Submitted:
25 February 2026
Posted:
27 February 2026
You are already at the latest version
Abstract
Mycoplasma pneumoniae is a significant pathogen responsible for community-acquired respiratory infections in children and adolescents, with the rising prevalence of macrolide-resistant M. pneumoniae (MRMP), particularly in Asia, presenting critical treatment challenges. Our previous study inferred that macrolide efflux pump may contribute to macrolide resistance in M. pneumoniae in addition to the common point mutations in 23S rRNA gene. This study aimed to define the specific pump and confirm its role. Through comparative genomic analysis, we identified a candidate gene, MPN_080, encoding an ABC transporter permease, which was further characterized using phylogenetic analysis, AlphaFold-based structural modeling, and biochemical assays. Overexpression of MPN_080 from an erythromycin-resistant isolate in the erythromycin-sensitive M129, and resulted in a significant increase in minimum inhibitory concentrations (MICs) from <0.125 µg/mL to 1 µg/mL, while similar overexpression of MPN_080 derived from M129 did not affect MICs. Notably, this resistance mechanism operates independently of M. pneumoniae virulence factors, as evidenced by unaltered colonization capacity in NCI-H292 cells and consistent immune response patterns across both strains. Our findings establish MPN_080 as a novel determinant of macrolide resistance functioning through an ATPase-dependent mechanism, distinct from classical 23S rRNA mutations. These insights into non-classical resistance mechanisms may guide future diagnostic and therapeutic strategies against MRMP.