
Submitted:
10 February 2026
Posted:
12 February 2026
You are already at the latest version
Abstract
Pulmonary arterial hypertension (PAH) is a life-threatening disease characterized primarily by sustained pulmonary vasoconstriction, pulmonary vascular remodeling, and elevated pulmonary artery pressure. Recent emerging studies have indicated that mitochondria-endoplasmic reticulum (ER) membrane (MAM) is increasingly implicated in pathogenesis of pulmonary hypertension (PH). Dysfunction of mitochondria or ER can trigger a series of pathological processes, including disrupted intracellular Ca2+ homeostasis, oxidative stress, ER stress, and inflammation, which in turn drive dysfunction of pulmonary artery smooth muscle (PASMCs) and endothelial cells (PAECs). This review article aims to systematically evaluates the pathogenic mechanisms of the MAM in PH, covering the latest research advances in mitochondrial dynamic disorders, Ca2+ homeostasis, ER stress, oxidative stress, cellular metabolic reprogramming, and inflammatory responses. Special focus is placed on the structural and functional regulatory mechanisms of MAMs and their roles in pulmonary vascular functional and structural dysfunction. Furthermore, the current review also discusses the potential of new and efficacious therapeutic strategies targeting mitochondrial dysfunctions and ER stress for treatments of PH.
Keywords:
pulmonary hypertension
; mitochondria-associated endoplasmic reticulum membrane
; mitochondrial dynamics
; endoplasmic reticulum stress
; Ca2+ homeostasis
; oxidative stress
1. Introduction
Pulmonary hypertension (PH) is a severe cardiovascular disorder. This disorder is mainly characterized by pulmonary vasocontraction and pulmonary vascular remodeling, thereby causing elevated blood flow resistance and right heart failure. PH is divided into five groups: Group I specifically refers to PH, which includes idiopathic, heritable, and medical conditions including congenital heart disease and other diseases; Group II occurs due to left heart disease; Group III results from chronic lung disease and/or hypoxia; Group IV is caused by chronic thromboembolic pulmonary hypertension; Group V is secondary to sarcoidosis, sickle cell anemia, chronic hemolytic anemia, and certain metabolic disorders[1]. PH represents a significant global health challenge, affecting individuals across all age groups. The current prevalence of PH is approximately 1% of the world population with its higher occurrence among people aged over 65 years[2]. The epidemiological characteristics of PH vary among countries. In developing countries, key contributors to PH include coronary heart disease, infectious diseases, HIV, and hypoxia[3]. In the UK, the observed prevalence of PH has doubled over the past decade and now stands at 125 cases per million inhabitants[3].
The histopathological features of PH include intimal and medial thickening, muscularization of distal pulmonary arteries, vascular occlusion, and complex plexiform lesions[4,5,6] . These histological changes are also observed in other groups of PH, although they are less prominent. Despite significant progress in the field, the molecular mechanisms underlying PH remain largely elusive. Currently, approved drug therapies for patients with PH mainly use non-specific vasodilators including phosphodiesterase type 5 inhibitors, soluble guanylate cyclase stimulators, endothelin receptor antagonists, and prostacyclin analogs. When used alone or in combination, these agents can improve pulmonary vascular function and hemodynamics, as well as reduce the number of PH-related hospitalizations[2], However, these vasodilators do not specifically target the key pathogenic features of PH. Such interventions have not been proven to reduce mortality rates, with the five-year mortality rate remaining at approximately 50%[7]. Thus, lung transplantation remains the ultimate therapeutic option, and exploring the precise pathogenesis of PH and identifying novel therapeutic targets represent an urgent need and challenging resolution.
Mitochondria-associated endoplasmic reticulum membranes(MAMs) have been implicated in PH, heart failure, and other cardiovascular diseases[8]. these critical structural platforms bridges the two organelles mitochondria and ER to play a central role in maintaining cellular homeostasis[9,10]. Functional impairments of MAMs may contribute to the pathogenesis of PH through multiple pathological pathways, including Ca2+ homeostasis disorders, oxidative stress, ER stress, lipid metabolism dysregulation, and abnormal cell proliferation[11,12].
As the physical contact sites, MAMs are involved in controlling various physiological cellular processes. Their formation and function are modulated by a range of proteins and play key roles in cellular stress responses. Structural and functional alterations of MAMs are closely linked to diverse inflammatory and metabolic diseases, thereby leading to PH pathogenesis[13,14]. For instance, MAM formation is tightly associated with ER stress responses, accordingly disrupting intracellular Ca2+ homeostasis and impairing mitochondrial function[15,16]. During PH progression, MAM-mediated Ca2+ signaling and crosstalk with cellular metabolism may drive the proliferation and migration of PASMCs, exacerbating pulmonary vasoconstriction and vasoremodeling [17,18].
MAM plays a crucial role in PAECs as well. In PH, the regulatory mechanisms of MAMs have garnered increasing attention. Mitochondrial and ER dysfunction can alter MAM formation and stability, subsequently affecting cell survival and proliferation[19,20]. Disrupting MAMs in PAECs alleviates mitochondrial dysfunction, reduces cell apoptosis and inflammatory responses, and enhances nitric oxide (NO) release[21]. Knocking down phosphofurin acidic cluster sorting protein 2 (PACS2), a MAM tethering protein, suppresses ox-LDL-triggered cell apoptosis, along with the associated mitochondrial Ca2+ elevation, ROS generation, and cytochrome c release[21]. Hypoxia promotes the formation of MAMs, which directly cause a damage of EC mitochondria, resulting in increased ROS production and mitophagy. Moreover, increasing MAM formation is involved in oxidized low-density lipoprotein (ox-LDL)-induced PAECs apoptosis.
Additionally, nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit the proliferation of vascular smooth muscle cells (VSMCs) not through their anti-inflammatory activity, but by depolarizing mitochondria, suppressing mitochondrial Ca²⁺ uptake, and thereby facilitating the Ca²⁺-dependent inactivation of calcium release-activated calcium (CRAC/Orai) channels[22]. This mechanism blocks store-operated Ca²⁺ entry (SOCE) in VSMCs, providing a novel explanation for the therapeutic effect of NSAIDs in treating vascular proliferative disorders. These results underscore the significance of MAMs in controlling PAEC and PASMSC functions and their involvement in related vascular diseases. Dysregulated Ca²⁺ and lipid metabolism in MAMs may also exacerbate cardiovascular pathological changes[23,24], this is due to intimate crosstalk between lipid metabolism and Ca²⁺ signaling at the MAM interface—impaired lipid homeostasis compromises membrane integrity, perturbs oxidative phosphorylation, and elicits aberrant inflammatory cascades in cardiomyocytes and vascular smooth muscle cells, ultimately accelerating cardiac hypertrophy, myocardial fibrosis, and atherosclerotic lesion formation. These findings offer an innovative perspective on PH pathogenesis, suggesting that targeting MAM function could emerge as a new therapeutic strategy for PH.
Exploring the contribution of MAMs in PH not only deepens on our understanding of its pathogenic mechanisms but also offers potential directions for developing new treatments. Modulating MAM function to restore intracellular Ca²⁺ homeostasis and improve mitochondria-ER crosstalk may alleviate PH pathological processes, offering more effective therapeutic options for patients [16,25]. MAM proteins are critical for maintaining MAM structure and function. Their abnormalities further contribute to the onset and progression of PH and other related diseases. In this review, we elaborate MAM structure and function, as well as the associated biological cellular processes. Additionally, we discuss key insights into proteins that influence MAMs and thereby participate in the pathophysiology of PH and other related diseases. Clarifying how MAM proteins and protein complexes regulate MAMs may offer new strategies for drug intervention and disease management.
2. Composition and Structural Characteristics of MAMs
MAMs, the contact sites between mitochondria and SR in cells, exert vital biological functions. Mass spectrometry analysis of MAM-enriched fractions has identified 1052 proteins, which are known to participate in MAM communication and contribute to the pathogenesis of certain diseases(Figure 1) [26].
MAMs are composed of diverse protein complexes, including mitochondrial fusion protein 2 (MFN2), voltage-dependent anion channel 1 (VDAC1) on the outer mitochondrial membrane, and inositol triphosphate receptor 3 (IP3R3) on the ER membrane. These proteins are critical for MAM structure and function. MFN2 not only mediates mitochondrial fusion but also acts as a bridge in MAM formation [27]; VDAC1 is responsible for metabolite exchange between mitochondria and the cytoplasm [28], IP3R3 plays a key role in controlling intracellular Ca²⁺ concentrations [29]. A number of other proteins are localized to MAMs, such as the Sigma-1 receptor (Sig-1R). During stress responses, Sig-1R dissociates from binding immunoglobulin protein (BiP) and then binds to IP3Rs, enabling sustained Ca²⁺ entry into mitochondria via the IP3R-Grp75-VDAC1 complex. Subsequently, Sig-1R is released from MAMs and redistributed across the ER[30].
Currently, 1052 proteins have been identified as essential components of MAMs. These proteins either participate in MAMs formation or are localized to MAMs domains to regulate core biological processes. Some typical members include,the tethering structure formed by the interaction between vesicle-associated membrane protein B (VAPB) and mitochondrial protein tyrosine phosphatase-interacting protein 51 (PTPIP51)[31], receptor expression-enhancing protein 1 at the mitochondria-ER interface[32], tumor suppressor phosphatase and tensin homolog (PTEN) and PTEN-induced kinase 1 (PINK1)[33], ER molecular chaperone calnexin (CNX)[34], ACS2, which governs ER vesicle sorting[35], Beclin 1 and syntaxin 17 (STX17), involved in autophagosome formation and maturation [36,37], and ER stress-related proteins such as ATF6, XBP1, and GRP78[38].
One key function of MAMs is to serve as a bridge for Ca²⁺ transport, controlling dynamic changes in intracellular Ca²⁺ signaling[39]. In intracellular Ca²⁺ signaling, MAMs facilitate Ca²⁺ transfer between the ER and mitochondria, ensuring cells maintain metabolic homeostasis and normal signal transduction. Studies have demonstrated that MAM-mediated Ca²⁺ signaling pathways play crucial roles in various cellular physiological and pathological processes, particularly in neurodegenerative and cardiovascular diseases, where MAM dysfunction is closely linked to cell death and tissue damage[8,29].
3. The Pathology of PH
3.1. Regulatory Mechanism of MAM in Mitochondrial Fission and Fusion
Mitochondrial fission and fusion are essential for preserving mitochondrial integrity, metabolic flexibility, and cellular homeostasis (Figure 2). Dysregulation of these processes has become recognized as a core pathological feature of PH, contributing to metabolic reprogramming, apoptosis resistance, and uncontrolled proliferation of PASMCs [40]. Central regulators including the fission mediator dynamin-related protein 1 (Drp1) and fusion proteins such as mitofusin-1/2 (MFN1/2) and optic atrophy protein 1 (OPA1) have been implicated in PH progression [41,42,43]. In PASMCs from patients with PH, Drp1 expression and activation are markedly increased, while levels of MFN1, MFN2, and OPA1 are reduced, resulting in fragmented mitochondrial networks, impaired oxidative metabolism, and enhanced proliferative and apoptosis-resistant phenotypes [44].
Growing experimental evidence further supports the pathogenic role of altered mitochondrial dynamics in PH. Excessive mitochondrial fission promotes PASMCs proliferation by ensuring sufficient bioenergetic support and sustaining pro-survival signaling pathways, whereas reduced fusion facilitates vascular remodeling and disease progression[16]. Regulatory pathways controlling these dynamics have also been identified. MicroRNAs (miR-125a and miR-140) reduce MFN1 expression, contributing to hypoxia-induced PASMC proliferation and right ventricular remodeling in SuHx models [45,46]. Upregulation of the Drp1-interacting proteins MiD49/51 drives pathological mitochondrial fission, enhancing PASMC proliferation and apoptosis resistance [47]. Reduced MFN2 and PGC-1α expression contributes to mitochondrial fragmentation in both human and experimental PH models, and restoring MFN2 improves disease manifestations [48]. Inhibiting Drp1, via Mdivi-1 or siDrp1, blocks pathological fission and induces cell-cycle arrest at G2/M, suppressing PASMCs proliferation [49]. Moreover, sentrin-specific protease 1 (SENP1) preserves endothelial mitochondrial morphology via modulation of FIS1 SUMOylation, protecting against hypoxia-induced PH [50]. These studies support mitochondrial dynamics, particularly enhancing fusion and limiting Drp1-dependent fission, as a promising therapeutic avenue in PH.
Beyond mitochondria-intrinsic mechanisms, emerging work demonstrates that the ER is an upstream coordinator of mitochondrial fission and fusion. Prior to Drp1 recruitment, ER tubules encircle mitochondria to pre-define and constrict future division sites[51]. ER-associated inverted formin-2 (INF2) facilitates this process by regulating actin polymerization, providing mechanical support for membrane constriction[52]. The positioning of mitochondrial fission sites is further influenced by mtDNA replication and mitochondrial metabolic status, suggesting that ER–mitochondria coupling integrates mitochondrial morphology with bioenergetic demand and genome maintenance[53]. ER signaling also modulates mitochondrial fusion by regulating MFN1/2 conformation and oligomerization. Under stress conditions such as unfolded protein response activation, ER stress suppresses MFN and OPA1 expression, impairing mitochondrial fusion and exacerbating dysfunction[53].
These regulatory interactions are spatially organized at MAMs, specialized ER–mitochondria contact domains enriched with proteins governing mitochondrial dynamics, calcium transfer, and metabolic signaling. Disruption of ER–mitochondria communication, particularly MAM dysfunction, extends pathological consequences to the right ventricle (RV). In SuHx-induced PH, cardiomyocytes exhibit reduced MFN2 and OPA1 expression, accompanied by mitochondrial fragmentation, hypertrophy, and impaired RV performance[43]. These findings position ER–mitochondria communication and MAM integrity as central determinants of mitochondrial dynamics in PH. Dysregulated Drp1-dependent fission, impaired MFN/OPA1-mediated fusion, and stress-driven disruption of ER–mitochondria signaling synergistically promote metabolic reprogramming, apoptosis resistance, and proliferative remodeling in both the pulmonary vasculature and right ventricle. Restoring mitochondrial dynamic balance, particularly through modulation of MAM-associated pathways, therefore represents a compelling therapeutic direction for both vascular and cardiac manifestations of PH.
