Submitted:
18 February 2026
Posted:
18 February 2026
You are already at the latest version
Abstract
Background/Objectives: Tinnitus and hyperacusis can occur together or in isolation, with hyperacusis being asso-ciated with tinnitus much more frequently than vice versa. This striking correlation be-tween tinnitus and hyperacusis prevalence implicates that there might be a common origin such as a (hidden) hearing loss and possibly interrelated neural mechanisms of pathological development of those two conditions. Here, we propose such interrelated pathological mechanisms. Methods: This is a theoretical work based solely on considerations and published data. Results: We propose a model localized in the dorsal cochlear nucleus (DCN) of the brain-stem, that is based on classical mechanisms of Hebbian and associative plasticity known from classical conditioning. Specifically, our model proposes that hyperacusis results from synaptic enhancement of cochlear input to the DCN, whereas chronic tinnitus re-sults from synaptic enhancement of somatosensory input to the DCN. Specific conditions leading to one or the other condition are discussed. Conclusions: Our model predicts that hearing loss leads to chronic tinnitus, while noise exposure (which may also cause hearing loss) leads to hyperacusis. We would like to emphasize that our aim with the proposed model is not to provide a self-contained theo-retical construct, but to stimulate thought regarding possible pathological causes of tinni-tus and hyperacusis that have not yet been investigated. Individual assumptions that cannot yet be substantiated by existing literature are intended to provide impetus for fu-ture experimental studies.
Keywords:
Hebbian plasticity
; classical conditioning
; hearing disorder
; phantom perception
; stochastic resonance
1. Introduction
Subjective tinnitus is defined as the perception of a sound in the absence of any physical sound source [1] and often leads to an enormous psychic burden [2]. In Europe the tinnitus prevalence ranges from 8,7 % to 28,3 %, with a prevalence of 12% in Germany [3]. Approximately 2% of tinnitus patients severely suffer from tinnitus and related comorbidities such as concentration issues, stress, and depression [2,4]. Interestingly, 30% to 80% of tinnitus patients suffer from another pathological condition, namely hyperacusis [5,6]. Hyperacusis refers to a reduced tolerance to sounds, which are perceived as normally loud by the majority of the population [7]. However, up to now there is no commonly accepted scientific definition [7]. In contrast to tinnitus patients where 8% to 28 % suffer from hyperacusis, only 5% to 10% of the general population suffers from hyperacusis [6]. Vice versa, 86% of the hyperacusis patients suffer from tinnitus [8]. In other words, patients may suffer exclusively from tinnitus, exclusively from hyperacusis or from both pathological conditions. The fact that both conditions can occur in isolation suggests that tinnitus and hyperacusis are caused by disjoint neural mechanisms [9]. However, the striking correlation between tinnitus and hyperacusis prevalence implicates that there might be a common origin such as a (hidden) hearing loss [10,11,12] and possibly interrelated neural mechanisms of pathological development [9].
In this theoretical paper, we propose such interrelated pathological mechanisms, localized in the brainstem, that are based on classical mechanisms of Hebbian and associative plasticity known from classical conditioning. Our model does not require any additional assumptions to explain the development of tinnitus and/or hyperacusis and therefore does not make any predictions or assumptions about possible mechanisms upstream the dorsal cochlear nucleus (DCN), like gating problems in the thalamus [13] or conscious cortical perception of a phantom sound [14,15,16]. Furthermore, we will use the term hyperacusis in the sense of recruitment [8,17], which refers to the characteristic feature that mild sounds are over-amplified along the auditory pathway and perceived as too loud [9,18]. The model presented here is a further development of the Erlangen Model of Tinnitus Development.
1.1. The Erlangen Model of Tinnitus Development
There is a broad consensus that tinnitus as well as hyperacusis are induced by some kind of hearing loss [11,12,19]. The hearing loss is based on a reduction in the innervation of the inner hair cells of the cochlea [20], which can be so slight that no increase in the hearing thresholds can be measured in the audiogram (“hidden hearing loss”, cf. [21]). However, the result is still a reduced input from the cochlea into the DCN, which can already lead to weak signals no longer triggering a supra-threshold reaction of the corresponding neurons in the DCN and thus no longer being transmitted to the subsequent auditory pathway. Additionally, tinnitus is characterized by an increased spontaneous neural activity along the auditory pathway starting at the [18,22]. However, tinnitus is not related to an increased stimulus evoked-activity, whereas hyperacusis in contrast is indeed characterized by increased stimulus-evoked activity [23,24]. Additionally, it is well known that tinnitus can occur within seconds after a short transient hearing loss [25], which is far too fast for plastic adaptations like homeostatic plasticity or so- called central gain mechanisms [10], which is a comparatively slow process on scales of hours or days [26,27]. In 2016, we proposed a mechanism based on a simple feedback loop that can cause the tinnitus related hyperactivity in the brainstem (The Erlangen Model of Tinnitus Development, [28,29]). The idea of that mechanism (cf. Figure 1) is that a signal coming from the cochlea (blue) which is subthreshold, e.g., due to hearing loss, is lifted above the detection threshold (dashes line) by means of stochastic resonance (SR), that is by adding neural noise which is believed to come from the somatosensory system (green, cf. [30]) to the cochlear input. By that, the transmission of weak signals into the auditory pathway is enabled again and thus hearing is improved overall. This improvement of hearing which is believed to be fine-tuned on a millisecond scale and to constantly operate even in the healthy auditory system. In case of tinnitus, the neuronal somatosensory noise that is added to the cochlear input itself is strong enough to cross the threshold of the fusiform cells (FC) of the DCN and by that is perceived as tinnitus. In other words, from the perspective of the Erlangen model, tinnitus is a by-product of a mechanism designed to constantly optimize the hearing process.
To achieve this optimization, the amplitude of the neural somatosensory noise that is added to the auditory input is fine-tuned through a feedback loop that maximizes information transmission into the auditory system by maximizing the autocorrelation function (ACo) and by that the information content of the DCN output [28,29,31]: If this amplitude is too low, the sum of cochlear and somatosensory input does not reach the threshold of the DCN neurons; if it is too high, the signal is masked by the noise. According to our model, the DCN uses so-called delay lines [32] for this purpose, which superimpose a signal on itself with a time delay and can thus recognize regularly occurring patterns at fixed time intervals (which are not to be expected in noise). Unlike Licklider's original model, the delay line in our context does not need to detect any delay (which would require a series of coincidence detectors representing different time delays), but only one coincidence detector per frequency channel, since the expected time delay corresponds to the characteristic frequency of that channel (cf. Figure 2). Therefore, the output of the yellow neuron is fed directly to a coincidence detector (red neuron) and with a frequency-specific time delay corresponding to the time interval to be detected (via the blue delay neuron). The red neuron only reacts if it simultaneously receives input from the direct and the time-delayed projection, i.e., exactly when two action potentials occur with the desired time interval, which corresponds to the characteristic frequency in the respective frequency channel.
