Submitted:
11 February 2026
Posted:
12 February 2026
You are already at the latest version
Abstract
Upregulation of glycolysis and resultant lactate production (hereafter referred to as fermentative glycolysis) even under normoxic conditions has been considered a hallmark of cancer. In recent years, however, it has become clear that fermentative glycolysis in tumors is not as all-inclusive as originally thought. Nevertheless, many tumor types at different stages of progression are characterized by a predominantly glycolytic metabolism. Fermentative glycolysis in tumors supports several different functions: energy production in form of ATP, maintenance and amplification of glycolytic metabolism itself, feeding of oxidative metabolism through the production of lactate, generation of metabolic intermediates for biomass production, execution of non-metabolic, non-canonical, so-called moonlighting functions. This knowledge, however, raises a number of different questions which, by and large, are still unanswered today. Are there different degrees of glycolysis upregulation in order to support the different functions? How is fermentative glycolysis maintained even under normoxic conditions? Why do moonlighting functions exist, given that they are unrelated to the metabolic steps of glycolysis? Moonlighting functions are generally discussed in the context of tumorigenesis, but do they exist also in non-transformed cells? Do they occur in a coordinated manner in all tumor cells or are they activated selectively depending on the tumor type, tumor stage, and on the inducing stimulus? While these issues are mostly unresolved, in this article we propose some tentative answers which, we hope, may promote new research directions which may further our understanding in this field.
Keywords:
glycolysis
; tumorigenesis
; lactate
; moonlighting
; danger
; stress
1. Introduction
Glycolysis converts glucose to pyruvate with a net yield of 2 adenosine triphosphate (ATP) molecules per glucose molecule. Under aerobic conditions pyruvate then enters oxidative metabolism [tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS)] upon conversion to acetyl-coenzyme A (CoA), which is then metabolized to CO2, reduced nicotinamide adenine dinucleotide (NADH) and reduced flavin adenine dinucleotide. In OXPHOS these molecules are further metabolized to generate ATP (up to 34 molecules of ATP per glucose molecule) [1]. Glucose is not the only molecule that fuels the TCA cycle. Fatty acids can also feed the TCA cycle upon β-oxidation to acetyl-CoA as well as glutamine, upon conversion to α-ketoglutarate. In anaerobiosis, i.e., in the absence of oxygen, pyruvate does not enter the TCA cycle, but is reduced (fermented) to lactate in a reaction catalyzed by lactate dehydrogenase (LDH). With the term fermentative glycolysis, we refer to the metabolization of glucose to pyruvate (glycolysis in the real sense of the word) plus the reduction of the pyruvate to lactate. The balance between glycolysis and oxidative metabolism is tightly regulated and this balance may become dysregulated in cancer [2].
About one century ago, Otto Warburg [3] discovered that tumor cells can undergo reprogramming from oxidative metabolism to fermentative glycolysis even under aerobic conditions. This phenomenon has been recognized as one of the hallmarks of cancer [4]. Subsequent studies found that fermentative glycolysis occurs in response to a large variety of stimuli, in addition to hypoxia [5]. These stimuli can be cell-autonomous or -nonautonomous (Figure 1).
Cell-autonomous stimuli that upregulate fermentative glycolysis are, for example, oncoproteins [6,7]. Thus, oncoproteins which have been shown to upregulate glycolysis include Kristen rat sarcoma virus [8], c-Myc [7], Src [9], EWS-FL1 [10], oncogenic viruses [11], the phosphoinositide 3-kinase/AKT/mechanistic target of rapamycin pathway [12], Wnt/ β-catenin [13], epidermal growth factor receptor [14], human epidermal growth factor receptor 2 [15]. Oncosuppressive proteins can also upregulate glycolysis if they lose their oncosuppressive function and/or acquire oncogenic functions [16]. Some of these oncoproteins (e.g., c-MYC) promote fermentative glycolysis by acting as transcription factors (TF) which increase the expression of one or more glycolytic enzymes [7,8,10].
As regards cell-nonautonomous stimuli that upregulate fermentative glycolysis, the first to be mentioned is hypoxia, the prototypic inducer of fermentative (anaerobic) glycolysis [17]. In addition, also mechanical stimuli (e.g., compressive stress), increased hyaluronan concentration in the extracellular matrix [18], too low or too high glucose levels in the extracellular milieu [19,20], extracellular acetate [21], bacterial infections [22], antitumor therapeutics [23], reactive oxygen species (ROS) [24] or cytokines [25] can upregulate fermentative glycolysis. Importantly, cell-autonomous and -nonautonomous stimuli can cooperate in inducing metabolic reprogramming towards glycolysis [2].
