Submitted:
13 February 2026
Posted:
13 February 2026
You are already at the latest version
Abstract
Chimeric antigen receptor (CAR)-T cell therapy has transformed the management of hematologic malignancies but faces obstacles, including severe treatment-related toxicities, highly suppressive tumor microenvironment (TME), inadequate long-term persistence, and poor trafficking/infiltration into solid tumor. This review summarizes recent genetic engineering strategies designed to overcome these barriers and to improve the safety, durability, and spatial effectiveness of CAR-T cell therapy. To mitigate cytokine release syndrome and neurotoxicity, approaches such as affinity-tuned and humanized scFvs, hinge/TM optimization, ITAM calibration have been developed alongside programmable “switch-off” and “switch-on” systems incorporating suicide genes, antibody-bridging switches, and optogenetic or hypoxia-gated circuits. TME remodelling strategies leverage nanomaterials for localized cytokine delivery, cell-surface “backpack” systems, and oncolytic viruses engineered to release cytokines or checkpoint-blocking biologics. Enhancing durability and resistance to exhaustion increasingly relies on precision genome engineering, including CRISPR-based editing and multiplexed shRNA platforms targeting inhibitory receptors and exhaustion-driving transcriptional programs. Finally, chemokine-receptor engineering and local biomaterial-based delivery systems are discussed as routes to improve CAR-T trafficking and intratumoral persistence. We also highlight the remaining translational challenges including checkpoint redundancy, in vivo payload dilution, vector capacity limits, and the safety of multiplex genome editing. Collectively, these interdisciplinary innovations point towards integrated, patient-tailored CAR-T platforms that combine safety control, metabolic and transcriptional resilience, and improved TME navigation to enable broader clinical application.
Keywords:
chimeric antigen receptor-T cells
; CAR-T cell therapy
; solid tumor
; tumor microenvironment
; immunotherapy
; cytokine release syndrome
; immune checkpoint regulation
; genome engineering
; chemokine trafficking
1. Introduction
Chimeric antigen receptor (CAR)-T cell therapy represents a groundbreaking advancement in immunotherapy, offering a highly personalized approach for the treatment of certain hematologic malignancies. CAR-T cells are genetically engineered T lymphocytes designed to recognize and eliminate the target cancer cells. This is achieved by modifying a patient’s autologous T cells to express synthetic receptors, known as CARs, which target specific tumor-associated antigens (TAAs). Unlike endogenous T cell receptors, CARs recognize antigens independently of major histocompatibility complex (MHC) presentation, enabling direct and potent immune responses against malignant cells [1]. The clinical success of this strategy is underscored by multiple U.S. Food and Drug Administration (FDA)-approved CAR-T cell products, which have demonstrated remarkable remission rates in patients with relapsed or refractory cancers, including leukemia, lymphoma, and multiple myeloma [2,3,4,5]. These milestones validate both the safety and therapeutic efficacy of CAR-T cell therapy and have accelerated further research and development in this rapidly evolving field.
The CAR molecule comprises several integrated domains that collectively enable T cells to recognize and eliminate tumor cells [6]. At the extracellular level, a single-chain variable fragment (scFv) derived from an antibody confers antigen specificity by directly binding to a TAA. This antigen-binding domain is linked to a hinge or spacer region, which provides structural flexibility and optimal spatial orientation for target engagement [7]. The transmembrane domain anchors the CAR into the T cell membrane and contributes to receptor stability. Intracellularly, the CAR contains a signalling domain (ICD), typically derived from CD3ζ, and, in more advanced designs, one or more co-stimulatory domains that enhance T cell activation, proliferation, and survival [8] (Figure 1A). The modular nature of CAR architecture has enabled systemic optimization, driving the evolution of successive CAR generations with progressively enhanced functionality.
First-generation CARs were the most basic, consisting solely of a CD3ζ signaling domain. Although they could initiate T cell activation upon antigen recognition, the lack of co-stimulatory signaling resulted in poor T cell proliferation, limited persistence, and ultimately weak clinical responses, particularly in solid tumors [9,10]. Second-generation CARs addressed these limitations by incorporating a single co-stimulatory domain, typically CD28 or 4-1BB (CD137), in addition to CD3ζ. This modification significantly improved CAR-T cell expansion, cytokine secretion, and in vivo persistence, leading to more robust and durable antitumor responses [11] (Figure 1B). Clinically, second-generation CARs form the backbone of most FDA-approved CAR-T cell therapies and have achieved significant success in B-cell malignancies (Figure 1C) [1,12].
Third-generation CARs further expanded upon this concept by integrating two co-stimulatory domains to amplify the intracellular signaling. While preclinical studies suggested enhanced antitumor potency and resistance to T cell exhaustion, clinical trials have yielded mixed results, with no consistent evidence of superiority over second-generation CARs to date [13,14,15]. Fourth-generation CARs, also known as T cells redirected for universal cytokine-mediated killing (TRUCKs), introduced an additional functional layer by incorporating genes encoding pro-inflammatory cytokines, such as interleukins (IL)-12. Beyond direct tumor cell killing, these CAR-T cells actively remodel the tumor microenvironment (TME) by recruiting and activating innate immune components. Although this strategy holds promise for overcoming immunosuppression in solid tumors, the risk of excessive inflammation and off-target toxicity remains a significant concern. More recently, fifth-generation CARs have been developed to integrate cytokine receptor signaling domains, thereby more closely mimicking physiological T cell activation pathways and further enhancing antitumor efficacy [11,16,17]. The defining features and current clinical status of each CAR generation are summarized in Table 1.
Despite these advances, the broader application of CAR-T cell therapy, particularly in solid tumors, remains limited by several critical challenges. These include severe treatment-related toxicities, inefficient trafficking and infiltration of CAR-T cells into tumor sites, the profoundly immunosuppressive nature of the TME, and insufficient long-term persistence of infused cells [18]. Addressing these barriers has become a central focus of current research. Increasingly, interdisciplinary approaches drawing on synthetic biology, materials science, and systems immunology are being leveraged to enhance CAR-T cell safety, functionality, and durability. This review highlights recent innovations aimed at overcoming these key limitations, with an emphasis on emerging strategies designed to break the biological barriers that currently constrain the full therapeutic potential of CAR-T cell therapy.
2. Toxicities Associated with CAR-T Cell Therapy
CAR-T cell therapy has revolutionized cancer treatment, yet its clinical applications are frequently limited by significant toxicities, which remain a major barrier to broader adoption, particularly in solid tumors. The most common complication is cytokine release syndrome (CRS), an acute systemic inflammatory response triggered by rapid activation and expansion of CAR-T cells, resulting in the massive release of cytokines such as IL-6, interferon-gamma (IFN-γ), and tumor necrosis factor-alpha (TNF-α) [19,20]. Severe CRS can progress to hypotension, multi-organ dysfunction, and even fatal outcomes if not promptly managed.
Neurological toxicities also pose substantial challenges to CAR-T cell therapy. Immune effector cell-associated neurotoxicity syndrome (ICANS) encompasses encephalopathy, seizures, and neuropsychiatric symptoms and is thought to arise from endothelial activation and cytokine-mediated blood-brain barrier disruption [19,21,22]. The risk and severity of both CRS and ICANS are closely linked to CAR-T cell dose, proliferative capacity, and cytokine production, particularly in patients with high tumor burdens [23]. Adaptive strategies such as split-dose infusion and patient-specific dosing are increasingly employed to mitigate these risks [24,25].
To improve CAR-T cell therapy, innovative strategies have emerged to mitigate toxicity while maintaining therapeutic potential. The self-regulating CARs, “switch-on/switch-off CARs,” have been developed by integrating active control mechanisms with external modulators, such as suicide genes and responsive sensors that detect light, sound, or oxygen levels, allowing clinicians to fine-tune CAR-T cell activity in real time, reducing toxicity while preserving antitumor efficacy [26]. These mechanistic advances underscore the potential to overcome toxicity as a critical barrier, paving the way for safer, more effective CAR-T therapies. In the following sections, we will focus on structural modifications and emerging strategies to manage these toxicities and improve therapeutic outcomes.
2.1. Self-Regulating CARs
One strategy to mitigate CAR-T cell toxicity is to fine-tune the CAR's binding affinity for TAAs. By lowering affinity, CAR-T cells preferentially target tumor cells with high antigen expression while sparing healthy tissues with low antigen levels. Recent research suggests that altering the binding affinity of the extracellular domain of CARs profoundly affects their activation threshold, cytokine release, and toxicity profiles [27].
In a study by Ghorashian et al., a CD19-targeted CAR with reduced antigen-binding affinity, termed CAT, was developed in contrast to the conventional FMC63-based construct. Despite its lower affinity, the CAT-CAR-T cells demonstrated enhanced antitumor activity, achieving complete remission in more than 80% of treated pediatric patients (12 of 14) with replaced or refractory B-cell acute lymphoblastic leukemia (B-ALL). Importantly, this enhanced efficacy was accompanied by an improved safety profile, with no reported cases of severe CRS and only one instance of ICANS [28]. These findings suggest that optimized CAR affinity can uncouple antitumor efficacy from excessive immune activation.
Humanizing CAR constructs is another approach that replaces murine-derived scFv regions with human counterparts, reducing immunogenicity, limiting immune-mediated rejection, and enhancing CAR-T cell persistence. In a small-scale clinical study involving patients with relapsed or refractory B-ALL, treatment with a humanized CD19-CAR-T cell product (huCART19) resulted in durable remission, with CAR-T cells remaining detectable for up to 24 months post-infusion [29]. Additionally, a humanized CAR containing the human-derived CD19-scFv with BAFFR single-variable domains showed excellent in vitro and in vivo efficacy. These CAR-T cells were less exhausted and had better tumor eradication abilities [30].
Beyond antigen-binding domains, the hinge and transmembrane domain (HD/TMD) play a pivotal role in regulating CAR-T cell activation and toxicity. Integration of a humanized scFv with a CD8α HD/TMD has been shown to synergistically reduce the treatment-related toxicities. For instance, Hu19-CD19ζ CAR-T cells exhibited substantially fewer ICANS cases (4%) than traditional FMC63-based CD19-CAR-T cells (50%). This enhanced safety was due to low levels of cytokines in the blood of patients receiving humanized CAR-T cells, which were dependent on the CAR's HD/TMD configuration [31,32,33]. Variations in the type and length of the HD/TMD may also influence toxicity levels [34]. CAR-T cells with longer CD8α-HD/TMD domains were associated with lower cytokine production and a lower incidence of severe CRS or ICANS among patients (n=25) with refractory B-cell lymphoma [35,36].
Intracellular signaling domains (ICD) also critically influence CAR-T cell function and toxicity. CARs incorporating the 4-1BB co-stimulatory domain generally induce more controlled and sustained activation compared with CD28-based CARs [37]. This difference may be attributed to the THEMIS-SHP1 complex, which decreases CD3ζ phosphorylation and limits excessive cytokine secretion, including IFN-γ [38,39]. Furthermore, reducing the number of immunoreceptor Tyrosine-based activation motifs (ITAMs) within the CD3ζ domain, or substituting CD3ζ with alternative single ITAM subunits such as CD3ε, has emerged as a promising strategy to constrain CAR-T cell hyperactivation and decrease CRS risk [40]. Figure 2 illustrates these strategies for self-regulating CAR design.
2.2. The ‘Switch Off’ CARs
Switch-off CARs are designed to enable conditional termination of CAR-T cell activity via internal or externally administered triggers, providing an additional layer of safety. These systems are crucial for preventing the severe, systemic side effects associated with related toxicities such as CRS and ICANS [41].
One of the earliest and most extensively studied switch-off strategies in CAR-T cell therapy relies on suicide gene systems, with inducible caspase-9 (iCasp9) being the most clinically advanced example. In this approach, CAR-T cells are engineered to express a modified human caspase-9 that can be rapidly activated by a small-molecule dimerizer, thereby inducing synchronized apoptosis in the engineered CAR-T cells. Compared with earlier viral suicide genes, iCasp9 offers faster activation kinetics, reduced immunogenicity, and more predictable in vivo performance as explained in Figure 3. Clinically, this system has demonstrated the capacity to rapidly control severe toxicities while allowing partial persistence of CAR-T cells, thereby enabling controlled debulking rather than irreversible elimination of the therapeutic cell population.
Another suicide switch is the Herpes simplex virus thymidine kinase (HSV-TK) system, in which HSV-TK converts the prodrug ganciclovir (GCV) into a toxic nucleotide analogue that inhibits DNA synthesis and thereby selectively eliminates dividing CAR-T cells [42]. This strategy provides a crucial safeguard against life-threatening complications, including CRS, neurotoxicity, and graft-versus-host disease, and demonstrated a favourable safety profile in multiple adoptive cell therapy studies [43]. In pre-clinical solid tumor models, such as small-cell lung cancer xenografts, GM2 ganglioside-targeted CAR-T cells engineered to express IL-7 and CCL-19 (anti-GM2-7×19) were infused with an HSV-TK safety switch, demonstrating excellent therapeutic efficacy following GCV administration. GCV treatment markedly reduced the circulating CAR-T cells within peripheral blood and reduced the tumor growth, confirming the functional on-demand ablation of the therapeutic CAR-T cells [44]. In another study, B7H3-targeted CAR-T cells co-expressing HSV-TK (B7H3-sr39tk) showed potent antitumor activity against osteosarcoma xenografts, while GCV administration rapidly reduced activated CAR-T cells without detectable damage to normal tissues [26,45].
Antibody-mediated switch-off strategies offer an alternative means of external control. In cetuximab-based systems, CAR-T cells are engineered to express receptors that recognize the Fc region of cetuximab, which binds to the epidermal growth factor receptor (EGFR) on tumor cells [46,47]. In this design, CAR-T cell activation is triggered only when cetuximab bridges CAR-T cells to tumor cells, allowing clinicians to finely tune the intensity and duration of CAR-T cell activity by adjusting cetuximab dosing [48]. Clinical relevance of this approach was demonstrated in a study of anti-CD5 CAR-T cells in which a truncated EGFR (tEGFR) safety switch was incorporated. Cetuximab administration eliminated more than 90% of the circulating CAR-T cells, and the remaining CAR-T cell population retained sufficient functional capacity to maintain the antitumor surveillance, indicating that antibody-mediated depletion can reduce toxicity without completely abolishing the therapeutic efficacy; therefore, this approach enables the controlled reduction rather than the complete termination of CAR-T cell activity [49].
A related antibody-mediated safety strategy exploits CD20 as an inducible elimination marker on CAR-T cells, enabling their depletion using the FDA-approved anti-CD20 antibody rituximab. Similar to the tEGFR-cetuximab system, this approach enables external pharmacologic control of CAR-T cell persistence, rather than relying on irreversible intracellular suicide mechanisms. In preclinical and early translational studies, CD20-expressing CAR-T cells retained full antitumor activity during the therapeutic phase and were subsequently cleared following rituximab administration to mitigate excessive toxicity [50]. However, in contrast to the more rapid debulking observed with cetuximab-mediated clearance, rituximab-induced CAR-T cell elimination depends on immune effector mechanisms, such as antibody-dependent cellular cytotoxicity and complement activation, which may delay the onset of CAR-T cell depletion [51]. A drawback of this approach is its delayed onset of action, which could limit its use in patients with severe cytokine-mediated toxicities who need immediate intervention.
