Submitted:
23 February 2026
Posted:
26 February 2026
You are already at the latest version
Abstract
Rheumatoid arthritis (RA) is increasingly recognized as a mucosa-initiated autoimmune disease rather than a disorder that begins within the joint itself. This review synthesizes epidemiological, mechanistic, and experimental evidence supporting a model in which mucosal dysbiosis, altered microbial metabolites, barrier dysfunction, and antigenic mimicry converge to prime systemic autoimmunity. Across gut, oral, and pulmonary sites, RA and at-risk individuals exhibit reproducible microbial shifts, reduced short-chain fatty acid production, increased succinate and indole signaling, and elevated permeability. These changes skew dendritic cell activation, amplify IL-1β/IL-6/IL-23–dependent Th17 responses, impair Treg stability, and lower systemic immune tolerance thresholds. In parallel, microbial citrullinated epitopes and cross-reactive peptides—particularly from Prevotella and Streptococcus species—provide antigenic substrates that may drive ACPA maturation and epitope spreading. While direct causality remains unresolved, we propose that RA initiation reflects cumulative immune conditioning at mucosal interfaces, with joint inflammation emerging as a secondary, tissue-focused consequence of a systemically primed inflammatory state.
Keywords:
rheumatoid arthritis
; mucosal immunity
; dysbiosis
; Th17/Treg imbalance
; molecular mimicry
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by persistent inflammatory arthritis and systemic inflammation (Guo et al., 2018; Alivernini, Firestein and McInnes, 2022). It is a heterogeneous disorder with multiple and highly individualized etiologies (Alivernini, Firestein and McInnes, 2022). Epidemiological studies estimate that approximately 0.5–1% of the adult population worldwide is affected by RA (Venetsanopoulou et al., 2023; Guo et al., 2018). According to the 2010 American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR) classification criteria for rheumatoid arthritis, RA is characterized by several features, including small joint involvement, the presence of autoantibodies (i.e., rheumatoid factor (RF) and anti-citrullinated peptide antibodies (ACPA)), elevated acute-phase reactants such as C-reactive protein (CRP) or erythrocyte sedimentation rate (ESR), and symptom duration (Taylor, 2020). Please note, RA is featured by chronic arthritis condition, not all of the above criteria are required for the diagnosis of RA, based on broad definition (Javed and Crowson, 2025). Hinted by serological test, RA can be categorized into seropositive RA and seronegative RA according to the presence or absence of RF and/or ACPA (Javed and Crowson, 2025). Significant progress has been made in understanding RA pathogenesis over the past 30 years; however, therapeutic outcomes remain suboptimal, partly due to interindividual differences in microbiome composition and lifestyle factors (Li and Kuhn, 2025). Temporally, the preclinical RA phase is characterized by the emergence of autoantibodies several years before the onset of clinical symptoms (Figure 1), during which mucosal sites may play an important role (Li and Kuhn, 2025). The precise mechanisms by which established RA symptoms are triggered remain poorly understood (Alivernini, Firestein and McInnes, 2022). This uncertainty is partly due to the lack of definitive evidence demonstrating a causal relationship between RA onset and the presence of ACPA or RF, as some individuals are ACPA/RF-positive without clinical RA, whereas others develop RA despite being seronegative (Smolen et al., 2018). Numerous studies have shown that dysbiosis of the gut, lung, or oral microbiome correlates with RA development, implicating mucosal immune responses in initiating systemic autoimmunity (Lu et al., 2025) (Holers et al., 2024). Therefore, this review integrates emerging evidence to examine how mucosal immune dysregulation contributes to the initiation of rheumatoid arthritis from a mechanistic perspective.
Main
- Mucosa Dysbiosis Contributes to RA Pathogenesis
Mucosal immunity plays a significant role in the constitution of the whole-body immune system. Mucosal immune responses are crucial for frontline defense against potential infection and for maintaining systemic immune homeostasis (Zhou et al., 2025). Based on a series of clinical studies, mucosal dysbiosis and dysfunction are associated with a range of remote and systemic diseases, including RA, inflammatory bowel disease (IBD), cardiovascular disease, and multiple sclerosis (MS) (Alivernini, Firestein and McInnes, 2022; Santana et al., 2022; Mohsenzadeh et al., 2025; Ordoñez-Rodriguez et al., 2023). From a holistic perspective, mucosa-associated lymphoid tissue (MALT) is the largest immune organ and acts as a first-line educator and activator of systemic immunity. It influences the systemic immune system through multiple mechanisms. Classically, mucosal tissues are routinely exposed to the external environment and pathogens (Velikova et al., 2024). They detect and initially respond to pathogens at mucosal surfaces, including respiratory, urogenital, and gastrointestinal mucosa, ideally before pathogens enter the bloodstream (Velikova et al., 2024). MALT generates dimeric secretory IgA (sIgA), which controls microbial translocation and limits the accumulation of microbes that induce infection and inflammation (Velikova et al., 2024). Studies also show that gut-associated lymphoid tissue (GALT) induces circulating peripheral Treg cells (Atarashi et al., 2010; Velikova et al., 2024). In murine studies, oral induction of gut commensal bacteria ameliorates colitis and systemic immunoglobulin E responses (Atarashi et al., 2010). MALT also contributes to barrier integrity as part of the immune response, preventing excessive systemic activation by microbes and microbial products, such as lipopolysaccharide (LPS) and peptidoglycan (Velikova et al., 2024). Altogether, mucosal immunity not only contributes to local immune defense and homeostasis but also plays a central role in systemic immune regulation.
A growing body of epidemiological and immunological evidence supports the concept that RA may originate at mucosal surfaces rather than in the joint itself. Several decades of research have identified mucosal abnormalities in individuals at risk for RA, along with longitudinal observations correlating lifestyle factors or biomarker changes prior to RA onset. Certain environmental exposures are known to increase RA risk by affecting mucosal surfaces. Cigarette smoking is predominantly associated with triple-positive RA (RF, ACPA, and anti-carbamylated protein antibodies) (Regueiro et al., 2020; Ishikawa and Terao, 2020). Affected patients show bronchiolar mucosal inflammation, skewed T helper (Th) cell subsets—including Th1, Th2, and Th17, varying among individuals—and increased levels of pro-inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1α, IL-1β, IL-5, IL-6, IL-8, IL-13, IL-15, IL-21, and interferon (IFN)-γ (Ishikawa and Terao, 2020). Smoking, as a representative example, illustrates how intrinsic and extrinsic factors interact in RA pathogenesis. The strongest association between cigarette smoking and RA is observed in individuals carrying HLA-DRB1*01:01, 01:02, or HLA-DRB110:01 shared-epitope (SE) alleles. In addition to smoking, periodontal disease, air pollution, and specific dietary factors have also been associated with RA onset (Bingham and Moni, 2013; Adami et al., 2021; Tedeschi et al., 2017; Maeda et al., 2016). Collectively, these observations suggest that mucosal tissues represent a critical immunological environment where RA-driving autoimmunity is initially primed.
Multiple high-throughput sequencing studies across mucosal sites demonstrate reproducible dysbiosis in RA and in at-risk individuals (RF⁺ or anti-CCP⁺). Across 29 studies, the gut microbiota of RA patients often shows enrichment of Prevotella, Megasphaera, Lactobacillus, and Escherichia, alongside depletion of SCFA-producing genera such as Roseburia and Bacteroides (Figure 2). Several gut microbial taxa have been reported to correlate with immunological parameters: Ruminococcus correlates with RF-IgA and anti-CCP titers, while Prevotella and Alloprevotella correlate with CRP levels and susceptibility to mucosa inflammation and RA onset (Iljazovic et al., 2020; Sun et al., 2019; Wu et al., 2024; Scher et al., 2013). In the oral cavity, expansion of Prevotella and Porphyromonadaceae—most notably P. gingivalis—is consistently observed. There is also expanded study identifying P. gingivalis as a potential trigger to RA onset (lucchino et al., 2019) Systemic microbial signatures are also detectable. In serum, taxa such as Achromobacter and Serratia, previously characterized as lymphoid-tissue-resident commensals, are reduced in RA cohorts (Hammad et al., 2020).
In addition, seronegative RA (defined as RA negative for serum RF and ACPA) is highly heterogeneous, less studied, and may involve distinct pathogenic mechanisms compared to seropositive RA (Paroli and Sirinian, 2023; Pratt and Isaacs, 2014). Microbiota differentiation has been observed between seropositive and seronegative RA subsets, suggesting that distinct mucosal–immune pathways may underlie these clinical phenotypes (Wu et al., 2025). Together, these findings highlight that RA and at-risk individuals display consistent mucosal dysbiosis with distinct microbial and immune signatures across disease subsets, providing direction for mechanistic studies.
Several interventional studies further reveal possible mechanisms linking mucosal immunity to RA pathogenesis. Häupl et al. (2023) demonstrated that short-term fasting rapidly reduced RA disease activity: clinical scores improved in 19 of 20 participants after 7–10 days of fasting, accompanied by declines in IL-6 and zonulin, critical markers of systemic inflammation and gut mucosal barrier dysfunction, respectively. Shifts in gut microbiota (reduced Actinobacteria and Firmicutes, increased Bacteroidetes and Akkermansia muciniphila) were also observed (Häupl et al., 2023). Concordantly, Th17 activation, reductions in Treg/Tfr populations, and increased intestinal permeability have been reported in early RA (Häupl et al., 2023; Wu et al., 2024; Luo et al., 2023). In vivo experiments show that collagen-induced arthritis (CIA) mice orally infected with P. gingivalis or Treponema denticola exhibit exacerbated arthritis scores (Kim et al., 2022). Furthermore, Parabacteroides distasonis, a taxon downregulated in RA patients, suppresses Th17 responses and promotes M2 macrophage polarization in murine experiments (Sun et al., 2023).
