
Submitted:
25 February 2026
Posted:
27 February 2026
You are already at the latest version
Abstract
1,2,4-Trisubstituted chiral homoallylic alcohols are valuable intermediates in natural product synthesis and complex molecular architectures; however, their asymmetric catalytic synthesis remains challenging due to the need for simultaneous and precise control of regioselectivity, diastereoselectivity, E/Z geometry, and enantioselectivity. Herein, we report a chiral transfer reaction based on a palladium-catalyzed three-component reaction of aldehydes, borylated allyl acetate, dimethylzinc for the efficient synthesis of 1,2,4-trisubstituted anti-(Z)-chiral homoallylic alcohol derivatives. Readily accessible borylated chiral homoallylic alcohols, prepared for example via Sharpless asymmetric epoxidation, serve as effective starting materials, allowing highly stereocontrolled formation of 1,2,4-trisubstituted anti-(Z)-chiral homoallylic alcohols.
Keywords:
three-component reaction
; palladium
; homoallylic alcohol
; chirality transfer
; enantiospecificity
1. Introduction
Chiral homoallylic alcohols constitute an important class of compounds that serve as key intermediates in the synthesis of a wide range of natural products and biologically active molecules, owing to their versatile reactivity and well-defined stereochemical features.[1,2,3] As such, considerable efforts have been devoted to the development of efficient and stereoselective methods for their preparation. To date, a variety of catalytic asymmetric approaches have been reported using chiral Lewis acid or chiral Lewis base catalysts.[4,5,6,7] In addition, palladium[8], ruthenium[9], iridium[8], or copper catalytic systems[10] have enabled the construction of substituted chiral homoallylic alcohols, allowing precise control over regio-, diastereo-, and enantioselectivity. Furthermore, notable recent examples include chromium-catalyzed asymmetric allylation of aldehydes with alkenes via allylic C(sp³)–H functionalization under photoredox conditions[11], as well as organophotoredox/nickel-cocatalyzed allylation of allenes, which enables diastereo- and enantioselective access to chiral homoallylic alcohols.[12] These catalytic methods significantly expand the synthetic toolbox for the stereoselective construction of substituted chiral homoallylic alcohols, although the development of more general, practical, and modular methods with broad substrate scope remains an important challenge. Despite these significant advances, the synthesis of 1,2,4-trisubstituted chiral homoallylic alcohols remains a formidable challenge, as it requires the simultaneous and precise control of regioselectivity, diastereoselectivity, E/Z geometry, and enantioselectivity; consequently, such examples remain limited.[13,14,15,16] In many cases, high enantioselectivity can be achieved through extensive ligand design and exhaustive screening of chiral ligands.
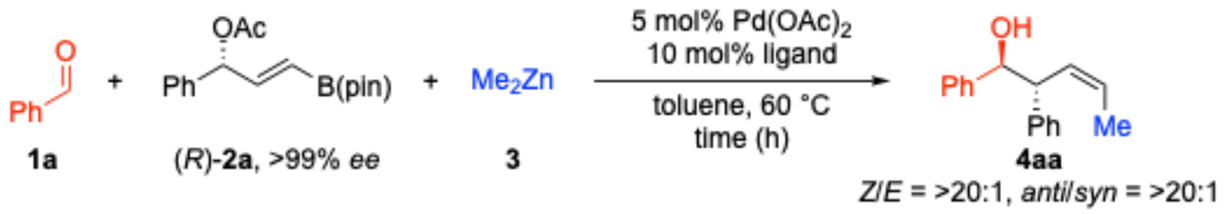
We have previously reported a three-component reaction for the synthesis of homoallylic alcohols that exhibits excellent regio- and diastereoselectivity (eqn 1)[17]. In this approach, carbonyl allylation reaction is initially performed using a borylated allyl acetate, for which regioselectivity concerns are inherently minimized, followed by a sequential coupling reaction with organoboranes. This sequence effectively circumvents the regioselectivity limitations commonly encountered in conventional allylation reactions.
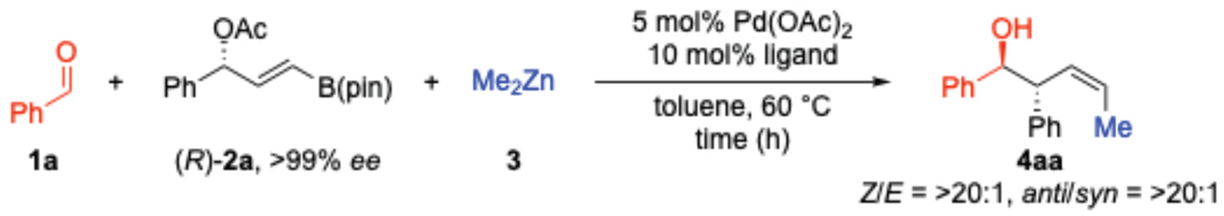
We herein report the extension of this three-component reaction to a chiral transfer reaction for the synthesis of 1,2,4-trisubstituted anti-(Z)-chiral homoallylic alcohol derivatives (eqn 2). Chiral transfer reactions represent an attractive approach to asymmetric synthesis[18] when optically active starting materials are readily accessible, stereochemical information is transferred with high fidelity, and the desired products are difficult to obtain by alternative asymmetric methods. The borylated chiral allyl acetates employed herein can be prepared efficiently, for example, via Sharpless-Katsuki asymmetric epoxidation[19]. Accordingly, their application as starting materials in chiral transfer reactions enables an efficient and highly stereocontrolled synthesis of 1,2,4-trisubstituted anti-(Z)-chiral homoallylic alcohols.