As CAR-T cell designs become more potent and are increasingly applied to solid tumors, the ability to actively modulate CAR-T cell activity is becoming essential. Switch-off CAR systems provide a practical means to control toxicity by allowing clinicians to reduce or terminate CAR-T cell persistence in response to emerging adverse events. Importantly, strategies that permit partial reduction in CAR-T cells, rather than complete elimination, may help balance toxicity control with ongoing antitumor surveillance. From a translational standpoint, the use of clinically approved antibodies or drugs offers a clear advantage, enabling predictable control and facilitating real-world clinical implementation [21].
2.3. The ‘Switch On’ CARs
To improve safety while preserving antitumor potency, switch-on CAR systems have been developed to restrict CAR-T cell activation to defined spatial, temporal, or environmental cues. Among these, optogenetic approaches enable external control of CAR signaling using light-sensitive molecular modules, allowing CAR-T cell activity to be confined to specific wavelengths of light and thereby enabling fine-tuned control of their activation, targeting, and deactivation [52]. Such spatiotemporal precision is particularly attractive for limiting off-tumor toxicity in solid tissues while maintaining robust local immune activation [52,53].
The light-inducible CAR (LiCAR) system exemplifies this strategy by physically separating key CAR signaling components that are reassembled only upon blue-light exposure [54]. The system relies on a light-sensitive interaction between two components: a bacterial small stable RNA A (SsrA) peptide fused to a light-oxygen-voltage 2 (LOV2) domain and its binding partner, stringent starvation protein B (SspB). Under dark conditions, the LOV2 domain maintains a conformation that keeps SsrA and SspB apart. However, when illuminated with blue light, LOV2 undergoes a structural change that allows SsrA to bind to SspB. This binding event reassembles the split CAR domains, thereby restoring CAR functionality and triggering T cell activation [55,56,57]. While effective, purely light-controlled systems may exhibit low-level background activity due to inadvertent exposure to ambient light, highlighting the need for additional regulatory safeguards [58].
To address this limitation, multi-input control systems, such as the TamPA-Cre platform, introduce an additional layer of regulation by integrating optical and pharmacological signals. This dual-control design operates as an AND-gated system, requiring simultaneous blue-light exposure (optical signal) and tamoxifen (pharmacological signal) administration to initiate CAR expression at the desired time and place [58]. The system uses a tamoxifen-dependent mutant estrogen receptor ligand-binding domain T2 (ERT2), which is fused to one of the two halves of a split Cre recombinase (ERT2-CreN-nMag). In the absence of tamoxifen, the ERT2 domain retains its Cre half in the cytoplasm, keeping the system inactive. A second split Cre half (NLS-pMag-CreC), which contains a nuclear localization sequence (NLS) and is already in the nucleus. The two Cre halves, CreN and CreC, are fused to interacting photoactivatable domains called nMag and pMag, which are derived from a fungal photoreceptor. When the system is primed with tamoxifen, blue light stimulation causes the nMag and pMag domains to dimerize (bind to each other), bringing the two Cre halves together to reconstitute a functional Cre recombinase [58,59]. The very first study using this system in the context of CAR-T cell therapy was published in 2019 by Wu et al, who demonstrated that this strategy enabled highly localized CAR-T cell activation, thereby reducing the risk of on-target, off-tumor toxicity in surrounding healthy tissues [58]. Such combinatorial control underscores the value of layered regulatory logic in enhancing the precision of CAR-T cells.
Beyond externally applied signals, endogenous tumor-associated cues have also been exploited to achieve switch-on control, such as a hypoxia-responsive CAR-T cell system. In this system, CAR expression is under the control of a tightly regulated hypoxia-sensing safety switch, which prevents on-target, off-tumor CAR-T cell activation while still enabling potent tumor-specific cytotoxicity [49]. Hypoxia-sensitive CAR-T cells are engineered to function only under low-oxygen conditions (characteristic of solid tumors), thereby limiting CAR expression or signaling in normoxic healthy tissues and reducing off-target toxicity [60]. Mechanistically, hypoxia-responsive CAR-T cells often incorporate hypoxia-responsive elements (HREs) that are activated upon stabilization of hypoxia-inducible factor 1α (HIF1α) in low-oxygen environments. The binding of HIF1α to HREs initiates CAR transcription, thereby restricting CAR-T cell activation to hypoxic tumor regions and sparing well-oxygenated normal tissues [61,62].
The HypoxiCAR platform further refines this concept through dual oxygen-dependent regulation, combining hypoxia-driven CAR transcription with oxygen-dependent degradation (ODD) of the CAR protein under normoxic conditions. This stringent control mechanism has demonstrated effective tumor targeting with minimal off-tumor toxicity in preclinical models, even when using broadly expressed antigens such as pan-ErbB, shown in Figure 4 [61].
3. Immunosuppression in the Tumor Microenvironment
Despite the transformative success of CAR-T cell therapy in hematological malignancies, its efficacy in solid tumors remains profoundly constrained by the immunosuppressive and structurally complex tumor microenvironment. The TME constitutes a multifaceted barrier that actively impairs CAR-T cell infiltration, persistence, and effector function, ultimately driving functional exhaustion. Key inhibitory cells, include regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), which suppress antitumor immunity through both contact-dependent mechanisms and the secretion of inhibitory cytokines. As well as, elevated levels of soluble immunosuppressive mediators, such as transforming growth factor-β (TGF-β) and interleukin-10 (IL-10), together with high expression of immune checkpoint ligands including programmed cell death ligand 1 (PD-L1), further attenuate CAR-T cell activation, proliferation, and cytotoxicity [26,63].
Not only immunological suppression, adverse metabolic and biophysical features of the TME, such as hypoxia, nutrient deprivation, and acidic pH, impose additional constraints on CAR-T cell survival and functionality. These hostile conditions disrupt T cell metabolism and signaling, contributing to reduced durability and diminished antitumor activity in solid tumors compared with hematological settings [64,65,66]. These barriers underscore the need for combinatorial strategies capable of reshaping the TME rather than relying solely on intrinsic CAR design. In this context, combining CAR-T cell therapy with auxiliary agents, such as engineered nanomaterials and oncolytic viruses (OVs) have emerged as promising tools to modulate the TME, enhance CAR-T cell trafficking and persistence, and restore antitumor immune activity [67,68].
3.1. Nanomaterials
Nanomaterials have emerged as versatile tools to enhance CAR-T cell therapy in solid tumors by modulating the TME, improving T cell persistence, and enabling localized delivery of immunomodulatory agents. Although nanotechnologies are broadly applied in CAR-T research for gene delivery, in vivo tracking, and functional augmentation [69]. This section focuses on strategies specifically designed to overcome the immunosuppressive TME.
One practical approach is to precondition the TME before CAR-T cell administration. In this context, tumor-targeting liposomes functionalized with the iRGD peptide were engineered to co-deliver phosphoinositide 3-kinase (PI3K) inhibitors and the immune agonist α-galactosylceramide (α-GalCer) [70]. This combination selectively depleted immunosuppressive cell populations while simultaneously enhancing antitumor immune activation. Repeated liposome pretreatment significantly sensitized tumors to subsequent CAR-T cell therapy, resulting in complete tumor regression in 50% of treated mice. Similarly, liposomal delivery of a TGF-β receptor inhibitor was used to counteract one of the dominant suppressive pathways (TGF-β) in the TME. By conjugating these liposomes to T cell surface receptors like CD90 or CD45 ex vivo, localized inhibitor release was achieved at the tumor site, thereby preserving CAR-T cell function without systemic exposure [71].
Beyond drug delivery, nanozymes, artificially engineered nanomaterials with enzyme-like activity, have been explored as tools to reshape the immunological landscape of solid tumors [72,73]. For instance, Copper (Cu)-based nanoparticles, acting as nanozymes, enhance treatment by employing photothermal therapy (PTT) to boost reactive oxygen species generation [74,75]. This combined action triggers immunogenic cell death, a process that releases TAAs, subsequently activating dendritic cells (DCs) and stimulating a robust adaptive effector T cell response [75]. Hyaluronic acid (HA)-modified Copper sulfide nanoparticles (termed PHCNs) were used to overcome the immunosuppressive TME in B7-H3 CAR-T cell therapy for non-small cell lung cancer (NSCLC). In preclinical models, the nanozyme-mediated TME modification and immune priming doubled the rate of complete tumor eradication and significantly prolonged the overall survival of mice compared to B7-H3 CAR-T monotherapy. This nanozyme-mediated reprograaming effectively converted the "cold" immunosuppressive TME into an "inflamed" immune-permissive environment, thereby overcoming a critical limitation of solid tumor CAR-T cell therapy [76].
A more integrated approach involves creating biohybrids in which nanomaterials are directly attached to CAR-T cells to enable simultaneous tumor targeting, imaging, and TME modulation. Using biorthogonal conjugation, indocyanine green nanoparticles (INPs) were attached to CAR-T cells to generate CAR-T-nanoparticle biohybrids (CT-INPs) [74,77]. Upon localized laser irradiation, these constructs produced mild photothermal effects that disrupted the extracellular matrix (ECM), enhanced vascular permeability, and promoted chemokine secretion, collectively improving CAR-T cell infiltration without impairing viability or cytotoxicity [74,77,78].
Nanomaterials have also been used to address challenges in cytokine delivery for adoptive cell therapy. While cytokines such as IL-15 and IL-21 are essential for T cell expansion and persistence, systemic administration is associated with significant toxicity [79,80]. Lipid nanoparticles (LNPs) (approx 300nm), decorated with malemide groups covalently attached to the thiol group on T cell membrane, provide a sustained, pseudo-autocrine cytokine supply, enhancing CAR-T cell durability and antitumor efficacy without detectable systemic adverse effects [81]. Similarly, CAR-T cells have been used as a carrier for IL-12-loaded nano-chaperone that rapidly breaks down upon T cell activation (when high levels of thiol groups are encountered), releasing their IL-12 to recruit additional immune cells and to amplify the antitumor response [82].
Despite their promises, cell-associated nanoparticle systems face limitations. Since the nanoparticle payload does not expand as T cells proliferate in vivo, the amount of drug carried per cell becomes diluted across successive cell divisions. Furthermore, these backpacks generally lack dynamic regulation, limiting their ability to adjust drug release in response to changes in TME. To address these challenges, localized delivery platforms such as injectable hydrogels have been developed. For example, a hyaluronic acid-hydrogel (HA hydrogel) encapsulating CAR-T cells, IL-15 nanoparticles, and anti-PD-L1-loaded platelets enabled sustained local immunomodulation following tumor resection, significantly enhancing CAR-T cell activity and reducing tumor recurrence in preclinical models, indicated in Figure 5 [83].
Schematic representation of complementary nanomaterial-based approaches designed to overcome immunosuppressive features of TME and improve CAR-T cell efficacy in solid tumors. Nanomaterials are used either in combination with or directly coupled to CAR-T cells to achieve localized immunomodulation. Shown strategies include: liposome-mediated TME priming and localized drug release to attenuate suppressive cytokine signaling; photothermal nanoparticle-CAR-T biohybrids that promote extracellular matrix remodeling, vascular dilation, and chemokine secretion; copper-based nanozymes that induce immunogenic tumor cell death and secondary immune activation; lipid nanoparticle systems providing pseudo-autocrine cytokine support to enhance CAR-T cell expansion and persistence; nanochaperone “backpacks” enabling activation-responsive cytokine release, and hydrogel-based depots for confined CAR-T cell delivery and sustained local immune support.
3.2. Oncolytic Viruses
Oncolytic viruses are either naturally occurring or genetically engineered viruses that selectively infect and replicate within malignant cells while leaving normal tissue unharmed [84]. These viruses replicate within tumor cells, causing them to burst (lyse) and release progeny virions, as well as TAAs and immunostimulatory danger signals. These properties make OVs potent immune modulators, capable of converting immunologically “cold” tumors into inflamed environments that are more permissive to adoptive T cell therapies. Combining the OVs and CAR-T cell therapy has emerged as a rational strategy to overcome the limitations of either approach alone. This synergy enhanced the treatment effectiveness, especially against solid tumors, including poor infiltration, immunosuppression, and insufficient activation [85,86,87].
Ongoing work demonstrates that OVs can enhance CAR-T cell expansion and function by providing endogenous T cell receptor (TCR) stimulation through viral or virally encoded epitopes. In immunocompetent mouse models of melanoma and glioma, systemically administered OVs augmented CAR-T cell activity by engaging native TCR signaling, thereby promoting proliferation, effector function, and memory differentiation [88]. Notably, preloading dual-specific CAR-T cells with oncolytic vesicular stomatitis virus (VSV) or reovirus enabled robust in vivo expansion and permitted CAR-T cell reactivation through subsequent viral boosting. Similarly, human CD19-CAR-T cells produced more cytokines when expanded in vitro with viral antigens, indicating improved TCR stimulation [88].
In addition to immune activation, OVs can be engineered to remodel physical barriers within the TME. A recombinant oncolytic vaccinia virus expressing hyaluronidase (Hyal1) was shown to degrade hyaluronic acid within tumors, thereby improving immune cell infiltration and enhancing overall antitumor efficacy [89]. This matrix-disruptive capability reduces tumors, where dense ECM components restrict CAR-T cell trafficking and persistence.
OV-mediated modulation of the immunosuppressive TME further supports CAR-T cell activity. Cancer cells shape the TME by actively recruiting Tregs, tumor-associated macrophages (TAMs), MDSCs, and stromal elements that collectively suppress effector T cell function. OVs counteract this suppression by inducing localized inflammation and reshaping cytokine and chemokine gradients, facilitating CAR-T cell infiltration and sustained activity [90,91]. In pancreatic cancer models, an oncolytic adenovirus engineered to express TNF-α and IL-2 (Onc.Ad-TNF-α/IL-2) significantly enhanced the antitumor efficacy of mesothelin-targeted CAR-T cells incorporating a 4-1BB co-stimulatory domain. This improved response was attributed to localized cytokine release of TNF-α and IL-2, combined with direct viral oncolysis, highlighting the complementary roles of viral and CAR-T-mediated cytotoxicity [92].
Immune checkpoint-mediated immunosuppression represents another major obstacle to CAR-T cell efficacy in solid tumors. To address this, OVs have been engineered to locally deliver immune checkpoint inhibitors or mini-antibodies. Shaw et al. developed an oncolytic adenovirus (CAd12-PDL1) that expresses both a PD-L1-blocking mini-antibody and the IL-12p70 cytokine. When combined with HER2-targeted CAR-T cells, this approach effectively suppressed primary tumor growth and metastatic spread, while limiting systemic toxicity associated with systemic checkpoint blockade [26,93].
From a signaling perspective, effective antitumor T cell responses require the integration of antigen recognition, co-stimulatory signaling, and inflammatory signals. While second- and third-generation CARs are designed to provide signals 1 and 2, the inflammatory “signal 3” is often insufficient in the solid TME. This inflammatory signal is typically supplied by cytokines such as IL-12 or type I interferons (IFNs). Type I IFNs are crucial in antiviral and antitumor responses, enhancing the host's adaptive immune system. Notably, OVs are potent inducers of type I interferons, which play a central role in antiviral immunity and support T cell survival, expansion, and cytotoxicity [94]. Type I interferon signaling in CAR-T cells may be amplified through 4-1BB-mediated activation of TNF receptor-associated factor 2 (TRAF2), suggesting that CAR-T products incorporating 4-1BB domains or chimeric co-stimulatory receptors (CCRs) may be particularly well suited for OV-based combination strategies [87]. Adjusting the signal-3 pathways, either through OV engineering or CAR design, substantially improves the performance of CAR-T cells once they are infused in vivo, shown in Figure 6.
