Submitted:
11 February 2026
Posted:
12 February 2026
You are already at the latest version
Abstract
The MADS-box transcription factor (TF) family constitutes a critical class of transcriptional regulators in plants, playing pivotal roles in diverse developmental processes. MIKC-type proteins represent Type II MADS-box TFs that widely function in regulating floral organ development and reproductive growth in plants. In this study, a total of 38 MIKC-type MADS TFs were identified from the sorghum genome, distributed across nine chromosomes. Based on sequence alignments and phylogenetic analysis, these 38 SbMIKC genes (SbMIKCs) were further classified into 10 distinct subfamilies. The expression profiles of these SbMIKCs across multiple tissues revealed five major patterns, among which SbMIKC17 exhibited relatively abundant transcript levels during grain development in sorghum. Further assays confirmed that the protein encoded by SbMIKC17 localizes to the nucleus without self-transactivation activity in yeast. Integrated results from DAP-seq, dual-luciferase assays, and yeast one-hybrid experiments demonstrated that SbMIKC17 binds to and activates the promoter of SbAGPS1, as well as enhancing the promoter activities of SbBt1, SbGBSSI, SbSSIIa, and SbISA1 simultaneously. Collectively, these findings suggest that the MIKC-type MADS gene SbMIKC17 may play a crucial role in starch biosynthesis in sorghum.
Keywords:
sorghum
; starch biosynthesis
; MIKC-type MADS-box
; sranscriptional regulation
; SbMIKC17
1. Introduction
Sorghum (Sorghum bicolor L.) is a versatile cereal crop widely cultivated in arid and semi-arid regions across the globe [1]. With an estimated annual planting area of approximately 40 million hectares and a production output of around 60 million metric tons, it ranks among the world’s top five cereal crops [1,2,3]. Like other cereals, starch is the major component in sorghum grain, constituting approximately 65% to 81% of its dry weight and existing primarily in the form of granules within the endosperm. Its composition significantly influences the physicochemical properties, production, and quality of the grain, thereby determining its diverse applications [3,4,5]. Although recent advances in synthetic biology have constructed cell-free chemoenzymatic starch synthesis from carbon dioxide [6], the vast majority of starch used by humans is still derived from photosynthetic processes in plants, which relies on the participation of a series of functional enzymes [7,8,9,10].
Starch biosynthesis in sorghum and other cereal crops is mediated by a coordinated array of functional enzymes [8,10]. For instance, ADP-glucose pyrophosphorylase (AGPase) catalyzes the synthesis of ADP-glucose (ADPG) and serves as the rate-limiting enzyme in starch production [11,12]. The ADPG synthesized in the cytoplasm relies on the Brittle1 (Bt1) protein for transport into the amyloplast, where it is utilized for the elongation of polyglucan chains [7,13]. Granule-bound starch synthase I (GBSSI) is responsible for the synthesis of amylose [14,15], while various soluble starch synthases (SSSs) catalyze chain elongation across different degrees of polymerization (DP) [10]. Starch branching enzymes (SBEs) facilitate the cleavage of α-1,4-glycosidic bonds and reattach the chain via an α-1,6-glucan linkage to form branch points [10,16]. Debranching enzyme (DBE) is not only involved in the removal of improper branch linkages but also implicated in the initiation of starch granule formation [17,18]. Starch phosphatases (SPs) have also been reported to participate in starch biosynthesis through multiple pathways [19,20]. Beyond the functional enzymes, transcriptional regulation is recognized as a pivotal mechanism governing starch biosynthesis [21,22]. For instance, several TFs, such as OsRSR1 [23], OsbZIP58 [24], OsbHLH144 [25], OsNAC20/26 [26], OsRISBZ1 [27], ZmNAC126/128/130 [28,29], ZmMYB14 [30], ZmbZIP91 [31] , ZmEREB167 [32], TaNAC019 [33], SbNAC22/68 [34,35], have been reported to participate in the regulation of starch biosynthesis [21].
The MADS-box TFs are widely distributed across fungi, animals, and plants [36], named for the four fabulous founder homeotic proteins: MCM1 from Saccharomyces cerevisiae, AGAMOUS from Arabidopsis thaliana, DEFICIENS from Antirrhinum majus, and SRF from Homo sapiens [37]. MADS-box genes encode a highly conserved MADS domain that facilitates DNA binding with the specific recognizing motif of CArG-box located within the cis-regulatory regions of target genes [36,38]. Based on the characteristics of conserved domains, plant-specific MADS-box TFs can be further classified into two types of Type I and Type II [39,40]. Type I proteins contain only the MADS (M) domain, whereas type II proteins exhibit a characteristic MIKC structure, comprising the M, intervening (I), keratin-like (K), and C-terminal domains [39,41,42]. Hence, type II proteins are also designated as MIKC-type MADS-box proteins [41,43], and can be further categorized into MIKCC and MIKC* based on variations within the I domain [44]. In addition, MIKCC-type MADS-box genes have been subdivided into a greater number of subfamilies in flowering plants, and sorghum comprises 15 of those subfamilies [45].
Plant MADS-box genes are central regulators that orchestrate multiple aspects of plant reproductive development, including flowering time, inflorescence architecture, floral organ identity, and seed development [46,47,48,49]. However, the functions of type I MADS-box genes are primarily implicated in plant reproduction such as female gametophyte, embryo, and seed development [50,51,52]. In contrast, MIKC-type MADS-box genes have been extensively characterized across diverse species, including Arabidopsis, rice, and wheat, and are established regulators of key developmental processes, notably floral morphogenesis through the ABCDE model, as well as root, embryo, and seed development and embryogenesis [48,53,54,55]. For instance, three MIKC*-Type MADS-box genes are involved in the process of pollen maturation in both Arabidopsis and rice [56]. SOC1, AGL15, AGL18, AGL24 are important regulatory factors for the floral organ development [48,57,58]. LcSVP2, PlSOC1, and DAM are reported to associate with bud dormancy in evergreen perennial litchi, herbaceous peony, and European plum [59,60,61]. Multiple MIKC*-Type MADS boxes are involved in pollen maturation and tube growth in Arabidopsis and rice [56,62]. Additionally, MADS-box TFs are also involved in root development [63], signal transduction [64], and diverse stress responses [35]. These findings collectively demonstrate that plant MADS-box TFs possess diverse and complex regulatory functions [55,65]. While the profiling of MADS-box genes in sorghum lags far behind that of other crops, with several documented family members and poorly understanding of the potential functions.
MADS TFs have also been demonstrated in both maize and rice to function as master regulators of starch biosynthesis, critically controlling the expression of key genes and metabolic networks involved in this process. For instance, ZmES22 from maize was reported to negatively regulate starch accumulation in rice grains [66], while ZmMADS1a acts as a positive regulator for the transcription of starch synthesis-related genes in maize [67]. The MIKC-type MADS-box protein SOC1 is considered as a prime candidate for genetic engineering aimed at boosting maize yield [68]. In rice, OsMADS14 cooperates with NF-YB1 to directly activate the expression of OsAGPL2 and Waxy during the process of starch synthesis in rice grains [69]. Additionally, MADS29 can interact with NAC25 to modulate cytoplasmic membrane integrity, degeneration, and starch biosynthesis in rice endosperm [70]. Furthermore, the suppressing of OsMADS7 stabilizes amylose content under high-temperature stress in rice endosperm [71]. Beyond rice and maize, the role of MADS-box TFs in governing the transcriptional regulation of genes involved in starch synthesis remains unelucidated in sorghum.
In the present study, we performed a comprehensive analysis using the sorghum genome (Sorghum_bicolor_NCBIv3, GCA_000003195) to elucidate the evolutionary history of MADS-box genes in sorghum and establish a foundation for functional studies of this TF family. A total of 38 MIKC-type MADS-box genes were identified and systematically characterized with respect to their phylogenetic relationships, gene structures, protein motifs, promoter cis-elements, gene duplication events, and chromosomal locations. Furthermore, we analyzed the expression patterns of all MIKC genes, cloned SbMIKC17, which is highly expressed in the endosperm, and provided preliminary evidence that it can regulate the transcription of SbAGPS1, a key gene involved in starch biosynthesis in sorghum grain. These findings provide a theoretical and technical basis for further functional dissection of MADS-box genes in sorghum.
2. Results
2.1. Identification of the MIKC-Type MADS Factors in Sorghum
Referred to the homologous queries of Arabidopsis and maize, a total of 38 MIKC-type MADS genes (SbMIKCs) were ultimately identified in sorghum across nine chromosomes (Chrs) except Chr05 by removing the redundant genes and alternative splicing transcripts (Table 1). The length of proteins encoded by these SbMIKCs ranges from 171 aa (SbMIKC12) to 476 aa (SbMIKC16), with molecular weights varying between 19.302 kDa (SbMIKC12) and 52.794 kDa (SbMIKC16) (Table 1). The isoelectric points of all SbMKKCs span from 4.98 (SbMIKC37) to 9.51 (SbMIKC3) (Table 1). Subcellular localization prediction revealed that all 38 SbMIKCs in sorghum were localized to the nucleus (Table S1).
2.2. Sequence Characterization of SbMIKCs
Neighbor-Joining (NJ) phylogenetic trees were constructed based on sequence alignments of 38 SbMIKCs, along with the full-length sequences of 42 MIKCs from Arabidopsis (AT), 44 from maize (GRMZM), and 37 from rice (LOC Os) (Figure 1A). The analysis revealed that the MIKC-type MADS proteins from these four species clustered into 11 distinct subgroups. Notably, subgroup VI consisted exclusively of Arabidopsis members without those from sorghum, rice, and maize (Figure 1A). Accordingly, MIKC-type MADS-box genes in sorghum can be classified into ten subgroups. Chromosomal localization analysis indicated that the 38 SbMIKCs were predominantly located near telomeric regions, which are characterized by high gene density (Figure 1B). Furthermore, collinearity analysis identified three collinear gene pairs of SbMIKC2 (Chr01)-SbMIKC7 (Chr02), SbMIKC3 (Chr01)-SbMIKC6 (Chr01), and SbMIKC3 (Chr01)-SbMIKC20 (Chr04) (Figure 1B).
Gene structure analysis revealed that SbMIKCs exhibit relatively conserved characteristics (Figure 1C). The results demonstrated that, with the exception of SbMIKC8 that contains only a single exon, other 37 SbMIKCs typically feature multiple intron-exon structures, generally comprising 4 to 11 exons. Exon analysis indicated that SbMIKCs possess a relatively large exon at the 3′ end along the sequence, while the intermediate exons are comparatively smaller (Figure 1C, 1D). Intron analysis further showed that several SbMIKCs contain a relatively large first or third intron (Figure 1D).
The analysis through MEME Suite (v5.5.7) identified 15 conserved motifs among 38 SbMIKCs. Phylogenetic clustering revealed that proteins within each subfamily displayed similar motif distribution patterns. Among these 15 identified motifs, motif 1 was shared by all 38 SbMIKCs, followed by motif 6 and motif 4 that correspondingly shared by 37 and 36 SbMIKCs (Figure 1E). Beyond, motif 2/3/5/7/8 were also observed in more than 10 SbMIKCs, while motif 9 to 15 were only sporadically distributed among a few SbMIKCs (Figure 1E).