While dysbiosis is observed in patients, enrichment of specific bacteria alone cannot establish causal immunopathology. Many RA-associated taxa are also present in healthy individuals and may vary due to diet, lifestyle, and other factors, making the causal relationship between specific microbes and RA pathogenesis ambiguous and heterogeneous. Most in vivo experiments, including oral administration of specific bacteria or fecal microbiota transplantation (FMT) in CIA models, demonstrate promotion of arthritis only in the presence of co-requisites such as pre-established joint inflammation or immune activation within the articular cavity. In contrast, such pre-established joint immune responses are not present in at-risk human individuals. This suggests that isolated introduction of susceptible taxa may not directly initiate RA pathogenesis. A second or third factor or pathway may be required for RA onset. Although recent research has explored several possible mechanisms, further studies are needed to clarify how microbes contribute to RA pathogenesis, particularly the mechanisms linking systemic inflammation to chronic joint inflammation. Based on recent efforts, several immunopathogenic pathways that may contribute to RA onset and initiation are summarized below.
- Integrated Mucosal Mechanisms in RA Initiation
A growing body of research in mucosal immunology and pathology suggests that the development of RA at mucosal sites is typically driven by multiple converging factors, rather than any single determinant such as bacterial infection or environmental exposure (Drago, 2019; Block et al., 2016; Wu et al., 2010). In recent years, work on microbe–host interactions has provided increasing evidence that many murine arthritis models, including the K/BxN model, show reduced clinical severity under germ-free conditions (Wu et al., 2010; Block et al., 2016). On the other hand, in many inducible arthritis models such as CIA, additional promoting factors—primarily mediated through disturbances in mucosal homeostasis—have been shown to be key drivers of the heightened inflammatory burden characteristic of arthritis.
However, most studies introduce specific microbial or environmental interventions into induced arthritis models and then assess changes in clinical and immunological readouts, thereby indicating the role of microbiota in promoting a pro-inflammatory shift associated with RA onset. In parallel, recent research inspired by metagenomic studies of clinical RA cohorts has revealed potential antigenic inputs for disease initiation derived from gut and oral microbiota (Brewer et al., 2023; Chriswell et al., 2022). Thus, dysbiosis may contribute to RA initiation through both the induction of a pro-inflammatory shift and the provision of antigenic stimuli.
- Microbe Initiated Pro-Inflammatory Skewing Milieu
Mucosal dysbiosis reprograms mucosal immune tone from a tolerance-favoring state to a pro-inflammatory one by (i) changing the repertoire and concentration of microbe-derived ligands that engage pattern-recognition receptors (PRRs), (ii) biasing antigen-presenting cells (APCs) toward a mature, co-stimulatory phenotype that preferentially primes inflammatory CD4⁺ programs, and (iii) reducing regulatory inputs that sustain FoxP3⁺ Tregs and tolerogenic DCs. These combined alterations lower the activation threshold of both innate and adaptive compartments, favoring the expansion of RORγt⁺ Th17 and IFN-γ/GM-CSF–producing Th1-like cells systemically.
Microbe exposure mediated reprogramming | Facilitated by increased permeability, dysbiotic communities expose epithelial and myeloid cells to an altered PAMP profile (e.g., lipoproteins, atypical LPS, peptidoglycan), increasing signaling through TLR2/TLR4–MyD88 and NOD-like receptors (Figure 3A). This repeatedly activates NF-κB and MAPK transcriptional programs in mucosal APCs and epithelial cells, resulting in the secretion of IL-1β, IL-6, and IL-23—cytokines that are canonical drivers of Th17 differentiation and systemic inflammatory skewing (Spindler et al., 2022; Mansouri, Akthar and Miyamoto, 2025). PRR-activated dendritic cells upregulate MHC-II and CD80/CD86, acquire migratory capacity, and traffic to draining lymph nodes, where they present antigen in a strongly co-stimulatory, polarizing context that preferentially induces Th17 or Th1 differentiation, thereby amplifying systemic inflammatory cytokine production (Spindler et al., 2022; Mansouri, Akthar and Miyamoto, 2025).
The pro-inflammatory contribution of dysbiotic taxa such as Megasphaera, Ruminococcus, and Bacteroides has been observed in both human cohorts and experimental models, where these taxa are associated with heightened innate activation and Th17 bias (Menofy et al., 2022). For P. copri, its expansion in new-onset RA correlates with higher systemic innate cytokine levels and a Th17-biased response, and P. copri–stimulated DCs prime naïve T cells to produce markedly more IL-17 than commensal controls (Maeda and Takeda, 2019; Huang et al., 2020). In addition, according to Häupl et al. (2023), short-term fasting in RA patients reduced disease scores and decreased serum IL-6 levels, which could be attributed to reduced microbiota burden. These innate and early adaptive effects are thought to lower the threshold for systemic autoimmunity, providing a pro-inflammatory background that may contribute to RA initiation in susceptible hosts.
Microbial metabolites mediated reprogramming | Microbial metabolites can also serve as important signals and modulators of immune homeostasis. Dysbiosis may contribute to pathogenic metabolite accumulation and loss of anti-inflammatory signals, thereby promoting local and systemic pro-inflammatory states (Figure 3B). For instance, succinate, a tricarboxylic acid (TCA) cycle intermediate, acts as an immunoregulatory signal that can amplify inflammatory responses through both intracellular metabolic reprogramming and extracellular receptor-mediated signaling (Wei et al., 2023). Bacteria are recognized as important producers of succinate; multiple taxa, including Bacteroides spp., Prevotella spp., and Firmicutes spp., contribute to succinate generation (Fernández-Veledo and Vendrell, 2019; Wei et al., 2023; Reichardt et al. 2014). Succinate binds to the SUCNR1 (GPR91) receptor and promotes IL-1β production in DCs, enhancing DC activation and migration, accelerating T cell priming and Th17 generation—phenotypes observed in CIA and IBD models (Saraiva et al., 2018; Connors, Dawe and Van Limbergen, 2018). In macrophages, succinate signaling appears more complex. A pro-inflammatory secretory pattern and M1 polarization have been observed (Harber et al., 2020; Wei et al., 2023; Huang, et al., 2024), whereas M2 polarization with regulatory and tissue-repair functions has also been reported (Wei et al., 2023). Thus, succinate may exert dual roles in promoting both M1 and M2 polarization, potentially forming a negative feedback loop that supports anti-inflammatory and tissue-repair processes (Harber et al., 2020). Specifically, pro-M2 signaling is mediated by the SUCNR1–PLC–IP3–Ca²⁺ pathway, accompanied by upregulated SUCNR1 expression on the macrophage plasma membrane, which is not observed in pro-M1 polarization signaling (Trauelsen et al., 2017; Trauelsen et al., 2021; Wei et al., 2023).
Tryptophan metabolites derived from the microbiome also play significant roles in mucosal immune modulation and disease progression. Gut microbes expressing tryptophanase (encoded by tnaA) convert dietary tryptophan into indole and its derivatives, such as indole-3-lactic acid (ILA) (Boya et al., 2021; Sakurai, Odamaki and Xiao, 2019). However, the immunoregulatory effects vary among indole and its derivatives. Seymour et al. (2023) reported that indole supplementation under low-tryptophan dietary conditions worsened arthritis development and increased serum IL-6 and TNF levels in vivo. Human colonic lymphocytes exposed to indole exhibited increased IL-17 signaling and plasma cell activation (Seymour et al., 2023). In vitro stimulation of bone marrow DCs with LPS supplemented with indole shifted the transcriptome toward a pro-Th17 profile compared to LPS alone (Kuhn et al., 2025). Conversely, in DSS-induced colitis models, cesarean section–delivered mice showed higher susceptibility and impaired group 3 innate lymphoid cell (ILC3) development, accompanied by reduced Lactobacillus abundance (Xia et al., 2023). Supplementation with L. acidophilus or its metabolite ILA attenuated intestinal inflammation and restored ILC3 frequency and IL-22 levels (Xia et al., 2023). Mechanistically, Xia et al. demonstrated that ILA activates ILC3 via aryl hydrocarbon receptor (AhR) signaling (Xia et al., 2023). Further studies are therefore required to clarify how Lactobacillus species and their metabolites contribute to immune system development and regulation. Dose-dependent effects and species-specific differences may underlie divergent immunological outcomes.
In addition, short-chain fatty acids (SCFAs), produced by microbes such as Roseburia, Bacteroides, and Parabacteroides distasonis, play important roles in regulating mucosal homeostasis and chronic inflammation (Fusco et al., 2023). GPCR-deficient mice exhibit severe DSS-induced colitis, asthma, and experimental arthritis (Sun et al., 2016; Maslowski et al., 2009). SCFAs inhibit BMDC activation by suppressing LPS-induced CD40 expression and IL-6/IL-12p40 secretion. They also facilitate Treg differentiation (Sun et al., 2016; Nastasi et al., 2015). SCFA deficiency–induced Treg suppression via GPR41/GPR43 signaling has been confirmed in colitis models (Smith et al., 2013). SCFAs further inhibit macrophage pro-inflammatory signaling, reducing CD40, PD-L2, and CD86 expression (Sun et al., 2016; Chang et al., 2014). However, SCFAs also contribute to early infection defense by promoting IL-6, IL-8, and IL-1β production (Sun et al., 2016), and they stimulate neutrophil migration through GPR43 activation (Vinolo et al., 2009; Sun et al., 2016). Butyrate and acetate may differentially modulate neutrophil activation (Vinolo et al., 2008; Sun et al., 2016). GPR41⁻/⁻ and GPR43⁻/⁻ mice show reduced acute inflammatory responses and impaired host defense (Kim et al., 2013). In adaptive immunity, GPR43⁻/⁻ mice exhibit decreased antibody responses and increased susceptibility to Citrobacter rodentium infection (Yang et al., 2019).