4. Limited Efficacy and Longevity of CAR-T Cell Responses
Although advances have been made in CAR-T cell design and manufacturing, the therapeutic efficacy and long-term persistence of CAR-T cells remain significant limitations, particularly in treating solid tumours. Various factors, including physical barriers, suppressive cellular components, suppressive cytokines, and inhibitory molecular signals, collectively impede CAR-T cell infiltration, survival, and sustained antitumor activity. Addressing these intrinsic limitations has therefore become a central focus in next-generation CAR-T cell engineering. Advances in genetic manipulation have enabled precise and durable reprogramming of CAR-T cells to resist these suppressive pressures. Genome-editing technologies, particularly the CRISPR-Cas system, allow targeted disruption of inhibitory receptors, transcriptional regulators, and metabolic checkpoints that constrain CAR-T cell function. Additionally, interference-based approaches, such as short-hairpin RNA (shRNA), offer a flexible, tunable strategy to silence genes that promote exhaustion, apoptosis, or suppressive signaling in CAR-T cells. These genetic strategies not only enhance immediate cytotoxicity but also aim to rewire CAR-T cell fate decisions, promoting sustained functionality, improved persistence, and long-term tumor control. In the following sections, we discuss how CRISPR-based genome editing and shRNA-mediated gene silencing are being used to address CAR-T cell exhaustion and durability in the immunosuppressive TME.
4.1. CRISPR Technology
CRISPR-Cas9 genome editing has revolutionized CAR-T cell engineering by enabling highly precise, permanent genetic modifications that directly address intrinsic mechanisms that limit CAR-T cell efficacy and durability within the TME. CRISPR-mediated gene editing induces permanent gene knockout, thereby allowing durable reprogramming of CAR-T cell fate, function, and persistence. Additionally, the integration of CRISPR into CAR-T manufacturing workflows has improved scalability and reproducibility, facilitating more effective cancer treatments [95,96].
One of the most prominent drivers of CAR-T cell dysfunction in solid tumors is chronic antigen stimulation, which induces sustained expression of inhibitory immune checkpoint receptors, leading to T cell exhaustion. The CRISPR-Cas9 technique has been extensively used by researchers to knock out genes encoding these inhibitory receptors, thereby preventing suppressive signaling and restoring CAR-T cell effector function [97]. Using CRISPR to knockout the programmed cell death protein 1 (PDCD1) gene prevents CAR-T cells from receiving inhibitory signals when they encounter PD-L1 on tumor cells. Preclinical studies consistently demonstrate that PD-1-deficient CAR-T cells exhibit enhanced cytokine production (including IFN-γ and TNF-α), increased proliferation, and better tumor control across multiple murine cancer models [98]. Early-phase I clinical trials, such as NCT03179943, have also confirmed the feasibility and safety of PD-1-edited CAR-T cells in humans, thus supporting the translational potential of this strategy [99,100].
Studies have confirmed that PD-1, as well as CRISPR-mediated deletion of other immune checkpoints, has further underscored the potential of this strategy. Deletion of cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) improves the proliferative capacity, persistence, and antitumor activity of CAR-T cells in preclinical models [101]. Inhibitory receptors such as T cell immunoglobulin mucin domain-containing protein 3 (TIM-3), Lymphocyte activation gene 3 protein (LAG-3), and 2B4 (CD244) play distinct, non-redundant roles in driving T-cell exhaustion under chronic stimulation. Using CRISPR-Cas9 to individually disrupt these genes in NY-ESO-1-specific T cells significantly improved resistance to functional exhaustion both in vitro and in vivo [102].
A significant advantage of CRISPR-Cas9 technology is its ability to simultaneously disrupt multiple genes within a single shot. Ren et al. demonstrated that a single CRISPR-Cas9 system could perform multiplex genome editing to generate universal, allogeneic CAR-T cells by knocking out endogenous TCR and HLA class I genes. The addition of PD-1 disruption further enhanced the antitumor activity of these engineered T cells in mouse models, indicating the synergistic benefit of multiplex genome editing for both immune evasion and functional enhancement [103].
In addition to surface inhibitory receptors, a specific set of transcription factors is also involved in T cell exhaustion [104]. Transcription factors such as TOX and the members of the NR4A family (NR4A1, NR4A2, NR4A3) play key roles in driving and maintaining the T cell exhaustion [105]. Using multiplex CRISPR editing to simultaneously delete all three NR4A genes, making human CAR-T cells highly resistant to exhaustion after repeated antigen exposure, both in vitro and in vivo. Comparative analysis revealed that triple knockout (TKO) cells outperformed single- or double-knockout cells, exhibiting superior persistence, enhanced antitumor activity across multiple donors, and increased mitochondrial oxidative phosphorylation, supporting sustained function within tumors [105]. Additionally, targeting genes like PR domain zinc finger protein 1 (PRDM1) and T cell factor 1 (TCF1) with CRISPR can enhance T cell ‘stemness,’ promoting a precursor exhausted T cell (TPEX) phenotype that remains responsive to immunotherapy [106].
CRISPR-based editing has also been applied to counteract extrinsic immunosuppressive signals within the TME. TGF-β is a dominant suppressive cytokine in solid tumors. CAR-T cells lacking TGF-β receptor II (TGFBR2) demonstrated significantly enhanced proliferation and tumor-killing capacity in vitro, even in the presence of high TGF-β concentrations. In vivo, TGFBR2-knockout CAR-T cells showed improved persistence and increased proportions of central and effector memory subsets, both of which are critical for durable antitumor immunity [107]. Combining TGFBR2 knockout with PD-1 disruption further enhanced efficacy in highly suppressive TMEs, underscoring the potential of multiplex gene-editing strategies.
Recent advances in CRISPR technology are further expanding its therapeutic potential. Double-strand break-free (DBS) approaches, including base editing and prime editing, enable precise genetic modifications with reduced genotoxicity and improved safety profiles [108]. So, CRISPR-Cas9-mediated multiplex knockouts of inhibitory receptors, transcriptional exhaustion drivers, and suppressive cytokine pathways offer a powerful strategy to overcome both intrinsic and extrinsic resistance mechanisms in CAR-T cell therapy. Integrating advanced gene-editing strategies into next-generation CAR-T cell therapies can improve treatment efficacy, reduce off-target effects, and increase accessibility through cost-effective, targeted interventions [109].
4.2. shRNA Technology
Short-hairpin RNA technology enables stable yet reversible gene silencing by targeting specific mRNAs for degradation, offering a regulatory flexibility that is particularly advantageous when complete gene knockout may compromise CAR-T cell viability or induce unintended toxicities [110]. Unlike CRISPR-based permanent genome editing, shRNA allows graded suppression of inhibitory pathways, making it well-suited for fine-tuning CAR-T cell function within the dynamic and immunosuppressive TME. This approach is especially relevant in solid tumors, where excessive genetic disturbance may impair exhaustion or persistence.
A major advantage of shRNA engineering is the ability to intrinsically silence immune checkpoint receptors within CAR-T cells, thereby enhancing activation, persistence, and cytotoxicity without the systemic toxicities associated with checkpoint-blocking antibodies [111]. Our group and others have demonstrated that shRNA-mediated suppression of negative regulatory pathways can substantially improve CAR-T cell performance in both hematological and solid tumor models [110,112,113,114]. For example, adrenergic stress, a key but underappreciated immunosuppressive axis in the solid TME. Beta-2 adrenergic receptor (ADRB2) functions as a stress-induced checkpoint receptor on T and NK cells, reducing antitumor immunity in catecholamine-rich tumor microenvironment [66,115]. Using shRNA technology, we developed ADRB2-downregulated CAR-T cells that are protected from adrenergic stress signaling. Incorporation of ADRB2 shRNA into second-generation CAR constructs significantly enhanced T cell activation and cytokine secretion. Importantly, these cells exhibited increased glucose transporter 1 (GLUT-1) expression and elevated Peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1-α) levels, reflecting improved metabolic fitness and mitochondrial biogenesis, which translated into superior antitumor activity in vitro against prostate and colorectal cancer models [66,113]. In another study, shRNA-mediated downregulation of the epigenetic checkpoint histone deacetylase 11 (HDAC11) enhanced CAR-T cell proliferation, reduced exhaustion markers (PD-1 and TIM-3), and promoted central memory T cell (TCM) differentiation via upregulation of Eomes [112]. HDAC11-silenced NKG2D-CAR-T cells achieved improved tumor control and prolonged survival in vivo compared with conventional CAR-T cells [112]. Similarly, other well-known immune checkpoints, such as PD-1, TIM-3, and LAG-3, have been intrinsically inhibited in CAR-T cells using shRNA technology, yielding enhanced CAR-T cell activity in preclinical studies [99,116,117].
However, a central limitation of single-gene silencing is the redundancy of checkpoints and compensatory upregulation. Tumors can adapt to the inhibition of one pathway by engaging alternative inhibitory receptors, thereby restoring immune suppression [118]. To address this, current shRNA strategies are shifting toward multiplexed checkpoint silencing, which simultaneously silences 2-4 immune checkpoint genes. A novel dual-function lentiviral vector was developed to enable efficient shRNA-mediated silencing of both PD-1 and T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domains (TIGIT) while maintaining strong CAR expression in CD19-CAR-T cells. These CAR-T cells, designated CRC01, demonstrated significantly enhanced antitumor activity both in vitro and in vivo compared with conventional CAR-T cells. Upon CD3/CD28 stimulation, analysis of CRC01 cells showed that PD-1 and TIGIT expression was markedly lower in CAR-positive T cells than in their CAR-negative counterparts, confirming effective and targeted knockdown of both immune checkpoint genes [119].
Advancing multiplex shRNA delivery introduces technical and biological challenges. Combining multiple shRNAs within a single vector and scaffolds that facilitate co-expression with therapeutic transgenes presents a promising strategy for creating CAR-T cells with customized phenotypes. However, efforts to multiplex shRNAs using the clinically approved miR196a2 scaffold have been linked to reduced vector titers. The reductions in vector titer varied among the tested duplex and triplex shRNA constructs; however, the overall titers remained too low for clinical application. To overcome this issue, a proprietary scaffold was developed to support the expression of duplexed and triplexed shRNAs while increasing vector titers by at least 2 to 3 times [120]. Further development of the multiplexed shRNA vector system is underway to assess its potential to enhance the therapeutic efficacy of CAR-T cells. In another study, four different shRNAs were expressed in a single vector, although the optimization remains ongoing [120].
As the number of targeted genes increases, careful systems-level evaluation becomes essential. Multiple immune checkpoints often converge on shared downstream signaling nodes, such as SHP-1 and SHP-2 phosphatases, meaning that not all combinations yield additive benefit. Future shRNA-based CAR-T design must therefore prioritize synergistic target selection, focusing on pathways that regulate distinct yet complementary inhibitory mechanisms. Furthermore, combining shRNA-engineered CAR-T cells with other immunotherapies creates multifaceted strategies that can neutralize immunosuppression, enhance T cell recruitment and survival, and boost cytotoxic and memory responses. This approach is particularly promising for treating solid tumors, where standalone CAR-T cell therapy has faced challenges due to physical barriers, antigen heterogeneity, and T cell exhaustion.
5. Trafficking Deficits and Infiltration Challenges
Effective antitumor activity of CAR-T cells requires not only potent cytotoxicity but also efficient trafficking to and infiltration into tumor sites. In solid tumors, this process is frequently impaired by dysregulated chemokine signaling. Many tumors either downregulate T cell-attracting chemokines or preferentially secrete chemokines that recruit immunosuppressive populations rather than effector lymphocytes, resulting in poor CAR-T cell homing [121]. Unlike hematologic malignancies, solid tumors are embedded within a highly structured and physically restrictive microenvironment. Dense extracellular matrix deposition, aberrant vasculature, and extensive networks of tumor-associated fibroblasts collectively form substantial barriers that limit T cell extravasation and intratumoral migration [122]. Even when CAR-T cells reach the tumor periphery, these structural constraints can prevent deep penetration into the tumor core, thereby limiting immune activity and incomplete tumor eradication. Addressing these trafficking and infiltration deficits has therefore emerged as a critical priority for extending CAR-T cell therapy to solid malignancies. Current strategies aim to reprogram CAR-T cells to better sense and respond to tumor-derived chemokines and to remodel the tumor stroma and vasculature to facilitate immune cell entry. In the following subsection, we focus on chemokine-based CAR-T cell engineering strategies to enhance tumor homing, infiltration, and distribution within solid tumors.
5.1. Chemokine-Based CAR-T Cell Advancements
Efficient trafficking of CAR-T cells to solid tumor sites remains a significant barrier to therapeutic success. Preclinical studies consistently show that only a tiny fraction of the transferred T cells, often around 1-2%, actually enter the tumor mass. In a prostate cancer model using PSMA-targeted CAR-T cells, fewer than 0.2% of transferred cells were detected within the tumor, while the majority accumulated in non-target organs such as the thyroid and salivary glands [121,123]. Optimizing CAR-T cell homing not only enhanced antitumor activity but also allowed dose reduction and minimized off-tumor toxicity.
Chemokine gradients orchestrate T cell migration, yet solid tumors frequently disrupt this system by suppressing effector T cell-attracting chemokines or by producing chemokines that preferentially recruit immunosuppressive populations. Rather than facilitating immune surveillance, distorted chemokine signals promote the accumulation of Tregs, MDSCs, and M2-type TAMs, effectively excluding cytotoxic lymphocytes from the tumor core [124]. One strategy to overcome this mismatch is to engineer CAR-T cells to express chemokine receptors that correspond to ligands enriched within the TME, thereby restoring chemotactic responsiveness and enhancing intratumoral accumulation [125].
CXCL8 (also known as IL-8) is a key chemokine abundantly expressed within many solid tumours, where it is typically associated with inflammation, angiogenesis, and immune suppression. Although CXCL8-rich environments are generally hostile to CAR-T cell therapy, this axis can be repurposed by engineering CAR-T cells to express its cognate receptors, CXCR1 or CXCR2. This modification converts a protumorigenic signal into a directional cue that enhances CAR-T cell trafficking and retention within tumors [126]. In preclinical models, CD70-directed CAR-T cells co-expressing CXCR1 or CXCR2 exhibited markedly improved migration toward IL-8 gradients and tumor-derived supernatants, leading to robust tumor regression and prolonged survival compared with unmodified CAR-T cells [127]. Another study in a pancreatic ductal adenocarcinoma (PDAC) model confirmed the benefits of CXCR1/2 engineering across CXCL8-rich malignancies, including improved CAR-T cell survival, reduced accumulation of myeloid suppressor cells, and inhibition of metastasis [128].
Similarly, elevated expression levels of CXCL1 and CXCL2 in several solid tumors have further motivated the use of CXCR1/2-modified CAR-T cells to broaden chemokine responsiveness [129]. Both CXCR1- and CXCR2-targeting CAR-T cells demonstrated dose-dependent migration abilities in vitro and enhanced tumor localization even 48 hours after CAR-T cell injection [127]. Engineering lymphocytes from the tumor ascites of ovarian cancer patients to express CXCR2 enhanced their migration toward both autologous and allogeneic ascites in vitro, and the ascites exhibited high levels of CXCL1 and CXCL8 expression [130]. CAR-T cells expressing CXCR2 and targeting integrin αvβ6 showed better tumor control in a human pancreatic tumor xenograft mouse model than CAR-T cells lacking CXCR2 [131].
