Submitted:
15 February 2026
Posted:
27 February 2026
You are already at the latest version
Abstract
Parvoviruses are small, non-enveloped DNA viruses, several of which have been adapted as vectors for gene therapy. Adeno-associated virus (AAV) is clinically established but constrained by limited genome capacity and pre-existing immunity. Human bocaviruses (HBoVs) possess larger packaging potential and airway tropism, motivating exploration of AAV-HBoV hybrid architectures. We modeled a chimeric construct (AAV-HB3) in which the α-helix and βH/βI strand of AAVGo.1 were replaced with the corresponding regions from HBoV3. AlphaFold2 predictions (pLDDT > 80, pTM > 0.75) confirmed reten-tion of the β-barrel scaffold, and RoseTTAFold2 refinement produced energetically stable conformations. 100 ns molecular-dynamics simulations showed distinct dynamic profiles: the AAVGo.1 VP1 control remained conformationally rigid (RMSD ≈ 0.03 nm), whereas AAV-HB3 exhibited increased flexibility at VR-VIII, VR-IV, and the HI loop, with loop dis-placements of ~43–54 Å localized near the three-fold and five-fold symmetry axes. These structure-based analyses define how cross-genus substitutions redistribute local flexibility within the parvoviral capsid, providing a predictive framework for engineering next-generation vectors with assessing structural tolerance relevant to capsid.
Keywords:
adeno-associated virus
; parvoviridae
; AlphaFold2
; RoseTTAFold2
; genome packaging
; chimeric capsid
1. Introduction
Parvoviruses are a family of small, non-enveloped viruses called Parvoviridae, characterized by linear, single-stranded DNA genomes of 4-6 kb, flanked by hairpin structures necessary for replication. The icosahedral capsids are 20-30 nm in diameter with a T=1 assembly and consist of 60 protein subunits [1]. Members of this family infect a wide range of hosts and can replicate independently or require helper viruses for productive infection [1]. The family is divided into three subfamilies: Parvovirinae, Densovirinae, and the most recently discovered family, Hamaparvovirinae, as shown in Figure 1 [1,2]. While some parvoviruses cause significant disease, others are non-pathogenic and have been adapted for biotechnological uses, especially in gene therapy [1,3].
Adeno-associated virus (AAV), a member of the genus Dependoparvovirus within the Parvovirinae family, is a replication-defective virus that is known for its ability to infect humans and other primates without causing disease [4,5]. Characterized by a diameter of 20-26 nm and a linear ssDNA genome of about 4.7 kb, the AAV genome is flanked by inverted terminal repeats (ITRs) that are essential for replication and packaging. The genome encodes two main open reading frames called a rep, which produces replication proteins (Rep78, Rep68, Rep52, and Rep40), and the cap, which encodes capsid proteins (VP1, VP2, VP3) forming the icosahedral capsid in a 1:1:10 ratio, along with an assembly-activating protein (AAP) that provides scaffolding for capsid assembly [4,5]. AAV’s ~4.7 kb packaging limit is a constraint in vector design and motivates capsid engineering strategies. AAVs exhibit a seroprevalence of 35-80% in humans, reflecting its non-pathogenic nature and suitability for biomedical applications [4,6].
This virus was first identified in the mid-1960s as a contaminant in adenovirus preparations during studies by researchers like Bob Atchison at the University of Pittsburgh and Wallace Rowe at the National Institutes of Health [5,7]. Initially classified as a dependoparvovirus that needs helper viruses for replication, serological studies verified its widespread presence in humans without causing illness [7]. The AAV2 serotype was cloned and sequenced in the 1980s, which opened the door for genetic analysis and vector development [5].
As a replication-deficient virus, AAV depends on helper viruses (i.e., adenovirus or herpesvirus) for a productive lytic cycle. Without helper virus function, AAV becomes latent, primarily persisting as episomal DNA or, less frequently, through specific site integration into the host genome at chromosome 19q13.4 in humans, with integration occurring in only 0.1% to 0.5% of infectious particles. Post-translational modifications of the capsid, including acetylation, methylation, phosphorylation, and deamidation, impact transduction efficiency and vector performance [4,5].
Over 13 AAV serotypes and more than 100 variants have been identified, with capsid protein differences determining tissue targeting and transduction efficiency [5,8]. For example, AAV2 has broad tropism for skeletal muscle, neurons, and hepatocytes through binding to heparan sulfate proteoglycan; AAV8 targets liver and kidney cells; AAV9 crosses the blood-brain barrier, allowing efficient delivery to the central nervous system (CNS) and heart; and AAV1/6 are effective for airway epithelia via sialic acid interactions [5,8]. Engineered variants, such as AAVDJ, a hybrid of eight serotypes, or AAV-PHP.B, created through directed evolution, improve immune evasion and targeted transduction, with AAV9 showing promise for widespread neuronal and non-neuronal cell targeting [8].
Animal-derived AAV variants, such as AAVGo.1, isolated from goats, expand the diversity of capsids available for engineering [9,10]. AAVGo.1 shares structural similarities with human AAV serotypes like AAV5, including an icosahedral capsid and a ~4.7 kb ssDNA genome. Still, its caprine origin provides unique surface topologies that may improve cross-species permissivity and transduction efficiency in specific tissues [10]. Like other AAVs, AAVGo.1 is replication-defective, requiring helper viruses for productive infection, and has been explored for gene therapy applications due to its potential for reduced immunogenicity and enhanced liver tropism in preclinical models [9,10]. (Figure 2b)
Another example, recombinant AAV (rAAV) vectors remain extra chromosomal in non-dividing cells, supporting long-term expression with minimal immunogenicity [4,5]. rAAV allows for gene replacement, silencing via microRNAs, and editing with CRISPR-Cas9, with over 250 clinical trials conducted by 2019 targeting monogenic disorders [5,8].
Clinically, the first successful long-term application was in inherited retinal disease, where subretinal delivery of an AAV2 vector restored RPE65 function in Leber congenital amaurosis, leading to the U.S. Food and Drug Administration (FDA) approval of Luxturna in 2017 [11]. This approval set a regulatory precedent for systemic and tissue-specific AAV gene therapy. Since then, additional AAV products have been approved for various monogenic disorders: Zolgensma; AAV9 for spinal muscular atrophy in 2019; Hemgenix; AAV5 for hemophilia B in 2022; Roctavian; AAV5 for hemophilia A in 2023; Elevidys; AAVrh74 for Duchenne muscular dystrophy, FDA accelerated approval in 2023, with full approval for ambulatory patients in 2024; and Beqvez; AAV-Rh74var for hemophilia B in 2024. More recently, Kebilidi, a different AAV-based construct, received approval in 2024, highlighting the expanding diversity of vector backbones in late-stage development [5,12].
Earlier, Glybera; AAV1 was approved by the European Medicines Agency in 2012 for lipoprotein lipase deficiency, though it was later withdrawn due to limited commercial viability rather than clinical failure [13,14]. Collectively, these approvals and ongoing trials highlight the translational maturity of AAV gene transfer and its potential to deliver long-term therapeutic benefits in previously intractable genetic diseases.
However, challenges remain, such as limited packaging capacity, pre-existing neutralizing antibodies in 30-60% of the population, potential genotoxicity at high doses, and scalability issues in manufacturing [15,16]. Immune responses, including hepatotoxicity and neurotoxicity, have been observed at high doses, prompting strategies such as capsid engineering and self-complementary designs to enhance efficacy and safety [17].
Human bocaparvoviruses (Bocaparvovirus, Parvovirinae) package 5.3 kb linear ssDNA genomes that encode NS1, NP1, and capsid proteins VP1–VP3 [18]. (Figure 2a) HBoV1 is associated with respiratory tract infections, while HBoV2–4 is more often linked to gastrointestinal disease [19,20]. HBoV1 exhibits efficient replication in airway epithelia and can package genomes up to 5.5 kb, indicating its potential as a vector system [18,21,22]. Recombinant methods have combined AAV genomes with HBoV1 capsids to produce rAAV2/HBoV1 chimeric particles. These constructs effectively package AAV genomes and demonstrate increased transduction in polarized airway epithelial cells compared to AAV2 and AAV1 [23]. Furthermore, platforms have been developed for high-yield production of rAAV/HBoV vectors that do not rely on bocavirus nonstructural proteins [21,24]. This established cross-genus compatibility motivates a mechanistic question about which capsid regions are permissive to substitution, and which are structurally constrained.
Computational methods have become essential for designing and evaluating AAV vectors. They allow for systematic prediction of how capsid modifications affect structure and function, supporting rational design strategies to overcome limitations in packaging, stability, tropism, and immune recognition [25]. One of the earliest computational approaches was computer-aided design (CADD), which used structural modeling and docking to modify AAV-receptor and AAV-antibody interactions. Initial efforts involved introducing point mutations and peptide insertions into capsid loops to change tissue tropism or decrease recognition by neutralizing antibodies [26,27]. More advanced CADD tools, like CapBuild, now support structure-based prediction of AAV assembly and receptor interaction [28]. Meanwhile,in silico frameworks have been developed for global liver de-targeting, along with the prediction of tissue-specific performance [29].
Molecular dynamics (MD) simulations are a well-established computational method for studying viral proteins and have been used to examine AAV capsid stability, conformational flexibility, and interactions with biological environments. Atomistic MD models assembled capsids under near-physiological conditions, offering insights into local flexibility of variable regions, stability of inter-subunit interfaces, and potential conformational changes relevant to genome packaging and uncoating [30,31]. Overall, these MD simulations enhance static CADD models by capturing conformational flexibility, stability, and biophysical interactions that are beyond the reach of docking alone.
Building on these early computational methods, rational design applies detailed structural knowledge to introduce targeted modifications with defined mechanistic objectives [32,33] such as engineered variants including capsids with altered glycosylation sites or PLA2-like motif mutations, which modified antigenic properties [34,35,36], and intra-articular AAV mutants designed to enhance joint-specific gene delivery in vivo [37]. Directed evolution, in contrast, employs iterative mutagenesis and selection to identify capsid variants with desirable traits, often guided by computational modeling to prioritize sequences for library design [38,39,40].
Machine learning (ML) has been increasingly used in AAV capsid engineering, particularly to predict relationships between sequence, structure, and function. [41,42]. Meanwhile, deep generative models have been developed to design synthetic AAV sequences with a high likelihood of assembly and functional performance, thereby expanding the sequence space beyond natural variants [43]. Other ML pipelines have sped up directed evolution by analyzing next-generation sequencing data from variant libraries to find common sequence motifs linked to tissue-specific targeting [44].
Accurate structure prediction has become essential for interpreting and refining ML-predicted capsid variants. AlphaFold2 (AF2) in 2021 introduced an end-to-end neural network framework combining multiple sequence alignments, template search, and transformer-based Evoformer modules to iteratively refine residue-level relationships before generating atomic coordinates [45]. AF2 has been applied to evaluate the structural impact of amino acid substitutions and insertions in AAV capsids, providing mechanistic insights into ML-prioritized variants [46]. AlphaFold3 (AF3) extends this capability by employing a diffusion-based generative model [47]. This enables the evaluation of chimeric constructs, such as AAV hybrids, and supports predictions of genome encapsidation and receptor engagement. RoseTTAFold2 (RF2) provides a complementary refinement framework, using a three-track network that integrates sequence, distance, and coordinate data [48]. RF2 has been applied to optimize predicted AAV structures by refining flexible loops, testing N-terminal truncations, and assessing strand rearrangements relevant to assembly [46].