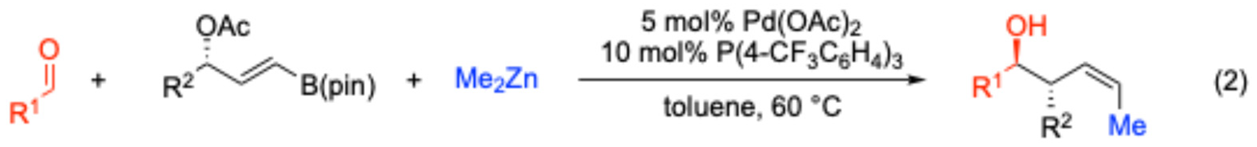
2. Results and Discussion
Initially, we selected the palladium-catalyzed reaction of 1a with benzaldehyde (2a) and dimethylzinc (3) as a model system and commenced optimization of the reaction conditions for the chiral transfer reaction (Table 1). 3 was prepared from commercially available MeLi (1 M in DME sol.) and ZnCl2 and used after filtration of the insoluble LiCl. The first examine using PPh3 gave 4aa in 44% yield with high diastereoselectivity and 88% es (entry 1). When the chiral transfer reaction was conducted using an electron-withdrawing ligand, P(4-CF₃C₆H₄)₃, or an electron-donating ligand, P(4-MeOC₆H₄)₃, the desired product was obtained with good chirality transfer of 88% es and 84% es, respectively, indicating that the electronic properties of the ligands had only a minor influence on the reaction outcome. Interestingly, regardless of the phosphine ligand employed, both the product yield and enantiospecificity were improved when the distilled 3 was used (entries 4–6).
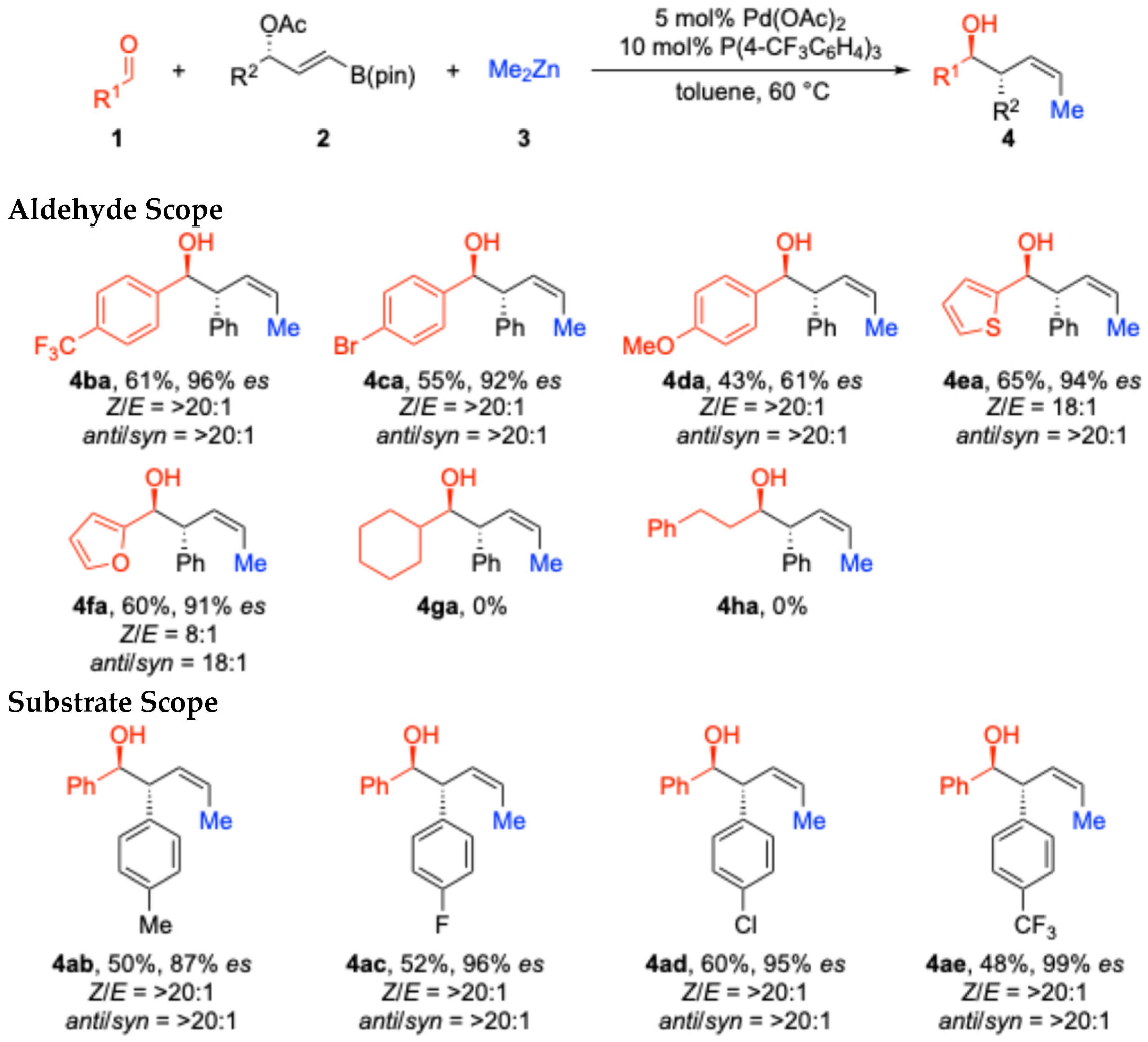
Under the optimized conditions, we next explored the applicability of the optimized conditions in the three-component reaction of 2a and 3 with a range of aldehyde electrophiles (Scheme 1). The reaction proved to be sensitive to the electronic nature of substituents on benzaldehyde derivatives. For example, 4-(trifluoromethyl)benzaldehyde (1b) furnished the desired product 4ba in 61% yield with 96% es, while 4-bromobenzaldehyde (1c) afforded 4ca in 55% yield with 92% es. In contrast, a marked decrease in enantiospecificity was observed for electron rich 4-methoxybenzaldehyde (1d), providing the (Z)-enynyl homoallylic alcohol 4da in 43% yield with 61% es. Heteroaryl aldehydes exhibited reactivity comparable to that of aromatic aldehydes. For example, the reaction of 2-thiophenecarboxaldehyde (1e) delivered 4ea in 69% yield with 94% es. Similarly, 2-furaldehyde (1f) afforded 4fa in 69% yield, albeit with slightly diminished enantiospecificity relative to 4ea. Although aromatic aldehydes were generally compatible with the reaction conditions, aliphatic aldehydes failed to afford the corresponding products 4ga and 4ha.
