
Submitted:
19 February 2026
Posted:
25 February 2026
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Natural product (NP)-based chemical libraries are a rich source of antiviral compounds that can serve as the basis for new therapeutic leads to treat viral diseases, particularly toward individuals lacking sustained access to existing vaccines and therapeutics. To identify new NP-based inhibitors of SARS-CoV-2 cellular entry and viral replication, we screened the pan-African Natural Products Library (p-ANAPL), a collection of over 500 physical pure compounds obtained from African medicinal plants, for NPs that can dis-rupt the in vitro interaction of the SARS-CoV-2 Spike receptor binding domain (RBD) with its host ACE2 entry receptor. This screen identified three compounds – oleanolic acid, poinsettifolin B, and rhuschalcone III – which disrupt RBD/ACE2 interactions with half-maximal inhibitory concentrations (IC50s) of 0.5 – 2.4 µM but do not disrupt an unre-lated PD-1/PD-L1 host ligand/receptor binding pair. Oleanolic acid and rhuschalcone III, but not poinsettifolin B, additionally inhibited SARS-CoV-2 replication in Vero cells without cytotoxicity at low micromolar concentrations. Computational modelling indi-cated that all three compounds interact with numerous residues of RBD and ACE2, in-cluding a subset of residues that remain conserved across SARS-CoV-2 variants of con-cern. Taken together, we identify three NPs that selectively interfere with factors involved in SARS-CoV-2 entry and/or viral replication, representing new antiviral leads for COVID-19 management in resource-limited areas.
Keywords:
natural products
; SARS-CoV-2
; viral entry
; antivirals
; p-ANAPL
1. Introduction
While SARS-CoV-2, the causative agent of COVID-19, has become increasingly managed in much of the world, access to SARS-CoV-2 vaccines and therapeutics in many low- and middle-income countries (LMICs) remains limited or inconsistent [1,2,3]. Due to this inaccessibility, as well as limited access to Western medicines more generally, many people in LMICs rely on local medicinal plants for their primary healthcare [4]. Identifying natural products (NPs), or naturally-produced chemical compounds, that are found in medicinal plants and inhibit SARS-CoV-2 replication is therefore important toward identifying locally-accessible options for management of COVID-19 and potentially other coronavirus-driven infections in the absence of sufficient availability of Western vaccines and therapeutics.
The pan-African Natural Products Library (p-ANAPL) represents one of the largest physical collections of NPs derived from African medicinal plants that is available for screening purposes [5]. This collection of over 500 compounds was previously screened to identify new inhibitors of HIV and influenza A virus replication as well as inhibitors of sirtuin activity [6,7,8,9,10]. As these studies all demonstrate the ability to obtain novel screening hits from p-ANAPL, we hypothesized that p-ANAPL also likely harbors NPs that may act on SARS-CoV-2 replication.
SARS-CoV-2 entry depends primarily on the binding of the receptor-binding domain (RBD) of the viral Spike protein to the host ACE2 receptor. This interaction facilitates proteolytic activation of Spike by host proteases, which, in turn, initiates membrane fusion and viral entry [11]. Our group and others have identified numerous NPs that selectively interfere with RBD/ACE2 interactions and/or inhibit SARS-CoV-2 replication in live virus or pseudovirus assays [11,12,13,14,15,16]. We therefore anticipated that NPs from p-ANAPL with activity against SARS-CoV-2 replication would be identified by initial screening for their ability to disrupt RBD/ACE2 protein-protein interactions. Here, we describe the results of this screen, which identified the previously-reported oleanolic acid, as well as new leads poinsettifolin B and rhuschalcone III, as inhibitors of RBD/ACE2 binding and/or SARS-CoV-2 replication.
2. Materials and Methods
2.1. Cells, Viruses, and Reagents
Vero-E6 cells were obtained from the American Tissue Culture Collection. Cells were cultured in D10+ medium (Dulbecco’s modified Eagle medium with 4.5 g/liter glucose and L-glutamine [Gibco, Gaithersburg, MD, USA], 10% fetal bovine serum [Gemini Bio Products, West Sacramento, CA, USA], 100 U/mL penicillin, and 100 µg/mL streptomycin [Sigma-Aldrich, St. Louis, MO, USA]) in a humidified incubator at 37 °C and 5% CO2. The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-related coronavirus 2, isolate USA-WA1/2020, NR-52281. Oleanolic acid, poinsettifolin B, and rhuschalcone III were obtained from p-ANAPL chemical stocks and as described previously [17,18,19]. (-)-Hopeaphenol was obtained as described previously [11].
2.2. AlphaScreen Assays
Protein-protein interaction assays were described previously [11]. Briefly, protein–protein interactions were assessed using AlphaScreen technology, where binding of SARS-CoV-2 spike RBD to human ACE2, and of PD-1 to PD-L1, were measured in 384-well white opaque plates under optimized buffer conditions. Recombinant Fc- and His-tagged proteins (Sino Biological, Chesterbrook, PA, USA) were incubated with donor and acceptor beads in the presence of test compounds dispensed in DMSO. Following 2 hours of incubation at room temperature, AlphaScreen signals were recorded on a ClarioStar plate reader (BMG Labtech, Cary, NC, USA). Data were normalized to percentage inhibition, with values expressed relative to positive (absence of His-tagged partner protein) and negative (both proteins plus DMSO vehicle) controls. All conditions were performed in duplicate.
2.3. Viral Cytopathic Effect Quantitative Assay
SARS-CoV-2 virus (USA-WA1/2020) was generated, and viral assays were performed in a biosafety level 3 facility as described previously [11]. Briefly, Vero-E6 cells were plated in D10+ to 20,000 cells per well in a 96-well format. Compounds were added to final concentrations and incubated for 2 hours before the addition of 150X tissue culture infectious dose 50% (TCID50) of virus. Cells were incubated for an additional 4 days, treated with 20 µg/mL resazurin for 4 hours (Sigma-Aldrich) to assess cell viability, fixed with paraformaldehyde to a final concentration of 4%, and incubated at room temperature for 30 minutes. Fluorescence intensity was then measured using a ClarioStar plate reader. Background fluorescence was subtracted from all wells based on the signal obtained from wells containing D10+ and resazurin and no cells. Data were then normalized to the signal obtained from wells containing uninfected cells.
2.4. Cell Viability Assays in Uninfected Cells
Assays were performed as described above in the absence of virus. Background fluorescence was subtracted from all wells based on the signal obtained from wells containing D10+ and resazurin and no cells. Data were then normalized to the signal obtained from wells containing cells with no drug treatment.
2.5. Target Proteins for Docking
All molecular modeling was conducted as previously reported [20,21,22,23,24]. The X-ray crystal structure (PDB ID: 6M0J) describing the RBD of SARS-CoV-2 spike in complex with the ACE2 human receptor protein, corresponding to the parental Wuhan strain, was retrieved from the Protein Data Bank (PDB; [25,26,27]).
2.6. Protein Preparation
Water molecules were first removed using the graphical user interface of the Molecular Operating Environment (MOE) suite [28] (version 2016.08; Chemical Computing Group, Montreal, QC, Canada). Next, the protein preparation for docking was carried out using the Protein Preparation Wizard integrated in the Schrödinger package software [29,30]. This was done by adding the missing H-bonds, assigning bond orders, and completing the missing side chains. This was followed by energy minimization to remove all atomic clashes and optimize the potential protein-ligand interactions during docking. Epik-tool was then used to predict the protonation states at a pH of 7.0 [31]. Finally, a restrained energy minimization step was conducted using the Optimized Potentials for Liquid Simulations 2005 (OPLS2005) forcefield [32], allowing a root mean square deviation (RMSD) of the displacement of the atoms to end with the minimization at 0.3 Å.
2.7. Ligand Preparation
The MOE builder module was used to generate the 3D models of compounds for docking. These were then energy-minimized using the MMFF94 force field [33,34,35,36,37]. Further preparation for docking was done with the LigPrep tool to generate all the plausible tautomers of each ligand, using Schrödinger’s Maestro software package [31] with the incorporated OPLS2005 force field [32] for energy minimization. The ConfGen tool was then used to compute 60 conformers for each ligand, by setting all other options to default except for the minimization of the output [38].
2.8. Docking, Scoring, and Selection of Binding Poses
Docking validation and docking of the prepared ligands were carried out using the Glide program incorporated in Schrödinger’s Maestro package [30] and as shown in our recent publications [20,21,22,23,24]. A docking grid box was generated for the spike/ACE2 complex around the following amino acid residues; Asp597, Thr598, Lys516, Val321, Gln121, Lys578, Ala283, Ser91, Asn746, Gln68, Pro744, Glu518 and Thr610. A total of 10 poses per ligand conformer were taken into consideration, and all other settings were allowed to default. The outputs were scored using the Standard Precision (SP) glide score (GScore) as the scoring function [39]. After the extraction of the docked outputs, the specific area ligands bound with the RBD/ACE2 complex were observed to search for the residues taking part in the interactions.