Collectively, these computational strategies provide a framework for rational AAV engineering and are increasingly coupled with experimental approaches to advance the development of next-generation vectors [25,46]. In this study, AlphaFold2-based structural prediction and molecular dynamics simulations were applied to evaluate whether the substitution of the α-helix and βH/βI strand in AAVGo.1 capsid with corresponding regions from Human Bocavirus 3 (HBoV3) preserves monomeric integrity and supports assembly-relevant conformations. Previous studies have shown that AAV genomes can be packaged into bocavirus capsids; however, the reciprocal question of whether bocavirus-derived structural elements can be introduced into AAV to extend its packaging potential remains unexplored. Given that HBoVs accommodate genomes up to ~5.5 kb compared with the ~4.7 kb limit of AAV, this work aims to determine whether such substitutions maintain structural stability while providing a strategy to overcome the packaging constraints of AAV.
2. Materials and Methods
Sequence Alignment and Chimeric Construct Design
The amino acid sequences of AAVGo.1 and HBoV3 VP proteins were obtained from Uniprot. Alignments were performed using SnapGene v8.1.0 to identify the α-helix and βH/βI strand regions targeted for substitution [49]. Homologous regions from HBoV3 were identified by pairwise alignment and substituted into the AAVGo.1 VP3 backbone to generate a chimeric construct. A wild-type AAVGo.1 VP1 control was also prepared to evaluate prediction accuracy relative to experimental PDB structures for further analysis of the N-terminal. The chimera was modeled as a VP3 protein to match available crystallographic data from the Protein Data Bank (PDB); 7TI4 [50].
Structural Prediction and Refinement
Structural predictions were performed using AF2, which was run through ChimeraX v1.7 [45,51]. Both the wild-type AAVGo.1 VP1 control and the AAVGo.1-HBoV3 (AAV-HB3) VP3 chimera were modeled using AF2 parameters. The multiple sequence alignments (MSAs) were generated against the UniRef90, MGnify, and PDB70 databases. For each sequence, five models were generated and ranked by predicted Local Distance Difference Test (pLDDT) scores. Additional model quality metrics included predicted Template Modeling score (pTM) and Predicted Aligned Error (PAE) matrices. For the AAVGo.1 VP1 control and AAV-HB3, predicted structures were compared with the experimentally resolved PDB structure to validate AF2 accuracy. Root mean standard deviation (RMSD) values between predicted and experimental structures were calculated in UCSF ChimeraX v1.7 [51,52,53]. The top-ranked AF2 models were refined using RF2 [48]. Refinement was conducted to reduce local backbone distortions and optimize side-chain packing. Output models were scored using the Rosetta all-atom energy function. Models were visually inspected for steric clashes and evaluated for loop stability before MD simulations.
The 60-mer capsid assemblies were generated in UCSF ChimeraX by applying established AAV icosahedral symmetry operations derived from cryo-EM capsid structures to the refined static monomer models. In parallel, experimentally resolved AAV capsid structures obtained from the Protein Data Bank were visualized directly in ChimeraX to examine native capsid surface topology and symmetry-related features. Monomer-level molecular dynamics simulations were used to identify regions of altered local flexibility, including VR-IV, VR-VIII, and the HI loop. These regions were then mapped onto their corresponding symmetry-related positions in the assembled capsid to visualize how localized conformational flexibility propagates across the 60-subunit capsid topology. The resulting models represent symmetry-based capsid assemblies informed by monomer-level dynamic behavior and are used to contextualize interface-adjacent flexibility within the assembled architecture.
Molecular Dynamics (MD) Simulations
Refined structures were prepared for MD simulations using GROMACS v2022.4 with the CHARMM27 force field [54,55], which was well-validated and widely used at the time the simulations were performed. Each protein was placed in a cubic box with at least 1.0 nm between protein atoms and the box edge. The systems were solvated with TIP3P water molecules and neutralized with counterions Na+ and Cl− to a physiological concentration of 0.15 M. Protonation states of ionizable residues were assigned assuming physiological pH (~7.0), with histidine protonation states set using standard GROMACS conventions unless otherwise indicated.
Energy minimization was performed using the steepest descent algorithm until the maximum force was <1000 kJ/mol/nm. Equilibration consisted of 500 ps under NVT conditions, using a Berendsen thermostat at 300 K, followed by 500 ps under NPT conditions, with a Parrinello–Rahman barostat at 1 bar. The Berendsen thermostat and Parrinello-Rahman barostat were applied using standard coupling schemes implemented in GROMACS [56,57]. LINCS constraints were applied to all hydrogen-containing bonds, using the LINCS algorithm [58], and long-range electrostatics were calculated using the Particle Mesh Ewald (PME) method with reciprocal-space treatment of long-range interactions [59,60] with a cutoff of 1.0 nm for Coulomb and van der Waals interactions. Production simulations were performed at 300 K for 100 ns with a 2 fs time step under periodic boundary conditions. Snapshots were saved every 10 ps for analysis. A single production trajectory was generated for each system. The 100 ns simulation length was selected to support a comparative assessment of monomer-level stability and local flexibility metrics, with convergence expectations interpreted as observable-dependent [61,62,63].
Trajectory Analysis
Cryo-EM structures of AAVGo.1 predominantly resolve the VP3 capsid protein; therefore, VP3-based structural comparison was used to ensure consistency with experimentally observed density while enabling direct comparison with the VP3-modeled chimera. VR and loop segments were defined based on established AAV capsid annotations, with residue numbering reported separately for AAVGo.1 VP1 and the AAV-HB3 VP3 chimera to account for differences in construct length and domain composition [64,65]. For the AAVGo.1 VP1 control, the VR-IV corresponds to residues 441–454, VR-VIII corresponds to residues 570–585, and the HI loop corresponds to residues 649–657. For the AAV-HB3 chimera, modeled as VP3, VR-IV corresponds to residues 249–262, VR-VIII corresponds to residues 378–393, and the HI loop corresponds to residues 457–465 (Figure 3).
Trajectory analysis was performed using built-in GROMACS utilities [54]. System stability was confirmed by monitoring temperature and potential energy across the production runs. Structural deviations were evaluated by calculating RMSD relative to the initial refined structure. RMSD calculations were performed on backbone Cα atoms after least-squares fitting to remove global translation and rotation. Global structural compactness was assessed by calculating the radius of gyration (Rg). Rg computed for each frame of the production trajectory after removal of translation and rotation, providing a measure of large conformational expansion and compaction at the monomeric level. Root mean square fluctuation (RMSF) was used to assess residue-level flexibility. RMSF was computed for Cα atoms over the production trajectory to quantify residue-level fluctuations. Principal component analysis (PCA) was conducted on backbone atomic coordinates to identify dominant modes of motion, with projections plotted along PC1, PC2, and PC3 to compare conformational sampling between AAVGo.1 VP1 and the AAV-HB3 chimera. PCA was performed on the covariance matrix of backbone Cα positional fluctuations after alignment to the reference structure.
Redistribution of mechanical flexibility was quantified as region-specific changes in RMSF magnitude and dominant PCA mode contributions, indicating a shift in the localization of conformational variability toward symmetry-interface-adjacent regions in the AAV-HB3 chimera relative to AAVGo.1. Loop displacements were quantified using Cα-Cα distance-based measurements for representative residues within VR-VIII, VR-IV, and the HI loop relative to their initial refined positions, and distances were monitored across the trajectory to distinguish sustained conformational shifts from transient excursions. Structural overlays and motion trajectories were examined to evaluate displacement in variable regions, particularly VR-IV, VR-VIII, and the HI loop. ChimeraX was used for structural visualization and comparison of the α-helix and βH/βI regions across models, as well as for alignment with the experimental PDB structure of AAVGo.1 [51]. All interpretations are restricted to comparative local flexibility and structural feasibility within the sampled conformational ensemble.
3. Results
Predicted Structural Features and Model Validation
AlphaFold2 generated models for both the wild-type AAVGo.1 VP1 control and the AAV-HB3 VP3 chimera. The AAVGo.1 VP1 prediction aligned with the experimentally determined structure (PDB ID: 7TI4), and both the control and chimeric models produced RMSD values < 0.8 when compared to the reference structures, supporting the reliability of the predicted folds. Model quality assessments showed mean pLDDT values exceeding 80 across the conserved β-barrel core, with lower values observed in surface loops and termini. pTM scores were > 0.75 for both constructs, indicating reliable global structural accuracy. PAE maps displayed low errors within structured domains and localized uncertainty in flexible loops. These results confirmed preservation of the β-barrel architecture while highlighting localized differences in the substituted α-helix and βH/βI strand of the chimera.
The top-ranked AlphaFold2 models for both constructs were further refined with RoseTTAFold2. Refinement produced consistent scores across the AAVGo.1 VP1 control and the AAV-HB3 VP3 chimera, with modest corrections to loop regions and removal of steric clashes. Rosetta energy scores remained stable before and after refinement, indicating that the backbone and global fold were not disrupted. Comparable score ranges across constructs indicate that refinement preserved backbone geometry and global fold integrity, supporting the suitability of the refined models for MD simulations. These refined monomer structures were additionally used to generate symmetry-expanded 60-mer AAV capsid assemblies in UCSF ChimeraX by applying established icosahedral symmetry operations derived from cryo-EM AAV capsid structures. Accordingly, Figure 4 shows the resulting 60-mer capsid models constructed from refined static monomers to visualize capsid topology and symmetry, contextualizing regions of altered local flexibility identified in the monomer-level simulations.
Stability Profiles from Molecular Dynamics
Both AAVGo.1 VP1 and the AAV-HB3 chimera maintained stable thermodynamic behavior over the sampled 100 ns trajectories (Figure 5a). The temperature remained constant at 298–301 K across trajectories for both systems, indicating stable thermostat performance without large fluctuations. Potential energy values stabilized early in the production runs and remained steady throughout, suggesting that the systems reached stable simulation conditions suitable for comparative analysis (Figure 5b)
RMSD trajectories revealed distinct stability patterns for the two systems. AAVGo.1 VP1 showed minimal backbone deviations, stabilizing rapidly within the first 10 ns at an average RMSD of ~0.03 nm and remaining consistent throughout the 100 ns trajectory. This behavior is consistent with a relatively rigid monomeric conformation over the sampled timescale, with limited structural drift. In contrast, AAV-HB3 displayed an initial rise in RMSD during the first 20 ns, reaching values greater than 0.7 nm before plateauing at elevated values for the remainder of the trajectory. This larger deviation indicates increased relative conformational flexibility, particularly in engineered secondary structure elements. The initial rise in RMSD during the first ~20 ns reflects structural relaxation from the refined starting model rather than continued drift, after which the trajectory stabilizes around a consistent mean value, indicating equilibration for local flexibility analysis. Together, these data indicate that while both systems exhibited stable behavior under simulation conditions, VP1 retained a compact fold, whereas AAV-HB3 sampled a broader range of conformations within the limits of the trajectory (Figure 6a).