Next, we examined the scope of borylated allyl acetates to evaluate the influence of the electronic nature of the aryl groups at the allylic position. When substrate 2b bearing a p-tolyl group was employed, the desired product 4ba was obtained with moderate chirality transfer (87% es). In contrast, substrates 2c–e bearing electron-withdrawing substituents, such as fluoro, chloro, and trifluoromethyl groups, were well suited to the present transformation, affording the corresponding products 4ac–ae with high levels of chirality transfer. Overall, electron-withdrawing substituents on the aryl group at the allylic position favored efficient chirality transfer, whereas an electron-donating substituent led to diminished stereochemical control
Considering the results of our previous study[19], we propose a tentative reaction mechanism as depicted in Scheme 2. π-Allylpalladium intermediate A is generated via oxidative addition of the borylated allyl acetate 2 to a Pd0 species. Subsequent transmetalation of A with 3 affords the π-allylpalladium intermediate B, in which the palladium atom in B coordinates to aldehyde to form a six-membered cyclic transition state C. Steric repulsion between B(pin) substituent and PdMeLn enforces the B(pin) group into a pseudoaxial position. Subsequent umpolung allylpalladation of the aldehyde generates the cis-vinylboronate intermediate D. The alkoxopalladium and cis-vinylboronate moieties in D can undergo intramolecular transmetalation to afford cis-vinylpalladium intermediate F via E. Reductive elimination of Pd species from F furnishes G. Meanwhile, anti-addition of another Pd0 species to A generates ent-A, and an equilibrium between A and ent-A forms accounts for the partial erosion of chirality transfer.[21] The decrease in enantiospecificity observed in the presence of lithium chloride (Table 1, entries 1–3) is likely attributable to the formation of a complex between lithium chloride and 3, which inhibits coordination to the acetoxy oxygen atom of A, thereby retarding the transmetalation step and reducing the efficiency of chiral transfer.