To give a rough estimate whether the computation of an autocorrelation function could plausibly be supported by dendritic processing in the human dorsal cochlear nucleus, we consider the following: The calculation of the autocorrelation value needed for the SR effect, requires for a signal frequency f, a time delay of 1/f, which means e.g., for a 1kHz tone a time delay of 1ms. Assuming an average dendritic conduction velocity of approximately 0.5 m/s, measured by Larkum and co-workers in 1996 [39], and that the time delay is exclusively generated through differences in dendritic path length, a length difference of approximately 8mm would permit to process frequencies down to 62 Hz. 8mm is approximately the size of the longest dendrites in the cat DCN [40]. For a 20 Hz tone a length difference of 25 mm would be needed. Thus, for lower frequency an intermediate neuron/synapse might be necessary. By contrast, calculating the autocorrelation of a 5 kHz signal would require length differences of only 100 µm, demonstrating that the upper frequency range is not limited by dendritic lengths but rather the precision of the spike timing of the input signal. AAN = auditory afferent nerve; FC = fusiform cell; PF = parallel fiber; SSC = superficial stellate cell. Figure adopted from [29].
For a characteristic frequency of 1 kHz, for example, this interval would be one millisecond. Gap junctions together with adjusted fiber length within this feedback loop allow for fine tuning to the desired interval. Finally, the coincidence detector in turn inhibits the somatosensory input. The greater the information content of the DCN output, the less noise from the somatosensory system has to be added to the cochlear input to ensure optimal information transmission.
1.2. Classical Conditioning
In classical conditioning, the association between and unconditioned stimulus (US) that is able to induce a naturally occurring unconditioned response (UR), and a conditioned stimulus (CS) is learned. In Pavlov’s original work the salivation of a dog is the UR that follows the presentation of food (US). The ringing of a bell (CS) is able to induce the same response if the CS is repeatedly presented prior to the US [41]. This response to the bell is therefore called conditioned response (CR). Here we now incorporate the mechanistic principles known from classical conditioning into our model, so that it is not only able to explain the initial development of acute tinnitus perception, but also the pathological conditions of chronic tinnitus and hyperacusis, at the level of the DCN. Here we make no statements on higher brain structures and top-down mechanisms.
2. Materials and Methods
This is a theoretical work based solely on considerations and published data.
3. Results
In this theoretical paper, we put forward the hypothesis that the Erlangen model for tinnitus development described in Figure 1 and Figure 2 is also able to explain the development of hyperacusis. Our hypothesis is based on the notion that the essential connectivity pattern of two converging inputs into the DCN, namely one from the cochlea and one from the somatosensory (trigeminal) system (cf. Figure 1), mimics the basic neuronal circuit known from classical conditioning. The central idea of the theory is that – analogously to classical conditioning – the weights of these inputs to the DCN can be enhanced by means of synaptic plasticity triggered by certain input conditions and especially the relative timing between them. As a result, amplification of the cochlear input to the DCN would result in hyperacusis (HA, Figure 3, upper right panel) while amplification of the somatosensory input would lead to chronic tinnitus (CT, Figure 3, lower right panel). While such plasticity has already been demonstrated for the somatosensory input, evidence for such plasticity in the auditory input is still pending [42].
3.1. Development of Chronic Tinnitus
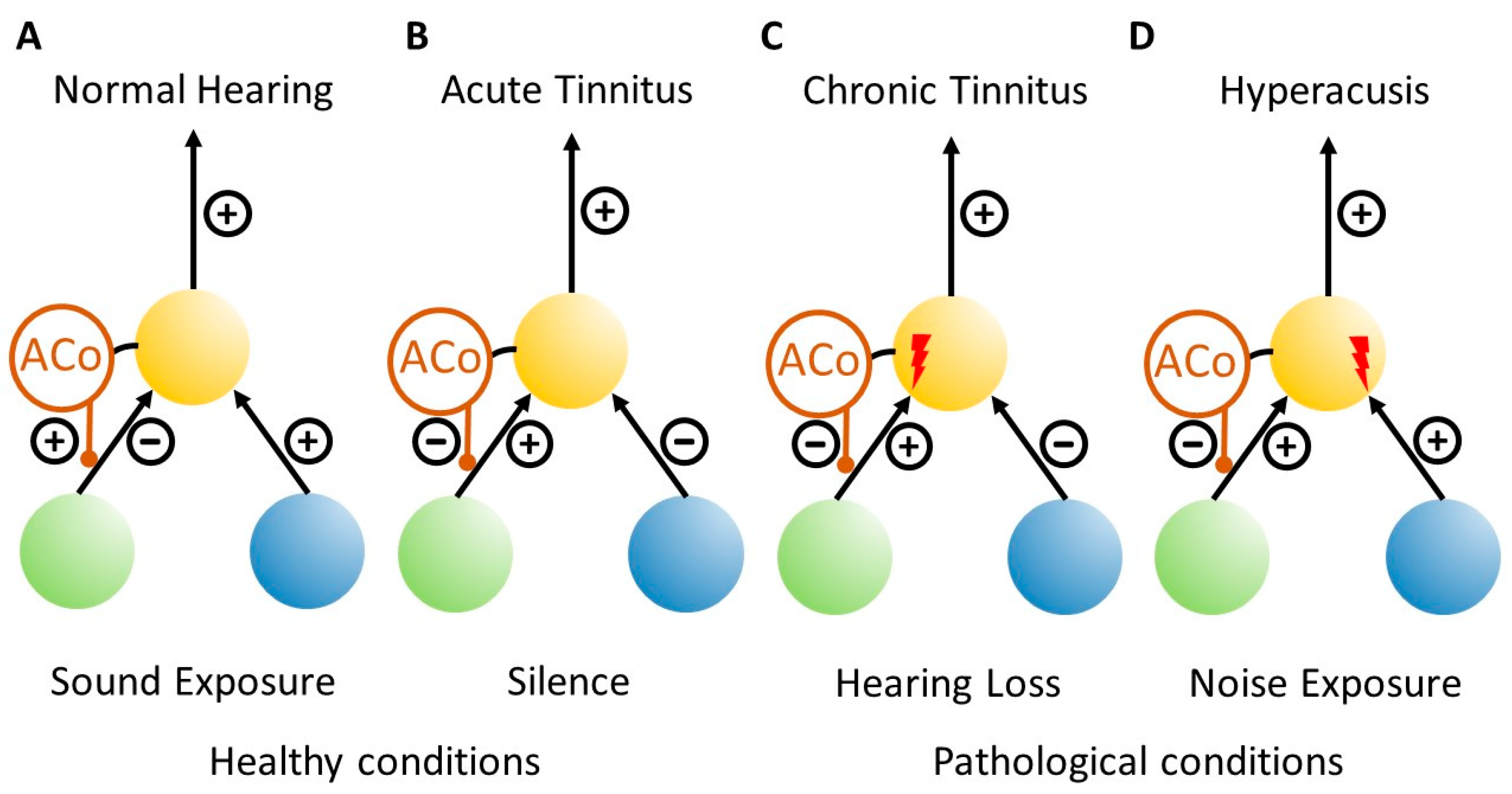
In our Erlangen model of tinnitus development, we have described how tinnitus-related hyperactivity develops by means of stochastic resonance (cf. Figure 1), but we have not yet explained how the initial occurrence of such hyperactivity, i.e., acute tinnitus, can eventually lead to the condition of chronic tinnitus. In our model, acute tinnitus (Figure 4B) occurs if the cochlear input to the DCN is reduced. This can be the case, for example, in complete silence in an anechoic chamber, where people experience transient tinnitus as long as they remain in silence. In the clinically relevant case of reduced cochlear input due to hearing loss, chronic tinnitus (Figure 4C) may develop if the hearing loss is permanent. In this case, according to our model, the reduced cochlear input leads to a reduced information transmission into the auditory system which is detected by the DCN circuit in the form of reduced autocorrelation of the DCN output (cf. Figure 1 and Figure 4C). The reduced autocorrelation causes a disinhibition of the somatosensory input to the DCN.