Domain prediction using TBtools-II identified three conserved domains among the 38 SbMIKCs proteins: MADS_MFF2_like, K-Box, and MADS super family (Figure 1F). Notably, the MADS_MFF2_like domain was present in 36 SbMIKCs except SbMIKC13 and SbMIKC36 (Figure 1F). In contrast, the MADS super family domain was primarily found in SbMIKC13 and SbMIKC36. Interestingly, SbMIKC36 and SbMIKC8 each contained only a single conserved domain: a MADS super family domain and a MADS_MFF2-like domain, respectively, and the remaining 36 SbMIKCs additionally harbored a conserved K-Box domain (Figure 1F).
2.3. Cis-Element Architecture and Expression of SbMIKCs
To investigate the potential functional and regulatory roles of the SbMIKCs, we conducted a cis-element analysis focused on the 2,000 bp within the promoter regions upstream TSS for all 38 SbMIKCs. A total of 56 distinct cis-elements were identified, which were classified into four functional categories, including phytohormone-responsive elements, growth and development-related elements, light-responsive elements, and stress-responsive elements (Figure 2A, Table S2).
Based on RNA-seq data from multiple sorghum tissues [83], we analyzed the expression patterns of 38 SbMIKCs (Figure 2B). The results showed that SbMIKCs could be classified into five distinct categories of Class I to V (Figure 2B). SbMIKCs in Class I were expressed across all tissues examined, with higher expression levels observed in vegetative organs, such as roots, stems, and leaves compared to floral organs and grains (Figure 2B). Members in Class II were predominantly highly expressed in floral organs and seeds, but transcripts were barely detectable in the endosperm (Figure 2B). Class III genes showed low transcript abundance across all tissues, although a few genes exhibited relatively high expression in floral organs (Figure 2B). Class IV genes exhibited moderate transcript levels in flowers, seeds, and endosperm, while their expression signals were almost undetectable in samples other than those related to inflorescence and seed (Figure 2B). Notably, SbMIKC17 in Class IV possessed relative higher expression level than the other members of this group (Figure 2B). Class V genes were consistently expressed at comparable levels across all tissues (Figure 2B).
According to RNA-seq data, nine SbMIKCs were identified with relatively high transcript abundance and further validated by qRT-PCR across developing sorghum grains (3-25 DAP), as well as floral, stem, root, and leaf tissues. The results revealed that SbMIKC4, 9, 20, 28, 30, and 32 exhibited higher transcript levels in inflorescences and developing grains, whereas SbMIKC21 was more abundant in roots, stems, and leaves. In contrast, SbMIKC14 and SbMIKC34 showed elevated expression during early stages of inflorescence and grain development (Figure 2C).
2.4. Functional Properties Analysis of SbMIKC17
Considering the extreme inflorescence- and seed-specific expression pattern, and relative higher expression levels within these tissues, SbMIKC17 was selected and independently characterized. The results of qRT-PCR revealed that SbMIKC17 was highly expressed within the seed samples at 3 DAP, while detectable transcript levels were also observed in root, stem, leaf, and floral tissues (Figure 3A). During grain development, the relative expression of SbMIKC17 gradually decreased. Nonetheless, its expression remained generally higher in developing grains compared to that in roots and other vegetative tissues throughout various developmental stages (Figure 3A).
The transactivation activity of SbMIKC17 was assessed using the GAL4-based Yeast Two-Hybrid (Y2H) system. The recombinant plasmid pGBKT7-SbMIKC17, along with the negative control pGBKT7 and the positive control pGBKT7-ZmMYB14 [30], were transformed into the yeast strain AH109. Successful transformations were confirmed by growth on nutrient-deficient media and validated by PCR screening. Upon addition of the X-α-Gal substrate, distinct degradation patterns were observed among the sample groups: the positive control clones displayed a clear blue coloration, whereas neither the negative control nor the experimental sample showed any colorimetric reaction. These results indicate that SbMIKC17 does not possess self-activating transactivation activity in yeast (Figure 3B). Furthermore, the subcellular localization of SbMIKC17 was examined in maize leaf protoplasts. The eGFP-tagged SbMIKC17 fusion protein consistently localized to the nucleus, suggesting that SbMIKC17 functions as a nuclear protein (Figure 3C).
2.5. Identification of the Target Gene Regulated by SbMIKC17
To confirm the direct binding sites and downstream target genes of SbMIKC17, a preliminary in vitro analysis was conducted through DAP-seq. A total of 56,996 shared binding sites were identified across two biological replicates (Figure 4A). Among these shared sites, 66.70% of the peaks were located in intergenic regions, while 33.30% were situated within genic regions and their flanking 2 kb regions (Figure 4B). Notably, 9.10% of the peaks were specifically distributed within the 2 kb upstream region of the TSS that correspondingly associated with 7,766 genes (Figure 4B, Table S3).
GO enrichment analysis of the promoter-associated peaks revealed significant enrichments across all three major categories of biological process, cellular component, and molecular function. The most highly enriched terms within each category were terpene biosynthetic process (biological process), CCAAT-binding factor complex (cellular component), and secondary active sulfate transmembrane transporter activity (molecular function) (Figure 4C). KEGG pathway analysis indicated significant enrichment in steroid biosynthesis, sesquiterpenoid and triterpenoid biosynthesis, nucleocytoplasmic transport, and carbon metabolism (Figure 4D). Additionally, starch and sucrose metabolism was identified as another prominently enriched pathway (Figure 4D).
By integrating transcriptomic data, expression pattern analysis was performed on key genes involved in starch and sucrose metabolism that contained binding peaks in their promoter regions. Several genes exhibited relatively high transcript levels during grain development (Figure 4E). Furthermore, based on the accession numbers of key sorghum SBRGs identified in a previous study [72], SbAGPS1 (SORBI_3007G101500), SbISA3 (SORBI_3002G233600), SbSSIIIb (SORBI_3006G221000), and SbSSIIc (SORBI_3001G239500) were confirmed to encode critical enzymes in sorghum starch biosynthesis. Notably, binding sites for SbMIKC17 were detected in the promoter regions of all these genes (Figure 4F). Expression analysis revealed that SbISA3, SbSSIIIb, and SbSSIIc possessed relatively low expression levels in sorghum grains, whereas SbAGPS1 exhibited significantly higher transcript abundance during grain development (Figure 4E). Consequently, SbAGPS1 was identified as a candidate downstream target of SbMIKC17 in the regulation of starch biosynthesis in grains.
2.6. SbMIKC17 Directly Regulates the Transcription of SbAGPS1
Through DAP-seq analysis, a 414 bp peak region was identified within the promoter region of SbAGPS1 (Figure 5A), located between positions 25,706,059 and 25,706,673 on chromosome 7 (Figure 5B). Motif analysis revealed that this region contains 10 predicted motifs (Figure 5C), which might serve as binding sites for SbMIKC17. Meanwhile, yeast one-hybrid assays demonstrated that SbMIKC17 could directly binds to the promoter fragment of SbAGPS1 (Figure 5D). Furthermore, dual-luciferase assays confirmed that SbMIKC17 significantly enhanced the promoter activity of SbAGPS1 (Figure 5E).
2.7. SbMIKC17 Regulates the Promoter Activities of Other Key SBRGs
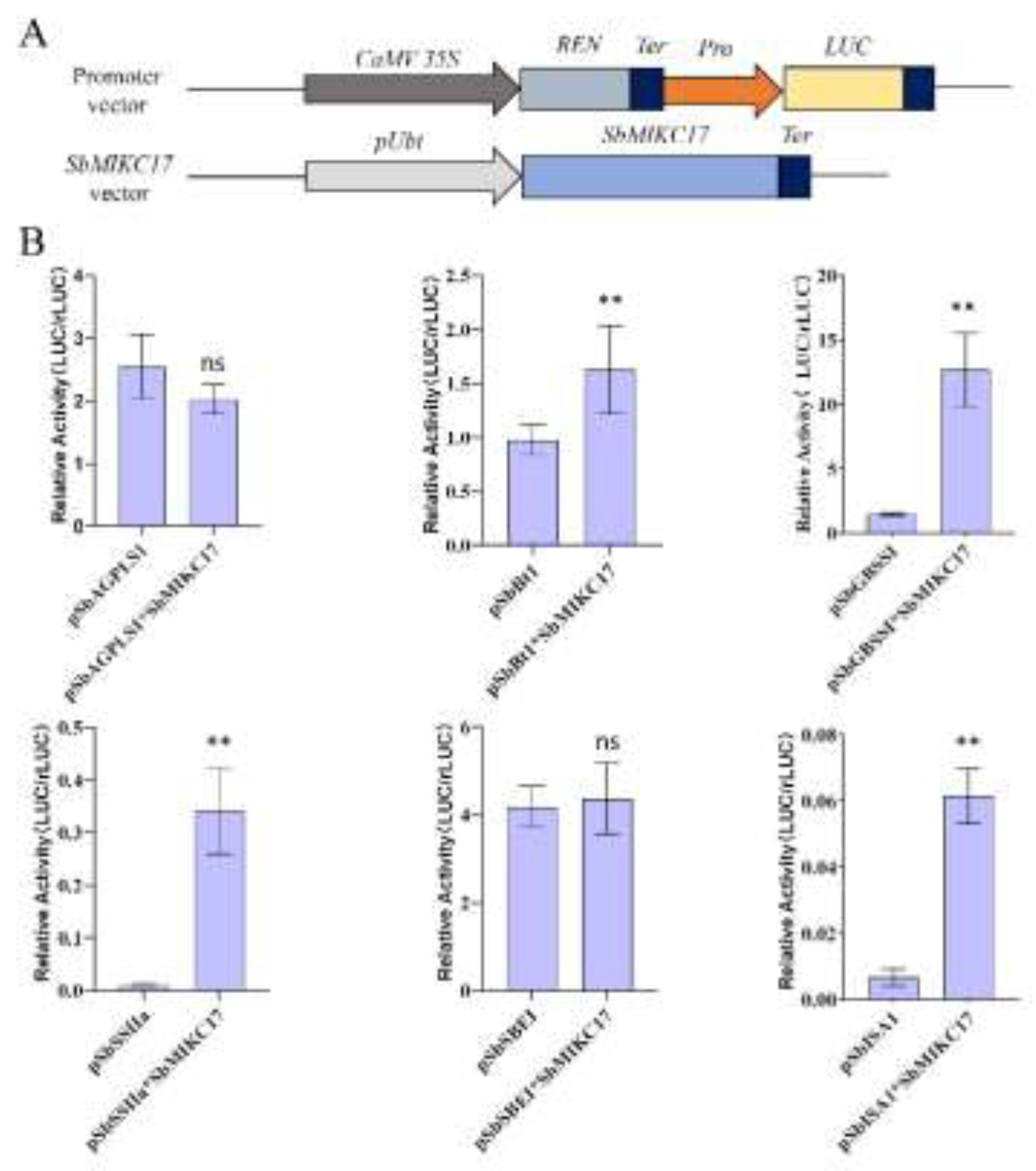
To further investigate the role of SbMIKC17 in the transcriptional regulation of starch synthesis in sorghum grains, we cloned the promoters of six SBRGs, including SbAGPLS1 (1377 bp), SbSSIIa (1917 bp), SbSBEI (1869 bp) 81, SbBt1 (1956 bp), SbGBSSI (1902 bp) [72], and SbISA1 (1320 bp) (Figure 6A). Then co-transformed the pUbi-SbMIKC17 and pGreenII0800-Pro-Luc constructs into maize leaf protoplasts. The activities of Renilla luciferase (rLUC) and firefly luciferase (LUC) were measured, and the LUC/rLUC ratio was calculated to assess whether SbMIKC17 regulates the promoter activity of the target SBRGs. The results demonstrated that SbMIKC17 significantly enhanced the promoter activities of SbBt1, SbGBSSI, SbSSIIa, and SbISA1, but had no significant effect on the promoters of SbAGPLS1 and SbSBEI (Figure 6B). However, the underlying regulatory mechanism requires further investigation.