3.2. Mechanisms of Ca²⁺ Homeostasis Imbalance
The Ca²⁺ signaling not only serves as central regulators of cellular metabolism, but also is essential for maintaining normal cellular functions, most notably in the proliferation and apoptosis of PASMCs[54,55]. In PH, the disruption of Ca²⁺ homeostasis represents a key pathophysiological mechanism. MAMs, the specialized membrane contact sites between mitochondria and the ER, exert a critical influence on Ca²⁺ signaling(Figure 3)[56,57]. These structures are intimately involved in Ca²⁺ transport and signal transduction processes. Studies have identified several key molecules in this context, ER Ca²⁺ release channels such as inositol triphosphate receptor 3 (IP3R3)[58] (inositol triphosphate receptor 3), VDAC1(Voltage-dependent anion channel 1)[59] , and MFN2(Mitofusin-2)[60]. IP3R3 activation facilitates Ca²⁺ release from the ER, while VDAC1 is critically involved in controlling mitochondrial Ca²⁺ influx and efflux[44]. MFN2, on the other hand, modulates mitochondrial morphology and function, thereby impacting Ca²⁺ signaling between the ER and mitochondria[61]. Beyond these MAM-localized molecules, the ER-resident transmembrane protein TMCO1 is also pivotal for ER Ca²⁺ homeostasis: it acts as a "Ca²⁺ load-activated Ca²⁺ (CLAC) channel" that senses ER Ca²⁺ overload, forms a Ca²⁺-selective channel via reversible homotetramerization, and mediates ER Ca²⁺ release to prevent excessive store filling[62]. This evolutionarily conserved protective mechanism, when lost, causes severe ER Ca²⁺ mishandling and is linked to developmental disorders like cerebrofaciothoracic (CFT) dysplasia spectrum[62]. By maintaining ER Ca²⁺ within a physiological range, TMCO1 indirectly stabilizes MAM-mediated Ca²⁺ transfer, avoiding ER Ca²⁺ overload that would disrupt ER-mitochondria Ca²⁺ signaling precision at MAMs. Thus, by governing Ca²⁺ signaling to influence PASMC proliferation and apoptosis, MAMs act as a critical bridge linking Ca²⁺ homeostasis imbalance to the progression of PH.
Interaction between Bap31 and Fis1 mediates the formation of the Bap31–Fis1 complex localized to MAMs[63,64,65]. Simmen et al. demonstrated that another protein called PACS2 modulates the contribution of Bap31 in tethering the two organelles. However, depletion of PACS2 was reported to cause Bap-31-dependent mitochondrial fragmentation and uncoupling from the ER along with inhibition of Ca2+ signal transmission[66]. The role of miR-25 as a mediator of apoptosis through the MCU has been confirmed in PH[67]. Bcl2 and Bclextra large (Bcl-XL) proteins exert part of their anti-apoptotic functions by directly binding to ER localized IP3Rs and to OMM localized VDAC1, respectively. The joining of Bcl-2 and Bcl-XL to these Ca2+ transport systems will negatively affect Ca2+ transfer and apoptosis[68].
Aberrant Ca²⁺ signaling drives the imbalance between excessive proliferation and insufficient apoptosis of PASMCs in PH. Studies have revealed that under hyperoxic or other pathological conditions, the intracellular Ca²⁺ concentration in PASMCs rises markedly; The following Ca²⁺ overload pushes PASMCs into a proliferative state, ultimately contributing to pulmonary artery remodeling[69]. Moreover, disrupted Ca²⁺ homeostasis can activate multiple intracellular signaling pathways including p38 MAPK and PI3K/Akt cascades, which are closely tied to cell proliferation and survival. For instance, enhanced Ca²⁺ signaling promotes PASMC proliferation and suppresses apoptosis by activating these pathways, a phenomenon particularly prominent in PH patients[70]. This process is further amplified by abnormal activation of the extracellular calcium-sensing receptor (CaSR), a G protein-coupled receptor that senses extracellular Ca²⁺ ([Ca²⁺]₀) and modulates intracellular Ca²⁺ homeostasis[71]. In PAH-PASMCs, CaSR expression is upregulated by hypoxia (via HIF-1α) and inflammation (via NF-κB), and its activation by elevated [Ca²⁺] triggers Gq/11-PLCβ-mediated ER Ca²⁺ release. Moreover, CaSR interacts with IP3R3 at MAMs, enhancing Ca²⁺ transfer to mitochondria and further exacerbating Ca²⁺ overload. This sustained Ca²⁺ signal activates CaMKII and ERK pathways, promoting Cyclin D1 expression and inhibiting mitochondrial apoptosis, thereby driving PASMCs hyperplasia[71]. Additionally, Ca²⁺ signaling abnormalities are strongly linked to cellular stress responses, reducing cells' ability to survive adverse environments like hypoxia[12]. Thus, aberrant Ca²⁺ signaling not only triggers excessive PASMCs proliferation but also disrupts apoptotic balance, collectively exacerbating PH pathogenesis.
A case in point is that MFN2 mediates mitochondrial Ca2+ transport via IP3R3, suppressing PASMCs proliferation and pulmonary vascular remodeling[72]. Researchers have found that inhibiting IP3R3 expression or restoring MFN2 levels significantly improves vascular function in PH models, suggesting that restoring Ca²⁺ homeostasis could serve as an effective therapeutic strategy for PH[9]. The mitochondria-ER axis also exerts a significant regulatory effect on the function of pulmonary artery endothelial cells. Studies have confirmed that Ca²⁺ signaling mediated by inositol triphosphate receptor 3 (IP3R3) can regulate the proliferation and migration of endothelial cells[73].
3.3. ER Stress and Oxidative Stress in PH
A close interplay exists between ERS and oxidative stress. Studies have demonstrated that in PH, forming a reciprocal vicious cycle that exacerbates vascular pathology. ERS can amplify cellular oxidative stress responses, further triggering inflammation and apoptosis. Conversely, oxidative stress increases ER load, forming a vicious cycle that ultimately leads to PASMCs dysfunction and vascular remodeling[20].
As a tightly interconnected pathogenic process with oxidative stress, ER stress in PH arises when the ER’s protein-folding capacity is overwhelmed, activating the unfolded protein response (UPR) via the PERK, IRE1, and ATF6 signaling pathways (Figure 4). These adaptive branches transiently reduce protein translation, increase molecular chaperone synthesis, and restore proteostasis. However, in PH, ER stress becomes chronic rather than adaptive. Hypoxia, inflammation, and viral infection, during PH development and progression, robustly activate ER stress in pulmonary artery endothelial cells (PAECs) and smooth muscle cells (PASMCs). Persistent UPR signaling induces CHOP-mediated apoptosis, disrupts endothelial barrier integrity, and promotes inflammatory cytokine expression (e.g., IL-6, TNF-α), contributing to vascular dysfunction and remodeling[9,15,74]. Notably, the ATF6–Nogo-B signaling axis has been selectively implicated in pulmonary vascular smooth muscle proliferation, emphasizing vascular-bed–specific stress responses[75,76].
Oxidative stress is tightly coupled to ER stress in this setting. Reactive oxygen and nitrogen species (ROS/RNS) intensify UPR activation, while unresolved ER stress increases ROS production, forming a feed-forward loop that promotes endothelial apoptosis, PASMC hyperproliferation, and inflammation[77,78]. Pharmacologic attenuation of ER stress, such as with the chemical chaperone 4-phenylbutyrate, has shown benefit in experimental PH, underscoring its therapeutic relevance[79,80]. Emerging evidence indicates that this interaction is spatially coordinated by mitochondria–ER contact sites, known as mitochondria-associated membranes (MAMs). Structural modules within MAMs, particularly the IP₃R–GRP75–VDAC1 axis, regulate Ca²⁺ transfer from the ER to mitochondria. In PH, enhanced ER stress tightens MAM coupling and increases Ca²⁺ flux, driving mitochondrial Ca²⁺ overload and excessive mitochondrial ROS (mtROS) generation (Figure 4). Elevated mtROS further disrupts ER proteostasis and alters Ca²⁺ channel function, thereby reinforcing MAM tethering and amplifying ER stress. This bidirectional amplification loop, ER stress, increased Ca²⁺ transfer, mtROS generation, and intensified ER stress, is a key driver of endothelial apoptosis and PASMCs hyperproliferation, central pathological features of PH vascular remodeling. Similar stress-coupling mechanisms have been reported in diabetic vasculopathy, liver fibrosis, and neurodegeneration, suggesting that this MAM-dependent circuit represents a conserved cellular stress program. Targeted disruption of this axis through modulation of MAM structure, inhibition of mitochondrial Ca²⁺ uptake, or activation of antioxidant programs such as the SIRT3/SOD2 pathway, reduces mitochondrial ROS, restores endothelial function, and attenuates vascular remodeling in PH models[81].
In summary, these findings position the mitochondria-ER axis, but not ER stress or oxidative stress in isolation, as a central pathogenic mechanism in PH. Therapeutically interrupting this MAM-driven stress–ROS–Ca²⁺ feedback loop represents a promising strategy now supported by emerging preclinical evidence.
3.4. Involvement of MAM in Inflammation, Autophagy and Lipid Metabolism in PH
Inflammation plays a pivotal role in the progression of PH, particularly in pulmonary vascular remodeling. Evidence indicates that inflammatory mediators drive PASMC proliferation and fibrosis through multiple mechanisms, exacerbating vascular remodeling. For instance, in chronic PH animal models, elevated levels of inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) correlate closely with PASMC proliferation and phenotypic transition toward a synthetic state[82,83]. Mitochondria act as central regulators of immune responses, and several mitochondria-associated membrane (MAM) proteins participate in this regulation (Figure 5). MAM-associated proteins are critical for initiating interferon and other inflammatory responses upon pathogen exposure or cellular stress[26,84,85]. While inflammation is a key defense mechanism against ischemia, hypoxia, excessive immune activation, and aging, it also constitutes a core pathological process in cardiovascular and cerebrovascular diseases, cancer, and metabolic disorders[86,87]. The nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) functions as a sensor of microbial infection and cellular damage, forming multi-protein inflammasomes to initiate inflammatory signaling[88]. MAMs not only facilitate inflammatory activation but also serve as a platform for NLRP3 inflammasome assembly. NLRP3 is widely expressed, with macrophages exhibiting the highest levels. In its inactive state, NLRP3 resides in the ER membrane and cytoplasm; upon activation, NLRP3 and its adaptor ASC translocate to MAMs to detect ROS from damaged mitochondria[89]. Respiratory chain inhibitors such as rotenone (complex I inhibitor) can activate the inflammasome, linked to mitochondrial membrane potential (ΔΨm) loss and ROS accumulation[90]. Excessive mitophagy, ER stress (ERS), and aberrant ER–mitochondria Ca²⁺ transfer can further impair mitochondrial function, increase ROS, and release mitochondrial DNA (mtDNA), collectively activating NLRP3 inflammasomes[26]. MAM-resident VDAC proteins are essential in this process; VDAC1 mediates mitochondrial Ca²⁺ uptake and ROS production, and VDAC knockout markedly suppresses IL-1β inflammasome formation[90]. In PH, ERS activation and the interplay of inflammatory signaling pathways, particularly NF-κB and NLRP3 inflammasomes, drive disease progression[91]. Activated NF-κB promotes transcription of pro-inflammatory cytokines such as TNF-α and IL-6, amplifying inflammation and inducing pulmonary artery endothelial cell apoptosis, ultimately contributing to vascular remodeling and hypertension[92,93,94,95]. NLRP3 inflammasomes thus act as a mechanistic bridge between ERS and PAH, suggesting that targeting ERS-related pathways could provide novel therapeutic strategies. Mitochondrial dysfunction, a hallmark of PH, disrupts cellular energy homeostasis and elevates oxidative stress, contributing further to inflammation. mtDNA and ROS serve as potent inflammatory triggers[96,97]. Impaired mitochondria release mtDNA into the cytoplasm, activating downstream interferon signaling via the cGAS-STING pathway and promoting inflammatory factor production[98]. Elevated mitochondrial ROS, observed in PH patients, is associated with pulmonary endothelial dysfunction and inflammatory responses[99]. Moreover, imbalances in mitochondrial fission and fusion exacerbate oxidative stress and inflammation[40].
MAMs are increasingly recognized for their roles in autophagy, with several autophagy-related proteins localized to MAMs[35]. In PINK/Parkin-dependent mitophagy, PINK accumulates on the outer mitochondrial membrane (OMM), phosphorylates ubiquitin at Ser65, recruits Parkin, and activates polyubiquitination of proteins such as VDAC1 and p62/SQSTM1[100,101]. MAMs serve as initiation sites for this mitophagy. Notably, PINK depletion hinders BECN1 accumulation independently of Parkin, indicating a unique regulatory role for PINK[33]. Parkin overexpression strengthens ER–mitochondria contacts, enhances ER-to-mitochondria Ca²⁺ transfer, and increases mitochondrial ATP production[102]. Similarly, under hypoxia, FUNDC1 accumulates in MAMs, highlighting MAM involvement in mitophagy regulation[103].
MAMs are integral to lipid metabolism. Lipid synthesis—including triacylglycerol (TAG), phosphatidylcholine (PC), and phosphatidylethanolamine (PE)—requires enzymatic activity spanning both the ER and mitochondria. Phosphatidylserine (PS), produced from PC by PSS1 in MAMs, is converted to PE by mitochondrial PS decarboxylase, and phosphatidylethanolamine N-methyltransferase 2 (PEMT2) completes PC synthesis[104]. In idiopathic PAH (IPAH), TAG, LPS, PA, PG, PE, PI, PS, SS, and St lipid levels are significantly elevated, suggesting potential pathogenic roles[105]. Cellular metabolic reprogramming is critical in PAH progression[106,107]. Mitochondria–ER crosstalk influences PASMC proliferation and apoptosis resistance, enabling survival under hypoxia and promoting vascular remodeling[108]. Hypoxia-inducible factor-2α (HIF-2α) is a key transcriptional regulator of EndMT in severe PAH: MAM dysfunction–induced Ca²⁺ imbalance activates CaMKII-mediated HIF-2α phosphorylation, enhancing its stability and nuclear translocation even under normoxic conditions. HIF-2α binds Snai1 and Twist1 promoters, suppressing endothelial markers (VE-cadherin) and upregulating mesenchymal markers (α-SMA), exacerbating remodeling[109]. Therapeutically, combining dichloroacetic acid (DCA) and atorvastatin alleviates PH by modulating mitochondrial metabolism[110,111,112,113], highlighting metabolic reprogramming as a potential therapeutic target.
Pulmonary vascular remodeling in PH involves PASMC proliferation, endothelial apoptosis, and small vessel restructuring, regulated by both growth factors and immune cells. Macrophages and neutrophils secrete inflammatory mediators that promote PASMC proliferation and migration. Oxidative stress further amplifies these effects by impairing endothelial function and enhancing smooth muscle cell migration and proliferation, partly via NF-κB signaling. Endothelial VEGFR2–VE-cadherin–PECAM-1 complexes respond to shear stress through p66Shc-mediated Gab2–PI3K signaling[114]. Epigenetic modifications of p66Shc (e.g., decreased methylation and increased acetylation) enhance its transcription, acetylation, and phosphorylation, promoting mitochondrial translocation and ROS production[115,116]. Mitochondrial dysfunction–induced lipid metabolism disorders exacerbate inflammation: chronic inflammation impairs fatty acid oxidation, generating excessive ROS and stimulating release of TNF-α and IL-1β[116]. Therapeutically, anti-inflammatory agents such as glucocorticoids alleviate PH symptoms by suppressing inflammatory mediator production, although current treatments cannot fully reverse vascular remodeling. Collectively, mitochondrial dysfunction and MAM dysregulation emerge as key contributors to PH pathogenesis, highlighting the potential of strategies targeting mitochondrial function, ER–mitochondria communication, and inflammatory signaling. A deeper understanding of MAM-mediated interactions among inflammation, autophagy, and lipid metabolism may inform more effective therapeutic approaches.
