Submitted:
14 February 2026
Posted:
14 February 2026
You are already at the latest version
Abstract
Breast cancer represents a highly heterogeneous malignancy encompassing multiple molecular subtypes, each with distinct therapeutic responses and clinical outcomes. Conventional treatment strategies—including surgery, chemotherapy, radiotherapy, endocrine, and targeted therapies—have improved survival rates but continue to face major limitations due to tumor relapse, metastasis, and therapy-induced resistance. In recent years, chimeric antigen receptor (CAR) T-cell therapy has emerged as a transformative modality in cancer immunotherapy, offering targeted and durable antitumor activity. While its efficacy in hematological malignancies is well established, translation to solid tumors such as breast cancer remains hindered by a complex interplay of tumor-intrinsic and micro environmental barriers. This review delineates the mechanistic underpinnings of CAR T-cell function and highlights the multifaceted challenges posed by solid tumors, including antigen heterogeneity, an immunosuppressive tumor microenvironment, inadequate trafficking and infiltration, limited T-cell persistence, and safety-related cytotoxicities. Finally, emerging strategies and innovations aimed at overcoming these barriers are discussed, highlighting the potential and future direction of CAR T-cell therapy in breast cancer management. Collectively, this review underscores the translational potential of CAR T-cell therapy for breast cancer and outlines the rational strategies required to enhance its clinical applicability and therapeutic efficacy in solid tumor settings.
Keywords:
CAR T-cell therapy
; tumor microenvironment
; breast cancer
; antigen heterogeneity
1. Introduction
Breast cancer is the most prevalent form of cancer affecting women worldwide [1,2]. This disease is broadly categorized into three molecular subtypes based on the expression of specific hormone receptors: hormone receptor-positive (HR-positive), HER2-enriched, and triple-negative breast cancer (TNBC). Each subtype differs significantly in terms of prognosis, treatment options, and biological behavior. Among these, TNBC is characterized by the absence of estrogen receptors, progesterone receptors, and HER2 expression, which contributes to its aggressive clinical course and poorer outcomes.
TNBC patients generally exhibit lower survival rates and reduced responsiveness to conventional treatments such as hormone therapy and HER2-targeted agents, which are effective in other breast cancer subtypes [3,4]. A major challenge in managing TNBC is the lack of well-defined molecular targets, which limits the effectiveness of targeted therapies currently available for other forms of breast cancer. Furthermore, TNBC is distinguished by a higher prevalence of circulating immunogenic cells, indicating an elevated level of immune system activity within the tumor microenvironment (TME).
This heightened immunogenicity positions TNBC as the most immunogenic among the breast cancer subtypes, thereby making immunotherapy an especially promising therapeutic approach [4]. Recent advances in immunotherapeutic strategies, including immune checkpoint inhibitors, have shown encouraging results by leveraging the body’s immune system to mount a robust anti-tumor response. Consequently, immunotherapy represents a hopeful avenue for improving treatment outcomes and survival rates in patients diagnosed with TNBC [5,6].
2. Existing Treatment Options
For early-stage invasive breast cancer, breast-conserving surgery (BCS) and mastectomy remain the primary treatment modalities [7]. Among these, BCS followed by adjuvant radiotherapy has demonstrated excellent long-term outcomes, with 10-year locoregional recurrence (LRR) rates reported to be approximately 2% for estrogen receptor-positive (ER-positive) and HER2-positive breast cancers, and around 5% for TNBC [8,9]. These findings underscore the efficacy of BCS combined with radiotherapy as the preferred treatment approach for early invasive breast cancers, offering the dual benefits of effective disease control and breast preservation.
However, certain patient populations may not be ideal candidates for radiotherapy following BCS. Contraindications to radiotherapy, such as prior radiation exposure or certain comorbidities, as well as the presence of suspicious micro-calcifications on imaging, increase the risk of treatment failure and recurrence, thereby necessitating consideration of alternative therapeutic strategies [10]. In instances where patients experience local recurrence after initial BCS, total mastectomy is generally regarded as the standard of care to achieve optimal disease control and reduce the risk of further recurrence.
Immunotherapy
Adoptive cell therapy (ACT) refers to a form of immunotherapy that involves the engineering and manipulation of the patient’s immune system to enhance its ability to recognize and eliminate malignant cells. Among the various approaches within ACT, chimeric antigen receptor (CAR) T-cell therapy has emerged as a ground-breaking strategy. This technique involves the genetic modification of T-cells to express CARs—synthetic receptors that enable these immune cells to specifically identify and target tumor-associated antigens presented on the surface of cancer cells [11]. Leukapheresis represents the initial stage in the production of CAR T-cells, encompassing the collection of blood from the patient to isolate T-cells, followed by the removal of myeloid cells through elutriation. Subsequently, T-lymphocytes are enriched, transgenes are introduced, and ex vivo expansion occurs [12].
CAR T-cell therapy harnesses the specificity and cytotoxic potential of T-lymphocytes, allowing for the selective targeting and destruction of malignant cells while sparing normal tissues. This precision is achieved through the engineered T-cells’ ability to recognize unique antigenic markers that are typically overexpressed on cancer cells. Figure 1 summarizes the various possible ways in which CAR T-cells act in building the immune response against cancer cells. The first clinical application of CAR T-cell therapy was reported in 2017, marking a significant milestone in the treatment of hematological malignancies such as certain leukemias and lymphomas [13].
Since its initial application, CAR T-cell therapy has revolutionized the field of cancer immunotherapy by demonstrating remarkable efficacy in refractory and relapsed blood cancers, offering new hope to patients who had limited treatment options. Ongoing research continues to expand the potential of this approach to a wider range of malignancies and to improve its safety and effectiveness.
3. The Evolution of CAR T-Cell Therapy
CAR T-cells are broadly classified into five generations, distinguished by the arrangement of their intracellular signaling domains, These domains are central to orchestrating a range of T-cell functions, including lineage differentiation, induction of cytotoxic activity, cytokine production, and recruitment of additional immune effectors [22]. Together, these mechanisms contribute not only to efficient tumor clearance but also to the capacity for tumor targeting in a manner that bypasses MHC restriction.
The conceptual foundation of CAR technology was first demonstrated by Kuwana and colleagues in 1987, who provided proof-of-principle that antibody-derived antigen recognition could be coupled with T-cell signalling. This was achieved by fusing the constant region of the T-cell receptor (TCR) to the variable regions of an antibody directed against a bacterial antigen. Building on this seminal work, single-chain variable fragments (scFvs)—consisting of the variable heavy (VH) and light (VL) domains of a monoclonal antibody linked by a short, flexible peptide—have since become the standard extracellular antigen-recognition modules employed in CAR design [23].
The first generation of CAR T-cells was designed with a relatively simple architecture, consisting of a single intracellular signaling domain—either the CD3ζ chain or the FcεRIγ motif (Figure 2)—and lacking additional costimulatory elements. Early reports in the 1990s established proof-of-concept for this design, along with demonstrating that fusing a tumor antigen–specific single-chain variable fragment (scFv), such as one recognizing human epidermal growth factor receptor 2 (HER2) to the CD3ζ domain, could confer antigen-specific cytotoxicity [24,25,26].
Despite these encouraging findings, the limitations of first-generation CARs quickly became evident. Although structurally analogous to endogenous TCRs, they failed to produce sufficient levels of interleukin-2 (IL-2), making them dependent on exogenous IL-2 supplementation to sustain activity. Moreover, these CAR T-cells exhibited poor proliferative capacity and limited in vivo persistence, both of which hindered their therapeutic efficacy in clinical settings [31,32,33]. Consequently, while first-generation CARs established the foundation for CAR-based immunotherapy, their restricted signaling capacity highlighted the need for further refinement, ultimately paving the way for second-generation designs incorporating costimulatory domains.
To overcome the limitations of first-generation CARs, researchers introduced intracellular costimulatory domains such as CD28, 4-1BB, and OX40 into CAR designs (Figure 2).
This modification significantly enhanced T-cell activation by providing a secondary signal upon antigen engagement, leading to improved proliferation, persistence, and cytotoxic function. Early comparative studies revealed that the addition of CD28 co-stimulation increased expansion and survival relative to CD3ζ-only constructs, while 4-1BB conferred superior persistence in the circulation, albeit with distinct effects on exhaustion and tonic signaling [34]. These insights laid the groundwork for the development of second- and third-generation CARs, which incorporated one or two costimulatory domains, respectively, reflecting the biological principle that endogenous T-cell receptor activation requires accessory signaling molecules [35]. Incorporation of CD28 or 4-1BB domains in particular was shown to enhance cytokine production, promote memory formation, and modulate metabolic pathways [36,37,38,39,40]. Subsequent iterations explored novel combinations of costimulatory modules to further optimize function: for instance, CARs engineered with both CD28 and OX40 suppressed CD28-driven interleukin-10 (IL-10) secretion, thereby countering immunosuppressive signaling [41], while ICOS paired with CD28 or 4-1BB improved in vivo persistence, and synthetic MyD88/CD40 signaling enhanced proliferation and antitumor activity [42,43]. Collectively, these advances not only established the clinical superiority of co-stimulation-enhanced CARs but also highlighted the importance of tailoring signaling domains to balance effector potency, persistence, and resistance to exhaustion. Refinements to third-generation CAR designs led to the emergence of fourth-generation CAR T-cells, which incorporated additional functional modules to enhance both efficacy and safety. These constructs often include transgenic elements—most commonly cytokines—that may be expressed either constitutively or inducibly upon antigen recognition. Some variants also integrate regulatory “safety switches,” such as inducible Caspase-9, to enable controlled elimination of CAR T-cells in the event of severe toxicity [44,45,46]. Further advances introduced protease-based CARs engineered for dose-dependent regulation, thereby allowing external control over CAR activity to improve safety and therapeutic precision [47]. A notable subset of fourth-generation constructs, termed “T-cells redirected for universal cytokine-mediated killing” (TRUCKs), harness a nuclear factor of activated T-cells (NFAT)-responsive promoter to drive inducible cytokine expression—such as interleukin-12 (IL-12)—directly within the TME. This design allows CAR T-cells to deliver immunomodulatory cytokines in situ, thereby enhancing antitumor efficacy while minimizing systemic toxicity [48,49]. Expanding on this principle, so-called “armored” CAR T-cells have been developed to secrete additional soluble factors, including IL-12, IL-15, and IL-18, which strengthen effector responses, counteract immunosuppressive cell populations such as regulatory T-cells (Tregs) and myeloid-derived suppressor cells (MDSCs), and promote sustained T-cell proliferation [44,48]. Other combinations, such as CCL19 with IL-7, have been explored to recruit endogenous immune subsets and support durable memory formation [50,51,52]. Collectively, these “armored” fourth-generation platforms represent a pivotal evolution in CAR design, equipping engineered T-cells not only with enhanced cytotoxic potential but also with the ability to actively remodel the tumor microenvironment.
Fifth-generation (5G) CAR T-cells represent a sophisticated evolution of cellular immunotherapy, building upon the foundational designs of second-generation (2G) CARs. While earlier iterations focused on enhancing T-cell activation through co-stimulatory domains, 5G CARs incorporate an additional dimension of signaling that closely mimics physiological T-cell activation. These next-generation constructs are specifically engineered to address critical limitations observed in earlier CAR generations—namely, T-cell exhaustion, poor persistence, and limited efficacy against solid tumors [53].
The central innovation of 5G CARs lies in their ability to engage the JAK-STAT signaling cascade in an antigen-dependent manner. This is achieved by incorporating a truncated cytoplasmic domain of the IL-2 receptor β-chain fused with a STAT3/STAT5-binding motif into a conventional 2G CAR backbone. Consequently, these cells simultaneously activate three key intracellular pathways: TCR signaling via the CD3ζ domain, co-stimulation via CD28, and cytokine signaling via JAK-STAT3/5. This design enables autonomous and sustained cytokine signaling without the need for exogenous cytokine support, thereby promoting T-cell persistence and functionality over time.
Preclinical studies have provided compelling evidence supporting the enhanced functionality of 5G CARs. Notably, Kagoya et al. demonstrated that JAK-STAT-augmented CAR T-cells outperformed earlier-generation counterparts in CD19+ leukemia xenograft models, exhibiting superior tumor control, increased expansion, and prolonged persistence [54]. These cells displayed a less differentiated, stem cell-like memory phenotype, reduced terminal differentiation, and lower rates of apoptosis. Enhanced secretion of effector cytokines such as IL-2, IFN-γ, and TNF-α contributed to improved cytotoxicity and offered a means of counteracting the immunosuppressive TME. In models of A375-CD19 melanoma, these CAR T-cells also showed improved tumor infiltration, underscoring their potential in addressing challenges associated with solid tumors [54].