Altogether, microbial dysbiosis alters metabolite profiles, increasing pro-inflammatory mediators such as succinate and certain indoles while reducing anti-inflammatory SCFAs, thereby disrupting immune homeostasis and promoting Th17 activation, impaired Treg function, and heightened inflammatory susceptibility.
Gastrointestinal epithelium integrity alteration | Mucosal barrier disruption and altered permeability represent another crucial factor contributing to the pro-inflammatory shift in RA. This may facilitate early microbial and metabolite translocation and is observed in multiple analyses of RA pathogenesis (Figure 3C). Under physiological conditions, the intestinal epithelium, tight junction complexes, mucus layer, and secretory IgA form a multilayered barrier that restricts luminal PAMPs and microbial metabolites from accessing the systemic immune compartment (Capaldo, Powell and Kalman, 2017). Clinically, patients with new-onset RA exhibit significantly elevated serum markers of gut permeability, including LPS, LBP, I-FABP, and zonulin, along with enrichment of the aforementioned taxa (Matei et al., 2021; Tajik et al., 2020). Certain dysbiotic taxa produce proteases or expose high PAMP loads that directly contact epithelial cells (Nonaka, Kadowaki and Nakanishi, 2022; Chen et al., 2016). Porphyromonas gingivalis gingipains cleave junctional proteins (JAM1, ZO-1, occludin), producing focal epithelial gaps (Nonaka, Kadowaki and Nakanishi, 2022). Collinsella aerofaciens and other RA-associated taxa reduce tight-junction gene expression (Chen et al., 2016).
Pathologically, luminal bacterial and gliadin exposure induces epithelial zonulin (pre-haptoglobin-2) release via MyD88-dependent CXCR3 signaling (Fasano, 2020). The precise signaling mechanisms by which microbes induce zonulin-mediated tight junction disassembly remain unclear, although the phenomenon is well documented (Serek and Oleksy-Wawrzyniak, 2021). Bacteria and viruses can increase permeability by secreting enterotoxins that directly disrupt tight junctions and the cytoskeleton or indirectly activate epithelial inflammatory signaling pathways (Paradis et al., 2021). Zonulin triggers MLCK-mediated contraction of perijunctional actomyosin and tight junction disassembly (Fasano, 2020). While this mechanism is physiologically transient, it becomes pathological when chronically engaged (Fasano, 2020). Translocated PAMPs activate mucosal DCs and macrophages (TLR/NOD → NF-κB, inflammasome), increasing IL-1β and IL-6 production and sustaining local inflammation (Li and Kuhn, 2025). Inflamed mucosa produces additional zonulin and recruits inflammatory cells, further impairing epithelial repair and creating a self-reinforcing loop absent in healthy individuals (Li and Kuhn, 2025; Matei et al., 2021). Consequently, translocated microbial components and metabolites promote a pro-inflammatory state and provide antigenic input, contributing holistically to RA pathogenesis. Outstanding questions remain: the precise role of permeability in RA pathogenesis is not fully defined; the mechanisms underlying barrier repair failure are unclear; and whether enriched junction-disrupting taxa uniquely drive chronic barrier dysfunction requires further investigation. Clinically, permeability markers strongly support barrier leakage as an early and convergent feature of RA pathogenesis, although temporal causality remains to be established.
Collectively, these findings support an RA initiation model in which mucosal dysbiosis, altered microbial metabolites, and barrier perturbation converge to establish a pro-inflammatory milieu within MALT and systemic immune compartments. This model emphasizes the lowering of activation thresholds across innate and adaptive immunity through enhanced Th17-skewing signals, reduced regulatory pathways, and increased systemic exposure to luminal PAMPs. However, such immunological conditioning does not directly determine antigen specificity; rather, it provides a permissive context in which autoreactive lymphocytes may become activated and expanded.
- Molecular Mimicry: Microbial Antigens Prime Cross-Reactive Adaptive Responses
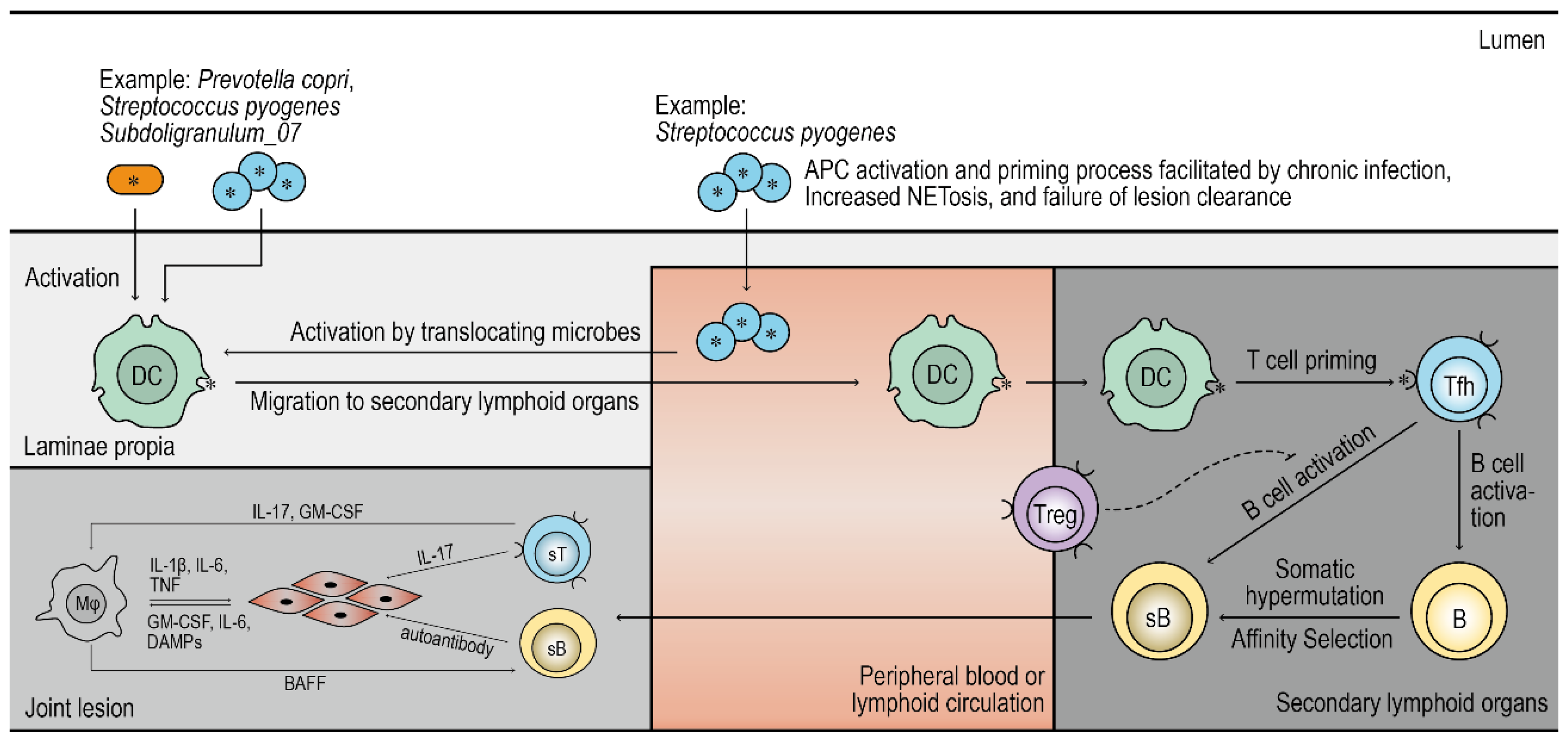
Beyond the pro-inflammatory shift, molecular mimicry phenomenon counts as another important mediator of joint inflammation in RA pathogenesis (Figure 4). In principle, potential sources—primarily infectious triggers—may induce the generation of self-reactive lymphocytes that subsequently target host molecules, particularly articular epitopes, thereby causing chronic inflammation.
Citrullination mediated molecular mimicry | One major pathway involves microbiota-driven citrullination. Citrullination is a physiological post-translational modification catalyzed by Ca2+ depended PAD enzymes and plays essential roles in histone modification and functionally linked to neutrophil NETosis formation, which consist of the normal antimicrobial defense (Pitter and Zou, 2025), (Yang et al., 2021). Citrullination is observed upregulated during infection and inflammation mediating NETosis formation (Ciesielski et al., 2022). In RA pathogenesis, a strong correlation between periodontal mucosa immune response initiated citrullination with RA severity is observed (González-Febles et al., 2020). Plus, PAD2 and PAD4 are highly upregulated in RA synovium, associated with increased NETosis and ACPA recognition in synovium (Foulquier et al., 2007). Pathogenetically, ACPA can amplify macrophage driven immune response by immune complexes targeting citrullinated proteins epitopes in joint tissue (Laurent et al., 2014).
The question of how orally initiated ACPA responses transition to joint-reactive ACPAs has recently been explored. As reported, “oral bacteria observed transiently in blood were broadly citrullinated in the mouth, and their in situ citrullinated epitopes were targeted by extensively somatically hypermutated ACPAs encoded by RA blood plasmablasts” (Brewer et al., 2023). Reactive citrullinated antigens were primarily identified from Streptococcus spp. (Brewer et al., 2023). These findings suggest that somatic hypermutation may expand epitope recognition toward human (neo)antigens. This mechanism could represent a promising therapeutic direction for ACPA- and RF-double-positive cases. However, the mediators and antigen-presenting mechanisms by which citrullinated peptides trigger adaptive immune responses remain to be fully elucidated. Joint (synovial) proteins are known to undergo citrullination in RA patients, as detected by mass spectrometry analysis of synovial fluid (Kinloch et al., 2008). Furthermore, sera from seropositive RA patients demonstrate ACPA binding to joint epitopes by ELISA (Ossipova et al., 2014). Nevertheless, it remains unclear whether synovial citrullination plays a primary, sufficient, or even necessary role in ACPA pathogenicity. There is insufficient evidence supporting the notion that joint-epitope–reactive ACPAs alone, together with downstream humoral responses, are sufficient to initiate arthritis.