3.5. MAM-Mediated Apoptosis in PH
Apoptosis, a form of programmed cell death (PCD) that maintains cellular homeostasis by regulating cell proliferation, development, and renewal, is tightly controlled in pulmonary vascular cells, with mitochondria-associated membranes (MAMs) serving as central hubs that mediate apoptotic signaling. Both mitochondria and the ER contribute to apoptotic regulation, and MAMs integrate multiple molecular cues to control cell fate.
The mitochondrial fission protein Fission 1 (Fis1) interacts with B cell receptor-associated protein 31 (Bap31) on the ER, cleaving Bap31 into the pro-apoptotic fragment p20Bap31. This Fis1–Bap31 complex is essential for activating pre-caspase-8, highlighting how MAMs mediate the transmission of apoptotic signals[65,117] (Figure 5).
ER-to-mitochondria Ca²⁺ transfer is another critical determinant of apoptosis. PTEN, an antagonist of Akt signaling, interacts with IP3R1 to enhance ER-to-mitochondria Ca²⁺ flux, thereby increasing cellular susceptibility to apoptosis[118]. Conversely, Bcl-xl, an anti-apoptotic Bcl-2 family member, translocates to MAMs during thapsigargin (Tg)-induced apoptosis, strengthening its interaction with IP3R3. Bcl-xl modulates mitochondrial Ca²⁺ uptake, limits cytosolic Ca²⁺ overload, supports cellular metabolism, and ultimately prevents apoptosis following Tg treatment[119]. Similarly, the translocation of Bcl2 from the ER to MAMs and then to mitochondria depends on MAM integrity and the Bcl2–TOM20 interaction, enhancing its anti-apoptotic function[120].
Pro-apoptotic Bax also relies on MAMs for translocation to the mitochondrial membrane, where it triggers cytochrome c release and apoptosis through mitochondrial Ca²⁺ accumulation and opening of the mitochondrial permeability transition pore (MPTP)[121]. Additional molecules localizing to MAMs mediate ER stress-induced apoptosis, including mitochondrial E3 ubiquitin ligase MITOL[122] , transcription factor C/EBP homologous protein (CHOP) [123], and FK506 binding protein 8 (FKBP8) [124].
Emerging evidence highlights the role of promyelocytic leukemia protein (PML) in apoptosis regulation via MAMs. PML modulates the ER machinery at ER–mitochondria contact sites, regulating Ca²⁺ signaling from ER to mitochondria and cytosol, thereby influencing cell survival and sensitivity to apoptotic stimuli[125]. Moreover, MAM-resident proteins such as p66Shc and tumor suppressor p53 can modulate ROS production and ER–mitochondria Ca²⁺ flux, amplifying apoptotic responses under stress conditions[126]. Structural dynamics of MAMs, including tethering protein expression and contact site density, further influence cellular susceptibility to apoptosis, highlighting MAMs as a highly dynamic signaling platform in PH.
These mechanisms underscore MAMs as central regulators of apoptosis, integrating Ca²⁺ signaling, pro- and anti-apoptotic protein interactions, ER stress, and oxidative cues to modulate vascular cell fate in PH.
4. Therapeutic Strategies Targeting Mitochondria-ER Axis
4.1. Mitochondrial Dynamics as A Potential Therapeutic Target
In recent years, a growing body of research has identified mitochondrial dynamics as a promising therapeutic target for PH. By targeting inhibitors of the mitochondrial fission protein Drp1 such as Mdivi-1. Researchers have observed symptom improvements in PH animal models. This inhibitory effect alleviates excessive mitochondrial fission, restores mitochondrial function and morphology, and thereby improves the proliferation and apoptosis balance of PASMCs[49]. Additionally, strategies to enhance mitochondrial fusion including the use of agonists for MFN1 or OPA1have also shown potential in ameliorating PH. Recently, researchers discovered that Drpitor1, a specific Drp1 GTPase inhibitor, reduces proliferation, induces apoptosis in PH human PASMCs (hPASMCs), and reverses monocrotaline-induced PH [41]. By controlling mitochondrial dynamic balance, researchers aim to reverse pulmonary artery remodeling and offer new therapeutic options for PH patients[16,127].
Activating mitochondrial fusion is equally crucial. MFN2, a crucial outer mitochondrial membrane fusion protein, can lead to mitochondrial fragmentation and dysfunction when functionally impaired. In PH, MFN2 activation promotes mitochondrial fusion, improving mitochondrial morphology and function. Research indicates that upcontrolling MFN2 enhances mitochondrial interactions, facilitates intracellular energy transport, and improves cellular metabolic status[43]. Targeted activation of MFN2 can effectively promote mitochondrial health, thereby countering PH progression. FUN14 domain-containing protein 1 (FUNDC1), an integral outer mitochondrial membrane protein, mediates the formation of MAMs. Silencing FUNDC1 reduces MAMs formation, which inhibits angiogenesis by decreasing VEGFR2 expression[128], suggesting that targeting FUNDC1-dependent MAMs could be a promising approach for PH treatment.
Together, mitochondrial dynamics regulators such as the Drp1 inhibitor Mdivi-1 and MFN2 activation strategies offer new targets and approaches for PH treatment. By modulating mitochondrial fission and fusion, these interventions can significantly improve mitochondrial function, reduce cellular oxidative stress, and slow PH pathological progression. Future research should further explore the clinical potential of these modulators to offer more effective therapeutic options for PH patients[16,127].
4.2. Antioxidant and Metabolic Regulating Drugs
The combination of antioxidant and metabolic regulatory drugs has shown significant potential in treating PH. Notably, the combination of dichloroacetic acid (DCA) and atorvastatin (ATO) is thought to synergistically improve the pathophysiological status of PH patients through distinct mechanisms.
DCA, a well-known metabolic regulator, activates pyruvate dehydrogenase (PDH) to promote aerobic metabolism, reduce lactate production, and enhance cellular energy metabolism. This is particularly critical for PH patients, especially those with hypoxia-induced metabolic disorders, since improving energy metabolism can alleviate right heart burden and enhance cardiac function[111].
Atorvastatin, a statin drug, primarily acts by lowering cholesterol levels and improving endothelial function. Studies have revealed that statins not only benefit cardiovascular health but also exhibit anti-inflammatory and antioxidant properties. Atorvastatin can slow PH progression by inhibiting endogenous oxidative stress and protecting endothelial function; additionally, it governs lipid metabolism to further promote pulmonary artery relaxation and reduce pulmonary arterial pressure[113].
The combination of DCA and ATO exerts positive effects on PH through multiple pathways. Firstly, their synergistic action effectively enhances cellular energy status, mitigates hypoxia-induced metabolic disturbances, and reduces oxidative damage. Moreover, atorvastatin’s anti-inflammatory effects complement DCA’s metabolic regulatory role, collectively alleviating PH pathological progression at the molecular level. Clinical data indicate that DCA-ATO combination therapy significantly improves heart function and quality of life in PH patients[129].
Noticeably, DCA-ATO combination therapy offers an innovative therapeutic strategy for PH, highlighting the importance of combining antioxidant and metabolic regulatory drugs in improving PH patient prognosis. However, further clinical trials are needed to verify its long-term efficacy and safety, laying a more robust theoretical foundation for clinical application.
The application prospects of antioxidant therapy in PH have garnered growing attention, particularly in the context of controlling ER-mitochondria crosstalk. Recent studies have highlighted that mitochondria-ER interactions play a pivotal role in cellular metabolism and oxidative stress, the processes closely linked to PH pathogenesis[130]. Since oxidative stress is recognized as a key pathological mechanism in PH, driving endothelial dysfunction and vascular remodeling, antioxidant-based interventions may offer innovative therapeutic insights for PH[130,131].
In antioxidant therapy research, activation of the SIRT3/SOD2 pathway has attracted widespread interest. SIRT3, a mitochondrial deacetylase, governs the activity of various antioxidant enzymes, most notably superoxide dismutase 2 (SOD2) via deacetylation, thereby reducing mitochondrial ROS levels. Studies have demonstrated that SIRT3 activation significantly lowers pulmonary artery pressure and alleviates right ventricular hypertrophy in PH model mice, underscoring its potential as a therapeutic target in PH[132].
The utility of natural antioxidants extends beyond direct free radical scavenging; they also modulate cellular signaling pathways and enhance cellular function. For instance, β-carotene has been shown to alleviate oxidative stress and ER stress in PH mouse models by controlling Ca²⁺ signaling between the ER and mitochondria, thereby exerting a protective effect against PH development[133,134]. Furthermore, some antioxidants can enhance cellular tolerance to oxidative stress by modulating autophagy and apoptosis pathways, which may positively impact PH pathophysiology.
Obviously, antioxidant therapy holds broad application prospects in PH, particularly regarding SIRT3/SOD2 pathway activation and natural antioxidant research. Future studies should further clarify the mechanisms of action of antioxidants and explore their efficacy and safety in clinical settings to offer more effective treatments for PH patients. A deeper understanding of Mitochondria-ER interactions and their regulatory roles in PH will also lay a crucial theoretical foundation for developing innovative antioxidant therapies.
4.3. Ca²⁺ Signaling Regulation-Related Functional Roles and Their Therapeutic Targets
Ca²⁺ signaling is critically involved in diverse cellular functions including cell proliferation, migration, and apoptosis, all of which are critical to PH pathogenesis. For instance, the application of Ca²⁺ channel inhibitors or specific signaling pathway antagonists reduces PASMC proliferative capacity under hyperoxic conditions[135]. Additionally, research has demonstrated that therapeutic strategies focusing on Ca²⁺ homeostasis such as agents that restore Ca²⁺ balance can significantly ameliorate PH in animal models, highlighting substantial therapeutic potential[17]. Thus, agents that modulate Ca²⁺ signaling pathways are regarded as promising therapeutic targets. In recent years, research on modulators of endogenous signaling molecules has grown, with a particular focus on inositol triphosphate receptor 3 (IP3R3) inhibitors and Ca²⁺ channel modulators.
As a key channel for intracellular Ca²⁺ release, abnormal IP3R function is closely linked to the development of various diseases. In PH, altered expression and activity of IP3R lead to excessive Ca²⁺ release in smooth muscle cells, driving their proliferation and migration, and exacerbating pulmonary artery remodeling and hypertension. Studies have demonstrated that IP3R3 lacking can effectively reduce migration, proliferation and mesenchymal transition of PAECs induced by hypoxia and improve physiological indicators in PH animal models, offering new insights for PH treatment[73].
Notably, while these targeted drugs have exhibited favorable efficacy in experimental and clinical studies, their long-term safety and effectiveness require further validation. Future research should focus on optimizing their administration and exploring combinations with other therapies to achieve better outcomes. Moreover, the identification of new biomarkers may enable more precise personalized strategies for Ca²⁺ signaling-targeted therapy.
Understandably, these findings not only offer critical insights into deepening our understanding of PH pathogenesis but also expand options for clinical intervention. IP3R3 inhibitors and Ca²⁺ channel modulators offer new targets and approaches for PH treatment. Ongoing research will further advance their clinical application, contributing to improved prognosis and quality of life for PH patients.
4.4. Autophagy Regulation-Related Functional Roles and Their Therapeutic Targets
Autophagy has been recognized as a crucial mechanism that sustains energy metabolism, thereby fueling cell proliferation, supporting cell survival, and suppressing apoptosis. This regulatory role of autophagy is not only well-documented in cancer[136,137], but also implicated in PH [77]. Study showed that treatment with ROC-325 was found to suppress autophagy in the lung tissues of rats exposed to monocrotaline (MCT). This inhibitory effect is supported by the observation that ROC-325 reversed the MCT-induced upregulation of LC3B and p62 expression levels[138]. The clinical application prospects of autophagy regulators in the treatment of PH are remarkably broad, with rapamycin, an inhibitor of mammalian target of rapamycin (mTOR), standing out as a particularly notable candidate. Rapamycin has been extensively studied and confirmed to promote autophagy by inhibiting the mTOR pathway, effectively improving pulmonary arterial hypertension and vascular remodeling in PH models[139]. Additionally, Research has shown that metformin promotes autophagy by activating the AMPK signaling pathway, thereby improving metabolic disorders and related cardiovascular pathological changes[140,141].
The treatment of PH targeting autophagy is a crucial breakthrough direction from "relieving symptoms" to "reversing pathological remodeling". Among them, AMPK activators (such as metformin) are currently the most promising candidate drugs for conversion due to their high safety and solid clinical foundation; Lung targeted mTOR inhibitors and Beclin-1 agonists are expected to achieve more efficient treatment by precisely controlling autophagy.
5. Conclusions
A deeper understanding of mitochondria-ER crosstalk, particularly MAMs as a critical cellular structure, has gradually emerged as an innovative therapeutic target in the treatment of PH. Evidently, MAMs not only play pivotal roles in Ca²⁺ signaling transduction, lipid metabolism, and autophagy, but also exert significant impacts on controlling cellular stress responses, cell survival, and cell death. Studies have confirmed that MAMs dysfunctions are closely linked to the pathogenesis of PH, other cardiovascular disorders, metabolic syndrome, and neurodegenerative diseases[128,142].
The role of MAMs in PH pathogenesis is increasingly recognized. PH development is closely associated with mitochondrial dysfunction and ER stress, which exacerbate the disease by impairing MAMs structure and function. Specifically, MAMs integrity is critical for maintaining cellular Ca²⁺ balance and energy metabolism. Its disruption may enhance ER stress responses, thereby promoting PASMC proliferation and migration, ultimately contributing to PH development. Innovative therapeutic strategies targeting MAMs primarily by modulating IP3R, GRP75, and VDAC can improve mitochondria-ER interactions, thereby restoring normal cellular function. In support, enhancing the expression and activity of these proteins effectively alleviates mitochondrial dysfunction and ER stress, ameliorating PH pathology[128,143]. Progress has been made in developing new drugs that act on MAMs. For instance, natural products and synthetic compounds have been found to mitigate intracellular stress responses by controlling MAMs formation and function. Their underlying mechanisms may involve in MAM-related signaling pathways, thereby improving cellular metabolic status and survival capacity[144]. Combining with other therapies (e.g., immunotherapy or gene therapy), MAM-based targeted strategies can substantially enhance therapeutic efficacy. Furthermore, combining MAM-function-improving agents with traditional PH drugs such as prostaglandins or endothelin receptor antagonists yields synergistic effects, thereby boosting therapeutic outcomes[145].