In parallel to the integration of cytokine signaling, researchers have explored novel synthetic regulatory strategies to improve safety and functional control of CAR T-cells. One promising avenue involves the inclusion of antigen-inducible membrane receptors—such as IL-2 receptor components—to conditionally activate the JAK/STAT pathway, thereby refining cytokine-driven responses while minimizing off-target effects [54].
Furthermore, the development of switchable CAR systems offers a significant leap toward safer and more controllable therapies. These include both ON-switch and OFF-switch mechanisms that allow for drug-mediated modulation of CAR activity. For instance, lenalidomide-gated CAR constructs exemplify the concept of external pharmacologic control, albeit with some trade-offs in in vitro potency [55]. Despite this, their improved safety profile renders them attractive candidates for further research.
Expanding on this concept, Li et al., introduced the VIPER (VersatIle ProtEase Regulatable) CAR platform, which utilizes viral protease domains to enable small molecule-inducible ON and OFF switches [55]. This modular system permits multi-antigen targeting and the design of sophisticated logic-gated CAR circuits. Importantly, VIPER CARs demonstrated limited systemic toxicity in a cytokine storm model, highlighting their translational potential for clinical use [56].
While these advancements mark significant progress in the field, several challenges remain. Ongoing research is essential to optimize dosing strategies, mitigate risks such as cytokine release syndrome (CRS), and broaden the applicability of 5G CAR T-cell therapies across diverse hematological and solid malignancies.
4. CAR T-Therapy and Cancer Stem Cells
Tumors consist of a heterogeneous array of cell types, among which cancer stem cells (CSCs) represent a small yet critically important subpopulation of undifferentiated cells capable of giving rise to the differentiated progeny that constitute the tumor bulk. According to the CSC hypothesis, tumor formation parallels normal tissue development in that a subset of “cancer-initiating cells” maintains itself through asymmetric division while simultaneously generating multiple differentiated lineages [57]. These tumorigenic CSCs exhibit distinct surface marker profiles—including CD29, CD34⁺, CD38⁻, CD166, CD133⁺/⁻, Lin, Sca-1, and EpCAM—many of which are associated with stem-like traits and have enabled their isolation via fluorescence-activated cell sorting or other immunoselection strategies [58,59,60]. However, the application of these markers requires caution, as several are also expressed by non-CSC populations [61,62].
Within breast cancer, a discrete CSC subset—termed breast cancer stem cells (BCSCs)—has been identified and is now recognized as a key driver of therapeutic resistance and disease recurrence. Despite the rapid expansion of breast cancer treatment modalities, persistent chemo resistance and adverse effects continue to underscore the need for strategies capable of targeting this resilient population. BCSCs have been repeatedly implicated in treatment survival and post-therapy tumor re-emergence, making them a major determinant of relapse [63] and high BCSC fractions correlate strongly with poor clinical outcomes [64].
Because CAR T-cells are designed to recognize tumor-associated surface antigens with high specificity and inhibit tumor proliferation, CSC-restricted or CSC-enriched markers represent promising targets for immunotherapeutic intervention. Owing to their capacity to recognize and eliminate tumor cells via tumor-associated antigen binding, CAR T-cells represent strong candidates for targeting CSCs. For example, Chen et al. showed that GD2-specific CAR T-cells effectively depleted side-population cells and eradicated established tumors in a neuroblastoma mouse model [65]. Multiple studies employing CAR T-cells engineered against established CSC markers have further demonstrated that these cells can efficiently eliminate CSC subsets while sparing normal stem cells or exhibiting only minimal cytotoxic effects on them [66,67,68,69,70,71]. A range of CSC-associated markers are therefore considered potentially targetable by CAR T-cell approaches. For instance, extensive studies have demonstrated that markers such as CD133, CD90, ALDH, and EpCAM are expressed across CSCs from multiple tumor types, providing molecular entry points for CAR-T–mediated elimination of CSCs and for the suppression of tumor recurrence and metastasis [72].
Current CSC-directed CAR T-cell strategies can be broadly divided into two categories. The first involves engineering CAR T-cells against CSC-specific antigens—such as CD133, EpCAM, or ALDH—and assessing their cytotoxic potential through in vitro assays and in vivo animal models. The second approach targets “general” antigens shared by both CSCs and bulk tumor cells. Although these CAR T-cell constructs are not originally designed for CSC specificity, the presence of the same markers on CSC surfaces enables collateral killing of CSCs during co-culture or in vivo treatment [73]. However, this broadened targeting specificity introduces risks: notable “on-target, off-tumor” toxicities have been reported, particularly within the hematopoietic compartment, an effect likely attributable to CD133 expression on CD34⁺ progenitor cells.
Identifying antigens appropriate for CSC targeting presents unique challenges distinct from those encountered when identifying conventional tumor antigens for platforms such as TCR-based therapies. Current hurdles include: (i) the requirement for strict tumor specificity to avoid on-tumor, off-target toxicity, which is generally more severe with CAR T-cells compared with monoclonal antibody-based therapies [74]; (ii) the necessity for antigens to be exclusively expressed on the surface of CSCs—rather than intracellularly—because CAR T-cells recognize only surface-exposed antigens in an HLA-independent manner; (iii) the frequent reliance on functional properties such as self-renewal rather than stable phenotypic markers to define CSCs; and (iv) the absence of surface antigen expression in certain CSC populations. Consequently, next-generation CAR T-cell designs (e.g., iCAR-expressing CAR T-cells) and combination strategies (e.g., differentiation therapy followed by CAR T-cell treatment) may help mitigate or overcome these limitations [75].
One of the most effective strategies for identifying CSCs within tumors is the use of CSC-specific biomarkers. Depending on their cellular localization, these markers can be categorized as intracellular or cell-surface markers (Table 1).
CD133, also referred to as prominin-1, is a pentaspan transmembrane glycoprotein encoded by the PROM1 gene [79]. High levels of CD133 expression have been reported across diverse CSC populations, including those derived from leukemia, brain, liver, breast, pancreatic, and ovarian tumors [80,81,82,83]. Owing to its broad involvement in CSC biology, CD133 has emerged as a promising therapeutic target, and in recent years multiple CD133-directed agents—most notably monoclonal antibodies—have been explored [84]. Preclinical investigations evaluating CD133-targeted CAR T-cells have demonstrated significant antitumor activity. In an orthotopic glioma mouse model, Hu et al. showed that CD133 CAR T-cells markedly suppressed tumor progression and improved survival [85]. Similarly, Zhu et al. reported that CD133-specific CAR T-cells effectively eliminated glioblastoma CSCs in both in vitro and in vivo settings [86]. Beyond T-cells, CD133-targeted CAR natural killer-cells have also been tested; one study demonstrated that such CAR-NK cells efficiently recognized and eradicated CD133⁺ primary ovarian cancer cells as well as established ovarian cancer cell lines [87]. Collectively, these data suggest that CD133-directed CAR-based therapies hold substantial promise for CSC eradication. However, clinical investigation remains limited; to date, only one study has evaluated CD133-targeted CAR T-cells against patient-derived glioblastoma stem cells [88]. Given that CD133 is expressed not only on malignant neural stem cells but also on normal neural stem cells, concerns regarding potential off-tumor toxicity remain. Approaches such as intratumoral delivery of CD133-targeted CAR T-cells have been proposed to mitigate these risks [89]. Mechanistically, CD133 (PROM1) can activate intracellular pathways including PI3K/AKT, Src, and β-catenin, thereby contributing to tumor progression [79]. Moreover, combinations of CSC markers may enhance specificity: for instance, CD44 and CD133 have been co-utilized to identify CSC populations in gallbladder cancer [90] ,while in colorectal cancer both CD44⁺/CD133⁻ and CD44⁻/CD133⁺ subpopulations have been shown to possess CSC properties [91,92].
EpCAM (CD326) is a type I transmembrane glycoprotein traditionally associated with epithelial cell–cell adhesion [93]. Beyond its structural role, EpCAM promotes epithelial–mesenchymal transition by suppressing E-cadherin expression, thereby enhancing migratory and invasive potential. Consistent with these functions, EpCAM contributes to cell signaling, differentiation, proliferation, and migration, and is recognized as a prominent CSC marker in hepatocellular carcinoma (HCC) and colorectal cancer [94,95]. Indeed, EpCAM⁺ HCC cell lines such as HuH1 and HuH7 display robust tumor-initiating capacity and generate sizeable tumors in SCID mouse models [94]. These biological properties have positioned EpCAM as an attractive target for adoptive cell therapies. Over the past decade, multiple investigations have shown that EpCAM-directed CAR T-cells exert potent antitumor activity.
For example, Zhang et al. observed that EpCAM CAR T-cells secrete cytotoxic mediators, including TNF-α and IFN-γ, enabling efficient elimination of EpCAM⁺ cancer cells in vitro and significant inhibition of tumor growth in colorectal xenograft models [96]. Parallel findings by Wu et al. demonstrated that these CAR T-cells eradicate PC3M prostate cancer cells in vitro and markedly suppress tumor progression in NOD/SCID mice [97]. Further supporting the therapeutic value of this antigen, Deng et al. reported that EpCAM-specific CAR T-cells not only eliminate PC3M prostate cells that overexpress EpCAM but also prolong survival in models using PC3 cells with comparatively low EpCAM expression [98]. Optimizing EpCAM-targeted CAR T-cells—particularly through tuning CAR affinity—may help achieve effective tumor control while minimizing off-tumor toxicity associated with physiological EpCAM expression [89].
5. Challenges Faced in the Case of Solid Tumors
Despite the transformative success of CAR T-cell therapies in hematologic malignancies, their translation to solid tumors has been met with substantial challenges. These limitations are primarily rooted in the complex and immunosuppressive architecture of the solid TME, which poses a multifactorial barrier to effective immunotherapy. Unlike blood cancers, where CAR T-cells have relatively unimpeded access to malignant targets, solid tumors exhibit a constellation of physiological and biochemical obstacles that restrict T-cell function and persistence [99,100,101].
The TME is a highly complex and dynamic ecosystem composed not only of malignant cells but also a diverse repertoire of non-neoplastic constituents, including fibroblasts, adipocytes, pericytes, immune cells, and other stromal elements [102]. This multifaceted milieu is further shaped by a dense stromal architecture, aberrant vasculature, immunosuppressive cellular networks, and a profoundly dysregulated chemokine landscape, which together impose formidable barriers to effective immune cell infiltration and antitumor immunity [103]. Upregulation of immune checkpoint ligands, secretion of pro-tumorigenic and anti-inflammatory cytokines, and physical exclusion of lymphocytes from tumor islets collectively limit CAR T-cell infiltration and cytotoxicity [104]. Moreover, antigenic heterogeneity and the scarcity of truly tumor-specific targets further compound the difficulty in achieving selective and sustained CAR T-cell activation [99].
These barriers do not operate in isolation but rather form a highly interconnected system of resistance. For example, the adaptive remodeling of the tumor microenvironment may simultaneously reduce antigen expression and modulate chemokine gradients, thereby hindering T-cell recruitment and activation. Such interactions underscore the need for a systems-level understanding of tumor-immune dynamics rather than piecemeal solutions targeting single mechanisms [105].
5.1. Immunosuppressive Tumor Microenvironment
Beyond its cellular makeup, the TME also incorporates extracellular matrix (ECM) components, abnormal vasculature, and a complex milieu of chemokines, cytokines, and other soluble factors that together orchestrate the pathophysiology of tumor progression [106,107].
Rather than serving as a passive backdrop, the TME functions as an active participant in tumor evolution, exerting a profound influence on the malignant phenotype of neoplastic cells. Reciprocal interactions between cancer cells and their microenvironment drive processes such as immune evasion, angiogenesis, metastasis, and resistance to therapy, thereby reinforcing the TME’s role as a dynamic enabler of carcinogenesis and disease progression [108].
A critical barrier to the success of CAR T-cell therapy in solid tumors lies in the immunosuppressive nature of this microenvironment. Studies have revealed that only 1–2% of infused CAR T-cells are capable of infiltrating the tumor core, severely limiting their cytotoxic activity against solid tumor cells [109]. This poor infiltration is partly due to the structural and biochemical obstacles embedded within the TME, including dense fibrotic stroma, ECM components such as glycoproteins, fibrous proteins, proteoglycans, and polysaccharides, all of which contribute to the formation of physical and immunological barriers [107,110].
Furthermore, the TME houses a heterogeneous population of immune and stromal cells that actively suppress T-cell function. These include tumor-associated macrophages, myeloid-derived suppressor cells, regulatory T-cells, dendritic cells, and natural killer cells, as well as myeloid progenitor and effector T-cells [111,112]. These cells release immunosuppressive mediators such as TGF-β and other cytokines that blunt CAR T-cell activity, while molecules such as TRIF-related adaptor molecule interfere with downstream signaling cascades crucial for T-cell function [112].