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Phylogenetic Analysis
2.3. Expression and Purification of the M. pneumoniae MPN_080 protein
2.4. Vector Construction and Bacterial Transformation
2.5. Western Blot Analysis
2.6. Minimum Inhibitory Concentration (MIC) Assays
2.7. Infection of NCI-H292 Cells with MPN_080 Overexpression Mutants
2.8. PCR
2.9. Real-Time Quantitative PCR
2.10. ATPase Activity Assay
2.11. Statistical Analysis
3. Results
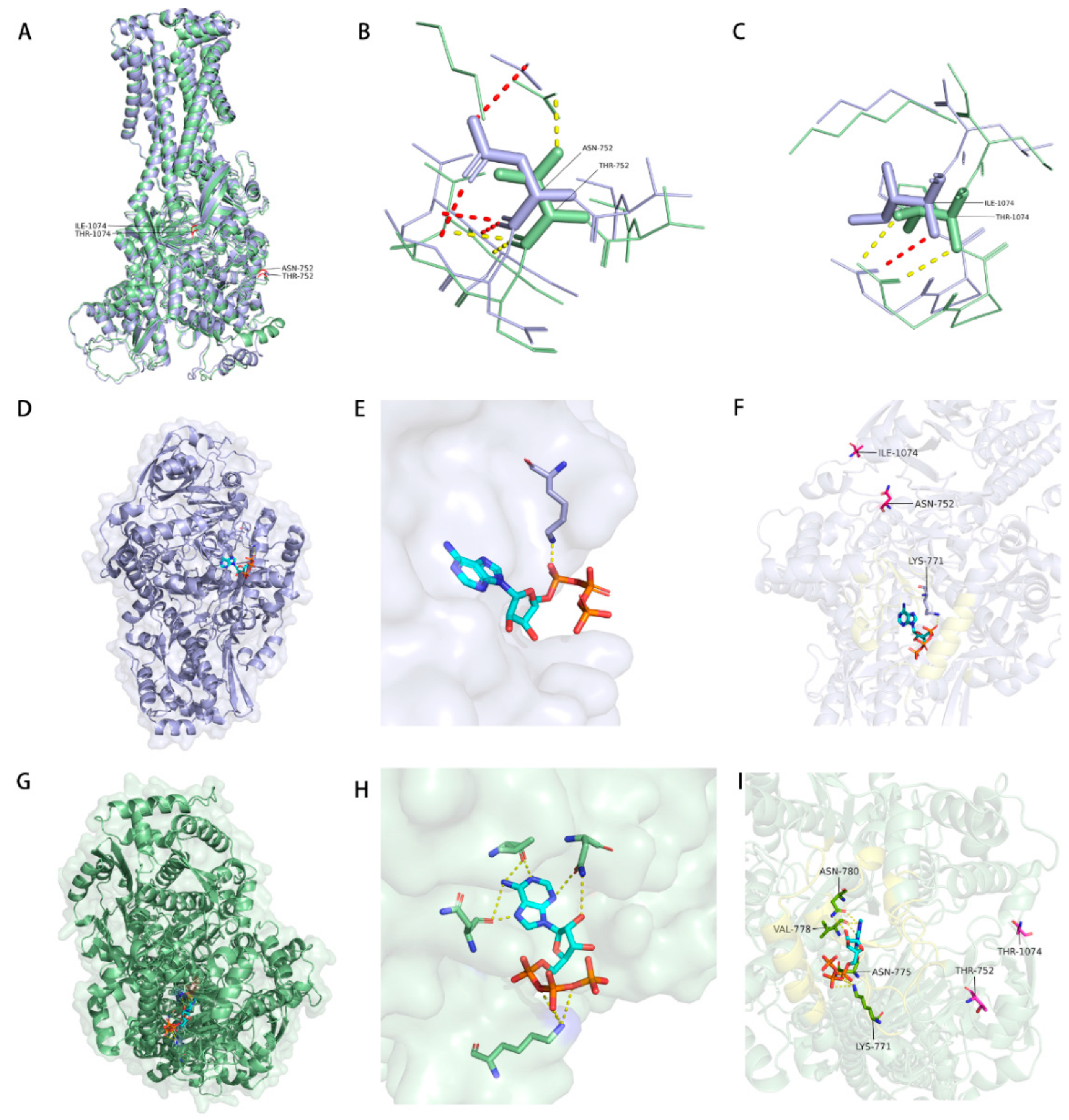
3.1. Phylogenetic Analysis and Protein Structure Comparison of RC267 and M129 Strain MPN_080 Genes
3.2. Structural Model of MPN_080 from RC267 and M129 and Comparison of Putative Interactions Within the ATP-Binding Pocket
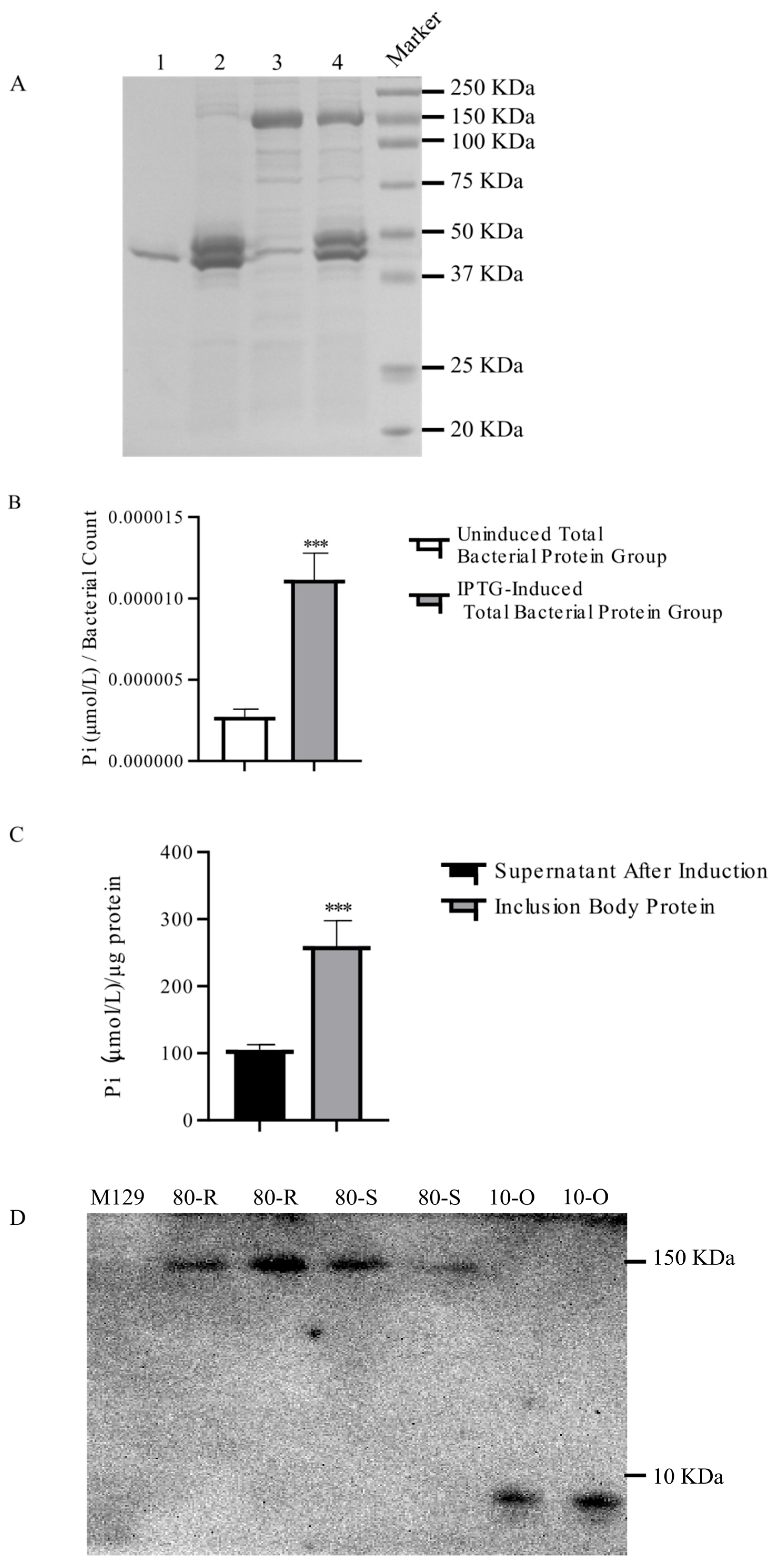
3.3. MPN_080 Exhibits ATPase Activity Following Refolding from Inclusion Bodies
3.4. RC267-derived MPN_080 Confers Increased Erythromycin MIC in M129
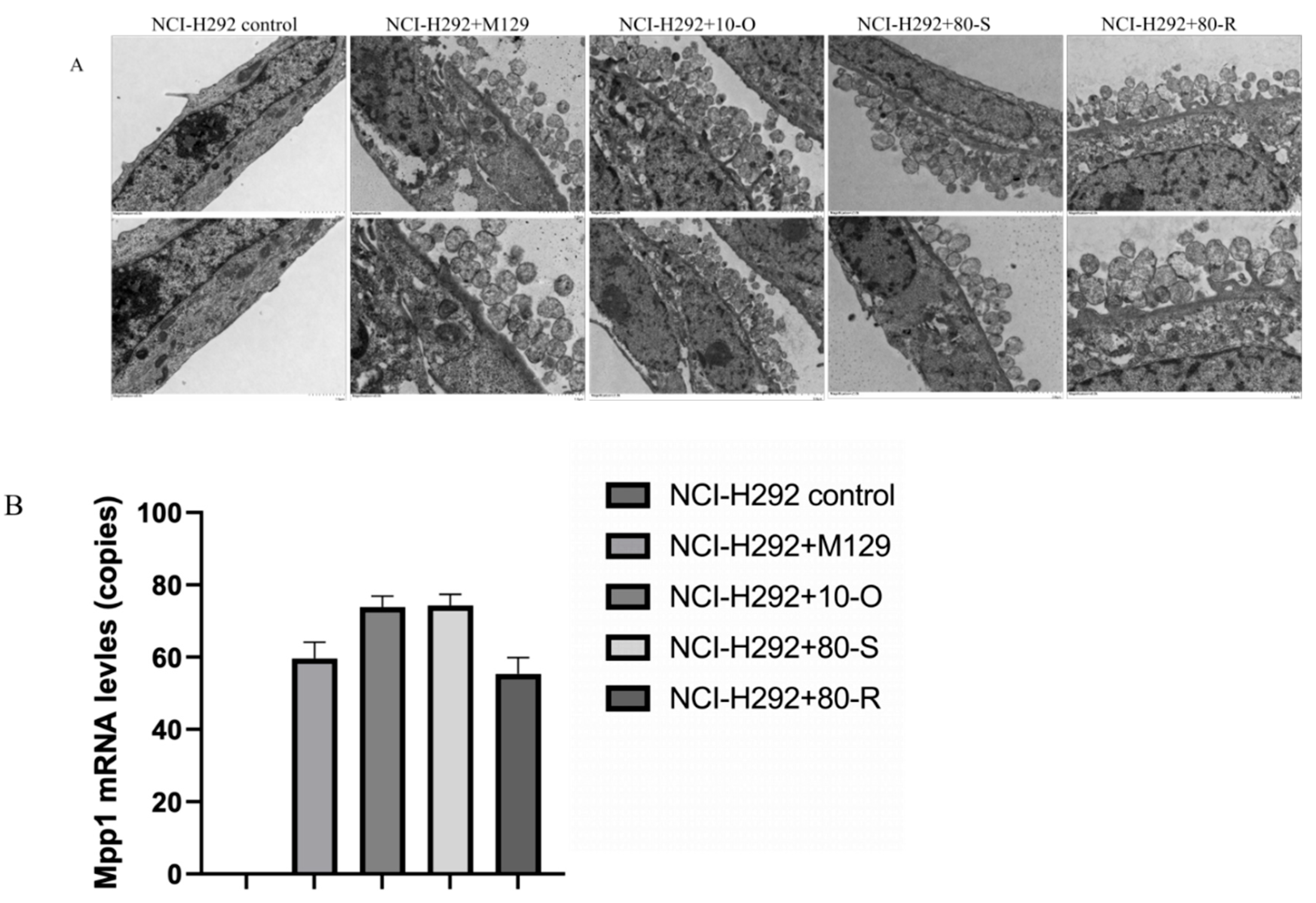
3.5. MPN_080 Does Not Affect the Colonization Ability of M.pneumoniae in NCI-H292 Cells
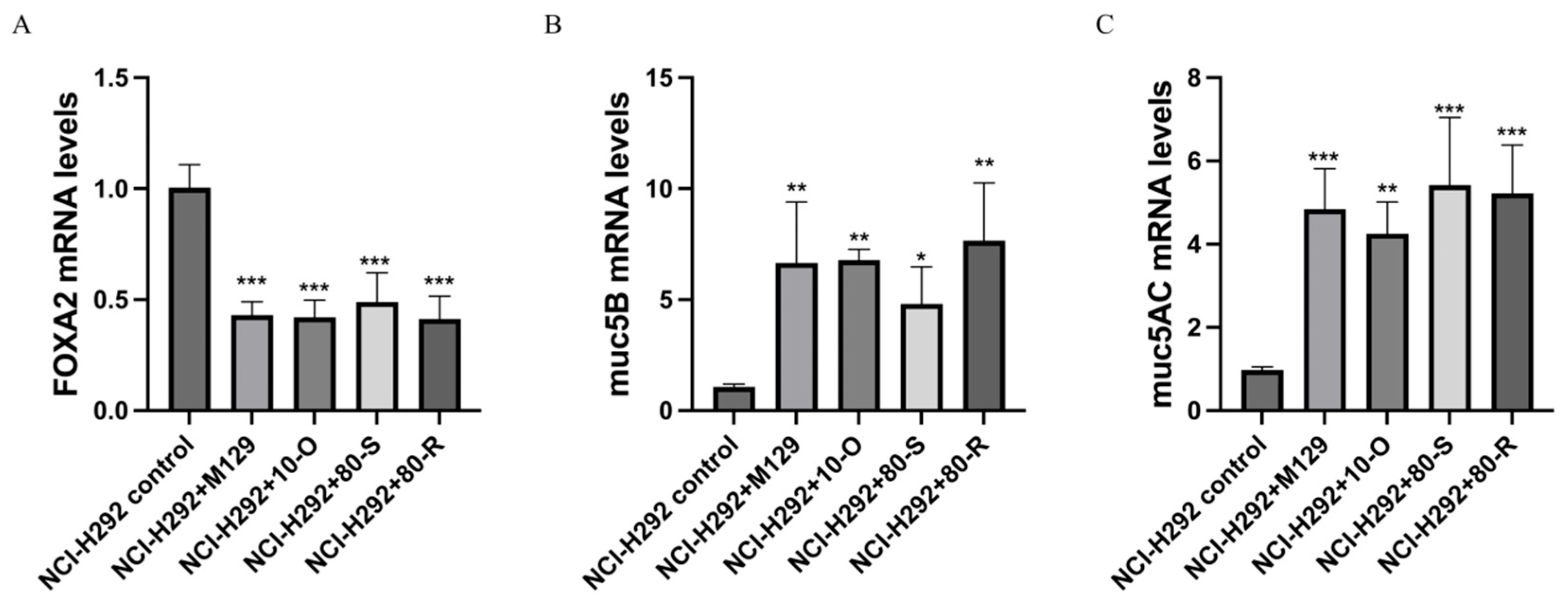
3.6. MPN_080 Does Not Affect the Immune Responses of M. pneumoniae
3.2. Figures, Tables and Schemes

4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Strains | Description and Genotype | Source |