Manifestly, innovative MAM-targeted therapeutic strategies offer new insights for PH treatments. These approaches not only improve mitochondria-ER interactions, but also alleviate PH pathological processes by controlling intracellular Ca²⁺ balance and energy metabolism. Future research should further explore MAM roles in PH and other related diseases to offer new ideas and directions for their novel and more effective clinical treatments and improve patient prognosis[16]. It is hoped that this intensive review article will shed new lights on the functional importance of mitochondria and ER interactions in PH and the discovery of new PH drugs, ultimately contributing to human health.
Funding Statement
This study received support from the National Key Research and Development Program of China (2019YFE0119400), the Natural Science Foundation of China (82370060, 82170057, 81970052, and 81770059), and Grant of State Key Laboratory of Respiratory Disease (SKLRD-OP-202301/202504).
Author Contributions
J.B. and H.S. drafted the manuscript; C.H., D.B., and J.Z. reviewed and edited the manuscript prior to submission; L.Y., C.B., and S.L. conducted literature search and screening; Q.W. and Y.W. provided suggestions for improving the language expression and logical coherence of the revised pre-submission manuscript; H.T. constructed the core conceptual framework of the review. All authors have read and approved the final published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest
Abbreviations
The following abbreviations are used in this manuscript:
| ER | Endoplasmic reticulum |
| ERS | Endoplasmic Reticulum Stress |
| IP3Rs | Inositol triphosphate receptors |
| MAMs | Mitochondria-associated endoplasmic reticulum membranes |
| MCU | mitochondrial Ca²⁺ uniporter |
| NO | Nitric oxide |
| PAH | Pulmonary arterial hypertension |
| PASMCs | pulmonary artery smooth muscle cells |
| PAECs | Pulmonary artery endothelial cells |
| ROS | Reactive Oxygen Species |
| VDAC1 | Voltage-dependent anion channel 1 |
References
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated Clinical Classification of Pulmonary Hypertension. Journal of the American College of Cardiology 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; Jing, Z.C.; Gibbs, J.S. A global view of pulmonary hypertension. Lancet Respir Med 2016, 4, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022, 43, 3618–3731. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M. Pathobiology of Pulmonary Hypertension. Annual Review of Pathology: Mechanisms of Disease 2007, 2, 369–399. [Google Scholar] [CrossRef]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. Journal of Clinical Investigation 2012, 122, 4306–4313. [Google Scholar] [CrossRef]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; Tuder, R.M. Modern Age Pathology of Pulmonary Arterial Hypertension. American Journal of Respiratory and Critical Care Medicine 2012, 186, 261–272. [Google Scholar] [CrossRef]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef]
- Gao, P.; Yan, Z.; Zhu, Z. Mitochondria-Associated Endoplasmic Reticulum Membranes in Cardiovascular Diseases. Frontiers in Cell and Developmental Biology 2020, 8. [Google Scholar] [CrossRef]
- Thenappan, T.; Al-Naamani, N.; Ghio, S.; Ghofrani, H.A.; Hassoun, P.M.; Pritzker, M.; Torbicki, A.; Nikkho, S.; Busse, D.; Preston, I.R. Effect of riociguat on pulmonary arterial compliance in the PATENT and CHEST studies. Pulmonary Circulation 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Huang, L.; Li, W.; Yang, T.; Xiong, C.; Ni, X.; Gu, Q.; He, J. Association between splenectomy and portal hypertension in the development of pulmonary hypertension. Pulmonary Circulation 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Sahay, S.; Tsang, Y.; Flynn, M.; Agron, P.; Dufour, R. Burden of pulmonary hypertension in patients with portal hypertension in the United States: a retrospective database study. Pulmonary Circulation 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Barnett, S.D.; King, C.S.; Provencher, S.; Barbera, J.A.; Pastre, J.; Shlobin, O.A.; Seeger, W. Impact of the new definition for pulmonary hypertension in patients with lung disease: an analysis of the United Network for Organ Sharing database. Pulmonary Circulation 2021, 11, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Chen, L.; Ye, F.; Tang, J.; Liu, B.; Lin, J.; Zhou, P.-H.; Lu, B.; Wu, M.; Lu, J.-H.; et al. Mic19 depletion impairs endoplasmic reticulum-mitochondrial contacts and mitochondrial lipid metabolism and triggers liver disease. Nature Communications 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Wu, S.; Wang, Z.; Shen, G.; Shi, X. Biotoxicity of paraquat to lung cells mediated by endoplasmic reticulum-mitochondria interaction. Journal of Molecular Histology 2024, 55, 1063–1077. [Google Scholar] [CrossRef]
- Rajagopal, S; Ghadimi, R.K.; Horn, K; Kelava, EM; Kudelko, M; Moreno-Duarte, KT; Preston, I; Rose Bovino, I; Smilowitz, LL; Vaidya, NR; A; American Heart Association Council on Cardiopulmonary; Critical Care; Perioperative and Resuscitation; and the Council on Cardiovascular and Stroke Nursing. Evaluation and Management of Pulmonary Hypertension in Noncardiac Surgery_ A Scientific Statement From the American Heart Association. Circulation 2023, 147, 1317–134. [CrossRef]
- Friedman, S.H.; Harley, R.A.; Williams, J.; Forcucci, J.A.; Ramakrishnan, V.; Strange, C.; Silver, R.M.; Feghali-Bostwick, C.; Argula, R.G. Pathologic differences between systemic sclerosis–associated and idiopathic pulmonary arterial hypertension. Journal of Scleroderma and Related Disorders 2024. [Google Scholar] [CrossRef]
- Mocumbi, A.; Humbert, M.; Saxena, A.; Jing, Z.-C.; Sliwa, K.; Thienemann, F.; Archer, S.L.; Stewart, S. Pulmonary hypertension. Nature Reviews Disease Primers 2024, 10. [Google Scholar] [CrossRef]
- Garry, J.D.; Kundu, S.; Annis, J.; Alcorn, C.; Eden, S.; Smith, E.; Greevy, R.; Maron, B.A.; Freiberg, M.S.; Brittain, E.L. Incidence of Pulmonary Hypertension in the Echocardiography Referral Population. Ann Am Thorac Soc 2025, 22. [Google Scholar] [CrossRef]
- Bonnet, D.; Szezepanski, I.; Delacourt, C.; Malkezadeh-Milani, S.; Lévy, M. Multifactorial pulmonary hypertension in infantile scimitar syndrome. Archives of Cardiovascular Diseases 2022, 115, 142–150. [Google Scholar] [CrossRef]
- Takano, R.; Aoki, T.; Asano, R.; Ueda, J.; Tsuji, A.; Omae, K.; Ogo, T. Recurrent pulmonary hypertension after balloon pulmonary angioplasty for inoperable chronic thromboembolic pulmonary hypertension. The Journal of Heart and Lung Transplantation 2024, 43, 737–744. [Google Scholar] [CrossRef]
- Yang, Y.D.; Li, M.M.; Xu, G.; Zhang, E.L.; Chen, J.; Sun, B.; Chen, D.W.; Gao, Y.Q. Targeting mitochondria-associated membranes as a potential therapy against endothelial injury induced by hypoxia. Journal of Cellular Biochemistry 2019, 120, 18967–18978. [Google Scholar] [CrossRef]
- Muñoz, E.; Valero, R.A.; Quintana, A.; Hoth, M.; Núñez, L.; Villalobos, C. Nonsteroidal Anti-inflammatory Drugs Inhibit Vascular Smooth Muscle Cell Proliferation by Enabling the Ca2+-dependent Inactivation of Calcium Release-activated Calcium/Orai Channels Normally Prevented by Mitochondria. Journal of Biological Chemistry 2011, 286, 16186–16196. [Google Scholar] [CrossRef] [PubMed]
- V, T. (2021 Mar 13). Life (Basel) 11. [CrossRef]
- V, T. Oligodendroglial Energy Metabolism and (re)Myelination. Life (Basel) 2021, 11, 238. [Google Scholar] [CrossRef]
- Luo, L.; Yin, H.; Gou, D. Gut Microbiota and Metabolome Changes in Three Pulmonary Hypertension Rat Models. Microorganisms 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death & Disease 2018, 9. [Google Scholar] [CrossRef]
- Dorn, G.W. Mitofusins as mitochondrial anchors and tethers. Journal of Molecular and Cellular Cardiology 2020, 142, 146–153. [Google Scholar] [CrossRef]
- Szabadkai, G.r.; Bianchi, K.; Várnai, P.t.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.s.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. The Journal of Cell Biology 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Peng, Y.; Zhou, L.; Jin, Y.; Wu, D.; Chen, N.; Zhang, C.; Liu, H.; Li, C.; Ning, R.; Yang, X.; et al. Calcium bridges built by mitochondria-associated endoplasmic reticulum membranes: potential targets for neural repair in neurological diseases. Neural Regeneration Research 2025, 20, 3349–3369. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Current Biology 2017, 27, 371–385. [Google Scholar] [CrossRef]
- Lim, Y.; Cho, I.T.; Schoel, L.J.; Cho, G.; Golden, J.A. Hereditary spastic paraplegia-linked REEP1 modulates endoplasmic reticulum/mitochondria contacts. Annals of Neurology 2015, 78, 679–696. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef]
- Gutiérrez, T.; Simmen, T. Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium 2018, 70, 64–75. [Google Scholar] [CrossRef]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proceedings of the National Academy of Sciences 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Yin, X.-M. Mitophagy: mechanisms, pathophysiological roles, and analysis. bchm 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. Journal of Hepatology 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Zhang, S.-S.; Zhou, S.; Crowley-McHattan, Z.J.; Wang, R.-Y.; Li, J.-P. A Review of the Role of Endo/Sarcoplasmic Reticulum-Mitochondria Ca2+ Transport in Diseases and Skeletal Muscle Function. International Journal of Environmental Research and Public Health 2021, 18. [Google Scholar] [CrossRef]
- Pokharel, M.D.; Garcia-Flores, A.; Marciano, D.; Franco, M.C.; Fineman, J.R.; Aggarwal, S.; Wang, T.; Black, S.M. Mitochondrial network dynamics in pulmonary disease: Bridging the gap between inflammation, oxidative stress, and bioenergetics. Redox Biology 2024, 70. [Google Scholar] [CrossRef]
- Wu, D.; Jansen-van Vuuren, R.D.; Dasgupta, A.; Al-Qazazi, R.; Chen, K.-H.; Martin, A.Y.; Mewburn, J.D.; Alizadeh, E.; Lima, P.D.A.; Jones, O.; et al. Novel Drp1 GTPase Inhibitor, Drpitor1a: Efficacy in Pulmonary Hypertension. Hypertension 2024, 81, 2189–2201. [Google Scholar] [CrossRef]
- Colpman, P.; Dasgupta, A.; Archer, S.L. The Role of Mitochondrial Dynamics and Mitotic Fission in Regulating the Cell Cycle in Cancer and Pulmonary Arterial Hypertension: Implications for Dynamin-Related Protein 1 and Mitofusin2 in Hyperproliferative Diseases. Cells 2023, 12. [Google Scholar] [CrossRef]
- Luo, F.; Fu, M.; Wang, T.; Qi, Y.; Zhong, X.; Li, D.; Liu, B. Down-regulation of the mitochondrial fusion protein Opa1/Mfn2 promotes cardiomyocyte hypertrophy in Su5416/hypoxia-induced pulmonary hypertension rats. Archives of Biochemistry and Biophysics 2023, 747. [Google Scholar] [CrossRef]
- Heresi, G.A.; Mey, J.T.; Bartholomew, J.R.; Haddadin, I.S.; Tonelli, A.R.; Dweik, R.A.; Kirwan, J.P.; Kalhan, S.C. Plasma metabolomic profile in chronic thromboembolic pulmonary hypertension. Pulmonary Circulation 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, C.; Ma, M.; Zhang, L.; Zhang, L.; Zhang, F.; Chen, Y.; Cao, F.; Li, M.; Wang, G.; et al. MiR-125a regulates mitochondrial homeostasis through targeting mitofusin 1 to control hypoxic pulmonary vascular remodeling. Journal of Molecular Medicine 2017, 95, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Potus, F.; Wu, D.; Dasgupta, A.; Chen, K.-H.; Mewburn, J.; Lima, P.; Archer, S.L. Increased Drp1-Mediated Mitochondrial Fission Promotes Proliferation and Collagen Production by Right Ventricular Fibroblasts in Experimental Pulmonary Arterial Hypertension. Frontiers in Physiology 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-H.; Dasgupta, A.; Lin, J.; Potus, F.; Bonnet, S.; Iremonger, J.; Fu, J.; Mewburn, J.; Wu, D.; Dunham-Snary, K.; et al. Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension. Circulation 2018, 138, 287–304. [Google Scholar] [CrossRef]
- Ryan, J.J.; Marsboom, G.; Fang, Y.-H.; Toth, P.T.; Morrow, E.; Luo, N.; Piao, L.; Hong, Z.; Ericson, K.; Zhang, H.J.; et al. PGC1α-mediated Mitofusin-2 Deficiency in Female Rats and Humans with Pulmonary Arterial Hypertension. American Journal of Respiratory and Critical Care Medicine 2013, 187, 865–878. [Google Scholar] [CrossRef]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.-H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-Related Protein 1–Mediated Mitochondrial Mitotic Fission Permits Hyperproliferation of Vascular Smooth Muscle Cells and Offers a Novel Therapeutic Target in Pulmonary Hypertension. Circulation Research 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, Y.; Wang, Y.; Fan, L.; Zhu, Y.; Chen, Y.; Wang, Y.; Zhu, Y.; Wang, H.; Pan, Z.; et al. Endothelial FIS1 DeSUMOylation Protects Against Hypoxic Pulmonary Hypertension. Circulation Research 2023, 133, 508–531. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Ma, L.; Chen, C.; Hai, S.; Wang, C.; Rahman, S.U.; Huang, W.; Zhao, C.; Feng, S.; Wang, X. Inhibition of Mitochondrial Fission Alleviates Zearalenone-Induced Mitochondria-Associated Endoplasmic Reticulum Membrane Dysfunction in Piglet Sertoli Cells. Toxins 2023, 15. [Google Scholar] [CrossRef]