It has also been proposed that fermentative glycolysis may become predominant over oxidative metabolism because the demand for oxidized nicotinamide adenine dinucleotide (NAD+) required to support oxidation reactions exceeds the rate of ATP turnover [26]. A mechanism of this kind may complement cell-autonomous or -nonautonomous stimuli that lead to an upregulation of glycolysis. Available NAD+ would then become limiting and this would promote fermentative glycolysis despite available oxygen.
Upregulation of glycolysis in response to these stimuli has two consequences. First, fermentative glycolysis becomes the predominant energy-producing pathway. Second, tumor glycolysis becomes upregulated compared to normal tissues [27]. Upregulation of glycolysis is the result of quantitative (e.g., overexpression of glucose transporters or glycolytic enzymes or overproduction of metabolites) and/or qualitative (e.g., PTMs or expression of embryonic isoforms of glycolytic enzymes) changes of glycolytic enzymes [28]. When such changes apply, initially, to an individual enzyme, this leads, in the following, to an overall upregulation of glycolysis [28,29,30]. Upregulated glycolysis can be transmitted to nearby cells by means of intercellular communications [31].
While fermentative glycolysis may become the main energy-producing pathway in tumors, this does not hold true for all tumors [32] and, importantly, even in tumors with a predominant glycolytic metabolism, oxidative metabolism is never completely inhibited. Thus, in all tumor cell lines that were investigated, fermentative glycolysis and oxidative metabolism coexisted, albeit at varying percentages depending on the tumor type and stage [33,34]. This metabolic heterogeneity has been confirmed in vivo, in non-small cell lung cancer patients that had been intraoperatively infused with 13C-glucose [27]. In fact, it has been claimed that the coexistence of glycolytic and oxidative metabolism is required to support full tumorigenicity [35]. This has been demonstrated for different tumor types [36,37].
2. The Outputs of Fermentative Glycolysis
After having considered the circumstances that lead to the upregulation of glycolysis in tumor cells allowing it to become the predominant energy-producing pathway, one is led to ask which are the outputs of fermentative glycolysis that influence tumor cell proliferation and tumor progression (Figure 2).
The first output is, obviously, the production of energy in form of ATP molecules. We have already discussed that oxidative metabolism is much more efficient in producing ATP than fermentative glycolysis. In fact, it has been shown that oxidative metabolism generates >90% of the overall ATP asset in aerobic conditions [38]. So, why do many, but not all tumor cells, recur to glycolysis if ATP production is a crucial item for cell survival and/or proliferation? One advantage of glycolytic ATP production is that ATP is produced at a faster rate than in oxidative metabolism [39,40]. In accordance with this view, an earlier study showed that ATP consumption can increase glycolytic flux [41]. Moreover, it has been reported that tumor cells relying predominantly on glycolysis have a growth advantage compared to similar cells relying predominantly on oxidative metabolism [42]. In fact, rapid onset of ATP production may be of particular utility for proliferating cells since it has been shown fermentative glycolysis is mainly used in the S phase of the cell cycle [43,44]. In fact, most, albeit not all, tumor cells undergo rapid proliferation. Rapid proliferation, however, is not an exclusive characteristic of tumor cells and, in fact, upregulation of fermentative glycolysis has been shown to occur also in non-transformed cells undergoing rapid proliferation like fibroblasts [45] or mitogen-stimulated lymphocytes [46].
The second reason as to why tumor cells may reprogram metabolism towards glycolysis is the production of metabolic intermediates that facilitate tumor cell proliferation and tumor growth. Thus, glycolysis upregulation in tumor cells can feed branching metabolic pathways to support synthesis of nucleotides, lipids and amino acids that are needed to support tumor cell proliferation [47]. In triple-negative breast cancer tissues it has been shown that glucose directly feeds ribose phosphate, amino acid synthesis, lactate and the TCA cycle [48]. These results suggest that oxidative and fermentative metabolism of glucose, collaborate in generating intermediates required to support the biological machinery of tumor cells.