The CXCL13-CXCR5 axis represents another promising route for improving CAR-T cell infiltration. CXCL13 is increasingly recognized as a chemokine enriched in specific solid tumors, where it influences lymphocyte organization and migration. Activation of CXCR5 has been linked to improved calcium signaling and enhanced motility in T cells [132,133]. Osteosarcoma is an often-overlooked cancer type, and our recent study demonstrated that CXCL13 was highly expressed in 143B and U-2OS osteosarcoma cell lines [134]. The co-expression of CXCR5 enhanced the migration and infiltration ability of second-generation NKG2D-based CAR-T cells in vitro and in mouse xenografts [134]. Similar strategies have been applied in non-small cell lung cancer, where EGFR-targeted CAR-T cells expressing CXCR5 achieved superior tumor infiltration and control without increasing off-tumor toxicity, while maintaining cytotoxic activity comparable to conventional CAR-T cells [135].
An alternative approach focuses on reshaping the chemokine milieu rather than solely modifying receptor expression. CCR7, a receptor critical for naïve and central memory T cell trafficking, has been leveraged in CAR-T cell engineering strategies that secrete CCL19 alongside homeostatic cytokines such as IL-7 [136]. CAR-T cells co-expressing IL-7 and CCL19 have demonstrated remarkable efficacy across multiple solid tumor models, including mesothelioma, pancreatic cancer, and glioblastoma. These cells not only achieved superior tumor control and prolonged survival but also promoted the recruitment of endogenous immune cells and reduced exhaustion marker expression on infiltrating T cells [137,138,139]. Importantly, long-term follow-up studies have supported the clinical safety of IL-7/CCL19-secreting CAR-T cells [31,140]. Glioblastoma is a tumor with a complex TME [141]. Apart from IL-7, IL-15 was also used, alongside CCL19, to produce CAR-T cells in a mouse model of glioblastoma, achieving encouraging results at the preclinical level [142]. The findings show that 15 × 19 CAR-T cells exhibit improved proliferation, enhanced chemotactic ability, and more favourable phenotypic traits than conventional CAR-T cells in vitro. This improved performance is partly due to IL-15 and CCL19, which promote greater T cell infiltration into the tumor and strengthen resistance to exhaustion within the TME.
6. Conclusions and Future Perspective
CAR-T cell therapy has demonstrated excellent efficacy in treating hematologic malignancies, and CAR-T-based personalised immunotherapy has led to notable remissions. However, this mode of immunotherapy is still in its developmental phase, and specific challenges remain. For example, the CAR-T-associated CRS and neurotoxicity are significant side effects. Additionally, noteworthy mentions include the limited persistence of CAR-T cells, their failure to target tumor sites, various inhibitory factors within the TME, exhaustion caused by metabolic stress, and inhibitory signals. Addressing these barriers is essential to translating CAR-T cell therapy into a durable and widely applicable cancer treatment.
Recent advances reviewed here highlight a shift toward rational, multi-layered engineering strategies. Self-regulating CAR designs and switch-on/switch-off CAR systems provide critical safety controls to mitigate CRS and neurotoxicity. Other strategies, such as integrating nanomaterials and OVs, offer powerful means to remodel the TME, enhance immune infiltration, and sustain CAR-T cell function under hostile metabolic and immunosuppressive conditions. At the cellular level, precision genome engineering using CRISPR-based editing and multiplexed shRNA has enabled targeted rewiring of inhibitory signaling pathways, exhaustion programs, and stress-response networks, thereby improving persistence and antitumor efficacy. Complementing these efforts, chemokine-based strategies realign CAR-T cell migration with tumor-specific cues, overcoming trafficking and infiltration deficits that have long limited efficacy in solid tumors. Although there is still a long journey ahead, these interdisciplinary innovations are paving the way for next-generation CAR-T products that combine vigorous antitumor activity with built-in safety mechanisms and improved durability.
Looking ahead, the field is poised to integrate these advancements into unified CAR-T platforms tailored to each patient’s unique tumor landscape. Future constructs are likely to combine safety switches, chemokine receptors, cytokine support, and resistance to suppressive signaling within single, modular designs, enabling potent yet tightly controllable immune responses. Synergistic treatment regimens that pair CAR-T cells with OVs, localized biomaterials, or metabolic modulators can reshape the TME and sustain T-cell activity in vivo. Advances in genome-editing technologies, including base and prime editing, offer the prospect of enhancing CAR-T cell fitness and memory formation while minimizing genomic risk. Concurrently, innovations in manufacturing, including off-the-shelf allogeneic products and automated production platforms, are expected to reduce costs and expand access beyond specialized medical centers.
Ultimately, the convergence of synthetic biology, materials science, and systems immunology is redefining CAR-T cell therapy from a highly effective but niche intervention into a versatile and adaptable treatment platform. As multi-omic biomarkers are increasingly integrated into clinical decision-making, CAR-T engineering can be tailored to the unique biological features of each tumor, bringing the field closer to truly personalized and durable cellular immunotherapy.
Author Contributions
Writing-original draft preparation, I.A. and M.A.F.; writing-review & editing, I.A., B.D., and M.A.F.; visualisation, B.D., N.H., Q.H., and D.J.; artwork, B.D. and D.J.; funding acquisition, I.A., and G.X.; and supervision: M.A.F. and G.X. All authors read and approved the final manuscript.
Funding
This research was funded by Guangdong Basic and Applied Basic Research Foundation (2024A1515110144, 2023A1515140148), High-level Talent Research Funding Program of the First Dongguan Affiliated Hospital of Guangdong Medical University (GCC2023004), Discipline Construction Project of Guangdong Medical University (20251K20220006), and Special Project for Clinical and Basic Sci&Tech Innovation of Guangdong Medical University (GDMULCJC2024098).
Institutional review board statement
Not applicable.
Informed consent statement
Not applicable.
Data availability statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of interest
The authors declare no conflict of interests.
Abbreviation
The following abbreviations are used in this manuscript:
ADRB2: Beta-2 adrenergic receptor
B-ALL: B cell acute lymphoblastic leukemia
CAR: Chimeric antigen receptor
CCRs: Chimeric co-stimulatory receptors
CRS: Cytokine release syndrome
CT-INPs: CAR-T cell-INP biohybrids
CTLA4: Cytotoxic T-lymphocyte associated antigen 4
Cu: Copper
DCs: Dendritic cells
DSB: Double-strand break
ECM: Extracellular matrix
EGFR: Epidermal growth factor receptor
ERT2: Estrogen receptor ligand-binding domain T2
FDA: U.S. Food and Drug Administration
GCV: Ganciclovir
GLUT-1: Glucose transporter 1
HA: Hyaluronic acid
HD/TMD: Hinge and transmembrane domain
HDAC11: Histone deacetylase 11
HIF1α: Hypoxia-inducible factor 1-alpha
HREs: Hypoxia-responsive elements
HSV-TK: Herpes simplex virus thymidine kinase
huCART19: Humanized CD19 chimeric antigen receptor T-cell product
HyaII: Hyaluronidase
ICANS: Immune effector cell-associated neurotoxicity syndrome
iCasp9: Inducible caspase 9
ICD: Intracellular co-stimulatory domains
IFNs: interferons
IFN-γ: Interferon gamma
IL: Interleukins
IL-10: Interleukin-10
INPs: Indocyanine green nanoparticles
ITAMs: Immunoreceptor Tyrosine-based activation motifs
LAG3: Lymphocyte activation gene 3 protein
LiCAR: Light-inducible CAR
LNPs: Lipid nanoparticles
LOV2: Light-oxygen-voltage 2
MDSCs: Myeloid-derived suppressor cells
MHC: Major histocompatibility complex
NLS: Nuclear localization sequence
NSCLC: Non-small cell lung cancer
ODD: Oxygen-dependent degradation
OVs: Oncolytic viruses
PDAC: Pancreatic ductal adenocarcinoma
PDCD1: Programmed cell death protein 1
PD-L1: Programmed cell death ligand 1
PGC1-α: Peroxisome proliferator-activated receptor gamma coactivator 1α
PI3K: Phosphoinositide 3-kinase
PRDM1: PR domain zinc finger protein 1
PSMA:
PTT: Photothermal therapy
scFv: Single-chain variable fragment
shRNA: Short-hairpin RNA
SspB: Stringent starvation protein B
SsrA: Small stable RNA A
TAA: Tumor-associated antigen
TAMs: Tumor-associated macrophages
TCF1: T cell factor 1
TCM: Central memory T cell
TCR: T cell receptor
TGFBR2: TGF-β receptor II
TGF-β: Transforming growth factor-beta
TIGIT: T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domains
TIM3: T cell immunoglobulin mucin domain-containing protein 3
TKOs: Triple knockouts
TME: Tumor microenvironment
TNF-α: Tumor necrosis factor-alpha
TPEX: Precursor exhausted T cell
TRAF2: TNF receptor-associated factor 2
Tregs: Regulatory T cells
TRUCKs: T cells redirected for universal cytokine-mediated killing
VSV: Vesicular stomatitis virus
α-GalCer: α-galactosylceramide
References
- Asmamaw, DT; Tiruneh, GM; Dessie, TG; Tadele, AF; Wale, TW; Chekol, AE. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum Vaccin Immunother. 2022, 18(6), 2114254. [Google Scholar] [CrossRef]
- Locke, FL; Ghobadi, A; Jacobson, CA; Miklos, DB; Lekakis, LJ; Oluwole, OO; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20(1), 31–42. [Google Scholar] [CrossRef]
- Maude, SL; Laetsch, TW; Buechner, J; Rives, S; Boyer, M; Bittencourt, H; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Eng J Med. 2018, 378(5), 439–48. [Google Scholar] [CrossRef]
- Munshi, NC; Anderson, LD; Shah, N; Madduri, D; Berdeja, J; Lonial, S; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Eng J Med. 2021, 384(8), 705–16. [Google Scholar] [CrossRef]
- Wang, M; Munoz, J; Goy, A; Locke, FL; Jacobson, CA; Hill, BT; et al. Three-Year Follow-Up of KTE-X19 in Patients With Relapsed/Refractory Mantle Cell Lymphoma, Including High-Risk Subgroups, in the ZUMA-2 Study. J Clin Oncol. 2023, 41(3), 555–67. [Google Scholar] [CrossRef]
- Kuwana, Y; Asakura, Y; Utsunomiya, N; Nakanishi, M; Arata, Y; Itoh, S; et al. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987, 149(3), 960–8. [Google Scholar] [CrossRef] [PubMed]
- Shirasu, N; Kuroki, M. Functional Design of Chimeric T-Cell Antigen Receptors for Adoptive Immunotherapy of Cancer: Architecture and Outcomes. Anticancer Res. 2012, 32(6), 2377–83. [Google Scholar]
- Honikel, MM; Olejniczak, SH. Co-Stimulatory Receptor Signaling in CAR-T Cells. Biomolecules 2022, 12(9), 1303. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M; Brentjens, R; Rivière, I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009, 21(2), 215–23. [Google Scholar] [CrossRef]
- Park, JH; Geyer, MB; Brentjens, RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood 2016, 127(26), 3312–20. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A; Barua, A; Huang, L; Ganguly, S; Feng, Q; He, B. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. 2023, 14, 1188049. [Google Scholar] [CrossRef]
- Leyfman, Y. Chimeric antigen receptors: unleashing a new age of anti-cancer therapy. Cancer Cell Int. 2018, 18, 182. [Google Scholar] [CrossRef]
- Cappell, KM; Kochenderfer, JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. 2021, 18(11), 715–27. [Google Scholar] [CrossRef]
- Smith, R; Shen, R. Complexities in comparing the impact of costimulatory domains on approved CD19 CAR functionality. J Transl Med. 2023, 21(1), 515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S; Gu, C; Huang, L; Wu, H; Shi, J; Zhang, Z; et al. The third-generation anti-CD30 CAR T-cells specifically homing to the tumor and mediating powerful antitumor activity. Sci Rep. 2022, 12(1), 10488. [Google Scholar] [CrossRef] [PubMed]
- Tang, L; Pan, S; Wei, X; Xu, X; Wei, Q. Arming CAR-T cells with cytokines and more: Innovations in the fourth-generation CAR-T development. Mol Ther. 2023, 31(11), 3146–62. [Google Scholar] [CrossRef] [PubMed]
- Minguet, S; Maus, MV; Schamel, WW. From TCR fundamental research to innovative chimeric antigen receptor design. Nat Rev Immunol. 2025, 25(3), 212–24. [Google Scholar] [CrossRef]
- Mehrabadi, AZ; Ranjbar, R; Farzanehpour, M; Shahriary, A; Dorostkar, R; Hamidinejad, MA; et al. Therapeutic potential of CAR T cell in malignancies: A scoping review. Biomed Pharmacother. 2022, 146, 112512. [Google Scholar] [CrossRef]
- Lin, M-Y; Nam, E; Shih, RM; Shafer, A; Bouren, A; Ayala Ceja, M; et al. Self-regulating CAR-T cells modulate cytokine release syndrome in adoptive T-cell therapy. J Exp Med. 2024, 221(6), e20221988. [Google Scholar] [CrossRef]
- Dong, J; Wu, J; Jin, Y; Zheng, Z; Su, T; Shao, L; et al. In-depth analysis of the safety of CAR-T cell therapy for solid tumors. Front Immunol. 2025, 16, 1548979. [Google Scholar] [CrossRef]
- Xiao, X; Huang, S; Chen, S; Wang, Y; Sun, Q; Xu, X; et al. Mechanisms of cytokine release syndrome and neurotoxicity of CAR T-cell therapy and associated prevention and management strategies. J Exp Clin Cancer Res. 2021, 40(1), 367. [Google Scholar] [CrossRef]