2.9. Rescoring by MM-GBSA
The Molecular Mechanics Generalized Born Solvation Area (MM-GBSA) model was employed as a means of re-scoring the selected protein-ligand docked poses, by implementing the PRIME tool incorporated in the Maestro package from Schrödinger [29]. The free energy of the binding (ΔGbind) for each ligand towards the spike/ACE2 complex was calculated by using the Prime MM-GBSA algorithm, using the default settings. Each docked pose was retrieved from the Glide docking output and input into the PRIME program for calculating several thermodynamic properties. The computed properties include the binding free energy (ΔGbind) and solvation free energy (ΔGsolv) values in kcal/mol. The binding pose of the complex structures was visually inspected by using the ligand interaction tool in MOE to gain insight into the binding mode of each docked ligand inside the angiotensin II binding site of the ACE2 protein.
2.9. Generative Modeling
The 3 active compounds were subsequently optimized through de novo design. This generative process began by obtaining suitable fragments for the target sites (the SARS-CoV-2 76 RBD/ACE2 interface and the HDAC1 receptor site). The BRICS (breaking of retrosynthetically interesting chemical substructures) module in RDKit [40,41,42] was used for fragmentation. These fragments then served as input for generative modeling using the REINVENT4 generators [43,44,45]. The Jupyter notebook detailing the fragmentation step is accessible on https://github.com/JudeBetow/brics-fragmentation. REINVENT4 was executed via the command line on Ubuntu 24.04 LTS. After generation, the compounds were visually assessed, and selected molecules proceeded to further evaluation, including docking and MM-GBSA analysis, to ensure proper selection.
3. Results
3.1. A Subset of p-ANAPL NPs Disrupts SARS-CoV-2 RBD / Host ACE2 Binding
To identify NPs with the potential to inhibit viral entry, we used an established binding assay where an SARS-CoV-2 RBD protein (USA-WA1/2020) with a C-terminal His tag, prebound to an acceptor bead, was co-incubated with a full-length ACE2 protein with a C-terminal Fc tag, prebound to a donor bead [11,46]. Following co-incubation, ligand/receptor binding events were measured by excitation at 680 nm, resulting in a single oxygen transfer between donor and receptor beads and luminescence at 615 nm. Using this assay, we then physically screened 213 compounds from p-ANAPL at 1 µg/mL. Three compounds were identified to inhibit at least 80% of RBD/ACE2 binding-dependent luminescence; these compounds included oleanolic acid, poinsettifolin B, and rhuschalcone III (Figure 1; Table 1). An additional seven compounds also inhibited binding between 70 and 80% (Table 1). Given the number of NPs with the potential to disrupt RBD/ACE2 binding, we prioritized the three most active compounds for study.
We next assessed the relative activity of these three compounds to disrupt RBD/ACE2 binding by repeating the binding assay in dose-response studies. Dose-response curves were obtained for all three compounds, allowing us to calculate half-maximal inhibitory concentrations (IC50s) of 2.4, 0.50, and 0.81 µM for oleanolic acid, poinsettifolin B, and rhuschalcone III, respectively (Figure 2A-C). In contrast, the previously-described positive control inhibitor (-)-hopeaphenol [11] disrupted binding in this assay with an IC50 of 0.39 µM. Thus, the three compounds identified here are roughly 1.3 to 6.2-fold less active than this established disruptor of RBD/ACE2 binding.
To determine whether these compounds may selectively disrupt RBD/ACE2 binding over other protein-protein interactions, we repeated the binding assay using the unrelated host PD-1/PD-L1 ligand/receptor pair [11]. While the positive control inhibitor BMS-1166 [47] was disrupted PD-1/PD-L1 binding in this assay with an IC50 of 0.0031 µM, oleanolic acid, poinsettifolin B, and rhuschalcone III had substantially weaker activities, with IC50s of 12.4, > 10.6, and > 19.1 µM, respectively (Figure 2). When comparing the ratios of IC50s of PD-1/PD-L1 and RBD/ACE2 binding for the three compounds, we observed selectivity indices of 5.2 for oleanolic acid, > 21.4 for poinsettifolin B, and > 23.5 for rhuschalcone III, indicating that all three NPs selectively disrupt RBD/ACE2 binding.
3.2. p-ANAPL NPs Inhibit In Vitro SARS-CoV-2 Replication Without Cellular Toxicity
To confirm whether the disruption of RBD/ACE2 binding by these NPs corresponded to the ability to inhibit virus replication, we infected Vero-E6 cells with SARS-CoV-2 virus (USA-WA1/2020) in the presence or absence of compounds and monitored cellular viability after 96 hours using a previously-established multicycle virus replication assay [11]. In parallel, we also treated uninfected Vero cells with compounds and measured viability after 96 hours to assess the intrinsic cytotoxicity of each NP. Of the three compounds, we observed that oleanolic acid was the most active, with 46.6 ± 7.1% (mean ± s.e.m.) block of virus replication at 10 µM, relative to infected cells without drug treatment (Figure 3A). This activity approached that of the positive control inhibitor remdesivir, which inhibited 85.8 ± 11.4% of virus replication at 10 µM. However, the antiviral activity of oleanolic acid at higher concentrations could not be determined due to off-target cytotoxicity caused by the compound (Figure 3A). In contrast, while poinsettifolin B did not inhibit SARS-CoV-2 replication at any concentration (Figure 3B), rhuschalcone III exhibited weak but consistent activity where, for example, it inhibited up to 19.9 ± 7.7% of SARS-CoV-2 replication at 3 µM, although less activity was seen at higher concentrations, possibility due to increased intrinsic cytotoxicity of this NP (Figure 3C). Taken together, these results indicate that oleanolic acid, and to a lesser extent, rhuschalcone III, have antiviral activity against SARS-CoV-2 in addition to their ability to disrupt RBD/ACE2 binding.
3.3. p-ANAPL NPs Interact with Amino Acid Residues Required for SARS-CoV-2 RBD and Host ACE2 Binding
To identify the mechanisms by which oleanolic acid, poinsettifolin B, and rhuschalcone III might disrupt the RBD/ACE2 complex, molecular docking was performed by targeting the angiotensin II binding site within the complex. This was followed by a rescoring step to estimate the binding free energy ΔGbind of the resulting ligand–receptor complexes. The docking scores obtained for oleanolic acid, poinsettifolin B, and rhuschalcone III were −4.312 kcal/mol, −4.703 kcal/mol, and −6.000 kcal/mol, respectively (Table 2). Accordingly, the predicted binding affinities for these ligands toward the RBD/ACE2 complex followed the order of rhuschalcone III > poinsettifolin B > oleanolic acid. The major components of the Glide docking score, namely the van der Waals (GScorevdw) and electrostatic (GScoreele) contributions, are presented in Table 2. For rhuschalcone III, poinsettifolin B, and oleanolic acid, GScorevdw and GScoreele were -38.585 and -16.285 kcal/mol, -38.583 and -5.516 kcal/mol, and -31.576 and -7.815 kcal/mol, respectively. In all cases, van der Waals interactions contribute more substantially to the total docking score than electrostatic interactions, suggesting that hydrophobic and steric complementarity may play a predominant role in the binding of these compounds to the RBD/ACE2 interface. Interestingly, rhuschalcone III exhibited a significantly higher electrostatic contribution compared to the other two compounds, which may indicate a greater involvement of polar or charged interactions in its binding mode. Rescoring of the docked complexes further supported the docking score trend, with calculated ΔGbind values of −29.29, −34.37, and −42.29 kcal/mol for oleanolic acid, poinsettifolin B, and rhuschalcone III, respectively (Table 2). The solvation energy contributions (ΔGsolv) were calculated to be +33.911, +37.503, and +32.360 kcal/mol for rhuschalcone III, poinsettifolin B, and oleanolic acid, respectively (Table 2). These positive values reflect the fact that, in the unbound state, solvation significantly stabilizes the charged or polar groups of both the ligand and the receptor through interactions with water molecules. Upon complex formation, this stabilization is reduced, resulting in a positive ΔGsolv. Notably, the solvation energies of rhuschalcone III and oleanolic acid are nearly identical and more favorable than that of poinsettifolin B, suggesting a lower desolvation penalty for these two compounds during binding.
We next analyzed the binding confirmations of the three ligands to the RBD/ACE2 complex directly within the active site. Figure 4 illustrates the superimposed structures and spatial orientation of oleanolic acid, poinsettifolin B, and rhuschalcone III within the binding site of the complex using MOE software. While oleanolic acid and rhuschalcone III occupy a similar region of the binding pocket, rhuschalcone III forms two key hydrogen binds with residues Ala387 and Asn33 of the 6M0J protein complex, while no hydrogen bonds are observed with oleanolic acid. In contrast, poinsettifolin B, perhaps due to greater conformational flexibility, partially occupies the same region as the other two ligands but also extends into an adjacent sub-pocket where it forms a key hydrogen bond with residue Gln37 of the ACE2 site of the 6M0J complex. Interestingly, the number of hydrogen bonds by each ligand also correlates with their docking scores and ΔGbind values (Table 2).
To investigate further the specific interactions between each ligand and the SARS-CoV-2 RBD/host ACE2 complex, we next generated 2D interaction diagrams using Discovery Studio. As shown in Figure 5A, oleanolic acid forms two conventional hydrogen bonds with Gln409 and Gln406 in addition to a single π–alkyl interaction with His34 of the ACE2 site of the 6M0J complex. The 2D interaction diagram of poinsettifolin B also reveals stabilization through three conventional hydrogen bonds involving residues His34, Arg403, and Gly37, along with a π–π T-shaped interaction with His34, and π–alkyl interactions with Tyr505, Pro389, and His34 (Figure 5B). In contrast, rhuschalcone III forms four conventional hydrogen bonds with residues Asn33, Ala387, Tyr421, and Tyr505. In addition, two π–cation interactions are observed with Arg393 and Lys417. Other notable interactions include a π–anion interaction with Asp30, an amide–π stacking interaction with Gln388, and a π–alkyl interaction with Lys26 (Figure 5C). These results are also consistent with rhuschalcone III having the strongest binding affinity with the RBD/ACE2 complex through the highest number and diversity of specific interactions with key residues in the binding site.