- Li, X.; Yang, Q.; Liu, S.; Song, S.; Wang, C. Mitochondria-associated endoplasmic reticulum membranes promote mitochondrial fission through AKAP1-Drp1 pathway in podocytes under high glucose conditions. Experimental Cell Research 2023, 424. [Google Scholar] [CrossRef]
- Dunham-Snary, K.J.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.G.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic Pulmonary Vasoconstriction: From Molecular Mechanisms to Medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Masson, B.; Saint-Martin Willer, A.; Dutheil, M.; Penalva, L.; Le Ribeuz, H.; El Jekmek, K.; Ruchon, Y.; Cohen-Kaminsky, S.; Sabourin, J.; Humbert, M.; et al. Contribution of transient receptor potential canonical channels in human and experimental pulmonary arterial hypertension. American Journal of Physiology-Lung Cellular and Molecular Physiology 2023, 325, L246–L261. [Google Scholar] [CrossRef] [PubMed]
- Masson, B.; Montani, D.; Humbert, M.; Capuano, V.; Antigny, F. Role of Store-Operated Ca2+ Entry in the Pulmonary Vascular Remodeling Occurring in Pulmonary Arterial Hypertension. Biomolecules 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Chen, J.; Wang, T.; Chen, B.; Yang, L.; Liao, J.; Chen, Y.; Wang, J; Tang, H.; Yi, J.; et al. PDGF_MEK_ERK axis represses Ca2+ clearance via decreasing the abundance of plasma membrane Ca2+ pump PMCA4 in pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 2021, 320, C66–C79. [Google Scholar] [CrossRef]
- Neumann, J.; Van Nieuwenhove, E.; Terry, L.E.; Staels, F.; Knebel, T.R.; Welkenhuyzen, K.; Ahmadzadeh, K.; Baker, M.R.; Gerbaux, M.; Willemsen, M.; et al. Disrupted Ca2+ homeostasis and immunodeficiency in patients with functional IP3 receptor subtype 3 defects. Cellular & Molecular Immunology 2022, 20, 11–25. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium 2018, 69, 81–100. [Google Scholar] [CrossRef]
- Chen, Y.; Csordás, G.; Jowdy, C.; Schneider, T.G.; Csordás, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-Containing Mitochondrial-Reticular Microdomains Direct Rapid Cardiomyocyte Bioenergetic Responses Via Interorganelle Ca2+Crosstalk. Circulation Research 2012, 111, 863–875. [Google Scholar] [CrossRef]
- Han, C.; Kim, J. Transcriptome profiling reveals novel insights into the regulation of calcium ion and detoxification genes driving chlorantraniliprole resistance in Spodoptera exigua. Heliyon 2024. [Google Scholar] [CrossRef]
- Wang, Q.-C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 Is an ER Ca 2+ Load-Activated Ca 2+ Channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. The Journal of Cell Biology 2003, 160, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Choy, G.; Deng, X.; Bhatia, B.; Daniel, P.; Tang, D.G. Association of Active Caspase 8 with the Mitochondrial Membrane during Apoptosis: Potential Roles in Cleaving BAP31 and Caspase 3 and Mediating Mitochondrion-Endoplasmic Reticulum Cross Talk in Etoposide-Induced Cell Death. Molecular and Cellular Biology 2023, 24, 6592–6607. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. The EMBO Journal 2011, 30, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.-H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum–mitochondria communication and Bid-mediated apoptosis. The EMBO Journal 2005, 24, 717–729. [Google Scholar] [CrossRef]
- Hong, Z.; Chen, K.-H.; DasGupta, A.; Potus, F.; Dunham-Snary, K.; Bonnet, S.; Tian, L.; Fu, J.; Breuils-Bonnet, S.; Provencher, S.; et al. MicroRNA-138 and MicroRNA-25 Down-regulate Mitochondrial Calcium Uniporter, Causing the Pulmonary Arterial Hypertension Cancer Phenotype. American Journal of Respiratory and Critical Care Medicine 2017, 195, 515–529. [Google Scholar] [CrossRef]
- Monaco, G.; Decrock, E.; Arbel, N.; van Vliet, A.R.; La Rovere, R.M.; De Smedt, H.; Parys, J.B.; Agostinis, P.; Leybaert, L.; Shoshan-Barmatz, V.; Bultynck, G. The BH4 Domain of Anti-apoptotic Bcl-XL, but Not That of the Related Bcl-2, Limits the Voltage-dependent Anion Channel 1 (VDAC1)-mediated Transfer of Pro-apoptotic Ca2+ Signals to Mitochondria. Journal of Biological Chemistry 2015, 290, 9150–9161. [Google Scholar] [CrossRef]
- Chen, J.; Rodriguez, M.; Miao, J.; Liao, J.; Jain, P.P.; Zhao, M.; Zhao, T.; Babicheva, A.; Wang, Z.; Parmisano, S.; et al. Mechanosensitive channel Piezo1 is required for pulmonary artery smooth muscle cell proliferation. American Journal of Physiology-Lung Cellular and Molecular Physiology 2022, 322, L737–L760. [Google Scholar] [CrossRef]
- Romito, O.; Guéguinou, M.; Raoul, W.; Champion, O.; Robert, A.; Trebak, M.; Goupille, C.; Potier-Cartereau, M. Calcium signaling: A therapeutic target to overcome resistance to therapies in cancer. Cell Calcium 2022, 108. [Google Scholar] [CrossRef]
- Tang, H.; Yamamura, A.; Yamamura, H.; Song, S.; Fraidenburg, D.R.; Chen, J.; Gu, Y.; Pohl, N.M.; Zhou, T.; Jiménez-Pérez, L.; et al. Pathogenic role of calcium-sensing receptors in the development and progression of pulmonary hypertension. American Journal of Physiology-Lung Cellular and Molecular Physiology 2016, 310, L846–L859. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Yu, J.; Li, Z.; Zhang, M.; Chen, Y.; Liu, F.; Jiang, D.; Guo, J.; Li, X.; Wu, Y. Mfn2 regulates calcium homeostasis and suppresses PASMCs proliferation via interaction with IP3R3 to mitigate pulmonary arterial hypertension. Journal of Translational Medicine 2025, 23. [Google Scholar] [CrossRef]
- Guo, X.; Meng, Y.; Wang, Y.; Nan, S.; Lu, Y.; Lu, D.; Yin, Y. Mice lacking 1,4,5-triphosphate inositol type III receptor demonstrate inhibition of hypoxic pulmonary hypertension. Biochemical and Biophysical Research Communications 2022, 629, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.-L.; Yi, J.; Cao, X.-Y.; Wang, F.-Y.; Xie, S.-L.; Zhou, L.-L.; Qin, L.; Dai, A.-G. Celastrol: The new dawn in the treatment of vascular remodeling diseases. Biomedicine & Pharmacotherapy 2023, 158. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Dromparis, P.; Wright, P.; Bonnet, S.; Haromy, A.; Hao, Z.; McMurtry, M.S.; Michalak, M.; Vance, J.E.; Sessa, W.C.; Michelakis, E.D. The Role of Nogo and the Mitochondria–Endoplasmic Reticulum Unit in Pulmonary Hypertension. Science Translational Medicine 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.P.; Zorzano, A. Endoplasmic reticulum stress enters a Nogo zone. Sci Transl Med 2011, 3, 88ps26. [Google Scholar] [CrossRef]
- Zhang, C.F.; Zhao, F.Y.; Xu, S.L.; Liu, J.; Xing, X.Q.; Yang, J. Autophagy in pulmonary hypertension: Emerging roles and therapeutic implications. Journal of Cellular Physiology 2019, 234, 16755–16767. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.-Y.; Shao, J.; Zhu, L.; Xie, T.-H.; Cai, J.; Wang, W.; Cai, M.-X.; Wang, Z.-L.; Yao, Y.; Wei, T.-T. GRP75 Modulates Endoplasmic Reticulum–Mitochondria Coupling and Accelerates Ca2+-Dependent Endothelial Cell Apoptosis in Diabetic Retinopathy. Biomolecules 2022, 12. [Google Scholar] [CrossRef]
- Koyama, M.; Furuhashi, M.; Ishimura, S.; Mita, T.; Fuseya, T.; Okazaki, Y.; Yoshida, H.; Tsuchihashi, K.; Miura, T. Reduction of endoplasmic reticulum stress by 4-phenylbutyric acid prevents the development of hypoxia-induced pulmonary arterial hypertension. American Journal of Physiology-Heart and Circulatory Physiology 2014, 306, H1314–H1323. [Google Scholar] [CrossRef]
- Victor, P.; Umapathy, D.; George, L.; Juttada, U.; Ganesh, G.V.; Amin, K.N.; Viswanathan, V.; Ramkumar, K.M. Crosstalk between endoplasmic reticulum stress and oxidative stress in the progression of diabetic nephropathy. Cell Stress and Chaperones 2021, 26, 311–321. [Google Scholar] [CrossRef]
- Ding, W.; Zhang, X.; Zhang, Q.; Dong, Y.; Wang, W.; Ding, N. Adiponectin ameliorates lung injury induced by intermittent hypoxia through inhibition of ROS-associated pulmonary cell apoptosis. Sleep and Breathing 2020, 25, 459–470. [Google Scholar] [CrossRef]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and Immunity in the Pathogenesis of Pulmonary Arterial Hypertension. Circulation Research 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Golembeski, S.M.; West, J.; Tada, Y.; Fagan, K.A. Interleukin-6 Causes Mild Pulmonary Hypertension and Augments Hypoxia-Induced Pulmonary Hypertension in Mice. Chest 2005, 128, 572S–573S. [Google Scholar] [CrossRef]
- Rieusset, J. Mitochondria-associated membranes (MAMs): An emerging platform connecting energy and immune sensing to metabolic flexibility. Biochemical and Biophysical Research Communications 2018, 500, 35–44. [Google Scholar] [CrossRef]
- Liu, Helene M.; Loo, Y.-M.; Horner, Stacy M.; Zornetzer, Gregory A.; Katze, Michael G.; Gale, M. The Mitochondrial Targeting Chaperone 14-3-3ε Regulates a RIG-I Translocon that Mediates Membrane Association and Innate Antiviral Immunity. Cell Host & Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nature Medicine 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. International Journal of Molecular Sciences 2019, 20. [Google Scholar] [CrossRef]
- Zhao, J.; Li, J.; Li, G.; Chen, M. The role of mitochondria-associated membranes mediated ROS on NLRP3 inflammasome in cardiovascular diseases. Frontiers in Cardiovascular Medicine 2022, 9. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2010, 469, 221–225. [Google Scholar] [CrossRef]
- Li, W.; Cao, T.; Luo, C.; Cai, J.; Zhou, X.; Xiao, X.; Liu, S. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Applied Microbiology and Biotechnology 2020, 104, 6129–6140. [Google Scholar] [CrossRef]
- Humbert M, M.G., Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. (1995). Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 151, 1628-1631. [CrossRef]
- Itoh T, N.N., Ishibashi-Ueda H, Kyotani S, Oya H, Sakamaki F, Kimura H, Nakanishi N. (2006). Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology 11, 158-163. [CrossRef] [PubMed]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 Overexpression Induces Pulmonary Hypertension. Circulation Research 2009, 104, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhao, Y.; Li, S.; Wu, H.; Ma, D.; Wan, C. TNF-α and IL-8 levels are positively correlated with hypobaric hypoxic pulmonary hypertension and pulmonary vascular remodeling in rats. Open Life Sciences 2023, 18. [Google Scholar] [CrossRef] [PubMed]
- Vringer, E.; Tait, S.W.G. Mitochondria and cell death-associated inflammation. Cell Death & Differentiation 2022, 30, 304–312. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxidants & Redox Signaling 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells 2021, 10. [Google Scholar] [CrossRef]
- Kazmirczak, F.; Vogel, N.T.; Prisco, S.Z.; Patterson, M.T.; Annis, J.; Moon, R.T.; Hartweck, L.M.; Mendelson, J.B.; Kim, M.; Calixto Mancipe, N.; et al. Ferroptosis-Mediated Inflammation Promotes Pulmonary Hypertension. Circulation Research 2024, 135, 1067–1083. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9. [Google Scholar] [CrossRef]
- Saraji, A.; Sydykov, A.; Schafer, K.; Garcia-Castro, C.F.; Henneke, I.; Alebrahimdehkordi, N.; Kosanovic, D.; Hadzic, S.; Guenther, A.; Hecker, M.; et al. PINK1-mediated Mitophagy Contributes to Pulmonary Vascular Remodeling in Pulmonary Hypertension. Am J Respir Cell Mol Biol 2021, 65, 226–228. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca2+ transfer to sustain cell bioenergetics. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2013, 1832, 495–508. [Google Scholar] [CrossRef]
- Wu, W.; Li, W.; Chen, H.; Jiang, L.; Zhu, R.; Feng, D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy 2016, 12, 1675–1676. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, H.; Li, J.; Xin, Y.; Li, J. Phosphatidylethanolamine N-methyltransferase: from Functions to Diseases. Aging and disease 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhao, H.; Kalionis, B.; Huai, X.; Hu, X.; Wu, W.; Jiang, R.; Gong, S.; Wang, L.; Liu, J.; et al. The Impact of Abnormal Lipid Metabolism on the Occurrence Risk of Idiopathic Pulmonary Arterial Hypertension. International Journal of Molecular Sciences 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, X.; Zhang, L.; Zhu, X.; Bai, J.; He, S.; Mei, J.; Jiang, J.; Guan, X.; Zheng, X.; et al. Super Enhancer-Associated Circular RNA-CircKrt4 Regulates Hypoxic Pulmonary Artery Endothelial Cell Dysfunction in Mice. Arterioscler Thromb Vasc Biol 2023, 43, 1179–1198. [Google Scholar] [CrossRef]