In support of this possibility and, somehow paradoxically given the reprogramming towards glycolysis, is that one consequence of the upregulation of fermentative glycolysis is the feeding of oxidative metabolism. In fact, the final metabolite of fermentative glycolysis, lactate and the ensuing lactic acidosis, are now recognized as a major fuel and stimulus for the upregulation of oxidative metabolism, respectively [49]. Once extruded, lactate can reenter cells through the transporter monocarboxylate transporter (MCT) 4 and inside the cytoplasm it is oxidized to pyruvate which then enters oxidative metabolism upon conversion to acetyl-CoA [50]. Lactate, however, upregulates OXPHOS also through several other mechanisms, including stimulation of mitochondrial biogenesis [51] and stimulation of the electron transport chain (ETC) upon entering the mitochondrial matrix [52]. On the other hand, it has also been reported that lactylation of some mitochondrial proteins leads to their inactivation and, consequently, to inhibition of OXPHOS because of reduced generation of acetyl-CoA [53]. This suggests that lactate can upregulate oxidative metabolism through different mechanisms but that feedback inhibitory mechanisms have also been put in place in order to safeguard metabolic heterogeneity. Eventually, also acidosis, the accompanying feature of lactate release has been shown to stimulate oxidative metabolism [54,55,56]. These observations reinforce the knowledge that cancer metabolism strives towards heterogeneity: when one energy-producing pathway predominates, from a certain point on mechanisms are at work that aim at reestablishing a more balanced equilibrium between the two pathways [2,32].
In contrast with the previous consequence, upregulated glycolysis puts in place also self-perpetuating mechanisms, i.e., it promotes the upkeeping of itself. Thus, glycolytic enzymes have been shown to mediate glycolysis upregulation in response to cell-autonomous or -nonautonomous stimuli [57,58] through a variety of different mechanisms, e.g., [30,59,60].
A further output of upregulated glycolysis are non-canonical, so-called moonlighting functions. These functions are mediated by glycolytic enzymes, either overexpressed or modified, or glycolytic metabolites such as lactate or pyruvate. Over the years a bewildering number of such functions has been reported in the literature. It would go beyond the scope of this article to list them all. Several recent reviews have described and summarized these functions [32,61,62,63]. Here, we briefly list the main classes of these functions [5,64]: induction of tumor-driving mutations, increased expression and/or activity of oncoproteins, epigenetic modifications including acetylation or lactylation of histone proteins, increased tumor cell proliferation and/or senescence, promotion of DNA damage response (DDR), antiapoptotic effects, induction of autophagy, reduction of drug accumulation and immunosuppressive effects.
3. The Molecular Changes That Underlie the Acquisition of Moonlighting Functions
Moonlighting functions appear in tumor cells as a result of upregulation of glycolysis. This leads to ask as to which molecular changes underlie the acquisition of these functions. One consequence of the modifications that will be discussed in the following is that glycolytic enzymes change their usual, cytoplasmic localization. In many cases, the modifications lead to the localization of glycolytic enzymes to the nucleus where they exert their non-canonical, moonlighting functions.
Moonlighting functions of glycolytic enzymes may become detectable in tumor cells due to increased expression of an individual glycolytic enzyme that will be the effector of this function (Figure 3). Even if this function is carried by an individual enzyme, this may occur in the context of an overall upregulation of glycolysis, e.g., [65,66,67].
In other cases, moonlighting functions are the result of PTMs of a glycolytic enzyme occurring in response to the stimuli that have been discussed in previous sections. These PTMs include phosphorylation, e.g., [68,69], acetylation [70,71], serotonylation [72], citrullination [30], O-GlcNAcylation [73], nitrosylation [74].
Still another possibility, non-exclusive with the previous ones, is represented by modifications of the quaternary structure of glycolytic enzymes. Thus, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) has been shown to undergo aggregation in response to oxidative stress, a change that led to the opening of permeability transition pores and subsequent mitochondrial dysfunction [75]. Pyruvate kinase isoform M2 (PKM2) is a PK isoform that exists as a highly active tetramer or a low-activity monomeric/dimeric forms. The latter forms localize to the cell nucleus where they perform non-canonical functions [76,77]. Nuclear localization of PKM2 is the consequence of the exposure of nuclear localization signals (NLS) upon tetramer-monomer/dimer conversion [78]. Interestingly, tetramer-monomer/dimer conversion of PKM2 has been suggested to endow it also with protein kinase activity which is absent in its tetrameric form [79,80]. An interesting example for nuclear localization of PKM2 upon tetramer-dimer/monomer conversion has been reported in a non-tumor system, i.e., lipopolysaccharide-stimulated dendritic cells [81]. Upon stimulation, PKM2 was acetylated at Lys433. This modification destabilized PKM2 and promoted its nuclear localization. Here, PKM2 associated with c-Rel and increased expression of Il12p35 which, in turn, facilitated T helper (Th) 1 cell differentiation.