3. Discussion
The MADS-box TF family represents a crucial class of transcriptional regulators in plants, playing pivotal roles in various developmental processes [46,55,65]. Previous studies have demonstrated that MADS-box TFs are involved in the regulation of multiple stages of plant development, from seed germination to the formation of vegetative organs, from floral to seed development [36,46,47,50,65]. Meanwhile, genome-wide identification of MADS-box family members has been accomplished in several crops, including maize [72,74], rice [47,75], Arabidopsis [75], barley [76], and sorghum [74]. These comprehensive and systematic identifications have laid a solid foundation for elucidating the functional diversity of MADS-box TFs in plants.
MIKC-type members are key components of the MADS-box family and participate extensively in the regulation of plant growth and development [56,59,77,78]. In this study, 38 SbMIKCs were identified as the candidates in the sorghum genome (Table S1). These 38 SbMIKCs formed 11 distinct subclades, differing in details from those of wheat, herbaceous peony [59], and peanut [78]. Proteins encoded by these SbMIKCs possessed family-specific conserved domains and motifs, even exhibited shared spatial distribution and organizational features of the family members from other plants [59,78]. Despite overall conservation, certain variations in gene expression and sequence features were also observed among sorghum MIKC_MADS members, suggesting potential functional divergence and their ability to participate broadly in sorghum development.
The tissue-specific expression patterns of genes are closely associated with their biological functions. In sorghum, expression analysis of SbMIKCs also revealed considerable diversity (Figure 2B). Notably, highly expressed genes in grains and floral organs are likely involved in grain development. For instance, in rice, MADS78 and MADS79, which are highly expressed in floral organs and grains, directly regulate early grain development [79]. Similarly, PlSOC1 from herbaceous peony and LcSVP2 from litchi, which are highly expressed in buds, also play crucial roles in bud dormancy transition [59,61]. Expression analysis of SbMIKCs also revealed that SbMIKC17 and SbMIKC38 exhibit abundant transcript levels in the endosperm, suggesting their potential involvement in metabolic regulation within this tissue (Figure 2B). Meanwhile, various cis-elements were identified in the promoters of SbMIKCs, including light-responsive, gibberellin-responsive, auxin-responsive, salicylic acid-responsive elements and defense/stress-related cis-elements (Figure 2A and Table S2), and similar cis-elements were also identified in the promoters of MIKC genes from other plant species [78,80]. The presence of these cis-elements in MIKC_MADS promoters suggests their potential roles in signal response. For example, the MIKC-type gene MADS31 in rice was reported to be involved in salinity tolerance, and MIKC-type MADS TFs in peanut were documented to be induced by abiotic stress [78,81].
Transcriptional regulation is also an important regulatory mode for starch biosynthesis in cereals, and numerous TFs involved in this pathway were documented in rice, maize, wheat, sorghum, and other crops [21,33,34,35,70]. Among all the reported TFs, MADS-box family members have been implicated in the regulation of starch biosynthesis and yield performances of cereals and other crops, such as rice [66], maize [68,82], blueberry [83]. In this study, preliminary DAP-seq analysis revealed that SbMIKC17 from MIKC-type MADS family could directly bind to the promoter region of SbAGPS1, significantly enhancing the promoter activity (Figure 5). Furthermore, SbMIKC17 was also found to influence the promoter activities of several other key sorghum SBRGs (Figure 6). Though the detail mechanisms remain undiscovered, summary of these findings in the present study suggest that SbMIKC17 might serve as potential TF involved in starch biosynthesis in sorghum grains.
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
The sequenced sorghum line of BTx623 used in this study was cultivated under general irrigation and fertilization practices on the college farm land of Xiema, Beibei (29° 48′ N, 106° 27′ E, Chongqing, China). Five distinct tissues of root, stem, leaf, inflorescence, and grains at 3, 5, 6, 9, 12, 15, 20, 25, and 30 days after pollination (DAP) were sampled across the plant’s life cycle. For each tissue, three biological replicates were collected, rapidly frozen in liquid nitrogen, and stored at -80℃ for subsequent analysis.
4.2. Identification of MIKC-Type TFs from the Sorghum Genome
To identify putative MIKC-type TFs in the sorghum genome, a conserved domain analysis was performed using the Pfam MADS domain profile (IPR002100) to screen the Sorghum bicolor reference genome (v3.1.1). Candidate genes were retrieved via blastp searches against NCBI (https://www.ncbi.nlm.nih.gov/) and Gramene (https://www.gramene.org/) databases using Arabidopsis and maize MIKC-type sequences as queries. Putative genes were further validated with SMART (https://smart.embl.de/smart/set_mode.cgi?NORMAL=1) and NCBI-CDD (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi), after which incomplete or redundant sequences were removed through alignment.
4.3. Bioinformatic Analysis of Sorghum MIKC-Type TFs
To elucidate the evolutionary relationships among sorghum MIKC-type TFs, a multiple sequence alignment was initially conducted using MUSCLE (v3.8.425). Neighbor-joining (NJ) phylogenetic trees were subsequently constructed in MEGA 11.0 with 1000 bootstrap replicates, employing the Poisson model and pairwise deletion of gaps [85]. Segmental and tandem duplication events were identified using MCScanX, and syntenic relationships were visualized with TBtools-II [84]. Genomic annotation data were analyzed through TBtools-II to characterize the exon-intron structures of sorghum MIKC-type genes (SbMIKCs) [84]. Conserved motifs in sorghum MIKC-type TFs were identified through the online MEME suite (http://meme.ebi.edu.au/meme/intro.html), and their distributions were visualized with TBtools-II [84]. The 2000 bp genomic sequences upstream the transcription start site (TSS) of all SbMIKCs were retrieved from the reference genome using TBtools-II [84]. Putative cis-elements in these promoter regions were predicted with the online tool PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html) and visualized using TBtools-II [84].
4.4. Transcriptome Data Analysis
4.5. RNA Extraction and Quantitative Real-Time PCR
Total RNA was isolated from various tissues with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and treated with DNase I (Invitrogen, Carlsbad, CA, USA) to eliminate genomic DNA contamination. First-strand cDNA was synthesized from 1.5 µg of total RNA using the PrimeScript™ RT Reagent Kit (TaKaRa, Kusatsu, Japan) according to the manufacturer’s protocol. Quantitative real-time PCR (qRT-PCR) was carried out in a 10 µL reaction mixture containing 1 µL of cDNA, using the Bio-Rad CFX96 Real-Time System. Sorghum bicolor eukaryotic translation initiation factor 4α (SbEif4α) was employed as an internal reference gene, and relative expression levels were calculated using the 2^(–∆∆CT) method normalized to SbEif4α expression [72]. All primers used are provided in Table S4.
4.6. Gene Cloning and Vector Constructions
KOD high-fidelity DNA polymerase (Toyobo, Osaka, Japan) was used to clone the SbMIKC17, and the PCR products were subsequently cloned into the pMD-19T vector (TaKaRa, Dalian, China). The cloned gene sequence was further confirmed by DNA sequencing. The primers used for gene cloning are listed in Table S4. The pBI221 plant expression vector was modified by replacing the original CaMV 35S promoter with the Ubiquitin (Ubi) promoter for transient overexpression assays. SbMIKC17 was subcloned into this modified pBI221 vector under the control of the Ubi promoter using BamHI and SacI restriction sites incorporated into the PCR primers to enable directional cloning. The vector pCAMBIA2300-35S-eGFP was employed for subcellular localization studies. SbMIKC17 was amplified via PCR using primers designed containing the restriction sites of BamHI and XbaI without the termination codon. The amplified product was subsequently cloned into the pCAMBIA2300-35S-eGFP vector to generate a C-terminal fusion with enhanced green fluorescent protein (eGFP). A GAL4-based yeast two-hybrid system was employed to examine the self-activation activity of SbMIKC17 in yeast. SbMIKC17 was subcloned into the pGBKT7 vector using primers containing the restriction sites of NdeI and BamHI. All primers used for vector construction are listed in Table S4. All constructs were generated using the ClonExpress® MultiS One Step Cloning Kit (Vazyme, Nanjing, China).
4.7. Functional Property Analysis of SbMIKC17
The subcellular localization of SbMIKC17 was analyzed in maize leaf protoplasts. The recombinant plasmid pCAMBIA2300-35S-SbMIKC17-eGFP was introduced into protoplasts through polyethylene glycol (PEG)-mediated transformation with Ca2+ co-treatment. Then the protoplasts were incubated in darkness for 16 hours to allow transgene expression prior to fluorescence imaging. Fluorescence signals were acquired using an LSM 800 confocal microscope (Zeiss, Jena, Germany) equipped with 488 nm blue light excitation.
To assess the transactivation activity of SbMIKC17, the recombinant plasmid pGBKT7-SbMIKC17 was transformed into the yeast strain AH109. Individual transformants were cultured in 2 mL of SD/-Trp liquid medium and grown to logarithmic phase (OD600 = 0.6-0.8) with shaking at 150 rpm. For transcriptional activation assays, the cultures were spotted onto quadruple-dropout medium (SD/-Trp-His-Ade-Leu) supplemented with 24 µg/mL X-α-Gal, followed by incubation at 28℃ in darkness for three days. In addition, the recombinant plasmids pGBKT7-SbMIKC17 and pGADT7-T were co-transformed into the yeast strain Y2H using the Super Yeast Competent Cell Preparation and Transformation Kit Plus (Coolaber, Beijing, China) to evaluate the transactivation activity of SbMIKC17. The plasmid combinations pGADT7-T with pGBKT7-53 and pGADT7-T with pGBKT7-lam were used as the positive and negative controls, respectively. Individual transformants were cultured in 2 mL of SD/-Leu/-Trp liquid medium and grown to logarithmic phase (OD600 = 0.6-0.8) with shaking at 150 rpm. For transcriptional activation assays, the cultures were spotted onto quadruple-dropout medium (SD/-Trp-His-Ade-Leu) supplemented with 24 µg/mL X-α-Gal, followed by incubation at 28℃ in darkness for three days.
4.8. DAP-Seq Library Preparation and Data Analysis
DAP-seq (DNA affinity purification sequencing) was performed according to Bartlett et al. (2017) and Cao et al. (2020). Recombinant SbMIKC17 protein fused to a HaloTag was heterologously expressed using the TnT SP6 High-Yield Wheat Germ Protein Expression System (Promega, L3260) and subsequently affinity-purified with Magne HaloTag Beads (Promega, G7281) in accordance with the manufacturer’s instructions. Genomic DNA (gDNA) library was prepared using the NEBNext® Ultra™ II DNA Library Prep Master Mix Set for Illumina® (NEB, #E6040S/L). High-molecular-weight genomic DNA was extracted via phenol-chloroform purification and ethanol precipitation, then mechanically sheared into fragments ranging from 200 to 500 bp. Illumina-compatible paired-end adapters were ligated to the fragmented DNA.