Compounding these cellular and molecular barriers are additional physiological constraints such as hypoxia, nutrient deprivation, and the buildup of metabolic waste products, all of which create a metabolically hostile environment that further compromises CAR T-cell efficacy [113]. Moreover, abnormal tumor vasculature and increased interstitial pressure hinder the effective trafficking and delivery of CAR T-cells or therapeutic agents to deeper tumor regions [110].
To address these formidable barriers, combinatorial strategies are being explored. Co-treatment with immune checkpoint inhibitors, for instance, offers a promising avenue to alleviate T-cell exhaustion and overcome the suppressive effects of the TME [114]. Engineering CAR T-cells with enhanced resistance to TME-derived inhibitory signals or improved capacity to degrade ECM components may also represent critical steps toward improving their performance in solid tumors.
5.2. Physical Barriers
The physical architecture of the tumor stroma presents a formidable barrier to the successful infiltration of CAR T-cells into solid tumors. Comprised of a dense ECM, blood and lymphatic vessels, fibroblasts, immune cells, and mesenchymal components, this structural network forms a compact and highly organized mesh that can significantly limit immune cell access [115]. In some instances, the degree of stromal compaction renders the tumor virtually inaccessible to therapeutic agents, including CAR T-cells—a major obstacle given the well-established correlation between T-cell infiltration and improved clinical outcomes [116].
This physical inaccessibility is further exacerbated by the dynamic interactions between stromal and malignant cells, which drive tumor evolution. These interactions induce a spectrum of metabolic, genetic, and morphological changes, transforming stromal elements into pro-tumorigenic agents that support cancer progression and metastasis. Key modulators such as proteoglycans and glycopeptides embedded in the ECM actively remodel immune responses during tumor development, thereby reinforcing immune evasion mechanisms [117].
Among the most influential stromal players are cancer-associated fibroblasts (CAFs), which arise through a phenotypic transition of normal fibroblasts. Once activated, CAFs express molecules such as fibroblast-activating protein and stromal-derived factor 1α (SDF1A), both of which contribute to ECM remodeling. By inducing collagen cross-linking and enzymatic degradation of matrix components, CAFs not only support tumor expansion but also fortify physical barriers that impede CAR T-cell infiltration [118].
In addition to local stromal barriers, anatomical structures such as the blood-brain barrier (BBB) present further challenges for CAR T-cell therapy, particularly in the treatment of central nervous system (CNS) tumors. Traditional intravenous delivery methods are often insufficient to enable CAR T-cells to cross the BBB, prompting exploration of alternative strategies including laser thermotherapy, electroporation, transcranial ultrasound, and other modalities designed to temporarily disrupt the barrier [119,120]. Direct delivery routes—such as intraventricular or intrathecal administration—are currently under investigation as targeted approaches capable of bypassing the BBB and delivering CAR T-cells directly into the CNS parenchyma [121].
However, efforts to modify or bypass the BBB are not without risk. Disruption of this critical interface can trigger serious CNS-related adverse effects, including cerebral edema and neuroinflammation [122]. Furthermore, even beyond the BBB, solid tumors harbor additional physical obstructions. Activated CAFs contribute to the generation of thicker, mechanically stressed collagen fibers that create a more rigid and supportive ECM structure conducive to tumor growth. Simultaneously, tumor basement membranes frequently exhibit breaches caused by both proteolytic degradation and mechanical realignment of ECM molecules, which complicates the orderly infiltration of therapeutic immune cells [123].
Altogether, these multilayered structural and physiological barriers necessitate innovative engineering of CAR T-cells and novel delivery strategies to optimize their efficacy in the context of solid tumors.
5.3. Antigen Escape
One of the foremost challenges limiting the efficacy of CAR T-cell therapy in solid tumors is the significant antigenic heterogeneity displayed by these malignancies. This heterogeneity often facilitates a rapid phenomenon known as antigen escape, whereby tumor cells evade immune targeting by either mutating the antigen or expanding from a subpopulation of antigen-negative cells already present prior to treatment [124,125]. Such escape mechanisms critically undermine both the persistence and the therapeutic effectiveness of CAR T-cells.
To address this limitation, multi-antigen targeting strategies have been developed. By engineering CAR T-cells to recognize two distinct tumor-associated antigens simultaneously—a double-antigen-targeted approach—researchers aim to mitigate antigen escape while improving specificity and reducing off-target effects [126]. These dual-targeted therapies can take various forms, including the administration of two separate CAR T-cell populations, bicistronic CAR T-cell constructs, or tandem bispecific CAR T-cells, each designed to enhance anti-tumor efficacy through diversified antigen recognition [127], (Figure 3).
Although single antigen-targeting CAR T-cells may initially elicit high response rates, tumor resistance often emerges because of partial or complete loss of the targeted antigen on malignant cells. This adaptive resistance, termed antigen escape, remains a significant barrier to durable responses and highlights the necessity for advanced CAR designs that can circumvent or pre-empt this mechanism.
5.4. Tumor Trafficking and Infiltration
CAR T-cell therapy for solid tumors faces considerable limitations compared to its application in hematological malignancies, primarily due to challenges in trafficking and infiltration. As previously mentioned, while CAR T-cells in hematologic cancers have direct access to tumor cells circulating in the bloodstream or residing in bone marrow, solid tumors present a more complex physical and immunosuppressive landscape that restricts CAR T-cell mobility and penetration [133,134]. The TME, combined with physical barriers such as the dense tumor stroma, impede effective CAR T-cell entry and function within solid tumor sites [135].
One key factor affecting CAR T-cell infiltration is the abnormal vasculature characteristic of solid tumors, which often displays reduced expression of endothelial adhesion molecules such as ICAM-1 and VCAM-1 that are crucial for lymphocyte extravasation [136,137]. This aberrant endothelium acts as a selective barrier, actively inhibiting immune cell entry by suppressing adhesion molecules and releasing factors such as ALCAM (activated leukocyte cell adhesion molecule), which modulate integrin-mediated arrest and impede T-cell trafficking. Additionally, the tumor-associated endothelial cells regulate cytokine production, further controlling immune cell migration and creating a hostile environment for CAR T-cell homing [136].
Despite occasional successful accumulation of CAR T-cells in the neoplastic stroma, their penetration into the tumor parenchyma remains notably limited. Vascular structures like high endothelial venules have been identified as facilitators of immune cell recruitment in certain cancers, including melanoma and breast cancer, highlighting potential routes for enhancing CAR T-cell infiltration [138,139]. The process of lymphocyte migration itself is tightly regulated by adhesion molecules—selectins, integrins, and chemokine receptors—that interact with chemokines to initiate lymphocyte rolling, firm adhesion, and eventual transendothelial migration toward the tumor [140].
However, the immunosuppressive TME often disrupts this coordinated trafficking by downregulating cytokine secretion and altering chemokine profiles, which diminishes T-cell recruitment and recognition [135,141,142]. Tumor and stromal cells in cancers such as pancreatic, ovarian, and breast carcinomas produce inhibitory chemokines likesuch as CXCL12 that restrict CAR T-cell proliferation and migration, effectively limiting their therapeutic delivery to tumor sites [143,144].
Moreover, manufacturing processes for CAR T-cells can inadvertently impair their homing capabilities. Extended ex vivo expansion may alter the expression of chemokine receptors necessary for effective trafficking, while loss of key enzymes such as heparanase—which degrades heparan sulfate in the tumor ECM—can reduce the ability of CAR T-cells to penetrate the dense stromal matrix and infiltrate tumor parenchyma [145].
Together, these physiological and technical hurdles underscore the complexity of achieving effective CAR T-cell localization and infiltration in solid tumors, necessitating innovative strategies to enhance trafficking, overcome stromal barriers, and modulate the tumor vasculature.
5.5. Target Antigen Selection
A major contributing factor to this difficulty is the inherent heterogeneity of tumor antigens, which varies markedly not only between different patients with the same tumor type but also among distinct cell populations within a single tumor. This variability complicates the process of antigen screening and selection, impeding the development of universally effective CAR T-cell therapies.
An additional critical challenge lies in ensuring target specificity to prevent “on-target off-tumor” toxicity. Many tumor-associated antigens are also expressed, albeit at varying levels, on normal healthy tissues. This nonspecific expression can inadvertently activate CAR T-cells against normal cells, causing collateral damage to essential tissues and posing potentially life-threatening risks to patients. The cytotoxic mechanisms underlying this adverse effect primarily involve the secretion of perforin and granzymes by CAR T-cells, which induce apoptosis in target cells, alongside the upregulation of T-cell surface molecules and the release of pro-inflammatory cytokines [146].
To maximize both the safety and efficacy of CAR T-cell therapies, it is imperative to identify novel antigens that exhibit exclusive expression on malignant cells while being absent from non-malignant tissues [146]. Approaches to mitigate off-tumor toxicity include fine-tuning CAR structural domains, designing logic-gated CAR T-cells that require multiple antigen signals for activation, incorporating suicide switches for controlled CAR T-cell elimination, regulating cytotoxicity levels and CAR expression, and utilizing localized delivery methods.

Table 2.
Next-Generation CAR T- Circuit Design and Function.
| Circuit Type | Core Logic |
Biological Function |
Solid Tumor Benefit |
Reference |
| Feedback-Controlled CAR | The CAR T-cell maintains internal activation balance. | Limits excessive CAR activation. | Minimizes activation toxicity, improving CAR T-cell safety | [147] |
| Hypoxia-responsive CAR | They detect hypoxia within tumor microenvironments. | Activates only within hypoxic tumor regions | Hypoxia-limited CAR T-cell activation reduces off-target effects | [148] |
| Kill-Switch Enhanced CAR |
Kill-switch prevents damage to normal tissues | Allows emergency CAR T- shutdown | Prevents normal tissue toxicity via CAR T-cell shutdown | [149] |
| Metabolic CAR | CAR T-cell performance improves under metabolic stress | Enhances CAR T-cell survival in hostile TME. | Survives hostile TME. |
[150] |
| Armored CAR T-cell (PD-1 scFv-secreting) |
CAR T-cell delivers checkpoint-blocking PD-1 scFv. | Overcomes PD-L1 immunosuppression, enhancing CAR T-cell function | Enhances CSC targeting and improves CAR T-cell persistence. |
[151] |
Furthermore, a promising strategy to circumvent the limitations posed by antigen expression on normal tissues involves targeting tumor-restricted post-translational modifications. Notably, solid tumors often overexpress truncated O-glycans such as Tn (GalNAcα1-O-Ser/Thr) and sialyl-Tn (STn) (NeuAcα2–6-GalNAcα1-O-Ser/Thr), which are largely absent in healthy cells. These tumor-specific glycoforms offer a refined target profile that may enhance selectivity and reduce off-tumor effects, thereby advancing the precision of CAR T-cell therapies for solid malignancies [152].
5.6. Adverse Reactions
While CAR T-cell therapy has undeniably transformed the landscape of cancer treatment, its adoption as a frontline option remains limited due to the high incidence of toxicities, some of which can be fatal. The occurrence and severity of adverse effects such as CRS, hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS), and immune effector cell-associated neurotoxicity syndrome (ICANS) are influenced by multiple factors including the CAR construct design, the specific antigen targeted, and the tumor type involved [153].
CAR T-cell therapy-related toxicities predominantly manifest as CRS and neurotoxicity, both of which can complicate treatment across hematological and solid tumors. CRS results from an exaggerated immune response characterized by excessive secretion of inflammatory cytokines, leading to a spectrum of clinical symptoms such as fever, fatigue, muscle pain, hypotension, hypoxia, coagulopathies, capillary leak syndrome, and even multi-organ failure, representing a significant risk of mortality [154]. Management of severe CRS often involves administration of immunosuppressive agents such as tocilizumab, either alone or in combination with corticosteroids [155]. ICANS represents a potentially life-threatening neurotoxic complication associated with CAR T-cell therapy. Patients may experience neurological symptoms including delirium, aphasia, encephalopathy, seizures, tremors, and in rare instances, rapid cerebral edema [156,157]. Proposed mechanisms underlying ICANS include disruption of the BBB and elevated production of cytokines such as IL-6, IL-8, IP-10, and MCP-1 [158].
The introduction of co-stimulatory domains in next-generation CAR designs, while enhancing efficacy, may also exacerbate the risk of severe toxicities. To address these challenges, innovative safety measures such as incorporating inducible suicide genes, such as iCaspase9, have been developed. These suicide switches enable timely termination of CAR T-cells, thereby mitigating cytotoxic damage and systemic adverse effects [159].