When the binding sites of each compound were overlayed with the canonical RBD protein sequence, we found that oleanolic acid interactions with RBD did not overlap with mutations arising in multiple SARS-CoV-2 variants including Alpha (B.1.1.7), Beta (B.1.351), Delta (B.1.617.2) and Omicron (BA.1; B.1.1.529.1). In contrast, poinsettifolin B interacted with Tyr505, which is mutated to His in Omicron variants, while rhuschalcone III interacted with both Tyr505 and Lys417, which is additionally mutated to Asn in both Beta and Omicron variants (Figure 5D).
The observations of selective RBD/ACE2 disruption and detectable antiviral activity by rhuschalcone III led us to ask whether additional bichalcones of this large chemical family may also target the SARS-CoV-2 spike/ACE2 interaction. To investigate this, we searched the PubChem repository for analogues exhibiting ≥ 95% structural similarity to the parent scaffold rhuschalcone III. This search yielded 132 candidate molecules, which were subjected to molecular docking under identical experimental conditions as the reference rhuschalcone III. Table 3 reports the docking outcomes for the top 12 scoring molecules from this collection. From these results, one observes that the docking scores of the selected twelve analogues span from −6.024 to −7.495 kcal/mol, compared to the docking score of the parent rhuschalcone III of −6.00 kcal/mol (Table 2). Thus, all twelve analogues exhibit more favorable predicted binding affinities relative to rhuschalcone III. Further dissection of the major energy-terms reveals that the van der Waals component (GScorevdw) ranges from −34.448 to −45.336 kcal/mol, whereas the coulombic component (GScoreele) spans −4.110 to −18.201 kcal/mol.
These data indicate that van der Waals (i.e., hydrophobic/steric) interactions markedly dominate over electrostatic (Coulomb) contributions for all compounds. Notably, this trend was also observed with the reference rhuschalcone III, suggesting that hydrophobic and steric complementarity may play a predominant role in the binding of these compounds to the spike/ACE2 interface. We further re-scored the resultant complexes using the MM-GBSA approach. The calculated binding free energies (ΔGbind) varied from −36.703 to −52.311 kcal/mol across the different analogues. Except for the two compounds registered under PubChem IDs 170221756 and 146036329, all molecules exhibited more favorable ΔGbind values than that of rhuschalcone III (−42.29 kcal/mol). The solvation energy contribution ranged from +21.895 to +43.037 kcal/mol for the various complexes; these positive values, as seen also for rhuschalcone III, are consistent with a binding mode dominated by hydrophobic (i.e., desolvation/van der Waals) rather than polar interactions. Taken together, these findings strongly support the hypothesis that analogues within the rhuschalcone III family may constitute promising inhibitors of the SARS-CoV-2 spike/ACE2 complex.
3.4. Generative Modeling of p-ANAPL Hits
Figure 6 shows selected virtual (de novo)-designed hits, derived from generative modeling, having lower binding free energy values than the 3 active compounds from the p-ANAPL library. Five analogs have been selected per compound and the docking scores and ΔGbind values have been summarized in Table 4.
4. Discussion
Here we probed the pan-African Natural Products Library, a collection of over 500 physical compounds obtained from African medicinal plants for screening purposes, to identify NPs that disrupt SARS-CoV-2 spike RBD / host ACE2 protein interactions and/or SARS-CoV-2 replication in vitro. The three most active NPs – oleanolic acid, poinsettifolin B, and rhuschalcone III – all disrupted RBD/ACE2 interactions at low or sub-micromolar activities with selectivity over the unrelated PD-1/PD-L1 protein-protein interaction. Oleanolic acid and rhuschalcone III, but not poinsettifolin B, also inhibited SARS-CoV-2 replication in infected Vero cells at concentrations that did not induce substantial cytotoxicity. These compounds represent useful biological probes for elucidating detailed mechanisms of disrupting SARS-CoV-2 entry and replication, as well as structural leads that can serve as the basis for chemical derivatives with improved preclinical properties against SARS-CoV-2 and potentially other coronaviruses.
Oleanolic acid is a triterpene compound found in many fruits and medicinal plants and is best characterized by its antioxidant and anti-inflammatory properties [48,49]. It has multiple total synthesis methods and natural and synthetic derivatives [48], making it a promising starting scaffold for future drug development. It is also previously reported to interact with both RBD and ACE2 [50,51], although the RBD and ACE2 residues bound by oleanolic acid in these studies differ from ours and from each other, suggesting multiple binding poses that can confer its mechanism of action. It was also found to disrupt entry of SARS-CoV-2 spike-expressing pseudoviruses as well as in vitro SARS-CoV-2 replication at low micromolar concentrations [52]. Our study supports these previous observations and further shows that oleanolic acid has selectivity for RBD/ACE2 disruption over unrelated ligand-receptor interactions. It also validates our strategy to identify new anti-SARS-CoV-2 agents from NP libraries.
On the other hand, this is the first report describing poinsettifolin B and rhuschalcone III as SARS-CoV-2 inhibitors. Poinsettifolin B is a chalcone-type flavonoid originally isolated from Dorstenia poinsettifolia, an herb found in forests of Cameroon and Gabon and reported to be traditionally used to treat yaws and infected wounds [18,53,54]. While it has reported cytotoxicity in some cell lines [55], its bioactivities, as well as those of its few known analogs, are largely unknown. In contrast, rhuschalcone III is a bichalcone originally isolated from the root bark of Searsia pyroides (previously named Rhus pyroides), which is traditionally used in South Africa to manage epilepsy [19,56]. While extracts of S. pyroides have in vitro activities consistent with gamma-aminobutyric acid agonists and/or NMDA antagonists [56,57], these activities have not been established for rhuschalcone III. However, we previously reported that rhuschalcone IV and other bichalcone analogues can inhibit sirtuins [7], indicating that this family of compounds may have important therapeutic potential for multiple diseases and human disorders. Taken together, our observation that both poinsettifolin B and rhuschalcone III are selective inhibitors of RBD/ACE2 binding and/or SARS-CoV-2 replication assigns new bioactive properties to these compounds. In addition, the identification of multiple rhuschalcone III analogs with predicted favorable binding affinities against the spike/ACE2 complex suggests that rhuschalcone III itself could serve not only as a direct inhibitor of spike/ACE2 binding but also as a scaffold for the rational design of novel anti-COVID-19 agents.
Binding confirmations and 2D interaction diagrams indicate that all three compounds interact at least in part with residues in the RBD that have not undergone substantial mutation from the parental Wuhan sequence to subsequent variants of concern. In particular, oleanolic acid does not interact with any residue with mutations in variants, indicating that it is likely to continue to maintain antiviral activity during RBD sequence evolution over time. This is supported by a study indicating that oleanolic acid continues to inhibit cellular entry of pseudoviruses bearing Beta mutations in the RBD, as well as pseudoviruses with spike sequences from SARS-CoV-1 and MERS-CoV [52]. In contrast, the predicted interactions of poinsettifolin B and rhuschalcone III with Tyr505 and/or Lys417 suggest that these compounds may have reduced ability to disrupt RBD/ACE2 interactions from subsequent SARS-CoV-2 variants of concern, although this hypothesis remains to be tested.
The docking and MM-GBSA analyses of compounds exhibiting 95% structural similarity to rhuschalcone III revealed that many members of this chemical family might effectively disrupt the SARS-CoV-2 spike/ACE2 complex. Almost all selected analogues showed more favorable docking scores and binding free energies than the parent rhuschalcone III, suggesting enhanced affinity and thermodynamic stability. Energy decomposition indicated that van der Waals interactions predominated over electrostatic contributions, implying that hydrophobic and steric complementarity are key determinants of binding at the spike/ACE2 interface. These findings are consistent with previous studies reporting flavonoid derivatives, such as quercetin and curcumin analogues, as effective spike/ACE2 interaction inhibitors [58,59]. Overall, rhuschalcone III and its close analogues represent promising scaffolds for the rational design of small-molecule inhibitors targeting viral entry.
The REINVENT4 generator employed proposes similar molecules to the input molecules without fragmentation and automatically removes invalid structures, i.e., compounds that are chemically correct and synthetically non-tractable and non-druglike. After filtering these out, the proposed analogs from Reinvent were then ranked based on the computed ΔGbind values derived from PRIME MM-GBSA. For the oleanolic acid, we can observe that the major suggested changes for the analog with the lowest ΔGbind value (OLA_037) result from the reduction of the number of rings to only 3 rings and the introduction of a tertbutylated amide functionality to increase the number of H-bonds possible at the docking site. OLA_019 and OLA_033 both show the same functionality at different positions, while OLA_026 shows an ester substituent at the same position, with a reduction of the number of rings to 4, as in its predecessors. This implies one way of reducing oleanolic acid to become medicinal chemistry amenable is by the reduction of the number of rings and the introduction of polar moieties.
