Submitted:
01 March 2026
Posted:
02 March 2026
You are already at the latest version
Abstract
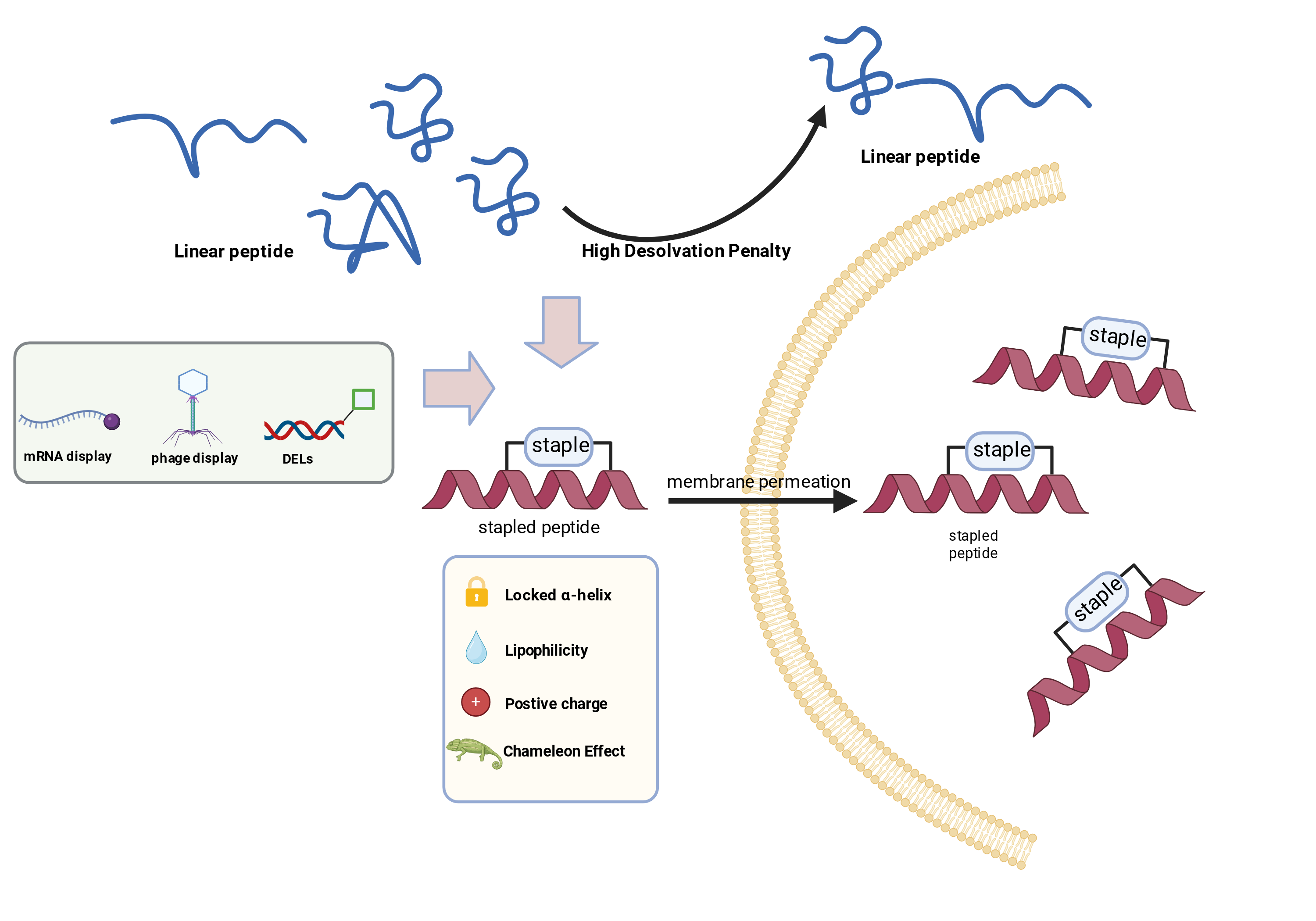
The optimization of membrane permeability is a decisive strategy for mitigating late-stage failures in peptide drug development. By leveraging linker chemical diver-sity, stapled peptides utilize linker engineering to precisely modulate key physico-chemical parameters—such as lipophilicity and conformational constraints—to over-come the desolvation energy penalty. This review systematically evaluates link-er-based strategies for enhancing the permeability of stapled peptides, categorized into two primary dimensions: (1) High-throughput screening (HTS) compatibility, focusing on the integration of functionalized linkers into mRNA display, phage display, and DNA-encoded libraries (DELs) to identify lead scaffolds with inherent permeability potential during early discovery ; and (2) Post-screening structural refinement, cover-ing rational design strategies including intramolecular hydrogen bond (IMHB) shield-ing, "chameleonic" adaptations, and stimuli-responsive reversible stapling . Further-more, we analyze the paradigm shift in assessment methodologies from qualitative imaging to quantitative cytosolic delivery assays, which have deepened our under-standing of mechanisms such as the charge/lipophilicity threshold balance and meta-bolic-driven trapping. Overall, linker engineering provides a robust technical roadmap for developing the next generation of cell-permeable stapled peptide therapeutics.
Keywords:
stapled peptides
; linker engineering
; membrane permeability
; high-throughput screening (HTS)
; intramolecular hydrogen bond (IMHB)
; chameleonicity
; intracellular delivery
; rational design
1. Introduction
Within the extensive scope of contemporary drug development, the modulation of protein–protein interactions (PPIs) stands as one of the most challenging and high-potential frontiers [1,2]. The topographical nature of PPI interfaces—typically expansive, flat, and devoid of deep hydrophobic pockets—precludes the effective binding of conventional small molecules that comply with the "Rule of Five” [3,4].Conversely, while macromolecular biologics like monoclonal antibodies offer exquisite target specificity, their clinical utility is fundamentally limited to the extracellular space by their inability to traverse the plasma membrane [5]. Peptides, by virtue of their distinctive molecular weight, exceptional selectivity, and expansive chemical space, have emerged as a compelling 'middle-ground' therapeutic modality. These macrocycles are currently spearheading a new paradigm in the targeting of historically PPIs [6,7,8,9,10].
Despite their therapeutic promise, the transition of linear peptide sequences into viable drug candidates is frequently thwarted by fundamental physicochemical barriers [11,12,13]. Natural peptides are inherently rich in polar backbone amides—serving as both hydrogen-bond donors and acceptors—and charged side chains. The energetic cost for these polar groups to shed aqueous solvation shells and partition into the hydrophobic lipid core, known as the "desolvation penalty", is prohibitively high [14]. Furthermore, linear peptides exist as high-entropy conformational ensembles in solution; constraining these flexible chains into specific, membrane-permeable conformations necessitates a significant entropic sacrifice, further impeding effective translocation [15].
To mitigate these limitations, researchers, inspired by natural structures such as Cyclosporin A, have extensively investigated N-methylation and related backbone modifications [16]. Although reducing the number of hydrogen-bond donors (HBDs) by substituting amide hydrogens can lower the energetic threshold for membrane entry, this strategy inevitably creates a critical trade-off: the disruption of internal hydrogen-bonding networks increases conformational flexibility. This added entropy frequently results in a precipitous drop in binding affinity, as the peptide fails to maintain the rigid, bioactive conformation required for target recognition [17,18,19,20].
Beyond these backbone modifications, macrocyclization—exemplified by head-to-tail and disulfide-bridged architectures—provides a more robust and systemic approach to surmounting the barriers of peptide membrane permeability [21,22]. By pre-organizing peptides into rigid conformations and eliminating solvent-exposed charged termini (elimination of charged termini), macrocyclization effectively minimizes conformational entropy loss and enhances metabolic stability. Furthermore, these macrocyclic frameworks frequently harness the "chameleonic property," employing trans-annular IMHBs to sequester residual polar groups from the surrounding environment, thereby significantly lowering the desolvation penalty [23,24]
As an advanced iteration of macrocyclization technology, peptide stapling primarily involves the introduction of chemical cross-links at the i, i+4 (spanning one helical turn) or i, i+7 (spanning two helical turns) positions to precisely induce and stabilize α-helical conformations. In recent years, a plethora of reviews has emerged focusing on their synthetic strategies, screening methodologies, and structural optimizations [25,26]. Traditionally, it has been posited that such robust conformational constraints not only markedly enhance the binding affinity of peptides toward target proteins but, more crucially, facilitate the physical shielding of polar backbone amides by reinforcing helicity [27]. Concurrently, the intrinsic physicochemical properties of the linkers—such as the introduction of hydrophobic moieties—play an indispensable role in modulating overall molecular polarity and strengthening interactions with lipid bilayers [28]. Nevertheless, despite the successive elucidation of various peptide transmembrane mechanisms, a systematic review focusing on strategies to optimize the membrane permeability of stapled peptides remains overdue.
Against this backdrop, this review provides a comprehensive overview of the pivotal role of linker engineering in optimizing peptide membrane permeability. The content is organized around two core dimensions: first, it discusses the integration of functionalized linkers into high-throughput display platforms—such as mRNA display, phage display, and DELs—to facilitate the "front-loading" of permeability traits during the early selection of macrocyclic leads. Second, it provides an in-depth analysis of sophisticated chemical modification strategies (e.g., leveraging chameleonicity, intramolecular hydrogen bonding, and charge balancing) designed to enhance both permeability and drug-like properties.
Furthermore, by incorporating recent advancements in permeability assays, this review systematically elucidates the molecular mechanisms underlying the transmembrane transport of stapled peptides. By bridging the gap between high-throughput discovery and rational post-screening refinement, this work aims to provide a robust technical roadmap and theoretical framework for developing next-generation peptide therapeutics against challenging "undruggable" intracellular targets.