For the binding reaction, Magne HaloTag Beads bound with SbMIKC1-HaloTag were incubated with 500 ng of the gDNA library in 40 µL of PBS buffer under slow rotation at 16℃ for 2 hours. The beads were then washed five times with 200 µL of PBS containing 0.005% (v/v) NP-40. Bound DNA was eluted in 25 µL of elution buffer (EB) by incubation at 98℃ for 10 minutes. The optimal concentration of the DAP-seq library for sequencing was determined based on the fragment size distribution. As a negative control, mock DAP-seq libraries were generated in parallel using the same procedure without the addition of recombinant protein.
Raw DAP-seq data were processed with Trimmomatic v0.36 to remove low-quality reads and adapter sequences. The resulting high-quality reads were aligned to the Sorghum bicolor reference genome (NVBIv3; Assembly: GCA_000003195.3) using BWA v0.7.12-r1039. Peak calling was performed with MACS2 v2.2.7.1, allowing duplicate reads and applying a q-value threshold of < 0.05 [86]. Target genes were defined as those harboring DAP-seq peaks within transcribed regions, 2 kb upstream of the transcription start site (TSS), 5′ UTR, exons, introns, 3′ UTR, or 2 kb downstream the TTS. The distribution of reads across gene bodies and flanking regions was analyzed using Deeptools v3.5.0. IGV_2.17.2 was employed for the visualization of peaks from DAP-seq data.
Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs) was conducted with GOATOOLS v0.9.9. P-values were adjusted using the false discovery rate (FDR) method, with an FDR ≤ 0.05 considered statistically significant. KEGG pathway enrichment was performed using KOBAS 3.0, with the Benjamini-Hochberg (BH) correction applied and a significance threshold of FDR ≤ 0.05. De novo motif discovery was carried out with MEME v5.0.5.
4.9. Dual-Luciferase Reporting Assay
Dual-luciferase assays were conducted in maize leaf protoplasts using the pGreenII0800-LUC vector system, following a previously described method [72]. The coding sequence (CDS) of SbMIKC17 was cloned into a modified plant expression vector under the control of the Ubi promoter. The promoter regions of sorghum starch biosynthesis related genes (SBRGs) were inserted into the pGreenII0800-LUC vector to drive the expression of the firefly luciferase (LUC) reporter gene. Plasmid constructs were co-transformed into maize protoplasts at predetermined ratios and incubated as described by Xiao et al. [29,72]. Luminescence signals from Renilla luciferase (REN) and firefly luciferase (LUC) were measured using the Dual Luciferase Reporter Gene Assay Kit (YEASEN, Shanghai, China) and quantified with a GloMax® 2020 luminometer (Thermo Fisher Scientific, Waltham, MA, USA). The Renilla luciferase activity was used as internal control, and the relative promoter activity was expressed as the ratio of LUC to REN luminescence. All experiments were independently repeated three independent biological replicates, and each replicate consisted of three technical replicates. The effect of SbMIKC17 on the promoter activity of SBRGs was assessed using Student’s t-test.
4.10. Yeast One-Hybrid Assay
The yeast one-hybrid assay was employed to examine the binding of SnMIKC17 to the promoter region of SbAGPS1 (pSbAGPS1). A 1956 bp fragment of pSbAGPS1 was amplified with primers containing restriction sites of EcoRI and MluI (Table S4) and subsequently subcloned into the yeast expression vector pHIS2 to form the pSbAGPS1-pHis2 plasmid. Meanwhile, the coding sequence of SbMIKC17 was amplified using primers SbMIKC17-Rec2F (sense) and SbMIKC17-Rec2R (antisense), which were designed with restriction sites of EcoRI and SacI (Table S4), respectively. The resulting product was cloned into the pGADT7-Rec2 vector to generate the pGADT7-Rec2-SbMIKC17 construct, which was then utilized in the yeast one-hybrid assays. The pGADT7-Rec2-SbMIKC17 with pSbAGPS1-pHis2 was co-transformed into the yeast strain Y187 by using the Super Yeast Competent Cell Preparation and Transformation Kit Plus (Coolaber, Beijing, China) as the experiment group, and the combinations of pGADT7-SbMIKC17 and pHis2 were used as negative controls.
5. Conclusions
A total of 38 MIKC-type MADS genes were identified in the sorghum genome, distributed across nine chromosomes and classified into ten distinct subfamilies. Promoter analysis revealed the presence of multiple cis-elements within the promoter regions of these SbMIKCs, and all genes exhibited five distinct expression patterns, among which SbMIKC17 showed inflorescence- and seed-specific expression pattern, and exhibited relatively higher transcript levels during grain development and in the endosperm. SbMIKC17 encodes a nuclear-localized protein without self-transactivation activity. Furthermore, SbMIKC17 could directly bind to the promoter of SbAGPS1 and significantly enhance its activity. Similar functions were also observed for some other sorghum SBRGs, including SbBt1, SbGBSSI, SbSSIIa, and SbISA1. Collectively, these results suggest that SbMIKC17 might possess critical role in regulating starch biosynthesis in sorghum grains.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Gene information and physicochemical properties of sorghum MIKC_MADS subfamily; Table S2: Cis-elements in the 2000-bp promoter sequence of the sorghum MIKC gene via PlantCARE; Table S3: Gene information associated with the SbMIKC17 peaks identified via DAP-seq; Table S4: All the primer sequence information used in this study.
Author Contributions
Conceptualization, Q.X. and Z.L.; methodology, Q.X. and Z.L.; software, H.M., M.H. and Y.L.; validation, J.Z., Z.Y., A.S. and X.G.; formal analysis, Q.X. and L.Z.; investigation, J.Z. and Z.L.; resources, Q.X. and L.Z.; data curation, H.M., M.H. and Y.L.; writing—original draft preparation, Q.X. and J.Z.; writing—review and editing, L.Z.; visualization, L.Z. and Q.X.; supervision, Q.X.; project administration, Q.X. and L.Z.; funding acquisition, Q.X. and L.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (32372076 to Q.X., 32401747 to L.Z.).
Data Availability Statement
The sequencing data can be downloaded at: https://www.ncbi.nlm.nih.gov/sra/PRJNA1416650. Other data are contained within the present article. All other relevant data are included within the article and its supplementary information Files.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Xin, Z.; Wang, M.; Cuevas, H.E.; Chen, J.; Harrison, M.; Pugh, N.A.; Morris, G. Sorghum genetic, genomic, and breeding resources. Planta 2021, 254, 114. [Google Scholar] [CrossRef]
- Hao, H.; Li, Z.; Leng, C.; Lu, C.; Luo, H.; Liu, Y.; Wu, X.; Liu, Z.; Shang, L.; Jing, H.C. Sorghum breeding in the genomic era: opportunities and challenges. Theor Appl Genet 2021, 134, 1899–1924. [Google Scholar] [CrossRef]
- Zahid, M.K.; Ahmad, D.; Amin, R.; Bao, J. Sorghum starch: Composition, structure, functionality, and strategies for its improvement. Compr Rev Food Sci Food Saf 2025, 24, e70101. [Google Scholar] [CrossRef]
- Khoddami, A.; Messina, V.; Vadabalija Venkata, K.; Farahnaky, A.; Blanchard, C.L.; Roberts, T.H. Sorghum in foods: Functionality and potential in innovative products. Crit Rev Food Sci Nutr 2023, 63, 1170–1186. [Google Scholar] [CrossRef]
- Haziman, M.L.; Ishaq, M.I.; Qonit, M.A.H.; Lestari, E.G.; Susilawati, P.N.; Widarsih, W.; Syukur, C.; Herawati, H.; Arief, R.; Santosa, B.; Purba, R.; Andoyo, R.; Yursak, Z.; Tan, S.S.; Musfal, M.; Mubarok, S. Sorghum starch review: Structural properties, interactions with proteins and polyphenols, and modification of physicochemical properties. Food Chem 2025, 463 Pt 1, 139810. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Sun, H.; Qiao, J.; Zhu, L.; Zhang, F.; Zhang, J.; Tang, Z.; Wei, X.; Yang, J.; Yuan, Q.; Wang, W.; Yang, X.; Chu, H.; Wang, Q.; You, C.; Ma, H.; Sun, Y.; Li, Y.; Li, C.; Jiang, H.; Wang, Q.; Ma, Y. Cell-free chemoenzymatic starch synthesis from carbon dioxide. Science 2021, 373, 1523–1527. [Google Scholar] [CrossRef]
- Bahaji, A.; Li, J.; Sánchez-López, Á.M.; Baroja-Fernández, E.; Muñoz, F.J.; Ovecka, M.; Almagro, G.; Montero, M.; Ezquer, I.; Etxeberria, E. Starch biosynthesis, its regulation and biotechnological approaches to improve crop yields. Biotechnol Advances 2014, 32, 87–106. [Google Scholar] [CrossRef]
- Zeeman, S.C.; Kossmann, J.; Smith, A.M. Starch: its metabolism, evolution, and biotechnological modification in plants. Annual Review in Plant Biology 2010, 61, 209–234. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.L.; Myers, A.M. Biochemistry and genetics of starch synthesis. Annual Review of Food Science and Technology 2010, 1, 271–303. [Google Scholar] [CrossRef]
- Jeon, J.S.; Ryoo, N.; Hahn, T.R.; Walia, H.; Nakamura, Y. Starch biosynthesis in cereal endosperm. Plant Physiology and Biochemistry 2010, 48, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Giroux, M.J.; Hannah, L.C. ADP-glucose pyrophosphorylase in shrunken-2 and brittle-2 mutants of maize. Mol Gen Genet 1994, 243, 400–408. [Google Scholar] [CrossRef]
- Stark, D.M.; Timmerman, K.P.; Barry, G.F.; Preiss, J.; Kishore, G.M. Regulation of the amount of starch in plant tissues by ADP Glucose pyrophosphorylase. Science 1992, 258, 287–292. [Google Scholar] [CrossRef]