- Newton, D.A.; Lottes, R.G.; Ryan, R.M.; Spyropoulos, D.D.; Baatz, J.E. Dysfunctional lactate metabolism in human alveolar type II cells from idiopathic pulmonary fibrosis lung explant tissue. Respiratory Research 2021, 22. [Google Scholar] [CrossRef]
- Leopold, J.; Maron, B. Molecular Mechanisms of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences 2016, 17. [Google Scholar] [CrossRef]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2α Contributes to Severe Pulmonary Hypertension by Inducing Endothelial-to-Mesenchymal Transition. American Journal of Physiology-Lung Cellular and Molecular Physiology 2017. [Google Scholar] [CrossRef]
- Plecitá-Hlavatá, L.; Tauber, J.; Li, M.; Zhang, H.; Flockton, A.R.; Pullamsetti, S.S.; Chelladurai, P.; D’Alessandro, A.; El Kasmi, K.C.; Ježek, P.; Stenmark, K.R. Constitutive Reprogramming of Fibroblast Mitochondrial Metabolism in Pulmonary Hypertension. American Journal of Respiratory Cell and Molecular Biology 2016, 55, 47–57. [Google Scholar] [CrossRef]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.B.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate Prevents and Reverses Pulmonary Hypertension by Inducing Pulmonary Artery Smooth Muscle Cell Apoptosis. Circulation Research 2004, 95, 830–840. [Google Scholar] [CrossRef]
- Guignabert, C.; Tu, L.; Izikki, M.; Dewachter, L.; Zadigue, P.; Humbert, M.; Adnot, S.; Fadel, E.; Eddahibi, S. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22α-targeted overexpression of the serotonin transporter. The FASEB Journal 2009, 23, 4135–4147. [Google Scholar] [CrossRef]
- Zeng, W.-J.; Xiong, C.-M.; Zhao, L.; Shan, G.-L.; Liu, Z.-H.; Xue, F.; Gu, Q.; Ni, X.-H.; Zhao, Z.-H.; Cheng, X.-S.; et al. Atorvastatin in Pulmonary Arterial Hypertension (APATH) study. European Respiratory Journal 2012, 40, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Haslem, L.; Hays, J.M.; Hays, F.A. p66Shc in Cardiovascular Pathology. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. P66Shc and vascular endothelial function. Bioscience Reports 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Freund-Michel, V.; Guibert, C.; Dubois, M.; Courtois, A.; Marthan, R.; Savineau, J.-P.; Muller, B. Reactive oxygen species as therapeutic targets in pulmonary hypertension. Therapeutic Advances in Respiratory Disease 2013, 7, 175–200. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Ge, L.; Sun, W.; Song, Z.; Lu, X.; Jin, C.; Wu, S.; Yang, J. Lanthanum decreased VAPB-PTPP51, BAP31-FIS1, and MFN2-MFN1 expression of mitochondria-associated membranes and induced abnormal autophagy in rat hippocampus. Food and Chemical Toxicology 2022, 161. [Google Scholar] [CrossRef]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death & Differentiation 2013, 20, 1631–1643. [Google Scholar] [CrossRef]
- Williams, A.; Hayashi, T.; Wolozny, D.; Yin, B.; Su, T.-C.; Betenbaugh, M.J.; Su, T.-P. The non-apoptotic action of Bcl-xL: regulating Ca2+ signaling and bioenergetics at the ER-mitochondrion interface. Journal of Bioenergetics and Biomembranes 2016, 48, 211–225. [Google Scholar] [CrossRef]
- Lalier, L.; Mignard, V.; Joalland, M.-P.; Lanoé, D.; Cartron, P.-F.; Manon, S.; Vallette, F.M. TOM20-mediated transfer of Bcl2 from ER to MAM and mitochondria upon induction of apoptosis. Cell Death & Disease 2021, 12. [Google Scholar] [CrossRef]
- Lee, G.-H.; Lee, H.-Y.; Li, B.; Kim, H.-R.; Chae, H.-J. Bax Inhibitor-1-Mediated Inhibition of Mitochondrial Ca2+ Intake Regulates Mitochondrial Permeability Transition Pore Opening and Cell Death. Scientific Reports 2014, 4. [Google Scholar] [CrossRef]
- Takeda, K.; Nagashima, S.; Shiiba, I.; Uda, A.; Tokuyama, T.; Ito, N.; Fukuda, T.; Matsushita, N.; Ishido, S.; Iwawaki, T.; et al. MITOL prevents ER stress-induced apoptosis by IRE 1α ubiquitylation at ER–mitochondria contact sites. The EMBO Journal 2019, 38. [Google Scholar] [CrossRef]
- Yang, H.; Niemeijer, M.; van de Water, B.; Beltman, J.B. ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. iScience 2020, 23. [Google Scholar] [CrossRef] [PubMed]
- Misaka, T.; Murakawa, T.; Nishida, K.; Omori, Y.; Taneike, M.; Omiya, S.; Molenaar, C.; Uno, Y.; Yamaguchi, O.; Takeda, J.; et al. FKBP8 protects the heart from hemodynamic stress by preventing the accumulation of misfolded proteins and endoplasmic reticulum-associated apoptosis in mice. Journal of Molecular and Cellular Cardiology 2018, 114, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Wieckowski, M.R.; Paolo Pandolfi, P.; Pinton, P. Mitochondria associated membranes (MAMs) as critical hubs for apoptosis. Communicative & Integrative Biology 2014, 4, 334–335. [Google Scholar] [CrossRef]
- Patergnani, S.; Missiroli, S.; Marchi, S.; Giorgi, C. Mitochondria-Associated Endoplasmic Reticulum Membranes Microenvironment: Targeting Autophagic and Apoptotic Pathways in Cancer Therapy. Frontiers in Oncology 2015, 5. [Google Scholar] [CrossRef]
- Kurakula, K.; Smolders, V.F.E.D.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.-J. Endothelial Dysfunction in Pulmonary Hypertension: Cause or Consequence? Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Wang, C.; Dai, X.; Wu, S.; Xu, W.; Song, P.; Huang, K.; Zou, M.-H. FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nature Communications 2021, 12. [Google Scholar] [CrossRef]
- Li, T.; Li, S.; Feng, Y.; Zeng, X.; Dong, S.; Li, J.; Zha, L.; Luo, H.; Zhao, L.; Liu, B.; et al. Combination of Dichloroacetate and Atorvastatin Regulates Excessive Proliferation and Oxidative Stress in Pulmonary Arterial Hypertension Development via p38 Signaling. Oxidative Medicine and Cellular Longevity 2020, 2020, 1–15. [Google Scholar] [CrossRef]
- Pokharel, M.D.; Marciano, D.P.; Fu, P.; Franco, M.C.; Unwalla, H.; Tieu, K.; Fineman, J.R.; Wang, T.; Black, S.M. Metabolic reprogramming, oxidative stress, and pulmonary hypertension. Redox Biology 2023, 64. [Google Scholar] [CrossRef]
- Xu, D.; Hu, Y.-H.; Gou, X.; Li, F.-Y.; Yang, X.-Y.-C.; Li, Y.-M.; Chen, F. Oxidative Stress and Antioxidative Therapy in Pulmonary Arterial Hypertension. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Paulin, R.; Dromparis, P.; Sutendra, G.; Gurtu, V.; Zervopoulos, S.; Bowers, L.; Haromy, A.; Webster, L.; Provencher, S.; Bonnet, S.; Michelakis, Evangelos D. Sirtuin 3 Deficiency Is Associated with Inhibited Mitochondrial Function and Pulmonary Arterial Hypertension in Rodents and Humans. Cell Metabolism 2014, 20, 827–839. [Google Scholar] [CrossRef]
- Meng, M.; Jiang, Y.; Wang, Y.; Huo, R.; Ma, N.; Shen, X.; Chang, G. β-carotene targets IP3R/GRP75/VDAC1-MCU axis to renovate LPS-induced mitochondrial oxidative damage by regulating STIM1. Free Radical Biology and Medicine 2023, 205, 25–46. [Google Scholar] [CrossRef]
- An, P.; Wan, S.; Luo, Y.; Luo, J.; Zhang, X.; Zhou, S.; Xu, T.; He, J.; Mechanick, J.I.; Wu, W.-C.; et al. Micronutrient Supplementation to Reduce Cardiovascular Risk. Journal of the American College of Cardiology 2022, 80, 2269–2285. [Google Scholar] [CrossRef]
- Carbone, E.A.; Menculini, G.; de Filippis, R.; D’Angelo, M.; De Fazio, P.; Tortorella, A.; Steardo, L. Sleep Disturbances in Generalized Anxiety Disorder: The Role of Calcium Homeostasis Imbalance. International Journal of Environmental Research and Public Health 2023, 20. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discovery 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. European Journal of Pharmaceutical Sciences 2019, 134, 116–137. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Liang, S.; Han, Y.; Yang, Z.; Liu, S.; Sun, Y.; Zheng, S.; Li, Y.; Wang, T.; Gu, Y.; et al. The Novel Lysosomal Autophagy Inhibitor (ROC-325) Ameliorates Experimental Pulmonary Hypertension. Hypertension 2023, 80, 70–83. [Google Scholar] [CrossRef]
- Houssaini A, A.S. mTOR_ A Key to Both Pulmonary Vessel Remodeling and Right Ventricular Function in Pulmonary Arterial Hypertension. Am J Respir Cell Mol Biol 2017, 57, 509–511. [Google Scholar] [CrossRef]
- Zhao, Q.; Song, P.; Zou, M.-H. AMPK and Pulmonary Hypertension: Crossroads Between Vasoconstriction and Vascular Remodeling. Frontiers in Cell and Developmental Biology 2021, 9. [Google Scholar] [CrossRef]
- McNair, B.D.; Polson, S.M.; Shorthill, S.K.; Yusifov, A.; Walker, L.A.; Weiser-Evans, M.C.M.; Kovacs, E.J.; Bruns, D.R. Metformin protects against pulmonary hypertension-induced right ventricular dysfunction in an age- and sex-specific manner independent of cardiac AMPK. American Journal of Physiology-Heart and Circulatory Physiology 2023, 325, H278–H292. [Google Scholar] [CrossRef]
- Zhang, J.; Tao, J.; Zhou, Z.; Pei, W.; Xiao, Y.; Guo, Y.; Gao, J.; Jiang, C.; Dai, L.; Zhang, G.; Tan, C. Current research on mitochondria-associated membranes in cardiovascular diseases (Review). Molecular Medicine Reports 2025, 31, 1–17. [Google Scholar] [CrossRef]
- Li, Z.; Hu, O.; Xu, S.; Lin, C.; Yu, W.; Ma, D.; Lu, J.; Liu, P. The SIRT3-ATAD3A axis regulates MAM dynamics and mitochondrial calcium homeostasis in cardiac hypertrophy. International Journal of Biological Sciences 2024, 20, 831–847. [Google Scholar] [CrossRef]
- Wang, H.; Yi, X.; Guo, S.; Wang, S.; Ma, J.; Zhao, T.; Shi, Q.; Tian, Y.; Wang, H.; Jia, L.; et al. The XBP1‒MARCH5‒MFN2 Axis Confers Endoplasmic Reticulum Stress Resistance by Coordinating Mitochondrial Fission and Mitophagy in Melanoma. Journal of Investigative Dermatology 2021, 141, 2932–2943.e2912. [Google Scholar] [CrossRef]
- Kalarikkal, M.; Saikia, R.; Oliveira, L.; Bhorkar, Y.; Lonare, A.; Varshney, P.; Dhamale, P.; Majumdar, A.; Joseph, J. Nup358 restricts ER-mitochondria connectivity by modulating mTORC2/Akt/GSK3β signalling. EMBO Reports 2024, 25, 4226–4251. [Google Scholar] [CrossRef]
Figure 1.
Function of MAM and its related proteins. The glucose regulatory protein 75 (Grp75), voltage dependent anion channel (VDAC), and inositol triphosphate receptor (IP3R) located in the mitochondrial associated endoplasmic reticulum membrane (MAM) connect the endoplasmic reticulum (ER) with the outer mitochondrial membrane (OMM) and participate in the transport of Ca²⁺ from the ER to the mitochondria. The Sigma-1 receptor (Sig-1R) serves as a dynamic multifunctional scaffold protein for ER mitochondrial communication, binding to the molecular chaperone protein immunoglobulin binding protein (BiP)/glucose regulatory protein 78 (Grp78) to form a complex at the MAM site. The binding of vesicle associated membrane protein B (VAPB) on MAM with protein tyrosine phosphatase interacting protein 51 (PTPIP51) forms a tethering structure. The ERMES complex consists of four core components: ER protein mitochondrial morphogenetic protein 1 (Mmm1), cytoplasmic protein mitochondrial distribution and morphogenetic protein 12 (Mdm12), and mitochondrial outer membrane barrel proteins Mdm10 and Mdm34. Mitochondrial fusion protein 2 (MFN2) connects ER and mitochondria through its homologous (MFN2-MFN2) or heterologous (MFN2-Mfn1) interactions, playing a crucial role in MAM, participating in mitochondrial fusion and division, and maintaining Ca²⁺ homeostasis. Protein 1 containing the FUN14 domain (FUNDC1) is enriched at the MAM site by binding to the ER membrane protein calnein.
Figure 1.
Function of MAM and its related proteins. The glucose regulatory protein 75 (Grp75), voltage dependent anion channel (VDAC), and inositol triphosphate receptor (IP3R) located in the mitochondrial associated endoplasmic reticulum membrane (MAM) connect the endoplasmic reticulum (ER) with the outer mitochondrial membrane (OMM) and participate in the transport of Ca²⁺ from the ER to the mitochondria. The Sigma-1 receptor (Sig-1R) serves as a dynamic multifunctional scaffold protein for ER mitochondrial communication, binding to the molecular chaperone protein immunoglobulin binding protein (BiP)/glucose regulatory protein 78 (Grp78) to form a complex at the MAM site. The binding of vesicle associated membrane protein B (VAPB) on MAM with protein tyrosine phosphatase interacting protein 51 (PTPIP51) forms a tethering structure. The ERMES complex consists of four core components: ER protein mitochondrial morphogenetic protein 1 (Mmm1), cytoplasmic protein mitochondrial distribution and morphogenetic protein 12 (Mdm12), and mitochondrial outer membrane barrel proteins Mdm10 and Mdm34. Mitochondrial fusion protein 2 (MFN2) connects ER and mitochondria through its homologous (MFN2-MFN2) or heterologous (MFN2-Mfn1) interactions, playing a crucial role in MAM, participating in mitochondrial fusion and division, and maintaining Ca²⁺ homeostasis. Protein 1 containing the FUN14 domain (FUNDC1) is enriched at the MAM site by binding to the ER membrane protein calnein.
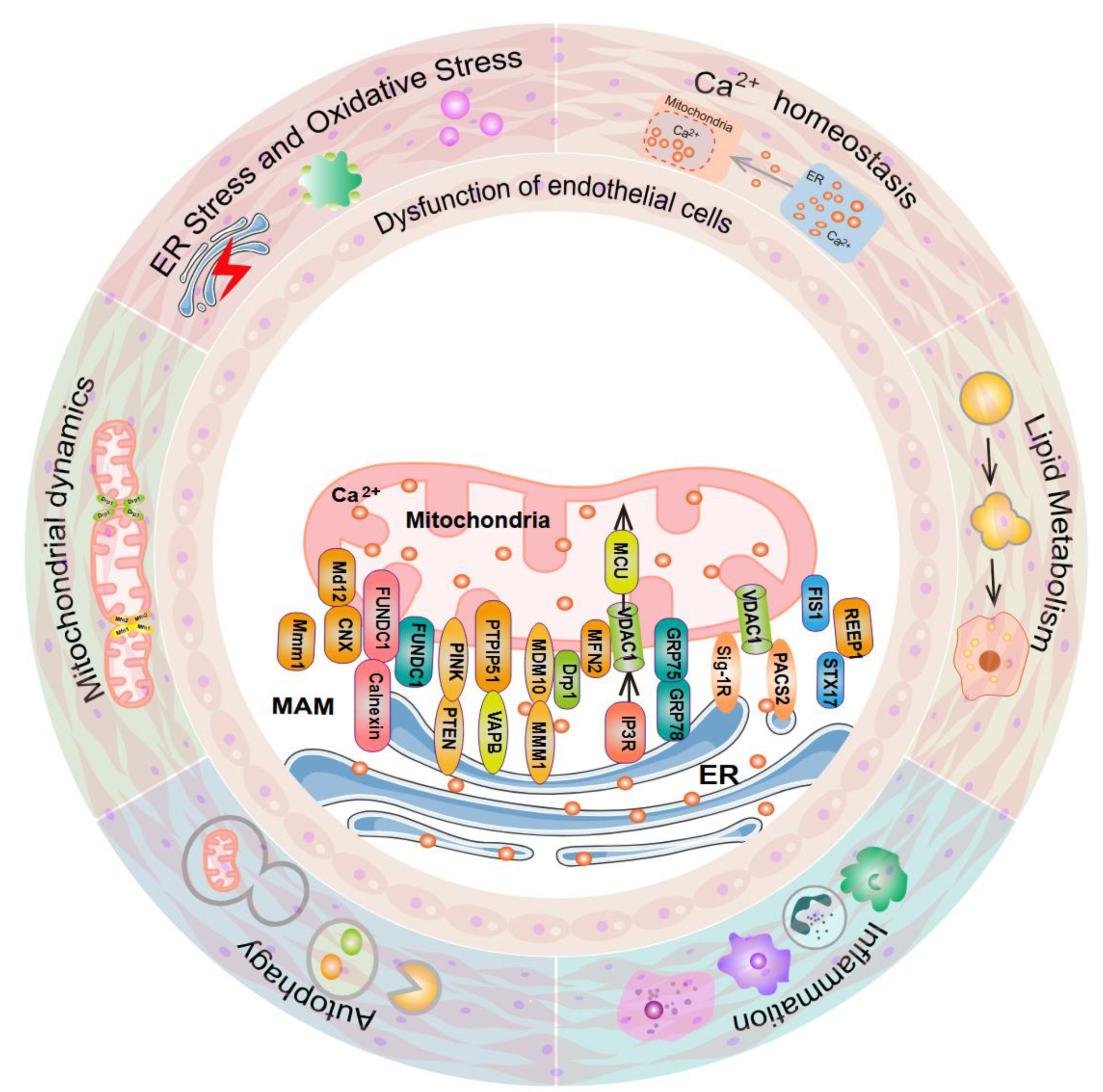
Figure 2.