2. Linkers Derived from In Vitro High-Throughput Screening
2.1. Arylene-Based Linkers via Thiol-Mediated Nucleophilic Substitution
To rapidly explore the vast chemical space of stapled peptides, in vitro HTS platforms—including mRNA display, phage display, and DELs—have become indispensable tools [29,30,31,32,33,34,35,36,37]. Unlike traditional rational design, these platforms allow for the simultaneous evaluation of billions of peptide sequences However, the success of such screenings relies heavily on the choice of linkers that are not only chemically robust but also compatible with aqueous, biocompatible conditions. Consequently, functionalized linkers that can be incorporated via efficient, bioorthogonal chemistries have emerged as the "bridge" connecting high-capacity selection with optimized pharmacological properties. The thiol group of cysteine, with its high selectivity and nucleophilicity, can readily undergo SN2 and SNAr reactions under biocompatible conditions, thus enabling efficient reactions with benzyl bromides and polyfluoroarenes, which has attracted significant attention
2.1.1. Aryl Linkers: Conformational Rigidification
Initially, all-hydrocarbon stapled peptides strongly demonstrated the contribution of conformational locking to the enhancement of cellular membrane permeability by stabilizing the α-helical structure [38]. However, ruthenium-catalyzed ring-closing metathesis (RCM), the core reaction on which they rely, exhibits poor compatibility with aqueous biological environments, rendering it incompatible with HTS platforms such as in vitro display systems. In contrast, although disulfide bonds possess excellent aqueous compatibility, their inherent instability in the reductive intracellular microenvironment makes them unable to meet the stringent requirements for intracellular targeting applications [39,40].
Against this backdrop, the CLIPS (Chemical Linkage of Peptides onto Scaffolds) technology introduced by Timmerman et al. spurred the development of aryl linkers, exemplified by m-xylene (mDBMB) [41]. By leveraging the SN2 nucleophilic substitution of cysteine residues under mild, biocompatible conditions, this strategy establishes chemically robust aryl-thioether bridges. This approach not only fundamentally mitigates the risks of instability inherent to traditional disulfide bonds within the intracellular reductive microenvironment but also offers a dependable chemical anchor for the precise conformational steering of the peptide backbone [39]. Although initial investigations primarily highlighted phenotypic improvements in proteolytic stability and target binding affinity, deeper biophysical scrutiny suggests that this α-helical constraint—analogous to that of all-hydrocarbon linkers—serves as the mechanistic cornerstone for traversing cellular barriers. By anchoring the peptide in its bioactive state through potent pre-organization, the significant entropic penalty typically incurred by conformational reorganization during translocation across the hydrophobic phospholipid bilayer is substantially diminished. This mechanism of "energetic pre-compensation" via structural rigidification provides a formidable theoretical framework for the design and evolution of stapled peptide architectures with superior cell-penetrating potential [42]. Representative chemical strategies for the construction of such membrane-permeable architectures—ranging from simple CLIPS-based monocyclization to complex bicyclic and backbone-shielding scaffolds—are summarized in Figure 1.
As screening technologies iterated, linker design evolved toward a strategic emphasis on hydrophobic shielding. The Qing Lin group demonstrated that biphenyl-based linkers, by providing an expanded hydrophobic patch, not only enhance binding affinity but also effectively compensate for the desolvation penalty of the peptide backbone [43]. Building on these monocyclic aryl linkers, the application of 1,3,5-tris(bromomethyl)benzene (TBMB), pioneered by Heinis and Winter, marked a significant evolution toward ultra-rigid bicyclic architectures [44]. Owing to its C3-symmetric scaffold and efficient SN2 reactivity, TBMB has established itself as a foundational tool for constructing vast libraries in both mRNA display (notably within the Suga group's RaPID system) and phage display platforms [45]. From a biophysical perspective, this "topological condensation" significantly compresses the molecular radius of gyration, minimizing the entropic penalty during membrane translocation through extreme conformational pre-organization [46]. The TBMB-linked bicyclic peptide CP21 by Slavoff group demonstrated rigorous intracellular targeting via FITC-labeled live-cell imaging and CETSA. By bypassing genetic compensation, this chemical probe identified 76 novel DCP2 substrates, highlighting its potential to target "undruggable" intracellular PPIs [47]. Furthermore, recent explorations of 1,2,3-TBMB isomers by the Lau group have further expanded the conformational landscape of macrocyclic peptides, driving the screening paradigm from "coarse-grained rigidification" toward "precision-tailored geometric topology" [48].
2.1.2. Perfluoroaromatic Linkers: Leveraging the "Fluorine Effect" for Translocation
Parallel to advancements in all-hydrocarbon and alkyl-aryl stapling technologies, the integration of perfluoroaromatic linkers represents a paradigm shift in leveraging the unique "Fluorine Effect" to enhance membrane permeability [49,50,51,52]. Pioneered by the Pentelute group, this strategy exhibits high selectivity for cysteine, in contrast to the limitation of bromoalkyl/benzyl chemistry that tends to produce over-alkylation products. Its core lies in constructing stable aryl thioether bonds via SNAr between cysteine thiols and electron-deficient perfluoroarenes (e.g., hexafluorobenzene, decafluorobiphenyl); flow cytometry and confocal microscopy have confirmed that perfluoroaryl-stapled peptides possess significantly superior cellular uptake capacity and protease stability compared to linear peptides [53,54]. This chemical system exhibits excellent chemoselectivity and biorthogonality under aqueous conditions compared with the Sₙ2 reaction of alkyl halides, and can be seamlessly adapted to HTS platforms such as phage display [55]. The chemical conjugation involving the SNAr reaction between cysteine thiols and these perfluoro-linkers is illustrated in Figure 2.
Recently, the research focus in this field has shifted to functional applications. Through 3D blood-brain barrier (BBB) sphere model penetration depth tests and in vivo biodistribution experiments in mice, the status of perfluoroaryl (ArF) linkers as "BBB shuttling carriers" has been established, with a penetration efficiency 8-12 times that of ordinary alkyl linkers, and it can successfully deliver macromolecular substances such as antisense oligonucleotides [56]. The latest research by Peng et al. further revealed that perfluoro linkers can drive peptides to accumulate in lipid raft microdomains of the cell membrane, thereby triggering caveolin-mediated transcytosis (CMT) [57]. This transport pathway is significantly upregulated in the aging brain due to a 44.2% decrease in pericyte coverage and a 51.3% decrease in Mfsd2a expression. Experimental data show that the optimized perfluoroalkyl-stapled peptide has a cellular uptake intensity in brain endothelial cells 7.73 times that of linear peptides, and its BBB crossing efficiency in the aged model is 1.4 times higher than that in the young model. Perfluoroaryl linkers still face safety challenges: rigid ArF is prone to causing hemolysis and cytotoxicity, while flexible perfluoroalkyl (AlkF) can maintain the steady state of cell membrane fluidity with significantly better biosafety. Therefore, flexible fluoro chains have become the future development direction of this field.
2.1.3. Bipyridine Linkers: IMHB Shielding
Although the hydrophobic scaffold of aromatic linkers offers certain advantages for improving membrane permeability, the extent of enhancement is often limited. In practical research and development, tedious N-methylation modifications are still commonly required to mask the backbone polarity, which significantly increases synthetic complexity and screening costs. In 2023, the Suga group proposed the use of bipyridine (BPy) as a macrocyclization unit for macrocyclic peptides (Figure 1d), providing a new approach to address this challenge [58].
This design enhances the overall hydrophobicity of the linker and utilizes the pyridine nitrogen atoms as potent hydrogen bond acceptors to form IMHBs with the exposed NH donors on the opposite side of the peptide chain, thereby effectively shielding the polar backbone and significantly improving transmembrane efficiency. Quantitative evaluation via the Chloroalkane Penetration Assay (CAPA) showed that the transmembrane efficiency of model peptides incorporating BPy units was approximately 40-fold higher than that of the control group containing standard acetyl thioether bonds; in uncharged sequences, BPy also exhibited a ~4.6-fold improvement compared to biphenyl (BPh) linkers [59]. Its membrane permeability even outperforms classic polycationic cell-penetrating peptides(CPPs) such as Tat and R9. Experimental results further confirmed that this performance is less affected by cyclization modes (e.g., macrocyclic or Lariat structures) and peptide chain length, demonstrating exceptional structural robustness. The BPy linker has been verified as a dominant factor driving the transmembrane transport of macrocyclic peptides, laying an important foundation for the subsequent development of other high-affinity heterocyclic macrocyclic peptide drugs targeting intracellular targets.
2.2. Heteroaromatic Linkers Constructed from Unnatural Amino Acids (UAAs)
Accompanied by the continuous enrichment of the chemical biology toolkit, a series of spontaneous, efficient, and biocompatible cyclization linkers has emerged. Beyond the aryl and polyfluoroaryl linkers widely employed in discovery stages, strategies capable of spontaneous cyclization under biocompatible conditions have garnered significant attention due to their exceptional capacity for physicochemical modulation [60,61]. These strategies typically circumvent the need for exogenous metal catalysts, not only simplifying the construction of high-throughput libraries but also reshaping the transmembrane properties of macrocyclic peptides at a fundamental molecular level through their unique heterocyclic architectures. Representative architectures of these heteroaromatic linkers, including triazole, thiazoline, and imidazopyridinium (IP+) scaffolds, are illustrated in Figure 3.