| E.coli Trans1-T1 | Used for plasmid construction | Laboratory stock |
| E.coli BL21-CodonPlus(DE3)-RIL | Used for protein expression | Laboratory stock |
| M. pneumoniae M129 | Wild-type strain | Laboratory stock |
| M.pneumoniae RC267 | Wild-type strain | Laboratory isolation |
| M. pneumoniae 10-O | M. pneumoniae M129+strepII operon | This study |
| M. pneumoniae 80-S | M. pneumoniae M129+ M. pneumoniae M129 MPN_080strepII operon | This study |
| M. pneumoniae 80-R | M. pneumoniae M129+ M. pneumoniae RC267 MPN_080strepII operon | This study |
| Plasmids | ||
| pET-32a(+) | expression vector for E.coli | Laboratory stock |
| pET-32a(+)-R | pET-32a(+) carrying codon optimization MPN_080 from RC267 | This study |
| pMT85 | Suicide vector; contains Tn4001 | Donated by Professor Guo |
| pMT85O | pMT85+strepII operon | This study |
| pMT85S | pMT85+M. pneumoniae M129 MPN_080strepII operon | This study |
| pMT85R | pMT85+M. pneumoniae RC267 MPN_080strepII operon | This study |
References
- Poddighe, D; Demirkaya, E; Sazonov, V; Romano, M. Mycoplasma pneumoniae Infections and Primary Immune Deficiencies. Int J Clin Pract. 2022, 2022, 6343818. [Google Scholar] [CrossRef]
- Ding, G; Zhang, X; Vinturache, A; van Rossum, AMC; Yin, Y; Zhang, Y. Challenges in the treatment of pediatric Mycoplasma pneumoniae pneumonia. Eur J Pediatr. 2024, 183, 3001–11. [Google Scholar] [CrossRef]