3. Materials and Methods
Unless otherwise noted, the reactions were carried out in flame-dried glassware under argon atmosphere. NMR spectra were recorded on Bruker AVANCE NEO 400 spectrometer. Chemical shifts (δ) are reported in ppm from the solvent resonance or tetramethylsilane (TMS) as the internal standard (CDCl3: 7.26 ppm, TMS: 0.00 ppm). Peak multiplicities are designated by the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad and coupling constants (J) are provided in Hz. 13C NMR spectra were recorded on Bruker AVANCE NEO 400 (100 MHz) spectrometer with complete proton decoupling. Chemical shifts are reported in ppm from the solvent resonance as the internal standard (CDCl3: 77.16 ppm) . Some reported spectra in CDCl3 include minor solvent impurities of water (1H NMR δ 1.56 ppm) and/or silicone grease (1H NMR δ 0.07 ppm, 13C NMR δ 1.19 ppm), which do not impact product assignments. Flash chromatography was performed with Fuji Silysia PSQ100B (100 μm). Analytical thin layer chromatography (TLC) was performed on Merck precoated TLC plates (silica gel 60 GF254, 0.25 mm). High-resolution mass (HRMS) spectral data were obtained on an Agilent 6546 LC/Q-TOF mass spectrometer.
3.1. General Method for Arylation Process
A 20 mL two-necked round-bottom flask was charged with Pd(OAc)₂ (3.37 mg, 0.015 mmol), P(4-CF3C6H4)₃ (14.0 mg, 0.03 mmol), and toluene (0.6 mL). The mixture was stirred at room temperature for 0.5 h, after which a solution of 2 (0.30 mmol) and aldehyde (1, 0.72 mmol) in toluene (0.6 mL) was added via cannula. After addition of dimethylzinc 3 (1 M in DME/Et2O = 1.8:1, 0.72 mmol), the reaction mixture was stirred at 60 °C for 4 h. After completion, the reaction was diluted with EtOAc (20 mL) and washed successively with saturated aqueous NH₄Cl (2 × 20 mL), saturated aqueous NaHCO₃ (2 × 20 mL), and brine (2 × 20 mL). The combined organic layers were dried over MgSO₄, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to afford 4.
Characterization Data
anti-(Z)-1,2-Diphenylpent-3-en-1-ol (4aa) was obtained as a yellow oil (99% ee of 2a was used. 42.9 mg, 60%, 93% ee, 93% es). 1H NMR (CDCl3, 400 MHz) δ 7.25-7.15 (m, 8H), 7.10-7.06 (m, 2H), 5.85 (m, 1H), 5.66 (dqd, J = 1.2, 6.8, 11.2 Hz, 1H), 4.82 (d, J = 7.6 Hz, 1H), 3.90 (dd, J = 7.6, 9.6 Hz, 1H), 2.27 (d, J = 2.4 Hz, 1H), 1.61 (dd, J = 1.6, 6.8 Hz, 3H). Spectroscopic data was consistent with the values reported in the literature.[10]
anti-(Z)-1-(4-Trifluoromethylphenyl)-2-phenylpent-3-en-1-ol (4ba) was obtained as a yellow oil (99% ee of 2a was used. 56.0 mg, 61% yield, 96% ee, 96% es, Rf = 0.53, AcOEt/Hex = 3/7). 1H NMR (CDCl3, 400 MHz) δ 7.46 (d, J = 8.0 Hz, 2H), 7.28–7.15 (m, 5H), 7.10-7.06 (m, 2H), 5.92–5.85 (m, 1H), 5.84–5.75 (m, 1H), 4.88 (dd, J = 2.0, 7.2 Hz, 1H), 3.86 (dd, J = 7.6, 9.6 Hz, 1H), 2.34 (d, J = 2.4 Hz, 1H), 1.60 (dd, J = 1.6, 6.8 Hz, 3H). Spectroscopic data was consistent with the values reported in the literature.[22]
anti-(Z)-1-(4-Bromophenyl)-2-phenylpent-3-en-1-ol (4ca) was obtained as a yellow oil (96.6% ee of 2a was used. 52.3 mg, 55% yield, 89% ee, 92% es, Rf = 0.48, AcOEt/Hex = 3/7). 1H NMR (CDCl3, 400 MHz) δ 7.35-7.30 (dm, J = 9.2 Hz, 2H), 7.25-7.10 (m, 3H), 7.08-7.00 (m, 4H), 5.87 (ddq, J = 1.6, 9.2, 10.8 Hz, 1H), 5.83-5.74 (m, 1H), 4.78 (dd, J = 2.0, 7.2 Hz, 1H), 3,83 (dd, J = 7.6, 9.6 Hz, 1H), 2.29 (d, J = 2.4 Hz, 1H), 1.62 (dd, J = 1.6, 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 141.0, 140.8, 131.0, 129.0, 128.5, 128.3, 128.34, 128.28, 126.7, 121.1, 77.5, 52.2, 13.3; HRMS-EI: [M-OH]+ calcd for C17H17BrO: 299.0430, found: 299.0400.