RMSF profiles showed elevated fluctuations at VR-VIII, VR-IV, and the HI loop, as well as at the engineered α-helix and βH/βI regions. These regions correspond to pronounced RMSF peaks in the AAV-HB3 chimera that are absent or substantially attenuated in the AAVGo.1 VP1 control, indicating a redistribution of residue-level flexibility toward specific surface-exposed loop regions rather than a uniform increase in global mobility (Figure 6b). These flexible segments underwent measurable outward displacements during the trajectory, with loop-loop separations expanding to as much as ~43-54 Å in late trajectory frames. The RMSF peaks spatially coincide with loop regions highlighted in structural overlays, demonstrating that the observed positional deviations arise from sustained local fluctuations rather than transient excursions. These displacements reflect localized flexibility within surface-exposed loop regions adjacent to the 3-fold and 5-fold symmetry axes, rather than large-scale global rearrangements of the capsid protein fold. Figure 7 provides a structural mapping of these RMSF-enriched regions, illustrating how enhanced residue-level fluctuations manifest as loop expansion and surface remodeling in the AAV-HB3 chimera.
Global Structural compactness was assessed using Rg analysis over the 100 ns trajectory. (Figure 8) The AAV-HB3 chimera exhibited consistently higher Rg values than the AAVGo.1 VP1 control, indicating an expanded monomeric conformational ensemble. Temporal fluctuations in Rg were correlated between the two systems, consistent with preservation of the underlying fold. However, the hybrid maintained a higher mean Rg and a wider range of values throughout the trajectory. These results indicate that the increased residue-level flexibility observed in VR-VIII, VR-IV, the HI loop, and engineered secondary structure elements contribute to global conformational breathing.
Comparative Structural Dynamics
Principal component analysis supported these observations, with dominant eigenvectors describing motions concentrated in the surface loops near the 3-fold and 5-fold symmetry interfaces. PCA was used here as a descriptive tool to compare dominant modes of motion sampled within each trajectory, and the resulting projections highlight relative differences in loop mobility. Structural overlays confirmed progressive deviations of these regions relative to the starting structure over the sampled timescale. (Figure 9)
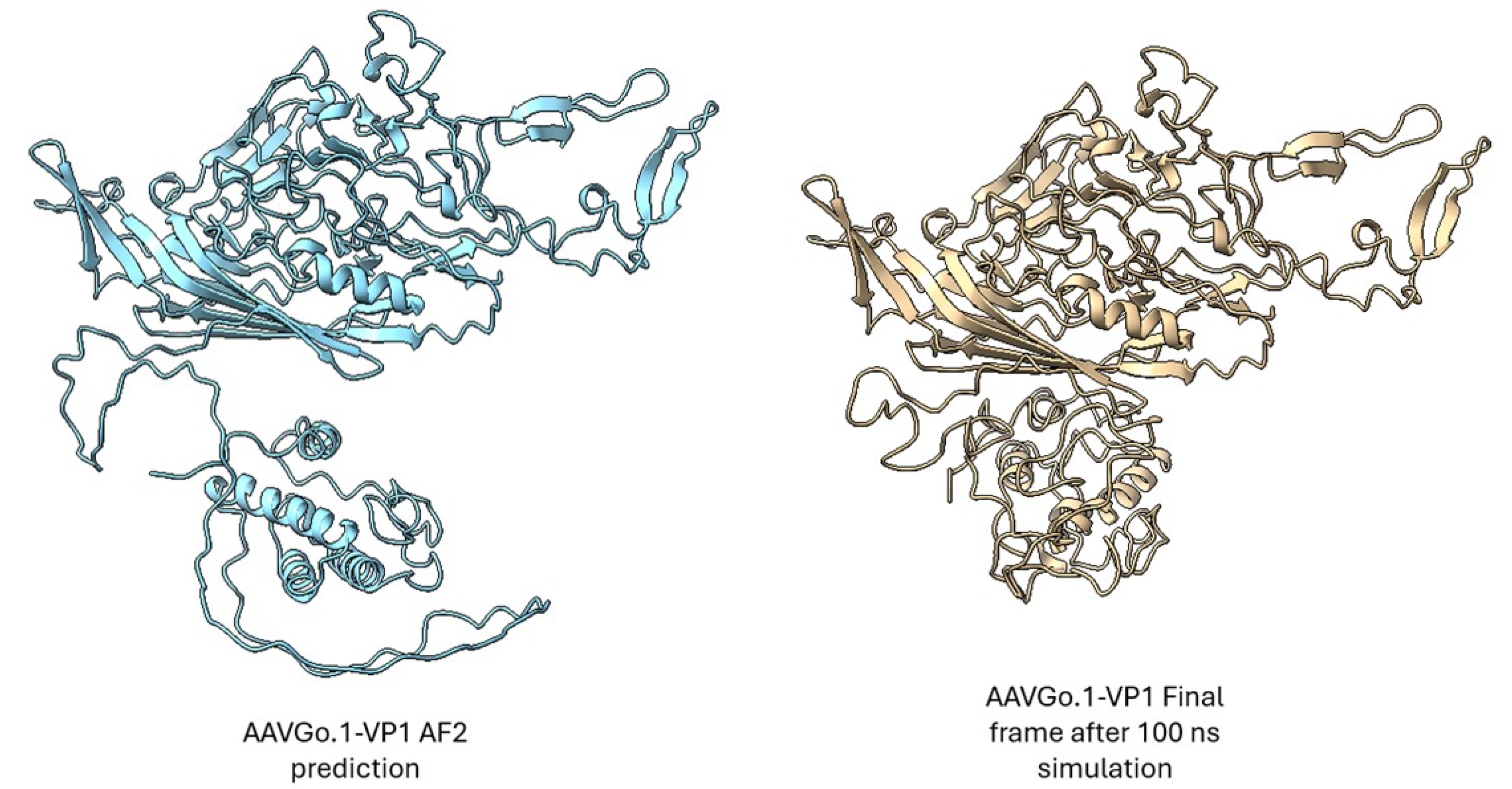
In comparison, AAVGo.1 VP1 showed uniformly low RMSF values across the monomer, with no pronounced peaks in variable regions, indicating comparatively preserved structural rigidity over the trajectory. PCA revealed limited conformational sampling relative to AAV-HB3, consistent with the low RMSD trace. A notable feature of VP1 was the N-terminal region folding inward toward the β-sheet core, a movement observed during the trajectory and aligned with electron microscopy reconstructions of genome-containing AAVs (AAV8, AAV11, AAV12, and AAV13), where VP1 N-terminal density contributes to an internal membrane-like structure [66]. (Figure 10) This observation suggests consistency between the sampled VP1 conformations and experimentally reported structural features. At the same time, AAV-HB3 introduces localized flexibility at VR-VIII, VR-IV, and the HI loop alters the relative dynamic profile of the monomer compared to VP1.
4. Discussion
Previous studies have demonstrated that incorporation of AAV genomes into bocavirus capsids can support efficient packaging and enhance transduction. For example, rAAV2/HBoV1 chimeras have been shown to package AAV genomes and increase transduction efficiency in polarized airway epithelial cells compared to parental AAV2 or AAV1 [21,23,24]. These findings established that bocavirus-derived capsid elements can accommodate AAV genomes and expand functional capacity, although they also raised concerns that incorporation of heterologous sequences may introduce structural liabilities [18,22]. In the present study, we extend this framework by asking a complementary mechanistic question: whether selected bocavirus-derived structural motifs can be introduced into an AAV capsid scaffold without compromising structural stability. Unlike previously described rAAV2/HBoV1 constructs, which rely on bocavirus capsids to package AAV genomes, our approach focuses on reciprocal HBoV3 motif substitution within a canonical AAV architecture, using AAVGo.1 as the scaffold. This distinction is critical, as it isolates the structural tolerance of conserved capsid elements rather than evaluating genome capsid compatibility alone.
Comparative simulations revealed that the AAVGo.1 VP1 control maintains a rigid fold with limited conformational sampling over the sampled timescale. Notably, the N-terminal domain of VP1 displayed an inward movement toward the β-sheet core, consistent with cryo-EM maps of genome-containing AAV particles [66]. This observation supports the role of VP1 in forming internal membrane-like density associated with genome packaging. By contrast, the AAV-HB3 chimera exhibited elevated fluctuations at VR-VIII, VR-IV, and the HI loop, as well as outward displacements in the engineered α-helix and βH/βI regions. These modeled displacements represent predicted flexibility at the 3-fold and 5-fold symmetry interfaces, reflecting localized motion in the computational trajectories. These observations show redistribution of mechanical flexibility across symmetry-related interfaces, defining structural determinants that may influence higher-order assembly and surface topology. Here, redistribution of mechanical flexibility refers to a shift in the relative magnitude and spatial localization of conformational variability, quantified by region-specific RMSF values and dominant principal component mode contributions, rather than changes in absolute mechanical stiffness or inter-subunit forces. Radius of gyration analysis provides a global measure of these effects. The AAV-HB3 chimera maintained higher Rg values than the AAVGo.1 VP1 control, indicating an expanded monomeric conformational ensemble. Correlated temporal fluctuations between systems suggest preservation of the underlying fold, while the higher mean Rg of the hybrid reflects propagation of localized loop flexibility into large-scale conformational breathing.
Limitations and relevance to 60-mer assembly. Although the AAV capsid forms a 60-mer icosahedral assembly, the present simulations were performed on a single capsid protein monomer to enable controlled, motif-level comparison between AAVGo.1 and the AAV-HB3 chimera. As a result, these trajectories do not directly model inter-subunit contacts, cooperative stabilization, or assembly energetics of the full capsid, and packaging capacity cannot be inferred quantitatively from monomer simulations alone. Nevertheless, VR-IV, VR-VIII, and the HI loop are surface-exposed elements positioned at or near symmetry-related interfaces in the assembled capsid, where they contribute to intersubunit complementarity and local curvature. Increased flexibility in these regions is therefore expected to propagate to the 60-mer context by altering the conformational tolerance and geometric matching required for stable interface formation. Consistent with this interpretation, the present results are limited to comparative local flexibility and structural feasibility and are intended to identify motif substitutions likely to perturb interface-adjacent dynamics that may influence downstream assembly and packaging behavior.
This study illustrates how computational modeling can be used to interrogate the structural consequences of capsid engineering strategies prior to experimental implementation. AlphaFold2 [45] and RoseTTAFold2 [48] accurately reproduced the conserved β-barrel scaffold while identifying regions of increased uncertainty in flexible surface elements, guiding targeted molecular dynamics simulations. At the time these simulations were performed, CHARMM27 represented a well-established and widely used force field for protein and viral capsid molecular dynamics, and its consistent application across all systems enabled direct comparative analysis of local flexibility without introducing force-field-dependent bias. Future studies will extend this framework using updated force fields such as CHARMM36m to further refine characterization of backbone and side-chain dynamics in flexible surface loops [67]. MD analysis quantified backbone stability, residue-level fluctuations, and correlated motions, enabling discrimination between globally preserved fold architecture and localized dynamic perturbations [30,31]. Rather than replacing experimental validation, these approaches provide a complementary framework for identifying design liabilities and prioritizing motifs for further testing. Importantly, the molecular dynamics results are interpreted as a comparative assessment of local flexibility and structural feasibility.
6. Conclusion
In summary, this study provides a computational feasibility assessment of cross-genus capsid engineering by examining the structural tolerance of conserved motif substitution within an AAV framework. The AAV-HB3 construct preserves the canonical parvoviral β-barrel scaffold but exhibits increased localized flexibility at the 3-fold and 5-fold symmetry interfaces following substitution of bocavirus-derived α-helix and βH/βI-associated regions.
These results indicate that conserved, scaffold-adjacent motifs are less permissive to cross-genus exchange than terminal or peripheral regions previously explored in chimeric designs. Rather than directly predicting expanded genome accommodation, the observed dynamics suggest that such substitutions may influence capsid assembly competence through altered symmetry-axis flexibility.