Further research is needed to clarify the upstream inducers of synovial protein citrullination (although studies conclude that synovial citrullination is PAD2- and PAD4-dependent, its origin remains unclear) and its pathogenetic significance (Badillo-Soto et al., 2016). Disordered neutrophil NETosis and impaired macrophage-mediated clearance may contribute, but definitive evidence is still lacking (Alivernini, Firestein and McInnes, 2022). Importantly, a critical unresolved question remains: what key factors drive the transition from an ACPA⁺ individual to the onset of ACPA⁺ joint inflammation?
Classical molecular mimicry | The classical pathway of molecular mimicry induced by enriched or commensal bacteria can mediate autoimmune responses in RA through cross-reactive epitopes that activate self-reactive T and B cell clones. P. copri is one of the most studied expanded taxa in early, untreated RA individuals (Scher et al., 2013). According to Pianta et al., two HLA-DR–presented autoantigens—acetylglucosamine-6-sulfatase (GNS) and filamin A (FLNA)—were identified and are highly expressed in synovial tissue of RA patients (Pianta et al., 2017). These proteins are homologous to proteins belonging to Prevotella spp. and Parabacteroides spp. (for GNS), and to Prevotella spp. and Butyricimonas spp. (for FLNA). Among them, GNS appears to be highly citrullinated (Pianta et al., 2017). HLA-DR molecules encoded by shared-epitope (SE) alleles have been shown to confer higher affinity binding to these microbial and self-antigens (Pianta et al., 2017). Between 2017 and 2021, five HLA-presented and T cell–reactive Prevotella copri peptides were gradually identified (Pianta et al., 2016; Pianta et al., 2021). Furthermore, mono-colonization of germ-free mice with specific strains from the Subdoligranulum genus demonstrated a greater capacity to stimulate arthritis than P. copri, accompanied by increased IgA production and Th17 skewing (Chriswell et al., 2022). Similarly, accumulating studies have identified molecular mimicry epitopes with the potential to generate autoantigens and elicit joint-specific immune responses. Examples include L-asparaginase from Streptococcus and type II collagen (Moten et al., 2022); ENO1 from Porphyromonas gingivalis and human enolase (Lee et al., 2015); and VtaA10755–766 from Glaesserella parasuis and human collagen II (Coll261–273) (Di Sante et al., 2021).
Together, molecular mimicry derived from invading or commensal microbes may redirect mucosal immune activation toward joint tissues, fostering systemic inflammation that ultimately evolves into persistent, articular-focused autoimmunity.
- Parallel Joint Inflammation Contributing to Mechanisms
Translocation of Microbes or Microbial Components | Limited but non-negligible evidence indicates that microbial components may reach the synovial compartment, providing a potential auxiliary pathway for joint-targeted inflammation in RA. This is supported by the verified presence of bacterial DNA and bacterial peptidoglycans in the joints of patients with rheumatoid arthritis, as well as bacterial peptidoglycan–loaded antigen-presenting cells in RA synovial tissues (Zhao et al., 2018; Arslan et al., 2000; Heijden et al., 2020; Hammad, Liyanapathirana and Tonge, 2019).
Zhao et al., using 16S rRNA sequencing, specifically confirmed the presence of Porphyromonas in both RA and OA synovial fluid samples, supporting the hypothesis of oral microbial participation in RA and other arthritis pathogenesis (Zhao et al., 2018). However, current evidence is insufficient to determine the extent to which microbial components at lesion sites contribute to RA or other arthritic diseases. It also remains unclear whether microbial components are exclusively present in synovial tissue or whether viable bacterial translocation occurs. Further research tracking the systemic distribution of bacterial components and the temporal development of lesions would help clarify these issues.
Immune Complex Kinetics and Deposition in Articular Tissue | Immune complex kinetics and deposition in articular tissue may represent a potential pathogenic process in RA. Following the generation of self-reactive antibodies, including RF and ACPA, immune complexes can infiltrate joint tissues and initiate a cascade of inflammatory responses (Kasiraja, Bakar and Suliman, 2025). Immune complex deposition has also been associated with other diseases, including models of diabetes and renal injury (Hickey and Martin, 2013; Xiao et al., 2009; Watanabe et al., 1987; Abrass, 1984).
Under normal physiological conditions, antibody deposition is typically prevented through clearance by mononuclear phagocytes via Fcγ receptor–mediated phagocytosis, promoted by complement activation (Esmail et al., 2018; Kasiraja, Bakar and Suliman, 2025). Studies suggest that when antibody levels exceed clearance capacity, immune complexes accumulate and deposit in organs where blood is filtered under high pressure to generate other fluids—such as synovial fluid—thereby promoting inflammation (Kumar, Abbas and Aster, 2012; Torres and Annamaraju, 2025; Kasiraja, Bakar and Suliman, 2025).
However, the phenomenon of immune complex deposition and its necessity in RA pathogenesis have not been specifically or extensively studied in RA patients or experimental disease models (Kasiraja, Bakar and Suliman, 2025). Altogether, immune complex deposition may contribute to rheumatoid arthritis–associated inflammation, but its mechanistic and causal significance remains insufficiently defined.
Conclusion
Collectively, current evidence supports a model in which mucosal dysbiosis, altered microbial metabolites, barrier dysfunction, and antigenic mimicry converge to establish a pro-inflammatory and tolerance-deficient immune landscape preceding joint inflammation. Rather than a single microbial trigger, RA initiation likely reflects the cumulative lowering of immune activation thresholds across mucosal and systemic compartments. However, several key questions remain unresolved. What constitutes the critical “second hit” that converts systemic autoimmunity into clinically manifest arthritis? Is gut barrier dysfunction causally upstream of joint inflammation, or merely a parallel consequence of systemic immune activation? Does peripheral immune tolerance failure occur at RA lesion sites, and how does it contribute to chronic inflammation? Finally, does immune complex deposition actively drive early synovitis, or does it amplify an already primed inflammatory niche? Addressing these questions will be essential to define the true initiating events in RA pathogenesis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
References
- Abrass, C.K. (1984) ’Evaluation of the presence of circulating immune complexes and their relationship to glomerular IgG deposits in streptozotocin-induced diabetic rats.,’ PubMed, 57(1), pp. 17–24. https://pubmed.ncbi.nlm.nih.gov/6378460.