2.2.1. Triazole Linkers: From Structural Stabilization to Decoupled Functionalization
Triazole-based linkers constructed via click chemistry have emerged as a pivotal strategy in both stapled peptide synthesis and mRNA display screening—facilitating, for instance, the direct competitive comparison of linear, monocyclic, and bicyclic libraries—owing to their exceptional reaction kinetics and high biorthogonality [62]. While simple triazole linkers can significantly enhance the helicity and proteolytic stability of peptides, their direct contribution to membrane permeability is often modest [54,63,64]. Nevertheless, the robust bioorthogonality of these linkers provides a versatile platform for functional modification to overcome this limitation. For instance, the conjugation of therapeutic peptides with membrane-permeable motifs, such as (CPPs), has become a prominent approach for enhancing cellular uptake(Enhancing the Cell Permeability of Stapled Peptides with a Cyclic Cell-Penetrating Peptide). Lau et al. have conducted a series of landmark studies in the field of triazole-linked two-component stapled peptides. By leveraging the efficient kinetics and high bioorthogonality of click chemistry, the team proposed an innovative "decoupling" strategy. This approach enables the independent optimization of membrane permeability and backbone binding affinity through the endogenous incorporation of cationic residues (e.g., Arg), the exogenous conjugation of cell-penetrating peptide sequences, or the utilization of diverse cross-linking toolboxes on the dialkynyl linkers. Confocal microscopy and reporter gene assays confirmed that these functionalized linker modifications significantly enhance cellular uptake efficiency. Notably, this strategy transcends the traditional dependence of stapling technologies on α-helical conformations, extending their application to non-helical/extended peptides (such as TNKS inhibitors) and significantly broadening the therapeutic potential of peptides for challenging intracellular targets [65,66,67].
2.2.2. Thiazoline Linkers: Spontaneous Formation and Intramolecular Hydrogen Bond Shielding
Inspired by the biosynthetic pathway of firefly luciferin, the thiazoline-bridged cyclization strategy has emerged as a highly efficient, "in-translation" approach compatible with high-throughput mRNA display platforms. Developed by the Hohsaka group, this strategy centers on an exceptionally mild and spontaneous condensation reaction between the N-terminal cysteine (Cys) and a side-chain nitrile group under physiological pH. Such spontaneous cyclization not only ensures the structural diversity of screening libraries but also, due to its excellent chemical orthogonality, provides robust technical support for the de novo selection of macrocyclic leads with intracellular targeting potential within mRNA display systems [68]. Experimental validation and physicochemical analyses have demonstrated that thiazoline-bridged peptides exhibit significantly superior membrane permeability compared to amide- or thioether-linked analogs of similar sequences. Tamura et al. confirmed via Parallel Artificial Membrane Permeability Assays (PAMPA) that thiazoline-based macrocycles achieve a substantial increase in apparent permeability Papp, even when compared to amide controls with higher or similar lipophilicity (c Log P/Log D).The biophysical mechanisms underlying this enhancement are two-fold: first, the spontaneous cyclization between the N-terminal cysteine and the side-chain nitrile group converts a hydrogen bond donor (HBD) into an acceptor, directly reducing the overall molecular polarity. Second, temperature-variable NMR and computational simulations revealed that the rigid thiazoline scaffold induces the formation of a stable IMHB network in hydrophobic environments. This creates a profound polar offset and hydrophobic shielding effect, effectively compressing the PSA, thereby lowering the desolvation penalty during transmembrane translocation [69].
2.2.3. Imidazopyridinium (IP+): Challenging Passive Diffusion Dogmas with Cationic Chameleons
Beyond the neutral linkers discussed above, the IP+ scaffold, as reported by Li et al, represents a transformative leap in macrocyclic permeability, fundamentally challenging the long-held dogma that charged moieties inherently preclude passive diffusion [70]. This "cationic chameleon" linker achieves exceptional cell permeability through a unique "charge-hydrophobicity synergy": its permanent, highly delocalized cationic center facilitates electrostatic enrichment at the anionic membrane surface, while the hydrophobic fused-ring system enables the molecule to "slide" through the lipid core as a pseudo-hydrophobic species. Validated by PAMPA and CAPA assays, IP+ linkers significantly augment the passive diffusion and cytosolic entry of high-molecular-weight macrocycles, effectively bypassing the endosomal entrapment common in traditional arginine-rich peptides. Furthermore, the IP+ macrocyclization exhibits extraordinary chemical robustness and modularity—proceeding efficiently in aqueous media and on miniaturized solid-phase resins with typical purities exceeding 85%— rendering it highly compatible with genetic encoding platforms such as mRNA display and DELs. This practical utility was exemplified by the HTS of a 480-member IP+-linked library, which yielded potent ligands with micromolar affinity, underscoring the platform's potential for the de novo discovery of cell-permeable leads against challenging intracellular targets.
In recent years, the expansion of synthetic methodologies and chemical biology toolboxes has led to the emergence of novel linkers featuring aromatic or heterocyclic scaffolds [71,72,73,74,75,76] Through the ongoing optimization of bioorthogonal reactivity and biocompatibility, these linkers are being progressively integrated into high-throughput platforms such as mRNA display and DELs. Their vast chemical diversity allows researchers to achieve the "front-loading" of favorable physicochemical traits during the earliest stages of selection. This shift from simple structural stabilization toward functional-driven design not only enhances the intrinsic quality of libraries but also provides a robust technical foundation for identifying cell-permeable leads with drug-like potential from trillion-scale chemical spaces.
3. Post-Screening Structural Refinement and Optimization Strategies
The bioactive sequences identified through high-throughput screening represent a starting point rather than a clinical finality. While display technologies excel at rapidly identifying high-affinity ligands, rational chemical refinement remains essential for addressing systemic hurdles, particularly membrane permeability. Linker engineering has evolved from basic structural bridging into a sophisticated strategy for precise permeability optimization [77]. In recent years, various functionalized linkers have demonstrated significant potential in facilitating cellular uptake. This section systematically reviews several representative linker strategies, focusing on their underlying mechanisms and recent breakthroughs in enhancing membrane permeability.
3.1. All-Hydrocarbon Staples: Refining the "Gold Standard" from Helical Stabilization to Metabolic Trapping
As the most established and widely used class of macrocyclic peptides, all-hydrocarbon stapled peptides have witnessed remarkable technological advancements to date, with their foundational research dating back to the late 1990s. Grubbs and Blackwell first utilized ruthenium-catalyzed ring-closing metathesis (RCM) to achieve side-chain cross-linking, establishing the chemical groundwork for the field. Subsequently [42,78,79]. In 2004, a landmark study by Walensky and Verdine published in Science demonstrated for the first time that all-hydrocarbon modification not only stabilizes the α-helical conformation but also confers exceptional cell-penetrating capabilities and induces apoptosis in vivo. This milestone marked the formal transition of stapled peptides into the era of intracellular therapeutic development [38].
In the ongoing evolution of the field, traditional design principles for membrane permeability—centered on lipophilicity, helicity, and charge distribution—are being continuously refined. Regarding the enhancement of peptide membrane permeability, the prevailing view, supported by numerous preceding studies, maintains that high lipophilicity and helicity promote cellular uptake by creating a "hydrophobic patch" and shielding the peptide backbone, thereby significantly reducing the desolvation energy penalty during translocation. While traditional perspectives held that positive charges were essential for initial electrostatic interactions with negatively charged cell membranes, and negative charges were viewed as barriers to entry, recent research has challenged these long-standing assumptions. Notably, in a landmark 2024 study, the Partridge, Brown, and Suga groups provided a robust revision of stapled peptide design rules based on a systematic analysis of over 350 variants [28]. To rigorously differentiate genuine cytosolic delivery from artifacts caused by membrane lysis, the researchers employed a comprehensive assay suite: the Nano-Click permeability assay for target-agnostic quantification, the LDH release assay for toxicity screening, and the Cell Ratio (EC50/KD) to evaluate translocation efficiency. Their data revealed a distinct "solubility cliff" for lipophilicity at Log D > 3 and a "threshold effect" for helicity (approximately 31%); furthermore, the strategic incorporation of anionic residues (Glutamic acid) rather than cationic residues proved critical for balancing solubility and safety. Most significantly, they unveiled a "metabolic-driven intracellular retention" mechanism: a hydrophobic poly-alanine (Poly-Ala) tail facilitates initial passive entry, followed by intracellular enzymatic cleavage to generate a negatively charged "trapped" metabolite, thereby achieving efficient cytosolic accumulation. This integration of quantitative design rules and metabolic orchestration marks a paradigm shift in the development of high-performance, cell-permeable peptide therapeutics.
Besides, hydrocarbon linkers have undergone a transformative evolution, shifting from simple sequence stabilization to the sophisticated orchestration of "advanced structural folding." Ma et al. (2024) introduced a heteroconformational double-stapled peptide (DSARTC), which, for the first time, simultaneously stabilizes both α-helix and β-sheet motifs within a single molecule [80]. This dual-stapling strategy leads to a highly compact conformation that physically shields the polar peptide backbone, thereby significantly reducing the desolvation energy penalty during membrane translocation. Experimental evidence demonstrated that DSARTC's permeability—validated by confocal microscopy and flow cytometry—surpassed that of traditional poly-arginine (PolyR) conjugation, transitioning the drug's activity from ineffective to the micromolar range (IC50 = 4.6 μM) against AR-V7. This highlights that structural pre-organization is often superior to simple cationic tagging for driving cellular uptake. Simultaneously, chemical innovations in linker architecture have provided non-destructive modalities for monitoring permeability. The "Diyne-Girder" technology developed by Jamieson et al. utilizes Glaser oxidative coupling to yield rigid, single-isomer linkers [81,82]. Beyond inducing high helicity (up to 59%) to minimize entropic loss, these linkers exhibit a strong Raman signal at 2252 cm-1 within the "cell-silent region." This allows for the direct tracking of peptide intracellular distribution via Raman microscopy without the interference of bulky fluorophores. Such bifunctional linkers not only enhance subtype selectivity (e.g., a 100-fold preference for MDM2 over MDMX) through extreme rigidity but also offer a unique tool for observing "authentic" peptide transport. The chemical structures of representative all-hydrocarbon linkers, including alkene and alkyne motifs, are shown in Figure 4.