Computational approaches integrating AI-based structure prediction and molecular dynamics simulations, therefore, provide a practical means to screen motif-level substitutions, identify scaffold-sensitive regions, and prioritize capsid designs for experimental evaluation. As hybrid and cross-genus capsid architecture continue to expand, such workflows offer a rational strategy for narrowing design space by identifying modifications most likely to preserve structural integrity.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com. Figure S1: Taxonomic organization of the family Parvoviridae; Figure S2: Comparative surface architecture of HBoV3 and AAVGo.1 capsids; Figure S3: Annotated amino acid sequences of AAV-HB3 VP3 chimera and AAVGo.1 VP1 control; Figure S4: Structural comparison of chimeric and control AAV capsids; Figure S5: Thermodynamic stability profiles during molecular dynamics simulations; Figure S6: RMSD and RMSF analyses of AAV-HB3 and AAVGo.1 VP1; Figure S7: Loop displacement overlays during MD simulation; Figure S8: Radius of gyration profiles; Figure S9: Principal component analysis (PCA) of MD trajectories; Figure S10: Structural comparison of AAVGo.1 VP1 before and after simulation.
Author Contributions
Conceptualization, H.A.N. and X.S.W.; methodology, H.A.N.; formal analysis, H.A.N.; investigation, H.A.N.; writing-original draft preparation, H.A.N.; writing-review and editing, H.A.N. and X.S.W.; supervision, X.S.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The input and output files for the AlphaFold2 prediction and RoseTTAFold2 refinement, along with the parameter files, input .gro files, output .pdb files, and md.mdp file used in the molecular dynamics simulations, are available in the Supporting Information. Peer-reviewer link is available through Zenodo DOI:10.5281/zenodo.18452491 (Reviewer link: https://zenodo.org/records/18452491?preview=1&token=eyJhbGciOiJIUzUxMiJ9.eyJpZCI6ImY3MjNkMGZkLWY1OTYtNDE1OS1hYTc3LTA4ZjcxNTk3MGY2MCIsImRhdGEiOnt9LCJyYW5kb20iOiIzZGMwZmIxNTI0ODZiMWI5MTczMDdmNGFmMTkxN2E0MyJ9.9ATg4xbfzvpB2i3-goVtsTj3A58QqW3zR882vjHnA8UltcUNafvDLKHO_9yNkhDCpAJo-wHFs47-bUbnONe1Fw).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; et al. ICTV virus taxonomy profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef]
- Pénzes, J.J.; Söderlund-Venermo, M.; Canuti, M.; Eis-Hübinger, A.M.; Hughes, J.; Cotmore, S.F.; et al. Reorganizing the family Parvoviridae: A revised taxonomy independent of the canonical approach based on host association. Arch. Virol. 2020, 165, 2133–2146. [Google Scholar] [CrossRef]
- Mohammadi, M. HBoV-1: Virus structure, genomic features, life cycle, pathogenesis, epidemiology, diagnosis and clinical manifestations. Front. Cell. Infect. Microbiol. 2023, 13, 1198127. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Li, T.; Liu, L.; Che, X.; Li, X.; Liu, C.; et al. Adeno-associated virus therapies: Pioneering solutions for human genetic diseases. Cytokine Growth Factor Rev. 2024, 80, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-H.; Gessler, D.J.; Zhan, W.; Gallagher, T.L.; Gao, G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct. Target. Ther. 2024, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Araujo, N.M. A dual role for adeno-associated virus in human health. Virol. J. 2023, 20, 228. [Google Scholar] [CrossRef]
- Hastie, E.; Samulski, R.J. Adeno-associated virus at 50: A golden anniversary of discovery, research, and gene therapy success—A personal perspective. Hum. Gene Ther. 2015, 26, 257–265. [Google Scholar] [CrossRef]
- Issa, S.S.; Shaimardanova, A.A.; Solovyeva, V.V.; Rizvanov, A.A. Various AAV serotypes and their applications in gene therapy: An overview. Cells 2023, 12, 785. [Google Scholar] [CrossRef]
- Arbetman, A.E.; Lochrie, M.; Zhou, S.; Wellman, J.; Scallan, C.; Doroudchi, M.M.; et al. Novel caprine adeno-associated virus (AAV-Go.1) capsid (AAV-Go.1) is closely related to the primate AAV-5 and has unique tropism and neutralization properties. J. Virol. 2005, 79, 15238–15245. [Google Scholar] [CrossRef] [PubMed]
- Large, E.E.; Silveria, M.A.; Weerakoon, O.; White, T.A.; Chapman, M.S. Cross-species permissivity: Structure of a goat adeno-associated virus and its complex with the human receptor AAVR. J. Virol. 2022, 96, e01484-22. [Google Scholar] [CrossRef]
- Irigoyen, C.; Amenabar Alonso, A.; Sanchez-Molina, J.; Rodríguez-Hidalgo, M.; Lara-López, A.; Ruiz-Ederra, J. Subretinal injection techniques for retinal disease: A review. J. Clin. Med. 2022, 11, 4717. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Approved cellular and gene therapy products. U.S. Department of Health and Human Services. 2025. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products.
- Ylä-Herttuala, S. Endgame: Glybera finally recommended for approval as the first gene therapy drug in the European Union. Mol. Ther. 2012, 20, 1831–1832. [Google Scholar] [CrossRef]
- Watanabe, N.; Yano, K.; Tsuyuki, K.; Okano, T.; Yamato, M. Re-examination of regulatory opinions in Europe: Possible contribution for the approval of the first gene therapy product Glybera. Mol. Ther. Methods Clin. Dev. 2015, 2, 14066. [Google Scholar] [CrossRef]
- Schulz, M.; Levy, D.I.; Petropoulos, C.J.; Bashirians, G.; Winburn, I.; Mahn, M.; et al. Binding and neutralizing anti-AAV antibodies: Detection and implications for rAAV-mediated gene therapy. Mol. Ther. 2023, 31, 616–630. [Google Scholar] [CrossRef]
- Zwi-Dantsis, L.; Mohamed, S.; Massaro, G.; Moeendarbary, E. Adeno-associated virus vectors: Principles, practices, and prospects in gene therapy. Viruses 2025, 17, 239. [Google Scholar] [CrossRef]
- Prasad, S.; Dimmock, D.P.; Greenberg, B.; Walia, J.S.; Sadhu, C.; Tavakkoli, F.; et al. Immune responses and immunosuppressive strategies for adeno-associated virus-based gene therapy for treatment of central nervous system disorders: Current knowledge and approaches. Hum. Gene Ther. 2022, 33, 1228–1245. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Keiser, N.W.; Song, Y.; Deng, X.; Cheng, F.; Qiu, J.; Engelhardt, J.F. A novel chimeric adeno-associated virus 2/human bocavirus 1 parvovirus vector efficiently transduces human airway epithelia. Mol. Ther. 2013, 21, 2181–2194. [Google Scholar] [CrossRef] [PubMed]
- Guido, M.; Tumolo, M.R.; Verri, T.; Romano, A.; Serio, F.; De Giorgi, M.; et al. Human bocavirus: Current knowledge and future challenges. World J. Gastroenterol. 2016, 22, 8684–8697. [Google Scholar] [CrossRef] [PubMed]
- Jartti, T.; Hedman, K.; Jartti, L.; Ruuskanen, O.; Allander, T.; Söderlund-Venermo, M. Human bocavirus—the first 5 years. Rev. Med. Virol. 2012, 22, 46–64. [Google Scholar] [CrossRef]
- Yan, Z.; Zou, W.; Feng, Z.; Shen, W.; Park, S.Y.; Deng, X.; et al. Establishment of a high-yield recombinant adeno-associated virus/human bocavirus vector production system independent of bocavirus nonstructural proteins. Hum. Gene Ther. 2019, 30, 556–570. [Google Scholar] [CrossRef]
- Fakhiri, J.; Schneider, M.A.; Puschhof, J.; Stanifer, M.; Schildgen, V.; Holderbach, S.; et al. Novel chimeric gene therapy vectors based on adeno-associated virus and four different mammalian bocaviruses. Mol. Ther. Methods Clin. Dev. 2019, 12, 202–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Deng, X.; Zou, W.; Engelhardt, J.F.; Yan, Z.; Qiu, J. Human bocavirus 1 is a novel helper for adeno-associated virus replication. J. Virol. 2017, 91, e00710-17. [Google Scholar] [CrossRef] [PubMed]
- Catalán-Tatjer, D.; Tzimou, K.; Nielsen, L.K.; Lavado-García, J. Unravelling the essential elements for recombinant adeno-associated virus (rAAV) production in animal cell-based platforms. Biotechnol. Adv. 2024, 72, 108370. [Google Scholar] [CrossRef] [PubMed]
- Nisanov, A.M.; Rivera de Jesús, J.A.; Schaffer, D.V. Advances in AAV capsid engineering: Integrating rational design, directed evolution and machine learning. Mol. Ther. 2025, 33, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Lin, L.F.; Oraskovich, S.V.; Rivera de Jesús, J.A.; Listgarten, J.; Schaffer, D.V.; et al. Computationally guided AAV engineering for enhanced gene delivery. Trends Biochem. Sci. 2024, 49, 457–469. [Google Scholar] [CrossRef]
- Vu Hong, A.; Suel, L.; Petat, E.; Dubois, A.; Le Brun, P.-R.; Guerchet, N.; Veron, P.; Poupiot, J.; Richard, I. An engineered AAV targeting integrin alpha V beta 6 presents improved myotropism across species. Nat. Commun. 2024, 15, 7965. [Google Scholar] [CrossRef]