- Adami, G. et al. (2021) ’Association between environmental air pollution and rheumatoid arthritis flares,’ Lara D. Veeken, 60(10), pp. 4591–4597. [CrossRef]
- Alivernini, S., Firestein, G.S. and McInnes, I.B. (2022) ’The pathogenesis of rheumatoid arthritis,’ Immunity, 55(12), pp. 2255–2270. [CrossRef]
- Arslan, L, Melief, M-J, Tak, PP, Hazenberg, MPH & Laman, J 2000, ’Antigen-presenting cells containing bacterial peptidoglycan in synovial tissues of rheumatoid arthritis patients coexpress costimulatory molecules and cytokines’, Arthritis & Rheumatism, vol. 43, no. 10, pp. 2160-2168. [CrossRef]
- Atarashi, K. et al. (2010) ’Induction of colonic regulatory T cells by indigenous clostridium species,’ Science, 331(6015), pp. 337–341. [CrossRef]
- Badillo-Soto, M.A. et al. (2016) ’Potential protein targets of the peptidylarginine deiminase 2 and peptidylarginine deiminase 4 enzymes in rheumatoid synovial tissue and its possible meaning,’ European Journal of Rheumatology, 3(2), pp. 44–49. [CrossRef]
- Bingham, C.O. and Moni, M. (2013) ’Periodontal disease and rheumatoid arthritis,’ Current Opinion in Rheumatology, 25(3), pp. 345–353. [CrossRef]
- Block, K.E. et al. (2016) ’Gut Microbiota Regulates K/BxN Autoimmune Arthritis through Follicular Helper T but Not Th17 Cells,’ The Journal of Immunology, 196(4), pp. 1550–1557. [CrossRef]
- Boya, B.R. et al. (2021) ’Diversity of the tryptophanase gene and its evolutionary implications in living organisms,’ Microorganisms, 9(10), p. 2156. [CrossRef]
- Brewer, R.C. et al. (2023) ’Oral mucosal breaks trigger anti-citrullinated bacterial and human protein antibody responses in rheumatoid arthritis,’ Science Translational Medicine, 15(684), p. eabq8476. [CrossRef]
- Capaldo, C.T., Powell, D.N. and Kalman, D. (2017) ’Layered defense: how mucus and tight junctions seal the intestinal barrier,’ Journal of Molecular Medicine, 95(9), pp. 927–934. [CrossRef]
- Chang, P.V. et al. (2014) ’The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition,’ Proceedings of the National Academy of Sciences, 111(6), pp. 2247–2252. [CrossRef]
- Chen, J. et al. (2016) ’An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis,’ Genome Medicine, 8(1), p. 43. [CrossRef]
- Chriswell, M.E. et al. (2022) ’Clonal IgA and IgG autoantibodies from individuals at risk for rheumatoid arthritis identify an arthritogenic strain of Subdoligranulum,’ Science Translational Medicine, 14(668), p. eabn5166. [CrossRef]
- Ciesielski, O. et al. (2022) ’Citrullination in the pathology of inflammatory and autoimmune disorders: recent advances and future perspectives,’ Cellular and Molecular Life Sciences, 79(2), p. 94. [CrossRef]
- Connors, J., Dawe, N. and Van Limbergen, J. (2018) ’The role of succinate in the regulation of intestinal inflammation,’ Nutrients, 11(1), p. 25. [CrossRef]
- Di Sante, G. et al. (2021) ’Haemophilus parasuis (Glaesserella parasuis) as a Potential Driver of Molecular Mimicry and Inflammation in Rheumatoid Arthritis,’ Frontiers in Medicine, 8, p. 671018. [CrossRef]
- Drago, L. (2019) ’Prevotella Copri and microbiota in rheumatoid arthritis: fully convincing evidence?,’ Journal of Clinical Medicine, 8(11), p. 1837. [CrossRef]
- Esmail, H. et al. (2018) ’Complement pathway gene activation and rising circulating immune complexes characterize early disease in HIV-associated tuberculosis,’ Proceedings of the National Academy of Sciences, 115(5), pp. E964–E973. [CrossRef]
- Fasano, A. (2020) ’All disease begins in the (leaky) gut: role of zonulin-mediated gut permeability in the pathogenesis of some chronic inflammatory diseases,’ F1000Research, 9, p. 69. [CrossRef]
- Fernández-Veledo, S. and Vendrell, J. (2019) ’Gut microbiota-derived succinate: Friend or foe in human metabolic diseases?,’ Reviews in Endocrine and Metabolic Disorders, 20(4), pp. 439–447. [CrossRef]
- Foulquier, C. et al. (2007) ’Peptidyl arginine deiminase type 2 (PAD-2) and PAD-4 but not PAD-1, PAD-3, and PAD-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation,’ Arthritis & Rheumatism, 56(11), pp. 3541–3553. [CrossRef]
- Fusco, W. et al. (2023) ’Short-Chain Fatty-Acid-Producing bacteria: key components of the human gut microbiota,’ Nutrients, 15(9), p. 2211. [CrossRef]
- González-Febles, J. et al. (2020) ’Association between periodontitis and anti-citrullinated protein antibodies in rheumatoid arthritis patients: a cross-sectional study,’ Arthritis Research & Therapy, 22(1), p. 27. [CrossRef]
- Guo, Q. et al. (2018) ’Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies,’ Bone Research, 6(1), p. 15. [CrossRef]
- Hammad, D.B.M., Liyanapathirana, V. and Tonge, D.P. (2019) ’Molecular characterisation of the synovial fluid microbiome in rheumatoid arthritis patients and healthy control subjects,’ PLoS ONE, 14(11), p. e0225110. [CrossRef]
- Harber, K.J. et al. (2020) ’Succinate is an Inflammation-Induced immunoregulatory metabolite in macrophages,’ Metabolites, 10(9), p. 372. [CrossRef]
- Hickey, F.B. and Martin, F. (2013) ’Diabetic kidney disease and immune modulation,’ Current Opinion in Pharmacology, 13(4), pp. 602–612. [CrossRef]
- Holers, V.M. et al. (2018) ’Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction,’ Nature Reviews Rheumatology, 14(9), pp. 542–557. [CrossRef]
- Holers, V.M. et al. (2024) ’Distinct mucosal endotypes as initiators and drivers of rheumatoid arthritis,’ Nature Reviews Rheumatology, 20(10), pp. 601–613. [CrossRef]
- Huang, H. et al. (2024) ’Cellular succinate metabolism and signaling in inflammation: implications for therapeutic intervention,’ Frontiers in Immunology, 15, p. 1404441. [CrossRef]
- Huang, Y. et al. (2020) ’Prevotella induces the production of TH17 cells in the colon of mice,’ Journal of Immunology Research, 2020(1), p. 9607328. [CrossRef]
- Iljazovic, A. et al. (2020) ’Perturbation of the gut microbiome by Prevotella spp. enhances host susceptibility to mucosal inflammation,’ Mucosal Immunology, 14(1), pp. 113–124. [CrossRef]
- Ishikawa, Y. and Terao, C. (2020) ’The Impact of cigarette smoking on risk of rheumatoid arthritis: A Narrative review,’ Cells, 9(2), p. 475. [CrossRef]
- Javed, I. and Crowson, C.S. (2025) ’The apprehension of seronegative rheumatoid arthritis,’ Nature Reviews Rheumatology, 21(10), pp. 575–576. [CrossRef]
- Kasiraja, V., Bakar, N.A.A. and Suliman, N.A. (2025) ’Rheumatoid arthritis unmasked: the immune complex as a key driver of disease progression,’ Exploration of Immunology, 5. [CrossRef]
- Kim, M.H. et al. (2013) ’Short-Chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice,’ Gastroenterology, 145(2), pp. 396-406.e10. [CrossRef]
- Kinloch, A. et al. (2008) ’Synovial fluid is a site of citrullination of autoantigens in inflammatory arthritis,’ Arthritis & Rheumatism, 58(8), pp. 2287–2295. [CrossRef]
- Kuhn, K.A. et al. (2025) ’Bacterial catabolism of dietary tryptophan into indole enhances DC-mediated pro-inflammatory responses in autoimmunity 3709,’ The Journal of Immunology, 214(Supplement_1). [CrossRef]
- Kumar, V., Abbas, A.K. and Aster, J.C. (2012) Robbins Basic Pathology E-Book. Elsevier Health Sciences.
- Laurent, L. et al. (2014) ’IgM rheumatoid factor amplifies the inflammatory response of macrophages induced by the rheumatoid arthritis-specific immune complexes containing anticitrullinated protein antibodies,’ Annals of the Rheumatic Diseases, 74(7), pp. 1425–1431. [CrossRef]
- Lee, J.Y. et al. (2015) ’Association between anti-Porphyromonas gingivalis or anti-α-enolase antibody and severity of periodontitis or rheumatoid arthritis (RA) disease activity in RA,’ BMC Musculoskeletal Disorders, 16(1), p. 190. [CrossRef]
- Li, J. and Kuhn, K.A. (2025) ’Microbial threads in the tapestry of rheumatoid arthritis,’ Journal of Clinical Investigation, 135(18). [CrossRef]
- Li, J. et al. (2020) ’Targeted Combination of Antioxidative and Anti-Inflammatory Therapy of Rheumatoid Arthritis using Multifunctional Dendrimer-Entrapped Gold Nanoparticles as a Platform,’ Small, 16(49), p. e2005661. [CrossRef]
- Lu, J. et al. (2025) ’Linking microbial communities to rheumatoid arthritis: focus on gut, oral microbiome and their extracellular vesicles,’ Frontiers in Immunology, 16, p. 1503474. [CrossRef]
- Lucchino, B. et al. (2019) ’Mucosa–Environment interactions in the pathogenesis of rheumatoid arthritis,’ Cells, 8(7), p. 700. [CrossRef]
- Maeda, Y. and Takeda, K. (2019) ’Host–microbiota interactions in rheumatoid arthritis,’ Experimental & Molecular Medicine, 51(12), pp. 1–6. [CrossRef]
- Maeda, Y. et al. (2016) ’Dysbiosis contributes to arthritis development via activation of autoreactive T cells in the intestine,’ Arthritis & Rheumatology, 68(11), pp. 2646–2661. [CrossRef]
- Mansouri, A., Akthar, I. and Miyamoto, A. (2025) ’TLR2 and TLR4 bridge physiological and pathological inflammation in the reproductive system,’ Communications Biology, 8(1), p. 1008. [CrossRef]
- Maslowski, K.M. et al. (2009) ’Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43,’ Nature, 461(7268), pp. 1282–1286. [CrossRef]
- Matei, D.E. et al. (2021) ’Intestinal barrier dysfunction plays an integral role in arthritis pathology and can be targeted to ameliorate disease,’ Med, 2(7), pp. 864-883.e9. [CrossRef]