With respect to Rhuschalcone III, the proposed analogs show that the diphenyl ether moiety is kept intact throughout for the analogs with the lowest predicted binding affinities. The substitutions are proposed on the terminal phenol groups that make the parent molecule toxic. These have been proposed to be methylated to give the equivalent methoxy substitutions, while the caffeol moiety remains unchanged in most cases, except for RS_098, where it has been proposed that this be replaced by a keto group.
For the Poinsettifolin B analogs, suggestions include the introduction of an O-atom before the prenyl, thereby increasing the chances of H-bond formation. This is the case with PS-017 and PS-024. Other suggestions include the removal of the bulkier and more hydrophobic prenyl group, while smaller hydrophobic halogen atoms like Cl and Br. In the case of PS_017, position isomers of the dihydroxy-substituted phenone moiety from ortho-para to ortho-meta, thereby placing the bulkier prenyl substituent at the para position, where it could address hydrophobic amino acid residues like Phe486 from the RBD of SARS-CoV-2 spike.
5. Conclusions
In summary, we demonstrate that NPs obtained from African medicinal plants continue to be a rich source of novel antivirals, including against SARS-CoV-2 cellular entry and replication. Three compounds identified from screening the p-ANAPL here (oleanolic acid, poinsettifolin B, and rhuschalcone III) represent new chemical leads to develop future therapies as well as to further characterize medicinal plants, which may represent locally available options to manage SARS-CoV-2 and potentially future CoV infections in the absence of accessible Western vaccines and therapies.
Author Contributions
Conceptualization: F.N.K., I.T.; Formal analysis: M.J.M.T., P.A.O., B.D.B., J,Y,B., C.V.S., Funding acquisition: J.M.S., L.J.M., F.N.K., I.T., Investigation: M.J.M.T., P.A.O., J,Y,B., B.D.B., C.V.S., J.C., I.T., Methodology: J.C., Resources: K.A.M., Supervision: J.M.S., L.J.M., F.N.K., I.T., Writing – original draft: M.J.M.T., F.N.K., I.T. All authors have read and agreed to the published version of the manuscript.
Funding
Funding was provided by the Wistar Science Discovery Fund (L.J.M. and J.S.), the Canadian Institutes for Health Research (PJT-153057) (I.T.), the Bill & Melinda Gates Foundation through the Calestous Juma Science Leadership Fellowship (INV-036848) (F.N.K.), and the Gates Foundation and LifeArc under the African Drug Discovery Accelerator Program (INV-055897 and 10646) (F.N.K.). F.N.K. also acknowledges funding from the Alexander von Humboldt Foundation for a Research Group Linkage Project (1156361 CMR-IP). This work was also supported by the following grants to L.J.M.: the Robert I Jacobs Fund of the Philadelphia Foundation and the Herbert Kean, M.D., Family Professorship.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
MDPI Research Data Policies at https://www.mdpi.com/ethics.
Acknowledgments
The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACE2 | Angiotensin-converting enzyme 2 receptor |
| MM-GBSA | Molecular Mechanics Generalized Born Surface Area |
| p-ANAPL | pan-African Natural Products Library |
| RBD | receptor binding domain |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome coronavirus 2 |
References
- Cheuyem, F. Z. L.; Amani, A.; Achangwa, C.; Ajong, B. N.; Minkandi, C. A.; Zeh, M. M. M. K.; Ntsek, L. L. E.; Essomba, J. P.; Jiogue, R. C.; Ndagijimana, O.; Nchanji, N. E.; Danwang, C. COVID- 19 vaccine uptake and its determinants in Cameroon: a systematic review and meta-analysis (2021-2024). BMC Infectious Diseases 2025, 25(1), 525. [Google Scholar] [CrossRef] [PubMed]
- Fousseni, S.; Ngangue, P.; Barro, A.; Ramde, S. W.; Bihina, L. T.; Ngoufack, M. N.; Bayoulou, S.; Kiki, G. M.; Salfo, O. Navigating the road to immunization equity: systematic review of challenges in introducing new vaccines into Sub-Saharan Africa's health systems. Vaccines 2025, 13(3), 269. [Google Scholar] [CrossRef]
- Tupps, C.; Curry, D.; Edwards, A.; Bazant, E.; Moen, A.; Mounts, A. W.; Bresee, J. COVID-19 vaccination implementation in six lower- and middle-income countries: Successes, challenges, and lessons for pandemic preparedness. PLOS Global Public Health 2025, 5(5), e0004417. [Google Scholar] [CrossRef]
- Kim, J. K.; Kim, K. H.; Shin, Y. C.; Jang, B. H.; Ko, S. G. Utilization of traditional medicine in primary health care in low- and middle-income countries: a systematic review. Health Policy and Planning 2020, 35(8), 1070–1083. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Amoa Onguéné, P.; Fotso, G. W.; Andrae-Marobela, K.; Bezabih, M.; Ndom, J. C.; Ngadjui, B. T.; Ogundaini, A. O.; Abegaz, B. M.; Meva'a, L. M. Virtualizing the p-ANAPL library: a step towards drug discovery from African medicinal plants. PloS One 2014, 9(3), e90655. [Google Scholar] [CrossRef]
- Tietjen, I.; Ntie-Kang, F.; Mwimanzi, P.; Onguéné, P. A.; Scull, M. A.; Idowu, T. O.; Ogundaini, A. O.; Meva'a, L. M.; Abegaz, B. M.; Rice, C. M.; Andrae-Marobela, K.; Brockman, M. A.; Brumme, Z. L.; Fedida, D. Screening of the Pan-African natural product library identifies ixoratannin A-2 and boldine as novel HIV-1 inhibitors. PloS One 2015, 10(4), e0121099. [Google Scholar] [CrossRef]
- Karaman, B.; Alhalabi, Z.; Swyter, S.; Mihigo, S. O.; Andrae-Marobela, K.; Jung, M.; Sippl, W.; Ntie-Kang, F. Identification of bichalcones as sirtuin inhibitors by virtual screening and in vitro testing. Molecules 2018, 23(2), 416. [Google Scholar] [CrossRef]
- Duncan, M. C.; Onguéné, P. A.; Kihara, I.; Nebangwa, D. N.; Naidu, M. E.; Williams, D. E.; Balgi, A. D.; Andrae-Marobela, K.; Roberge, M.; Andersen, R. J.; Niikura, M.; Ntie-Kang, F.; Tietjen, I. Virtual screening identifies chebulagic acid as an inhibitor of the M2(S31N) viral ion channel and influenza A virus. Molecules 2020, 25(12), 2903. [Google Scholar] [CrossRef] [PubMed]
- Richard, K.; Schonhofer, C.; Giron, L. B.; Rivera-Ortiz, J.; Read, S.; Kannan, T.; Kinloch, N. N.; Shahid, A.; Feilcke, R.; Wappler, S.; Imming, P.; Harris, M.; Brumme, Z. L.; Brockman, M. A.; Mounzer, K.; Kossenkov, A. V.; Abdel-Mohsen, M.; Andrae-Marobela, K.; Montaner, L. J.; Tietjen, I. The African natural product knipholone anthrone and its analogue anthralin (dithranol) enhance HIV-1 latency reversal. The Journal of Biological Chemistry 2020, 295(41), 14084–14099. [Google Scholar] [CrossRef] [PubMed]
- Schonhofer, C.; Yi, J.; Sciorillo, A.; Andrae-Marobela, K.; Cochrane, A.; Harris, M.; Brumme, Z. L.; Brockman, M. A.; Mounzer, K.; Hart, C.; Gyampoh, K.; Yuan, Z.; Montaner, L. J.; Tietjen, I. Flavonoid-based inhibition of cyclin-dependent kinase 9 without concomitant inhibition of histone deacetylases durably reinforces HIV latency. Biochemical Pharmacology 2021, 186, 114462. [Google Scholar] [CrossRef]
- Dong, M.; Galvan Achi, J. M.; Du, R.; Rong, L.; Cui, Q. Development of SARS-CoV-2 entry antivirals. Cell Insight 2024, 3(1), 100144. [Google Scholar] [CrossRef]