Nuclear localization, however, can also result from overexpression [82] or PTM of a glycolytic enzyme [83,84]. In addition to nuclear localization, also mitochondrial localization of glycolytic enzymes has been reported [85]. Thus, mitochondrial phosphoglycerate kinase 1 phosphorylated and activated pyruvate dehydrogenase kinase 1, thereby reducing pyruvate utilization in the TCA cycle and increasing fermentative glycolysis and lactate production [85]. This represents a feed-forward mechanism whereby a glycolytic enzyme amplifies glycolysis through a non-canonical mechanism in a non-canonical localization. PKM2 has also been shown to translocate to mitochondria in response to oxidative stress [86]. Here, PKM2 interacted with and phosphorylated the antiapoptotic protein B-cell lymphoma 2 (Bcl2), thereby preventing the degradation of Bcl2 and inhibiting apoptosis.
While most attention has been focused on the moonlighting functions of glycolytic enzymes [87], also glycolytic metabolites can exert non-canonical functions, unrelated to their role in metabolism.
One example are the non-canonical functions of pyruvate. Pyruvate enters the TCA cycle upon transformation to acetyl-CoA or, anaplerotically, upon transformation to oxalacetate or upon transamination. A non-canonical function of pyruvate was suggested by its inhibitory effect on the anti-neoplastic activity of carnosine on glioblastoma multiforme (GBM) cells even when oxidative ATP production was blocked [88]. In fact, pyruvate may exert antioxidant effects as shown by the protection afforded to human neuroblastoma cells from hydrogen peroxide toxicity [89]. These effects were the consequence of scavenging H2O2-induced ROS, of suppressing superoxide production by submitochondrial particles, and reducing oxidative stress-effected decrease of mitochondrial membrane potential. More recently, it has been shown that increased mitochondrial pyruvate levels prevent growth in non-transformed cells by increasing the NADH/NAD+ ratio, which rewired metabolism towards gluconeogenesis and suppressed glycolysis [90]. Decreased amino acid synthesis caused reduced protein synthesis and inhibited cell growth. The precursor of pyruvate, phosphoenolpyruvate (PEP), was also found to perform non-canonical functions [91]. Glucose-poor tumor microenvironment (TME) decreased glycolytic metabolism in tumor-infiltrating lymphocytes, thereby limiting the tumoricidal function of these cells. Such inhibition of T cell effector functions was due to reduced levels of PEP in T cells. Overexpression in these cells of PEP carboxykinase 1, which converts oxalacetate into PEP, stimulated the effector functions of T cells, thereby prolonging the survival of melanoma-bearing mice. Later, PEP has been shown to inhibit differentiation of non-transformed Th17 cells through binding to the TF JunB [92]. This caused inhibition of DNA binding of the JunB/basic leucine zipper transcription factor ATF-like (BATF)/interferon regulatory factor 4 (IRF4) complex, thereby regulating the transcriptional program of Th17 cells.
Lactate, the final metabolite of fermentative glycolysis, and its acid, have raised great interest in recent years. Lactate has long been considered a waste product of fermentative glycolysis. In the following, however, it was found to serve as a fuel for oxidative metabolism upon transformation into pyruvate in a reaction catalyzed preferentially by LDH isoform B [49,93,94,95]. Given that this effect falls within the definition of metabolic activities, we will refer to it as the canonical activity of lactate. In addition, lactate has several other non-canonical activities. Beforehand, it should be recalled that lactate can enter or exit cells through MCTs: MCT1 is used mainly for cell exit and MCT4 for cell entry [96]. In addition, extracellular lactate can bind to the proton-sensitive cell surface receptors G-protein-coupled receptor (GPR) 81/hydrocarboxylic acid receptor 1 and GPR132 [97]. A prominent non-canonical activity of lactate that has been described in recent years is the lactylation of proteins, both histone and non-histone proteins. The plethora of biological activities, including tumor-promoting effects [98], that are modulated in response to this PTM in cancer and non-cancer settings have been reviewed [50,99,100]. As regards the other non-canonical activities of lactate, it would go beyond the scope of this article to enter a detailed discussion them. Nevertheless, we will briefly mention some of the most relevant ones. Thus, lactate and lactate acidosis exert immunosuppressive effects that support tumor growth and dissemination through different mechanisms of action, e.g., [101,102,103]. Moreover, lactate has been shown to regulate the cell cycle [104], to promote resistance to different antitumor drugs, e.g., [105,106], to accelerate tumor growth [107], to induce EMT [108,109]. Interestingly, a link between histone lactylation and immunosuppressive activity has been recently described [110], as it was shown that histone lactylation induced a transcriptional program in microglia/macrophages in GBM that led to the upregulation of CD47, a cell surface-molecule that acts as a “don’t eat me” signal that suppresses phagocytosis.