anti-(Z)-1-(4-Methoxyphenyl)-2-phenylpent-3-en-1-ol (4da) was obtained as a yellow oil (99% ee of 2a was used. 34.6 mg, 43% yield, 61% ee, 61% es, Rf = 0.53, AcOEt/Hex = 3/7). 1H NMR (CDCl3, 400 MHz) δ 7.23-7.17 (m, 2H), 7.16-7.11 (m, 1H), 7.10-7.05 (m, 4H), 6.77-6.72(m, 2H), 5.93-5.85 (m, 1H), 5.82-5.72 (m, 1H), 4.78 (dd, J = 2.0, 7.6 Hz, 1H), 3,87 (dd, J = 7.6, 10.0 Hz, 1H), 3.76 (s, 3H), 2.21 (d, J = 2.4 Hz, 1H), 1.62 (dd, J = 1.6, 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 158.8, 141.4, 134.2, 129.7, 128.3, 128.0, 127.8, 126.4, 113.3, 77.7, 55.2, 52.2, 13.3; HRMS (ESI-TOF) [M-OH]+ calcd for C18H20O2: 251.1430, found: 251.1428.
anti-(Z)-2-Phenyl-1-(thiophen-2-yl)pent-3-en-1-ol (4ea) was obtained as a yellow oil (96.6% ee of 2a was used. 47.6 mg, 65% yield, 90.3% ee, 94% es, Rf = 0.51, AcOEt/Hex = 3/7). 1H NMR (CDCl3, 400 MHz) δ 7.27-7.21 (m, 2H), 7.20-7.12 (m, 4H), 6.82 (dd, J = 3.2, 4.8 Hz, 1H), 6.65 (ddd, J = 0.8, 1.2, 3.2 Hz, 1H), 5.90 (ddq, J = 1.6, 9.2, 11.2 Hz, 1H), 5.80 (m, 1H), 5.11 (dd, J = 2.4, 7.6 Hz, 1H), 3.95 (dd, J = 7.6, 9.6 Hz, 1H), 2.41 (d, J = 2.4 Hz, 1H), 1.68 (dd, J = 1.6, 6.4 Hz, 3H). Spectroscopic data was consistent with the values reported in the literature.[10]
anti-(Z)-1-(Furan-2-yl)-2-phenylhex-3-en-1-ol (4fa) was obtained as a yellow oil (96.6% ee of 2a was used. 41.1 mg, 60% yield, 87.3% ee, 91% es, Rf = 0.40, AcOEt/Hex = 3/7). 1H NMR (CDCl3, 400 MHz) δ 7.33 (dd, J = 0.8, 1.6 Hz, 1H), 7.27-7.20 (m, 2H), 7.20-7.14 (m, 3H), 6.22 (dd, J = 1.6, 3.2 Hz, 1H), 6.06 (d, J = 3.2 Hz, 1H), 5.86 (ddq, J = 1.6, 9.2, 11.2 Hz, 1H), 5.77 (m, 1H), 4.86 (d, J = 7.6 Hz, 1H), 4.17 (dd, J = 7.6, 9.6 Hz, 1H), 2.24 (br s, 1H), 1.68 (dd, J = 1.6, 6.8 Hz, 3H). Spectroscopic data was consistent with the values reported in the literature.[10]
anti-(Z)-2-(4-Methylphenyl)-1-phenylpent-3-en-1-ol (4ab) was obtained as a yellow oil (75% ee of 2b was used. 37.9 mg, 50% yield, 65.3% ee, 87% es, Rf = 0.57, AcOEt/Hex = 3/7). 1H NMR (CDCl3, 400 MHz) δ7.25-7.16 (m, 5H), 7.05-6.96 (m, 4H), 5.91-5.84 (m, 1H), 5.78-5.71 (m, 1H), 4.82 (dd, J = 2.0, 7.2 Hz, 1H), 3.88 (dd, J = 7.2, 10.0 Hz, 1H), 2.28 (s, 3H), 2.23 (d, J = 2.4 Hz, 1H), 1.60 (dd, J = 2.0, 6.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 142.2, 138.4, 136.1, 129.6, 129.2, 128.2, 127.99, 127.96, 127.4, 126.8, 78.2, 51.7, 21.1, 13.3; HRMS-EI: [M-OH]+ calcd for C18H20O: 235.1481, found: 235.1487.