- Tietjen, I.; Cassel, J.; Register, E. T.; Zhou, X. Y.; Messick, T. E.; Keeney, F.; Lu, L. D.; Beattie, K. D.; Rali, T.; Tebas, P.; Ertl, H. C. J.; Salvino, J. M.; Davis, R. A.; Montaner, L. J. The natural stilbenoid (-)-hopeaphenol inhibits cellular entry of SARS-CoV-2 USA-WA1/2020, B.1.1.7, and B.1.351 variants. Antimicrobial Agents and Chemotherapy 2021, 65(12), e0077221. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, L.; Moyo, P.; Cassel, J.; Isaacs, F. J.; Salvino, J. M.; Montaner, L. J.; Tietjen, I.; Maharaj, V. Use of hyphenated analytical techniques to identify the bioactive constituents of Gunnera perpensa L.; a South African medicinal plant, which potently inhibit SARS-CoV-2 spike glycoprotein-host ACE2 binding. Analytical and Bioanalytical Chemistry 2022, 414(13), 3971–3985. [Google Scholar] [CrossRef]
- Williams, D. E.; Cassel, J.; Zhu, J. L.; Yang, J. X.; de Voogd, N. J.; Matainaho, T.; Salvino, J. M.; Wang, Y. A.; Montaner, L. J.; Tietjen, I.; Andersen, R. J. Thorectidiol A isolated from the marine sponge Dactylospongia elegans disrupts interactions of the SARS-CoV-2 spike receptor binding domain with the host ACE2 Receptor. Journal of Natural Products 2023, 86(3), 582–588. [Google Scholar] [CrossRef]
- Matos-Hernández, M. L.; Samples, R.; Dyer, G.; Casimir Montán, V. M.; Morales-Colón, C. A.; Salvino, J. M.; Montaner, L. J.; Cassel, J. A.; Messick, T. E.; Tietjen, I.; Caro-Diaz, E. J. E. Metabolomic analysis and antiviral screening of a marine algae library yield jobosic acid (2,5-dimethyltetradecanoic acid) as a selective inhibitor of SARS-CoV-2. Journal of Natural Products 2024, 87(6), 1513–1520. [Google Scholar] [CrossRef]
- Szabó, D.; Crowe, A.; Mamotte, C.; Strappe, P. Natural products as a source of Coronavirus entry inhibitors. Frontiers in Cellular and Infection Microbiology 2024, 14, 1353971. [Google Scholar] [CrossRef]
- Abegaz, B.; Tecle, B. A new triterpenoid glycoside from the seeds of Glinus lotoides. Phytochemistry 1980, 19, 1553–1554. [Google Scholar] [CrossRef]
- Ngadjui, B.T.; Kapche, G.W.F.; Tamboue, H.; Abegaz, B.M.; Connolly, J.D. Prenylated flavonoids and a dihydro-4-phenylcoumarin from Dorstenia poinsettifolia. Phytochemistry 1999, 51, 119–123. [Google Scholar] [CrossRef]
- Mdee, L.K.; Yeboah, S.O.; Abegaz, B.M. Rhuschalcones II-VI, five new bichalcones from the root bark of Rhus pyroides. Journal of Natural Products 2003, 66, 599–604. [Google Scholar] [CrossRef]
- Divsalar, D.N.; Simoben, C.V.; Schonhofer, C.; Richard, K.; Sippl, W.; Ntie-Kang, F.; Tietjen, I. Novel histone deacetylase inhibitors and HIV-1 latency-reversing agents identified by large-scale virtual screening. Frontiers in Pharmacology 2020, 11, 905. [Google Scholar] [CrossRef] [PubMed]
- Eni, D. B.; Cassel, J.; Namba-Nzanguim, C. T.; Simoben, C. V.; Tietjen, I.; Akunuri, R.; Salvino, J. M.; Ntie-Kang, F. Design, synthesis, and biochemical and computational screening of novel oxindole derivatives as inhibitors of Aurora A kinase and SARS-CoV-2 spike/host ACE2 interaction. Medicinal Chemistry Research 2024, 33(4), 620–634. [Google Scholar] [CrossRef]
- Majoumo-Mbe, F.; Sangbong, N. A.; Tadjong Tcho, A.; Namba-Nzanguim, C. T.; Simoben, C. V.; Eni, D. B.; Alhaji Isa, M.; Poli, A. N. R.; Cassel, J.; Salvino, J. M.; Montaner, L. J.; Tietjen, I.; Ntie-Kang, F. 5-chloro-3-(2-(2,4-dinitrophenyl) hydrazono)indolin-2-one: synthesis, characterization, biochemical and computational screening against SARS-CoV-2. Chemical Papers 2024, 78(6), 3431–3441. [Google Scholar] [CrossRef]
- Namba-Nzanguim, C.T.; Simoben, C.V.; Bekono, B.D.; Tietjen, I.; Cassel, J.; Salvino, J.M.; Montaner, L.J.; Davis, R.A.; Ntie-Kang, F. Investigation of some plant stilbenoids and their fragments for the identification of inhibitors of SARS-CoV-2 viral spike/ACE2 protein binding. The Microbe 2024, 3, 100059. [Google Scholar] [CrossRef]
- Simoben, C. V.; Ghazy, E.; Zeyen, P.; Darwish, S.; Schmidt, M.; Romier, C.; Robaa, D.; Sippl, W. Binding free energy (BFE) calculations and quantitative structure-activity relationship (QSAR) analysis of Schistosoma mansoni histone deacetylase 8 (smHDAC8) inhibitors. Molecules 2021, 26(9), 2584. [Google Scholar] [CrossRef] [PubMed]
- Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E. The Protein Data Bank. Nucleic Acids Research 2000, 28(1), 235–242. [Google Scholar] [CrossRef]
- Burley, S. K.; Berman, H. M.; Kleywegt, G. J.; Markley, J. L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The single global macromolecular structure archive. Methods in Molecular Biology 2017, 1607, 627–641. [Google Scholar] [CrossRef]
- Burley, S. K.; Berman, H. M.; Christie, C.; Duarte, J. M.; Feng, Z.; Westbrook, J.; Young, J.; Zardecki, C. RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Science 2018, 27(1), 316–330. [Google Scholar] [CrossRef]
- Chemical Computing Group, Molecular Operating Environment (MOE) suite (version 2016.08; Montreal, QC, Canada).
- Schrödinger, Maestro, Glide release (version 2017-2; New York, NY, USA).
- Sastry, G. M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-aided Molecular Design 2013, 27(3), 221–234. [Google Scholar] [CrossRef]
- Shelley, J. C.; Cholleti, A.; Frye, L. L.; Greenwood, J. R.; Timlin, M. R.; Uchimaya, M. Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. Journal of Computer-aided Molecular Design 2007, 21(12), 681–691. [Google Scholar] [CrossRef]
- Banks, J. L.; Beard, H. S.; Cao, Y.; Cho, A. E.; Damm, W.; Farid, R.; Felts, A. K.; Halgren, T. A.; Mainz, D. T.; Maple, J. R.; Murphy, R.; Philipp, D. M.; Repasky, M. P.; Zhang, L. Y.; Berne, B. J.; Friesner, R. A.; Gallicchio, E.; Levy, R. M. Integrated Modeling Program, Applied Chemical Theory (IMPACT). Journal of Computational Chemistry 2005, 26(16), 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular Force Field. I. Basis, form, scope, parameterization, and performance of MMFF94. Journal of Computational Chemistry 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. Journal of Computational Chemistry 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. III. Molecular geometries and vibrational frequencies for MMFF94. Journal of Computational Chemistry 1996, 17, 553–586. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. Journal of Computational Chemistry 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, N.B. Merck Molecular Force Field. IV. Conformational energies and geometries for MMFF94. Journal of Computational Chemistry 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Watts, K. S.; Dalal, P.; Murphy, R. B.; Sherman, W.; Friesner, R. A.; Shelley, J. C. ConfGen: a conformational search method for efficient generation of bioactive conformers. Journal of Chemical Information and Modeling 2010, 50(4), 534–546. [Google Scholar] [CrossRef]
- Halgren, T. A.; Murphy, R. B.; Friesner, R. A.; Beard, H. S.; Frye, L. L.; Pollard, W. T.; Banks, J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. Journal of Medicinal Chemistry 2004, 47(7), 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- REINVENT4, available at https://github.com/MolecularAI/REINVENT4.