3.2. Fluorine-Thiol Displacement Reaction (FTDR): Leveraging Chameleonic Adaptability for Dynamic Shielding
The Fluorine-Thiol Displacement Reaction (FTDR) stapling technology, developed by the Wang and Yang groups, offers a sophisticated chemical modality to surpass the permeability constraints of traditional hydrocarbon staples [83,84]. The conjugation chemistry and the resulting scaffold of FTDR-mediated linkers are illustrated in Figure 5. This strategy hinges on a "chameleonic adaptability" mediated by aromatic dithiol linkers (e.g., 1,3-benzenedimethanethiol). In silico modeling and environmental studies demonstrate that FTDR-peptides maintain innate flexibility in aqueous media but undergo a dramatic conformational shift upon membrane contact, with α-helicity surging from ~12% to ~89%, effectively masking the PSA during translocation. To rigorously quantify this uptake, researchers employed confocal microscopy and flow cytometry, utilizing Trypan Blue quenching to eliminate extracellular background and confirm genuine cytosolic internalization. Quantitative analysis revealed that FTDR-stapled peptides achieve up to a 5-fold increase in cellular uptake compared to RCM-based analogs. Mechanistic elucidation through inhibitor panels (e.g., chlorpromazine, nystatin) and competition assays indicated that the FTDR scaffold bypasses the singular, Heparan Sulfate Proteoglycans(HSPG)-dependent pathway characteristic of hydrocarbon staples. Instead, it recruits multiple endocytic machineries, including clathrin-, caveolin-, and actin-mediated pathways, particularly when optimized with an L,D-stereochemical configuration that facilitates nuclear enrichment. This synergy between multi-channel endocytosis and dynamic conformational shielding translates into potent intracellular inhibition of challenging targets like the Wnt signaling pathway and ERα-SRC2.
3.3. Self-Tracing Linkers: Eliminating Experimental Artifacts via Intrinsic Fluorescence
Beyond the previously mentioned strategies of utilizing diyne groups for Raman spectroscopy detection (located in the cell silent region) to achieve non-destructive observation, chemical innovations in linkers have also catalyzed the development of molecular tools with intrinsic autofluorescent properties. The Fluorescent Isoindole Crosslinking (FlICk) technology, developed by the research groups of Perrin, Li, and Chen, provides a more intuitive "what you see is what you get" solution. This technique employs ortho-phthalaldehyde (OPA) or its derivatives (ArKBCHOs) to undergo site-specific conjugation with Lysine and Cysteine residues on peptide side chains [85]. The site-specific conjugation chemistry and the resulting isoindole architecture are illustrated in Figure 6. This process constructs a characteristic fluorescent isoindole bridge in situ at the i, i+4 positions. The core advantage of this design is that it fundamentally resolves the "experimental artifact" problem common in traditional membrane permeability studies—where the mandatory attachment of bulky hydrophobic dyes (such as FITC) can bias results. Consequently, FlICk provides authentic intracellular transport data that is undisturbed by exogenous charges or additional molecular weight. Experimental evidence confirms that FlICk-stapled peptides exhibit significant intracellular accumulation in cell lines such as HeLa. The underlying mechanism is that the linker potently induces an α-helical conformation and physically shields the PSA of the peptide backbone. These linkers, which combine structural constraint with self-tracing functionality, not only replicate the biological activity of traditional hydrocarbon-stapled peptides but also—due to their mild reaction conditions and potential for HTS—offer a precise chemical platform for the development of theranostic peptide drugs.
3.4. Stimuli-Responsive Reversible Staples: Harmonizing Permeability with Pharmacophore Regeneration
To harmonize membrane permeability with native bioactivity, environment-responsive reversible stapling utilizes stimulus-driven "unstapling" to mitigate the potency loss associated with permanent modifications. Li, Yin, and colleagues developed a methionine bis-alkylation strategy (70%-93% conversion) incorporating dual sulfonium cations into the linker to enhance electrostatic enrichment [86]. Their subsequent work applied this technology to target the hepatitis B virus (HBV) HBx/Bcl-2 protein-protein interaction [87]. Experimental data revealed that the stapled peptide FAM-2b exhibited significantly higher uptake in HepG2.2.15 cells (MFI = 226) compared to its linear parent FAM-1 (MFI = 126), with confocal microscopy confirming strong co-localization with intracellular Bcl-2. Mechanistically, stapling improved the binding affinity to Bcl-2 from 45.6 μM (linear) to 5.3 μM. This dual enhancement in permeability and affinity translated into potent antiviral effects, with stapled analogs 2a and 2b effectively inhibiting the secretion of HBsAg without significant cytotoxicity. The sulfonium linkage undergoes traceless reduction triggered by intracellular glutathione (GSH), ensuring in situ pharmacophore regeneration.
Wan and coworkers introduced a carbamate-based "one-trigger, two-releases" platform leveraging a dual 1,4-elimination mechanism responsive to GSH or H2O2 [88]. In the design of LSD1 inhibitors, although stapling weakened in vitro Ki values by 16–36 fold, 4T1 cell viability assays demonstrated superior potency over the linear parent, underscoring the synergy between enhanced passive permeability and intracellular release. Similarly, Qian et al. developed reversible bicyclic scaffolds using disulfide bonds to achieve stimulus-responsive cargo release. Validated by flow cytometry and dual-luciferase reporter assays against the NEMO-IKK interaction, this system exhibited 3-fold higher uptake than standard monocyclic peptides and extended the serum half-life to 10 hours [89]. By integrating charge-induced uptake, passive diffusion, and environment-activated release, these dynamic linkers offer intelligent solutions for the precision targeting of intracellular proteins (Figure 7).
Beyond the aforementioned mainstream strategies, several novel linker architectures have recently emerged, including bis-urea bridges, double-guanidinium crosslinks, and foldamer-4MP hybrid topologies [90,91,92]. Although these structures possess diverse chemical natures, their core design principles are highly consistent: they aim to lower the transmembrane energy barrier through conformational rigidification, polar shielding, charge modulation, or environmental responsiveness. However, these cases also serve as a cautionary tale; blind charge accumulation or excessive lipophilicity often carries risks of hemolytic toxicity or endosomal trapping. This further underscores the urgency of transitioning evaluation frameworks from "apparent cellular entry" toward the establishment of more physiologically relevant platforms for "effective cytosolic quantification".
Notably, the demarcation between "biocompatible post-modification" and "HTS-compatible chemistry" is becoming increasingly blurred. For instance, ortho-phthalaldehyde (OPA)-based FlICk chemistry, originally viewed as a specific tool for rational structural modification, is currently being explored for broader genetic encoding platforms due to its exceptional reaction efficiency and in situ fluorogenic properties [93]. Similarly, ruthenium-catalyzed RCM for all-hydrocarbon stapling—once considered unsuitable for aqueous high-throughput screening—has recently achieved significant breakthroughs in its compatibility with DELs [94,95,96]. This trend suggests that linker selection is no longer a binary opposition between "discovery screening" and "rational design." Instead, it is an integrated process that deeply unifies high-capacity screening capabilities with complex chemical functionalities, aimed at collectively conquering the frontier of intracellular targeting
4. Discussion
The evaluation framework for stapled peptide membrane permeability has undergone a paradigmatic leap from "apparent cellular uptake observation" to "effective cytosolic quantification." Early research exhibited an over-reliance on Confocal Laser Scanning Microscopy and Flow Cytometry— paradigms frequently prone to membrane enrichment-induced false positives. These errors often stem from the hydrophobic synergistic effects between linkers and exogenous fluorescent probes (such as FITC), which can lead to an overestimation of a molecule's true permeability potential.
As the field matures, evaluation tools are evolving into integrated platforms with higher physiological relevance. While PAMPA facilitates the high-throughput screening of passive diffusion potential, its nature as a purely physical model precludes it from reflecting the compensatory effects of conformational pre-organization on the transmembrane desolvation penalty; furthermore, it cannot account for active transport mechanisms, such as caveolae-mediated endocytosis. Consequently, the concurrent application of Caco-2 models, CAPA, and LDH release assays has emerged as the new gold standard for pinpointing "effective cytosolic concentration." At the heart of this transition lies a profound understanding of the complex, non-linear relationship between physicochemical properties and biological outcomes. The structural features, mechanisms, and evaluation methods of the various linkers discussed in this review are summarized in Table 1.
For decades, high helicity, lipophilicity, and positive charge density have been regarded as the 'golden rules' for enhancing permeability. However, these simplistic physicochemical metrics are increasingly being challenged. Our analysis suggests that an ideal linker should not merely act as a hydrophobic or cationic 'patch'; instead, it should function as a dynamic regulator. By inducing 'Chameleonicity' through spontaneous intramolecular hydrogen bond (IMHB) formation, the peptide can expose polar groups in aqueous environments to maintain solubility while shielding backbone amides in lipid phases to reduce PSA, thereby lowering the transmembrane energy barrier without relying on extreme lipophilicity. Furthermore, the discovery of 'negative charge safety design' has corrected the traditional 'cation-driven' logic, proving that strategic placement of acidic residues (e.g., Glu) can balance amphiphilicity and mitigate membrane-disruption risks while maintaining permeability. This environment-adaptive approach provides a systematic mechanistic framework to resolve the 'Impossible Triangle' of solubility, permeability, and safety. Finally, advanced strategies such as stimulus-responsive reversible stapling and conjugation with cyclic cCPPs have expanded this chemical toolbox. By establishing a design paradigm that decouples the delivery engine from the target-binding domain, these strategies not only reconcile the intrinsic conflict between 'rigid permeation' and 'flexible binding' but also enable the 'traceless' intracellular release of active payloads, driving the evolution of stapled peptides from static structural constraints into intelligent, dynamic therapeutic entities.