- Hughes, AD; Teachey, DT; Diorio, C. Riding the storm: managing cytokine-related toxicities in CAR-T cell therapy. Semin Immunopathol 2024, 46(3-4), 5. [Google Scholar] [CrossRef]
- Ventin, M; Cattaneo, G; Maggs, L; Arya, S; Wang, X; Ferrone, CR. Implications of High Tumor Burden on Chimeric Antigen Receptor T-Cell Immunotherapy: A Review. JAMA Oncol. 2024, 10(1), 115. [Google Scholar] [CrossRef]
- Frigault, M; Rotte, A; Ansari, A; Gliner, B; Heery, C; Shah, B. Dose fractionation of CAR-T cells. A systematic review of clinical outcomes. J Exp Clin Cancer Res. 2023, 42(1), 11. [Google Scholar] [CrossRef]
- Rotte, A; Frigault, MJ; Ansari, A; Gliner, B; Heery, C; Shah, B. Dose-response correlation for CAR-T cells: a systematic review of clinical studies. J Immunother Cancer 2022, 10(12), e005678. [Google Scholar] [CrossRef]
- Wang, Z; Li, P; Zeng, X; Guo, J; Zhang, C; Fan, Z; et al. CAR-T therapy dilemma and innovative design strategies for next generation. Cell Death Dis. 2025, 16(1), 211. [Google Scholar] [CrossRef] [PubMed]
- Drent, E; Poels, R; Ruiter, R; van de Donk, NWCJ; Zweegman, S; Yuan, H; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin Cancer Res. 2019, 25(13), 4014–25. [Google Scholar] [CrossRef]
- Ghorashian, S; Kramer, AM; Onuoha, S; Wright, G; Bartram, J; Richardson, R; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. 2019, 25(9), 1408–14. [Google Scholar] [CrossRef] [PubMed]
- Myers, RM; Li, Y; Barz Leahy, A; Barrett, DM; Teachey, DT; Callahan, C; et al. Humanized CD19-Targeted Chimeric Antigen Receptor (CAR) T Cells in CAR-Naive and CAR-Exposed Children and Young Adults With Relapsed or Refractory Acute Lymphoblastic Leukemia. J Clin Oncol. 2021, 39(27), 3044–55. [Google Scholar] [CrossRef] [PubMed]
- Wu, S; Luo, Q; Li, F; Zhang, S; Zhang, C; Liu, J; et al. Development of novel humanized CD19/BAFFR bicistronic chimeric antigen receptor T cells with potent antitumor activity against B-cell lineage neoplasms. Br J Haematol. 2024, 205(4), 1361–73. [Google Scholar] [CrossRef]
- Brudno, JN; Lam, N; Vanasse, D; Shen, Y; Rose, JJ; Rossi, J; et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. 2020, 26(2), 270–80. [Google Scholar] [CrossRef] [PubMed]
- Zenere, G; Wu, C; Midkiff, CC; Johnson, NM; Grice, CP; Wimley, WC; et al. Extracellular domain, hinge, and transmembrane determinants affecting surface CD4 expression of a novel anti-HIV chimeric antigen receptor (CAR) construct. PLos One 2024, 19(8), e0293990. [Google Scholar] [CrossRef]
- Gu, T; Hu, K; Si, X; Hu, Y; Huang, H. Mechanisms of immune effector cell-associated neurotoxicity syndrome after CAR-T treatment. WIREs Mech Dis. 2022, 14(6), e1576. [Google Scholar] [CrossRef]
- Fujiwara, K; Tsunei, A; Kusabuka, H; Ogaki, E; Tachibana, M; Okada, N; et al. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9(5), 1182. [Google Scholar] [CrossRef]
- Jayaraman, J; Mellody, MP; Hou, AJ; Desai, RP; Fung, AW; Pham, AHT; et al. CAR-T design: Elements and their synergistic function. eBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef] [PubMed]
- Ferreras, C; Fernández, L; Clares-Villa, L; Ibáñez-Navarro, M; Martín-Cortázar, C; Esteban-Rodríguez, I; et al. Facing CAR T Cell Challenges on the Deadliest Paediatric Brain Tumours. Cells 2021, 10(11), 2940. [Google Scholar] [CrossRef] [PubMed]
- Wu, L; Chen, J; Cai, R; Wang, X; Liu, Y; Zheng, Q; et al. Difference in Efficacy and Safety of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy Containing 4-1BB and CD28 Co-Stimulatory Domains for B-Cell Acute Lymphoblastic Leukemia. Cancers (Basel) 2023, 15(10), 2767. [Google Scholar] [CrossRef]
- Sun, C; Shou, P; Du, H; Hirabayashi, K; Chen, Y; Herring, LE; et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell. 2020, 37(2), 216–25.e6. [Google Scholar] [CrossRef]
- Chuan, Tong; Yao, Wang; Weidong, Han. Structural optimization and prospect of chimeric antigen receptor T cells. Zhonghua Xueyexue Zazhi 2021, 42(9), 771–7. [Google Scholar] [CrossRef]
- Feucht, J; Sun, J; Eyquem, J; Ho, Y-J; Zhao, Z; Leibold, J; et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019, 25(1), 82–8. [Google Scholar] [CrossRef]
- Moghanloo, E; Mollanoori, H; Talebi, M; Pashangzadeh, S; Faraji, F; Hadjilooei, F; et al. Remote controlling of CAR-T cells and toxicity management: Molecular switches and next generation CARs. Translational Oncology 2021, 14(6), 101070. [Google Scholar] [CrossRef]
- Koehne, G; Doubrovin, M; Doubrovina, E; Zanzonico, P; Gallardo, HF; Ivanova, A; et al. Serial in vivo imaging of the targeted migration of human HSV-TK-transduced antigen-specific lymphocytes. Nat Biotechnol. 2003, 21(4), 405–13. [Google Scholar] [CrossRef] [PubMed]
- Greco, R; Oliveira, G; Stanghellini, MTL; Vago, L; Bondanza, A; Peccatori, J; et al. Improving the safety of cell therapy with the TK-suicide gene. Front Pharmacol. 2015, 6, 95. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T; Sakoda, Y; Adachi, K; Tokunaga, Y; Tamada, K. Therapeutic effects of anti-GM2 CAR-T cells expressing IL-7 and CCL19 for GM2-positive solid cancer in xenograft model. Cancer Med. 2023, 12(11), 12569–80. [Google Scholar] [CrossRef] [PubMed]
- Murty, S; Labanieh, L; Murty, T; Gowrishankar, G; Haywood, T; Alam, IS; et al. PET Reporter Gene Imaging and Ganciclovir-Mediated Ablation of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2020, 80(21), 4731–40. [Google Scholar] [CrossRef]
- Haist, C; Poschinski, Z; Bister, A; Hoffmann, MJ; Grunewald, CM; Hamacher, A; et al. Engineering a single-chain variable fragment of cetuximab for CAR T-cell therapy against head and neck squamous cell carcinomas. Oral Oncol. 2022, 129, 105867. [Google Scholar] [CrossRef]
- Shabaneh, TB; Moffett, HF; Stull, SM; Derezes, T; Tait, LJ; Park, S; et al. Safety switch optimization enhances antibody-mediated elimination of CAR T cells. Front Mol Med. 2022, 2, 1026474. [Google Scholar] [CrossRef]
- Maus, MV. Designing CAR T cells for glioblastoma. OncoImmunology 2015, 4(12), e1048956. [Google Scholar] [CrossRef]
- Lin, H; Cheng, J; Zhu, L; Zeng, Y; Dai, Z; Zhang, Y; et al. Anti-CD5 CAR-T cells with a tEGFR safety switch exhibit potent toxicity control. Blood Cancer J 2024, 14(1), 98. [Google Scholar] [CrossRef]
- Lungova, K; Putman, M. Barriers to CAR T-cell therapy in rheumatology. Lancet Rheumatol. 2025, 7(3), e212-6. [Google Scholar] [CrossRef]
- Philip, B; Kokalaki, E; Mekkaoui, L; Thomas, S; Straathof, K; Flutter, B; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124(8), 1277–87. [Google Scholar] [CrossRef]
- McKee, B; Liu, S; Cai, PX; Yang, Z; Lan, T-H; Zhou, Y. Optogenetic control of T cells for immunomodulation. Essays Biochem. 2025, 69(2), 33–46. [Google Scholar] [CrossRef]
- Flugel, CL; Majzner, RG; Krenciute, G; Dotti, G; Riddell, SR; Wagner, DL; et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol. 2023, 20(1), 49–62. [Google Scholar] [CrossRef]
- Russell, GC; Hamzaoui, Y; Rho, D; Sutrave, G; Choi, JS; Missan, DS; et al. Synthetic biology approaches for enhancing safety and specificity of CAR-T cell therapies for solid cancers. Cytotherapy 2024, 26(8), 842–57. [Google Scholar] [CrossRef]
- Chandrasekar, S; Beach, JR; Oakes, PW. Shining a light on RhoA: Optical control of cell contractility. Int J Biochem Cell Biol. 2023, 161, 106442. [Google Scholar] [CrossRef]
- Wang, W; Wildes, CP; Pattarabanjird, T; Sanchez, MI; Glober, GF; Matthews, GA; et al. A light- and calcium-gated transcription factor for imaging and manipulating activated neurons. Nat Biotechnol. 2017, 35(9), 864–71. [Google Scholar] [CrossRef]
- Zhu, C; Wu, Q; Sheng, T; Shi, J; Shen, X; Yu, J; et al. Rationally designed approaches to augment CAR-T therapy for solid tumor treatment. Bioact Mater. 2024, 33, 377–95. [Google Scholar] [CrossRef]
- Allen, ME; Zhou, W; Thangaraj, J; Kyriakakis, P; Wu, Y; Huang, Z; et al. An AND-Gated Drug and Photoactivatable Cre-loxP System for Spatiotemporal Control in Cell-Based Therapeutics. ACS Synth Biol. 2019, 8(10), 2359–71. [Google Scholar] [CrossRef]
- Zhou, Y; Wei, Y; Yin, J; Kong, D; Li, W; Wang, X; et al. A rapid and efficient red-light-activated Cre recombinase system for genome engineering in mammalian cells and transgenic mice. Nucleic Acids Res. 2025, 53(15), gkaf758. [Google Scholar] [CrossRef]
- Juillerat, A; Marechal, A; Filhol, JM; Valogne, Y; Valton, J; Duclert, A; et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. 2017, 7, 39833. [Google Scholar] [CrossRef]
- Liao, Q; He, H; Mao, Y; Ding, X; Zhang, X; Xu, J. Engineering T cells with hypoxia-inducible chimeric antigen receptor (HiCAR) for selective tumor killing. Biomark Res. 2020, 8(1), 56. [Google Scholar] [CrossRef]
- Basheeruddin, M; Qausain, S. Hypoxia-Inducible Factor 1-Alpha (HIF-1α): An Essential Regulator in Cellular Metabolic Control. Cureus 2024, 16(7), e63852. [Google Scholar] [CrossRef]
- Kankeu Fonkoua, LA; Sirpilla, O; Sakemura, R; Siegler, EL; Kenderian, SS. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. MolTher Oncolytics 2022, 25, 69–77. [Google Scholar] [CrossRef]
- Zhu, X; Chen, J; Li, W; Xu, Y; Shan, J; Hong, J; et al. Hypoxia-Responsive CAR-T Cells Exhibit Reduced Exhaustion and Enhanced Efficacy in Solid Tumors. Cancer Res. 2024, 84(1), 84–100. [Google Scholar] [CrossRef]
- Zheng, S; Che, X; Zhang, K; Bai, Y; Deng, H. Potentiating CAR-T cell function in the immunosuppressive tumor microenvironment by inverting the TGF-β signal. Mol Ther. 2025, 33(2), 688–702. [Google Scholar] [CrossRef]
- Farooq, MA; Ajmal, I; Hui, X; Chen, Y; Ren, Y; Jiang, W; et al. β2-Adrenergic Receptor Mediated Inhibition of T Cell Function and Its Implications for CAR-T Cell Therapy. Int J Mol Sci. 2023, 24(16), 12837. [Google Scholar] [CrossRef]
- Balakrishnan, PB; Sweeney, EE. Nanoparticles for Enhanced Adoptive T Cell Therapies and Future Perspectives for CNS Tumors. Front Immunol. 2021, 12, 600659. [Google Scholar] [CrossRef]
- Ponterio, E; Haas, TL; De Maria, R. Oncolytic virus and CAR-T cell therapy in solid tumors. Front Immunol. 2024, 15, 1455163. [Google Scholar] [CrossRef]
- Lafuente-Gómez, N; Kang, S; Mooney, DJ. Nanotechnology for CAR T cells and tumour-infiltrating lymphocyte therapies. Nat Nanotechnol. 2025, 20(9), 1186–98. [Google Scholar] [CrossRef]
- Zhang, F; Stephan, SB; Ene, CI; Smith, TT; Holland, EC; Stephan, MT. Nanoparticles That Reshape the Tumor Milieu Create a Therapeutic Window for Effective T-cell Therapy in Solid Malignancies. Cancer Res. 2018, 78(13), 3718–30. [Google Scholar] [CrossRef]
- Zheng, Y; Tang, L; Mabardi, L; Kumari, S; Irvine, DJ. Enhancing Adoptive Cell Therapy of Cancer through Targeted Delivery of Small-Molecule Immunomodulators to Internalizing or Noninternalizing Receptors. ACS Nano 2017, 11(3), 3089–100. [Google Scholar] [CrossRef]
- Wang, Z; Li, Z; Sun, Z; Wang, S; Ali, Z; Zhu, S; et al. Visualization nanozyme based on tumor microenvironment “unlocking” for intensive combination therapy of breast cancer. Sci Adv. 2020, 6(48), eabc8733. [Google Scholar] [CrossRef]
- Zeng, Q; Liu, Z; Niu, T; He, C; Qu, Y; Qian, Z. Application of nanotechnology in CAR-T-cell immunotherapy. Chinese Chemical Letters 2023, 34(3), 107747. [Google Scholar] [CrossRef]
- Chen, Z; Pan, H; Luo, Y; Yin, T; Zhang, B; Liao, J; et al. Nanoengineered CAR-T Biohybrids for Solid Tumor Immunotherapy with Microenvironment Photothermal-Remodeling Strategy. Small 2021, 17(14), 2007494. [Google Scholar] [CrossRef]
- Phan, NM; Nguyen, TL; Kim, J. Nanozyme-Based Enhanced Cancer Immunotherapy. Tissue Eng Regen Med. 2022, 19(2), 237–52. [Google Scholar] [CrossRef]
- Zhu, L; Liu, J; Zhou, G; Liu, T-M; Dai, Y; Nie, G; et al. Remodeling of Tumor Microenvironment by Tumor-Targeting Nanozymes Enhances Immune Activation of CAR T Cells for Combination Therapy. Small 2021, 17(43), 2102624. [Google Scholar] [CrossRef]
- Wu, W; Li, H; Chen, W; Hu, Y; Wang, Z; She, W; et al. CAR T Cell Membrane Camouflaged Nanocatalyst Augments CAR T Cell Therapy Efficacy Against Solid Tumor. Small 2024, 20(37), 2401299. [Google Scholar] [CrossRef]
- Huang, S; Zhao, Y; Shen, J. Advances in integrating biomaterials with CAR-T cells for enhancing solid tumor therapy. Chin J Cancer Res. 2025, 37(5), 742–58. [Google Scholar] [CrossRef]
- Bhagwat, AS; Torres, L; Shestova, O; Shestov, M; Mellors, PW; Fisher, HR; et al. Cytokine-mediated CAR T therapy resistance in AML. Nat Med. 2024, 30(12), 3697–708. [Google Scholar] [CrossRef]
- Biery, DN; Turicek, DP; Diorio, C; Schroeder, BA; Shah, NN. Need for standardization of cytokine profiling in CAR T cell therapy. Mol Ther. 2024, 32(9), 2979–83. [Google Scholar] [CrossRef]
- Stephan, MT; Moon, JJ; Um, SH; Bershteyn, A; Irvine, DJ. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med. 2010, 16(9), 1035–41. [Google Scholar] [CrossRef]
- Luo, Y; Chen, Z; Sun, M; Li, B; Pan, F; Ma, A; et al. IL-12 nanochaperone-engineered CAR T cell for robust tumor-immunotherapy. Biomaterials 2022, 281, 121341. [Google Scholar] [CrossRef]
- Hu, Q; Li, H; Archibong, E; Chen, Q; Ruan, H; Ahn, S; et al. Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets. Nat Biomed Eng. 2021, 5(9), 1038–47. [Google Scholar] [CrossRef]
- Fukuhara, H; Ino, Y; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107(10), 1373–9. [Google Scholar] [CrossRef]
- Lawler, SE; Speranza, M-C; Cho, C-F; Chiocca, EA. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3(6), 841–9. [Google Scholar] [CrossRef]