- Menofy, N.G.E. et al. (2022) ’Bacterial Compositional Shifts of Gut Microbiomes in Patients with Rheumatoid Arthritis in Association with Disease Activity,’ Microorganisms, 10(9), p. 1820. [CrossRef]
- Mohsenzadeh, A. et al. (2025) ’The gut microbiota and cardiovascular disease: Exploring the role of microbial dysbiosis and metabolites in pathogenesis and therapeutics,’ Life Sciences, 381, p. 123981. [CrossRef]
- Moten, D. et al. (2022) ’Molecular Mimicry of the Rheumatoid Arthritis-Related Immunodominant T-Cell Epitope within Type II Collagen (CII260-270) by the Bacterial L-Asparaginase,’ International Journal of Molecular Sciences, 23(16), p. 9149. [CrossRef]
- Nastasi, C. et al. (2015) ’The effect of short-chain fatty acids on human monocyte-derived dendritic cells,’ Scientific Reports, 5(1), p. 16148. [CrossRef]
- Nonaka, S., Kadowaki, T. and Nakanishi, H. (2022) ’Secreted gingipains from Porphyromonas gingivalis increase permeability in human cerebral microvascular endothelial cells through intracellular degradation of tight junction proteins,’ Neurochemistry International, 154, p. 105282. [CrossRef]
- Ordoñez-Rodriguez, A. et al. (2023) ’Changes in gut microbiota and multiple sclerosis: A Systematic review,’ International Journal of Environmental Research and Public Health, 20(5), p. 4624. [CrossRef]
- Ossipova, E. et al. (2014) ’Affinity purified anti-citrullinated protein/peptide antibodies target antigens expressed in the rheumatoid joint,’ Arthritis Research & Therapy, 16(4), p. R167. [CrossRef]
- Paradis, T. et al. (2021) ’Tight junctions as a key for pathogens invasion in intestinal epithelial cells,’ International Journal of Molecular Sciences, 22(5), p. 2506. [CrossRef]
- Paroli, M. and Sirinian, M.I. (2023) ’When autoantibodies are missing: The challenge of seronegative rheumatoid arthritis,’ Antibodies, 12(4), p. 69. [CrossRef]
- Pianta, A. et al. (2016) ’Evidence of the Immune Relevance of Prevotella copri, a Gut Microbe, in Patients With Rheumatoid Arthritis,’ Arthritis & Rheumatology, 69(5), pp. 964–975. [CrossRef]
- Pianta, A. et al. (2017) ’Two rheumatoid arthritis–specific autoantigens correlate microbial immunity with autoimmune responses in joints,’ Journal of Clinical Investigation, 127(8), pp. 2946–2956. [CrossRef]
- Pianta, A. et al. (2021) ’Identification of Novel, Immunogenic HLA–DR-Presented Prevotella copri Peptides in Patients With Rheumatoid Arthritis,’ Arthritis & Rheumatology, 73(12), pp. 2200–2205. [CrossRef]
- Pitter, M.R. and Zou, W. (2025) ’Citrullination in tumor immunity and therapy,’ Journal of Clinical Investigation, 135(20). [CrossRef]
- Pratt, A.G. and Isaacs, J.D. (2014) ’Seronegative rheumatoid arthritis: Pathogenetic and therapeutic aspects,’ Best Practice & Research Clinical Rheumatology, 28(4), pp. 651–659. [CrossRef]
- Van der Heijden, I M et al. “Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides.” Arthritis and rheumatism vol. 43,3 (2000): 593-8. [CrossRef]
- Regueiro, C. et al. (2020) ’A predominant involvement of the triple seropositive patients and others with rheumatoid factor in the association of smoking with rheumatoid arthritis,’ Scientific Reports, 10(1), p. 3355. [CrossRef]
- Reichardt, N. et al. (2014) ’Phylogenetic distribution of three pathways for propionate production within the human gut microbiota,’ The ISME Journal, 8(6), pp. 1323–1335. [CrossRef]
- Sakurai, T., Odamaki, T. and Xiao, J.-Z. (2019) ’Production of Indole-3-Lactic acid by bifidobacterium strains isolated from Human Infants,’ Microorganisms, 7(9), p. 340. [CrossRef]
- Santana, P.T. et al. (2022) ’Dysbiosis in inflammatory bowel Disease: Pathogenic role and potential therapeutic targets,’ International Journal of Molecular Sciences, 23(7), p. 3464. [CrossRef]
- Saraiva, A.L. et al. (2018) ’Succinate receptor deficiency attenuates arthritis by reducing dendritic cell traffic and expansion of T h 17 cells in the lymph nodes,’ The FASEB Journal, 32(12), pp. 6550–6558. [CrossRef]
- Scher, J.U. et al. (2013) ’Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis,’ eLife, 2, p. e01202. [CrossRef]
- Serek, P. and Oleksy-Wawrzyniak, M. (2021) ’The effect of bacterial infections, probiotics and zonulin on intestinal barrier integrity,’ International Journal of Molecular Sciences, 22(21), p. 11359. [CrossRef]
- Seymour, B.J. et al. (2023) ’Microbiota-dependent indole production stimulates the development of collagen-induced arthritis in mice,’ Journal of Clinical Investigation, 134(4). [CrossRef]
- Seymour, B.J., Allen, B.E. and Kuhn, K.A. (2024) ’Microbial mechanisms of rheumatoid arthritis pathogenesis,’ Current Rheumatology Reports, 26(4), pp. 124–132. [CrossRef]
- Smith, P.M. et al. (2013) ’The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic T reg Cell Homeostasis,’ Science, 341(6145), pp. 569–573. [CrossRef]
- Smolen, J.S. et al. (2018) ’Rheumatoid arthritis,’ Nature Reviews Disease Primers, 4(1), p. 18001. [CrossRef]
- Spindler, M.P. et al. (2022) ’Human gut microbiota stimulate defined innate immune responses that vary from phylum to strain,’ Cell Host & Microbe, 30(10), pp. 1481-1498.e5. [CrossRef]
- Sun, M. et al. (2016) ’Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases,’ Journal of Gastroenterology, 52(1), pp. 1–8. [CrossRef]
- Tajik, N. et al. (2020) ’Targeting zonulin and intestinal epithelial barrier function to prevent onset of arthritis,’ Nature Communications, 11(1), p. 1995. [CrossRef]
- Taylor, P.C. (2020) ’Update on the diagnosis and management of early rheumatoid arthritis,’ Clinical Medicine, 20(6), pp. 561–564. [CrossRef]
- Tedeschi, S.K. et al. (2017) ’Diet and rheumatoid arthritis symptoms: Survey results from a rheumatoid arthritis registry,’ Arthritis Care & Research, 69(12), pp. 1920–1925. [CrossRef]
- Torres, J.S.S. and Annamaraju, P. (2025) Type III hypersensitivity reaction. https://www.ncbi.nlm.nih.gov/books/NBK559122/.
- Trauelsen, M. et al. (2017) ’Receptor structure-based discovery of non-metabolite agonists for the succinate receptor GPR91,’ Molecular Metabolism, 6(12), pp. 1585–1596. [CrossRef]
- Trauelsen, M. et al. (2021) ’Extracellular succinate hyperpolarizes M2 macrophages through SUCNR1/GPR91-mediated Gq signaling,’ Cell Reports, 35(11), p. 109246. [CrossRef]
- Velikova, T. et al. (2024) ’Mucosal immunity and trained innate immunity of the gut,’ Gastroenterology Insights, 15(3), pp. 661–675. [CrossRef]
- Venetsanopoulou, A.I. et al. (2023) ’Epidemiology and risk factors for rheumatoid arthritis development,’ Mediterranean Journal of Rheumatology, 34(4), p. 404. [CrossRef]
- Vinolo, M. a. R. et al. (2008) ’Effects of short chain fatty acids on effector mechanisms of neutrophils,’ Cell Biochemistry and Function, 27(1), pp. 48–55. [CrossRef]
- Vinolo, M. a. R. et al. (2009) ’Short-chain fatty acids stimulate the migration of neutrophils to inflammatory sites,’ Clinical Science, 117(9), pp. 331–338. [CrossRef]
- Watanabe, S. et al. (1987) ’Detection of immunoglobulins and/or complement in kidney tissues from non-obese diabetic (NOD) mice.,’ PubMed, 12(3), pp. 201–8. https://pubmed.ncbi.nlm.nih.gov/3454080.
- Wei, Y.-H. et al. (2023) ’Succinate metabolism and its regulation of host-microbe interactions,’ Gut Microbes, 15(1), p. 2190300. [CrossRef]
- Wu, C.-Y., Yang, H.-Y. and Lai, J.-H. (2020) ’Anti-Citrullinated Protein Antibodies in Patients with Rheumatoid Arthritis: Biological Effects and Mechanisms of Immunopathogenesis,’ International Journal of Molecular Sciences, 21(11), p. 4015. [CrossRef]
- Wu, H.-J. et al. (2010) ’Gut-Residing segmented filamentous bacteria drive autoimmune arthritis via T Helper 17 cells,’ Immunity, 32(6), pp. 815–827. [CrossRef]
- Wu, J. et al. (2025) ’Investigating correlation between gut microbiota and rheumatoid arthritis subtypes by Mendelian randomization,’ Pathogens, 14(4), p. 385. [CrossRef]
- Wu, R. et al. (2024) ’Impaired immune tolerance mediated by reduced Tfr cells in rheumatoid arthritis linked to gut microbiota dysbiosis and altered metabolites,’ Arthritis Research & Therapy, 26(1), p. 21. [CrossRef]
- Xia, Y. et al. (2023) ’Lactobacillus-derived indole-3-lactic acid ameliorates colitis in cesarean-born offspring via activation of aryl hydrocarbon receptor,’ iScience, 26(11), p. 108279. [CrossRef]
- Xiao, X. et al. (2009) ’Cellular and humoral immune responses in the early stages of diabetic nephropathy in NOD mice,’ Journal of Autoimmunity, 32(2), pp. 85–93. [CrossRef]
- Yang, M.-L. et al. (2021) ’Citrullination and PAD enzyme biology in Type 1 diabetes – regulators of inflammation, autoimmunity, and pathology,’ Frontiers in Immunology, 12, p. 678953. [CrossRef]
- Yang, W. et al. (2019) ’Microbiota Metabolite Short-Chain Fatty Acids Facilitate Mucosal Adjuvant Activity of Cholera Toxin through GPR43,’ The Journal of Immunology, 203(1), pp. 282–292. [CrossRef]
- Zhao, Y. et al. (2018) ’Detection and characterization of bacterial nucleic acids in culture-negative synovial tissue and fluid samples from rheumatoid arthritis or osteoarthritis patients,’ Scientific Reports, 8(1), p. 14305. [CrossRef]
- Zhou, X. et al. (2025b) ’Mucosal immune response in biology, disease prevention and treatment,’ Signal Transduction and Targeted Therapy, 10(1), p. 7. [CrossRef]
Figure 1.