- Riniker, S.; Landrum, G. A. Better informed distance geometry: using what we know to improve conformation generation. Journal of Chemical Information and Modeling 2015, 55(12), 2562–2574. [Google Scholar] [CrossRef]
- Degen, J.; Wegscheid-Gerlach, C.; Zaliani, A.; Rarey, M. On the art of compiling and using 'drug-like' chemical fragment spaces. ChemMedChem 2008, 3(10), 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, H. H.; He, J.; Tibo, A.; Janet, J. P.; Voronov, A.; Mervin, L. H.; Engkvist, O. Reinvent 4: Modern AI-driven generative molecule design. Journal of Cheminformatics 2024, 16(1), 20. [Google Scholar] [CrossRef]
- Fialková, V.; Zhao, J.; Papadopoulos, K.; Engkvist, O.; Bjerrum, E. J.; Kogej, T.; Patronov, A. LibINVENT: Reaction-based generative scaffold decoration for in silico library design. Journal of Chemical Information and Modeling 2022, 62(9), 2046–2063. [Google Scholar] [CrossRef]
- Guo, J.; Knuth, F.; Margreitter, C.; Janet, J.P.; Papadopoulos, K.; Engkvist, O.; Patronov, A. Link-INVENT: generative linker design with reinforcement learning. Digital Discovery 2023, 2(1), 392–408. [Google Scholar] [CrossRef]
- Yasgar, A.; Jadhav, A.; Simeonov, A.; Coussens, N. P. AlphaScreen-based assays: ultra-high-throughput screening for small-molecule inhibitors of challenging enzymes and protein-protein interactions. Methods in Molecular Biology 2016, 1439, 77–98. [Google Scholar] [CrossRef]
- Skalniak, L.; Zak, K. M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; Krzanik, S.; Pyrc, K.; Dömling, A.; Dubin, G.; Holak, T. A. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8(42), 72167–72181. [Google Scholar] [CrossRef]
- Verma, N.; Raghuvanshi, D. S.; Singh, R. V. Recent advances in the chemistry and biology of oleanolic acid and its derivatives. European Journal of Medicinal Chemistry 2024, 276, 116619. [Google Scholar] [CrossRef]
- Günther, A.; Bednarczyk-Cwynar, B. Oleanolic acid: a promising antioxidant-sources, mechanisms of action, therapeutic potential, and enhancement of bioactivity. Antioxidants 2025, 14(5), 598. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Li, P. Screening of potential spike glycoprotein / ACE2 dual antagonists against COVID-19 in silico molecular docking. Journal of Virological Methods 2022, 301, 114424. [Google Scholar] [CrossRef]
- Avelar, M.; Pedraza-González, L.; Sinicropi, A.; Flores-Morales, V. Triterpene derivatives as potential inhibitors of the RBD spike protein from SARS-CoV-2: an in silico approach. Molecules 2023, 28(5), 2333. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Liu, Y.; Zhou, M.; Dong, S.; Hou, Y.; Jia, X.; Lan, X.; Zhang, Y.; Guo, J.; Xiao, G.; Wang, W. Screening of botanical drugs against SARS-CoV-2 entry reveals novel therapeutic agents to treat COVID-19. Viruses 2022, 14(2), 353. [Google Scholar] [CrossRef] [PubMed]
- Tsopmo, A.; Tene, M.; Kamnaing, P.; Ngnokam, D.; Ayafor, J.F.; Sterner, O. Geranylated flavonoids from Dorstenia poinsettifolia. Phytochemistry 1998, 48(2), 345–348. [Google Scholar] [CrossRef]
- Pollard, B.J.; Cheek, M.; Bygrave, P. New Dorstenia (Moraceae) discoveries in Western Cameroon. Kew Bulletin 2003, 58, 185–193. [Google Scholar] [CrossRef]
- Kuete, V.; Mbaveng, A. T.; Zeino, M.; Ngameni, B.; Kapche, G. D.; Kouam, S. F.; Ngadjui, B. T.; Efferth, T. Cytotoxicity of two naturally occurring flavonoids (dorsmanin F and poinsettifolin B) towards multi-factorial drug-resistant cancer cells. Phytomedicine 2015, 22(7-8), 737–743. [Google Scholar] [CrossRef]
- Stafford, G. I.; Pedersen, M. E.; van Staden, J.; Jäger, A. K. Review on plants with CNS-effects used in traditional South African medicine against mental diseases. Journal of Ethnopharmacology 2008, 119(3), 513–537. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Gavazzo, P.; Stafford, G. I.; Van Staden, J. South African plants used in traditional medicine to treat epilepsy have an antagonistic effect on NMDA receptor currents. Journal of Ethnopharmacology 2011, 137(1), 382–388. [Google Scholar] [CrossRef] [PubMed]
- Shanmugarajan, D.; P, P.; Kumar, B. R. P.; Suresh, B. Curcumin to inhibit binding of spike glycoprotein to ACE2 receptors: computational modelling, simulations, and ADMET studies to explore curcuminoids against novel SARS-CoV-2 targets. RSC Advances 2020, 10(52), 31385–31399. [Google Scholar] [CrossRef] [PubMed]
- Rosal, R. J. Z.; Paderes, M. C. Inhibiting SARS-CoV-2 viral entry by targeting spike:ACE2 interaction with O-modified quercetin derivatives. RSC Medicinal Chemistry 2024, 15(9), 3212–3222. [Google Scholar] [CrossRef]
Figure 1.
Structures of three NPs isolated from p-ANAPL which disrupt > 80% of RBD/ACE2 binding at 1 µg/mL.
Figure 1.
Structures of three NPs isolated from p-ANAPL which disrupt > 80% of RBD/ACE2 binding at 1 µg/mL.
Figure 2.
Dose-response curves of oleanolic acid (A), poinsettifolin B (B), and rhuschalcone III (C) indicating ability to disrupt binding of RBD and ACE2 (blue) or PD-1 and PD-L1 proteins (red).
Figure 2.
Dose-response curves of oleanolic acid (A), poinsettifolin B (B), and rhuschalcone III (C) indicating ability to disrupt binding of RBD and ACE2 (blue) or PD-1 and PD-L1 proteins (red).
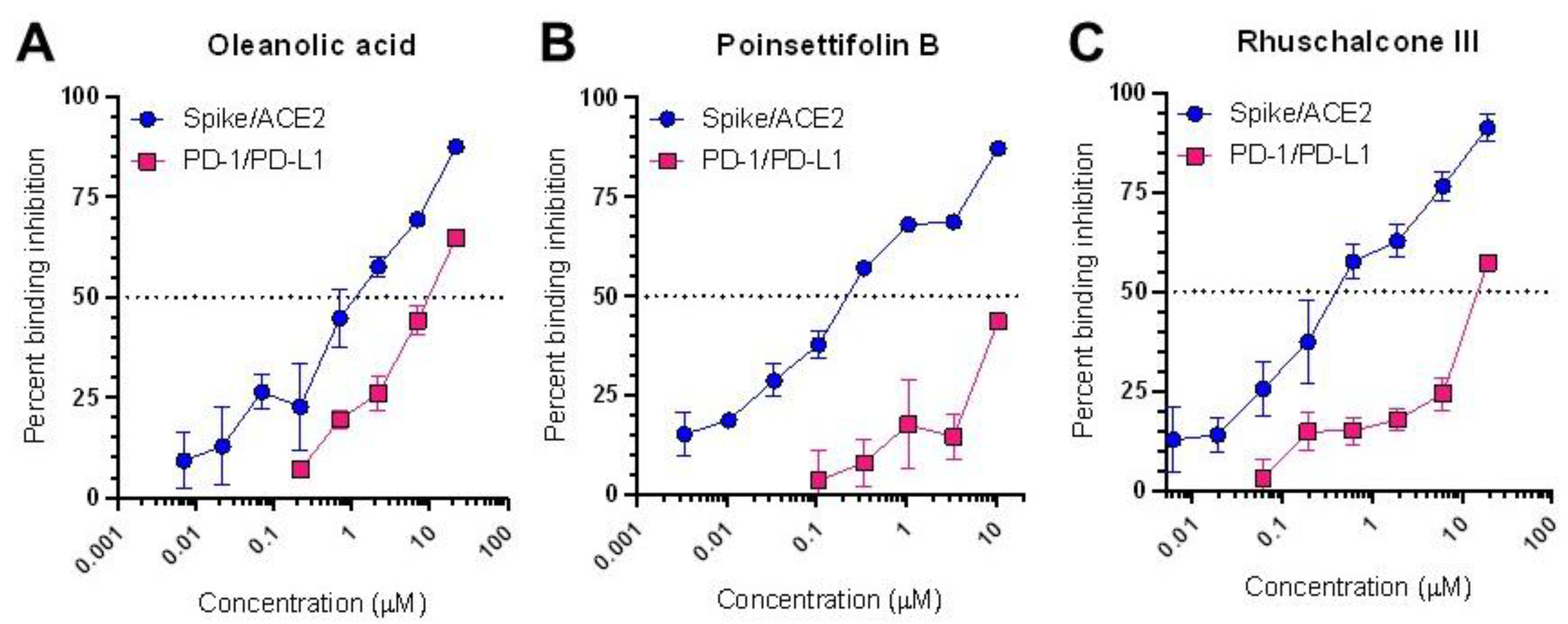
Figure 3.
Dose-response curves of oleanolic acid (A), poinsettifolin B (B), and rhuschalcone III (C) indicating ability to inhibit SARS-CoV-2 replication in infected Vero cells (green) and cell viability in uninfected Vero cells (violet).
Figure 3.
Dose-response curves of oleanolic acid (A), poinsettifolin B (B), and rhuschalcone III (C) indicating ability to inhibit SARS-CoV-2 replication in infected Vero cells (green) and cell viability in uninfected Vero cells (violet).
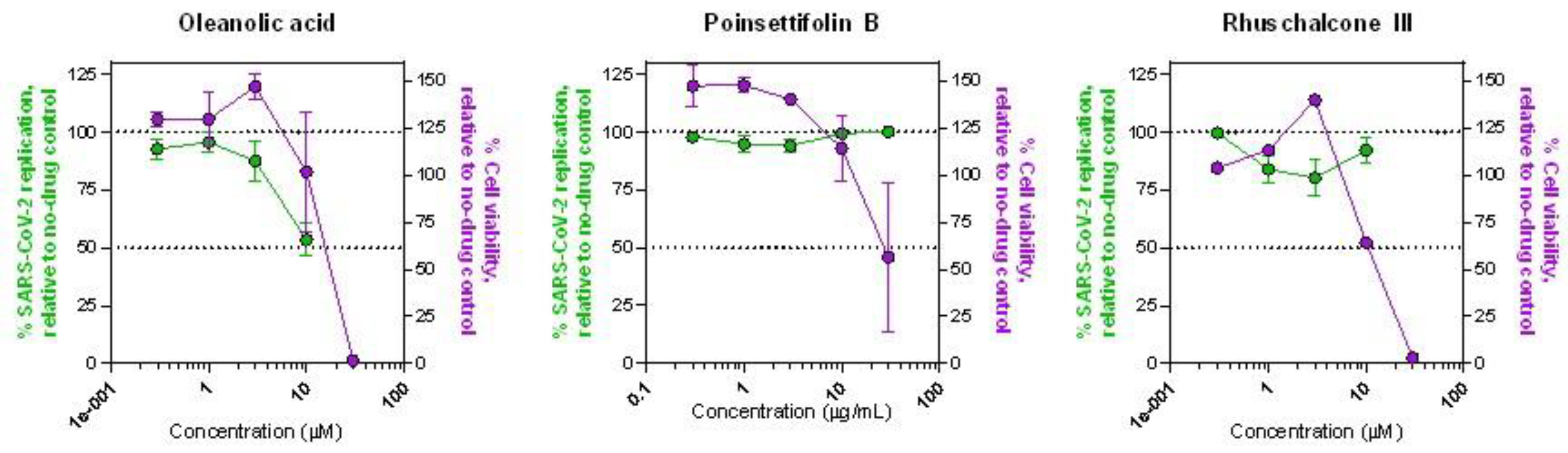
Figure 4.