6. Challenges Faced with CAR T-Cell Targeting of Cancer Stem Cells
CSC-targeted CAR T-cell therapy holds considerable promise but faces substantial challenges across multiple functional domains. Toxicity remains a primary barrier, as many CSC markers—including CD133 and ALDH—are also expressed on normal progenitor cells, creating significant on-target/off-tumour risks [160,161,162]. Strategies to address this include the use of safer antigens, dual-targeted CAR constructs to enhance tumour specificity [163], intratumoral delivery to reduce systemic exposure [164], and incorporation of suicide genes or inhibitory receptors as built-in safety switches [165,166]. Additional adverse events, such as CRS and ICANS, remain clinically important and are managed with IL-6 blockade, corticosteroids, and supportive interventions [167,168,169,170].
Limited persistence and potency—particularly in solid tumours—further restrict therapeutic efficacy [171]. Enhancements have been achieved through optimized co-stimulatory domains such as: 4-1BB, ICOS, OX40, and CD27,71–74 integration of immune checkpoint blockade elements targeting PD-1, CTLA-4, TIM3, LAG3; and A2AR,74 and cytokine-armoured CAR T-cells engineered to express IL-12, IL-18, IL-7, IL-15, or IL-21 [172,173,174,175]. More recently, CARs incorporating JAK-STAT signalling domains demonstrated superior proliferative and anti-tumour capacity [54].
Overcoming poor trafficking into solid tumours remains essential, as CSCs are often shielded by dense stroma [176]. Approaches include local delivery [177],chemokine receptor engineering (CCR4, CCR2b, CXCR2) [178,179,180], and combining CAR T-cell therapy with cytoreductive treatments [181]. CAR T-cell infiltration is further hindered by ECM barriers, driving interest in FAP-targeted CAR T-cells [182], VEGFR2-targeted vascular disruption [183], and heparanase-expressing CAR T-cells [143].
Within the immunosuppressive TME, inhibitory cytokines, suppressive immune cells, and PD-L1 expression on CSCs reduce CAR T-cell functionality [184,185,186,187,188,189,190]. Supporting CAR T-cells with cytokine secretion [175], neutralizing TGF-β or IL-10 [187], in combination with PD-1/PD-L1 or CTLA-4 blockade offer promising strategies [114,188,189,190,191]. Finally, pronounced heterogeneity within CSC populations contributes to antigen escape [192,193,194,195,196]. Bispecific or multi-target CAR T-cells [197] as well as CAR T-cells engineered to secrete BiTEs that recruit bystander T-cells [198,199,200] represent key innovations to overcome this limitation.
7. Conclusions
In response to the challenges presented in the preceding sections, recent strategies have focused on engineering CAR T-cells to better navigate and resist the immunosuppressive cues of the TME. Approaches such as the incorporation of synthetic receptors responsive to tumor-derived signals, armored CAR constructs that secrete pro-inflammatory cytokines, and combination therapies targeting immune checkpoints have shown promise in enhancing T-cell infiltration, proliferation, and durability. These innovations aim not only to improve therapeutic efficacy but to fundamentally reshape the immunological landscape within solid tumors. Ultimately, overcoming the multifaceted resistance mechanisms of the TME holds the potential to catalyze a paradigm shift in the treatment of solid malignancies. A comprehensive and integrated approach—grounded in an in-depth understanding of tumor biology and immune evasion—will be essential for unlocking the full potential of CAR T-cell therapy in these complex settings.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30, . [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J Clin. 2023, 73, 17–48. doi:10.3322/caac.21763.
- Makhoul I, Atiq M, Alwbari A, Kieber-Emmons T. Breast Cancer Immunotherapy: An Update. Breast Cancer (Auckl). 2018, 12, 1178223418774802. doi: 10.1177/1178223418774802.
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 1–13, . [CrossRef]
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520, . [CrossRef]
- Cao, Y.; Chen, C.; Tao, Y.; Lin, W.; Wang, P. Immunotherapy for Triple-Negative Breast Cancer. Pharmaceutics 2021, 13, 2003, . [CrossRef]
- Wang, J.; Wu, S.-G. Breast Cancer: An Overview of Current Therapeutic Strategies, Challenge, and Perspectives. Breast Cancer: Targets Ther. 2023, ume 15, 721–730, . [CrossRef]
- Zumsteg, Z.S.; Morrow, M.; Arnold, B.; Zheng, J.; Zhang, Z.; Robson, M.; Traina, T.; McCormick, B.; Powell, S.; Ho, A.Y. Breast-Conserving Therapy Achieves Locoregional Outcomes Comparable to Mastectomy in Women with T1-2N0 Triple-Negative Breast Cancer. Ann. Surg. Oncol. 2013, 20, 3469–3476, . [CrossRef]
- van Maaren, M.C.; de Munck, L.; Strobbe, L.J.; Sonke, G.S.; Westenend, P.J.; Smidt, M.L.; Poortmans, P.M.; Siesling, S. Ten-year recurrence rates for breast cancer subtypes in the Netherlands: A large population-based study. Int. J. Cancer 2018, 144, 263–272, . [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300, doi:10.1001/jama.2018.19323.
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398, . [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9, . [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554, . [CrossRef]
- Dejenie, T.A.; G/Medhin, M.T.; Terefe, G.D.; Admasu, F.T.; Tesega, W.W.; Abebe, E.C. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum. Vaccines Immunother. 2022, 18, 2114254, . [CrossRef]
- Gao, Y.; Liu, S.; Huang, Y.; Wang, H.; Zhao, Y.; Cui, X.; Peng, Y.; Li, F.; Zhang, Y. CAR T Cells Engineered to Secrete IFNκ Induce Tumor Ferroptosis via an IFNAR/STAT1/ACSL4 Axis. Cancer Immunol. Res. 2024, 12, 1691–1702, . [CrossRef]
- Montalvo, M.J.; Bandey, I.N.; Rezvan, A.; Wu, K.-L.; Saeedi, A.; Kulkarni, R.; Li, Y.; An, X.; Sefat, K.M.S.R.; Varadarajan, N. Decoding the mechanisms of chimeric antigen receptor (CAR) T cell-mediated killing of tumors: insights from granzyme and Fas inhibition. Cell Death Dis. 2024, 15, 1–14, . [CrossRef]
- Shi, Y.; Kotchetkov, I.S.; Dobrin, A.; Hanina, S.A.; Rajasekhar, V.K.; Healey, J.H.; Sadelain, M. GLUT1 overexpression enhances CAR T cell metabolic fitness and anti-tumor efficacy. Mol. Ther. 2024, 32, 2393–2405, . [CrossRef]
- Pourzia, A.L.; Olson, M.L.; Bailey, S.R.; Boroughs, A.C.; Aryal, A.; Ryan, J.; Maus, M.V.; Letai, A. Quantifying requirements for mitochondrial apoptosis in CAR T killing of cancer cells. Cell Death Dis. 2023, 14, 1–14, . [CrossRef]
- Liu, Y.; Fang, Y.; Chen, X.; Wang, Z.; Liang, X.; Zhang, T.; Liu, M.; Zhou, N.; Lv, J.; Tang, K.; et al. Gasdermin E–mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 2020, 5, . [CrossRef]
- Conde, E.; Vercher, E.; Soria-Castellano, M.; Suarez-Olmos, J.; Mancheño, U.; Elizalde, E.; Rodriguez, M.L.; Glez-Vaz, J.; Casares, N.; Rodríguez-García, E.; et al. Epitope spreading driven by the joint action of CART cells and pharmacological STING stimulation counteracts tumor escape via antigen-loss variants. J. Immunother. Cancer 2021, 9, e003351, . [CrossRef]
- Xiong, W.; Chen, Y.; Kang, X.; Chen, Z.; Zheng, P.; Hsu, Y.-H.; Jang, J.H.; Qin, L.; Liu, H.; Dotti, G.; et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Mol. Ther. 2021, 29, 1349–1351, . [CrossRef]
- Khan, S.H.; Choi, Y.; Veena, M.; Lee, J.K.; Shin, D.S. Advances in CAR T cell therapy: antigen selection, modifications, and current trials for solid tumors. Front. Immunol. 2025, 15, 1489827, . [CrossRef]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968, . [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors.. Proc. Natl. Acad. Sci. 1993, 90, 720–724, . [CrossRef]
- Moritz, D.; Wels, W.; Mattern, J.; Groner, B. Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2-expressing tumor cells.. Proc. Natl. Acad. Sci. 1994, 91, 4318–4322, . [CrossRef]
- Stancovski, I.; Schindler, D.G.; Waks, T.; Yarden, Y.; Sela, M.; Eshhar, Z. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors.. J. Immunol. 1993, 151, 6577–6582, . [CrossRef]
- Tomasik, J.; Jasiński, M.; Basak, G.W. Next generations of CAR-T cells - new therapeutic opportunities in hematology?. Front. Immunol. 2022, 13, 1034707, . [CrossRef]
- Sun, D.; Shi, X.; Li, S.; Wang, X.; Yang, X.; Wan, M. CAR-T cell therapy: A breakthrough in traditional cancer treatment strategies (Review). Mol. Med. Rep. 2024, 29, 1–9, . [CrossRef]
- Celichowski, P.; Turi, M.; Charvátová, S.; Radhakrishnan, D.; Feizi, N.; Chyra, Z.; Šimíček, M.; Jelínek, T.; Bago, J.R.; Hájek, R.; et al. Tuning CARs: recent advances in modulating chimeric antigen receptor (CAR) T cell activity for improved safety, efficacy, and flexibility. J. Transl. Med. 2023, 21, 1–23, . [CrossRef]
- Zheng, Z.; Li, S.; Liu, M.; Chen, C.; Zhang, L.; Zhou, D. Fine-Tuning through Generations: Advances in Structure and Production of CAR-T Therapy. Cancers 2023, 15, 3476, . [CrossRef]
- Brocker, T. Chimeric Fv-Zeta or Fv-Epsilon Receptors Are Not Sufficient to Induce Activation or Cytokine Production in Peripheral T Cells. Blood 2000, 96, 1999–2001.
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115, doi:10.1158/1078-0432.ccr-06-1183.
- Park, J.R.; DiGiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.-C.; Ostberg, J.R.; et al. Adoptive Transfer of Chimeric Antigen Receptor Re-directed Cytolytic T Lymphocyte Clones in Patients with Neuroblastoma. Mol. Ther. 2007, 15, 825–833, . [CrossRef]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826, doi:10.1172/jci46110.
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242, doi:10.1038/nri3405, Erratum in Nat. Rev. Immunol. 2013, 13, 542.
- Haynes, N.M.; Trapani, J.A.; Teng, M.W.L.; Jackson, J.T.; Cerruti, L.; Jane, S.M.; Kershaw, M.H.; Smyth, M.J.; Darcy, P.K. Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood 2002, 100, 3155–3163, . [CrossRef]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684, . [CrossRef]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 2011, 3, 95ra73–95ra73, . [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; Mcgettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390, doi:10.1016/j.immuni.2016.01.021.
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590, . [CrossRef]
- Hombach, A.A.; Heiders, J.; Foppe, M.; Chmielewski, M.; Abken, H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+T cells. OncoImmunology 2012, 1, 458–466, . [CrossRef]
- Collinson-Pautz, M.R.; Chang, W.-C.; Lu, A.; Khalil, M.; Crisostomo, J.W.; Lin, P.-Y.; Mahendravada, A.; Shinners, N.P.; Brandt, M.E.; Zhang, M.; et al. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia 2019, 33, 2195–2207, . [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976, doi:10.1172/jci.insight.96976.
- Pegram, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Curran, K.J.; A Giralt, S.; Barker, J.N.; Brentjens, R.J. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 2014, 29, 415–422, . [CrossRef]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266, . [CrossRef]
- Guercio, M.; Manni, S.; Boffa, I.; Caruso, S.; Di Cecca, S.; Sinibaldi, M.; Abbaszadeh, Z.; Camera, A.; Ciccone, R.; Polito, V.A.; et al. Inclusion of the Inducible Caspase 9 Suicide Gene in CAR Construct Increases Safety of CAR.CD19 T Cell Therapy in B-Cell Malignancies. Front. Immunol. 2021, 12, . [CrossRef]
- Labanieh, L.; Majzner, R.G.; Klysz, D.; Sotillo, E.; Fisher, C.J.; Vilches-Moure, J.G.; Pacheco, K.Z.B.; Malipatlolla, M.; Xu, P.; Hui, J.H.; et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 2022, 185, 1745–1763.e22, . [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors Can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut Down Tumor Antigen Expression. Cancer Res. 2011, 71, 5697–5706, . [CrossRef]
- Liu, Y.; Di, S.; Shi, B.; Zhang, H.; Wang, Y.; Wu, X.; Luo, H.; Wang, H.; Li, Z.; Jiang, H. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3–Targeted Chimeric Antigen Receptor–Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019, 203, 198–207, . [CrossRef]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170, doi:10.1038/leu.2010.75.