- Shannon, J.C.; Pien, F.M.; Cao, H.; Liu, K.C. Brittle-1, an adenylate translocator, facilitates transfer of extraplastidial synthesized ADP-glucose into amyloplasts of maize endosperms. Plant Physiology 1998, 117, 1235–1252. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Zheng, F.Q.; Shen, G.Z.; Gao, J.P.; Snustad, D.P.; Li, M.G.; Zhang, J.L.; Hong, M.M. The amylose content in rice endosperm is related to the post-transcriptional regulation of the waxy gene. Plant J 1995, 7, 613–622. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamamori, M.; Hirano, H.; Hidaka, S.; Nagamine, T. Production of Waxy (Amylose-Free) Wheats. Mol Gen Genet 1995, 248, 253–259. [Google Scholar] [CrossRef]
- Seo, B.S.; Kim, S.; Scott, M.P.; Singletary, G.W.; Wong, K.S.; James, M.G.; Myers, A.M. Functional interactions between heterologously expressed starch-branching enzymes of maize and the glycogen synthases of Brewer’s yeast. Plant Physiol 2002, 128, 1189–1199. [Google Scholar] [CrossRef]
- Hussain, H.; Mant, A.; Seale, R.; Zeeman, S.; Hinchliffe, E.; Edwards, A.; Hylton, C.; Bornemann, S.; Smith, A.M.; Martin, C. Three isoforms of isoamylase contribute different catalytic properties for the debranching of potato glucans. The Plant Cell 2003, 15, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Sim, L.; Beeren, S.R.; Findinier, J.; Dauvillee, D.; Ball, S.G.; Henriksen, A.; Palcic, M.M. Crystal structure of the Chlamydomonas starch debranching enzyme isoamylase ISA1 reveals insights into the mechanism of branch trimming and complex assembly. J Biol Chem 2014, 289, 22991–23003. [Google Scholar] [CrossRef] [PubMed]
- Orawetz, T.; Malinova, I.; Orzechowski, S.; Fettke, J. Reduction of the plastidial phosphorylase in potato (Solanum tuberosum L.) reveals impact on storage starch structure during growth at low temperature. Plant Physiol Biochem 2016, 100, 141–149. [Google Scholar] [CrossRef]
- Subasinghe, R.M.; Liu, F.; Polack, U.C.; Lee, E.A.; Emes, M.J.; Tetlow, I.J. Multimeric states of starch phosphorylase determine protein-protein interactions with starch biosynthetic enzymes in amyloplasts. Plant Physiol Biochem 2014, 83, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tan, H.; Zhang, C.; Li, Q.; Liu, Q. Starch biosynthesis in cereal endosperms: An updated review over the last decade. Plant Commun 2021, 2, 100237. [Google Scholar] [CrossRef]
- Ahmad, D.; Ying, Y.; Bao, J. Understanding starch biosynthesis in potatoes for metabolic engineering to improve starch quality: A detailed review. Carbohydr Polym 2024, 346, 122592. [Google Scholar] [CrossRef]
- Fu, F.; Xue, H. Coexpression analysis identifies Rice Starch Regulator1, a rice AP2/EREBP family transcription factor, as a novel rice starch biosynthesis regulator. Plant Physiology 2010, 154, 927–938. [Google Scholar] [CrossRef]
- Wang, J.; Xu, H.; Zhu, Y.; Liu, Q.; Cai, X. OsbZIP58, a basic leucine zipper transcription factor, regulates starch biosynthesis in rice endosperm. Journal of Experimental Botany 2013, 64, 3453–3466. [Google Scholar] [CrossRef] [PubMed]
- Bello, B.K.; Hou, Y.; Zhao, J.; Jiao, G.; Jian, Z. NF-YB1-YC12-bHLH144 complex directly activates Wx to regulate grain quality in rice (Oryza sativa L.). Plant Biotechnol J 2019, 17, 1222–1235. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Z.; Zhang, Q.; Meng, S.; Wei, C. The NAC transcription factors OsNAC20 and OsNAC26 regulate starch and storage protein synthesis. Plant physiol 2020, 184, 1775–1791. [Google Scholar] [CrossRef]
- Sun, Q.; Duan, E.; Jing, R.; Ren, Y.; Xu, H.; Gu, C.; Lv, W.; Jiang, X.; Chen, R.; Wang, Q.; Zhang, Y.; Zhang, R.; Xu, H.; Zhang, Y.; Chi, J.; Fu, Y.; Zhu, Y.; Zhang, Y.; Zhang, B.; Teng, X.; Dong, H.; Yang, X.; Zhou, L.; Tian, Y.; Liu, X.; Liu, S.; Guo, X.; Lei, C.; Jiang, L.; Wang, Y.; Wan, J. The bZIP transcription factor RISBZ1 balances grain filling and ER stress response in rice grains. Plant Commun 2025, 6, 101458. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, J.; Ji, C.; Wu, Y.; Messing, J. NAC-type transcription factors regulate accumulation of starch and protein in maize seeds. Proc Natl Acad Sci U S A 2019, 116, 11223–11228. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Wang, Y.; Li, H.; Zhang, C.; Wei, B.; Wang, Y.; Huang, H.; Li, Y.; Yu, G.; Liu, H.; Zhang, J.; Liu, Y.; Hu, Y.; Huang, Y. Transcription factor ZmNAC126 plays an important role in transcriptional regulation of maize starch synthesis-related genes. Crop Journal 2021, 9, 192–203. [Google Scholar] [CrossRef]
- Xiao, Q.; Wang, Y.; Du, J.; Li, H.; Wei, B.; Wang, Y.; Li, Y.; Yu, G.; Liu, H.; Zhang, J.; Liu, Y.; Hu, Y.; Huang, Y. ZmMYB14 is an important transcription factor involved in the regulation of the activity of the ZmBT1 promoter in starch biosynthesis in maize. Febs J 2017, 284, 3079–3099. [Google Scholar] [CrossRef]
- Chen, J.; Yi, Q.; Cao, Y.; Wei, B.; Zheng, L.; Xiao, Q.; Xie, Y.; Gu, Y.; Li, Y.; Huang, H.; Wang, Y.; Hou, X.; Long, T.; Zhang, J.; Liu, H.; Liu, Y.; Yu, G.; Huang, Y. ZmbZIP91 regulates expression of starch synthesis-related genes by binding to ACTCAT elements in their promoters. J Exp Bot 2016, 67, 1327–1338. [Google Scholar] [CrossRef]
- Qing, X.; Li, J.; Lin, Z.; Wang, W.; Yi, F.; Chen, J.; Liu, Q.; Song, W.; Lai, J.; Chen, B.; Zhao, H.; Yang, Z. Maize transcription factor ZmEREB167 negatively regulates starch accumulation and kernel size. Journal of Genetics and Genomics 2025, 52, 411–421. [Google Scholar] [CrossRef]
- Gao, Y.; An, K.; Guo, W.; Chen, Y.; Zhang, R.; Zhang, X.; Chang, S.; Vincenzo, R.; Jin, F.; Cao, X.; Xin, M.; Peng, H.; Hu, Z.; Guo, W.; Du, J.; Ni, Z.; Sun, Q.; Yao, Y. The endosperm-specific transcription factor TaNAC019 regulates glutenin and starch accumulation and its elite allele improves wheat grain quality. Plant Cell 2021, 33, 603–622. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gong, X.; Li, J.; Duan, H.; Dai, T.; Wei, W.; Yin, M.; Zheng, Y.; Gul, H.; Wang, J.; Liu, C.; Xiao, Q. Discovering of the transcriptional regulatory network involved in starch biosynthesis in sorghum grains. Plant Diversity 2025. [Google Scholar] [CrossRef]
- Yin, M.; Zheng, Y.; Zeng, H.; Yan, Z.; Sun, A.; Ma, K.; Gong, X.; Li, J.; Xiao, Q.; Liu, Z. SbNAC22 coordinates starch biosynthesis in sorghum grain by regulating pathway related functional genes. Journal of Integrative Agriculture 2025. [Google Scholar] [CrossRef]
- Shore, P.; Sharrocks, A.D. The MADS-Box Family of Transcription Factors. European Journal of Biochemistry 1995, 229, 1–13. [Google Scholar] [CrossRef]
- Adhikari, P.B.; Kasahara, R.D. An Overview on MADS Box Members in Plants: A Meta-Review. Int J Mol Sci 2024, 25, 8233. [Google Scholar] [CrossRef]
- Tilly, J.J.; Allen, D.W.; Jack, T. The CArG boxes in the promoter of the Arabidopsis floral organ identity gene APETALA3 mediate diverse regulatory effects. Development 1998, 125, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Ribas de Pouplana, L.; Martínez-Castilla, L.; Yanofsky, M.F. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc Natl Acad Sci U S A 2000, 97, 5328–5333. [Google Scholar] [CrossRef] [PubMed]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol 2010, 11, 216. [Google Scholar] [CrossRef]
- Smaczniak, C.; Immink, R.G.H.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef]
- Han, J.P.; Wan, J.N.; Guan, Z.L.; Xu, H.; Wang, Q.F.; Wan, T. The origin, evolution, and diversification of MADS-box transcription factors in green plants. Plant Commun 2025, 6, 101462. [Google Scholar] [CrossRef]
- Kaufmann, K.; Melzer, R.; Theißen, G. MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene 2005, 347, 183–198. [Google Scholar] [CrossRef]
- Henschel, K.; Kofuji, R.; Hasebe, M.; Saedler, H.; Munster, T.; Thei?En, G. Two Ancient Classes of MIKC-type MADS-box Genes are Present in the Moss Physcomitrella patens. Molecular Biology Evolution 2002, 19, 801–814. [Google Scholar] [CrossRef]
- Gramzow, L.; Theissen, G. Phylogenomics reveals surprising sets of essential and dispensable clades of MIKC(c)-group MADS-box genes in flowering plants. J Exp Zool B Mol Dev Evol 2015, 324, 353–362. [Google Scholar] [CrossRef]
- Schilling, S.; Pan, S.; Kennedy, A.; Melzer, R. MADS-box genes and crop domestication: the jack of all traits. J Exp Bot 2018, 69, 1447–1469. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Hirano, H.Y. Function and diversification of MADS-box genes in rice. ScientificWorldJournal 2006, 6, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wu, Y.; Li, R.; Cao, H.; Li, Z.; Li, Q.; Zhou, L. Research Progress on the Regulation of Plant Floral Organ Development by the MADS-box Gene Family. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef]
- Colombo, M.; Masiero, S.; Vanzulli, S.; Lardelli, P.; Kater, M.M.; Colombo, L. AGL23, a type I MADS-box gene that controls female gametophyte and embryo development in Arabidopsis. Plant J 2008, 54, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Bemer, M.; Heijmans, K.; Airoldi, C.; Davies, B.; Angenent, G.C. An atlas of type I MADS box gene expression during female gametophyte and seed development in Arabidopsis. Plant Physiol 2010, 154, 287–300. [Google Scholar] [CrossRef]
- Masiero, S.; Colombo, L.; Grini, P.E.; Schnittger, A.; Kater, M.M. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 2011, 23, 865–872. [Google Scholar] [CrossRef]