- Chih-Cheng, L; Hsueh, CC; Hsu, CK; Tsai, YW; Hsueh, PR. Disease burden and macrolide resistance of Mycoplasma pneumoniae infection in adults in the Asia-Pacific region. Int J Antimicrob Agents 2024, 64, 107205. [Google Scholar] [CrossRef] [PubMed]
- Wang, N; Xu, X; Xiao, L; Liu, Y. Novel mechanisms of macrolide resistance revealed by in vitro selection and genome analysis in Mycoplasma pneumoniae. Front Cell Infect Microbiol. 2023, 13, 1186017. [Google Scholar] [CrossRef]
- Zhao, F; Liu, J; Shi, W; Huang, F; Liu, L; Zhao, S; Zhang, J. Antimicrobial susceptibility and genotyping of Mycoplasma pneumoniae isolates in Beijing, China, from 2014 to 2016. Antimicrob Resist Infect Control 2019, 8, 18. [Google Scholar] [CrossRef]
- Li, SL; Sun, HM; Zhu, BL; Liu, F; Zhao, HQ. Whole Genome Analysis Reveals New Insights into Macrolide Resistance in Mycoplasma pneumoniae. Biomed Environ Sci. 2017, 30, 343–50. [Google Scholar]
- Big Mohammadi, H; Pouladi, I; Zolfaghari, MR; Niakan, M. The Prevalence of 23S rRNA Mutations in ML-Resistant M. pneumoniae Isolates to Clarithromycin in Patients with Respiratory Infections. Rep Biochem Mol Biol. 2020, 9, 156–62. [Google Scholar]
- Waites, KB; Bade, DJ; Bebear, C; Brown, SD; Davidson, MK; Duffy, LB; et al. Methods for Antimicrobial Susceptibility Testing for Human Mycoplasmas; Approved Guideline; Clinical and Laboratory Standards Institute: Wayne (PA), Oct 2011; Report No. M43-A. [Google Scholar]