5. Conclusions
This review systematically delineates the logical trajectory of stapled peptide linkers designed to enhance transmembrane permeability, tracing their evolution from early empirical all-hydrocarbon modifications to modern HTS and precision chemical entities. Supported by standardized evaluation frameworks, the academic understanding of "chameleonicity" and the mechanistic balance between charge and lipophilic thresholds has shifted from qualitative observation to quantitative analysis, effectively eliminating the experimental artifacts prevalent in earlier studies.
While current drug discovery and development efforts are still dominated by chemical modification strategies, stapled peptide design is undergoing a clear strategic transformation from "empirical trial-and-error" toward a "rational data-driven" approach. In the future, by integrating high-throughput biological data generated via mRNA display and leveraging Artificial Intelligence (AI) and machine learning for deep mining of multidimensional chemical spaces, we anticipate achieving automated prediction of linker performance and multidimensional synergistic optimization. This interdisciplinary fusion will fundamentally resolve the permeability and bioavailability bottlenecks hindering the clinical translation of stapled peptides, laying a robust foundation for the development of innovative drugs targeting challenging, intracellular "undruggable" targets.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created.
Acknowledgments
We appreciate the financial support from the Doctoral Science Foundation of Xinjiang Second Medical College (XGYK2025-102) and the Karamay Science and Technology Plan Project (2025BA0090).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| PPI | Protein–protein interaction |
| HTS | High-throughput screening |
| HBDs | hydrogen-bond donors |
| DEL | DNA-encoded library |
| ArF | Perfluoroaryl |
| AlkF | perfluoroalkyl |
| IMHB | Intramolecular hydrogen bond |
| PSA | Polar surface area |
| RCM | Ring-closing metathesis |
| CPPs | cell-penetrating peptides |
| CLIPS | Chemical Linkage of Peptides onto Scaffolds |
| TBMB | 1,3,5-Tris(bromomethyl)benzene |
| BBB | Blood–brain barrier |
| BPy | Bipyridine |
| UAA | Unnatural amino acid |
| HSPG | Heparan Sulfate Proteoglycans |
| IP+ | Imidazopyridinium |
| PAMPA | Parallel Artificial Membrane Permeability Assay |
| CAPA | Chloroalkane Penetration Assay |
| FTDR | Fluorine–Thiol Displacement Reaction |
| OPA | ortho-phthalaldehyde |
| FlICk | Fluorescent Isoindole Crosslinking |
| AI | Artificial Intelligence |
References
- Greenblatt, J.F.; Alberts, B.M.; Krogan, N.J. Discovery and significance of protein-protein interactions in health and disease. Cell 2024, 187, 6501–6517. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Ejaz, W.; Dutta, K.; Thayumanavan, S. Antibody Delivery for Intracellular Targets: Emergent Therapeutic Potential. Bioconjugate Chem 2019, 30, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the 'undruggable' cancer targets. Nature reviews. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Hong, S.H.; Nguyen, T.; Ongkingco, J.F.; Nazzaro, A.; Arora, P.S. From Concepts to Inhibitors: A Blueprint for Targeting Protein-Protein Interactions. Chem Rev 2025, 125, 6819–6869. [Google Scholar] [CrossRef]
- Chan, A.; Tsourkas, A. Intracellular Protein Delivery: Approaches, Challenges, and Clinical Applications. BME frontiers 2024, 5, 35. [Google Scholar] [CrossRef]
- Buyanova, M.; Pei, D. Targeting intracellular protein-protein interactions with macrocyclic peptides. Trends Pharmacol Sci 2022, 43, 234–248. [Google Scholar] [CrossRef]
- Wang, S.; Faucher, F.F.; Bertolini, M.; Kim, H.; Yu, B.; Cao, L.; Roeltgen, K.; Lovell, S.; Shanker, V.; Boyd, S.D.; et al. Identification of Covalent Cyclic Peptide Inhibitors Targeting Protein-Protein Interactions Using Phage Display. bioRxiv: the preprint server for biology 2024, 2011–2024. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L. The latest trends in peptide drug discovery and future challenges. Expert Opin Drug Dis 2024, 19, 869–872. [Google Scholar] [CrossRef]
- Anand, U.; Bandyopadhyay, A.; Jha, N.K.; Pérez De La Lastra, J.M.; Dey, A. Translational aspect in peptide drug discovery and development: An emerging therapeutic candidate. BioFactors (Oxford, England) 2023, 49, 251–269. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nature reviews. Drug discovery 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Pei, D. Designing Cell-Permeable Peptide Therapeutics That Enter the Cell by Endocytosis. ACS symposium series. American Chemical Society 2022, 1417, 179–197. [Google Scholar] [CrossRef]
- Kremsmayr, T.; Aljnabi, A.; Blanco-Canosa, J.B.; Tran, H.N.T.; Emidio, N.B.; Muttenthaler, M. On the Utility of Chemical Strategies to Improve Peptide Gut Stability. J Med Chem 2022, 65, 6191–6206. [Google Scholar] [CrossRef]
- Rezai, T.; Yu, B.; Millhauser, G.L.; Jacobson, M.P.; Lokey, R.S. Testing the conformational hypothesis of passive membrane permeability using synthetic cyclic peptide diastereomers. J Am Chem Soc 2006, 128, 2510–2511. [Google Scholar] [CrossRef]
- Mathiowetz, A.M. Design Principles for Intestinal Permeability of Cyclic Peptides. Methods in molecular biology (Clifton, N.J.) 2019, 2001, 1–15. [Google Scholar] [CrossRef]
- Jiang, F.; Geng, H. Computational Methods for Studying Conformational Behaviors of Cyclic Peptides. Methods in molecular biology (Clifton, N.J.) 2019, 2001, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, W.; Xu, Z. Improvement on Permeability of Cyclic Peptide/Peptidomimetic: Backbone N-Methylation as A Useful Tool. Mar Drugs 2021, 19, 311. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.F.; Zehfus, M.H. The effect of N-methylation on helical peptides. Biopolymers 1996, 40, 609–616. [Google Scholar] [CrossRef]
- Chatterjee, J.; Rechenmacher, F.; Kessler, H. N-methylation of peptides and proteins: an important element for modulating biological functions. Angewandte Chemie (International ed. in English) 2013, 52, 254–269. [Google Scholar] [CrossRef]
- Ghosh, P.; Raj, N.; Verma, H.; Patel, M.; Chakraborti, S.; Khatri, B.; Doreswamy, C.M.; Anandakumar, S.R.; Seekallu, S.; Dinesh, M.B.; et al. An amide to thioamide substitution improves the permeability and bioavailability of macrocyclic peptides. Nat Commun 2023, 14, 6050. [Google Scholar] [CrossRef] [PubMed]
- Hosono, Y.; Uchida, S.; Shinkai, M.; Townsend, C.E.; Kelly, C.N.; Naylor, M.R.; Lee, H.; Kanamitsu, K.; Ishii, M.; Ueki, R.; et al. Amide-to-ester substitution as a stable alternative to N-methylation for increasing membrane permeability in cyclic peptides. Nat Commun 2023, 14, 1416. [Google Scholar] [CrossRef]
- Wang, C.K.; Swedberg, J.E.; Harvey, P.J.; Kaas, Q.; Craik, D.J. Conformational Flexibility Is a Determinant of Permeability for Cyclosporin. The journal of physical chemistry. B 2018, 122, 2261–2276. [Google Scholar] [CrossRef]
- Corbett, K.M.; Ford, L.; Warren, D.B.; Pouton, C.W.; Chalmers, D.K. Cyclosporin Structure and Permeability: From A to Z and Beyond. J Med Chem 2021, 64, 13131–13151. [Google Scholar] [CrossRef] [PubMed]
- Mathiowetz, A.M. Design Principles for Intestinal Permeability of Cyclic Peptides. Methods in molecular biology (Clifton, N.J.) 2019, 2001, 1–15. [Google Scholar] [CrossRef]
- Tang, X.; Kokot, J.; Waibl, F.; Fernández-Quintero, M.L.; Kamenik, A.S.; Liedl, K.R. Addressing Challenges of Macrocyclic Conformational Sampling in Polar and Apolar Solvents: Lessons for Chameleonicity. J Chem Inf Model 2023, 63, 7107–7123. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Pang, W.; Xuan, S.; Chan, W.; Leung, K.C. Recent advances in peptide macrocyclization strategies. Chem Soc Rev 2024, 53, 11725–11771. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.H.; Mazzola, E.; Opoku-Nsiah, K.; Lammert, M.A.; Godes, M.; Neuberg, D.S.; Walensky, L.D. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat Chem Biol 2016, 12, 845–852. [Google Scholar] [CrossRef]
- Li, L.; Li, R.; Jiang, Y.; Chao, J.; Chen, S.; Liao, H.; Li, X. Advances in Hydrocarbon Stapled Peptides via Ring-Closing Metathesis: Synthetic Strategies, Structural Diversity, and Therapeutic Applications. Chembiochem: a European journal of chemical biology 2025, 26, e202500527. [Google Scholar] [CrossRef]
- Chandramohan, A.; Josien, H.; Yuen, T.Y.; Duggal, R.; Spiegelberg, D.; Yan, L.; Juang, Y.A.; Ge, L.; Aronica, P.G.; Kaan, H.Y.K.; et al. Design-rules for stapled peptides with in vivo activity and their application to Mdm2/X antagonists. Nat Commun 2024, 15, 489. [Google Scholar] [CrossRef]