MAMs in mitochondrial dynamics. Mitofusin 2 (MFN2) localized at MAMs initiates mitochondrial fusion through physical interaction with Mfn1 on the outer mitochondrial membrane. Prior to mitochondrial fission, dynamin-related protein 1 (DRP1) and its adaptors, the mitochondrial dynamics proteins 49 and 51 (MiD49 and MiD51), are specifically recruited to mitochondria-ER contact sites. Inverted formin 2 (INF2) on the ER, together with the mitochondria-localized actin nucleation protein Spire1C, coordinately governs DRP1 recruitment at these contact sites and drives the contraction of mitochondrial tubules. miR-214, miR-125a, miR-140 and PGC1α regulate the expression of Mfn1/2 and affect mitochondrial homeostasis, leading to pulmonary arterial hypertension;Reduced expression of miR-34a-3p leads to upregulation of MiD. Increased expression of MiD in PASMCs with PH accelerates Drp1 mediated mitosis, increases cell proliferation, and reduces apoptosis;MFN2 binds to IP3R3 to mediate mitochondrial Ca2+transport, inhibit PASMCs proliferation and pulmonary vascular remodeling; Short-term hypoxia induces SENP1 translocation to endothelial mitochondria to regulate the reversible process of SUMOylation of mitochondrial fission protein FIS1, which protects against pulmonary arterial hypertension;Mitochondrial mitosis mediated by motor protein-related protein 1 allows for excessive proliferation of vascular smooth muscle cells. Both DRP1 inhibitors Mdivi-1 and siDRP1 can prevent mitosis and block PH PASMCs in the G2/M interphase.
Figure 2.
MAMs in mitochondrial dynamics. Mitofusin 2 (MFN2) localized at MAMs initiates mitochondrial fusion through physical interaction with Mfn1 on the outer mitochondrial membrane. Prior to mitochondrial fission, dynamin-related protein 1 (DRP1) and its adaptors, the mitochondrial dynamics proteins 49 and 51 (MiD49 and MiD51), are specifically recruited to mitochondria-ER contact sites. Inverted formin 2 (INF2) on the ER, together with the mitochondria-localized actin nucleation protein Spire1C, coordinately governs DRP1 recruitment at these contact sites and drives the contraction of mitochondrial tubules. miR-214, miR-125a, miR-140 and PGC1α regulate the expression of Mfn1/2 and affect mitochondrial homeostasis, leading to pulmonary arterial hypertension;Reduced expression of miR-34a-3p leads to upregulation of MiD. Increased expression of MiD in PASMCs with PH accelerates Drp1 mediated mitosis, increases cell proliferation, and reduces apoptosis;MFN2 binds to IP3R3 to mediate mitochondrial Ca2+transport, inhibit PASMCs proliferation and pulmonary vascular remodeling; Short-term hypoxia induces SENP1 translocation to endothelial mitochondria to regulate the reversible process of SUMOylation of mitochondrial fission protein FIS1, which protects against pulmonary arterial hypertension;Mitochondrial mitosis mediated by motor protein-related protein 1 allows for excessive proliferation of vascular smooth muscle cells. Both DRP1 inhibitors Mdivi-1 and siDRP1 can prevent mitosis and block PH PASMCs in the G2/M interphase.
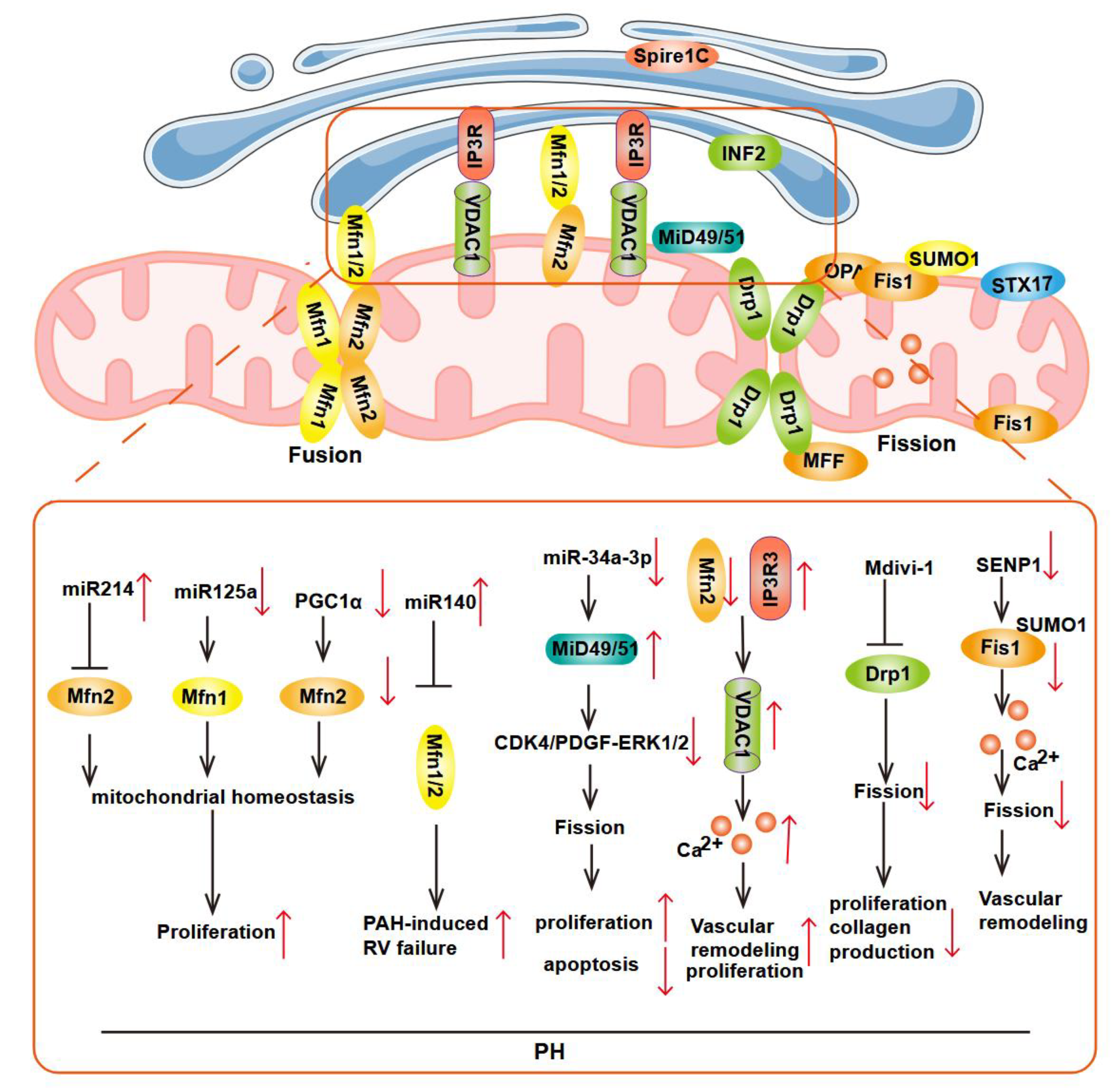
Figure 3.
MAMs in Ca²⁺ homeostasis. Mitochondrial Ca2+ elevation increases the activity of Ca2+-sensitive dehydrogenases, which diminishes the NAD+/NADH ratio. The NAD+-dependent deacetylase activity of sirtuin 3 (SIRT3) deacetylates superoxide dismutase 2 (SOD2), and the deacetylated and active form of SOD2 decreases ROS levels. Low levels of NAD+, caused by Ca2+-sensitive dehydrogenases, inhibit SIRT3 activity, and the inactive form of SOD2 allows accumulation of ROS, which activates the c-Jun N-terminal kinase pathway; IP3R3 bind Bcl-xL inhibiting apoptosis; VDAC1 bind Bcl-2 inhibiting apoptosis; MiR-25 binding to MCU mRNA promotes a decrease in mitochondrial Ca²⁺, an increase in cytoplasmic Ca²⁺ ions, and promotes cell proliferation; The Fis1-Bap31 complex (ARCosome), which spans the mitochondria-ER junction, acts as a platform for activating the initiator procaspase-8, thus connecting these two key organelles in apoptotic signaling. Caspase-3 activation and induction of PARP cleavage than induce apoptosis.
Figure 3.
MAMs in Ca²⁺ homeostasis. Mitochondrial Ca2+ elevation increases the activity of Ca2+-sensitive dehydrogenases, which diminishes the NAD+/NADH ratio. The NAD+-dependent deacetylase activity of sirtuin 3 (SIRT3) deacetylates superoxide dismutase 2 (SOD2), and the deacetylated and active form of SOD2 decreases ROS levels. Low levels of NAD+, caused by Ca2+-sensitive dehydrogenases, inhibit SIRT3 activity, and the inactive form of SOD2 allows accumulation of ROS, which activates the c-Jun N-terminal kinase pathway; IP3R3 bind Bcl-xL inhibiting apoptosis; VDAC1 bind Bcl-2 inhibiting apoptosis; MiR-25 binding to MCU mRNA promotes a decrease in mitochondrial Ca²⁺, an increase in cytoplasmic Ca²⁺ ions, and promotes cell proliferation; The Fis1-Bap31 complex (ARCosome), which spans the mitochondria-ER junction, acts as a platform for activating the initiator procaspase-8, thus connecting these two key organelles in apoptotic signaling. Caspase-3 activation and induction of PARP cleavage than induce apoptosis.
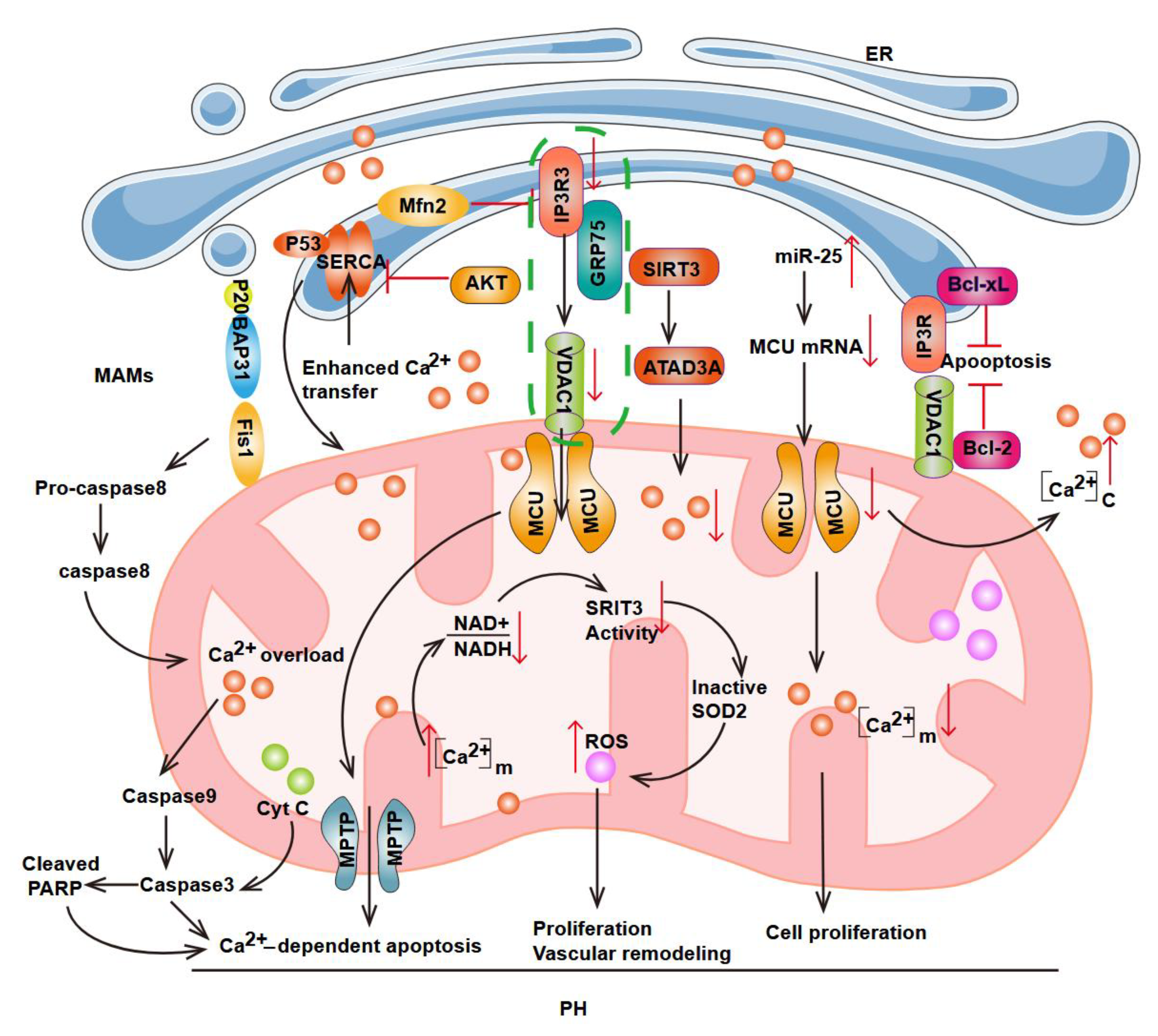
Figure 4.