anti-(Z)-1-Phenyl-2-[4-(fluoromethyl)phenyl]pent-3-en-1-ol (4ac) was obtained as a light yellow oil (91% ee of 2c was used. 40.0 mg, 52% yield, 91% ee, 96% es, Rf = 0.60, AcOEt/Hex = 3/7). 1H NMR (CDCl3, 400 MHz) δ 7.25-7.18 (m, 3H), 7.16-7.12 (m, 2H), 7.05-6.98 (m, 2H), 6.92-6.86 (m, 2H), 5.87 (m, 1H), 5.77 (m, 1H), 4.77 (dd, J = 2.0, 7.6 Hz, 1H), 3.88 (dd, J = 7.6, 9.6 Hz, 1H), 2.24 (d, J = 2.0 Hz, 1H), 1.61 (dd, J = 1.6, 6.8 Hz, 3H). Spectroscopic data was consistent with the values reported in the literature. [10]
anti-(Z)-1-Phenyl-2-[4-(chloromethyl)phenyl]pent-3-en-1-ol (4ad) was obtained as a brown oil (93% ee of 2d was used. 49.0 mg, 60% yield, 88% ee, 95% es, Rf = 0.55, AcOEt/Hex = 3/7). 1H NMR (CDCl3, 400 MHz) δ 7.25-7.19 (m, 3H), 7.18-7.13 (m, 4H), 7.02-6.98 (m, 2H), 5.86 (m, 1H), 5.77 (m, 1H), 4.78 (dd, J = 2.0, 7.6 Hz, 1H), 3.87 (dd, J = 7.6, 9.6 Hz, 1H), 2.27 (d, J = 2.4 Hz, 1H), 1.60 (dd, J = 1.6, 6.4 Hz, 3H). Spectroscopic data was consistent with the values reported in the literature. [10]
anti-(Z)-1-Phenyl-2-[4-(trifluoromethyl)phenyl]pent-3-en-1-ol (4ae) was obtained as a brown oil (94% ee of 2e was used. 44.1 mg, 48% yield, 92.7% ee, 99% es, Rf = 0.50, AcOEt/Hex = 3/7). 1H NMR (CDCl3, 400 MHz) δ 7.46 (d, J = 8.0 Hz, 2H),7.23-7.14 (m, 7H), 5.90 (m, 1H), 5.79 (m, 1H), 4.84 (dd, J = 2.0, 7.2 Hz, 1H), 3.96 (dd, J = 7.6, 9.6 Hz, 1H), 2.23 (d, J = 2.4 Hz, 1H), 1.59 (dd, J = 1.6, 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 145.7, 141.7, 128.9, 128.8 (q, J C-F = 31.2 Hz), 128.8, 128.7, 128.2, 127.8, 126.7, 125.3 (q, J C-F = 3.7 Hz), 124.3 (q, J C-F = 270.2 Hz), 78.1, 52.0, 13.4; HRMS (ESI-TOF) m/z: [M-OH]+ Calcd for C18H16F3O+ : 311.1341, found: 311.1432.
4. Conclusions
We have developed a palladium-catalyzed chiral transfer three-component reaction that enables the efficient and highly stereocontrolled synthesis of 1,2,4-trisubstituted chiral anti-(Z)-homoallylic alcohols. By employing readily accessible chiral homoallylic alcohol derivatives as starting materials, this strategy circumvents the challenges associated with regioselectivity, diastereoselectivity, E/Z geometry, and enantioselectivity that are inherent to conventional allylation reactions of unsymmetrical allyl alcohol derivatives.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. 1H and 13C NMR spectra for 4.
Author Contributions
Conceptualization, Y.H.; investigation, A.N., M.I., Y.H., and M.A.; writing—original draft preparation, Y.H..; writing—review and editing, Y.H. All authors have read and agreed to the published version of the manuscript.
Funding
This study was financially supported by JSPS KAKENHI Grant Numbers JP15K05496.
Data Availability Statement
The data presented in this study are available in the Supplementary Material.