Superposition of three NPs within the binding site of the RBD/ACE2 complex following molecular docking. Oleanolic acid is shown in cyan, poinsettifolin B is in green, and rhuschalcone III is in violet.
Figure 4.
Superposition of three NPs within the binding site of the RBD/ACE2 complex following molecular docking. Oleanolic acid is shown in cyan, poinsettifolin B is in green, and rhuschalcone III is in violet.
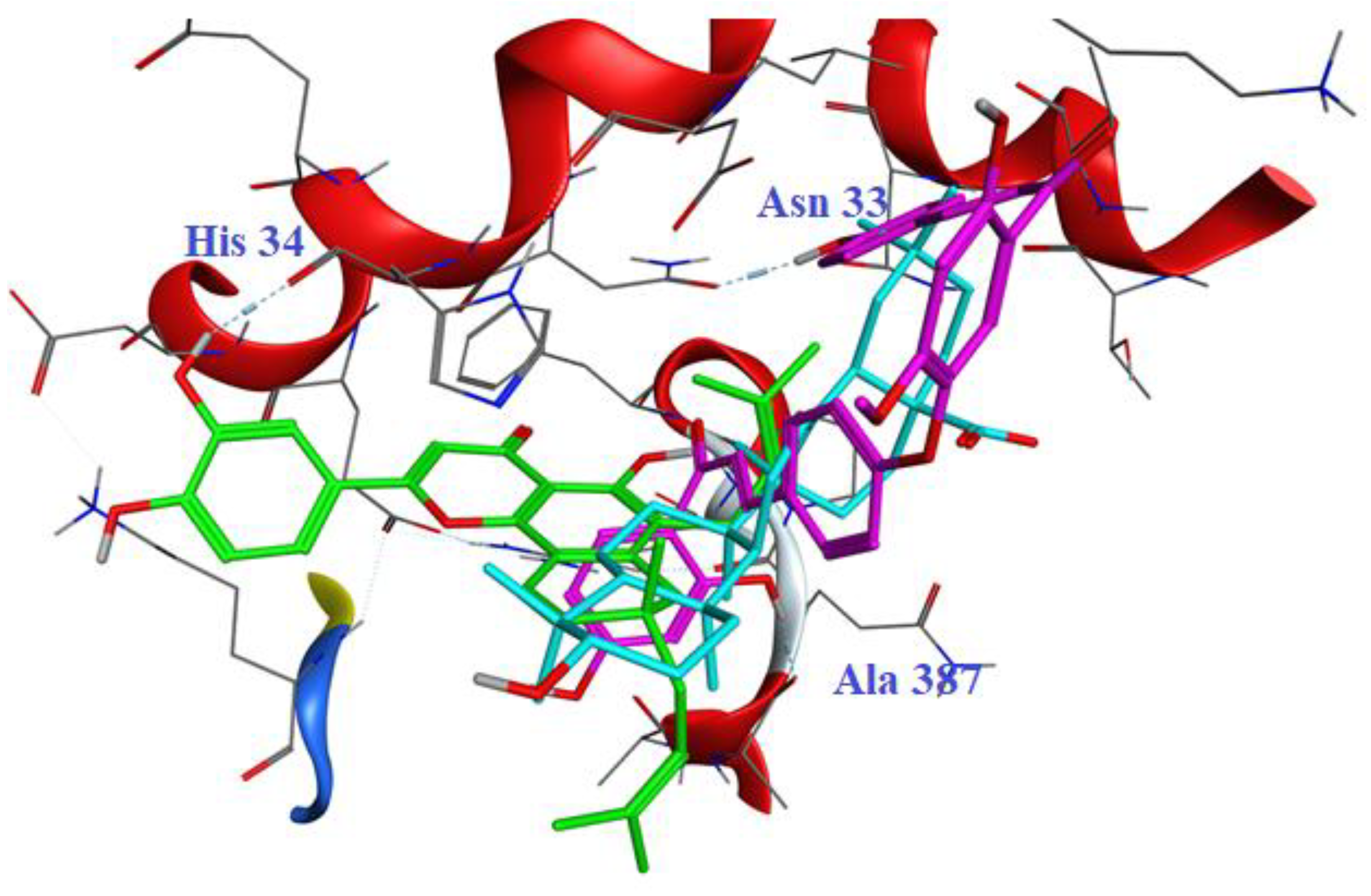
Figure 5.
2D interaction diagrams of oleanolic acid (A), poinsettifolin B (B), and rhuschalcone III (C) within the binding site of the spike RBD/ACE2 complex. D, Summary of binding contacts of three NPs within the canonical sequence of the parental SARS-CoV-2 Spike RBD (USA-WA1/2020). Interactions by oleanolic acid, poinsettifolin B, and rhuschalcone III with the RBD are highlighted in cyan, green, and violet, respectively. Sequence changes in variants of concern are highlighted in gray.
Figure 5.
2D interaction diagrams of oleanolic acid (A), poinsettifolin B (B), and rhuschalcone III (C) within the binding site of the spike RBD/ACE2 complex. D, Summary of binding contacts of three NPs within the canonical sequence of the parental SARS-CoV-2 Spike RBD (USA-WA1/2020). Interactions by oleanolic acid, poinsettifolin B, and rhuschalcone III with the RBD are highlighted in cyan, green, and violet, respectively. Sequence changes in variants of concern are highlighted in gray.
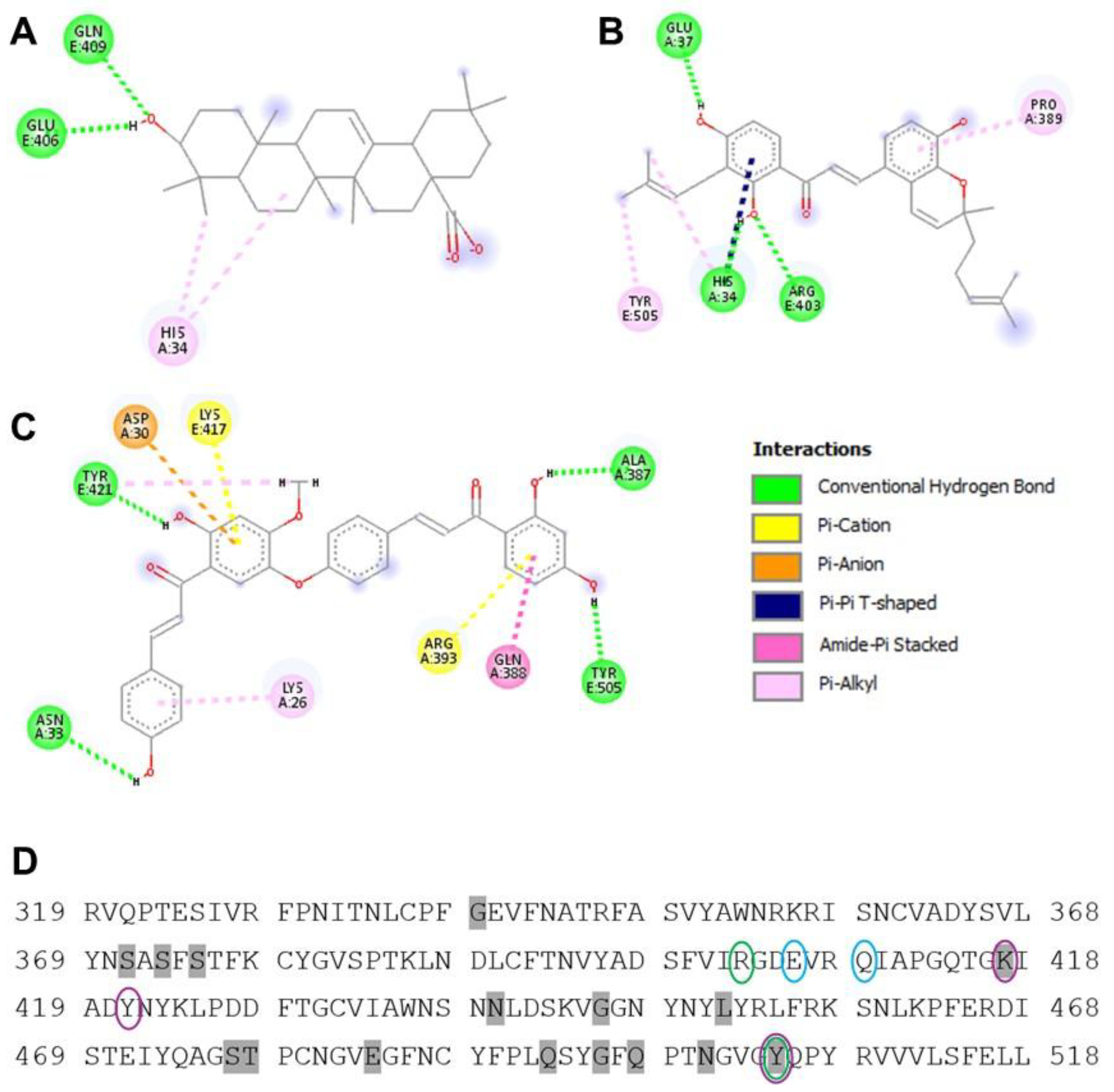
Figure 6.