- Colas, K.; Bindl, D.; Suga, H. Selection of Nucleotide-Encoded Mass Libraries of Macrocyclic Peptides for Inaccessible Drug Targets. Chem Rev 2024, 124, 12213–12241. [Google Scholar] [CrossRef]
- Wang, X.S.; Chen, P.C.; Hampton, J.T.; Tharp, J.M.; Reed, C.A.; Das, S.K.; Wang, D.; Hayatshahi, H.S.; Shen, Y.; Liu, J.; et al. A Genetically Encoded, Phage-Displayed Cyclic-Peptide Library. Angewandte Chemie (International ed. in English) 2019, 58, 15904–15909. [Google Scholar] [CrossRef]
- Yao, X.; Aphicho, K.; Pani, S.; Rupanya, A.; Lan, T.; Dickinson, B.C. Discovery of Macrocyclic Peptide Binders, Covalent Modifiers, and Degraders of a Structured RNA by mRNA Display. J Am Chem Soc 2025, 147, 34256–34270. [Google Scholar] [CrossRef]
- Petrov, D.; Plais, L.; Schira, K.; Cai, J.; Keller, M.; Lessing, A.; Bassi, G.; Cazzamalli, S.; Neri, D.; Gloger, A.; et al. Flexibility-tuning of dual-display DNA-encoded chemical libraries facilitates cyclic peptide ligand discovery. Nat Commun 2025, 16, 3273. [Google Scholar] [CrossRef]
- Bai, L.; Dan, T.; Cheng, P.; Yang, X.; Xiang, H.; Zhai, W.; Chen, Y.; Huang, R.; Wang, Q.; Li, K.; et al. Isothiocyanate-mediated cyclization of phage-displayed peptides enables discovery of macrocyclic binders. Sci Adv 2026, 12, b7086. [Google Scholar] [CrossRef]
- Colas, K.; Bindl, D.; Suga, H. Selection of Nucleotide-Encoded Mass Libraries of Macrocyclic Peptides for Inaccessible Drug Targets. Chem Rev 2024, 124, 12213–12241. [Google Scholar] [CrossRef]
- Zhao, H.; Li, X.; Yao, X.; Zhang, S.; Bao, Y.; Lu, W.; Xing, M.; Wang, X.; Wang, X.; Zhao, Y.; et al. Influence of Macrocyclization Strategies on DNA-Encoded Cyclic Peptide Libraries. JACS Au 2025, 5, 3399–3407. [Google Scholar] [CrossRef] [PubMed]
- Chavali, S.S.; Mali, S.M.; Bonn, R.; Saseendran Anitha, A.; Bennett, R.P.; Smith, H.C.; Fasan, R.; Wedekind, J.E. Cyclic peptides with a distinct arginine-fork motif recognize the HIV trans-activation response RNA in?vitro and in cells. The Journal of biological chemistry 2021, 297, 101390. [Google Scholar] [CrossRef] [PubMed]
- Anananuchatkul, T.; Chang, I.V.; Miki, T.; Tsutsumi, H.; Mihara, H. Construction of a Stapled α-Helix Peptide Library Displayed on Phage for the Screening of Galectin-3-Binding Peptide Ligands. ACS omega 2020, 5, 5666–5674. [Google Scholar] [CrossRef]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science (New York, N.Y.) 2004, 305, 1466–1470. [Google Scholar] [CrossRef]
- Colas, K.; Bindl, D.; Suga, H. Selection of Nucleotide-Encoded Mass Libraries of Macrocyclic Peptides for Inaccessible Drug Targets. Chem Rev 2024, 124, 12213–12241. [Google Scholar] [CrossRef]
- Chen, F.; Pinnette, N.; Gao, J. Strategies for the Construction of Multicyclic Phage Display Libraries. Chembiochem: a European journal of chemical biology 2024, 25, e202400072. [Google Scholar] [CrossRef]
- Timmerman, P.; Beld, J.; Puijk, W.C.; Meloen, R.H. Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem: a European journal of chemical biology 2005, 6, 821–824. [Google Scholar] [CrossRef]
- Li, Y.; Wu, M.; Fu, Y.; Xue, J.; Yuan, F.; Qu, T.; Rissanou, A.N.; Wang, Y.; Li, X.; Hu, H. Therapeutic stapled peptides: Efficacy and molecular targets. Pharmacol Res 2024, 203, 107137. [Google Scholar] [CrossRef] [PubMed]
- Muppidi, A.; Zhang, H.; Curreli, F.; Li, N.; Debnath, A.K.; Lin, Q. Design of antiviral stapled peptides containing a biphenyl cross-linker. Bioorg Med Chem Lett 2014, 24, 1748–1751. [Google Scholar] [CrossRef]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol 2009, 5, 502–507. [Google Scholar] [CrossRef]
- Villequey, C.; Zurmühl, S.S.; Cramer, C.N.; Bhusan, B.; Andersen, B.; Ren, Q.; Liu, H.; Qu, X.; Yang, Y.; Pan, J.; et al. An efficient mRNA display protocol yields potent bicyclic peptide inhibitors for FGFR3c: outperforming linear and monocyclic formats in affinity and stability. Chem Sci 2024, 15, 6122–6129. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O.; Lombardi, L.; Williams, D.R.; Albericio, F. Strategies for Improving Peptide Stability and Delivery. Pharmaceuticals (Basel, Switzerland) 2022, 15, 1283. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Schofield, J.A.; Na, Z.; Hann, T.; Simon, M.D.; Slavoff, S.A. Discovery of cellular substrates of human RNA-decapping enzyme DCP2 using a stapled bicyclic peptide inhibitor. Cell Chem Biol 2021, 28, 463–474. [Google Scholar] [CrossRef]
- Krishna Sudhakar, H.; Yau, J.T.K.; Alcock, L.J.; Lau, Y.H. Accessing diverse bicyclic peptide conformations using 1,2,3-TBMB as a linker. Org Biomol Chem 2024, 22, 6095–6102. [Google Scholar] [CrossRef] [PubMed]
- Kadota, K.; Kohata, A.; Sando, S.; Morimoto, J.; Aikawa, K.; Okazoe, T. Comparison of the effects of perfluoroalkyl and alkyl groups on cellular uptake in short peptides. Rsc Adv 2025, 15, 8189–8194. [Google Scholar] [CrossRef]
- Ni, C.; Hu, J. The unique fluorine effects in organic reactions: recent facts and insights into fluoroalkylations. Chem Soc Rev 2016, 45, 5441–5454. [Google Scholar] [CrossRef]
- Dognini, P.; Chaudhry, T.; Scagnetti, G.; Assante, M.; Hanson, G.S.M.; Ross, K.; Giuntini, F.; Coxon, C. R. 5,10,15,20-Tetrakis(pentafluorophenyl)porphyrin as a Functional Platform for Peptide Stapling and Multicyclisation. Chemistry (Weinheim an der Bergstrasse, Germany) 2023, 29, e202301410. [Google Scholar] [CrossRef]
- Miles, S.A.; Nillama, J.A.; Hunter, L. Tinker, Tailor, Soldier, Spy: The Diverse Roles That Fluorine Can Play within Amino Acid Side Chains. Molecules (Basel, Switzerland) 2023, 28, 6192. [Google Scholar] [CrossRef]
- Spokoyny, A.M.; Zou, Y.; Ling, J.J.; Yu, H.; Lin, Y.; Pentelute, B.L. A perfluoroaryl-cysteine S(N)Ar chemistry approach to unprotected peptide stapling. J Am Chem Soc 2013, 135, 5946–5949. [Google Scholar] [CrossRef]
- de Araujo, A.D.; Lim, J.; Wu, K.; Hoang, H.N.; Nguyen, H.T.; Fairlie, D.P. Landscaping macrocyclic peptides: stapling hDM2-binding peptides for helicity, protein affinity, proteolytic stability and cell uptake. RSC chemical biology 2022, 3, 895–904. [Google Scholar] [CrossRef]
- Kalhor-Monfared, S.; Jafari, M.R.; Patterson, J.T.; Kitov, P.I.; Dwyer, J.J.; Nuss, J.M.; Derda, R. Rapid biocompatible macrocyclization of peptides with decafluoro-diphenylsulfone. Chem Sci 2016, 7, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Fadzen, C.M.; Wolfe, J.M.; Cho, C.; Chiocca, E.A.; Lawler, S.E.; Pentelute, B.L. Perfluoroarene-Based Peptide Macrocycles to Enhance Penetration Across the Blood-Brain Barrier. J Am Chem Soc 2017, 139, 15628–15631. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, Q.; Li, Z.; Tian, Y.; Zhou, S. A Chemically Engineered Macrocyclic Peptide Shuttle Overcomes Blood-Brain Barrier in Aging Brain. Angewandte Chemie (International ed. in English) 2026, 65, e20279. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Katoh, T.; Suga, H. Macrocyclic Peptides Closed by a Thioether-Bipyridyl Unit That Grants Cell Membrane Permeability. ACS bio & med chem Au 2023, 3, 429–437. [Google Scholar] [CrossRef]
- Peraro, L.; Deprey, K.L.; Moser, M.K.; Zou, Z.; Ball, H.L.; Levine, B.; Kritzer, J.A. Cell Penetration Profiling Using the Chloroalkane Penetration Assay. J Am Chem Soc 2018, 140, 11360–11369. [Google Scholar] [CrossRef]
- Zhan, W.; Duan, H.; Li, C. Recent Advances in Metal-Free Peptide Stapling Strategies. Chem & bio engineering 2024, 1, 593–605. [Google Scholar] [CrossRef]
- He, J.; Ghosh, P.; Nitsche, C. Biocompatible strategies for peptide macrocyclisation. Chem Sci 2024, 15, 2300–2322. [Google Scholar] [CrossRef]
- Hacker, D.E.; Abrigo, N.A.; Hoinka, J.; Richardson, S.L.; Przytycka, T.M.; Hartman, M.C.T. Direct, Competitive Comparison of Linear, Monocyclic, and Bicyclic Libraries Using mRNA Display. Acs Comb Sci 2020, 22, 306–310. [Google Scholar] [CrossRef]
- Kawamoto, S.A.; Coleska, A.; Ran, X.; Yi, H.; Yang, C.; Wang, S. Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J Med Chem 2012, 55, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, V.; Choudhury, A.R.; Chakrabarti, R. Decoding the dynamics of BCL9 triazole stapled peptide. Biophys Chem 2024, 307, 107197. [Google Scholar] [CrossRef]
- Xu, W.; Lau, Y.H.; Fischer, G.; Tan, Y.S.; Chattopadhyay, A.; de la Roche, M.; Hyv Nen, M.; Verma, C.; Spring, D.R.; Itzhaki, L.S. Macrocyclized Extended Peptides: Inhibiting the Substrate-Recognition Domain of Tankyrase. J Am Chem Soc 2017, 139, 2245–2256. [Google Scholar] [CrossRef]