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033, . [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351, . [CrossRef]
- Alsaieedi, A.A.; Zaher, K.A. Tracing the development of CAR-T cell design: from concept to next-generation platforms. Front. Immunol. 2025, 16, 1615212, . [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; O Butler, M.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359, . [CrossRef]
- Jan, M.; Scarfò, I.; Larson, R.C.; Walker, A.; Schmidts, A.; Guirguis, A.A.; Gasser, J.A.; Słabicki, M.; Bouffard, A.A.; Castano, A.P.; et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci. Transl. Med. 2021, 13, eabb6295, doi:10.1126/scitranslmed.abb6295.
- Li, H.-S.; Wong, N.M.; Tague, E.; Ngo, J.T.; Khalil, A.S.; Wong, W.W. High-performance multiplex drug-gated CAR circuits. Cancer Cell 2022, 40, 1294–1305.e4, . [CrossRef]
- Lawson, J.C.; Blatch, G.L.; Edkins, A.L. Cancer stem cells in breast cancer and metastasis. Breast Cancer Res. Treat. 2009, 118, 241–254, . [CrossRef]
- Xia, P. Surface Markers of Cancer Stem Cells in Solid Tumors. Curr. Stem Cell Res. Ther. 2014, 9, 102–111, . [CrossRef]
- Klonisch, T.; Wiechec, E.; Hombach-Klonisch, S.; Ande, S.R.; Wesselborg, S.; Schulze-Osthoff, K.; Los, M. Cancer stem cell markers in common cancers – therapeutic implications. Trends Mol. Med. 2008, 14, 450–460, . [CrossRef]
- Han, S.; Hwang, P.G.; Chung, D.H.; Kim, D.; Im, S.; Kim, Y.T.; Kim, T.; Heo, D.S.; Bang, Y.; Kim, N.K. Epidermal growth factor receptor (EGFR) downstream molecules as response predictive markers for gefitinib (Iressa®, ZD1839) in chemotherapy-resistant non-small cell lung cancer. Int. J. Cancer 2004, 113, 109–115, . [CrossRef]
- Karsten, U.; Goletz, S. What makes cancer stem cell markers different?. SpringerPlus 2013, 2, 301, . [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111, doi:10.1038/35102167.
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.-F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. JNCI J. Natl. Cancer Inst. 2008, 100, 672–679, . [CrossRef]
- Zhou, L.; Jiang, Y.; Yan, T.; Di, G.; Shen, Z.; Shao, Z.; Lu, J. The prognostic role of cancer stem cells in breast cancer: a meta-analysis of published literatures. Breast Cancer Res. Treat. 2010, 122, 795–801, . [CrossRef]
- Chen, Y.; Sun, C.; Landoni, E.; Metelitsa, L.S.; Dotti, G.; Savoldo, B. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin. Cancer Res. 2019, 25, 2915–2924, . [CrossRef]
- Drent, E.; Groen, R.W.; Noort, W.A.; Themeli, M.; van Bueren, J.J.L.; Parren, P.W.H.I.; Kuball, J.; Sebestyen, Z.; Yuan, H.; de Bruijn, J.; et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica 2016, 101, 616–625, . [CrossRef]
- Drent, E.; Poels, R.; Ruiter, R.; Van De Donk, N.W.C.J.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor–engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025, doi:10.1158/1078-0432.ccr-18-2559.
- Laborda, E.; Mazagova, M.; Shao, S.; Wang, X.; Quirino, H.; Woods, A.K.; Hampton, E.N.; Rodgers, D.T.; Kim, C.H.; Schultz, P.G.; et al. Development of A Chimeric Antigen Receptor Targeting C-Type Lectin-Like Molecule-1 for Human Acute Myeloid Leukemia. Int. J. Mol. Sci. 2017, 18, 2259, . [CrossRef]
- Pizzitola, I.; Anjos-Afonso, F.; Rouault-Pierre, K.; Lassailly, F.; Tettamanti, S.; Spinelli, O.; Biondi, A.; Biagi, E.; Bonnet, D. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 2014, 28, 1596–1605, . [CrossRef]
- Huang, J.; Yang, Y.; Fang, F.; Liu, K. MALAT1 modulates the autophagy of retinoblastoma cell through miR-124-mediated stx17 regulation. J. Cell. Biochem. 2017, 119, 3853–3863, . [CrossRef]
- Tettamanti, S.; Marin, V.; Pizzitola, I.; Magnani, C.F.; Giordano Attianese, G.M.; Cribioli, E.; Maltese, F.; Galimberti, S.; Lopez, A.F.; Biondi, A.; et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br. J. Haematol. 2013, 161, 389–401, . [CrossRef]
- Gzil, A.; Zarębska, I.; Bursiewicz, W.; Antosik, P.; Grzanka, D.; Szylberg, Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol. Biol. Rep. 2019, 46, 6629–6645, . [CrossRef]
- Cui, X.; Liu, R.; Duan, L.; Cao, D.; Zhang, Q.; Zhang, A. CAR-T therapy: Prospects in targeting cancer stem cells. J. Cell. Mol. Med. 2021, 25, 9891–9904, . [CrossRef]
- A Morgan, R.; Yang, J.C.; Kitano, M.; E Dudley, M.; Laurencot, C.M.; A Rosenberg, S. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851, . [CrossRef]
- Masoumi, J.; Jafarzadeh, A.; Abdolalizadeh, J.; Khan, H.; Philippe, J.; Mirzaei, H.; Mirzaei, H.R. Cancer stem cell-targeted chimeric antigen receptor (CAR)-T cell therapy: Challenges and prospects. Acta Pharm. Sin. B 2021, 11, 1721–1739, . [CrossRef]
- Erler, P.; Kurcon, T.; Cho, H.; Skinner, J.; Dixon, C.; Grudman, S.; Rozlan, S.; Dessez, E.; Mumford, B.; Jo, S.; et al. Multi-armored allogeneic MUC1 CAR T cells enhance efficacy and safety in triple-negative breast cancer. Sci. Adv. 2024, 10, eadn9857, . [CrossRef]
- Li, D.; Guo, X.; Yang, K.; Yang, Y.; Zhou, W.; Huang, Y.; Liang, X.; Su, J.; Jiang, L.; Li, J.; et al. EpCAM-targeting CAR-T cell immunotherapy is safe and efficacious for epithelial tumors. Sci. Adv. 2023, 9, eadg9721, . [CrossRef]
- Seitz, C.M.; Schroeder, S.; Knopf, P.; Krahl, A.-C.; Hau, J.; Schleicher, S.; Martella, M.; Quintanilla-Martinez, L.; Kneilling, M.; Pichler, B.; et al. GD2-targeted chimeric antigen receptor T cells prevent metastasis formation by elimination of breast cancer stem-like cells. OncoImmunology 2019, 9, 1683345, . [CrossRef]
- Moreno-Londoño, A.P.; Robles-Flores, M. Functional Roles of CD133: More than Stemness Associated Factor Regulated by the Microenvironment. Stem Cell Rev. Rep. 2023, 20, 25–51, . [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323, . [CrossRef]
- Ma, S.; Lee, T.K.; Zheng, B.-J.; Chan, K.W.; Guan, X.-Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758, doi:10.1038/sj.onc.1210811.
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 1–67, . [CrossRef]
- Kryczek, I.; Liu, S.; Roh, M.; Vatan, L.; Szeliga, W.; Wei, S.; Banerjee, M.; Mao, Y.; Kotarski, J.; Wicha, M.S.; et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer 2011, 130, 29–39, . [CrossRef]
- Schmohl, J.U.; Vallera, D.A. CD133, Selectively Targeting the Root of Cancer. Toxins 2016, 8, 165, . [CrossRef]
- Hu, B.; Zou, Y.; Zhang, L.; Tang, J.; Niedermann, G.; Firat, E.; Huang, X.; Zhu, X. Nucleofection with Plasmid DNA for CRISPR/Cas9-Mediated Inactivation of Programmed Cell Death Protein 1 in CD133-Specific CAR T Cells. Hum. Gene Ther. 2019, 30, 446–458, . [CrossRef]
- Zhu, X.; Prasad, S.; Gaedicke, S.; Hettich, M.; Firat, E.; Niedermann, G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget 2014, 6, 171–184, . [CrossRef]
- Klapdor, R.; Wang, S.; Hacker, U.; Büning, H.; Morgan, M.; Dörk, T.; Hillemanns, P.; Schambach, A. Improved Killing of Ovarian Cancer Stem Cells by Combining a Novel Chimeric Antigen Receptor–Based Immunotherapy and Chemotherapy. Hum. Gene Ther. 2017, 28, 886–896, . [CrossRef]
- Zhu, X.; Chen, J.; Li, W.; Xu, Y.; Shan, J.; Hong, J.; Zhao, Y.; Xu, H.; Ma, J.; Shen, J.; et al. Hypoxia-Responsive CAR-T Cells Exhibit Reduced Exhaustion and Enhanced Efficacy in Solid Tumors. Cancer Res. 2023, 84, 84–100, . [CrossRef]
- Ruella, M.; Barrett, D.M.; Shestova, O.; Perazzelli, J.; Posey, A.D.; Hong, S.J.; Kozlowski, M.; Lacey, S.F.; Melenhorst, J.J.; June, C.H.; et al. A cellular antidote to specifically deplete anti-CD19 chimeric antigen receptor–positive cells. Blood 2020, 135, 505–509, . [CrossRef]
- Shi, C.; Tian, R.; Wang, M.; Wang, X.; Jiang, J.; Zhang, Z.; Li, X.; He, Z.; Gong, W.; Qin, R. CD44+CD133+population exhibits cancer stem cell-like characteristics in human gallbladder carcinoma. Cancer Biol. Ther. 2010, 10, 1182–1190, . [CrossRef]
- Wang, C.; Xie, J.; Guo, J.; Manning, H.C.; Gore, J.C.; Guo, N. Evaluation of CD44 and CD133 as cancer stem cell markers for colorectal cancer. Oncol. Rep. 2012, 28, 1301–1308, . [CrossRef]
- Zhang, X.; Yang, L.; Lei, W.; Hou, Q.; Huang, M.; Zhou, R.; Enver, T.; Wu, S. Single-cell sequencing reveals CD133+CD44−-originating evolution and novel stemness related variants in human colorectal cancer. EBioMedicine 2022, 82, 104125, . [CrossRef]
- Litvinov, S.V.; Velders, M.P.; A Bakker, H.; Fleuren, G.J.; O Warnaar, S. Ep-CAM: a human epithelial antigen is a homophilic cell-cell adhesion molecule.. J. Cell Biol. 1994, 125, 437–446, . [CrossRef]
- Yamashita, T.; Honda, M.; Nakamoto, Y.; Baba, M.; Nio, K.; Hara, Y.; Zeng, S.S.; Hayashi, T.; Kondo, M.; Takatori, H.; et al. Discrete nature of EpCAM+ and CD90+ cancer stem cells in human hepatocellular carcinoma. Hepatology 2012, 57, 1484–1497, . [CrossRef]
- Dalerba, P.; Dylla, S.J.; Park, I.-K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163, doi:10.1073/pnas.0703478104.