- Zhao, F; Cao, B; He, LH; Yin, YD; Tao, XX; Song, SF; et al. Evaluation of a new real-time PCR assay for detection of Mycoplasma pneumoniae in clinical specimens. Biomed Environ Sci. 2012, 25(1), 77–81. [Google Scholar] [PubMed]
- Ma, Y; Gu, Y; Zhang, X; Gu, W; Wang, T; Sun, H; et al. High Expression of MUC5AC, MUC5B, and Layilin Plays an Essential Role in Prediction in the Development of Plastic Bronchitis Caused by MPP. Front Microbiol. 2022, 13, 911228. [Google Scholar] [CrossRef]
- Bebear, C; Pereyre, S; Peuchant, O. Mycoplasma pneumoniae: susceptibility and resistance to antibiotics. Future Microbiol. 2011, 6, 423–31. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y; Dou, H; Xu, B; Xu, B; Zhou, W; Wang, H; et al. Macrolide resistance of Mycoplasma pneumoniae in several regions of China from 2013 to 2019. Epidemiol Infect. 2024, 152, e75. [Google Scholar] [CrossRef]
- Kawakami, N; Namkoong, H; Saito, F; Ishizaki, M; Yamazaki, M; Mitamura, K. Epidemiology of macrolide-resistant Mycoplasma pneumoniae by age distribution in Japan. J Infect Chemother. 2021, 27, 45–8. [Google Scholar] [CrossRef]
- Kim, K; Jung, S; Kim, M; Park, S; Yang, HJ; Lee, E. Global Trends in the Proportion of Macrolide-Resistant Mycoplasma pneumoniae Infections: A Systematic Review and Meta-analysis. JAMA Netw Open 2022, 5, e2220949. [Google Scholar] [CrossRef]
- Rothstein, TE; Cunningham, SA; Rieke, RA; Mainella, JM; Mutchler, MM; Patel, R. Macrolide Resistance in Mycoplasma pneumoniae, Midwestern United States, 2014 to 2021. Antimicrob Agents Chemother. 2022, 66, e0243221. [Google Scholar] [CrossRef]