- Bente, H.; Kohler, C. Molecular basis and evolutionary drivers of endosperm-based hybridization barriers. Plant Physiol 2024, 195, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Malabarba, J.; Buffon, V.; Mariath, J.E.A.; Gaeta, M.L.; Dornelas, M.C.; Margis-Pinheiro, M.; Pasquali, G.; Revers, L.F. The MADS-box gene Agamous-like 11 is essential for seed morphogenesis in grapevine. J Exp Bot 2017, 68, 1493–1506. [Google Scholar] [CrossRef]
- Silva, C.S.; Sriharsha, P.; Adam, R.; Martha, B.; Agnès, J.; François, P.; Veronique, H.; Chloe, Z. Evolution of the Plant Reproduction Master Regulators LFY and the MADS Transcription Factors: The Role of Protein Structure in the Evolutionary Development of the Flower. Frontiers in Plant Science 2016, 6, 1193. [Google Scholar] [CrossRef]
- Tripathi, A.; Vishwakarma, K.; Tripathi, S.; Jadaun, J.S.; Nayak, A.K. Utilization of MADS-Box genes for agricultural advancement: current insights and future prospects. Mol Biol Rep 2025, 53, 20. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, S.; Wu, F.; Yan, S.; Lin, X.; Du, X.; Chong, K.; Schilling, S.; Theissen, G.; Meng, Z. Functional conservation of MIKC*-Type MADS box genes in Arabidopsis and rice pollen maturation. Plant Cell 2013, 25, 1288–1303. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J Exp Bot 2010, 61, 2247–2254. [Google Scholar] [CrossRef]
- Adamczyk, B.J.; Lehti-Shiu, M.D.; Fernandez, D.E. The MADS domain factors AGL15 and AGL18 act redundantly as repressors of the floral transition in Arabidopsis. Plant J 2007, 50, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chen, X.; Zhong, S.; Wu, S.; Guo, J.; Wang, Q.; Li, J.; Li, D.; Xia, Y.; Zhang, J.; Wang, X. MIKC-Type MADS-Box Gene Analysis Reveals the Role of PlSOC1 in Bud Dormancy Transition in Herbaceous Peony. Plants (Basel) 2025, 14, 928. [Google Scholar] [CrossRef]
- Quesada-Traver, C.; Guerrero, B.I.; Badenes, M.L.; Rodrigo, J.; Rios, G.; Lloret, A. Structure and Expression of Bud Dormancy-Associated MADS-Box Genes (DAM) in European Plum. Front Plant Sci 2020, 11, 1288. [Google Scholar] [CrossRef]
- Ma, M.M.; Zhang, H.F.; Tian, Q.; Wang, H.C.; Zhang, F.Y.; Tian, X.; Zeng, R.F.; Huang, X.M. MIKC type MADS-box transcription factor LcSVP2 is involved in dormancy regulation of the terminal buds in evergreen perennial litchi (Litchi chinensis Sonn.). Hortic Res 2024, 11, uhae150. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.J.; Fernandez, D.E. MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol 2009, 149, 1713–1723. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Garcia-Ponce, B.; Sanchez, M.P.; Espinosa-Soto, C.; Garcia-Gomez, M.L.; Pineyro-Nelson, A.; Garay-Arroyo, A. MADS-box genes underground becoming mainstream: plant root developmental mechanisms. New Phytol 2019, 223, 1143–1158. [Google Scholar] [CrossRef]
- Griebel, T.; Zeier, J. Light regulation and daytime dependency of inducible plant defenses in Arabidopsis: phytochrome signaling controls systemic acquired resistance rather than local defense. Plant Physiol 2008, 147, 790–801. [Google Scholar] [CrossRef]
- Zhang, Z; Zou, W; Lin, P; Wang, Z; Chen, Y; Yang, X; Zhao, W; Zhang, Y; Wang, D; Que, Y; Wu, Q. Evolution and Function of MADS-Box Transcription Factors in Plants. International Journal of Molecular Sciences 2024b, 25. [Google Scholar] [CrossRef] [PubMed]
- Zha, K.; Xie, H.; Ge, M.; Wang, Z.; Wang, Y.; Si, W.; Gu, L. Expression of Maize MADS Transcription Factor ZmES22 Negatively Modulates Starch Accumulation in Rice Endosperm. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Dong, Q.; Wang, F.; Kong, J.; Xu, Q.; Li, T.; Chen, L.; Chen, H.; Jiang, H.; Li, C.; Cheng, B. Functional analysis of ZmMADS1a reveals its role in regulating starch biosynthesis in maize endosperm. Sci Rep 2019, 9, 3253. [Google Scholar] [CrossRef]
- Song, G.Q.; Han, X.; Ryner, J.T.; Thompson, A.; Wang, K. Utilizing MIKC-type MADS-box protein SOC1 for yield potential enhancement in maize. Plant Cell Rep 2021, 40, 1679–1693. [Google Scholar] [CrossRef]
- Feng, T.; Wang, L.; Li, L.; Liu, Y.; Chong, K.; Theissen, G.; Meng, Z. OsMADS14 and NF-YB1 cooperate in the direct activation of OsAGPL2 and Waxy during starch synthesis in rice endosperm. New Phytol 2022, 234, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wu, M.W.; Liu, J.; Xu, X.; Bao, Y.; Liu, C.M. NAC25 transcription factor regulates the degeneration of cytoplasmic membrane integrity and starch biosynthesis in rice endosperm through interacting with MADS29. Front Plant Sci 2025, 16, 1563065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.; Feng, M.; Zhu, Y. Suppression of OsMADS7 in rice endosperm stabilizes amylose content under high temperature stress. Plant Biotechnol J 2018, 16, 18–26. [Google Scholar] [CrossRef]
- Xiao, Q.; Huang, T.; Cao, W.; Ma, K.; Liu, T.; Xing, F.; Ma, Q.; Duan, H.; Ling, M.; Ni, X.; Liu, Z. Profiling of transcriptional regulators associated with starch biosynthesis in sorghum (Bicolor sorghum L.). Frontiers in Plant Science 2022, 13, 999747. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Chen, Z.; Xu, L.; Zhang, L.; Zou, Q. Genome-Wide Analysis of the MADS-Box Gene Family in Maize: Gene Structure, Evolution, and Relationships. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, X.; Chen, W.; Peng, X.; Cheng, X.; Zhu, S.; Cheng, B. Whole-genome survey and characterization of MADS-box gene family in maize and sorghum. Plant Cell, Tissue and Organ Culture (PCTOC) 2010, 105, 159–173. [Google Scholar] [CrossRef]
- Abdullah-Zawawi, M.R.; Ahmad-Nizammuddin, N.F.; Govender, N.; Harun, S.; Mohd-Assaad, N.; Mohamed-Hussein, Z.A. Comparative genome-wide analysis of WRKY, MADS-box and MYB transcription factor families in Arabidopsis and rice. Sci Rep 2021, 11, 19678. [Google Scholar] [CrossRef]
- Wang, F.; Zhou, Z.; Zhu, L.; Gu, Y.; Guo, B.; Lv, C.; Zhu, J.; Xu, R. Genome-wide analysis of the MADS-box gene family involved in salt and waterlogging tolerance in barley (Hordeum vulgare L.). Front Plant Sci 2023, 14, 1178065. [Google Scholar] [CrossRef]
- Zhang, L; Ma, F; Duan, G; Ju, Y; Yu, T; Zhang, Q; Sodmergen. MIKC*-type MADS transcription factors control JINGUBANG expression and the degree of pollen dormancy in Arabidopsis. Plant Physiol 2024a, 197. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Yuan, C.; Sun, Q.; Yan, C.; Zhao, X.; Wang, J.; Wang, Q.; Shan, S.; Li, C. MIKC-type MADS-box transcription factor gene family in peanut: Genome-wide characterization and expression analysis under abiotic stress. Front Plant Sci 2022, 13, 980933. [Google Scholar] [CrossRef]
- Paul, P.; Dhatt, B.K.; Miller, M.; Folsom, J.J.; Wang, Z.; Krassovskaya, I.; Liu, K.; Sandhu, J.; Yu, H.; Zhang, C.; Obata, T.; Staswick, P.; Walia, H. MADS78 and MADS79 Are Essential Regulators of Early Seed Development in Rice. Plant Physiol 2020, 182, 933–948. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, T.; Li, Y.; Hou, J.; He, J.; Ma, N.; Zhou, X. Genome-wide identification and expression analysis of MIKC(C) genes in rose provide insight into their effects on flower development. Front Plant Sci 2022, 13, 1059925. [Google Scholar] [CrossRef]
- Yin, X.; Gao, Q.; Wang, F.; Liu, W.; Yu, S.; Zhong, S.; Feng, J.; Bai, R.; Luo, Y.; Chen, L.; Dai, X.; Liang, M. MIKC-type MADS-box transcription factor OsMADS31 positively regulates salinity tolerance in rice. Front Plant Sci 2025, 16, 1628305. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Zhong, S.; Huo, Q.; Wang, Q.; Shi, Y.; Liu, H.; Liu, J.; Song, Y.; Fang, X.; Lin, Z. MADS-box encoding gene Tunicate1 positively controls maize yield by increasing leaf number above the ear. Nat Commun 2024, 15, 9799. [Google Scholar] [CrossRef]
- Song, G.Q.; Chen, Q. Overexpression of the MADS-box gene K-domain increases the yield potential of blueberry. Plant Sci 2018, 276, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; Xia, R. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; Liu, X.S. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
Figure 1.
Sequence Characterization of Sorghum MIKC-Type MADS-Box Transcription Factors. (A) Phylogenetic tree of MIKC-type MADS proteins from Arabidopsis, maize, rice, and sorghum. (B) Chromosomal localization and collinearity analysis of sorghum MIKC-type MADS genes. Gray lines indicate collinear gene pairs associated with the sorghum MIKC-type MADS gene family. (C-D) Phylogenetic analysis (C) and gene structure (D) of SbMIKCs, blue and yellow boxes correspondingly represent UTRs and exons, while black lines indicate introns. (E-F) Motif composition (E) and domain architecture (F) among the proteins of SbMIKCs.
Figure 1.
Sequence Characterization of Sorghum MIKC-Type MADS-Box Transcription Factors. (A) Phylogenetic tree of MIKC-type MADS proteins from Arabidopsis, maize, rice, and sorghum. (B) Chromosomal localization and collinearity analysis of sorghum MIKC-type MADS genes. Gray lines indicate collinear gene pairs associated with the sorghum MIKC-type MADS gene family. (C-D) Phylogenetic analysis (C) and gene structure (D) of SbMIKCs, blue and yellow boxes correspondingly represent UTRs and exons, while black lines indicate introns. (E-F) Motif composition (E) and domain architecture (F) among the proteins of SbMIKCs.