In classical conditioning, the conditioned stimulus (CS) must precede the unconditioned stimulus (US) in order to induced synaptic plasticity that results in conditioned responses (CR), i.e., strengthens the synaptic weight of the CS input [41]. Applied to our model, this means that if cochlear input (analogue the US) that is reduced due to permanent hearing loss reaches the DCN neuron that is already activated by the disinhibited somatosensory input (analogue the CS) then the synaptic weight of the somatosensory input would be increased (Figure 3, xm; Figure 4C, red flash). As a result, this input alone becomes strong enough to drive the DCN neuron above threshold, generating an output that is transmitted upstream into the auditory pathway and can finally be perceived as tinnitus (analogue the CR).
3.2. Development of Hyperacusis
If we now take this model further, hyperacusis (Figure 4D) can develop if the cochlear input to the DCN is amplified rather than the somatosensory input (Figure 3, xn; Figure 4D, red flash). For this to happen, the relative timing of the two inputs is crucial [41]. This means that if the cochlear input is to be amplified, it must precede the somatosensory input. This would occur in case of continuous noise exposure, where the cochlear input is constantly activated, but due to the uncorrelated nature of the noise, the autocorrelation of the DCN output would still be low, which also leads to disinhibition of the somatosensory input. Therefore, in contrast to the permanent hearing loss condition described above, where activation of somatosensory input precedes cochlear input, this relative timing of inputs to the DCN would be (statistically) reversed in the case of continuous noise exposure, resulting in increased synaptic weight of cochlear input and consequently hyperacusis.
4. Discussion
Tinnitus and hyperacusis can occur separately or together, which is easily explained by our model as it links the two pathologies to the plastic amplification of two separate synapses: according to the model, amplification of the cochlear input to the DCN leads to hyperacusis, while amplification of the somatosensory input to the DCN leads to tinnitus. In tinnitus, these changes during an acute phase depend on the autocorrelation of the output activity of the fusiform cells, which reflects information content and which is calculated within the DCN to modulate the input from the somatosensory system via a feedback loop (cf. Figure 1). Once this somatosensory input is permanently amplified by LPT, as described in our extension of the Erlangen model presented here, acute tinnitus would turn into chronic tinnitus.
In cases where both inputs to the DCN, the somatosensory and the auditory, are amplified, tinnitus and hyperacusis occur together. In such cases, interestingly, when both conditions co-occur, increases in sound-evoked activity can be found throughout the auditory pathway [23,43]. This phenomenon is also consistent with our model, as the DC shift and the greater steepness of the rate-intensity-functions (Figure 3, insets on the right) in tinnitus and hyperacusis are superimposed here. In patients who only have tinnitus but no hyperacusis, this superimposition and thus the additional amplification factor of the cochlear input is missing. As our model predicts, the two pathologies may reinforce each other, which is consistent with clinical data [44,45].
Interestingly, a recent paper on tinnitus and hyperacusis in rats [46] reported that tinnitus symptoms were typically correlated with responses to low-intensity tones, while neural correlates of hyperacusis were found in response to moderate- and high-intensity tones. This observation—although made at the level of the auditory cortex—would be consistent with our model, since tinnitus there is based on stochastic resonance in the DCN, which raises low-level signals above the neural response threshold of fusiform cells, while hyperacusis presumably further amplifies signals that would be above the threshold anyway.
Central to the unified theory for tinnitus and hyperacusis development presented here is the hypothesis that both the development of tinnitus and hyperacusis are based on synaptic reinforcements - i.e. long term potentiation (LTP). In favor of this view is the finding, that in bimodal neurons in the DCN, that is, those (fusiform) neurons that receive both auditory as well as somatosensory input, an increase in response amplitudes to somatosensory input has been observed after noise-induced hearing loss in guinea pigs [47]. Furthermore, the same study reported an enhancement of bimodal integration in those cells.