- Zhang, B.-L.; Li, D.; Gong, Y.-L.; Huang, Y.; Qin, D.-Y.; Jiang, L.; Liang, X.; Yang, X.; Gou, H.-F.; Wang, Y.-S.; et al. Preclinical Evaluation of Chimeric Antigen Receptor–Modified T Cells Specific to Epithelial Cell Adhesion Molecule for Treating Colorectal Cancer. Hum. Gene Ther. 2019, 30, 402–412, . [CrossRef]
- Wu, Y.; Deng, Z.; Tang, Y.; Zhang, S.; Zhang, Y.-Q. Over-expressing Akt in T cells to resist tumor immunosuppression and increase anti-tumor activity. BMC Cancer 2015, 15, 603, . [CrossRef]
- Deng, Z.; Wu, Y.; Ma, W.; Zhang, S.; Zhang, Y.-Q. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015, 16, 1–9, . [CrossRef]
- Jindal, V.; Arora, E.; Gupta, S. Challenges and prospects of chimeric antigen receptor T cell therapy in solid tumors. Med Oncol. 2018, 35, 87, . [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022, . [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912, . [CrossRef]
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133, . [CrossRef]
- Dugnani, E.; Pasquale, V.; Bordignon, C.; Canu, A.; Piemonti, L.; Monti, P. Integrating T cell metabolism in cancer immunotherapy. Cancer Lett. 2017, 411, 12–18, . [CrossRef]
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front. Immunol. 2017, 8, 1850, . [CrossRef]
- Castellarin, M.; Watanabe, K.; June, C.H.; Kloss, C.C.; Posey, A.D., Jr. Driving cars to the clinic for solid tumors. Gene Ther. 2018, 25, 165–175, . [CrossRef]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48, . [CrossRef]
- Huang, R.; Zhu, J.; Fan, R.; Tang, Y.; Hu, L.; Lee, H.; Chen, S. Extracellular vesicle-based drug delivery systems in cancer. Extracell. Vesicle 2024, 4, . [CrossRef]
- Overchuk, M.; Zheng, G. Overcoming obstacles in the tumor microenvironment: Recent advancements in nanoparticle delivery for cancer theranostics. Biomaterials 2018, 156, 217–237, . [CrossRef]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T Cells into Tumors. Cancer Res. 2014, 74, 7168–7174, . [CrossRef]
- Kaushik, S.; Pickup, M.W.; Weaver, V.M. From transformation to metastasis: deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016, 35, 655–667, . [CrossRef]
- Chen, W.; Li, Y.; Liu, C.; Kang, Y.; Qin, D.; Chen, S.; Zhou, J.; Liu, H.; Ferdows, B.E.; Patel, D.N.; et al. In situ Engineering of Tumor-Associated Macrophages via a Nanodrug-Delivering-Drug (β-Elemene@Stanene) Strategy for Enhanced Cancer Chemo-Immunotherapy. Angew. Chem. Int. Ed. Engl. 2023, 62, e202308413, . [CrossRef]
- Hou, A.J.; Chen, L.C.; Chen, Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021, 20, 531–550, . [CrossRef]
- Cheever, A.; Townsend, M.; O’neill, K. Tumor Microenvironment Immunosuppression: A Roadblock to CAR T-Cell Advancement in Solid Tumors. Cells 2022, 11, 3626, . [CrossRef]
- Ai, L.; Chen, J.; Yan, H.; He, Q.; Luo, P.; Xu, Z.; Yang, X. Research Status and Outlook of PD-1/PD-L1 Inhibitors for Cancer Therapy. Drug Des. Dev. Ther. 2020, ume 14, 3625–3649, . [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773, . [CrossRef]
- Abken, H. Driving CARs on the Highway to Solid Cancer: Some Considerations on the Adoptive Therapy with CAR T Cells. Hum. Gene Ther. 2017, 28, 1047–1060, . [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548, . [CrossRef]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118, . [CrossRef]
- Guzman, G.; Pellot, K.; Reed, M.R.; Rodriguez, A. CAR T-cells to treat brain tumors. Brain Res. Bull. 2023, 196, 76–98, . [CrossRef]
- Rodriguez, A.; Brown, C.; Badie, B. Chimeric antigen receptor T-cell therapy for glioblastoma. Transl. Res. 2017, 187, 93–102, . [CrossRef]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84, . [CrossRef]
- Huang, J.; Li, Y.B.; Charlebois, C.; Nguyen, T.; Liu, Z.; Bloemberg, D.; Zafer, A.; Baumann, E.; Sodja, C.; Leclerc, S.; et al. Application of blood brain barrier models in pre-clinical assessment of glioblastoma-targeting CAR-T based immunotherapies. Fluids Barriers CNS 2022, 19, 1–15, . [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [CrossRef]
- Rennert, P.; Wu, L.; Su, L.; Lobb, R.; Ambrose, C. 160 Evaluation and development of dual and triple antigen targeting CAR-T Engager proteins for Her2-positive CNS metastases and solid tumors. SITC 36th Anniversary Annual Meeting (SITC 2021) Abstracts. LOCATION OF CONFERENCE, COUNTRYDATE OF CONFERENCE; pp. A170–A170.
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226, doi:10.1158/2159-8290.cd-18-0442.
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. - Nucleic Acids 2013, 2, e105, . [CrossRef]
- Jiang, Y.; Wen, W.; Yang, F.; Han, D.; Zhang, W.; Qin, W. Prospect of Prostate Cancer Treatment: Armed CAR-T or Combination Therapy. Cancers 2022, 14, 967, . [CrossRef]
- Hawkins, E.R.; D’Souza, R.R.; Klampatsa, A. Armored CAR T-Cells: The Next Chapter in T-Cell Cancer Immunotherapy. Biol. Targets Ther. 2021, ume 15, 95–105, . [CrossRef]
- Chinnasamy, D.; Yu, Z.; Kerkar, S.P.; Zhang, L.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Local Delivery of lnterleukin-12 Using T Cells Targeting VEGF Receptor-2 Eradicates Multiple Vascularized Tumors in Mice. Clin. Cancer Res. 2012, 18, 1672–1683, . [CrossRef]
- Qin, Y.; Xu, G. Enhancing CAR T-cell therapies against solid tumors: Mechanisms and reversion of resistance. Front. Immunol. 2022, 13, 1053120, . [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737, doi:10.1038/s41598-017-00462-8.
- Hamieh, M.; Mansilla-Soto, J.; Rivière, I.; Sadelain, M. Programming CAR T Cell Tumor Recognition: Tuned Antigen Sensing and Logic Gating. Cancer Discov. 2023, 13, 829–843, . [CrossRef]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86, . [CrossRef]
- Nakagawa, Y.; Negishi, Y.; Shimizu, M.; Takahashi, M.; Ichikawa, M.; Takahashi, H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol. Lett. 2015, 167, 72–86, . [CrossRef]
- Nakagawa, Y.; Negishi, Y.; Shimizu, M.; Takahashi, M.; Ichikawa, M.; Takahashi, H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol. Lett. 2015, 167, 72–86, . [CrossRef]
- van der Zijpp, Y.J.; Poot, A.A.; Feijen, J. ICAM-1 and VCAM-1 expression by endothelial cells grown on fibronectin-coated TCPS and PS. J. Biomed. Mater. Res. Part A 2003, 65A, 51–59, . [CrossRef]
- Stock, S.; Schmitt, M.; Sellner, L. Optimizing Manufacturing Protocols of Chimeric Antigen Receptor T Cells for Improved Anticancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 6223, . [CrossRef]
- Brown, M.H.; Dustin, M.L. Steering CAR T Cells into Solid Tumors. New Engl. J. Med. 2019, 380, 289–291, . [CrossRef]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740, . [CrossRef]
- Ager, A.; Watson, H.A.; Wehenkel, S.C.; Mohammed, R.N. Homing to solid cancers: a vascular checkpoint in adoptive cell therapy using CAR T-cells. Biochem. Soc. Trans. 2016, 44, 377–385, . [CrossRef]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131, doi:10.1158/2326-6066.cir-14-0160.
- Waldmann, T.A. Cytokines in Cancer Immunotherapy. Cold Spring Harb. Perspect. Biol. 2017, 10, a028472, . [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529, . [CrossRef]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting Migration of T Cells to Chemokine Secreted from Tumors by Genetic Modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980, . [CrossRef]
- EBMT. EBMT Registry. 2023. Available Online: Https://Www.Ebmt.Org/Registry/Ebmt-Car-t-Data- Collection-Initiative (Accessed on 21 December 2023).
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-El-Enein, M. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat. Rev. Clin. Oncol. 2022, 20, 49–62, . [CrossRef]
- Lu, L.; Xie, M.; Yang, B.; Zhao, W.-B.; Cao, J. Enhancing the safety of CAR-T cell therapy: Synthetic genetic switch for spatiotemporal control. Sci. Adv. 2024, 10, eadj6251, . [CrossRef]
- Kosti, P.; Opzoomer, J.W.; Larios-Martinez, K.I.; Henley-Smith, R.; Scudamore, C.L.; Okesola, M.; Taher, M.Y.; Davies, D.M.; Muliaditan, T.; Larcombe-Young, D.; et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep. Med. 2021, 2, 100227, . [CrossRef]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2017, 25, 580–592, . [CrossRef]
- Frlic, T.; Pavlin, M. Metabolic reprogramming of CAR T cells: a new frontier in cancer immunotherapy. Front. Immunol. 2025, 16, 1688995, . [CrossRef]
- Yang, C.; You, J.; Pan, Q.; Tang, Y.; Cai, L.; Huang, Y.; Gu, J.; Wang, Y.; Yang, X.; Du, Y.; et al. Targeted delivery of a PD-1-blocking scFv by CD133-specific CAR-T cells using nonviral Sleeping Beauty transposition shows enhanced antitumour efficacy for advanced hepatocellular carcinoma. BMC Med. 2023, 21, 1–19, . [CrossRef]
- Steentoft, C.; Migliorini, D.; King, T.R.; Mandel, U.; June, C.H.; Posey, A.D. Glycan-directed CAR-T cells. Glycobiology 2018, 28, 656–669, . [CrossRef]
- Roex, G.; Timmers, M.; Wouters, K.; Campillo-Davo, D.; Flumens, D.; Schroyens, W.; Chu, Y.; Berneman, Z.N.; Lion, E.; Luo, F.; et al. Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J. Hematol. Oncol. 2020, 13, 1–14, . [CrossRef]
- Wang, Z.; Han, W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark. Res. 2018, 6, 1–10, . [CrossRef]
- Lee, K.; Paek, H.; Ai, L.; Liu, Z.; Jin, L.; Li, M.; Jun, T.; Higashi, M.K.; Onel, K.; Oh, W.K.; et al. Treatment Profile of CAR-T Cell Therapy Induced Cytokine Release Syndrome and Neurotoxicity: Insights from Real-World Evidence. Blood 2022, 140, 12750–12752, . [CrossRef]
- Sterner, R.C.; Sterner, R.M. Immune effector cell associated neurotoxicity syndrome in chimeric antigen receptor-T cell therapy. Front. Immunol. 2022, 13, 879608, . [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419, doi:10.1158/2159-8290.cd-17-0698.
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971, . [CrossRef]
- Xiong, X.; Yu, Y.; Jin, X.; Xie, D.; Sun, R.; Lu, W.; Wei, Y.; Guo, R.; Zhao, M. Functional Validation of the RQR8 Suicide /Marker Gene in CD19 CAR-T Cells and CLL1CAR-T Cells. Ann. Hematol. 2023, 102, 1523–1535, . [CrossRef]
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-Modified T Cells: Toxicities and Overcoming Strategies. J. Immunol. Res. 2018, 2018, 1–10, . [CrossRef]
- Peh, G.S.-L.; Lang, R.J.; Pera, M.F.; Hawes, S.M. CD133 Expression by Neural Progenitors Derived from Human Embryonic Stem Cells and Its Use for Their Prospective Isolation. Stem Cells Dev. 2009, 18, 269–282, . [CrossRef]
- Schuurhuis, G.J.; Meel, M.H.; Wouters, F.; A Min, L.; Terwijn, M.; A De Jonge, N.; Kelder, A.; Snel, A.N.; Zweegman, S.; Ossenkoppele, G.J.; et al. Normal Hematopoietic Stem Cells within the AML Bone Marrow Have a Distinct and Higher ALDH Activity Level than Co-Existing Leukemic Stem Cells. PLOS ONE 2013, 8, e78897, . [CrossRef]
- Zhang, E.; Yang, P.; Gu, J.; Wu, H.; Chi, X.; Liu, C.; Wang, Y.; Xue, J.; Qi, W.; Sun, Q.; et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J. Hematol. Oncol. 2018, 11, 1–14, . [CrossRef]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020, 26, 712–719, . [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1– and CTLA-4–Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172–215ra172, . [CrossRef]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235, . [CrossRef]
- Singh, A.K.; McGuirk, J.P. CAR T cells: continuation in a revolution of immunotherapy. Lancet Oncol. 2020, 21, e168–e178, . [CrossRef]
- Chou, C.K.; Turtle, C.J. Assessment and management of cytokine release syndrome and neurotoxicity following CD19 CAR-T cell therapy. Expert Opin. Biol. Ther. 2020, 20, 653–664, . [CrossRef]
- Si, S.; Teachey, D.T. Spotlight on Tocilizumab in the Treatment of CAR-T-Cell-Induced Cytokine Release Syndrome: Clinical Evidence to Date. 2020, 16, 705–714, . [CrossRef]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 433–444, . [CrossRef]
- McLellan, A.D.; Rad, S.M.A.H. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019, 97, 664–674, . [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 2013, 13, 525–541, . [CrossRef]
- Pachella, R.L.A.; adsen, P.L.T.; Dains, D.J.E. The Toxicity and Benefit of Various Dosing Strategies for Interleukin-2 in Metastatic Melanoma and Renal Cell Carcinoma. J. Adv. Pr. Oncol. 2015, 6, 212–21, . [CrossRef]
- Conlon, K.C.; Lugli, E.; Welles, H.C.; Rosenberg, S.A.; Fojo, A.T.; Morris, J.C.; Fleisher, T.A.; Dubois, S.P.; Perera, L.P.; Stewart, D.M.; et al. Redistribution, Hyperproliferation, Activation of Natural Killer Cells and CD8 T Cells, and Cytokine Production During First-in-Human Clinical Trial of Recombinant Human Interleukin-15 in Patients With Cancer. J. Clin. Oncol. 2015, 33, 74–82, doi:10.1200/jco.2014.57.3329.