Structures of the top de novo-generated hits based on MM-GBSA re-scoring.
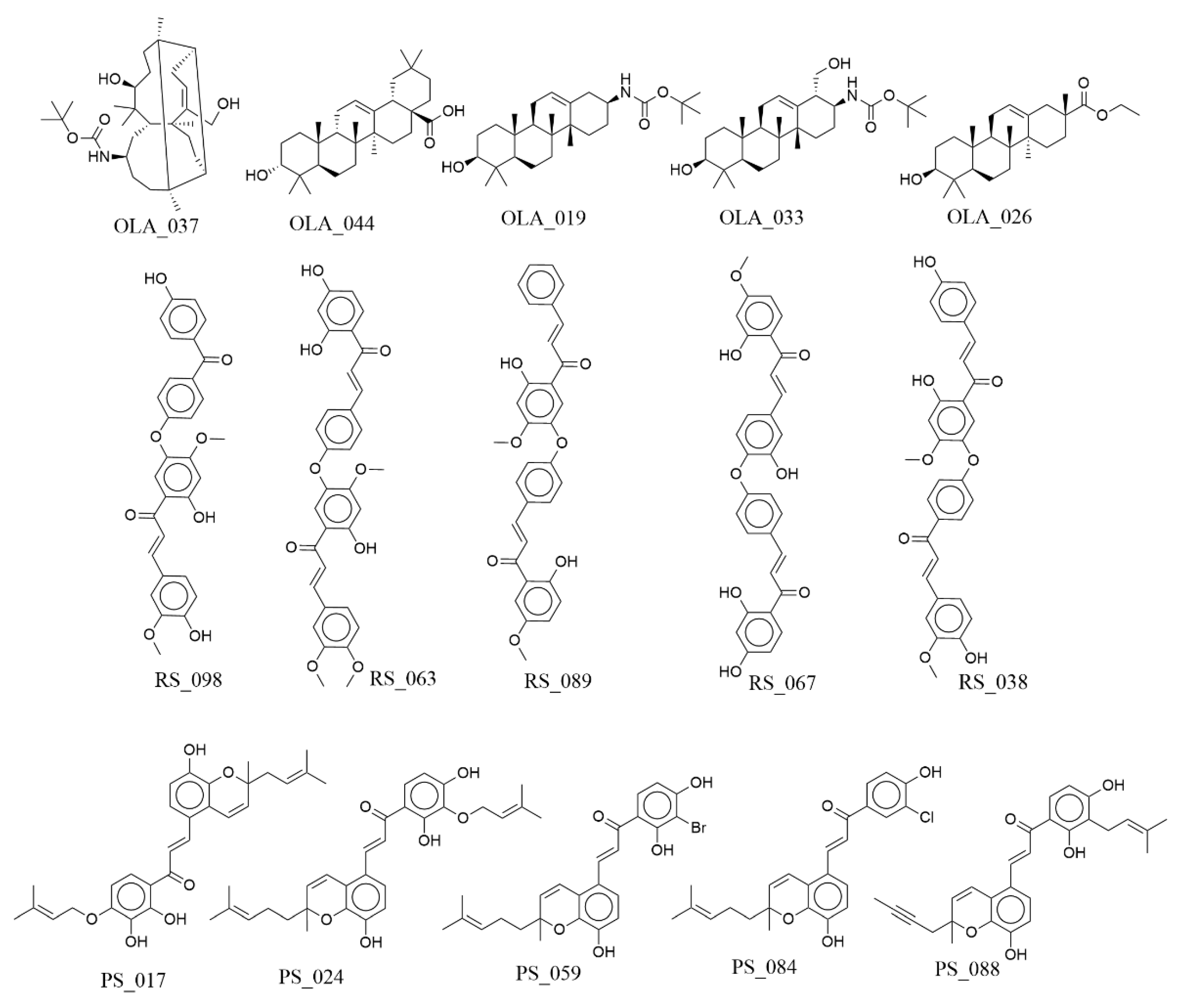

Table 1.
Compounds from p-ANAPL that inhibit at least 70% of SARS-CoV-2 Spike RBD/ACE2 binding at 1 µg/mL
Table 1.
Compounds from p-ANAPL that inhibit at least 70% of SARS-CoV-2 Spike RBD/ACE2 binding at 1 µg/mL
| Compound | Percent inhibition |
|---|---|
| Oleanolic acid | 83.5 |
| Poinsettifolin B | 81.7 |
| Rhuschalcone III | 80.8 |
| 4-Hydroxylonchocarpin | 78.0 |
| Agathisflavone | 76.9 |
| Pelargonidin chloride | 76.3 |
| 4’-Demethylknipholone | 75.5 |
| Knipholone 6-methylether | 72.2 |
| Trans-Hinokiresinol | 70.6 |
| 6,8-Diprenyleeriodictyol | 70.4 |
| Oleanolic acid | 83.5 |
| Poinsettifolin B | 81.7 |
Table 2.
Docking (GScore) score and ΔGbind (Prime MM-GBSA) values of NPs with the SARS-CoV-2 spike/ACE2 complex.
Table 2.
Docking (GScore) score and ΔGbind (Prime MM-GBSA) values of NPs with the SARS-CoV-2 spike/ACE2 complex.
| Complexes | Docking score (kcal/mol) | GScoreele | GScorevdW | ΔGbind (kcal/mol) |
|---|---|---|---|---|
| 6MOJ/oleanolic acid | -4.312 | -31.576 | -7.815 | -29.29 |
| 6MOJ/poinsettifolin B | -5.421 | -38.583 | -5.516 | -30.48 |
| 6MOJ/rhuschalcone III | -6.00 | -39.585 | -16.285 | -42.29 |
Table 3.
Docking (GScore) score and ΔGbind (Prime MM-GBSA) values for analogues exhibiting ≥ 95% structural similarity to the parent scaffold rhuschalcone III toward the SARS-CoV-2 spike/ACE2 complex.
Table 3.
Docking (GScore) score and ΔGbind (Prime MM-GBSA) values for analogues exhibiting ≥ 95% structural similarity to the parent scaffold rhuschalcone III toward the SARS-CoV-2 spike/ACE2 complex.
| Compounds (Pubchem ID) | Docking score (kcal/mol) | GScoreele (kcal/mol) | GScorevdw (kcal/mol) | ΔGbind (kcal/mol) | ΔGsolv (kcal/mol) |
|---|---|---|---|---|---|
| 170221756 | -7.495 | -16.388 | -41.448 | -36.836 | 39.572 |
| 13873817 | -6.642 | -11.028 | -40.496 | -52.311 | 32.504 |
| 13963933 | -6.637 | -8.429 | -40.146 | -48.614 | 30.961 |
| 118898656 | -6.516 | -10.908 | -39.159 | -49.981 | 32.289 |
| 173625139 | -6.516 | -10.908 | -39.159 | -49.981 | 32.289 |
| 163103777 | -6.365 | -18.201 | -36.598 | -44.766 | 40.004 |
| 162966847 | -6.217 | -10.216 | -39.451 | -46.213 | 43.037 |
| 10816879 | -6.210 | -5.267 | -45.338 | -47.241 | 38.689 |
| 142830944 | -6.175 | -4.796 | -41.924 | -48.102 | 29.427 |
| 404554 | -6.168 | -16.619 | -36.871 | -46.404 | 40.818 |
| 5472700 | -6.168 | -16.619 | -36.871 | -46.404 | 40.818 |
| 146036329 | -6.024 | -4.110 | -34.448 | -36.703 | 21.895 |
Table 4.
Docking scores and ΔGbind values of the top de novo-generated hits compared to the NP hits.
Table 4.
Docking scores and ΔGbind values of the top de novo-generated hits compared to the NP hits.
| Compound code | docking score (kcal/mol) | ΔGbind (kcal/mol) |
|---|---|---|
| OLA_037 | -4.012 | -36.47 |
| OLA_044 | -3.809 | -36.18 |
| OLA_019 | -4.974 | -36.16 |
| OLA_033 | -4.254 | -33.44 |
| OLA_026 | -4.812 | -31.48 |
| Oleanolic acid | -4.312 | -29.29 |
| RS_098 | -7.04 | -64.18 |
| RS_063 | -7.382 | -55.8 |
| RS_089 | -6.226 | -55.41 |
| RS_067 | -6.27 | -54.7 |
| RS_038 | -6.425 | -53.09 |
| Rhuschalcone III | -6.00 | -42.29 |
| PS_017 | -5.74 | -52.16 |
| PS_024 | -3.959 | -45.97 |
| PS_059 | -5.242 | -45.28 |
| PS_084 | -5.089 | -45.27 |
| PS_088 | -4.496 | -44.79 |
| Poinsettifolin B | -5.421 | -30.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