A direct and testable prediction from our model is that no tinnitus should develop if the development of LTP in the DCN were to be prevented. In particular, tinnitus should not develop if the LTP at the somatosensory input synapses were blocked. In fact, it has already been shown that this does indeed appear to be the case: Tagoe and colleagues [48] have demonstrated that blocking LTP at parallel fiber input to DCN by a NMDA receptor antagonist or increasing extracellular calcium concentration reduces behavioral signs of tinnitus in rats after acoustic over-exposure. The fact that NMDA receptor blockers have not yet proven effective in clinical tinnitus therapy [49,50,51] may be due to the fact that, according to our model, their use should only be effective in an early acute phase of tinnitus development and that local administration to the DCN might be necessary.
Finally, we would like to point out that our model makes no assumptions about what happens along the auditory pathway beyond the DCN, since there are other models published that deal with these higher-order phenomena, such as the predictive coding model [14,52] or gating models that involve the limbic system [13,53]. Our model therefore only explains how the pathophysiological neuronal activity initially develops in the periphery, which may then be perceived as tinnitus (or hyperacusis) when it is propagated to the auditory cortex. If it is not propagated to the auditory cortex, e.g. due to some kind of gating mechanism, tinnitus is not perceived even in the presence of the pathophysiological activity in the peripheral auditory pathway. This view also explains why not all patients with hearing loss experience tinnitus. An overview of how the various peripheral and central mechanisms involved in tinnitus may be related to each other can be found in the work of Schilling and colleagues [9].
5. Conclusions
Our model offers a new, very simple explanation for the development of both chronic tinnitus and hyperacusis and is able to describe possible interactions between the two pathologies. To the best of our knowledge, our theory is the first that links and mechanistically explains LTP in the DCN with principles of classical conditioning and the development of tinnitus and hyperacusis. As far as we know, it is also the first model that describes a developmental mechanism purely in the DCN and without the involvement of higher brain areas. The fact that the plastic synaptic changes postulated here are based on mechanisms that are analogous to known learning phenomena such as classical conditioning potentially opens up the possibility to specifically reverse the pathophysiological processes described and thus contribute to a genuine cure for tinnitus and hyperacusis. One conceivable approach here would be to revive well-known concepts such as Jastreboff's tinnitus retraining strategy [54] in an adapted form or pharmacological interventions aimed at blocking LTP in the DCN in a critical time window after noise trauma or inducing LTD at the respective DCN input synapses after tinnitus and / or hyperacusis has become chronic.
Author Contributions
Conceptualization, H.S. and A.Swriting—original draft preparation, H.S.; writing—review and editing, H.S. and A.S.; visualization, H.S.; funding acquisition, A.S. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This work was funded by the Deutsche Forschungsgemeinschaft (DFG) grant SCHI 1482/6-1 (pro-ject number 563909707) to AS.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Acknowledgments
During the preparation of this manuscript, the author(s) used deepl.com for the purposes of linguistic revision. The authors have reviewed and edited the output and take full responsibility for the content of this publication. We would like to thank Dr. Konstantin Tziridis for his assistance in formatting the references.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AAN | auditory afferent nerve |
| ACo | autocorrelation |
| CR | conditioned response |
| CS | conditioned stimulus |
| CT | chronic tinnitus |
| DCN | dorsal cochlear nucleus |
| FC | fusiform cells |
| HA | hyperacusis |
| LTD | Long term depression |
| LTP | long term potentiation |
| NMDA | N-methyl-D-aspartate |
| PF | parallel fiber |
| SR | stochastic resonance |
| SSC | superficial stellate cell |
| UR | unconditioned response |
| US | unconditioned stimulus |
References
- Prengel, J.; Dobel, C.; Guntinas-Lichius, O. Tinnitus. Laryngo-Rhino-Otologie 2023, 102, 132-145. [CrossRef]
- Boecking, B.; von Sass, J.; Sieveking, A.; Schaefer, C.; Brueggemann, P.; Rose, M.; Mazurek, B. Tinnitus-related distress and pain perceptions in patients with chronic tinnitus–Do psychological factors constitute a link? PloS one 2020, 15, e0234807. [CrossRef]
- Biswas, R.; Lugo, A.; Akeroyd, M.A.; Schlee, W.; Gallus, S.; Hall, D.A. Tinnitus prevalence in Europe: a multi-country cross-sectional population study. The Lancet Regional Health - Europe 2022, 12, 100250. [CrossRef]
- Neff, P.; Simões, J.; Psatha, S.; Nyamaa, A.; Boecking, B.; Rausch, L.; Dettling-Papargyris, J.; Funk, C.; Brueggemann, P.; Mazurek, B. The impact of tinnitus distress on cognition. Scientific reports 2021, 11, 1-9. [CrossRef]
- Cederroth, C.R.; Lugo, A.; Edvall, N.K.; Lazar, A.; Lopez-Escamez, J.-A.; Bulla, J.; Uhlen, I.; Hoare, D.J.; Baguley, D.M.; Canlon, B. Association between hyperacusis and tinnitus. Journal of Clinical Medicine 2020, 9, 2412. [CrossRef]
- Pienkowski, M. Rationale and efficacy of sound therapies for tinnitus and hyperacusis. Neuroscience 2019, 407, 120-134. [CrossRef]
- Bigras, C.; Villatte, B.; Duda, V.; Hébert, S. The electrophysiological markers of hyperacusis: a scoping review. International journal of audiology 2023, 62, 489-499. [CrossRef]
- Baguley, D.M. Hyperacusis: an overview. In Proceedings of the Seminars in Hearing, 2014; pp. 074-083.
- Schilling, A.; Sedley, W.; Gerum, R.; Metzner, C.; Tziridis, K.; Maier, A.; Schulze, H.; Zeng, F.-G.; Friston, K.J.; Krauss, P. Predictive coding and stochastic resonance as fundamental principles of auditory phantom perception. Brain 2023. [CrossRef]
- Schaette, R.; McAlpine, D. Tinnitus with a normal audiogram: physiological evidence for hidden hearing loss and computational model. J Neurosci 2011, 31, 13452-13457. [CrossRef]
- Noreña, A.J.; Chery-Croze, S. Enriched acoustic environment rescales auditory sensitivity. Neuroreport 2007, 18, 1251-1255. [CrossRef]
- Knipper, M.; Van Dijk, P.; Nunes, I.; Ruttiger, L.; Zimmermann, U. Advances in the neurobiology of hearing disorders: recent developments regarding the basis of tinnitus and hyperacusis. Prog Neurobiol 2013, 111, 17-33. [CrossRef]
- Rauschecker, J.P. The Frontostriatal Gating Model of Tinnitus. In Textbook of Tinnitus; Springer: 2024; pp. 221-230.