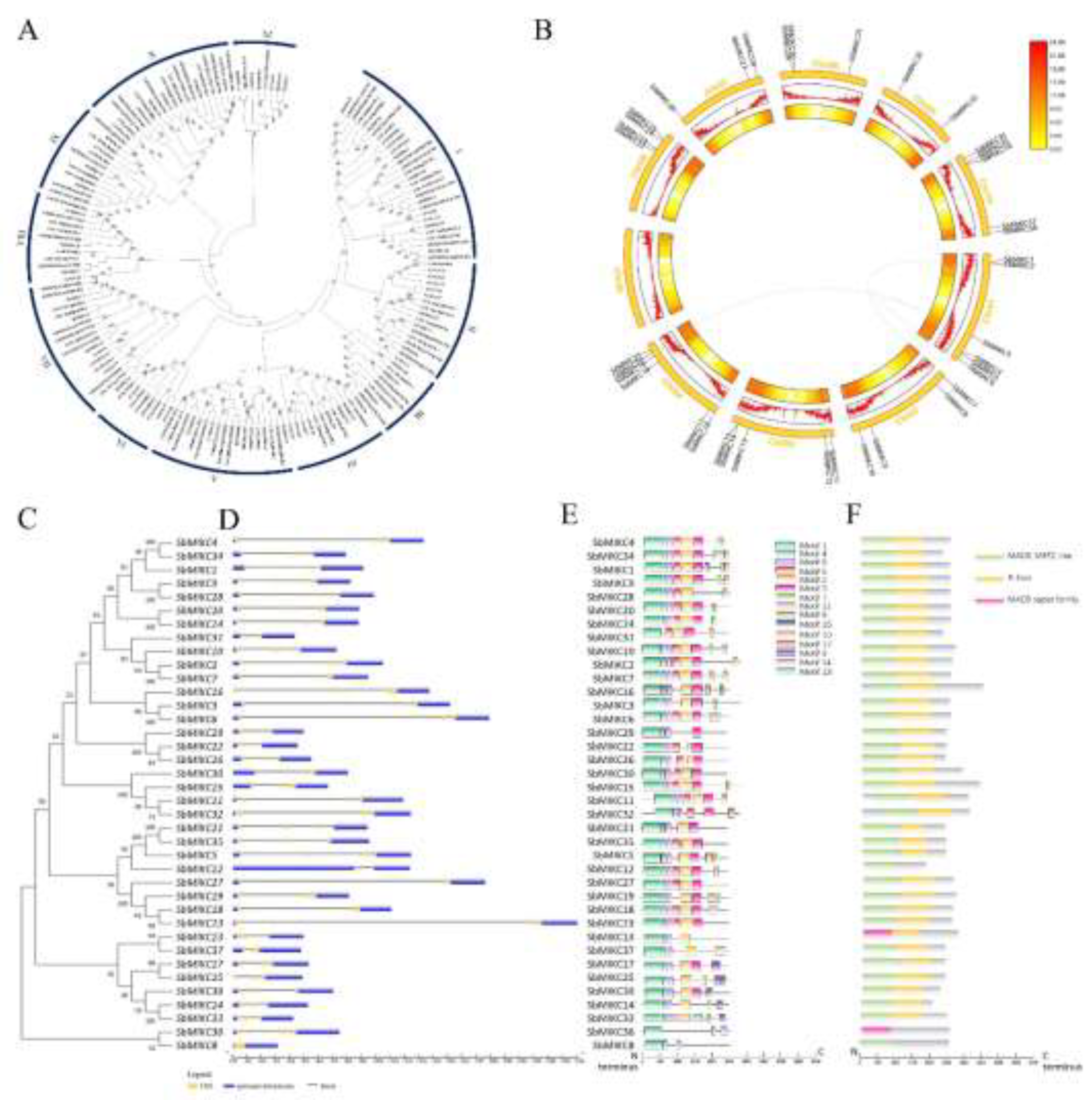
Figure 2.
Composition of cis-acting elements in the promoters and transcriptional profiles of sorghum MIKC-type MADS-box genes. (A) Composition of cis-elements within the promoters of SbMIKCs. (B) The expression pattern and clustering analysis of SbMIKCs among different tissues based on the RNA-seq data. (C) The expression analysis of nine SbMIKCs among different tissues through qRT-PCR.
Figure 2.
Composition of cis-acting elements in the promoters and transcriptional profiles of sorghum MIKC-type MADS-box genes. (A) Composition of cis-elements within the promoters of SbMIKCs. (B) The expression pattern and clustering analysis of SbMIKCs among different tissues based on the RNA-seq data. (C) The expression analysis of nine SbMIKCs among different tissues through qRT-PCR.
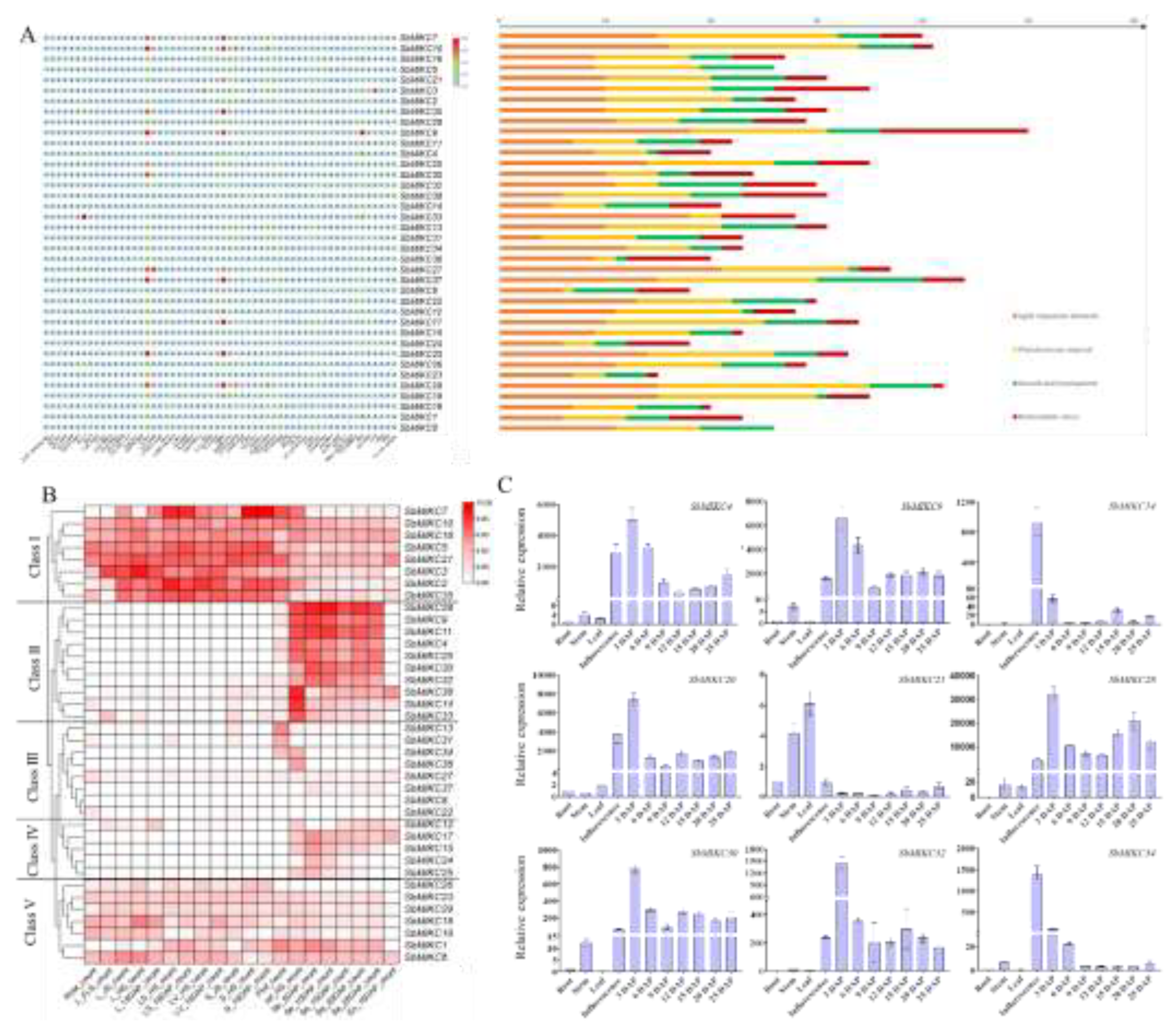
Figure 3.
The functional properties analysis of SbMIKC17. (A) The expression pattern of SbMIKC17 among different tissues in sorghum. (B-C) The self-activation analysis (B) and sub-cellular location (C) of SbMIKC17.
Figure 3.
The functional properties analysis of SbMIKC17. (A) The expression pattern of SbMIKC17 among different tissues in sorghum. (B-C) The self-activation analysis (B) and sub-cellular location (C) of SbMIKC17.
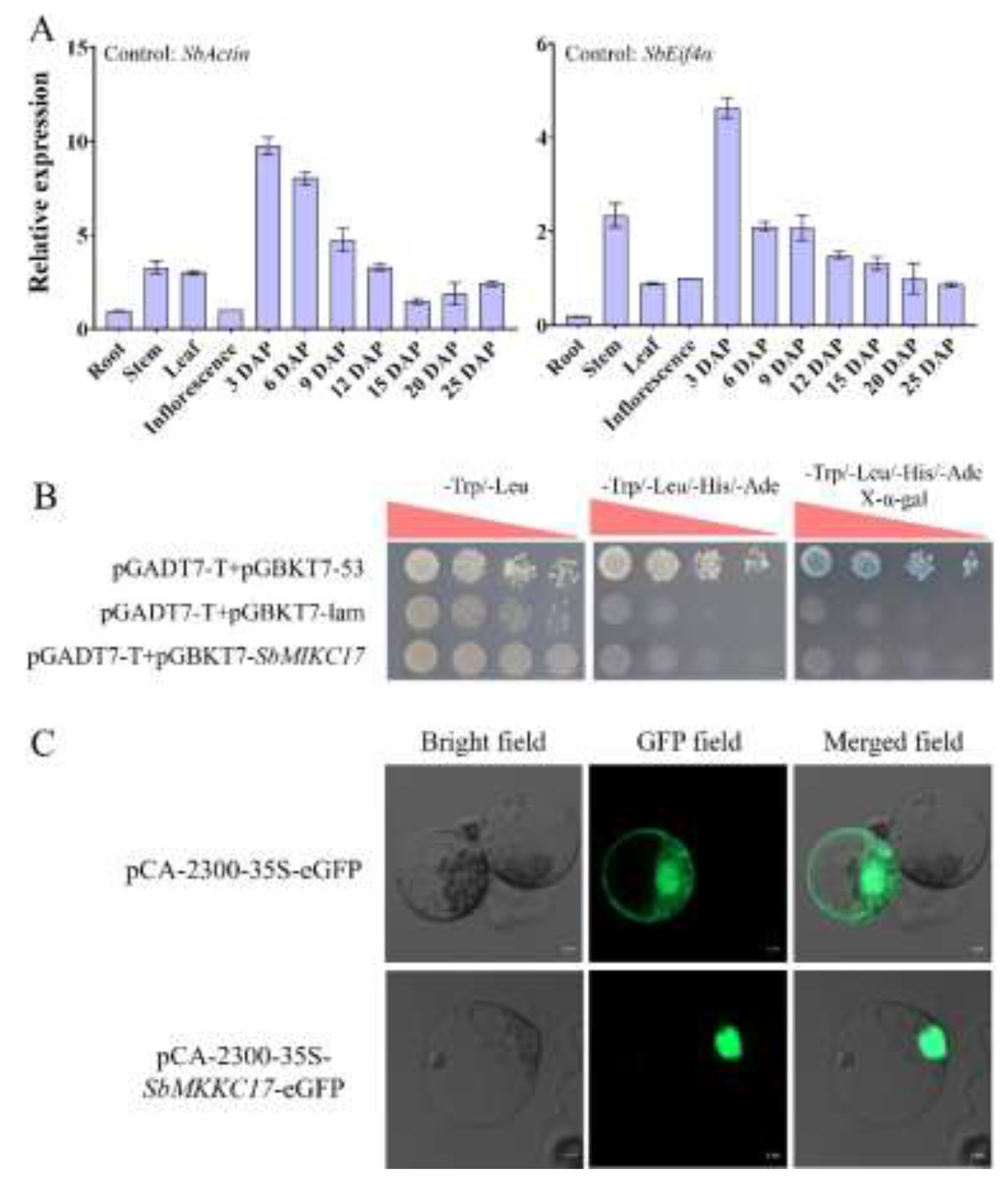
Figure 4.