- Wu, Y.; Kaur, A.; Fowler, E.; Wiedmann, M.M.; Young, R.; Galloway, W.R.J.D.; Olsen, L.; Sore, H.F.; Chattopadhyay, A.; Kwan, T.T.; et al. Toolbox of Diverse Linkers for Navigating the Cellular Efficacy Landscape of Stapled Peptides. Acs Chem Biol 2019, 14, 526–533. [Google Scholar] [CrossRef]
- Lau, Y.H.; De Andrade, P.; Quah, S.T.; Rossmann, M.; Laraia, L.; Sköld, N.; Sum, T.J. Functionalised staple linkages for modulating the cellular activity of stapled peptides. Chem Sci 2014, 5, 1804–1809. [Google Scholar] [CrossRef]
- Liu, M.; Morewood, R.; Yoshisada, R.; Pascha, M.N.; Hopstaken, A.J.P.; Tarcoveanu, E.; Poole, D.A.R.; de Haan, C.A.M.; Nitsche, C.; Jongkees, S.A.K. Selective thiazoline peptide cyclisation compatible with mRNA display and efficient synthesis. Chem Sci 2023, 14, 10561–10569. [Google Scholar] [CrossRef] [PubMed]
- Takashi Tamura, M.I.Y.Y.; Kyosuke Tsumura, K.S.T.W. Selected Paper Chemical Synthesis and Cell-Free Expression of Thiazoline Ring-Bridged Cyclic Peptides and Their Properties on Biomembrane Permeability. B Chem Soc Jpn 2021, 95, 359–366. [Google Scholar] [CrossRef]
- Li, B.; Parker, J.; Tong, J.; Kodadek, T. Synthesis of Membrane-Permeable Macrocyclic Peptides via Imidazopyridinium Grafting. J Am Chem Soc 2024, 146, 14633–14644. [Google Scholar] [CrossRef]
- Nie, Q.; Xu, T.; Fang, X.; Dan, Y.; Zhang, G.; Li, Y.; Li, J.; Li, Y. The Furan-Thiol-Amine Reaction Facilitates DNA-Compatible Thiopyrrole-Grafted Macrocyclization and Late-Stage Amine Transformation. Org Lett 2025, 27, 498–503. [Google Scholar] [CrossRef]
- Bao, Y.; Xing, M.; Matthew, N.; Chen, X.; Wang, X.; Lu, X. Macrocyclizing DNA-Linked Peptides via Three-Component Cyclization and Photoinduced Chemistry. Org Lett 2024, 26, 2763–2767. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Arico-Muendel, C.C.; Ding, Y.; Pollastri, M.P.; Scott, S.; Mantell, M.A.; Yao, G. Synthesis of a DNA-Encoded Macrocyclic Library Utilizing Intramolecular Benzimidazole Formation. Bioconjugate Chem 2023, 34, 988–993. [Google Scholar] [CrossRef]
- Brown, S.P.; Smith, A.B.R. Peptide/protein stapling and unstapling: introduction of s-tetrazine, photochemical release, and regeneration of the peptide/protein. J Am Chem Soc 2015, 137, 4034–4037. [Google Scholar] [CrossRef]
- Morewood, R.; Nitsche, C. Bioinspired peptide stapling generates stable enzyme inhibitors. Chemical communications (Cambridge, England) 2022, 58, 10817–10820. [Google Scholar] [CrossRef]
- Rivas, M.; Gevorgyan, V. Advances in Selected Heterocyclization Methods. Synlett: accounts and rapid communications in synthetic organic chemistry 2023, 34, 1554–1562. [Google Scholar] [CrossRef]
- Merz, M.L.; Habeshian, S.; Li, B.; David, J.G.L.; Nielsen, A.L.; Ji, X.; Il Khwildy, K.; Duany Benitez, M.M.; Phothirath, P.; Heinis, C. De novo development of small cyclic peptides that are orally bioavailable. Nat Chem Biol 2024, 20, 624–633. [Google Scholar] [CrossRef]
- Li, L.; Li, R.; Jiang, Y.; Chao, J.; Chen, S.; Liao, H.; Li, X. Advances in Hydrocarbon Stapled Peptides via Ring-Closing Metathesis: Synthetic Strategies, Structural Diversity, and Therapeutic Applications. Chembiochem: a European journal of chemical biology 2025, 26, e202500527. [Google Scholar] [CrossRef]
- Blackwell, H.E.; Grubbs, R.H. Highly Efficient Synthesis of Covalently Cross-Linked Peptide Helices by Ring-Closing Metathesis. Angewandte Chemie (International ed. in English) 1998, 37, 3281–3284. [Google Scholar] [CrossRef]
- Ma, B.; Liu, D.; Zheng, M.; Wang, Z.; Zhang, D.; Jian, Y.; Ma, J.; Fan, Y.; Chen, Y.; Gao, Y.; et al. Development of a Double-Stapled Peptide Stabilizing Both α-Helix and β-Sheet Structures for Degrading Transcription Factor AR-V7. JACS Au 2024, 4, 816–827. [Google Scholar] [CrossRef]
- Morgan, D.C.; McDougall, L.; Knuhtsen, A.; Jamieson, A.G. Development of Bifunctional, Raman Active Diyne-Girder Stapled α-Helical Peptides. Chemistry (Weinheim an der Bergstrasse, Germany) 2023, 29, e202300855. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.C.; McDougall, L.; Knuhtsen, A.; Buetow, L.; Steven, C.F.; Shepperson, O.A.; Huang, D.T.; Hulme, A.N.; Jamieson, A.G. Raman active diyne-girder conformationally constrained p53 stapled peptides bind to MDM2 for visualisation without fluorophores. RSC chemical biology 2025, 6, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Junod, S.L.; Zhang, S.; Buuh, Z.Y.; Guan, Y.; Zhao, M.; Kaneria, K.H.; Kafley, P.; Cohen, C.; Maloney, R.; et al. Unprotected peptide macrocyclization and stapling via a fluorine-thiol displacement reaction. Nat Commun 2022, 13, 350. [Google Scholar] [CrossRef]
- Maloney, R.; Junod, S.L.; Hagen, K.M.; Lewis, T.; Cheng, C.; Shajan, F.J.; Zhao, M.; Moore, T.W.; Truong, T.H.; Yang, W.; et al. Flexible fluorine-thiol displacement stapled peptides with enhanced membrane penetration for the estrogen receptor/coactivator interaction. The Journal of biological chemistry 2024, 300, 107991. [Google Scholar] [CrossRef]
- Dayanara, N.L.; Froelich, J.; Roome, P.; Perrin, D.M. Chemoselective, regioselective, and positionally selective fluorogenic stapling of unprotected peptides for cellular uptake and direct cell imaging. Chem Sci 2024, 16, 584–595. [Google Scholar] [CrossRef]
- Shi, X.; Zhao, R.; Jiang, Y.; Zhao, H.; Tian, Y.; Jiang, Y.; Li, J.; Qin, W.; Yin, F.; Li, Z. Reversible stapling of unprotected peptides via chemoselective methionine bis-alkylation/dealkylation. Chem Sci 2018, 9, 3227–3232. [Google Scholar] [CrossRef]
- Cai, X.; Zheng, W.; Shi, X.; Chen, L.; Liu, Z.; Li, Z. HBx-Derived Constrained Peptides Inhibit the Secretion of Hepatitis B Virus Antigens. Mol Pharmaceut 2018, 15, 5646–5652. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhu, J.; Deng, X.; Chen, H.; Jin, Y.; Miclet, E.; Alezra, V.; Wan, Y. Customized Reversible Stapling for Selective Delivery of Bioactive Peptides. J Am Chem Soc 2022, 144, 23614–23621. [Google Scholar] [CrossRef]
- Qian, Z.; Rhodes, C.A.; McCroskey, L.C.; Wen, J.; Appiah-Kubi, G.; Wang, D.J.; Guttridge, D.C.; Pei, D. Enhancing the Cell Permeability and Metabolic Stability of Peptidyl Drugs by Reversible Bicyclization. Angewandte Chemie (International ed. in English) 2017, 56, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Chen, P.; Liu, J.; Liao, Y.; Tang, M.; Zhao, J. Synthesis of Bis-urea-Bridged Cyclopeptides via Stapling of Unprotected Peptides. Org Lett 2025, 27, 14230–14235. [Google Scholar] [CrossRef] [PubMed]
- Neuville, M.; Bornez, M.M.; Bourgeais, M.; Samueli, H.; Mauran, L.; Goudreau, S.R.; Khatib, A.; Guichard, G.; Pasco, M. A General Synthesis Approach to Double-Guanidinium Stapled Peptides and Foldamers. Chemistry (Weinheim an der Bergstrasse, Germany) 2025, 31, e2273. [Google Scholar] [CrossRef]
- Neuville, M.; Bourgeais, M.; Buratto, J.; Saragaglia, C.; Li, B.; Galeano-Otero, I.; Mauran, L.; Varajao, L.; Goudreau, S.R.; Kauffmann, B.; et al. Optimal Stapling of a Helical Peptide-Foldamer Hybrid Using a C-Terminal 4-Mercaptoproline Enhances Protein Surface Recognition and Cellular Activity. Chemistry (Weinheim an der Bergstrasse, Germany) 2025, 31, e202403330. [Google Scholar] [CrossRef] [PubMed]
- Bakhshinejad, B.; Ghiasvand, S. A Beautiful Bind: Phage Display and the Search for Cell-Selective Peptides. Viruses 2025, 17, 975. [Google Scholar] [CrossRef] [PubMed]
- Monty, O.B.C.; Nyshadham, P.; Bohren, K.M.; Palaniappan, M.; Matzuk, M.M.; Young, D.W.; Simmons, N. Homogeneous and Functional Group Tolerant Ring-Closing Metathesis for DNA-Encoded Chemical Libraries. Acs Comb Sci 2020, 22, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Blanco, C.O.; Cormier, S.K.; Koller, A.J.; Boros, E.; Fogg, D.E. Olefin Metathesis in Water: Speciation of a Leading Water-Soluble Catalyst Pinpoints Challenges and Opportunities for Chemical Biology. J Am Chem Soc 2025, 147, 9441–9448. [Google Scholar] [CrossRef]
- Zou, Z.; Kalvet, I.; Lozhkin, B.; Morris, E.; Zhang, K.; Chen, D.; Ernst, M.L.; Zhang, X.; Baker, D.; Ward, T.R. De novo design and evolution of an artificial metathase for cytoplasmic olefin metathesis. Nature catalysis 2025, 8, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemical strategies for the construction of stapled and multicyclic peptides using aromatic linkers. (a) CLIPS technology utilizing mDBMB for monocyclization; (b) Biphenyl-based linkers for hydrophobic shielding; (c) Construction of bicyclic architectures via C3-symmetric TBMB; (d) Bipyridine (BPy) linkers for backbone shielding via intramolecular hydrogen bonding.