- Sedley, W.; Friston, K.J.; Gander, P.E.; Kumar, S.; Griffiths, T.D. An integrative tinnitus model based on sensory precision. Trends in neurosciences 2016, 39, 799-812. [CrossRef]
- Schilling, A.; Krauss, P. The Bayesian brain: world models and conscious dimensions of auditory phantom perception. 2024, 132, 317-318. [CrossRef]
- Yasoda-Mohan, A.; Chen, F.; Ó Sé, C.; Allard, R.; Ost, J.; Vanneste, S. Phantom perception as a Bayesian inference problem: a pilot study. Journal of Neurophysiology 2024, 131, 1311-1327. [CrossRef]
- Zeng, F.-G. An active loudness model suggesting tinnitus as increased central noise and hyperacusis as increased nonlinear gain. Hearing research 2013, 295, 172-179. [CrossRef]
- Kaltenbach, J.A. The dorsal cochlear nucleus as a contributor to tinnitus: mechanisms underlying the induction of hyperactivity. Progress in brain research 2007, 166, 89-106. [CrossRef]
- Krauss, P.; Schilling, A.; Tziridis, K.; Schulze, H. Models of tinnitus development: From cochlea to cortex. HNO 2019. [CrossRef]
- Tziridis, K.; Forster, J.; Buchheidt-Dorfler, I.; Krauss, P.; Schilling, A.; Wendler, O.; Sterna, E.; Schulze, H. Tinnitus development is associated with synaptopathy of inner hair cells in Mongolian gerbils. Eur J Neurosci 2021, 54, 4768-4780. [CrossRef]
- Kujawa, S.G.; Liberman, M.C. Adding insult to injury: cochlear nerve degeneration after "temporary" noise-induced hearing loss. J Neurosci 2009, 29, 14077-14085. [CrossRef]
- Kaltenbach, J.A.; Afman, C.E. Hyperactivity in the dorsal cochlear nucleus after intense sound exposure and its resemblance to tone-evoked activity: a physiological model for tinnitus. Hearing research 2000, 140, 165-172. [CrossRef]
- Hofmeier, B.; Wertz, J.; Refat, F.; Hinrichs, P.; Saemisch, J.; Singer, W.; Rüttiger, L.; Klose, U.; Knipper, M.; Wolpert, S. Functional biomarkers that distinguish between tinnitus with and without hyperacusis. Clinical and Translational Medicine 2021, 11, e378. [CrossRef]
- Koops, E.; van Dijk, P. Hyperacusis in tinnitus patients relates to enlarged subcortical and cortical responses to sound except at the tinnitus frequency. Hearing research 2021, 401, 108158. [CrossRef]
- Almond, L.M.; Patel, K.; Rejali, D. Transient auditory dysfunction: a description and study of prevalence. Ear, Nose & Throat Journal 2013, 92, 352-356. [CrossRef]
- Turrigiano, G.G. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends in neurosciences 1999, 22, 221-227. [CrossRef]
- Zenke, F.; Gerstner, W.; Ganguli, S. The temporal paradox of Hebbian learning and homeostatic plasticity. Current opinion in neurobiology 2017, 43, 166-176. [CrossRef]
- Krauss, P.; Tziridis, K.; Metzner, C.; Schilling, A.; Hoppe, U.; Schulze, H. Stochastic Resonance Controlled Upregulation of Internal Noise after Hearing Loss as a Putative Cause of Tinnitus-Related Neuronal Hyperactivity. Front Neurosci 2016, 10, 597. [CrossRef]
- Schulze, H.; Schilling, A.; Krauss, P.; Tziridis, K. The Erlangen model of tinnitus development-New perspective and treatment strategy. HNO 2023, 71, 662-668. [CrossRef]
- Shore, S.E.; Zhou, J. Somatosensory influence on the cochlear nucleus and beyond. Hearing research 2006, 216, 90-99. [CrossRef]
- Krauss, P.; Metzner, C.; Schilling, A.; Schutz, C.; Tziridis, K.; Fabry, B.; Schulze, H. Adaptive stochastic resonance for unknown and variable input signals. Sci Rep 2017, 7, 2450. [CrossRef]
- Licklider, J.C.R. A duplex theory of pitch perception. Experientia 1951, 7, 128-134. [CrossRef]
- Apostolides, P.F.; Trussell, L.O. Superficial stellate cells of the dorsal cochlear nucleus. Frontiers in Neural Circuits 2014, 8, 63. [CrossRef]
- Balmer, T.S.; Trussell, L.O. Descending axonal projections from the inferior colliculus target nearly all excitatory and inhibitory cell types of the dorsal cochlear nucleus. Journal of Neuroscience 2022, 42, 3381-3393. [CrossRef]
- Trussell, L.O.; Oertel, D. Microcircuits of the dorsal cochlear nucleus. In The Mammalian Auditory Pathways: Synaptic Organization and Microcircuits; Springer: 2018; pp. 73-99.