- Klein, A.H.; Kuiper, M.J.; Burgess, M.; Wickramarachchi, A.; Jain, Y.; Bauer, D.C.; et al. CapBuild: A cloud-native tool for adeno-associated virus capsid engineering. Nucleic Acids Res. 2025, 53, W110–W117. [Google Scholar] [CrossRef]
- Brittain, T.J.; Jang, S.; Coughlin, G.M.; Barcelona, B.H.; Giriat, I.; Ristic, F.; et al. Structural basis of liver de-targeting and neuronal tropism of CNS-targeted AAV capsids. bioRxiv 2025, preprint. [Google Scholar] [CrossRef]
- Tarasova, E.; Farafonov, V.; Khayat, R.; Okimoto, N.; Komatsu, T.S.; Taiji, M.; et al. All-atom molecular dynamics simulations of entire virus capsid reveal the role of ion distribution in capsid’s stability. J. Phys. Chem. Lett. 2017, 8, 779–784. [Google Scholar] [CrossRef]
- Pipatpadungsin, N.; Chao, K.; Rouse, S.L. Coarse-grained simulations of adeno-associated virus and its receptor reveal influences on membrane lipid organization and curvature. J. Phys. Chem. B 2024, 128, 10139–10153. [Google Scholar] [CrossRef]
- Sen, D. Improving clinical efficacy of adeno-associated vectors by rational capsid bioengineering. J. Biomed. Sci. 2014, 21, 103. [Google Scholar] [CrossRef]
- Lee, E.J.; Guenther, C.M.; Suh, J. Adeno-associated virus (AAV) vectors: Rational design strategies for capsid engineering. Curr. Opin. Biomed. Eng. 2018, 7, 58–63. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Sellnow, R.C.; Boye, S.L.; Coberly, B.; Bennett, A.; Agbandje-McKenna, M.; et al. Rationally engineered AAV capsids improve transduction and volumetric spread in the CNS. Mol. Ther. Nucleic Acids 2017, 8, 184–197. [Google Scholar] [CrossRef]
- Zhong, L.; Li, B.; Mah, C.S.; Govindasamy, L.; Agbandje-McKenna, M.; Cooper, M.; et al. Next generation of adeno-associated virus 2 vectors: Point mutations in tyrosines lead to high-efficiency transduction at lower doses. Mol. Ther. 2008, 16, 1717–1722. [Google Scholar] [CrossRef]
- Han, J.; Zhu, L.; Zhang, J.; Guo, L.; Sun, X.; Huang, C.; et al. Rational engineering of adeno-associated virus capsid enhances human hepatocyte tropism and reduces immunogenicity. Cell Prolif. 2022, 55, e13339. [Google Scholar] [CrossRef]
- Li, W.; Feng, S.L.; Herrschaft, L.; Samulski, R.J.; Li, C. Rationally engineered novel AAV capsids for intra-articular gene delivery. Mol. Ther. Methods Clin. Dev. 2024, 32, 101211. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Amaran, L.; Song, L.; Tretiakova, A.P.; Mikhail, S.A.; Samulski, R.J. AAV vector development, back to the future. Mol. Ther. 2025, 33, 1903–1936. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Lagerborg, K.A.; Stanton, A.; King, E.M.; Ye, S.; Tellez, L.; et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 2021, 184, 4919–4938.e22. [Google Scholar] [CrossRef]
- Tan, F.; Dong, Y.; Qi, J.; Yu, W.; Chai, R. Artificial intelligence-based approaches for AAV vector engineering. Adv. Sci. 2025, 12, 2411062. [Google Scholar] [CrossRef] [PubMed]
- Eid, F.-E.; Chen, A.T.; Chan, K.Y.; Huang, Q.; Zheng, Q.; Tobey, I.G.; et al. Systematic multi-trait AAV capsid engineering for efficient gene delivery. Nat. Commun. 2024, 15, 6602. [Google Scholar] [CrossRef]
- Wec, A.Z.; Lin, K.S.; Kwasnieski, J.C.; Sinai, S.; Gerold, J.; Kelsic, E.D. Overcoming immunological challenges limiting capsid-mediated gene therapy with machine learning. Front. Immunol. 2021, 12, 674021. [Google Scholar] [CrossRef]
- Zhu, D.; Brookes, D.H.; Busia, A.; Carneiro, A.; Fannjiang, C.; Popova, G.; et al. Optimal trade-off control in machine learning-based library design, with application to adeno-associated virus (AAV) for gene therapy. Sci. Adv. 2024, 10, eadj3786. [Google Scholar] [CrossRef]
- Jang, M.J.; Coughlin, G.M.; Jackson, C.R.; Chen, X.; Chuapoco, M.R.; Vendemiatti, J.L.; et al. Spatial transcriptomics for profiling the tropism of viral vectors in tissues. Nat. Biotechnol. 2023, 41, 1272–1286. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Noriega, H.A.; Wang, Q.; Yu, D.; Wang, X.S. Structural studies of Parvoviridae capsid assembly and evolution: Implications for novel AAV vector design. Front. Artif. Intell. 2025, 8, 1559461. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Baek, M.; Anishchenko, I.; Humphreys, I.R.; Cong, Q.; Baker, D.; DiMaio, F. Efficient and accurate prediction of protein structure using RoseTTAFold2. bioRxiv 2023, preprint. [Google Scholar] [CrossRef]
- Weber, S.; Ramirez, C.; Doerfler, W. Signal hotspot mutations in SARS-CoV-2 genomes evolve as the virus spreads and actively replicates in different parts of the world. Virus Res. 2020, 289, 198170. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; et al. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. GROMACS: Analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Benson, N.C.; Daggett, V. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 8722–8731. [Google Scholar] [CrossRef]
- May, E.R. Recent developments in molecular simulation approaches to study spherical virus capsids. Mol. Simul. 2014, 40, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.E.; Pérez-Segura, C.; Bryer, A.J.; Perilla, J.R.; Hadden-Perilla, J.A. Molecular dynamics of the viral life cycle: Progress and prospects. Curr. Opin. Virol. 2021, 50, 128–138. [Google Scholar] [CrossRef]
- Ito, M.; Takino, N.; Nomura, T.; Kan, A.; Muramatsu, S.-i. Engineered adeno-associated virus 3 vector with reduced reactivity to serum antibodies. Sci. Rep. 2021, 11, 9322. [Google Scholar] [CrossRef]
- DiPrimio, N.; Asokan, A. Surface loop dynamics in adeno-associated virus capsid assembly. J. Virol. 2008, 82, 5178–5189. [Google Scholar] [CrossRef]
- Mietzsch, M.; Barnes, C.; Hull, J.A.; Chipman, P.; Xie, J.; Bhattacharya, N.; et al. Comparative analysis of the capsid structures of AAVrh.10, AAVrh.39, and AAV8. J. Virol. 2020, 94, e01769-19. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Taxonomic organization of the family Parvoviridae. Parvoviridae is divided into three subfamilies: Densovirinae, Parvovirinae, and Hamaparvovirinae, with representative genera and viruses shown, including AAV (Dependoparvovirus) and HBoV (Bocaparvovirus). Capsid genome sizes range from 3.5 to 6.5 kb, as indicated by the scale bar.
Figure 1.
Taxonomic organization of the family Parvoviridae. Parvoviridae is divided into three subfamilies: Densovirinae, Parvovirinae, and Hamaparvovirinae, with representative genera and viruses shown, including AAV (Dependoparvovirus) and HBoV (Bocaparvovirus). Capsid genome sizes range from 3.5 to 6.5 kb, as indicated by the scale bar.
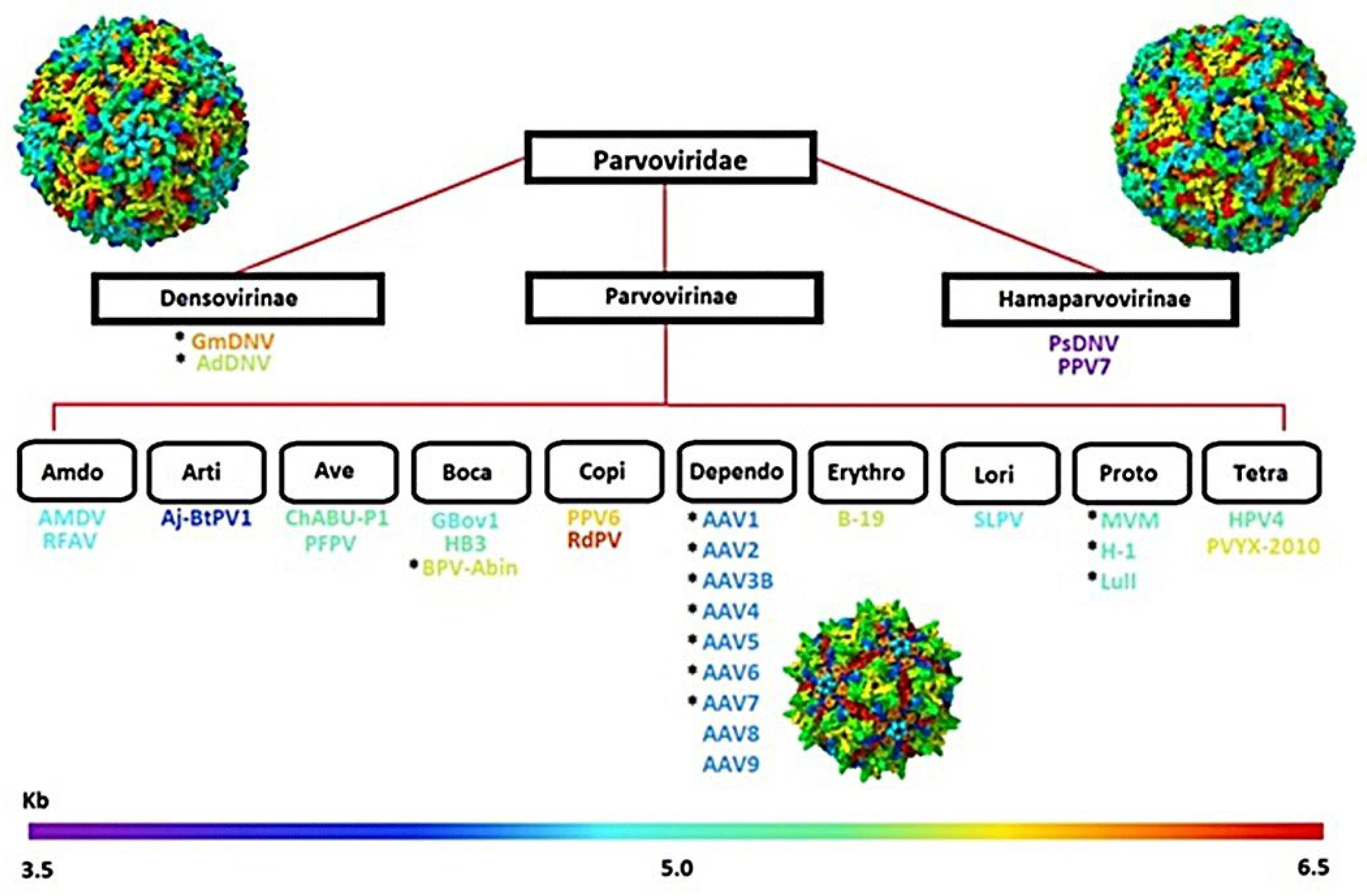
Figure 2.
Comparative surface architecture of Human Bocavirus 3 (HBoV3) and AAVGo.1 capsids. (A) Cryo-EM-derived surface representation of HBoV3 (outer diameter ≈ 282 Å; genome ≈ 5.5 kb) showing a relatively expanded shell with prominent surface protrusions at the three-fold and five-fold symmetry axes.(B) Modeled AAVGo.1 capsid (outer diameter ≈ 278 Å; genome ≈ 4.7 kb) displaying a more compact topology typical of Dependoparvoviruses. Both capsids exhibit the conserved icosahedral β-barrel framework characteristic of the Parvoviridae family. At the same time, distinct loop contours at symmetry interfaces account for lineage-specific differences in particle size and genome capacity. The structure was visualized in UCSF ChimeraX to evaluate capsid surface topology and the spatial organization of variable regions in the assembled particle.
Figure 2.
Comparative surface architecture of Human Bocavirus 3 (HBoV3) and AAVGo.1 capsids. (A) Cryo-EM-derived surface representation of HBoV3 (outer diameter ≈ 282 Å; genome ≈ 5.5 kb) showing a relatively expanded shell with prominent surface protrusions at the three-fold and five-fold symmetry axes.(B) Modeled AAVGo.1 capsid (outer diameter ≈ 278 Å; genome ≈ 4.7 kb) displaying a more compact topology typical of Dependoparvoviruses. Both capsids exhibit the conserved icosahedral β-barrel framework characteristic of the Parvoviridae family. At the same time, distinct loop contours at symmetry interfaces account for lineage-specific differences in particle size and genome capacity. The structure was visualized in UCSF ChimeraX to evaluate capsid surface topology and the spatial organization of variable regions in the assembled particle.

Figure 3.