- Shalhout, SZ; Miller, DM; Emerick, KS; Kaufman, HL. Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol. 2023, 20(3), 160–77. [Google Scholar] [CrossRef]
- Ajina, A; Maher, J. Prospects for combined use of oncolytic viruses and CAR T-cells. J Immunother Cancer 2017, 5(1), 90. [Google Scholar] [CrossRef]
- Evgin, L; Kottke, T; Tonne, J; Thompson, J; Huff, AL; van Vloten, J; et al. Oncolytic virus–mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci Transl Med. 2022, 14(640), eabn2231. [Google Scholar] [CrossRef]
- Wang, S; Li, Y; Xu, C; Dong, J; Wei, J. An oncolytic vaccinia virus encoding hyaluronidase reshapes the extracellular matrix to enhance cancer chemotherapy and immunotherapy. J Immunother Cancer 2024, 12(3), e008431. [Google Scholar] [CrossRef]
- McGrath, K; Dotti, G. Combining Oncolytic Viruses with Chimeric Antigen Receptor T Cell Therapy. Human Gene Ther. 2021, 32(3-4), 150–7. [Google Scholar] [CrossRef]
- Munn, DH; Bronte, V. Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol. 2016, 39, 1–6. [Google Scholar] [CrossRef]
- Watanabe, K; Luo, Y; Da, T; Guedan, S; Ruella, M; Scholler, J; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3(7), e99573. [Google Scholar] [CrossRef]
- Shaw, AR; Porter, CE; Watanabe, N; Tanoue, K; Sikora, A; Gottschalk, S; et al. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol Ther. 2017, 25(11), 2440–51. [Google Scholar] [CrossRef]
- Zarezadeh Mehrabadi, A; Roozbahani, F; Ranjbar, R; Farzanehpour, M; Shahriary, A; Dorostkar, R; et al. Overview of the pre-clinical and clinical studies about the use of CAR-T cell therapy of cancer combined with oncolytic viruses. World J Surg Onc 2022, 20(1), 16. [Google Scholar] [CrossRef]
- Li, C; Mei, H; Hu, Y. Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy. Brief Funct Genomics 2020, 19(3), 175–82. [Google Scholar] [CrossRef] [PubMed]
- Razeghian, E; Nasution, MKM; Rahman, HS; Gardanova, ZR; Abdelbasset, WK; Aravindhan, S; et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res Ther. 2021, 12(1), 428. [Google Scholar] [CrossRef] [PubMed]
- Tao, R; Han, X; Bai, X; Yu, J; Ma, Y; Chen, W; et al. Revolutionizing cancer treatment: enhancing CAR-T cell therapy with CRISPR/Cas9 gene editing technology. Front Immunol. 2024, 15, 1354825. [Google Scholar] [CrossRef] [PubMed]
- Abdoli Shadbad, M; Hemmat, N; Khaze Shahgoli, V; Derakhshani, A; Baradaran, F; Brunetti, O; et al. A Systematic Review on PD-1 Blockade and PD-1 Gene-Editing of CAR-T Cells for Glioma Therapy: From Deciphering to Personalized Medicine. Front Immunol. 2022, 12, 788211. [Google Scholar] [CrossRef]
- McGowan, E; Lin, Q; Ma, G; Yin, H; Chen, S; Lin, Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomed Pharmacother. 2020, 121, 109625. [Google Scholar] [CrossRef]
- Wang, Z; Li, N; Feng, K; Chen, M; Zhang, Y; Liu, Y; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. 2021, 18(9), 2188–98. [Google Scholar] [CrossRef]
- Shi, L; Meng, T; Zhao, Z; Han, J; Zhang, W; Gao, F; et al. CRISPR knock out CTLA-4 enhances the anti-tumor activity of cytotoxic T lymphocytes. Gene 2017, 636, 36–41. [Google Scholar] [CrossRef]
- Cianciotti, BC; Magnani, ZI; Ugolini, A; Camisa, B; Merelli, I; Vavassori, V; et al. TIM-3, LAG-3, or 2B4 gene disruptions increase the anti-tumor response of engineered T cells. Front Immunol. 2024, 15, 1315283. [Google Scholar] [CrossRef]
- Ren, J; Liu, X; Fang, C; Jiang, S; June, CH; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res. 2017, 23(9), 2255–66. [Google Scholar] [CrossRef]
- Jenkins, E; Whitehead, T; Fellermeyer, M; Davis, SJ; Sharma, S. The current state and future of T-cell exhaustion research. Oxf Open Immunol 2023, 4(1), iqad006. [Google Scholar] [CrossRef]
- Nakagawara, K; Ando, M; Srirat, T; Mise-Omata, S; Hayakawa, T; Ito, M; et al. NR4A ablation improves mitochondrial fitness for long persistence in human CAR-T cells against solid tumors. J Immunother Cancer 2024, 12(8), e008665. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M; Li, H; Zhan, Y; Ma, D; Gao, Q; Fang, Y. Functional CRISPR screens in T cells reveal new opportunities for cancer immunotherapies. Mol Cancer 2024, 23(1), 73. [Google Scholar] [CrossRef] [PubMed]
- Tang, N; Cheng, C; Zhang, X; Qiao, M; Li, N; Mu, W; et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5(4), e133977. [Google Scholar] [CrossRef]
- Merlin, JPJ; Abrahamse, H. Optimizing CRISPR/Cas9 precision: Mitigating off-target effects for safe integration with photodynamic and stem cell therapies in cancer treatment. Biomed Pharmacother. 2024, 180, 117516. [Google Scholar] [CrossRef] [PubMed]
- Dimitri, A; Herbst, F; Fraietta, JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer 2022, 21(1), 78. [Google Scholar] [CrossRef]
- Su, Q; Yao, J; Farooq, MA; Ajmal, I; Duan, Y; He, C; et al. Modulating Cholesterol Metabolism via ACAT1 Knockdown Enhances Anti-B-Cell Lymphoma Activities of CD19-Specific Chimeric Antigen Receptor T Cells by Improving the Cell Activation and Proliferation. Cells 2024, 13(6), 555. [Google Scholar] [CrossRef]
- Grosser, R; Cherkassky, L; Chintala, N; Adusumilli, PS. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell. 2019, 36(5), 471–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H; Yao, J; Ajmal, I; Farooq, MA; Jiang, W. shRNA-mediated gene silencing of HDAC11 empowers CAR-T cells against prostate cancer. Front Immunol. 2024, 15, 1369406. [Google Scholar] [CrossRef]
- Ajmal, I; Farooq, MA; Duan, Y; Yao, J; Gao, Y; Hui, X; et al. Intrinsic ADRB2 inhibition improves CAR-T cell therapy efficacy against prostate cancer. Mol Ther. 2024, 32(10), 3539–57. [Google Scholar] [CrossRef]
- Gao, Y; Lin, H; Guo, D; Cheng, S; Zhou, Y; Zhang, L; et al. Suppression of 4.1R enhances the potency of NKG2D-CAR T cells against pancreatic carcinoma via activating ERK signaling pathway. Oncogenesis 2021, 10(9), 62. [Google Scholar] [CrossRef]
- Ajmal, I; Farooq, MA; Abbas, SQ; Shah, J; Majid, M; Jiang, W. Isoprenaline and salbutamol inhibit pyroptosis and promote mitochondrial biogenesis in arthritic chondrocytes by downregulating β-arrestin and GRK2. Front Pharmacol. 2022, 13, 996321. [Google Scholar] [CrossRef]
- Zhou, J; Yu, J; Wang, Y; Wang, H; Wang, J; Wang, Y; et al. ShRNA-mediated silencing of PD-1 augments the efficacy of chimeric antigen receptor T cells on subcutaneous prostate and leukemia xenograft. Biomed Pharmacother. 2021, 137, 111339. [Google Scholar] [CrossRef]
- Jafarzadeh, L; Masoumi, E; Mirzaei, HR; Alishah, K; Fallah-Mehrjardi, K; Khakpoor-Koosheh, M; et al. Targeted knockdown of Tim3 by short hairpin RNAs improves the function of anti-mesothelin CAR T cells. Mol Immunol 2021, 139, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S; Akbay, EA; Li, YY; Herter-Sprie, GS; Buczkowski, KA; Richards, WG; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Park, Y; Jeon, D-H; Yi, H; Lee, S-H; Kim, HC. Characteristics of CRC01, an investigational CD19 CAR-T product with PD-1 and TIGIT genes silenced by shRNAs. J Clin Oncol. 2022, 40, e14514–e14514. [Google Scholar] [CrossRef]
- Steklov, M; Lecalve, B; Marijse, J; Huberty, F; Ramelot, N; Jacques-Hespel, C; et al. 146 Evolving Mutliplexed shRNA to generate tailored CAR T cell therapy. J Immunother Cancer 2021, 9, A154–154. [Google Scholar] [CrossRef]
- Foeng, J; Comerford, I; McColl, SR. Harnessing the chemokine system to home CAR-T cells into solid tumors. Cell Rep Med. 2022, 3(3), 100543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P-F; Wang, C; Zhang, L; Li, Q. Reversing Chemokine/Chemokine Receptor Mismatch to Enhance the Antitumor Efficacy of CAR-T Cells. Immunotherapy 2022, 14(6), 459–73. [Google Scholar] [CrossRef]
- Emami-Shahri, N; Foster, J; Kashani, R; Gazinska, P; Cook, C; Sosabowski, J; et al. Clinically compliant spatial and temporal imaging of chimeric antigen receptor T-cells. Nat Commun. 2018, 9(1), 1081. [Google Scholar] [CrossRef] [PubMed]
- Chen, T; Wang, M; Chen, Y; Liu, Y. Current challenges and therapeutic advances of CAR-T cell therapy for solid tumors. Cancer Cell Int. 2024, 24(1), 133. [Google Scholar] [CrossRef]
- Zhang, Y; Guan, X; Jiang, P. Cytokine and Chemokine Signals of T-Cell Exclusion in Tumors. Front Immunol. 2020, 11, 594609. [Google Scholar] [CrossRef]
- Liu, G; Rui, W; Zhao, X; Lin, X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol Immunol. 2021, 18(5), 1085–95. [Google Scholar] [CrossRef]
- Jin, L; Tao, H; Karachi, A; Long, Y; Hou, AY; Na, M; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019, 10(1), 4016. [Google Scholar] [CrossRef]
- Dai, Z; Lin, X; Wang, X; Zou, X; Yan, Y; Wang, R; et al. Ectopic CXCR2 expression cells improve the anti-tumor efficiency of CAR-T cells and remodel the immune microenvironment of pancreatic ductal adenocarcinoma. Cancer Immunol Immunother. 2024, 73(4), 61. [Google Scholar] [CrossRef]
- Girbl, T; Lenn, T; Perez, L; Rolas, L; Barkaway, A; Thiriot, A; et al. Distinct Compartmentalization of the Chemokines CXCL1 and CXCL2 and the Atypical Receptor ACKR1 Determine Discrete Stages of Neutrophil Diapedesis. Immunity 2018, 49(6), 1062–1076.e6. [Google Scholar] [CrossRef] [PubMed]
- Idorn, M; Skadborg, SK; Kellermann, L; Halldórsdóttir, HR; Holmen Olofsson, G; Met, Ö; et al. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. OncoImmunology 2018, 7(8), e1450715. [Google Scholar] [CrossRef]
- Whilding, LM; Halim, L; Draper, B; Parente-Pereira, AC; Zabinski, T; Davies, DM; et al. CAR T-Cells Targeting the Integrin αvβ6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers. (Basel) 2019, 11(5), 674. [Google Scholar] [CrossRef]
- Gu, X; Li, D; Wu, P; Zhang, C; Cui, X; Shang, D; et al. Revisiting the CXCL13/CXCR5 axis in the tumor microenvironment in the era of single-cell omics: Implications for immunotherapy. Cancer Lett. 2024, 605, 217278. [Google Scholar] [CrossRef]
- Han, D; Jeong, B-K; Hong, JM; Seo, J-H; Lee, G; Kim, K; et al. Optimal chemokine receptors for enhancing immune cell trafficking in adoptive cell therapy. Immunol Res. 2025, 73(1), 36. [Google Scholar] [CrossRef]
- Hui, X; Farooq, MA; Chen, Y; Ajmal, I; Ren, Y; Xue, M; et al. A novel strategy of co-expressing CXCR5 and IL-7 enhances CAR-T cell effectiveness in osteosarcoma. Front Immunol. 2024, 15, 1462076. [Google Scholar] [CrossRef]
- Li, G; Guo, J; Zheng, Y; Ding, W; Han, Z; Qin, L; et al. CXCR5 guides migration and tumor eradication of anti-EGFR chimeric antigen receptor T cells. Mol Ther Oncolytics 2021, 22, 507–17. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A; Dagur, PK; Biancotto, A. Multiparametric Flow Cytometry Analysis of Naïve, Memory, and Effector T Cells. Methods Mol Biol. 2019, 2032, 129–40. [Google Scholar] [CrossRef] [PubMed]
- Goto, S; Sakoda, Y; Adachi, K; Sekido, Y; Yano, S; Eto, M; et al. Enhanced anti-tumor efficacy of IL-7/CCL19-producing human CAR-T cells in orthotopic and patient-derived xenograft tumor models. Cancer Immunol Immunother. 2021, 70(9), 2503–15. [Google Scholar] [CrossRef]
- Hu, J; Wang, Z; Liao, C; Chen, Z; Kang, F; Lin, C; et al. Induced expression of CCL19 promotes the anti-tumor ability of CAR-T cells by increasing their infiltration ability. Front Immunol. 2022, 13, 958960. [Google Scholar] [CrossRef]
- Ohta, K; Sakoda, Y; Adachi, K; Shinozaki, T; Nakajima, M; Yasuda, H; et al. Therapeutic Efficacy of IL7/CCL19-Expressing CAR-T Cells in Intractable Solid Tumor Models of Glioblastoma and Pancreatic Cancer. Cancer Res Commun. 2024, 4(9), 2514–24. [Google Scholar] [CrossRef]
- Qian, W; Liu, H; Lei, W; Wang, L; Chen, P. Long-Term Efficacy and Safety of CD19-Specific CAR-T Cells Armed with IL-7 and CCL19 in Patients with Relapsed or Refractory Large B-Cell Lymphoma: A Multi-Center Prospective Study. Blood 2024, 144, 2087. [Google Scholar] [CrossRef]
- Bhutani, B; Sharma, V; Ganguly, NK; Rana, R. Unravelling the modified T cell receptor through Gen-Next CAR T cell therapy in Glioblastoma: Current status and future challenges. Biomed Pharmacother. 2025, 186, 117987. [Google Scholar] [CrossRef] [PubMed]
- Chen, W; Hong, L; Lin, S; Xian, N; Yan, C; Zhao, N; et al. Enhanced anti−tumor efficacy of “IL−15 and CCL19” −secreting CAR−T cells in human glioblastoma orthotopic xenograft model. Front Oncol. 2025, 15, 1539055. [Google Scholar] [CrossRef]
Figure 1.
Structure, evolution, and clinical translation of CAR-T cell therapy. (A) Schematic overview of the modular CAR structure, including the antigen-binding scFv, hinge/spacer, transmembrane domain, and intracellular signaling modules. (B) Progressive evolution of CAR designs from first- to fifth-generation constructs, reflecting increasing signaling complexity and functional refinement. (C) Overview of FDA-approved CAR-T cell therapies and their primary indications in hematologic malignancies.
Figure 1.
Structure, evolution, and clinical translation of CAR-T cell therapy. (A) Schematic overview of the modular CAR structure, including the antigen-binding scFv, hinge/spacer, transmembrane domain, and intracellular signaling modules. (B) Progressive evolution of CAR designs from first- to fifth-generation constructs, reflecting increasing signaling complexity and functional refinement. (C) Overview of FDA-approved CAR-T cell therapies and their primary indications in hematologic malignancies.