- Tzounopoulos, T.; Rubio, M.E.; Keen, J.E.; Trussell, L.O. Coactivation of pre-and postsynaptic signaling mechanisms determines cell-specific spike-timing-dependent plasticity. Neuron 2007, 54, 291-301. [CrossRef]
- Wu, Q.-W.; Tang, Z.-Q. Focusing on the emerging role of kainate receptors in the Dorsal Cochlear Nucleus (DCN) and cerebellum. International Journal of Molecular Sciences 2023, 24, 1718. [CrossRef]
- Wever, E.G.; Bray, C.W. The perception of low tones and the resonance-volley theory. The Journal of Psychology 1937, 3, 101-114. [CrossRef]
- Larkum, M.E.; Rioult, M.G.; Luscher, H.-R. Propagation of action potentials in the dendrites of neurons from rat spinal cord slice cultures. Journal of neurophysiology 1996, 75, 154-170. [CrossRef]
- Blackstad, T.; Osen, K.; Mugnaini, E. Pyramidal neurones of the dorsal cochlear nucleus: a Golgi and computer reconstruction study in cat. Neuroscience 1984, 13, 827-854. [CrossRef]
- Bi, G.-q.; Poo, M.-m. Synaptic modification by correlated activity: Hebb's postulate revisited. Annual review of neuroscience 2001, 24, 139-166. [CrossRef]
- Oertel, D.; Young, E.D. What's a cerebellar circuit doing in the auditory system? Trends in neurosciences 2004, 27, 104-110. [CrossRef]
- Knipper, M.; Mazurek, B.; van Dijk, P.; Schulze, H. Too Blind to See the Elephant? Why Neuroscientists Ought to Be Interested in Tinnitus. J Assoc Res Otolaryngol 2021, 22, 609-621. [CrossRef]
- Refat, F.; Wertz, J.; Hinrichs, P.; Klose, U.; Samy, H.; Abdelkader, R.M.; Saemisch, J.; Hofmeier, B.; Singer, W.; Rüttiger, L. Co-occurrence of hyperacusis accelerates with tinnitus burden over time and requires medical care. Frontiers in Neurology 2021, 12, 627522. [CrossRef]
- Vielsmeier, V.; Santiago Stiel, R.; Kwok, P.; Langguth, B.; Schecklmann, M. From acute to chronic tinnitus: pilot data on predictors and progression. Frontiers in Neurology 2020, 11, 997. [CrossRef]
- Wake, N.; Shiramatsu, T.I.; Takahashi, H. Map plasticity following noise exposure in auditory cortex of rats: implications for disentangling neural correlates of tinnitus and hyperacusis. Frontiers in Neuroscience 2024, 18, 1385942. [CrossRef]
- Shore, S.E.; Koehler, S.; Oldakowski, M.; Hughes, L.F.; Syed, S. Dorsal cochlear nucleus responses to somatosensory stimulation are enhanced after noise-induced hearing loss. European Journal of Neuroscience 2008, 27, 155-168. [CrossRef]
- Tagoe, T.; Deeping, D.; Hamann, M. Saturation of long-term potentiation in the dorsal cochlear nucleus and its pharmacological reversal in an experimental model of tinnitus. Experimental Neurology 2017, 292, 1-10. [CrossRef]
- Suckfüll, M.; Althaus, M.; Ellers-Lenz, B.; Gebauer, A.; Görtelmeyer, R.; Jastreboff, P.J.; Moebius, H.J.; Rosenberg, T.; Russ, H.; Wirth, Y. A randomized, double-blind, placebo-controlled clinical trial to evaluate the efficacy and safety of neramexane in patients with moderate to severe subjective tinnitus. BMC Ear, Nose and Throat Disorders 2011, 11, 1. [CrossRef]
- Salembier, L.; De Ridder, D.; Van de Heyning, P. The use of flupirtine in treatment of tinnitus. Acta Oto-Laryngologica 2006, 126, 93-95. [CrossRef]
- Figueiredo, R.R.; Langguth, B.; de Oliveira, P.M.; de Azevedo, A.A. Tinnitus treatment with memantine. Otolaryngology—Head and Neck Surgery 2008, 138, 492-496. [CrossRef]
- Sedley, W.; Alter, K.; Gander, P.E.; Berger, J.; Griffiths, T.D. Exposing pathological sensory predictions in tinnitus using auditory intensity deviant evoked responses. Journal of Neuroscience 2019, 39, 10096-10103. [CrossRef]
- Rauschecker, J.P.; May, E.S.; Maudoux, A.; Ploner, M. Frontostriatal gating of tinnitus and chronic pain. Trends in cognitive sciences 2015, 19, 567-578. [CrossRef]
- Jastreboff, P.J. Tinnitus retraining therapy. Progress in brain research 2007, 166, 415-423. [CrossRef]
Figure 1.
The Erlangen Model of Tinnitus development. AAN = auditory afferent nerve; ACo = Autocorrelation; FC = fusiform cell; PF = parallel fiber; SR = stochastic resonance. Figure adopted from [29]. For explanation refer to the text.
Figure 1.
The Erlangen Model of Tinnitus development. AAN = auditory afferent nerve; ACo = Autocorrelation; FC = fusiform cell; PF = parallel fiber; SR = stochastic resonance. Figure adopted from [29]. For explanation refer to the text.
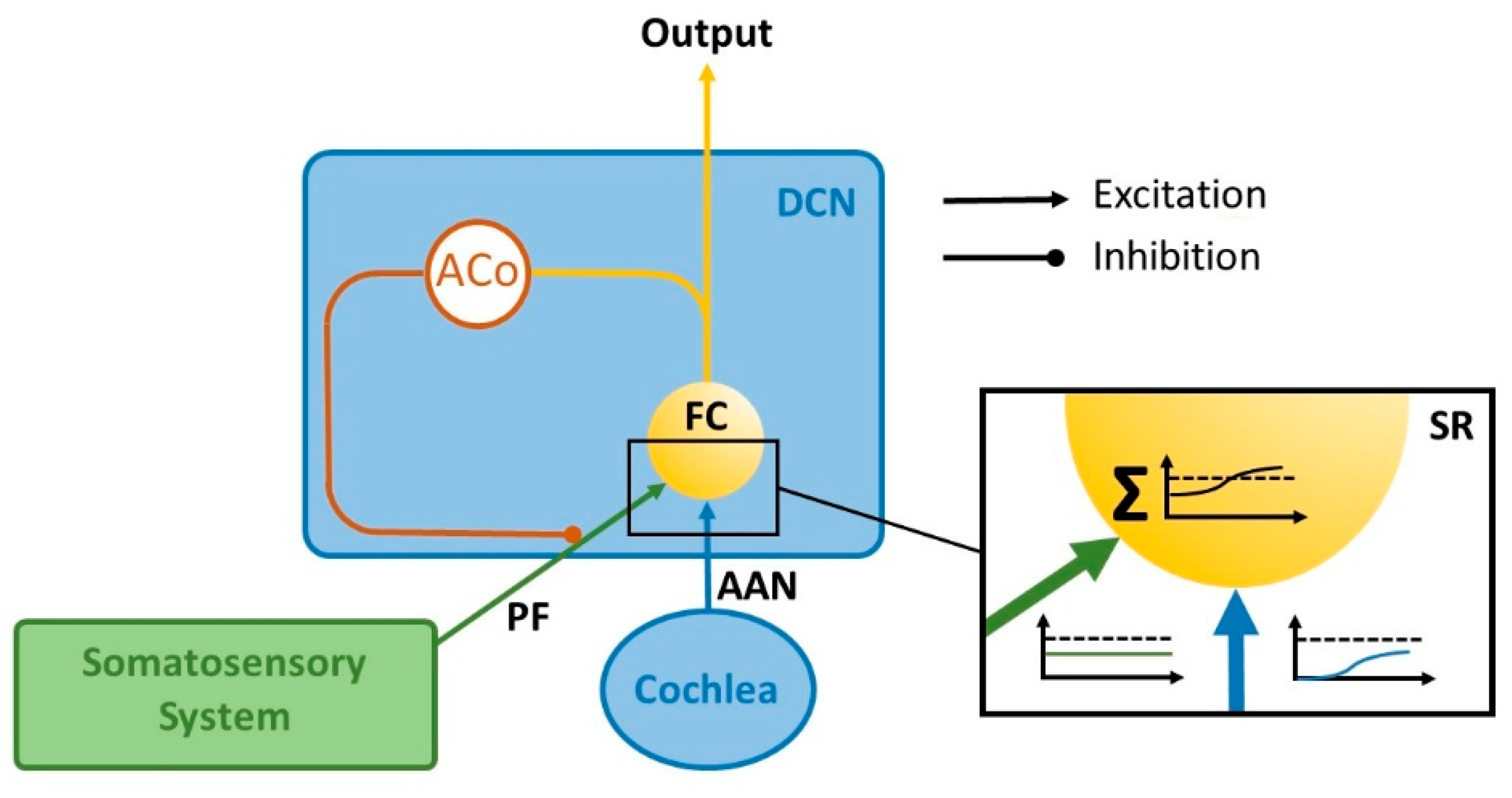
Figure 2.
