)b | 1395 | 46 |
| TPA-azo (AA’serr)b | 1911 | 28 |
| TPA-azoxy (AA)b | 1686 | 38 |
| TPA-azoxy (AB)b | 2051 | 169 |
| TPA-azodioxy (AA)b | 1501 | 48 |
| TPA-azodioxy (AB)b | 1570 | 177 |
| TPP-azo (AA)b | 1880 | 18 |
| TPP-azo (AB)b | 1894 | 31 |
| TPP-azo (AA’incl)b | 132 | 37 |
| TPP-azo (AA’serr)b | 1783 | 18 |
| TPP-azoxy (AA)b | 1588 | 21 |
| TPP-azoxy (AA’incl)b | 194 | 37 |
| TPP-azodioxy (AA)b | 1636 | 27 |
| TPP-azodioxy (AA’incl)b | 342 | 68 |
| AZO-Bc | 1957 | 20 |
| AZO-B-PPDc | 2237 | 19 |
| AZO-B-BZDc | 2306 | 19 |
| AZO-Tc | 1828 | 14 |
| AZO-T-PPDc | 2127 | 14 |
| AZO-T-BZDc | 2207 | 15 |
| APP-1d | 1969 | 13 |
| APP-2d | 2246 | 18 |
| APP-3d | 1931 | 15 |
| APP-4d | 1934 | 20 |
| APP-5d | 6777 | 49 |
| APP-6d | 7567 | 49 |
| APP-7d | 1332 | 23 |
| APP-8d | 1332 | 23 |
| APP-BP-OH (AA)e | 1750 | 25 |
| APP-BP-OH (AB)e | 2716 | 85 |
| APP-BP-OH (AA’ serr)e | 1894 | 213 |
| APP-AQe | 1699 | 23 |
| APP-BP-Mee | 1805 | 20 |
| APP-Ph-Mee | 1660 | 23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