DAP-seq analysis of SbMIKC17. (A) Venn analysis of binding sites between two biological replicates via DAP-seq. (B): The genomic distribution of SbMIKC17 binding peaks identified by DAP-seq. (C-D) GO annotation (C) and KEGG analysis (D) of genes associated with the genomic loci identified through the DAP-seq analysis of SbMIKC17. (E) Expression analysis of key starch and sucrose metabolism genes downstream of SbMIKC17 identified by DAP-Seq. (F) IGV screenshot showed four SbMIKC17 DAP-seq enriched peaks located in the genes of SbAGPS1 (SORBI_3007G101500), SbISA3 (SORBI_3002G233600), SbSSIIIb (SORBI_3006G221000), and SbSSIIc (SORBI_3001G239500).
Figure 4.
DAP-seq analysis of SbMIKC17. (A) Venn analysis of binding sites between two biological replicates via DAP-seq. (B): The genomic distribution of SbMIKC17 binding peaks identified by DAP-seq. (C-D) GO annotation (C) and KEGG analysis (D) of genes associated with the genomic loci identified through the DAP-seq analysis of SbMIKC17. (E) Expression analysis of key starch and sucrose metabolism genes downstream of SbMIKC17 identified by DAP-Seq. (F) IGV screenshot showed four SbMIKC17 DAP-seq enriched peaks located in the genes of SbAGPS1 (SORBI_3007G101500), SbISA3 (SORBI_3002G233600), SbSSIIIb (SORBI_3006G221000), and SbSSIIc (SORBI_3001G239500).
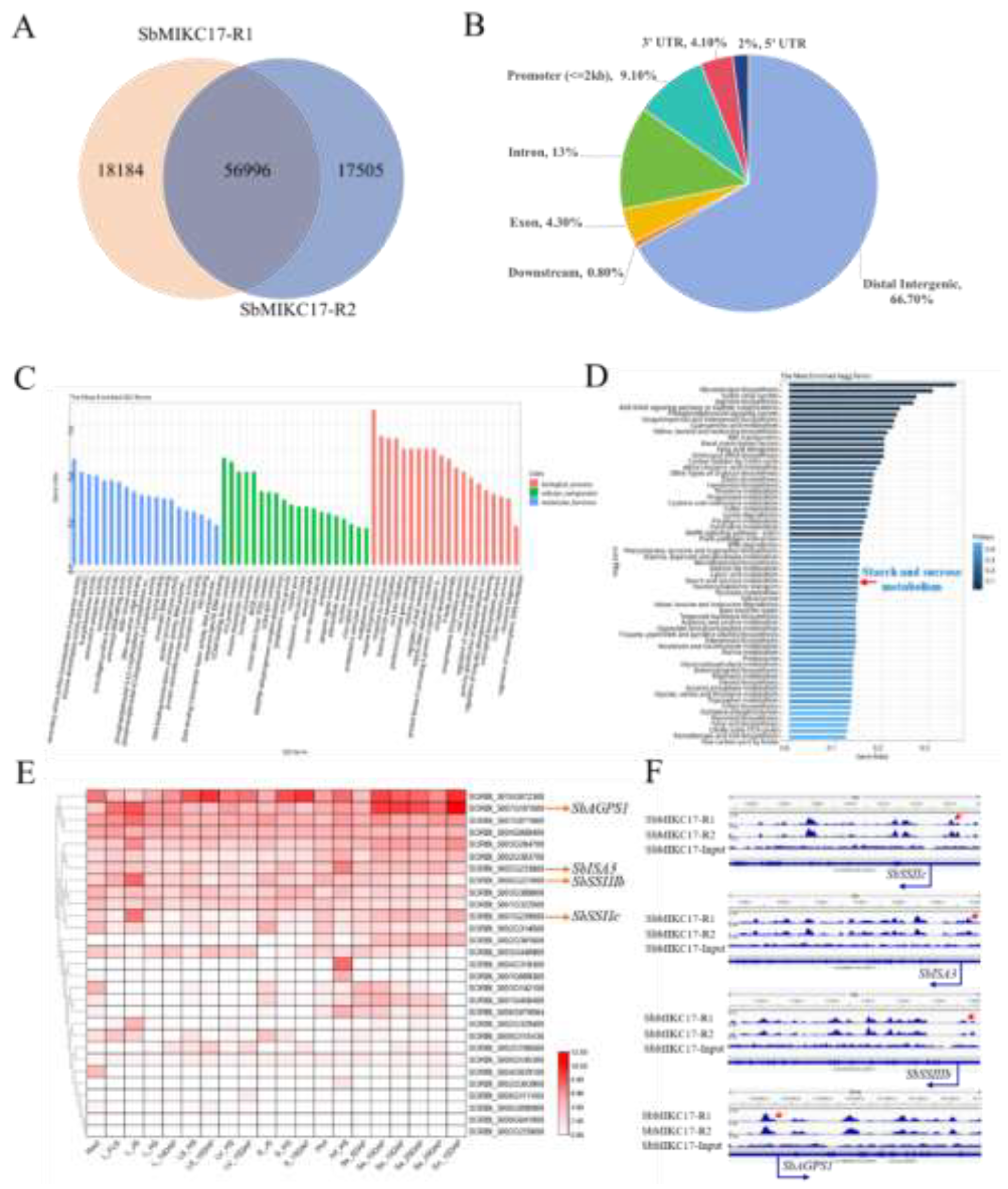
Figure 5.
Transcriptional Regulation of SbAGPS1 by SbMIKC17. (A) The IGV screenshot reveals the precise peak location in the SbAGPS1 gene. (B) The genomic coordinates of the peak identified within the SbAGPS1 promoter region, as determined by DAP-seq analysis. The differentially colored blocks represented the locations of predicted motifs across the peak interval. (C) Sequence information of the motifs in the peak region of SbAGPS1 predicted by DAP-Seq. (D) Yeast one-hybrid assay demonstrating the binding characteristics between SbMIKC17 and the promoter region of the SbAGPS1. (E) The effect of SbMIKC17 on the promoter activity of SbAGPS1. Data are given as means and S.D., significant differences are determined by Student’s t-test (n = 6), ** refers to the significance level of p < 0.01.
Figure 5.
Transcriptional Regulation of SbAGPS1 by SbMIKC17. (A) The IGV screenshot reveals the precise peak location in the SbAGPS1 gene. (B) The genomic coordinates of the peak identified within the SbAGPS1 promoter region, as determined by DAP-seq analysis. The differentially colored blocks represented the locations of predicted motifs across the peak interval. (C) Sequence information of the motifs in the peak region of SbAGPS1 predicted by DAP-Seq. (D) Yeast one-hybrid assay demonstrating the binding characteristics between SbMIKC17 and the promoter region of the SbAGPS1. (E) The effect of SbMIKC17 on the promoter activity of SbAGPS1. Data are given as means and S.D., significant differences are determined by Student’s t-test (n = 6), ** refers to the significance level of p < 0.01.
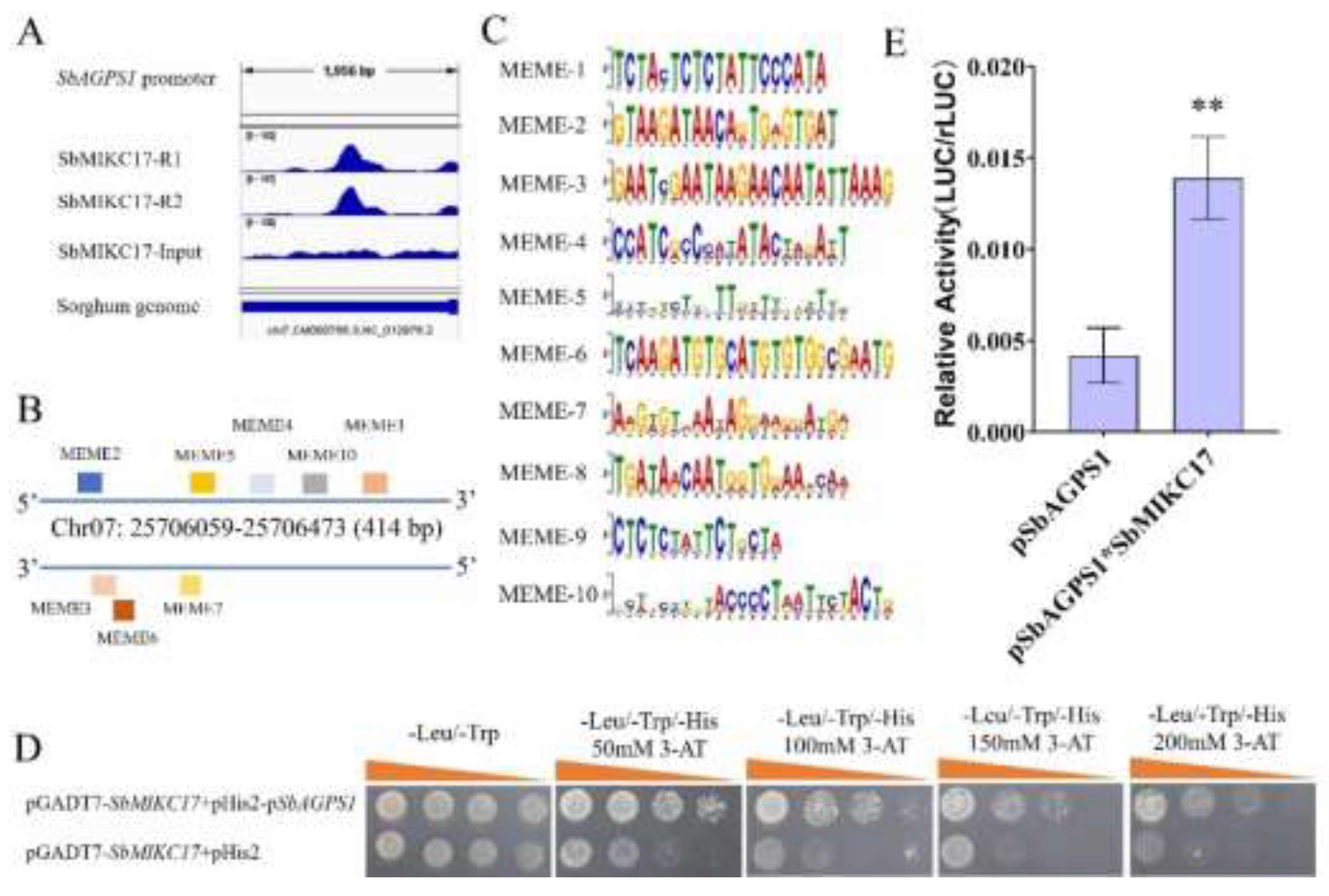
Figure 6.
The transcription activity of starch synthesis related genes by SbMIKC17. (A) Schematic of experimental vector construction. (B) Effect of SbMIKC17 on promoter activity of SBRGs in sorghum, ns and ** refer to non-significance and significance at p < 0.01.
Figure 6.
The transcription activity of starch synthesis related genes by SbMIKC17. (A) Schematic of experimental vector construction. (B) Effect of SbMIKC17 on promoter activity of SBRGs in sorghum, ns and ** refer to non-significance and significance at p < 0.01.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