Possible implementations (A to C) of delay lines for calculating the autocorrelation function within the DCN. A: Anatomically plausible DCN neuronal network to detect a given delay. References for the proposed circuitry: [33,34,35,36,37]. Note that the autocorrelation function may in principle also be calculated on the dendrites of a SSC (cf. B, C): If Δt1 (time difference between to spikes) equals Δt2, spatial summation of EPSPs would occur that may drive the SSC. This can be achieved by either copying the input of a single FC to two different locations on the same dendrite (B) or by converging input of several FCs within a given frequency channel to the same SSC dendrite (C). Note that in the latter case even Wever‘s Volley Theory could allow for Δt-intervals below 1ms [38].
Figure 2.
Possible implementations (A to C) of delay lines for calculating the autocorrelation function within the DCN. A: Anatomically plausible DCN neuronal network to detect a given delay. References for the proposed circuitry: [33,34,35,36,37]. Note that the autocorrelation function may in principle also be calculated on the dendrites of a SSC (cf. B, C): If Δt1 (time difference between to spikes) equals Δt2, spatial summation of EPSPs would occur that may drive the SSC. This can be achieved by either copying the input of a single FC to two different locations on the same dendrite (B) or by converging input of several FCs within a given frequency channel to the same SSC dendrite (C). Note that in the latter case even Wever‘s Volley Theory could allow for Δt-intervals below 1ms [38].
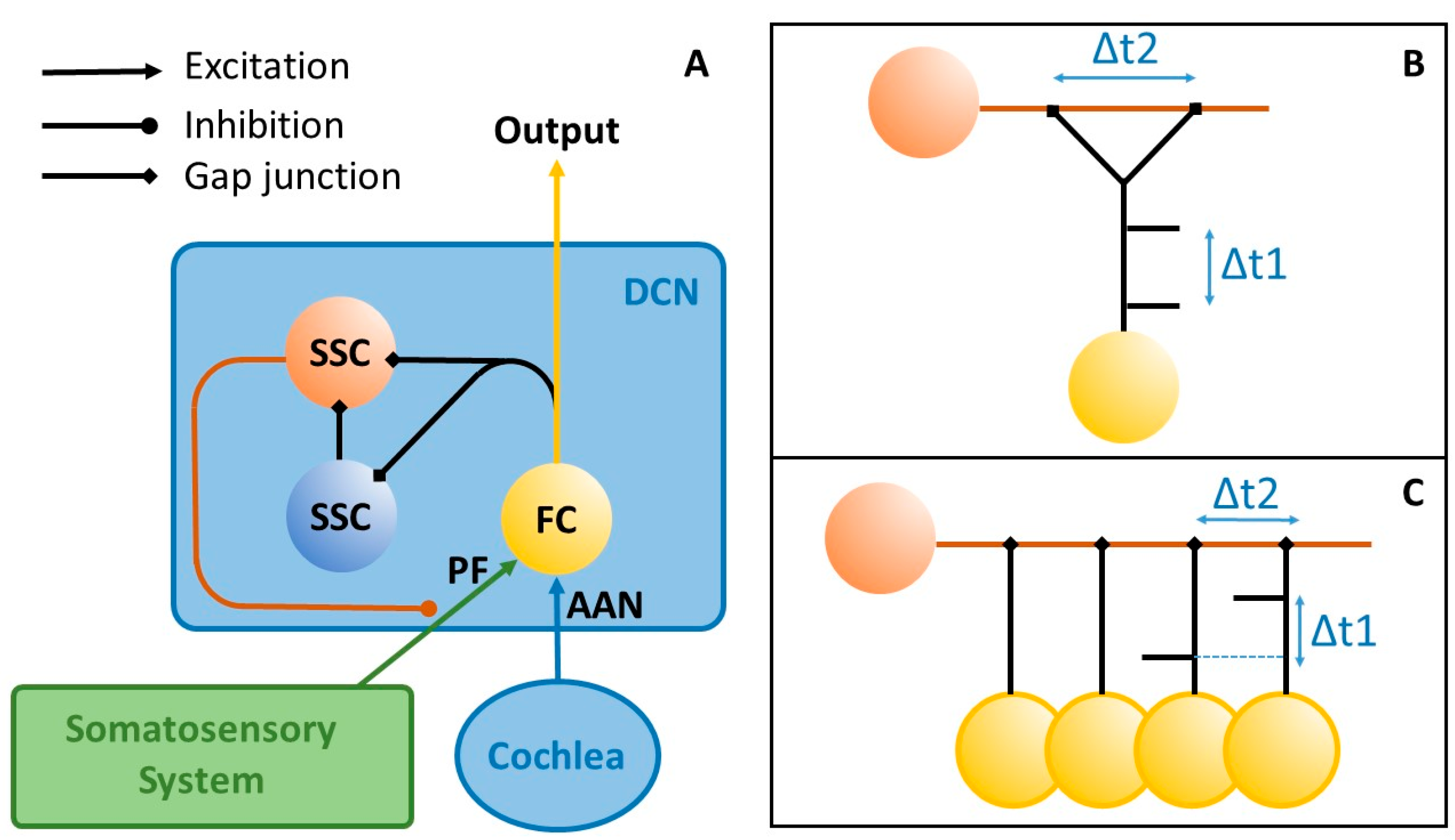
Figure 3.