Acknowledgments
This study was conducted at the Chitose Institute of Science and Technology, supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, Grant Number JPMXP1225CT0139.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yeung, K.; Paterson, I. Advances in the Total Synthesis of Biologically Important Marine Macrolides; 2005; pp. 4237–4313. [Google Scholar] [CrossRef]
- Florence, G.J.; Gardner, N.M.; Paterson, I. Development of Practical Syntheses of the Marine Anticancer Agents Discodermolide and Dictyostatin. Nat. Prod. Rep. 2008, 25, 342–375. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Verboom, K.L.; Krische, M.J. Catalytic Enantioselective C–C Coupling of Alcohols for Polyketide Total Synthesis beyond Chiral Auxiliaries and Premetalated Reagents. Chem. Rev. 2024, 124, 13715–13735. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Fu, J. Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev. 2003, 103, 2763–2794. [Google Scholar] [CrossRef] [PubMed]
- Yus, M.; Gonz, C.; Foubelo, F. Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines; 2011; pp. 7774–7854. [Google Scholar] [CrossRef]
- Yus, M.; González-Gómez, J.C.; Foubelo, F. Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev. 2011, 111, 7774–7854. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, D.; Grayson, M.; Fustero, S.; Barrio, P. Recent Developments and Applications of the Chiral Brønsted Acid Catalyzed Allylboration of Carbonyl Compounds. Synthesis 2018, 50, 1935–1957. [Google Scholar] [CrossRef]
- Spielmann, K.; Niel, G.; de Figueiredo, R.M.; Campagne, J.-M. Catalytic Nucleophilic 'Umpoled' π-Allyl Reagents. Chem. Soc. Rev. 2018, 47, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.; Schwartz, L.A.; Krische, M.J. Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev. 2018, 118, 6026–6052. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Chong, Q.; Meng, F. Cu-Catalyzed Enantioselective Reductive Coupling of 1,3-Dienes and Carboxylic Acid Anhydrides. Asian J. Org. Chem. 2023, 12, e202300436. [Google Scholar] [CrossRef]
- Mitsunuma, H.; Kanai, M. Catalytic Asymmetric Allylation of Aldehydes with Alkenes through Allylic C(sp3)–H Functionalization Mediated by Organophotoredox and Chiral Chromium Hybrid Catalysis. Chem. Sci. 2019, 10, 3459–3465. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Breit, B. Organophotoredox/Ni-Cocatalyzed Allylation of Allenes: Regio- and Diastereoselective Access to Homoallylic Alcohols. ACS Catal. 2022, 12, 3249–3255. [Google Scholar] [CrossRef]
- Hesse, M.J.; Essafi, S.; Watson, C.G.; Harvey, J.N.; Hirst, D.; Willis, C.L.; Aggarwal, V.K. Highly Selective Allylborations of Aldehydes Using α,α-Disubstituted Allylic Pinacol Boronic Esters. Angew. Chem. Int. Ed. 2014, 53, 6145–6149. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.L.; Adili, A.; Shen, H.C.; Han, Z.Y.; Gong, L.Z. Catalytic Enantioselective Assembly of Homoallylic Alcohols from Dienes, Aryldiazonium Salts, and Aldehydes. Angew. Chem. Int. Ed. 2016, 55, 4322–4326. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.C.; Wang, P.S.; Tao, Z.L.; Han, Z.Y.; Gong, L.Z. An Enantioselective Multicomponent Carbonyl Allylation of Aldehydes with Dienes and Alkynyl Bromides Enabled by Chiral Palladium Phosphate. Adv. Synth. Catal. 2017, 359, 2383–2389. [Google Scholar] [CrossRef]
- Lv, Y.-F.; Shi, Z.; Liu, G.; Shen, H.; Wang, Z. Stereoselective Synthesis of Chiral Homoallylic Alcohols with Functionalized Alkenes via Radical-Involved Triple Catalysis. ACS Catal. 2025, 15, 20157–20169. [Google Scholar] [CrossRef]
- Horino, Y.; Aimono, A.; Abe, H. Pd-Catalyzed Three-Component Reaction of 3-(Pinacolatoboryl)Ally Acetates, Aldehydes, and Organoboranes: A New Entry to Stereoselective Synthesis of (Z)-anti-Homoallylic Alcohols. Org. Lett. 2015, 17, 2824–2827. [Google Scholar] [CrossRef] [PubMed]
- Schmidtmann, E.S.; Oestreich, M. Enantiospecific Synthesis and Allylation of All-Carbon-Substituted α-Chiral Allylic Stannanes. Angew. Chem. Int. Ed. 2009, 48, 4634–4638. [Google Scholar] [CrossRef] [PubMed]
- Katsuki, T.; Sharpless, K.B. The First Practical Method for Asymmetric Epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- Horino, Y.; Ishibashi, M.; Sakamoto, J.; Murakami, M.; Korenaga, T. Palladium-Catalyzed Diastereoselective Synthesis of (Z)-Conjugated Enynyl Homoallylic Alcohols. Adv. Synth. Catal. 2021, 363, 3592–3599. [Google Scholar] [CrossRef]
- Oono, M.; Yamada, A.; Kimura, M.; Kanomata, K.; Akai, S. Lipase-Palladium Co-Catalyzed Dynamic Kinetic Resolution of Racemic Allylic Esters. Chem. Eur. J. 2025, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cooze, C.; Dada, R.; Lundgren, R.J. Direct Formic Acid Mediated Z-Selective Reductive Coupling of Dienes and Aldehydes. Angew. Chem. Int. Ed. 2019, 58, 12246–12251. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Scope of palladium-catalyzed three-component reaction.
Scheme 2.
Plausible reaction pathway.