(A) Annotated amino acid sequence of the AAV-HB3 VP3 chimera showing the locations of substituted HBoV3-derived structural motifs (α-helix and βH/βI strand) and defined AAV variable regions. Measurement positions used for loop displacement analyses are indicated. (B) Annotated amino acid sequence of the AAVGo.1 VP1 control highlighting corresponding variable regions and structural elements. Residue numbering is reported separately for VP3 and VP1 constructs to account for differences in protein length and domain composition. This figure provides the sequence-based framework used for defining flexible regions analyzed in subsequent MD simulations.
Figure 3.
(A) Annotated amino acid sequence of the AAV-HB3 VP3 chimera showing the locations of substituted HBoV3-derived structural motifs (α-helix and βH/βI strand) and defined AAV variable regions. Measurement positions used for loop displacement analyses are indicated. (B) Annotated amino acid sequence of the AAVGo.1 VP1 control highlighting corresponding variable regions and structural elements. Residue numbering is reported separately for VP3 and VP1 constructs to account for differences in protein length and domain composition. This figure provides the sequence-based framework used for defining flexible regions analyzed in subsequent MD simulations.
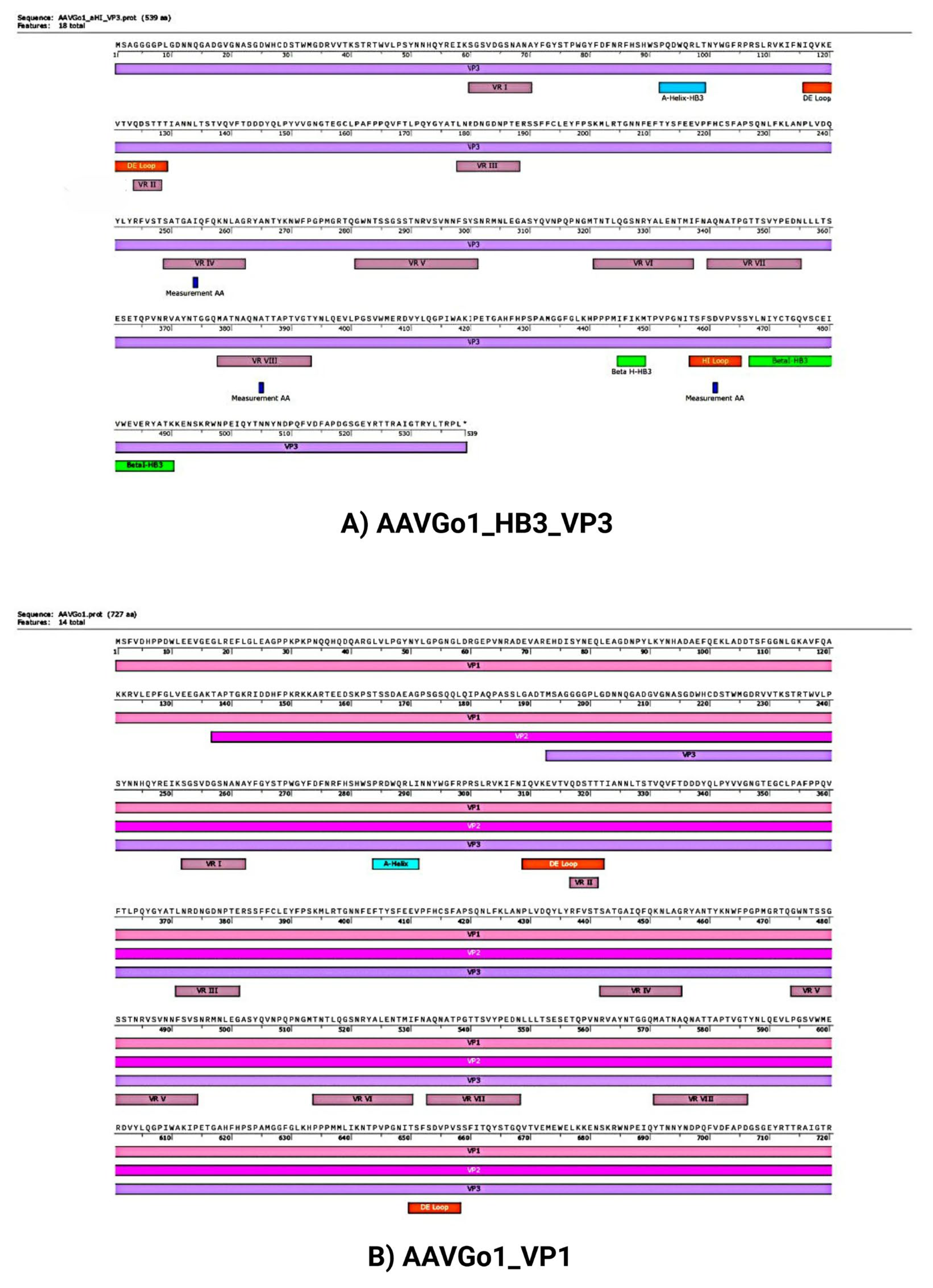
Figure 4.
Structural comparison of chimeric and control AAV capsids. Predicted surface reconstructions of the AAV-HB3 chimera and wild-type AAVGo.1 VP1 highlights conserved icosahedral organization while revealing localized topological differences introduced by substitution of HBoV3-derived regions. The 60-mer capsid models are static assemblies generated from refined monomer structures, with highlighted regions corresponding to flexible segments identified from monomer-level MD simulations. Monomeric superimpositions of the experimental AAVGo.1 structure (PDB: 7TI4) with the 100 ns MD-derived conformations of AAV-HB3 VP3 and AAVGo.1 VP1 are shown to illustrate localized conformational deviations relative to the reference fold.
Figure 4.
Structural comparison of chimeric and control AAV capsids. Predicted surface reconstructions of the AAV-HB3 chimera and wild-type AAVGo.1 VP1 highlights conserved icosahedral organization while revealing localized topological differences introduced by substitution of HBoV3-derived regions. The 60-mer capsid models are static assemblies generated from refined monomer structures, with highlighted regions corresponding to flexible segments identified from monomer-level MD simulations. Monomeric superimpositions of the experimental AAVGo.1 structure (PDB: 7TI4) with the 100 ns MD-derived conformations of AAV-HB3 VP3 and AAVGo.1 VP1 are shown to illustrate localized conformational deviations relative to the reference fold.
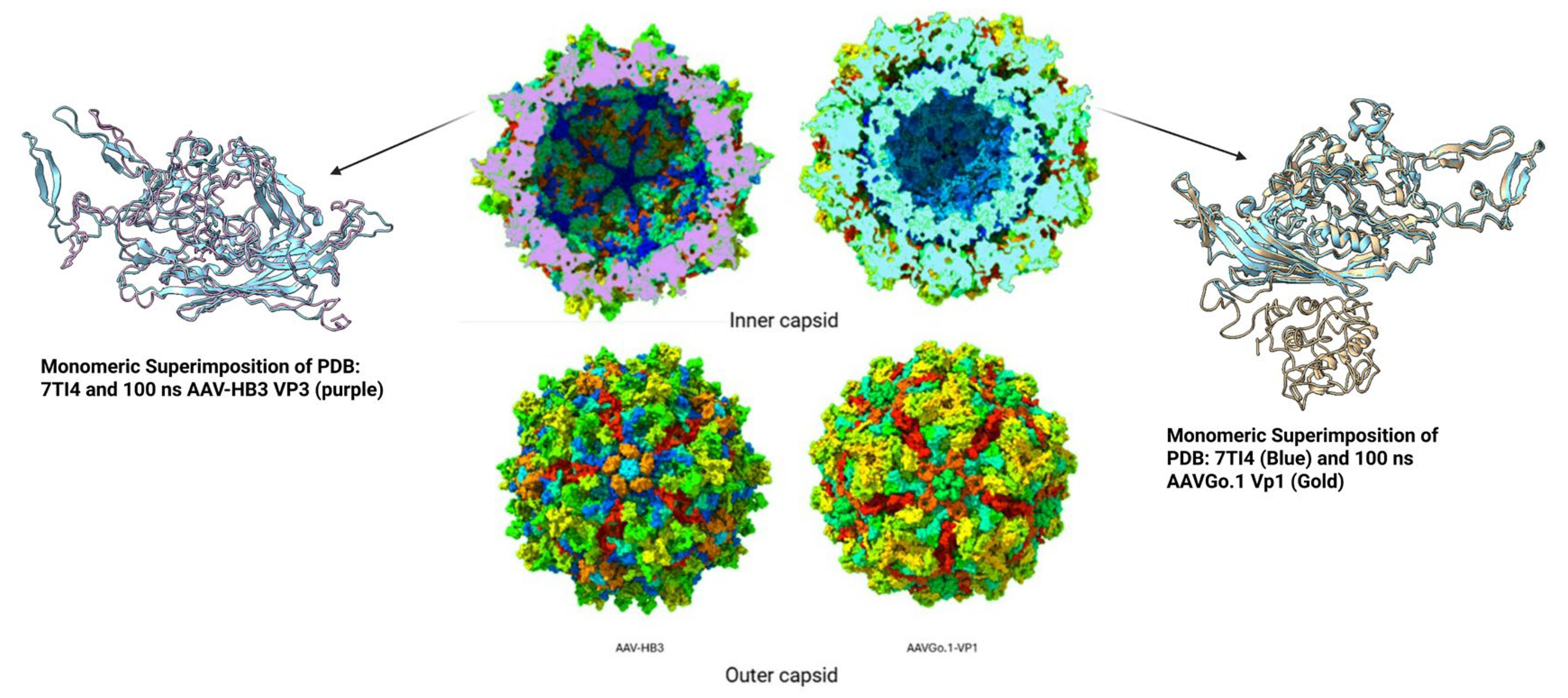
Figure 5.
Figure 5. Thermodynamic stability of AAV-HB3 and AAVGo.1 VP1 during molecular dynamics simulations. Temperature and potential energy profiles over 100 ns indicate both systems-maintained equilibrium conditions, with stable trajectories at ~300 K and constant potential energy values, confirming suitability for comparative structural analyses.
Figure 5.
Figure 5. Thermodynamic stability of AAV-HB3 and AAVGo.1 VP1 during molecular dynamics simulations. Temperature and potential energy profiles over 100 ns indicate both systems-maintained equilibrium conditions, with stable trajectories at ~300 K and constant potential energy values, confirming suitability for comparative structural analyses.