MAMs in ER stress and oxidative stress (ROS production). The three ER transmembrane proteins involved in UPR including ATF6, PERK, and IRE1 are all distributed in MAMs. Oxidative conditions induce serine 36 phosphorylation of p66Shc, prompting its translocation to MAMs and subsequent ROS production. This triggers ER stress signaling via the PERK-EIF2α-ATF4-CHOP pathway. Notably, the interaction between sigma-1 receptor (Sig-1R) and IRE1 at MAMs can mitigate this stress state. Nup358 in controlling ERMCSs through the modulation of the mTORC2/Akt/GSK3β axis; Elevating VAPB-PTPIP51 integration repairs damaged mitochondria-associated ER membranes and inhibits lung fibroblasts activation. Enhancing the interaction between VAPB and PTPIP51 repairs impaired mitochondria-associated ER membranes and suppresses the activation of lung fibroblasts. Under ERS, ERO1α oxidizes IP3R1, resulting in the dissociation of ERP44. ERP44, which is present in MAMs, can bind to IP3R1 to inhibit Ca²⁺ transfer from IP3R1 to mitochondria. This process promotes Ca²⁺ release from the ER, ultimately leading to excessive mitochondrial reactive oxygen species (mtROS) production. ERMCSs: ER–mitochondria contact sites; PDI: protein disulfide isomerase.
Figure 4.
MAMs in ER stress and oxidative stress (ROS production). The three ER transmembrane proteins involved in UPR including ATF6, PERK, and IRE1 are all distributed in MAMs. Oxidative conditions induce serine 36 phosphorylation of p66Shc, prompting its translocation to MAMs and subsequent ROS production. This triggers ER stress signaling via the PERK-EIF2α-ATF4-CHOP pathway. Notably, the interaction between sigma-1 receptor (Sig-1R) and IRE1 at MAMs can mitigate this stress state. Nup358 in controlling ERMCSs through the modulation of the mTORC2/Akt/GSK3β axis; Elevating VAPB-PTPIP51 integration repairs damaged mitochondria-associated ER membranes and inhibits lung fibroblasts activation. Enhancing the interaction between VAPB and PTPIP51 repairs impaired mitochondria-associated ER membranes and suppresses the activation of lung fibroblasts. Under ERS, ERO1α oxidizes IP3R1, resulting in the dissociation of ERP44. ERP44, which is present in MAMs, can bind to IP3R1 to inhibit Ca²⁺ transfer from IP3R1 to mitochondria. This process promotes Ca²⁺ release from the ER, ultimately leading to excessive mitochondrial reactive oxygen species (mtROS) production. ERMCSs: ER–mitochondria contact sites; PDI: protein disulfide isomerase.
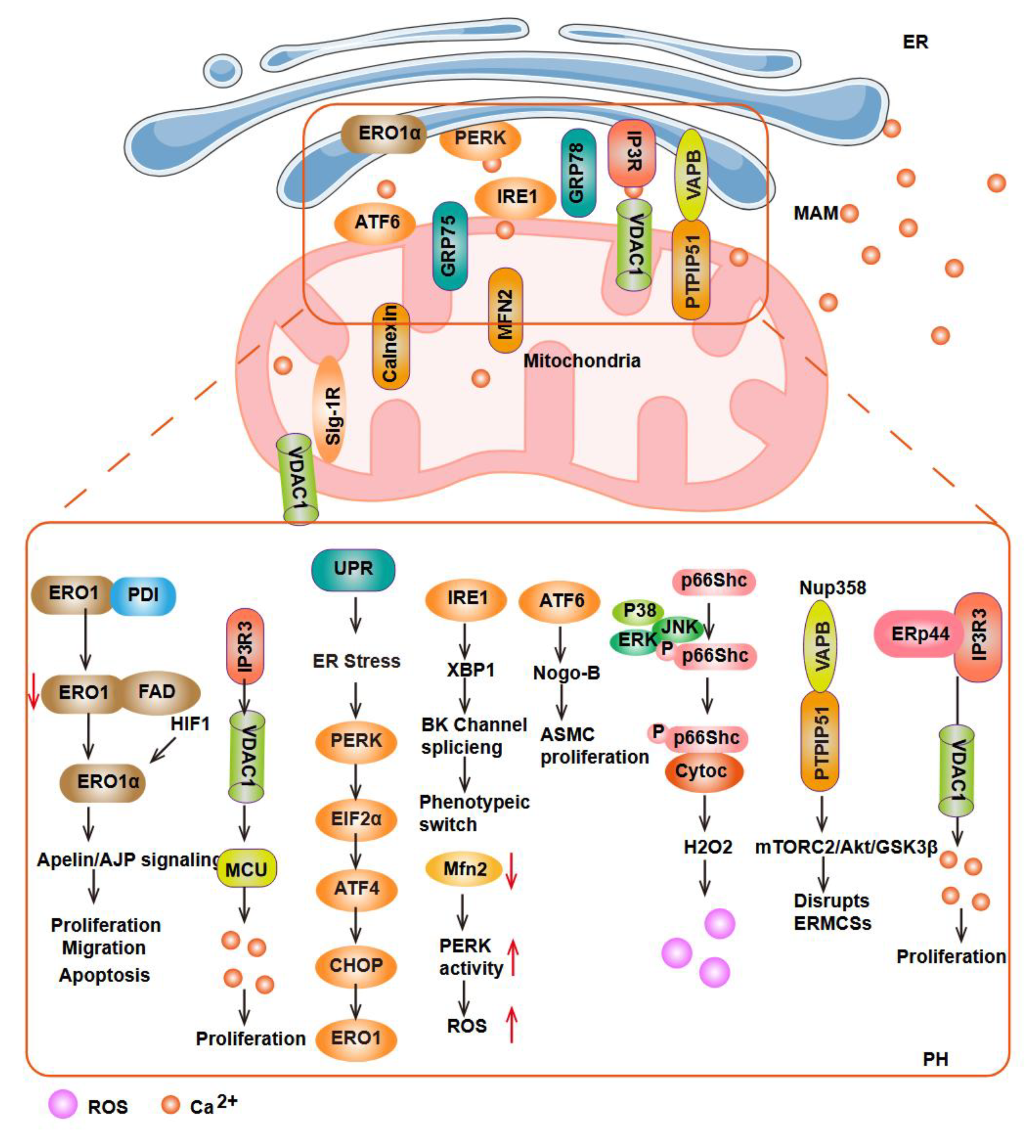
Figure 5.
Role of MAMs in inflammation, autophagy and Lipid Metabolism. In response to ROS stimulation, NLRP3 translocates from the ER to MAMs, where it interacts with its adaptor ASC. This interaction activates caspase-1, leading to the generation of mature IL-1β and IL-18 and initiating inflammasome formation; p66Shc-mediated inflammation via Gab2-PI3K signaling; Within the ER, phosphatidic acid (PA) is catalyzed into phosphatidylserine (PS) by the enzymes PSS1 and PSS2. Subsequently, PS is transported to mitochondria, where it is converted to phosphatidylethanolamine (PE) via phosphatidylserine decarboxylase (PSD). The PE synthesized in mitochondria is then transported back to the ER, where phosphatidylethanolamine N-methyltransferase 2 (PEMT2) catalyzes its conversion to phosphatidylcholine (PC); PC is ultimately transported from the ER to mitochondria. Furthermore, acyl-coenzyme cholesterol acyltransferase 1 (ACAT1), which is localized in MAMs, catalyzes the conversion of intracellular free cholesterol to cholesterol esters, thereby maintaining the dynamic balance between bound and free cholesterol under resting conditions. Under physiological conditions, syntaxin 17 (STX17) interacts with dynamin-related protein 1 (DRP1) to regulate mitochondrial fission. During starvation, the STX17-DRP1 interaction becomes abnormal, leading to mitochondrial elongation. STX17 then binds to ATG14 and recruits it to MAMs to participate in autophagosome formation. The TOM70-TOM40 complex recruits ATG2A to MAMs, which further binds to ATG9 to promote membrane structure expansion and autophagy progression. In mitophagy, PINK1 and Parkin are enriched at MAMs, facilitating mitochondria-ER contact and autophagosome formation. FUN14 domain-containing protein 1 (FUNDC1) binds to calnexin and localizes to MAMs. PGAM5-mediated dephosphorylation at Ser13 and ULK1-mediated phosphorylation at Ser17 facilitate the binding of FUNDC1 to LC3, thereby triggering the initiation of mitophagy. During mitophagy, the interaction between FUNDC1 and calnexin is weakened, and FUNDC1 recruits DRP1 to MAMs through direct interaction, thereby mediating mitochondrial fission. Additionally, Rab32 interacts with the ER-phagy receptor reticulon-3 (RTN3L), both localized at MAMs to induce ER phagy.
Figure 5.
Role of MAMs in inflammation, autophagy and Lipid Metabolism. In response to ROS stimulation, NLRP3 translocates from the ER to MAMs, where it interacts with its adaptor ASC. This interaction activates caspase-1, leading to the generation of mature IL-1β and IL-18 and initiating inflammasome formation; p66Shc-mediated inflammation via Gab2-PI3K signaling; Within the ER, phosphatidic acid (PA) is catalyzed into phosphatidylserine (PS) by the enzymes PSS1 and PSS2. Subsequently, PS is transported to mitochondria, where it is converted to phosphatidylethanolamine (PE) via phosphatidylserine decarboxylase (PSD). The PE synthesized in mitochondria is then transported back to the ER, where phosphatidylethanolamine N-methyltransferase 2 (PEMT2) catalyzes its conversion to phosphatidylcholine (PC); PC is ultimately transported from the ER to mitochondria. Furthermore, acyl-coenzyme cholesterol acyltransferase 1 (ACAT1), which is localized in MAMs, catalyzes the conversion of intracellular free cholesterol to cholesterol esters, thereby maintaining the dynamic balance between bound and free cholesterol under resting conditions. Under physiological conditions, syntaxin 17 (STX17) interacts with dynamin-related protein 1 (DRP1) to regulate mitochondrial fission. During starvation, the STX17-DRP1 interaction becomes abnormal, leading to mitochondrial elongation. STX17 then binds to ATG14 and recruits it to MAMs to participate in autophagosome formation. The TOM70-TOM40 complex recruits ATG2A to MAMs, which further binds to ATG9 to promote membrane structure expansion and autophagy progression. In mitophagy, PINK1 and Parkin are enriched at MAMs, facilitating mitochondria-ER contact and autophagosome formation. FUN14 domain-containing protein 1 (FUNDC1) binds to calnexin and localizes to MAMs. PGAM5-mediated dephosphorylation at Ser13 and ULK1-mediated phosphorylation at Ser17 facilitate the binding of FUNDC1 to LC3, thereby triggering the initiation of mitophagy. During mitophagy, the interaction between FUNDC1 and calnexin is weakened, and FUNDC1 recruits DRP1 to MAMs through direct interaction, thereby mediating mitochondrial fission. Additionally, Rab32 interacts with the ER-phagy receptor reticulon-3 (RTN3L), both localized at MAMs to induce ER phagy.
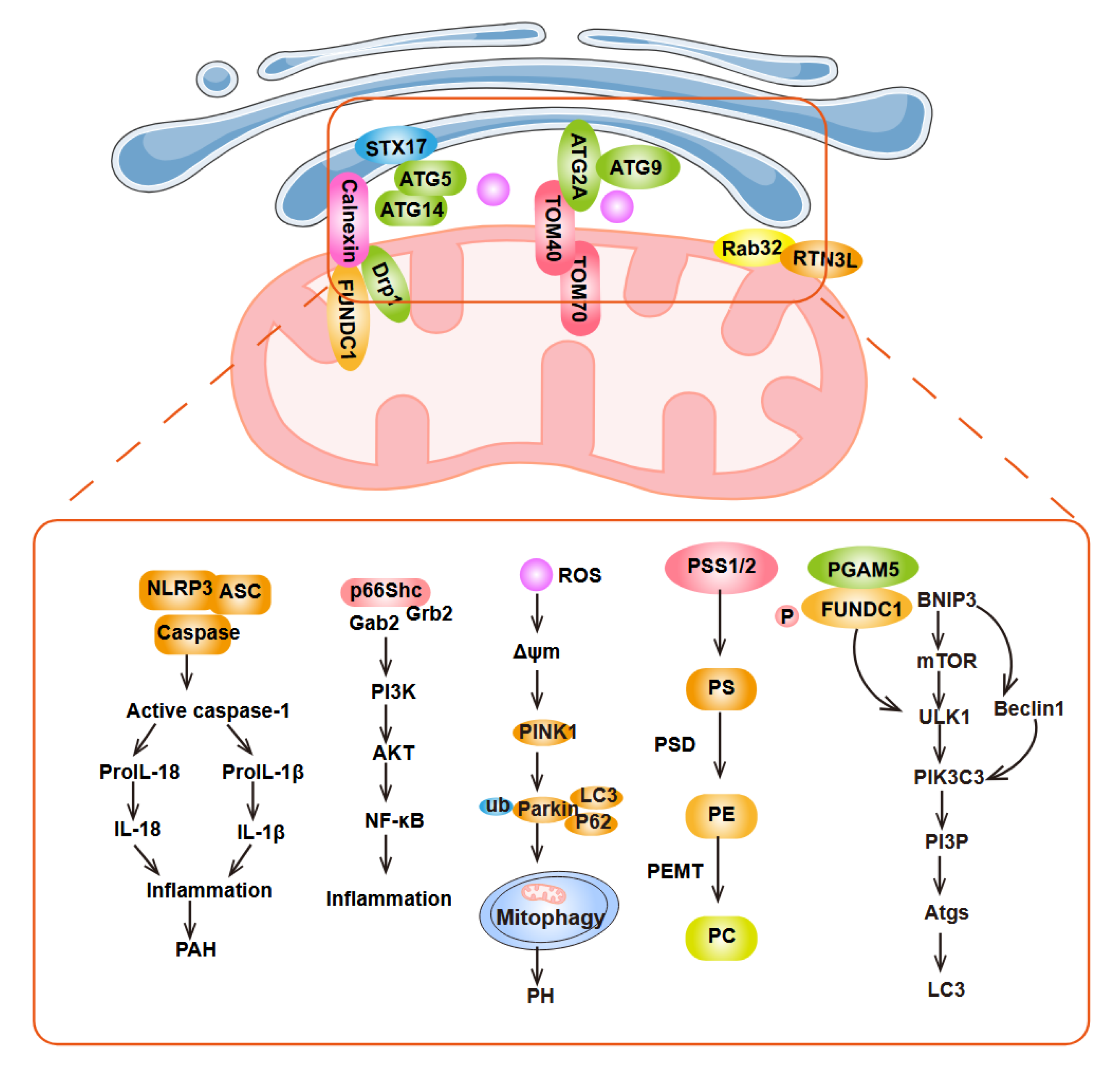
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