Figure 1.
Chemical strategies for the construction of stapled and multicyclic peptides using aromatic linkers. (a) CLIPS technology utilizing mDBMB for monocyclization; (b) Biphenyl-based linkers for hydrophobic shielding; (c) Construction of bicyclic architectures via C3-symmetric TBMB; (d) Bipyridine (BPy) linkers for backbone shielding via intramolecular hydrogen bonding.

Figure 2.
Schematic representation of the SNAr reaction between cysteine thiols. This strategy utilizes the "Fluorine Effect" to drive lipid raft accumulation and trigger caveolin-mediated transcytosis (CMT).
Figure 2.
Schematic representation of the SNAr reaction between cysteine thiols. This strategy utilizes the "Fluorine Effect" to drive lipid raft accumulation and trigger caveolin-mediated transcytosis (CMT).

Figure 3.
Representative heteroaromatic linkers derived from UAAs for cell-permeable macrocyclic peptides. (A) Triazole linkers featuring a modular R group (e.g., CPPs or cationic clusters) to enable a "decoupling strategy" for independent optimization of permeability and affinity; (B) Thiazoline linkers facilitating intramolecular hydrogen bond (IMHB) networks to effectively reduce the polar surface area (PSA); and (C) Imidazopyridinium (IP+) scaffolds acting as "cationic chameleons" to enhance passive diffusion via charge-hydrophobicity synergy.
Figure 3.
Representative heteroaromatic linkers derived from UAAs for cell-permeable macrocyclic peptides. (A) Triazole linkers featuring a modular R group (e.g., CPPs or cationic clusters) to enable a "decoupling strategy" for independent optimization of permeability and affinity; (B) Thiazoline linkers facilitating intramolecular hydrogen bond (IMHB) networks to effectively reduce the polar surface area (PSA); and (C) Imidazopyridinium (IP+) scaffolds acting as "cationic chameleons" to enhance passive diffusion via charge-hydrophobicity synergy.

Figure 4.
Representative all-hydrocarbon linkers for peptide stapling. (A) Alkene-based staples generated via ring-closing metathesis (RCM); (B) Alkyne-based linkers (Diyne-Girder) formed through oxidative coupling.
Figure 4.
Representative all-hydrocarbon linkers for peptide stapling. (A) Alkene-based staples generated via ring-closing metathesis (RCM); (B) Alkyne-based linkers (Diyne-Girder) formed through oxidative coupling.

Figure 5.
FTDR-mediated stapling using aromatic dithiols. This strategy leverages "chameleonic adaptability" to shield polar surfaces through environment-induced helicity shifts.
Figure 5.
FTDR-mediated stapling using aromatic dithiols. This strategy leverages "chameleonic adaptability" to shield polar surfaces through environment-induced helicity shifts.

Figure 6.
Schematic representation of the Fluorescent Isoindole Crosslinking (FlICk) strategy. The reaction between ortho-phthalaldehyde (OPA) and the side chains of Lysine and Cysteine residues forms a fluorescent isoindole bridge at the i, i+4 positions, providing both structural stabilization and intrinsic self-tracing functionality.
Figure 6.
Schematic representation of the Fluorescent Isoindole Crosslinking (FlICk) strategy. The reaction between ortho-phthalaldehyde (OPA) and the side chains of Lysine and Cysteine residues forms a fluorescent isoindole bridge at the i, i+4 positions, providing both structural stabilization and intrinsic self-tracing functionality.

Figure 7.
General mechanism of environment-responsive reversible stapling.(A) Phase I: In vitro peptide locking via site-specific conjugation with trigger-sensitive linkers. (B) Phase II: Stimuli-driven (e.g., GSH, H2O2) intracellular "unstapling." This decoupling strategy enables traceless pharmacophore regeneration and reconciles the trade-off between membrane permeability and target affinity.
Figure 7.
General mechanism of environment-responsive reversible stapling.(A) Phase I: In vitro peptide locking via site-specific conjugation with trigger-sensitive linkers. (B) Phase II: Stimuli-driven (e.g., GSH, H2O2) intracellular "unstapling." This decoupling strategy enables traceless pharmacophore regeneration and reconciles the trade-off between membrane permeability and target affinity.


Table 1.
Summary of representative linker engineering strategies, their underlying membrane permeation mechanisms, and corresponding evaluation methodologies for stapled peptides.
Table 1.
Summary of representative linker engineering strategies, their underlying membrane permeation mechanisms, and corresponding evaluation methodologies for stapled peptides.
| Linker Strategy & Examples | Primary Permeation Mechanism | Key Evaluation Assays | Ref. |
| Arylene-based (mDBMB, TBMB, BPh) | Enhances permeability via conformational rigidification; "topological condensation" reduces the molecular radius of gyration and entropic penalty. | FITC imaging, CETSA, Live-cell imaging. | [43] [47] |
| Perfluoro-aromatic (ArF, AlkF) | Leverages the "Fluorine Effect" to increase lipophilicity; drives lipid raft accumulation and triggers CMT. | 3D BBB sphere model, Flow cytometry, Confocal microscopy. | [53,54] [56,57] |
| Bipyridine (BPy) (BPy unit) | Pyridine nitrogens act as H-bond acceptors to form IMHBs with backbone amides, providing effective shielding and increasing lipophilicity. | CAPA, PAMPA. | [58] |
| Triazole-based (Dialkynyl linkers) | Employs a "decoupling" strategy to independently optimize permeability (via cationic motifs or CPPs) and binding affinity. | Confocal microscopy, Reporter gene assays. | [54,63,64] |
| Thiazoline (Cys-Nitrile condensation) | Converts an HBD into an HBA; induces a stable IMHB network in hydrophobic environments to compress the PSA and lower desolvation energy. | PAMPA, Temp-variable NMR, Computational simulations. | [69] |
| Imidazo-pyridinium (IP+ scaffold) | Acts as a "cationic chameleon" where the delocalized cation facilitates membrane enrichment while the fused-ring system enables "pseudo-hydrophobic sliding". | PAMPA, CAPA. | [70] |
| All-hydrocarbon (RCM-based staples) | Stabilizes α-helical structures to shield polar amides; utilizes metabolic-driven trapping where enzymatic cleavage of a Poly-Ala tail leads to cytosolic accumulation. | Nano-Click, LDH release, Cell Ratio (EC50/KD). | [28] [80,81,82] |
| FTDR-based (Aromatic dithiols) | Exhibits "chameleonic adaptability" with dynamic helicity increases (from 12% to 89%) upon membrane contact; bypasses HSPG-dependence via multiple endocytic pathways. | Flow cytometry, Trypan Blue quenching, Inhibitor panels. | [83,84] |
| Self-tracing (Isoindole, Diyne) | Eliminates artifacts from external dyes using intrinsic fluorescence (FlICk) or Raman signals in the "cell-silent" region. | Raman microscopy, Live-cell fluorescence imaging. | [85] |
| Stimuli-responsive (Sulfonium, Carbamate) | Enhances enrichment via cationic linkers followed by intracellular "unstapling" (GSH or H2O2 triggered) to regenerate the pharmacophore. | MFI, Co-localization, Flow cytometry. | [86,87,88,89] |
Abbreviations: BBB, blood–brain barrier; CAPA, chloroalkane penetration assay; CETSA, cellular thermal shift assay; CMT, caveolin-mediated transcytosis; CPP, cell-penetrating peptide; FITC, fluorescein isothiocyanate; GSH, glutathione; HBA, hydrogen bond acceptor; HBD, hydrogen bond donor; HSPG, heparan sulfate proteoglycan; IMHB, intramolecular hydrogen bond; LDH, lactate dehydrogenase; MFI, mean fluorescence intensity; PAMPA, parallel artificial membrane permeability assay; PSA, polar surface area.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