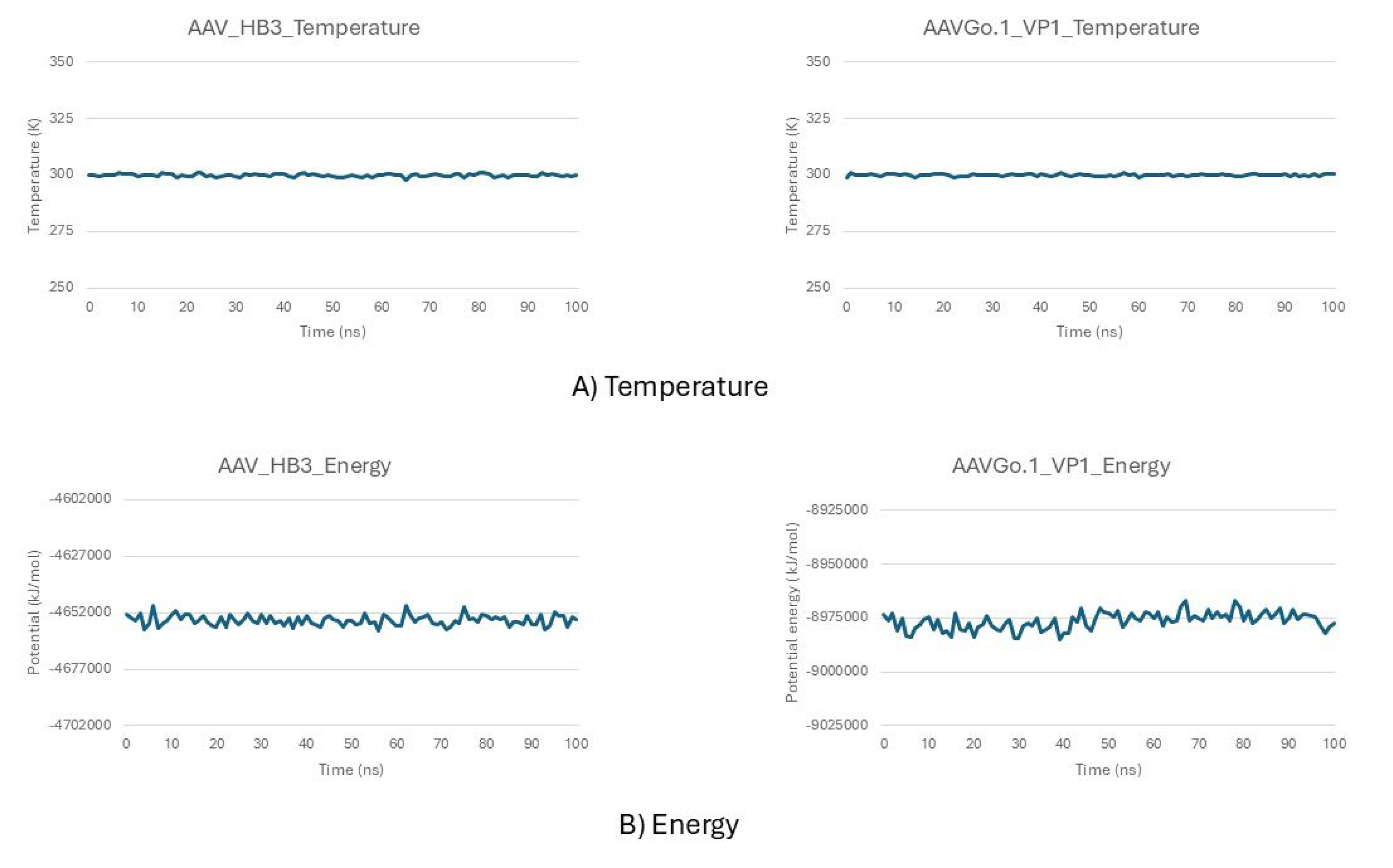
Figure 6.
Structural stability and residue-level flexibility of AAV-HB3 and AAVGo.1 VP1 during 100 ns simulations. (A) RMSD traces show AAVGo.1 VP1 rapidly stabilized at ~0.03 nm, while AAV-HB3 exhibited larger deviations, reaching ~0.8–1.0 nm by the end of the trajectory. (B) RMSF profiles reveal localized fluctuations in AAV-HB3 at VR-VIII, VR-IV, and the HI loop, compared to uniformly low flexibility across AAVGo.1 VP1, consistent with its preserved structural rigidity.
Figure 6.
Structural stability and residue-level flexibility of AAV-HB3 and AAVGo.1 VP1 during 100 ns simulations. (A) RMSD traces show AAVGo.1 VP1 rapidly stabilized at ~0.03 nm, while AAV-HB3 exhibited larger deviations, reaching ~0.8–1.0 nm by the end of the trajectory. (B) RMSF profiles reveal localized fluctuations in AAV-HB3 at VR-VIII, VR-IV, and the HI loop, compared to uniformly low flexibility across AAVGo.1 VP1, consistent with its preserved structural rigidity.
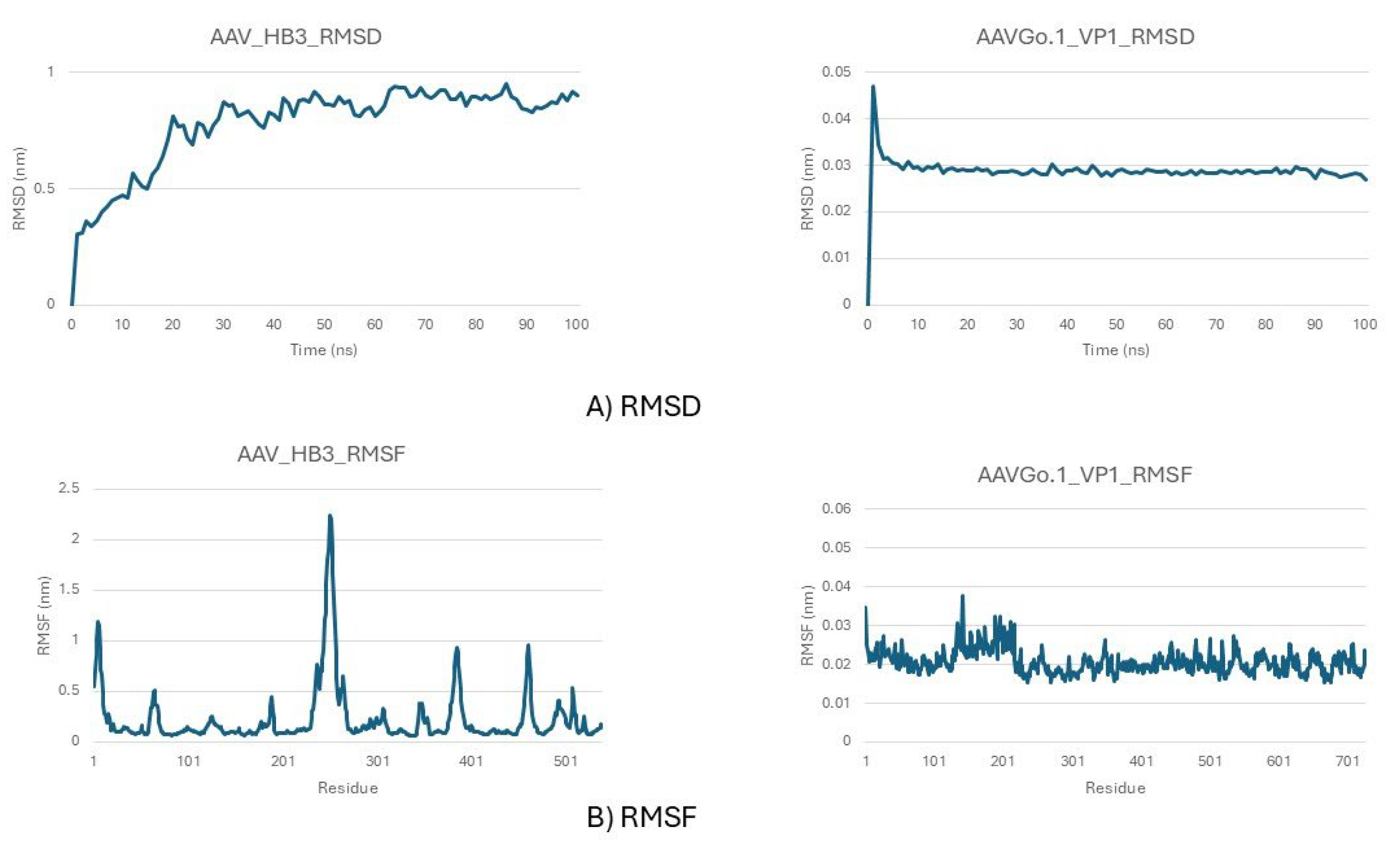
Figure 7.
Structural overlays illustrating loop dynamics of the AAV-HB3 chimera relative to the AAVGo.1 VP1 reference during molecular dynamics simulations. The dark blue structure represents the reference conformation, while colored structures correspond to MD snapshots sampled at successive time points: (A) time step 20 (teal), (B) time step 45 (yellow), and (C) time step 90 (light purple). Panel (D) shows the final conformation at 100 ns (purple). Measured loop–loop separations reach up to ~54 Å over the trajectory. Highlighted regions indicate variable regions VR-VIII, VR-IV, and the HI loop, which undergo progressive outward displacement relative to the reference structure. These conformational changes localize to surface-exposed regions adjacent to the 3-fold and 5-fold symmetry axes and are consistent with residue-level flexibility and principal component analyses.
Figure 7.
Structural overlays illustrating loop dynamics of the AAV-HB3 chimera relative to the AAVGo.1 VP1 reference during molecular dynamics simulations. The dark blue structure represents the reference conformation, while colored structures correspond to MD snapshots sampled at successive time points: (A) time step 20 (teal), (B) time step 45 (yellow), and (C) time step 90 (light purple). Panel (D) shows the final conformation at 100 ns (purple). Measured loop–loop separations reach up to ~54 Å over the trajectory. Highlighted regions indicate variable regions VR-VIII, VR-IV, and the HI loop, which undergo progressive outward displacement relative to the reference structure. These conformational changes localize to surface-exposed regions adjacent to the 3-fold and 5-fold symmetry axes and are consistent with residue-level flexibility and principal component analyses.
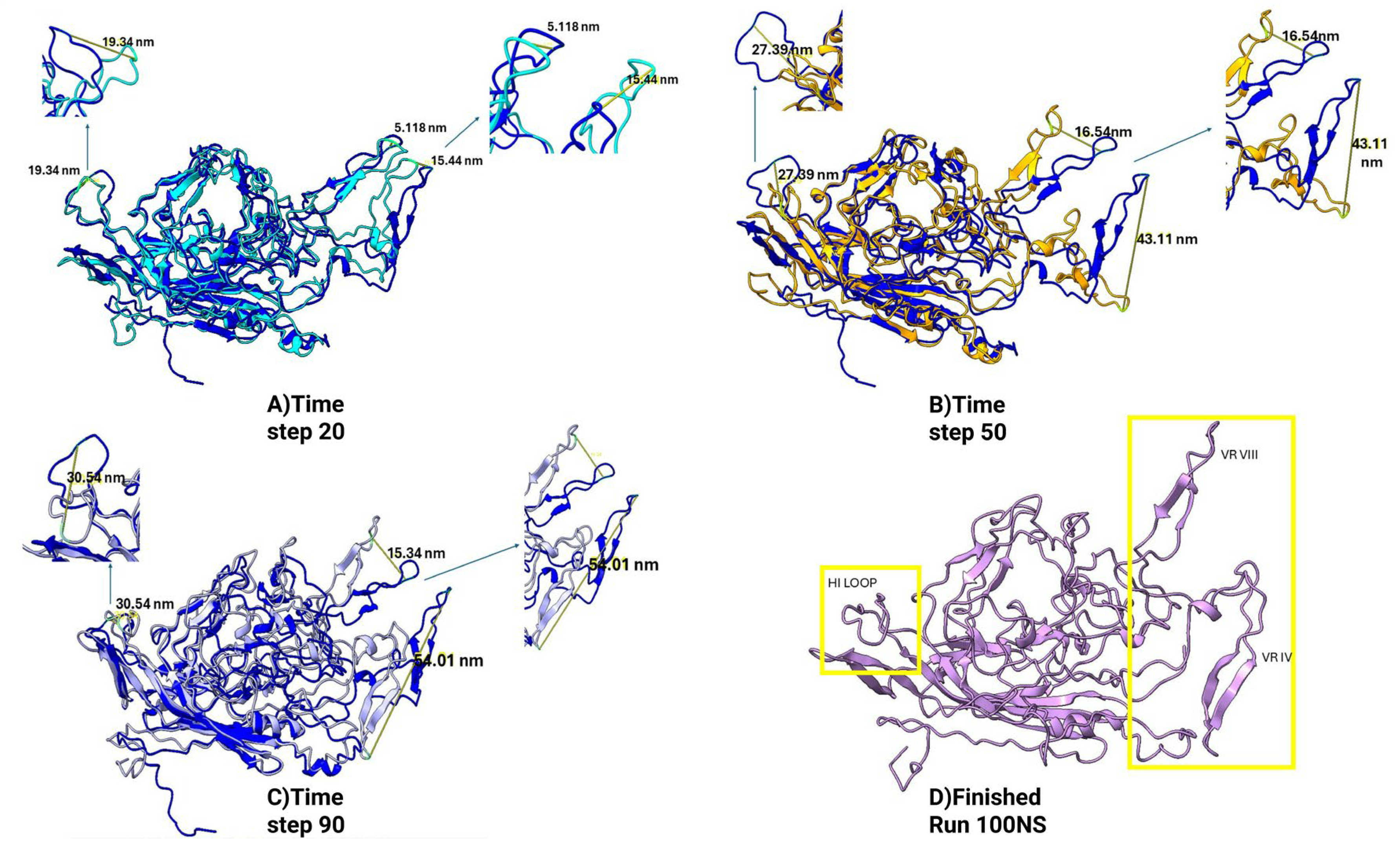
Figure 8.