Graphical abstract of the theory proposed. The theory is based on activity dependent plastic changes of synaptic weights of inputs from the somatosensory system (i.e., the trigeminal ganglion and spinal trigeminal nucleus, [30]) and the cochlear nerve to the DCN. Increased synaptic strength (red circles) of the cochlear input is responsible for HA, while increased synaptic strength of the somatosensory input is responsible for CT. DCN = Dorsal cochlear nucleus; SR = Stochastic resonance; HA = Hyperacusis; CT = Chronic tinnitus. For further explanations, refer to the text.
Figure 3.
Graphical abstract of the theory proposed. The theory is based on activity dependent plastic changes of synaptic weights of inputs from the somatosensory system (i.e., the trigeminal ganglion and spinal trigeminal nucleus, [30]) and the cochlear nerve to the DCN. Increased synaptic strength (red circles) of the cochlear input is responsible for HA, while increased synaptic strength of the somatosensory input is responsible for CT. DCN = Dorsal cochlear nucleus; SR = Stochastic resonance; HA = Hyperacusis; CT = Chronic tinnitus. For further explanations, refer to the text.
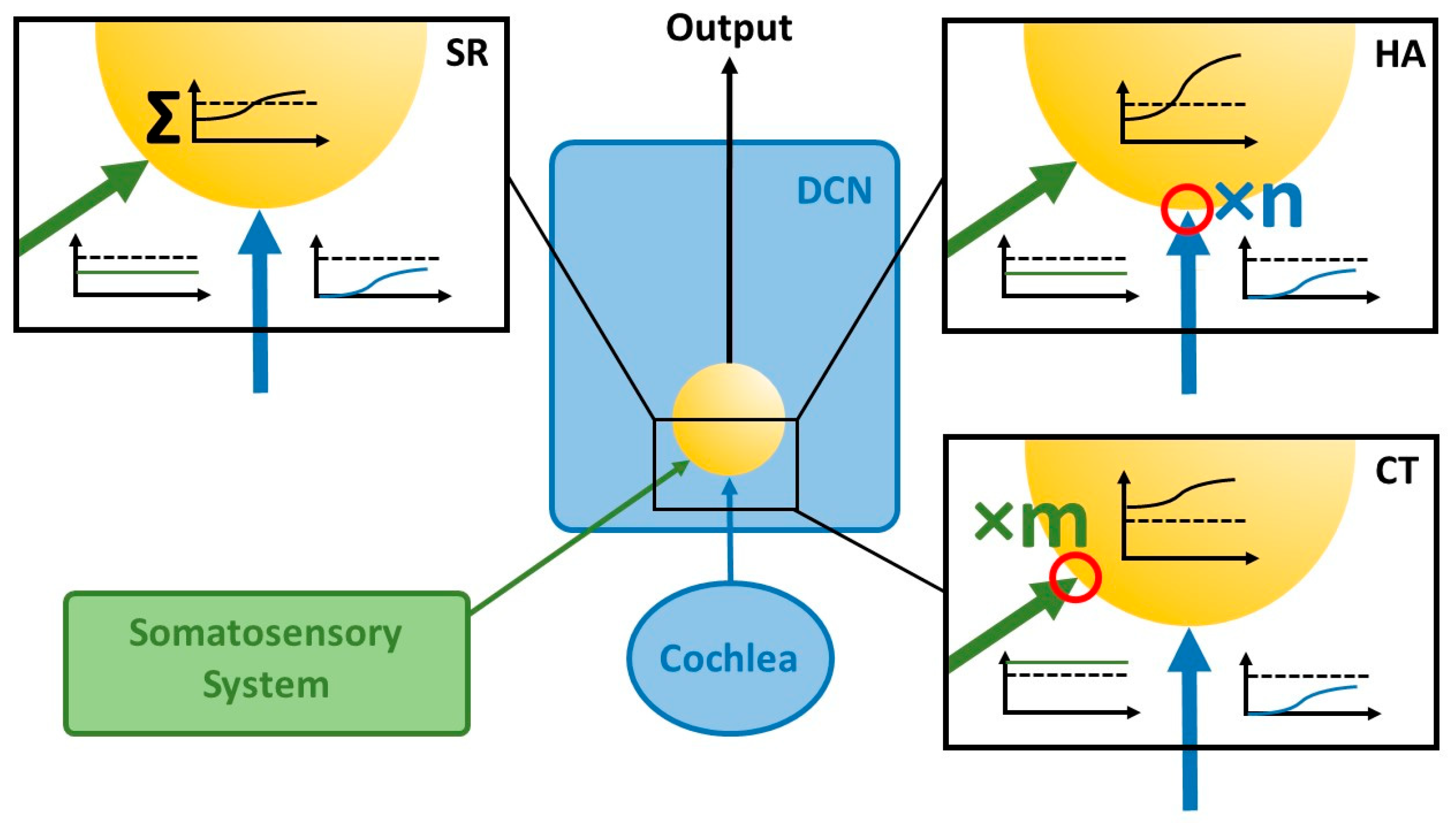
Figure 4.
The Erlangen Unified Model of Tinnitus and Hyperacusis Development. The panels show simplified sketches of the connectivity scheme presented in Figure 1: Yellow ball: DCN input neuron. Blue ball: Cochlear input to the DCN. Green ball: Somatosensory input to the DCN. ACo: Autocorrelation detector. Black lines: Excitatory connections. Red lines: Inhibitory connections. Red flash: Synaptic strengthening. For further explanation, refer to the text.
Figure 4.
The Erlangen Unified Model of Tinnitus and Hyperacusis Development. The panels show simplified sketches of the connectivity scheme presented in Figure 1: Yellow ball: DCN input neuron. Blue ball: Cochlear input to the DCN. Green ball: Somatosensory input to the DCN. ACo: Autocorrelation detector. Black lines: Excitatory connections. Red lines: Inhibitory connections. Red flash: Synaptic strengthening. For further explanation, refer to the text.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