Conceptual model of mucosal-driven rheumatoid arthritis pathogenesis. Pathogenesis phases of RA initiation are displayed from left to right and top to bottom. Environmental risk factors such as smoking, air pollution, dietary habits predispose to mucosal dysbiosis characterized by altered microbial composition, metabolite imbalance, epithelial barrier dysfunction. These perturbations promote hyperactivation of mucosa-associated lymphoid tissue (MALT), with enhanced TLR2/TLR4/NOD signaling, reduced activation thresholds, impaired Treg induction, and Th17/Th1 skewing. Facilitated by HLA-DRB polymorphisms etc. genetic risk factors, the pro-inflammatory and possibly tolerance-deficient milieu facilitates the generation of autoreactive T and B cells through mechanisms including molecular mimicry and aberrant citrullination, leading to ACPA/RF production. Systemic dissemination of inflammatory mediators and probably immune complexes dynamics establishes a primed inflammatory niche in joint finally promote clinically manifest RA and memory formation.
Figure 1.
Conceptual model of mucosal-driven rheumatoid arthritis pathogenesis. Pathogenesis phases of RA initiation are displayed from left to right and top to bottom. Environmental risk factors such as smoking, air pollution, dietary habits predispose to mucosal dysbiosis characterized by altered microbial composition, metabolite imbalance, epithelial barrier dysfunction. These perturbations promote hyperactivation of mucosa-associated lymphoid tissue (MALT), with enhanced TLR2/TLR4/NOD signaling, reduced activation thresholds, impaired Treg induction, and Th17/Th1 skewing. Facilitated by HLA-DRB polymorphisms etc. genetic risk factors, the pro-inflammatory and possibly tolerance-deficient milieu facilitates the generation of autoreactive T and B cells through mechanisms including molecular mimicry and aberrant citrullination, leading to ACPA/RF production. Systemic dissemination of inflammatory mediators and probably immune complexes dynamics establishes a primed inflammatory niche in joint finally promote clinically manifest RA and memory formation.
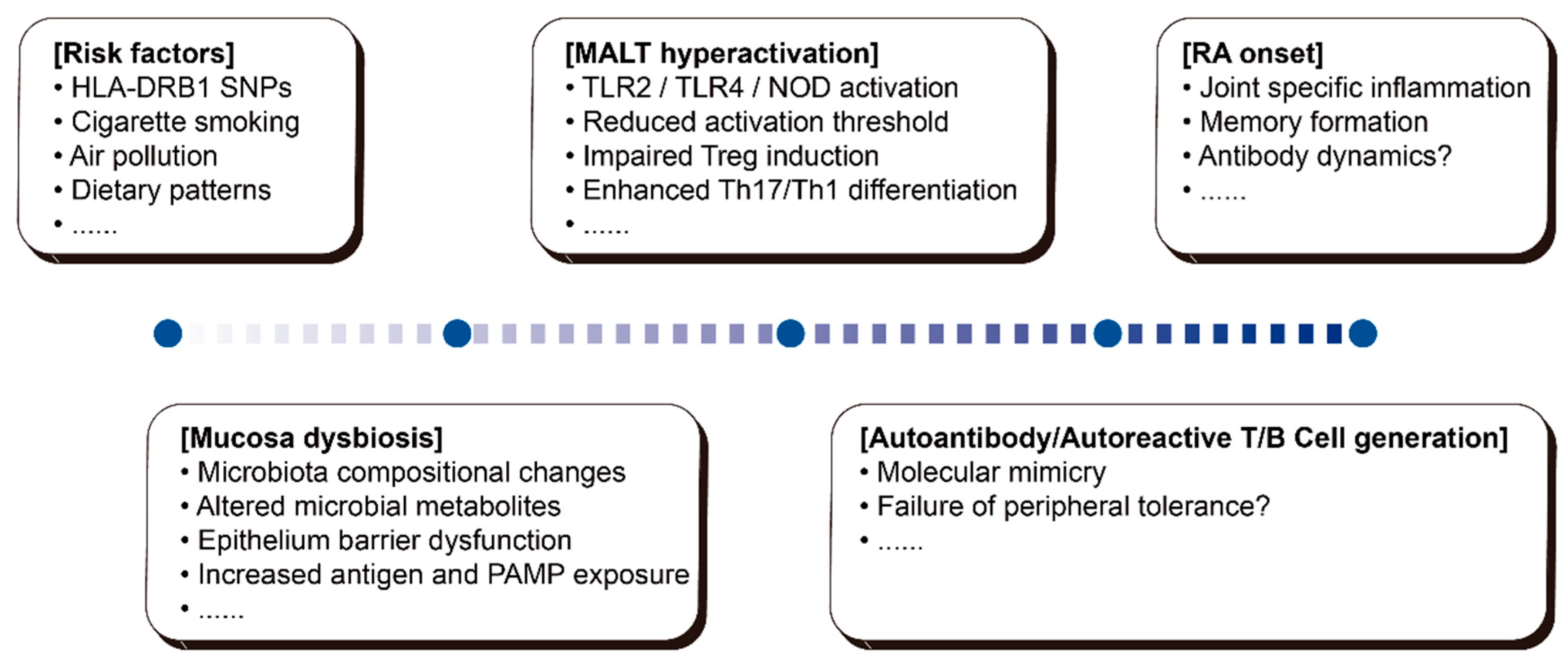
Figure 2.
Literature screening workflow and distribution of RA–microbiota sequencing studies. (A) Flow diagram of the systematic literature selection process. PubMed was utilized applying conditions “rheumatoid arthritis” AND “microbiota” AND “sequencing” for studies published between 2004 and 2025 (n = 304). After exclusion of irrelevant or non–RA-focused studies, 40 records remained. Further exclusion of review articles, meta-analyses and Mendelian randomization studies yielded 29 eligible original research articles for plotting and downstream analysis. (B) Bubble plot summarizing characteristics of the included studies. Each dot represents an individual study, stratified by sampling site and/or experimental category. Bubble size reflects relative study weight (e.g., cohort size or frequency), and color denotes study grouping as defined in the analysis.
Figure 2.
Literature screening workflow and distribution of RA–microbiota sequencing studies. (A) Flow diagram of the systematic literature selection process. PubMed was utilized applying conditions “rheumatoid arthritis” AND “microbiota” AND “sequencing” for studies published between 2004 and 2025 (n = 304). After exclusion of irrelevant or non–RA-focused studies, 40 records remained. Further exclusion of review articles, meta-analyses and Mendelian randomization studies yielded 29 eligible original research articles for plotting and downstream analysis. (B) Bubble plot summarizing characteristics of the included studies. Each dot represents an individual study, stratified by sampling site and/or experimental category. Bubble size reflects relative study weight (e.g., cohort size or frequency), and color denotes study grouping as defined in the analysis.
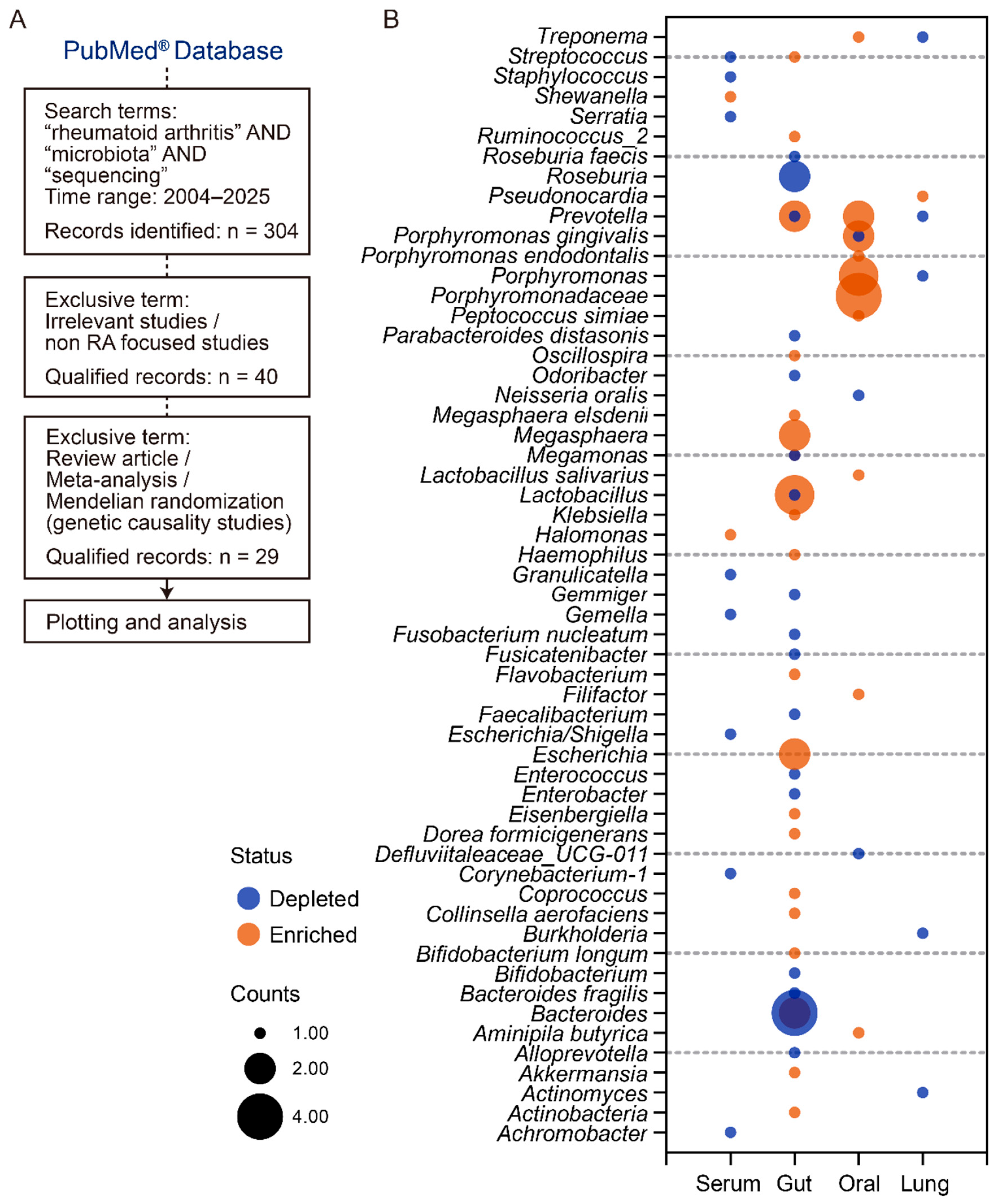
Figure 3.