Figure 8. Radius of gyration (Rg) profiles for the AAVGo.1 VP1 control and the AAV-HB3 chimera over a 100 ns molecular dynamics trajectory. The AAV-HB3 chimera exhibits consistently higher Rg values relative to AAVGo.1 VP1, indicating an expanded monomeric conformational ensemble. Correlated temporal fluctuations between the two systems reflect preservation of the underlying fold, while differences in mean Rg indicate altered global compactness associated with localized flexibility in engineered structural regions.
Figure 8.
Figure 8. Radius of gyration (Rg) profiles for the AAVGo.1 VP1 control and the AAV-HB3 chimera over a 100 ns molecular dynamics trajectory. The AAV-HB3 chimera exhibits consistently higher Rg values relative to AAVGo.1 VP1, indicating an expanded monomeric conformational ensemble. Correlated temporal fluctuations between the two systems reflect preservation of the underlying fold, while differences in mean Rg indicate altered global compactness associated with localized flexibility in engineered structural regions.
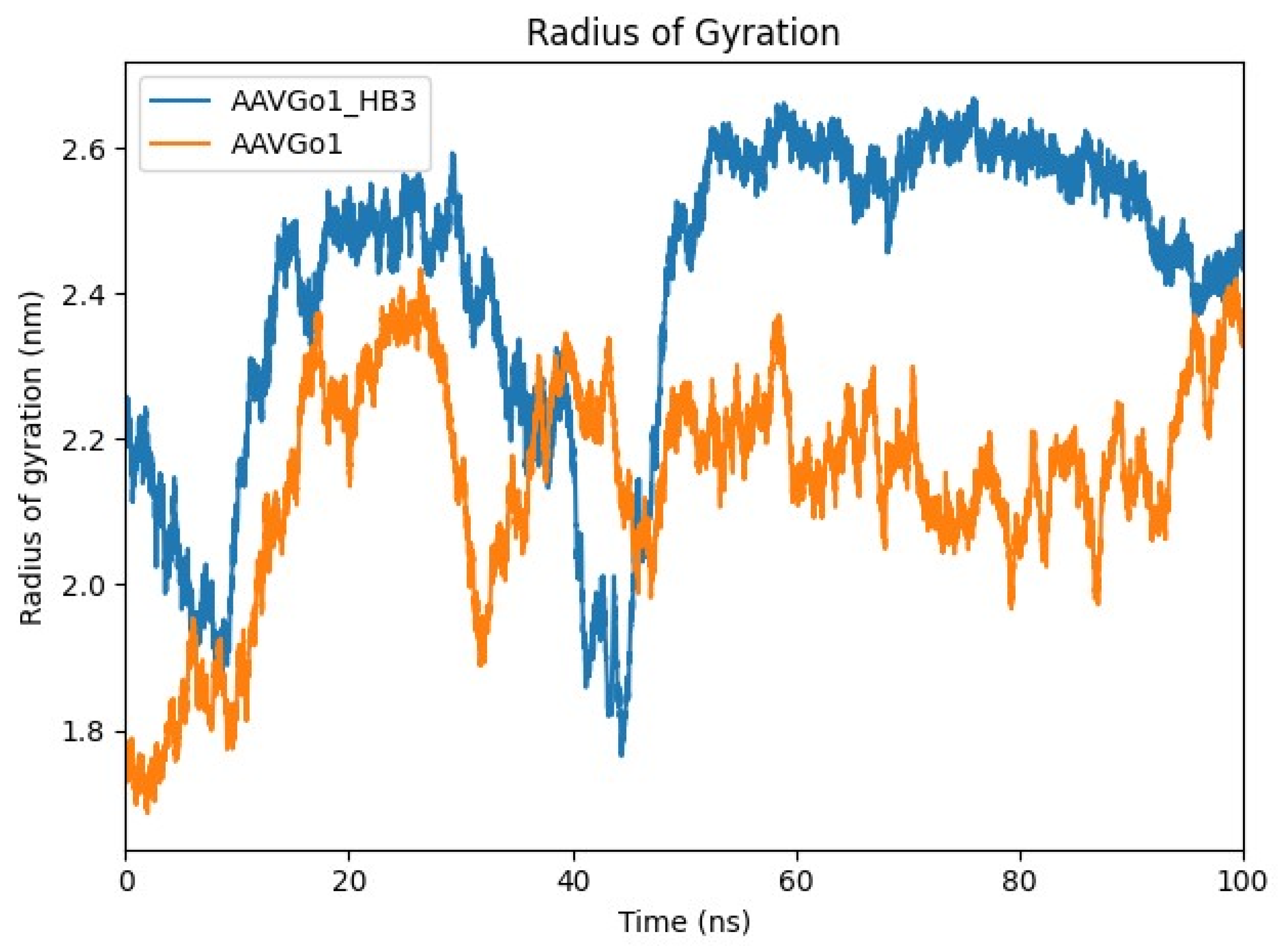
Figure 9.
Principal component analysis (PCA) of molecular dynamics trajectories for (A) the AAV-HB3 chimera and (B) AAVGo.1 VP1 over 100 ns simulations. For each system, scree plots report the variance explained by the top 10 principal components. Scatter plots show conformational sampling projected onto PC1 versus PC2, PC1 versus PC3, and PC2 versus PC3. Each point represents an individual trajectory frame, colored by simulation time. The AAV-HB3 chimera samples a broader region of principal component space, reflecting increased conformational variability dominated by surface-exposed regions, whereas AAVGo.1 VP1 exhibits more confined sampling consistent with a comparatively stable monomeric fold.
Figure 9.
Principal component analysis (PCA) of molecular dynamics trajectories for (A) the AAV-HB3 chimera and (B) AAVGo.1 VP1 over 100 ns simulations. For each system, scree plots report the variance explained by the top 10 principal components. Scatter plots show conformational sampling projected onto PC1 versus PC2, PC1 versus PC3, and PC2 versus PC3. Each point represents an individual trajectory frame, colored by simulation time. The AAV-HB3 chimera samples a broader region of principal component space, reflecting increased conformational variability dominated by surface-exposed regions, whereas AAVGo.1 VP1 exhibits more confined sampling consistent with a comparatively stable monomeric fold.
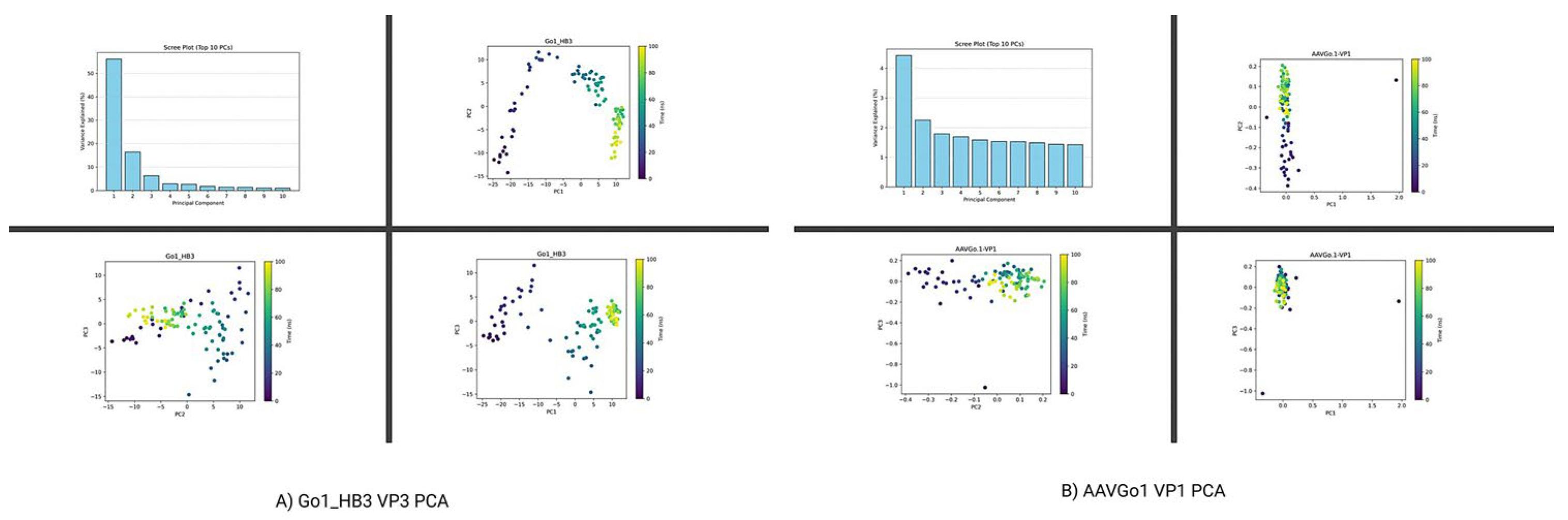
Figure 10.
Structural comparison of AAVGo.1 VP1 before and after simulation. AlphaFold2-predicted structure of AAVGo.1 VP1 (left) is shown alongside the final frame after 100 ns of molecular dynamics simulation (right). The conserved β-barrel fold was maintained, with only minor adjustments in loop regions. Notably, the N-terminal region exhibited an inward shift toward the β-sheet core during the simulation, consistent with cryo-EM observations of genome-containing AAV capsids where the VP1 N-terminus contributes to internal membrane formation.
Figure 10.
Structural comparison of AAVGo.1 VP1 before and after simulation. AlphaFold2-predicted structure of AAVGo.1 VP1 (left) is shown alongside the final frame after 100 ns of molecular dynamics simulation (right). The conserved β-barrel fold was maintained, with only minor adjustments in loop regions. Notably, the N-terminal region exhibited an inward shift toward the β-sheet core during the simulation, consistent with cryo-EM observations of genome-containing AAV capsids where the VP1 N-terminus contributes to internal membrane formation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