- Tian, Y.; Li, Y.; Shao, Y.; Zhang, Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J. Hematol. Oncol. 2020, 13, 1–16, . [CrossRef]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86, doi:10.3389/fcell.2019.00086.
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569, doi:10.1056/nejmoa1610497.
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683, doi:10.1056/nejmoa1106152.
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724, . [CrossRef]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J. Immunother. 2010, 33, 780–788, . [CrossRef]
- Yu, W.-D.; Sun, G.; Li, J.; Xu, J.; Wang, X. Mechanisms and therapeutic potentials of cancer immunotherapy in combination with radiotherapy and/or chemotherapy. Cancer Lett. 2019, 452, 66–70, . [CrossRef]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting Fibroblast Activation Protein in Tumor Stroma with Chimeric Antigen Receptor T Cells Can Inhibit Tumor Growth and Augment Host Immunity without Severe Toxicity. Cancer Immunol. Res. 2013, 2, 154–166, . [CrossRef]
- Chinnasamy, D.; Yu, Z.; Theoret, M.R.; Zhao, Y.; Shrimali, R.K.; Morgan, R.A.; Feldman, S.A.; Restifo, N.P.; Rosenberg, S.A. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J. Clin. Investig. 2010, 120, 3953–3968, . [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells — a clinical update. Nat. Rev. Clin. Oncol. 2019, 17, 204–232, . [CrossRef]
- Najafi, M.; Mortezaee, K.; Majidpoor, J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019, 234, 116781, . [CrossRef]
- Badrinath, N.; Yoo, S.Y. Recent Advances in Cancer Stem Cell-Targeted Immunotherapy. Cancers 2019, 11, 310, . [CrossRef]
- Takahashi, H.; Sakakura, K.; Kudo, T.; Toyoda, M.; Kaira, K.; Oyama, T.; Chikamatsu, K. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 2016, 8, 8633–8647, . [CrossRef]
- Polónia, A.; Pinto, R.; Cameselle-Teijeiro, J.F.; Schmitt, F.C.; Paredes, J. Prognostic value of stromal tumour infiltrating lymphocytes and programmed cell death-ligand 1 expression in breast cancer. J. Clin. Pathol. 2017, 70, 860–867, . [CrossRef]
- Inaguma, S.; Lasota, J.; Wang, Z.; Czapiewski, P.; Langfort, R.; Rys, J.; Szpor, J.; Waloszczyk, P.; Okoń, K.; Biernat, W.; et al. Expression of ALCAM (CD166) and PD-L1 (CD274) independently predicts shorter survival in malignant pleural mesothelioma. Hum. Pathol. 2018, 71, 1–7, . [CrossRef]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2018, 68, 365–377, . [CrossRef]
- Zheng, H.; Pomyen, Y.; Hernandez, M.O.; Li, C.; Livak, F.; Tang, W.; Dang, H.; Greten, T.F.; Davis, J.L.; Zhao, Y.; et al. Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology 2018, 68, 127–140, doi:10.1002/hep.29778.
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 1–9, . [CrossRef]
- Hirata, A.; Hatano, Y.; Niwa, M.; Hara, A.; Tomita, H. Heterogeneity in Colorectal Cancer Stem Cells. Cancer Prev. Res. 2019, 12, 413–420, . [CrossRef]
- Hirata, A.; Hatano, Y.; Niwa, M.; Hara, A.; Tomita, H. Heterogeneity of Colon Cancer Stem Cells. In Stem Cells Heterogeneity in Cancer; Birbrair, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Swit-zerland, 2019; Volume 1139, pp. 115–126, ISBN 978-3-030-14365-7.
- Zheng, F.; Dang, J.; Zhang, H.; Xu, F.; Ba, D.; Zhang, B.; Cheng, F.; Chang, A.E.; Wicha, M.S.; Li, Q. Cancer Stem Cell Vaccination With PD-L1 and CTLA-4 Blockades Enhances the Eradication of Melanoma Stem Cells in a Mouse Tumor Model. J. Immunother. 2018, 41, 361–368, . [CrossRef]
- Sousa, B.; Ribeiro, A.S.; Paredes, J. Heterogeneity and Plasticity of Breast Cancer Stem Cells. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; in press.
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. Hematol. Oncol. 2020, 13, 1–10, . [CrossRef]
- Kailayangiri, S.; Altvater, B.; Wiebel, M.; Jamitzky, S.; Rossig, C. Overcoming Heterogeneity of Antigen Expression for Effective CAR T Cell Targeting of Cancers. Cancers 2020, 12, 1075, . [CrossRef]
- Iwahori, K.; Kakarla, S.; Velasquez, M.P.; Yu, F.; Yi, Z.; Gerken, C.; Song, X.-T.; Gottschalk, S. Engager T Cells: A New Class of Antigen-specific T Cells That Redirect Bystander T Cells. Mol. Ther. 2015, 23, 171–178, . [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058, . [CrossRef]
Figure 1.
Killing mechanisms of CAR T-cells: CAR T- cells eliminate tumor cells through multiple complementary cytotoxic mechanisms within the tumor microenvironment. CAR T-cells induce direct cytolysis via perforin–granzyme–grnulysin axis, mediating pore formation and caspase-dependent apoptosis. Death-receptor signaling through Fas–FasL, TRAIL–DR4/DR5, and TNF-alpha activates extrinsic apoptotic cascades in antigen-expressing tumor cells. TNF-α–mediated apoptosis and cytokine-driven bystander killing extend cytotoxicity beyond direct cell–cell contact. Mitochondrial apoptosis via BID/BAX activation and cytochrome-c release amplifies intrinsic death signaling. Granulysin secretion contributes to membrane damage and mitochondrial dysfunction in susceptible tumor cells. Pyroptosis mediated by granzyme-B–dependent GSDME cleavage in susceptible tumor cells. Ferroptosis can be induced through IFN-driven lipid peroxidation pathways, sensitizing tumor cells to oxidative death. Metabolic competition within the TME restricts tumor growth while metabolically optimized CAR T- cells can preserve functional fitness. Mechanical disruption at the immunological synapse causes membrane rupture and cytoskeletal collapse. CAR T–cell derived cytokines activate macrophages, promoting phagocytosis and ROS/NO-dependent tumor killing. Epitope spreading recruits endogenous T-cells, NK cells, and dendritic cells, enabling multi-antigen immune surveillance and control of antigen-escape variants [14,15,16,17,18,19,20,21].
Figure 1.
Killing mechanisms of CAR T-cells: CAR T- cells eliminate tumor cells through multiple complementary cytotoxic mechanisms within the tumor microenvironment. CAR T-cells induce direct cytolysis via perforin–granzyme–grnulysin axis, mediating pore formation and caspase-dependent apoptosis. Death-receptor signaling through Fas–FasL, TRAIL–DR4/DR5, and TNF-alpha activates extrinsic apoptotic cascades in antigen-expressing tumor cells. TNF-α–mediated apoptosis and cytokine-driven bystander killing extend cytotoxicity beyond direct cell–cell contact. Mitochondrial apoptosis via BID/BAX activation and cytochrome-c release amplifies intrinsic death signaling. Granulysin secretion contributes to membrane damage and mitochondrial dysfunction in susceptible tumor cells. Pyroptosis mediated by granzyme-B–dependent GSDME cleavage in susceptible tumor cells. Ferroptosis can be induced through IFN-driven lipid peroxidation pathways, sensitizing tumor cells to oxidative death. Metabolic competition within the TME restricts tumor growth while metabolically optimized CAR T- cells can preserve functional fitness. Mechanical disruption at the immunological synapse causes membrane rupture and cytoskeletal collapse. CAR T–cell derived cytokines activate macrophages, promoting phagocytosis and ROS/NO-dependent tumor killing. Epitope spreading recruits endogenous T-cells, NK cells, and dendritic cells, enabling multi-antigen immune surveillance and control of antigen-escape variants [14,15,16,17,18,19,20,21].
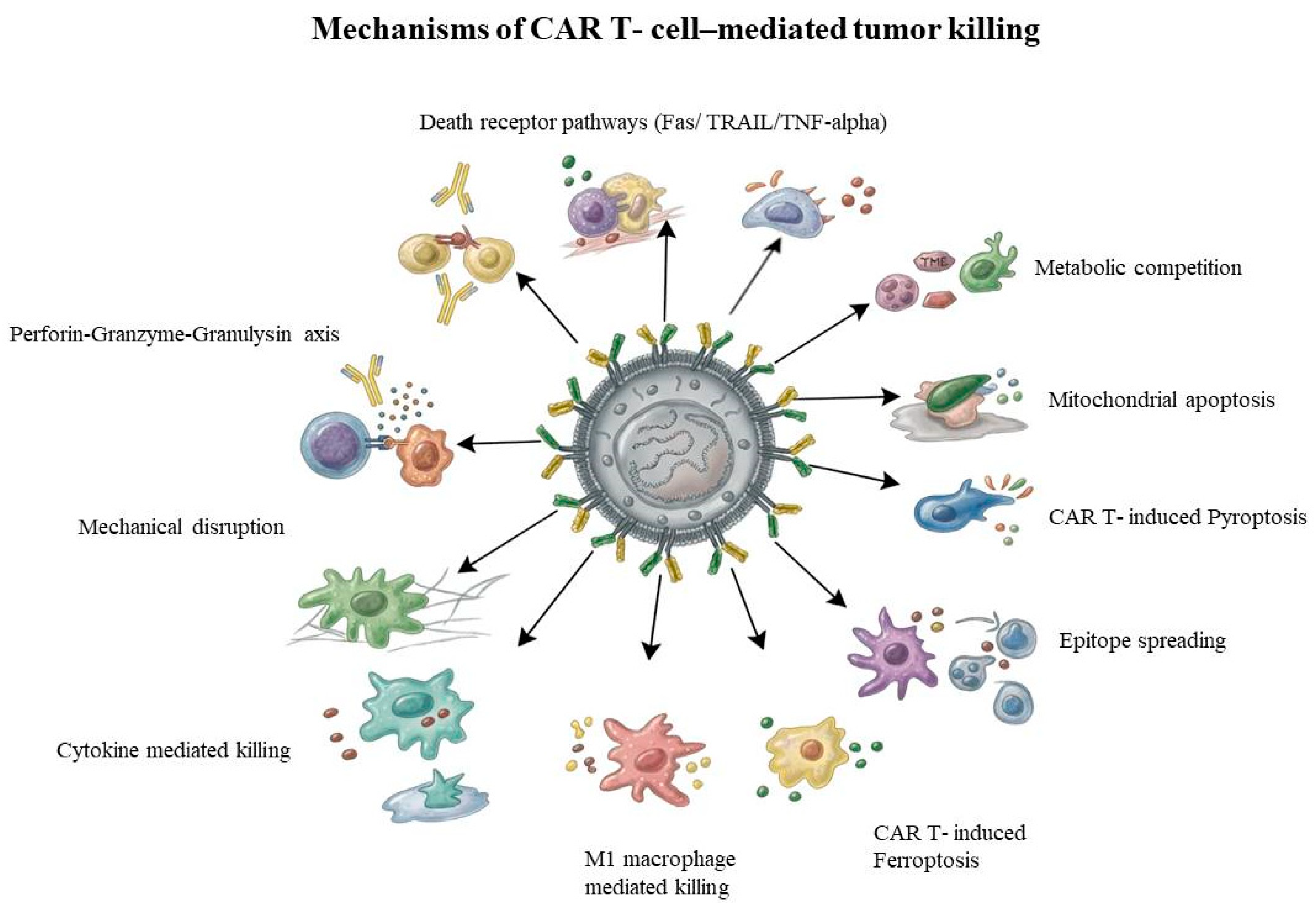
Figure 2.