Microbe, microbial metabolites and barrier integrity in mucosal-driven RA pathogenesis. (A) Pattern recognition and epithelial sensing induced inflammatory milieu in intestine mucosa. Microbial components activate epithelial and innate immune signaling pathways, including NOD1/2–NF-κB in enterocytes and MyD88- and NLRP6-associated pathways in goblet cells. Dendritic cell (DC) activation is also facilitated by goblet cell associated antigen passages. These events promote IL-6, TNF-α, and IL-8 production, neutrophil recruitment, Th17-favoring DC polarization, and establishment of a pro–rheumatoid arthritis (RA) inflammatory milieu in the lamina propria and influence systematically. (B) Imbalanced microbial metabolites alter immune homeostasis. Succinate signaling through SUCNR1 enhances DC activation, macrophage (Mφ) polarization, and antigen presenting cell priming ability. Indole and its derivatives converted by tnaA-positive bacteria influence immune system in varying ways. Indole promotes DC activation and migration ability and cause antibody isotype switching. ILA promote IL-22 signaling and ILC3 development. SCFA exposure plays dual function for both pro- and anti-inflammatory. SCFA promote acute inflammation and B cell inflammation. It also inhibits DC and macrophage CD40 expression and promotes Treg differentiation. (C) Barrier disruption and systemic inflammation. Gliadin and microbe up-regulates zonulin, stimulates PAR2 and EGFr mediated tight junction (TJ) disassembly. Inflammatory cytokines like TNF-α contributes to TJ dysfunction by increase claudin-2 expression and activates myosin light chain kinase (MLCK). Certain pathogens can pathogenically destroy intestine epithelium permeability, though not identified as the major etiology of RA. SCFA and increased PAMP sensing may increase TJ integrity by promote tight junction molecules expression and assembly to prevent infection.
Figure 3.
Microbe, microbial metabolites and barrier integrity in mucosal-driven RA pathogenesis. (A) Pattern recognition and epithelial sensing induced inflammatory milieu in intestine mucosa. Microbial components activate epithelial and innate immune signaling pathways, including NOD1/2–NF-κB in enterocytes and MyD88- and NLRP6-associated pathways in goblet cells. Dendritic cell (DC) activation is also facilitated by goblet cell associated antigen passages. These events promote IL-6, TNF-α, and IL-8 production, neutrophil recruitment, Th17-favoring DC polarization, and establishment of a pro–rheumatoid arthritis (RA) inflammatory milieu in the lamina propria and influence systematically. (B) Imbalanced microbial metabolites alter immune homeostasis. Succinate signaling through SUCNR1 enhances DC activation, macrophage (Mφ) polarization, and antigen presenting cell priming ability. Indole and its derivatives converted by tnaA-positive bacteria influence immune system in varying ways. Indole promotes DC activation and migration ability and cause antibody isotype switching. ILA promote IL-22 signaling and ILC3 development. SCFA exposure plays dual function for both pro- and anti-inflammatory. SCFA promote acute inflammation and B cell inflammation. It also inhibits DC and macrophage CD40 expression and promotes Treg differentiation. (C) Barrier disruption and systemic inflammation. Gliadin and microbe up-regulates zonulin, stimulates PAR2 and EGFr mediated tight junction (TJ) disassembly. Inflammatory cytokines like TNF-α contributes to TJ dysfunction by increase claudin-2 expression and activates myosin light chain kinase (MLCK). Certain pathogens can pathogenically destroy intestine epithelium permeability, though not identified as the major etiology of RA. SCFA and increased PAMP sensing may increase TJ integrity by promote tight junction molecules expression and assembly to prevent infection.
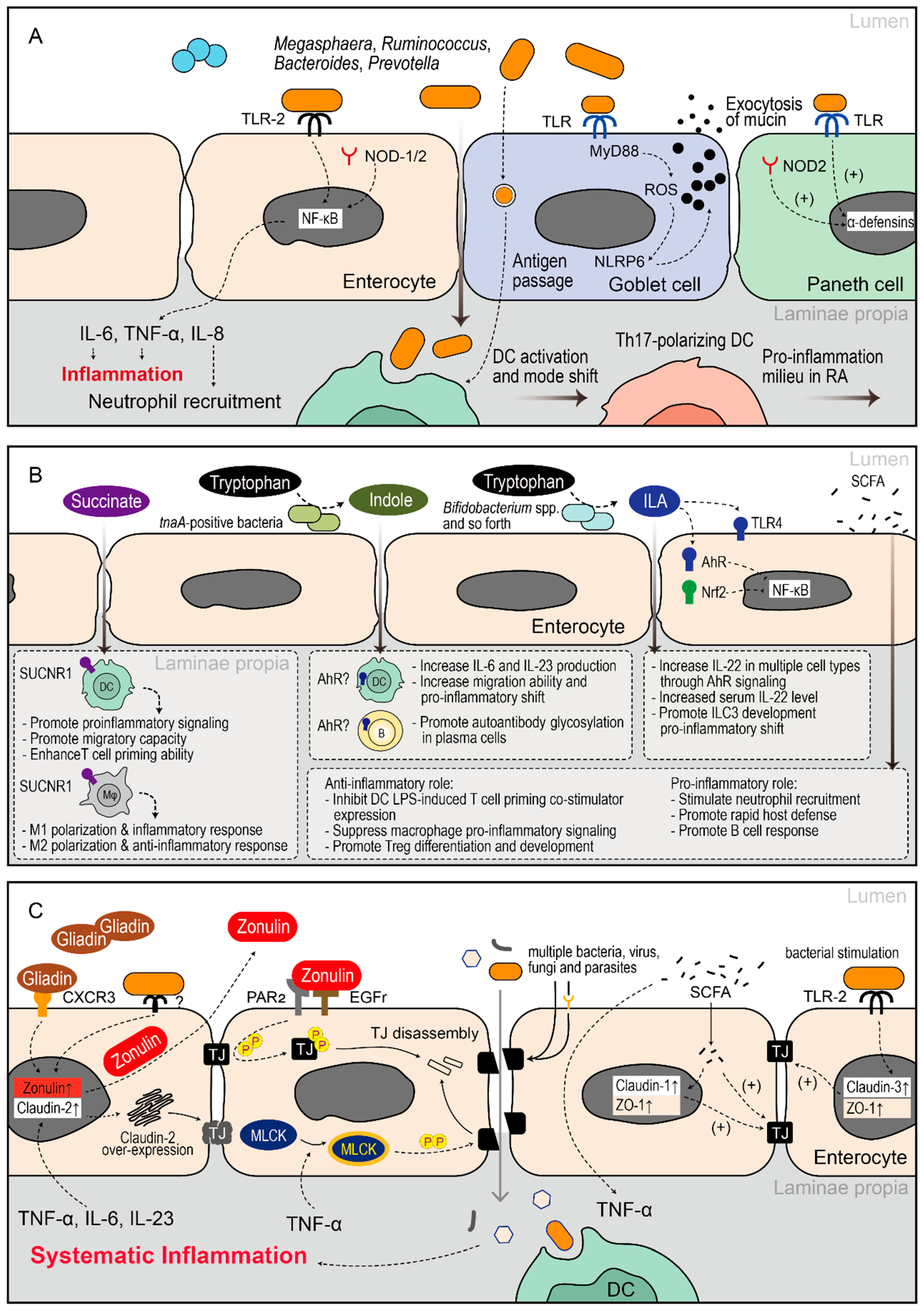
Figure 4.
Molecular mimicry—microbial antigens prime cross-reactive adaptive immune responses in RA. Facilitated by chronic infection, continuous NETosis, and altered macrophage (Mφ) clearance function, microbial antigen is presented by DC cell to prime T cell. For some pathogens like Group A Streptococcus, they can penetrate mucosa barrier and invade circulation system, thereby possibly inducing a greater activation and exposure of antigen. Antigen presented to T cell can raise a auto-reactive immune response in RA by majorly two pathways: direct mimicry to autoantigens or neoantigens; only becoming self-reactive after undergoing somatic hypermutation and affinity selection. Potential failure or delay of peripheral tolerance mechanism may contribute to self-reactive response towards joint tissue but not clearly explored in causal etiology perspective. Self-reactive T cell and B cell migrates to synovium and raise an inflammatory response with the assistance of phagocytes and granulocyte towards fibroblasts.
Figure 4.
Molecular mimicry—microbial antigens prime cross-reactive adaptive immune responses in RA. Facilitated by chronic infection, continuous NETosis, and altered macrophage (Mφ) clearance function, microbial antigen is presented by DC cell to prime T cell. For some pathogens like Group A Streptococcus, they can penetrate mucosa barrier and invade circulation system, thereby possibly inducing a greater activation and exposure of antigen. Antigen presented to T cell can raise a auto-reactive immune response in RA by majorly two pathways: direct mimicry to autoantigens or neoantigens; only becoming self-reactive after undergoing somatic hypermutation and affinity selection. Potential failure or delay of peripheral tolerance mechanism may contribute to self-reactive response towards joint tissue but not clearly explored in causal etiology perspective. Self-reactive T cell and B cell migrates to synovium and raise an inflammatory response with the assistance of phagocytes and granulocyte towards fibroblasts.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