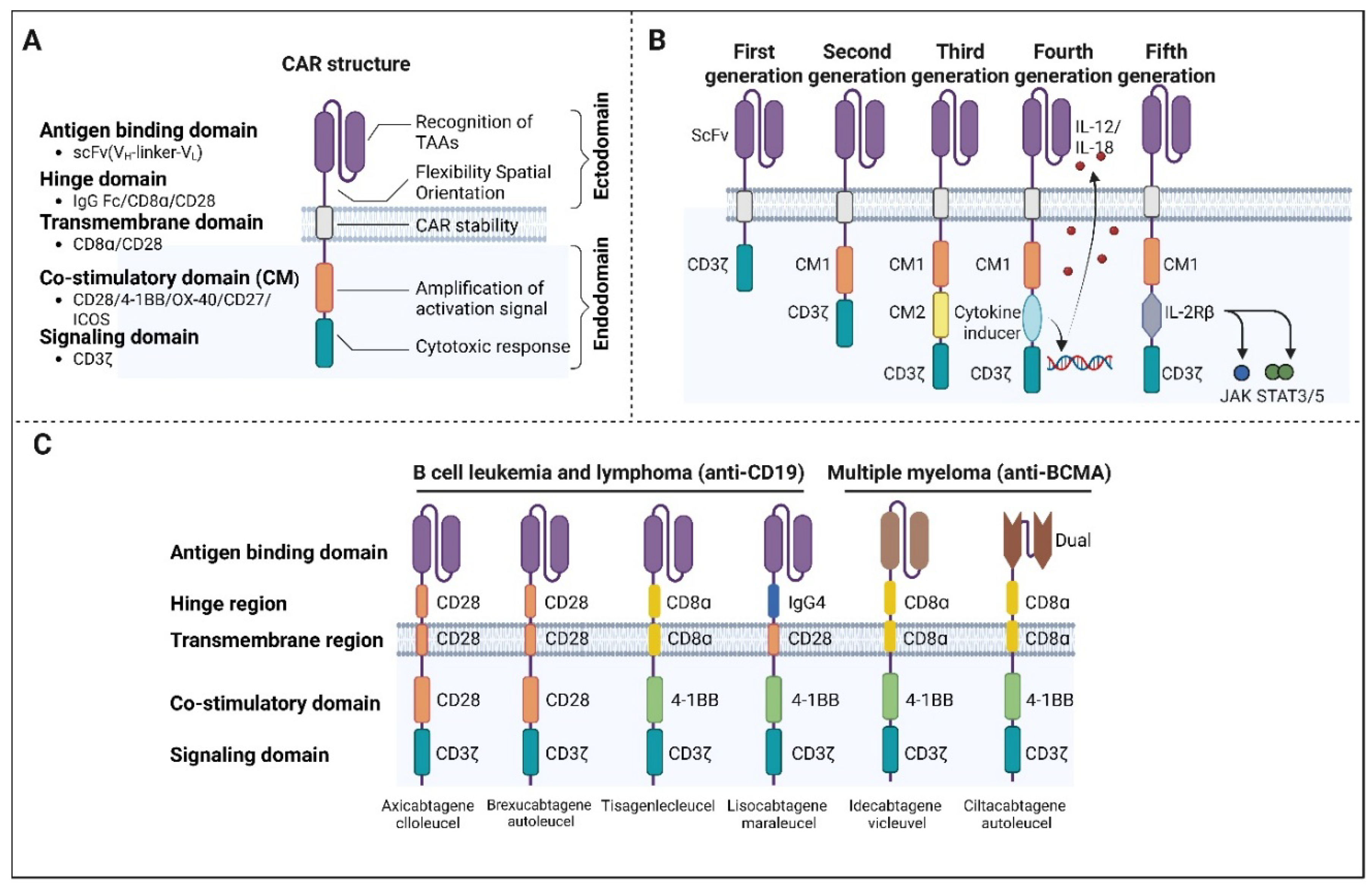
Figure 2.
Structural strategies for self-regulating CAR-T cell activity. Schematic representations of CAR design features that modulate strength and toxicity. (A) Reduced antigen-binding affinity enhances discrimination between high-antigen-expressing tumor cells and low-antigen-expressing normal cells. (B) Humanized scFv reduces immunogeneicity and supports CAR-T cell persistence. (C) Hing and transmembrane domain variants alter signaling intensity and cytokine output. (D) Modification of the CD3 signaling domain, including reduction in ITAM number, constrains excessive activation and cytokine release.
Figure 2.
Structural strategies for self-regulating CAR-T cell activity. Schematic representations of CAR design features that modulate strength and toxicity. (A) Reduced antigen-binding affinity enhances discrimination between high-antigen-expressing tumor cells and low-antigen-expressing normal cells. (B) Humanized scFv reduces immunogeneicity and supports CAR-T cell persistence. (C) Hing and transmembrane domain variants alter signaling intensity and cytokine output. (D) Modification of the CD3 signaling domain, including reduction in ITAM number, constrains excessive activation and cytokine release.
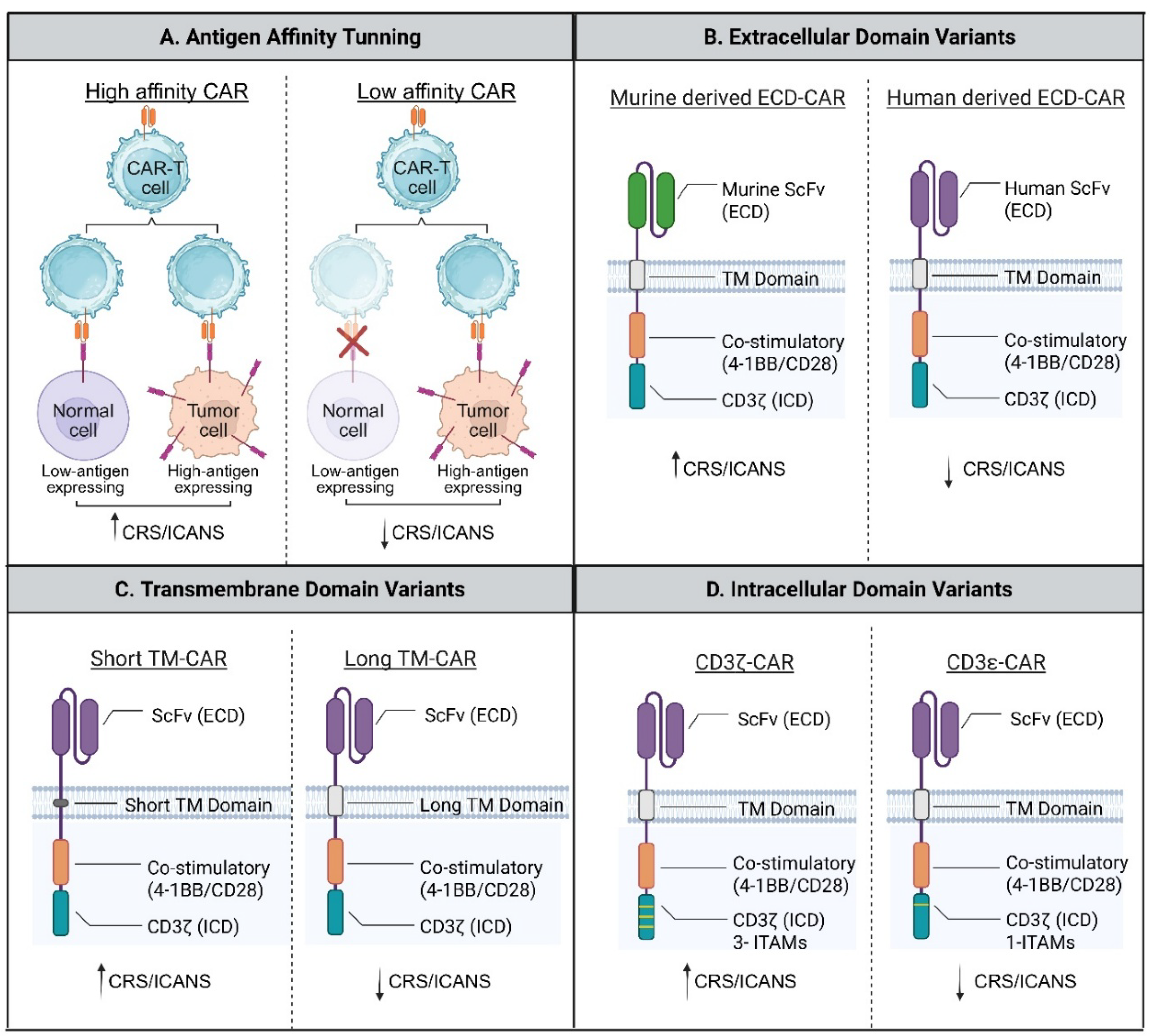
Figure 3.
Switch-off CAR strategies for controlled regulation of CAR-T cell activity. Schematic representation of switch-off CAR designs, including early suicide gene-based systems and more recent reversible control strategies. (A) Inducible caspase-9 suicide switch activated by a small-molecule dimerizer. (B) HSV-TK suicide gene system triggered by prodrug administration. (C) Antibody-mediated elimination of CAR-T cells via co-expressed surface safety markers (CD20 or truncated EGFR). (D) Pharmacologic inhibition of proximal CAR signaling to achieve transient and reversible CAR-T cell inactivation.
Figure 3.
Switch-off CAR strategies for controlled regulation of CAR-T cell activity. Schematic representation of switch-off CAR designs, including early suicide gene-based systems and more recent reversible control strategies. (A) Inducible caspase-9 suicide switch activated by a small-molecule dimerizer. (B) HSV-TK suicide gene system triggered by prodrug administration. (C) Antibody-mediated elimination of CAR-T cells via co-expressed surface safety markers (CD20 or truncated EGFR). (D) Pharmacologic inhibition of proximal CAR signaling to achieve transient and reversible CAR-T cell inactivation.
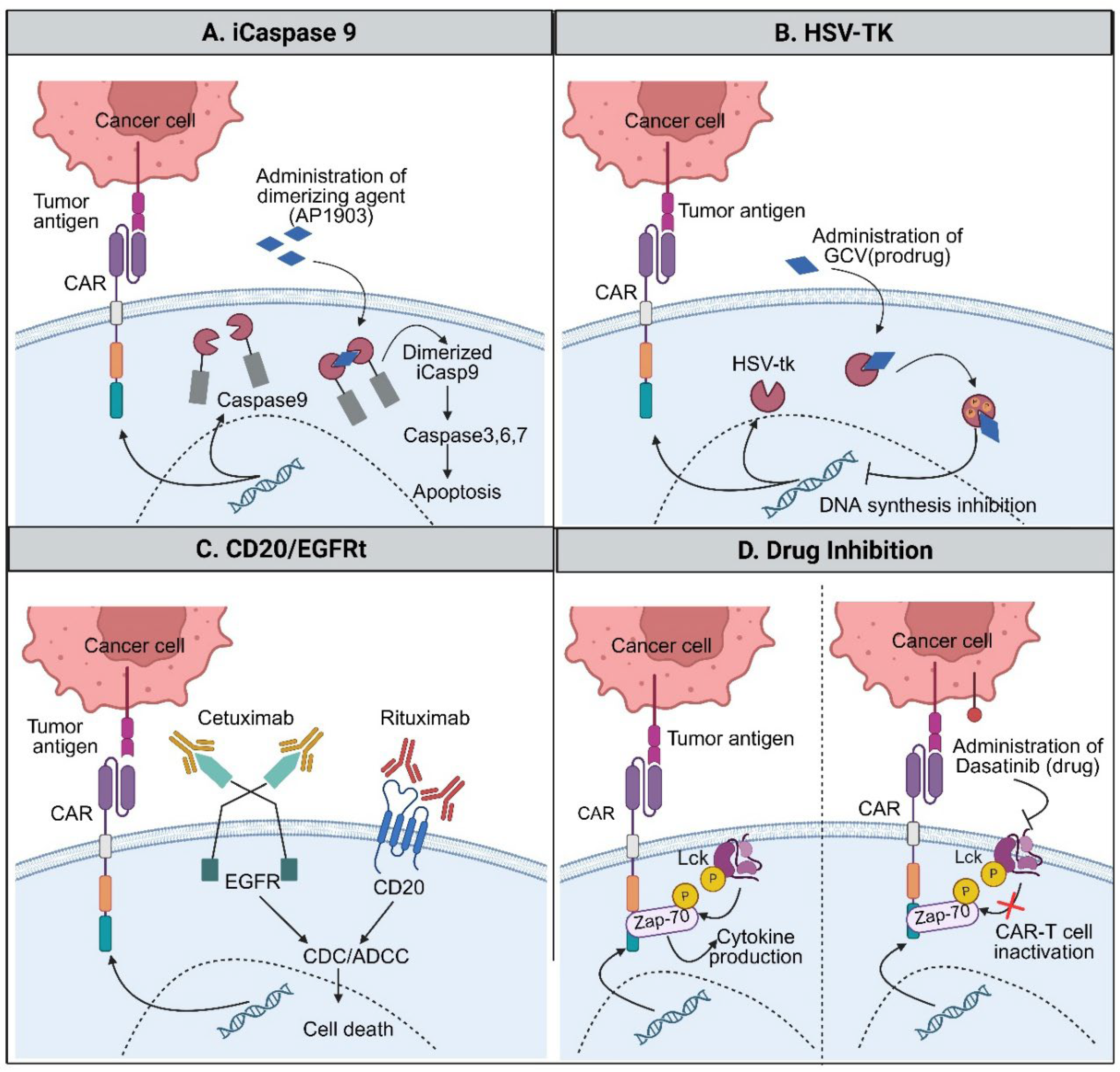
Figure 4.
Switch-on CAR systems enabling conditional and localized CAR-T cell activation. Schematic overview of representative strategies in which CAR-T cell activity is selectively enabled by defined environmental or external cues. (A) LiCAR remain inactive under dark conditions and are transiently activated upon blue-light exposure through reversible optogenetic domain interactions, allowing spatial control of signaling. (B) Hypoxia-responsive CARs restrict CAR expression and/or stability to low-oxygen tumor regions via hypoxia-dependent transcriptional and post-translational regulation, thereby limiting activity in normoxic tissues. (C) AND-gated CAR systems require the simultaneous presence of multiple inputs-such as dual antigens combined with exogenous signals (e.g., tamoxifen and light)-to trigger CAR expression or activation, enhancing specificity and reducing unintended targeting. Together, these designs illustrate complementary approaches for achieving precise temporal, spatial, and contextual control of CAR-T cell function.
Figure 4.
Switch-on CAR systems enabling conditional and localized CAR-T cell activation. Schematic overview of representative strategies in which CAR-T cell activity is selectively enabled by defined environmental or external cues. (A) LiCAR remain inactive under dark conditions and are transiently activated upon blue-light exposure through reversible optogenetic domain interactions, allowing spatial control of signaling. (B) Hypoxia-responsive CARs restrict CAR expression and/or stability to low-oxygen tumor regions via hypoxia-dependent transcriptional and post-translational regulation, thereby limiting activity in normoxic tissues. (C) AND-gated CAR systems require the simultaneous presence of multiple inputs-such as dual antigens combined with exogenous signals (e.g., tamoxifen and light)-to trigger CAR expression or activation, enhancing specificity and reducing unintended targeting. Together, these designs illustrate complementary approaches for achieving precise temporal, spatial, and contextual control of CAR-T cell function.