Evolution of chimeric antigen receptor (CAR) T-cell generations and signaling architecture. First-generation CARs comprise an extracellular single-chain variable fragment (scFv) linked solely to the intracellular CD3ζ activation domain, resulting in limited T-cell activation due to the absence of co-stimulation. Second-generation CARs incorporate an additional costimulatory domain (e.g., CD28 or 4-1BB) alongside CD3ζ, enabling dual signaling and improved effector function. Third-generation CARs further augment signaling by including two costimulatory domains in tandem with CD3ζ to enhance T-cell activation following antigen engagement. There are several different iterations categorized as Fourth-generation CAR T-cells. One well-described subtype is referred to as TRUCKs (T-cells Redirected for Universal Cytokine-mediated Killing), where CARs are based on second-generation constructs and incorporate an NFAT-responsive gene cassette that is activated following CAR engagement with its target antigen, resulting in localized cytokine release that acts as an additional downstream signaling and immune-modulatory mechanism following CAR activation. Fifth-generation CARs build on the second-generation framework by integrating a cytokine receptor signaling domain, typically IL-2Rβ (Interleukin-2 receptor beta chain) This additional intracellular module enables activation of the JAK–STAT pathway, leading to STAT3 and STAT5 signaling upon antigen engagement, resulting in synergistic triple signaling through CD3ζ activation, co-stimulation, and intracellular cytokine receptor-mediated pathways [14,27,28,29,30].
Figure 2.
Evolution of chimeric antigen receptor (CAR) T-cell generations and signaling architecture. First-generation CARs comprise an extracellular single-chain variable fragment (scFv) linked solely to the intracellular CD3ζ activation domain, resulting in limited T-cell activation due to the absence of co-stimulation. Second-generation CARs incorporate an additional costimulatory domain (e.g., CD28 or 4-1BB) alongside CD3ζ, enabling dual signaling and improved effector function. Third-generation CARs further augment signaling by including two costimulatory domains in tandem with CD3ζ to enhance T-cell activation following antigen engagement. There are several different iterations categorized as Fourth-generation CAR T-cells. One well-described subtype is referred to as TRUCKs (T-cells Redirected for Universal Cytokine-mediated Killing), where CARs are based on second-generation constructs and incorporate an NFAT-responsive gene cassette that is activated following CAR engagement with its target antigen, resulting in localized cytokine release that acts as an additional downstream signaling and immune-modulatory mechanism following CAR activation. Fifth-generation CARs build on the second-generation framework by integrating a cytokine receptor signaling domain, typically IL-2Rβ (Interleukin-2 receptor beta chain) This additional intracellular module enables activation of the JAK–STAT pathway, leading to STAT3 and STAT5 signaling upon antigen engagement, resulting in synergistic triple signaling through CD3ζ activation, co-stimulation, and intracellular cytokine receptor-mediated pathways [14,27,28,29,30].
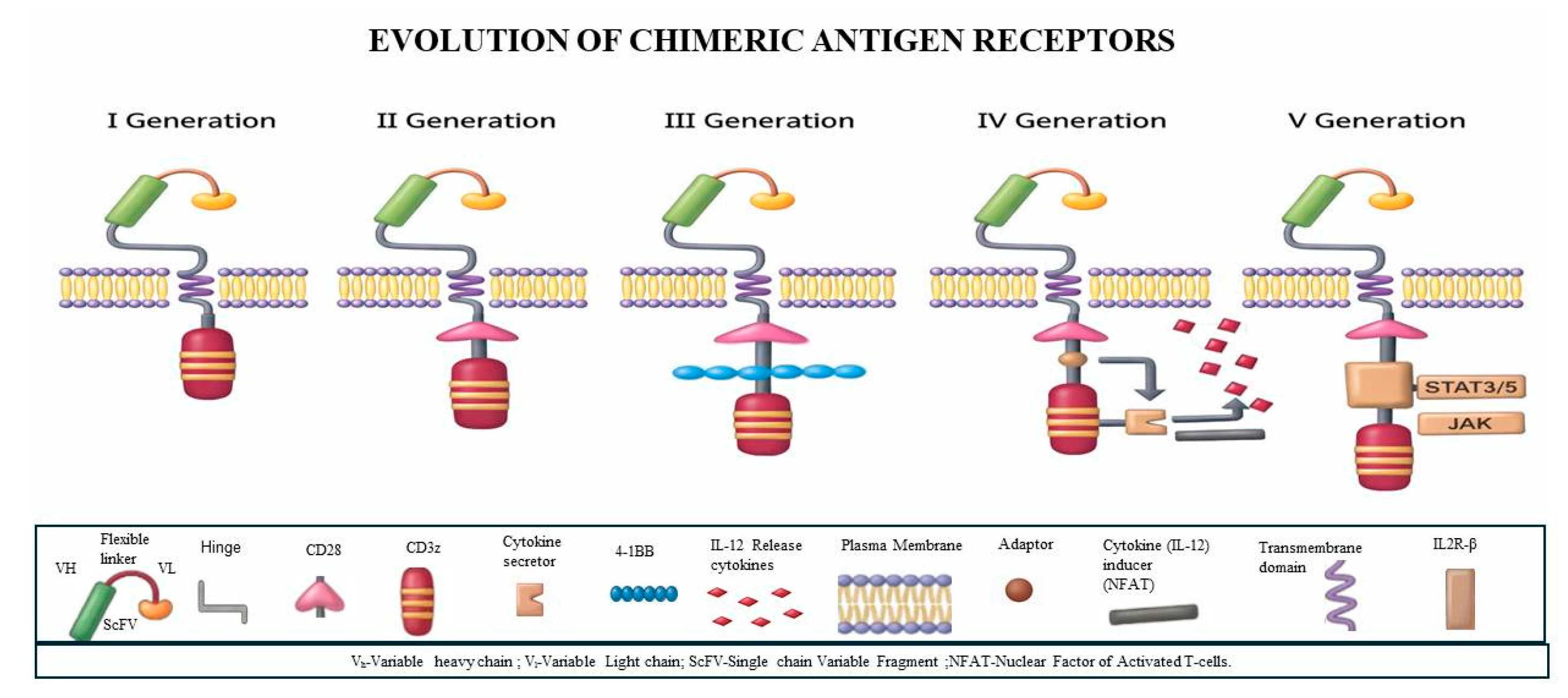
Figure 3.
Schematic representation of advanced CAR T-cell engineering strategies designed to overcome major biological barriers in solid tumors. Cytokine-armored CAR T-cells are engineered to secrete immunomodulatory cytokines, enhancing local immune activation and antitumor efficacy in hostile tumor niches. VEGFR-2–targeted CAR T-cells exploit tumor vasculature targeting to disrupt tumor angiogenesis and enable indirect eradication of vascularized solid tumors. Dual CAR T-cells incorporate multiple antigen-recognition domains to address antigen heterogeneity and reduce tumor escape. CCR-2(Chemokine Receptor Type 2) engineered CAR T-cells express chemokine receptors that enhance trafficking and infiltration into chemokine-rich solid tumors. PD-1 knockout CAR T-cells, generated using genome-editing approaches, attenuate immune checkpoint–mediated suppression and restore T-cell effector function. Logic-gated CAR T-cells employ synthetic antigen-sensing circuits to refine tumor recognition, improve specificity, and limit off-tumor toxicity. Collectively, these engineered strategies aim to enhance CAR T-cell persistence, tumor infiltration, and antitumor activity in solid tumors [128,129,130,131,132].
Figure 3.
Schematic representation of advanced CAR T-cell engineering strategies designed to overcome major biological barriers in solid tumors. Cytokine-armored CAR T-cells are engineered to secrete immunomodulatory cytokines, enhancing local immune activation and antitumor efficacy in hostile tumor niches. VEGFR-2–targeted CAR T-cells exploit tumor vasculature targeting to disrupt tumor angiogenesis and enable indirect eradication of vascularized solid tumors. Dual CAR T-cells incorporate multiple antigen-recognition domains to address antigen heterogeneity and reduce tumor escape. CCR-2(Chemokine Receptor Type 2) engineered CAR T-cells express chemokine receptors that enhance trafficking and infiltration into chemokine-rich solid tumors. PD-1 knockout CAR T-cells, generated using genome-editing approaches, attenuate immune checkpoint–mediated suppression and restore T-cell effector function. Logic-gated CAR T-cells employ synthetic antigen-sensing circuits to refine tumor recognition, improve specificity, and limit off-tumor toxicity. Collectively, these engineered strategies aim to enhance CAR T-cell persistence, tumor infiltration, and antitumor activity in solid tumors [128,129,130,131,132].
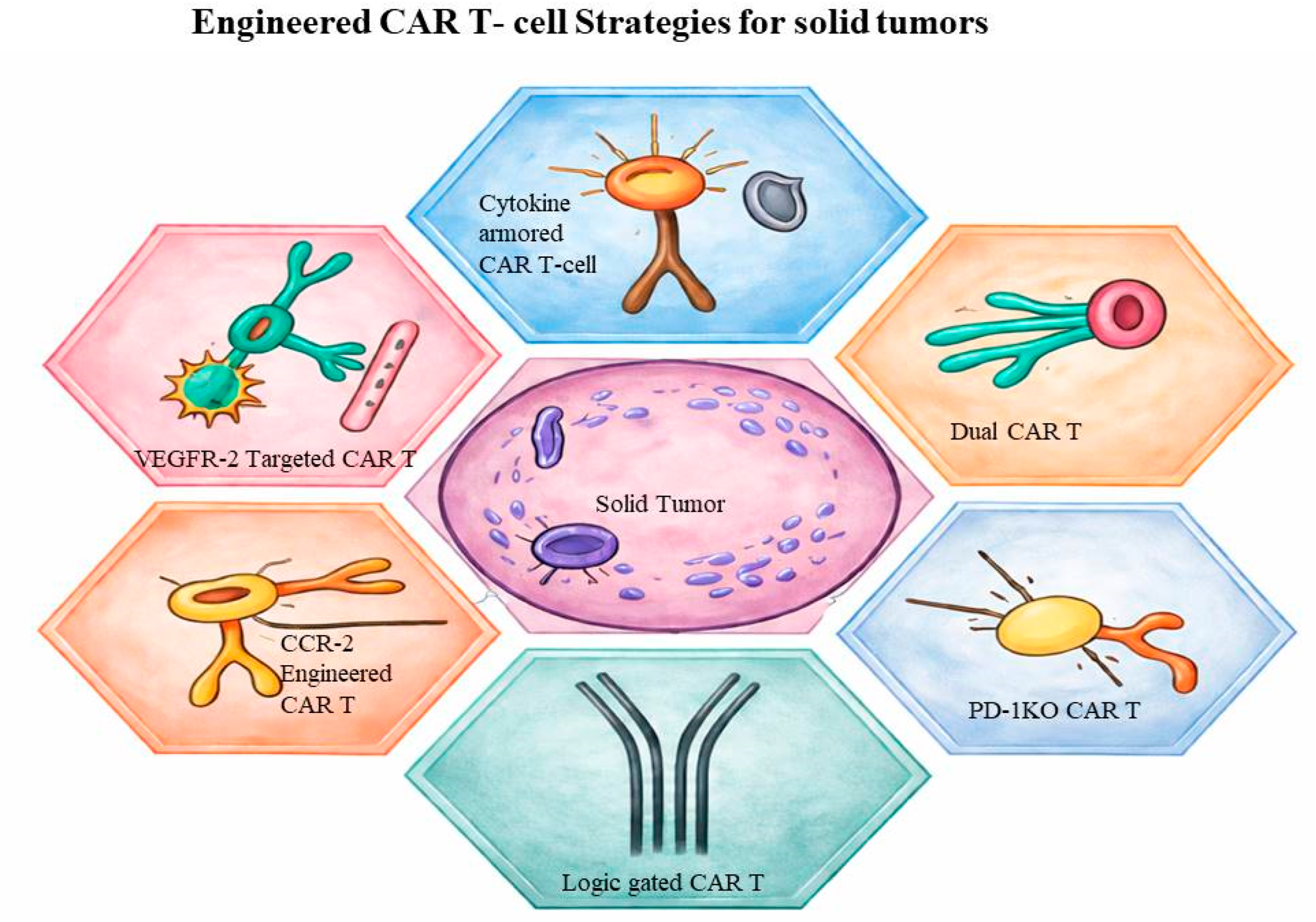
Table 1.
Some identified BCSC markers.
| BCSC Marker | Targeting Mechanism | Preclinical Safety | Therapeutic Rationale | References |
| MUC1-C / MUC1 | Induces tumor cell death through disruption of oncogenic MUC1-C signaling. | Built-in tumor specificity reduces damage to normal tissues. | Enhanced efficacy and limited off-tumor activation. | [76] |
| EpCAM | Eliminates epithelial tumor bulk through surface adhesion molecule targeting | Antigen-density–dependent targeting due to EpCAM expression on normal epithelium. | Marked reduction in epithelial tumor burden. | [77] |
| GD2 | Eradicates stem-like metastatic initiators to block tumor spread | Favorable preclinical safety through selective targeting of stem-like metastatic cells. | Disrupts metastatic seeding in TNBC models. | [78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