- Xu, M; Li, Y; Shi, Y; Liu, H; Tong, X; Ma, L; et al. Molecular epidemiology of Mycoplasma pneumoniae pneumonia in children, Wuhan, 2020-2022. BMC Microbiol 2024, 24, 23. [Google Scholar] [CrossRef]
- Zhang, M; Wen, W; Ma, Y; Hua, Y; Cao, Q; Lai, H; et al. Multicenter analysis of the Mycoplasma pneumoniae resurgence in China 2023-2024. Sci Rep. 2025, 16, 76. [Google Scholar] [CrossRef]
- Vazquez-Laslop, N; Thum, C; Mankin, AS. Molecular mechanism of drug-dependent ribosome stalling. Mol Cell. 2008, 30, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, X; Jiang, Y; Chen, X; Li, J; Shi, D; Xin, D. Drug resistance mechanisms of Mycoplasma pneumoniae to macrolide antibiotics. Biomed Res Int. 2014, 2014, 320801. [Google Scholar] [PubMed]
- Fyfe, C; Grossman, TH; Kerstein, K; Sutcliffe, J. Resistance to Macrolide Antibiotics in Public Health Pathogens. Cold Spring Harb Perspect Med. 2016, 6(10), a025395. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, KD; Nisbet, R; Stephens, DS. Macrolide efflux in Streptococcus pneumoniae is mediated by a dual efflux pump (mel and mef) and is erythromycin inducible. Antimicrob Agents Chemother. 2005, 49(10), 4203–9. [Google Scholar] [CrossRef]