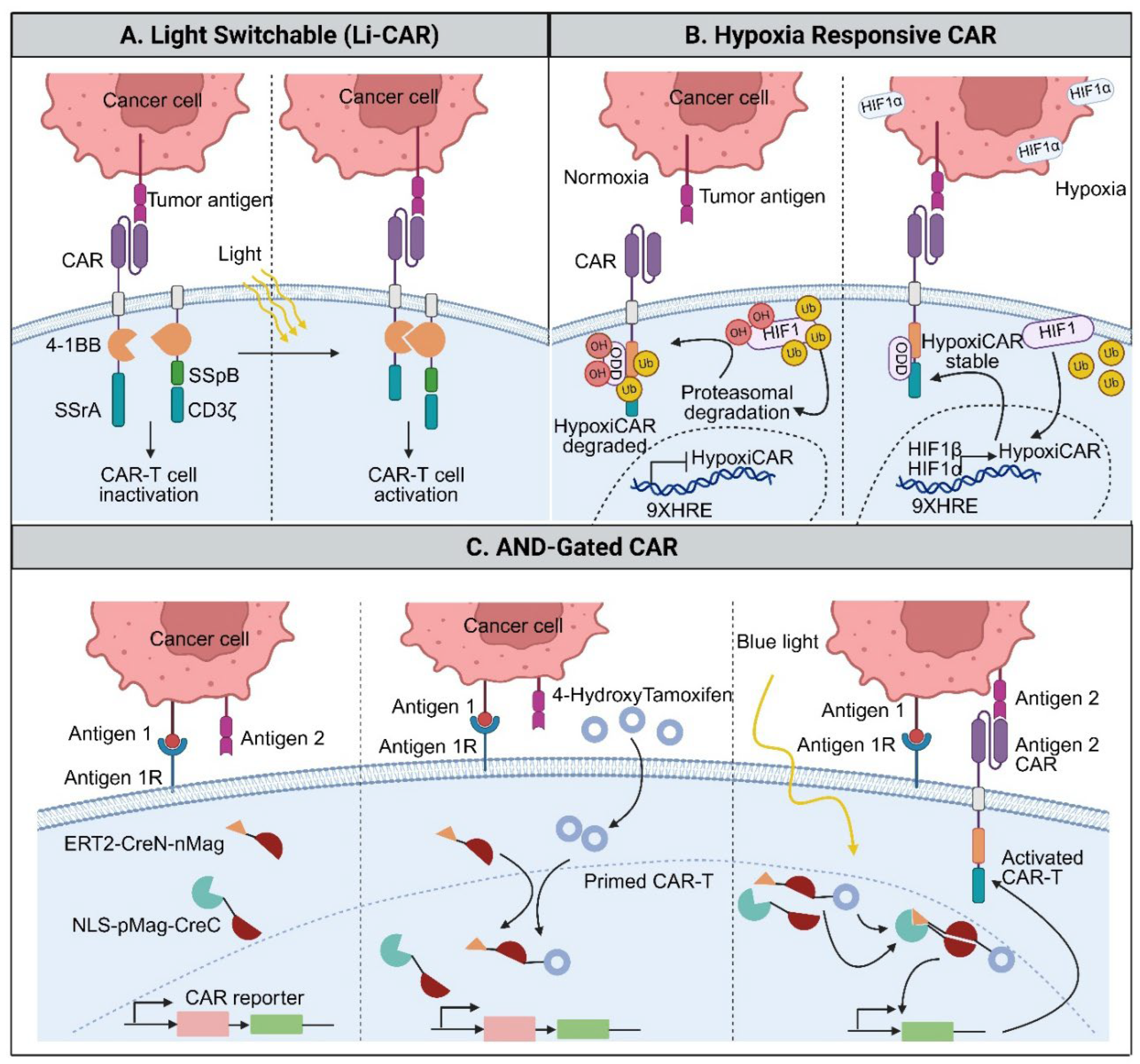
Figure 5.
Nanomaterial-enabled strategies to remodel the tumor microenvironment and enhance CAR-T cell therapy.
Figure 5.
Nanomaterial-enabled strategies to remodel the tumor microenvironment and enhance CAR-T cell therapy.
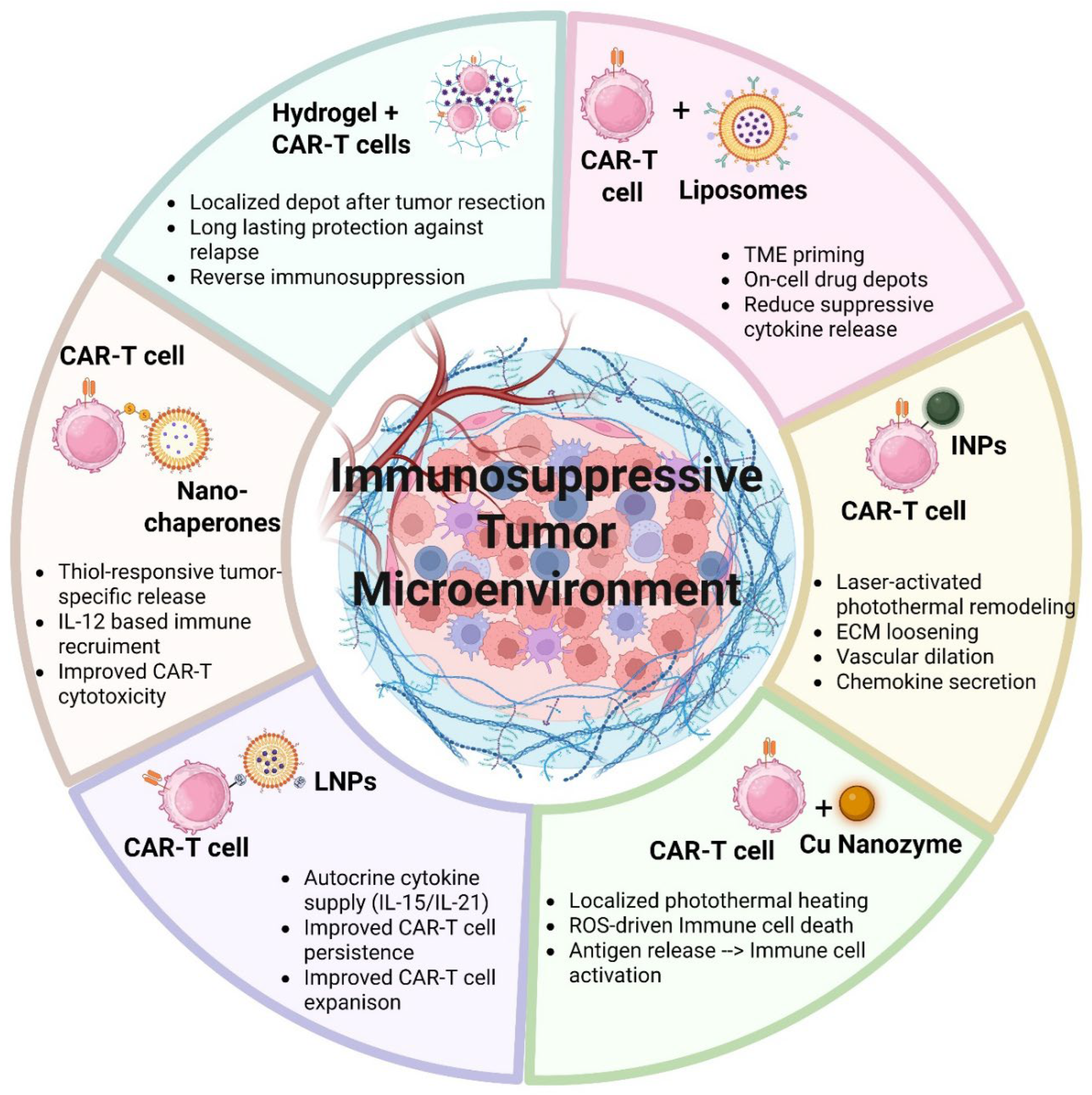
Figure 6.
Synergistic mechanisms of oncolytic virus and CAR-T cell combination strategies in solid tumors. Conceptual illustration comparing conventional and engineered oncolytic virus + CAR-T cell combination approaches. (Left) Co-administration of CAR-T cells with unmodified OVs promotes direct tumor debulking through complementary cytolytic mechanisms and induces immunogenic tumor cell death, resulting in the release of tumor- and virus-derived antigens that enhance CAR-T cell activation and infiltration. This inflammatory milieu partially alleviates local immunosuppression and supports antitumor immune responses. (Right) Engineered OVs further amplify therapeutic efficacy by delivering immunomodulatory transgenes, including immune checkpoint inhibitors, cytokines, and chemokines, directly within the tumor microenvironment. Localized expression of these factors enhances CAR-T cell trafficking, activation, proliferation, and persistence while reducing exhaustion and suppressive signaling.
Figure 6.
Synergistic mechanisms of oncolytic virus and CAR-T cell combination strategies in solid tumors. Conceptual illustration comparing conventional and engineered oncolytic virus + CAR-T cell combination approaches. (Left) Co-administration of CAR-T cells with unmodified OVs promotes direct tumor debulking through complementary cytolytic mechanisms and induces immunogenic tumor cell death, resulting in the release of tumor- and virus-derived antigens that enhance CAR-T cell activation and infiltration. This inflammatory milieu partially alleviates local immunosuppression and supports antitumor immune responses. (Right) Engineered OVs further amplify therapeutic efficacy by delivering immunomodulatory transgenes, including immune checkpoint inhibitors, cytokines, and chemokines, directly within the tumor microenvironment. Localized expression of these factors enhances CAR-T cell trafficking, activation, proliferation, and persistence while reducing exhaustion and suppressive signaling.
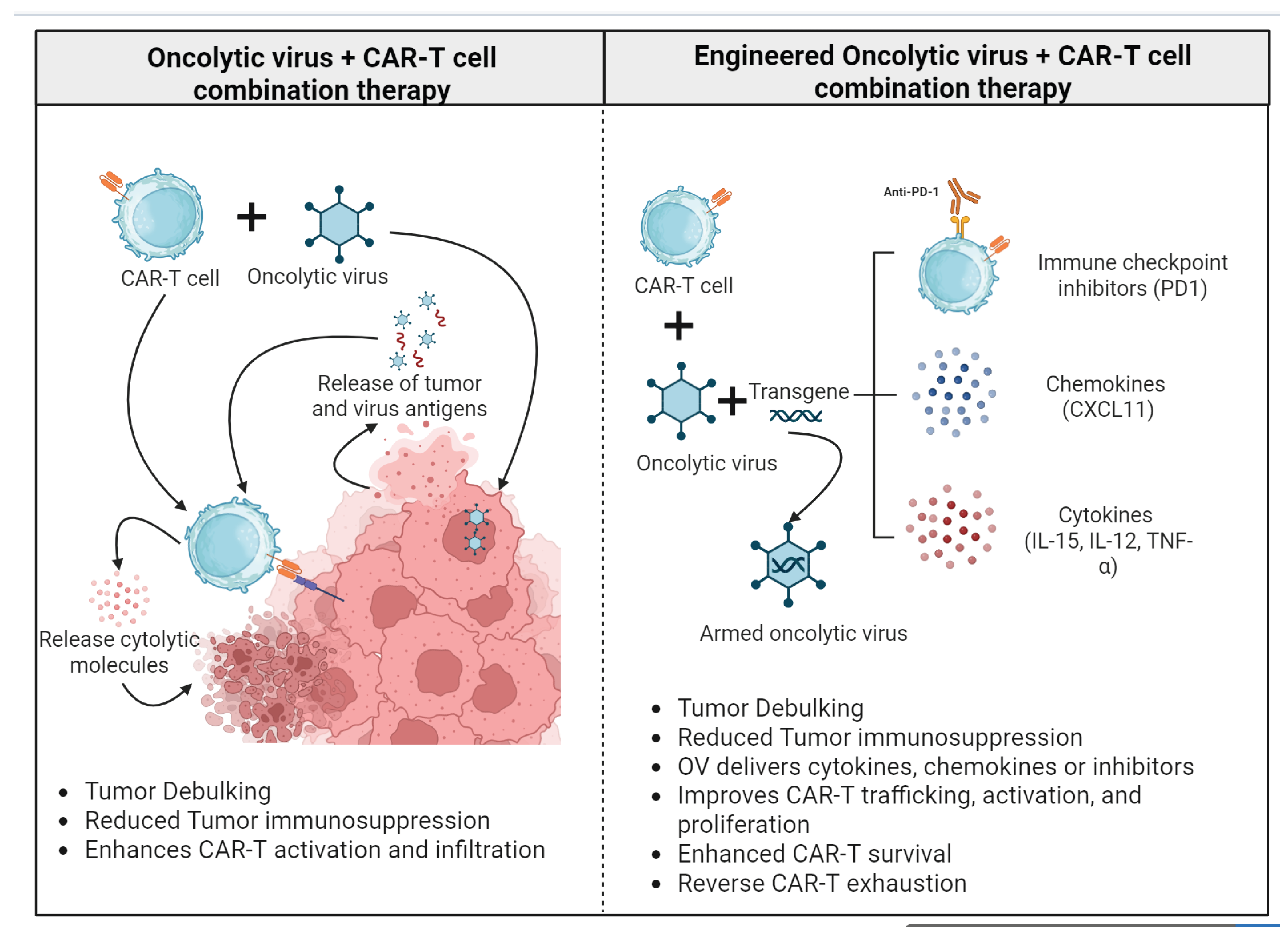

Table 1.
Structural features, functional characteristics, and clinical status of CAR-T cell generations.
Table 1.
Structural features, functional characteristics, and clinical status of CAR-T cell generations.
| CAR Generation | Key Structural Components | Primary Signaling Domains | Functional Advantages | Major Limitations | Clinical Status |
| 1st generation | scFv + hinge/spacer + transmembrane domain | CD3ζ | Antigen-specific T cell activation | Poor proliferation and persistence; limited cytokine production; weak antitumor efficacy | Early clinical trials; no FDA-approved products |
| 2nd generation | scFv + hinge/spacer + transmembrane domain | CD3ζ + one co-stimulatory CD28 or 4-1BB | Enhanced activation, expansion, cytokine secretion, and in vivo persistence | Risk of CRS and neurotoxicity; limited efficacy in solid tumors | FDA-approved products for B-ALL, DLBCL, MM |
| 3rd generation | scFv + hinge/spacer + transmembrane domain | CD3ζ + two co-stimulatory domains (e.g., CD28 + 4-1BB) | Strong signaling; improved resistance to exhaustion in preclinical models | No consistent clinical superiority over the second generation; increased signaling complexity | Clinical trials ongoing; limited adoption |
| 4th generation | scFv + hinge/spacer + transmembrane domain + inducible cytokine cassette | CD3ζ + co-stimulatory molecule + cytokine gene (e.g. IL-12) | Local cytokine release (e.g., IL-12) remodels TME; recruits innate immunity | Risk of systemic inflammation; safety concerns | Primarily preclinical and early clinical development |
| 5th generation | scFv + hinge/spacer + transmembrane domain + cytokine receptor motifs | CD3ζ + co-stimulatory molecule + JAK/STAT signaling domain | Mimics physiological T cell activation; enhanced persistence and antitumor activity | Early-stage development; safety and durability under evaluation | Preclinical and early-phase clinical studies |
B-ALL: B-cell acute lymphoblastic leukemia, DLBCL: Diffuse large B cell lymphoma, MM: Multiple myeloma, JAK/STAT: Janus kinase/signal transducers and activators of transcription.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