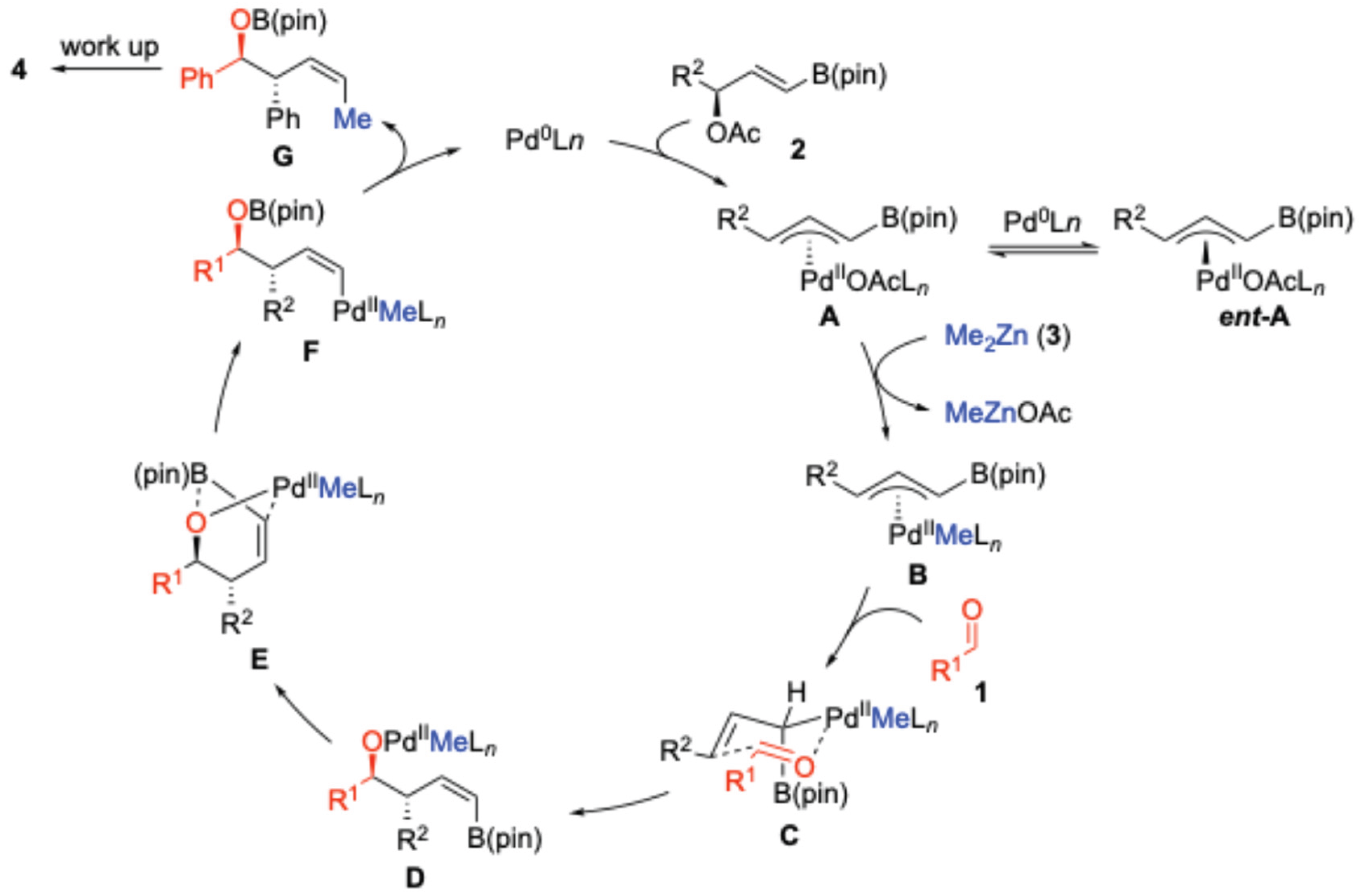

Table 1.
Reaction Optimization.1.
 | |||||
| Entry | Ligand | Time (h) | 4aa (%) | ee (%)2 | es (%)3 |
| 1 | PPh3 | 4 | 44 | 88 | 88 |
| 2 | P(4-MeOC6H4)3 | 6 | 43 | 84 | 84 |
| 3 | P(4-CF3C6H4)3 | overnight | 44 | 88 | 88 |
| 44 | PPh3 | 4 | 56 | 93 | 93 |
| 54 | P(4-MeOC6H4)3 | 4 | 57 | 93 | 93 |
| 64 | P(4-CF3C6H4)3 | overnight | 60 | 93 | 93 |
1 Conditions: 1a (0.72 mmol), 2a (0.3 mmol), 3 (1 M in DME/Et2O, 0.72 mmol), Pd(OAc)2 (0.015 mmol), and ligand (0.03 mmol). 2 Enantiomeric excess (ee) was determined by HPLC. 3 es (enantiospecificity) = (ee % of product)/(ee % of starting material). 4 Distilled Me2Zn was used.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