- Okada, U; Yamashita, E; Neuberger, A; Morimoto, M; van Veen, HW; Murakami, S. Crystal structure of tripartite-type ABC transporter MacB from Acinetobacter baumannii. Nat Commun. 2017, 8, 1336. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, MS; Nakane, D; Toyonaga, T; Kawamoto, A; Kato, T; Namba, K; et al. Refined Mechanism of Mycoplasma mobile Gliding Based on Structure, ATPase Activity, and Sialic Acid Binding of Machinery. mBio 2019, 10. [Google Scholar] [CrossRef]
- Hlavacek, O; Vachova, L. ATP-dependent proteinases in bacteria. Folia Microbiol (Praha) 2002, 47, 203–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.W; L.P., Z; F. H. SPECIFICITY OF ANTI-ATPase antibody against Chlamydia leieri membrane. Chinese Science Bulletin (in Chinese). 1989, 34, 3. [Google Scholar]
- Hopfe, M; Henrich, B. OppA, the substrate-binding subunit of the oligopeptide permease, is the major Ecto-ATPase of Mycoplasma hominis. J Bacteriol. 2004, 186, 1021–928. [Google Scholar] [CrossRef]
- Shi, J; Ma, C; Hao, X; Luo, H; Li, M. [Dickkopf-1 inhibits the secretion of MUC5AC induced by Mycoplasma pneumoniae P1-C in mouse lung epithelial cells]. Sheng Wu Gong Cheng Xue Bao 2023, 39, 248–61. [Google Scholar]

| Primer Name | Sequence |
| Homo GAPDHF | 5‘-TCAAGAAGGTGGTGAAGCAGG-3 |
| Homo GAPDHR | 5‘-TCAAAGGTGGAGGAGTGGGT-3 |
| Homo MUC5ACF | 5‘-CGACCTGTGCTGTGTACCAT-3’, |
| Homo MUC5ACR | 5‘-GTGCAGGGTCACATTCCTCA-3’ |
| Homo MUC5BF | 5‘-AACTGCACCGTGTACCTCTG-3’ |
| Homo MUC5BR | 5‘-TCGTGTTGATGCGGACTTGA-3’ |
| Homo FOXA2F | 5‘-TGTTCGAGAACGGCTGCTAC-3’ |
| Homo FOXA2R | 5‘-GAGTGAGGCGACTCGGTG-3’ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