4. Several Unanswered Questions About the Generation and the Function of Fermentative Glycolysis in Tumor Cells
The previous sections have briefly summarized the fundamentals of fermentative glycolysis in tumors. A particular attention has been devoted to some new insights concerning the non-canonical functions of glycolytic enzymes and metabolites. Yet, current knowledge raises several questions on tumor glycolysis that are still unanswered. In the following, some of these questions will be addressed as well as some tentative answers that will require experimental verification.
4.1. How Much Glycolysis to Do What?
As already discussed, rapidly proliferating cells, either normal or tumor cells, recur to fermentative glycolysis in order to satisfy several metabolic requirements. In addition, also a large number of cell-autonomous and -nonautonomous stimuli can upregulate glycolysis. This raises the question whether constitutive metabolic needs on one hand, and stimuli on the other hand, give rise to additive or synergistic upregulation of glycolysis or if each one induces a maximal upregulation that cannot be further increased. Another possibility, non-exclusive with the previous one, is that said stimuli select tumor cells that have an upregulated baseline level of glycolysis compared to non-transformed cells. Evidence for this latter possibility has been brought [20]. In any case, one is led to ask whether there is a correlation between the level of upregulated glycolysis and one or more of the outputs that have been discussed before. Thus, it is possible that a minimum level of upregulated glycolysis might be required to support rapid proliferation, but a higher level might be necessary to perform moonlighting functions. In order to validate this hypothesis, precise, quantitative investigations on different conditions or stimuli that upregulate fermentative glycolysis are required.
4.2. How Is Fermentative Glycolysis, the Warburg Effect, Maintained Under Normoxic Conditions?
The reasons as to why fermentative glycolysis is upregulated in tumor cells have been discussed in a previous section. Many of the stimuli that upregulate glycolysis are self-limiting, i.e., they vanish after a certain period of time. If these stimuli disappear, then how is fermentative glycolysis preserved once the stimulus is no longer present? This is the case, for example, when tumor cells upregulate glycolysis in the presence of anaerobiosis, and continue to rely, predominantly, on fermentative glycolysis once they return back to aerobic conditions. We suggest that in this and similar cases, epigenetic changes induced by self-limiting stimuli can be transmitted to the cellular offspring for a prolonged, undefined period of time.
Several observations support this view. Thus, in non-transformed cells (C2C12 mouse skeletal myoblasts) glycolysis was upregulated following low-temperature stress (35 °C) [111]. This was paralleled by epigenetic changes, including expression of the methyltransferase Dnmt1 as well as the histone acetyltransferase Gcn5. Other epigenetic changes have been shown to upregulate, either directly or indirectly, the expression of genes encoding glycolytic enzymes [112,113]. Epigenetic changes of this kind can be inherited through different mechanisms [114]. This suggests that one mechanism allowing to establish the Warburg effect in normoxic tumor cells is through inheritance of epigenetic changes in response to the cell-autonomous or -nonautonomous stimuli that have been discussed in previous sections. Interestingly, lactate itself, through lactylation of histone proteins, and consequent activation of the transcription of genes encoding glycolytic enzymes, has been shown to be an epigenetic regulator of glycolysis in a non-tumor system [115]. To this regard, it is interesting to note that lactylation of histone H3, mainly at distal regulatory regions, characterizes trained monocytes/macrophages which respond with enhanced production of cytokines to a secondary stimulation [116]. Thus, also fermentative glycolysis itself, through lactate production and consequent lactylation may be at the origin of epigenetic changes which promote self-maintenance of glycolysis.
4.3. The Mystery of the Moonlighting Functions—The Many Unresolved Issues
As already mentioned, the number of non-canonical functions of glycolytic enzymes and metabolites, i.e., functions unrelated to their role in metabolism, is very large, up to a point that it is becoming difficult to list them in a single, even if extensive, article. Not surprisingly then, they raise many questions.
Thus, when considering the moonlighting functions as a whole, one is led to ask whether they are induced coordinately altogether, or in subgroups or individually in response to some stimuli but not others and/or if their induction depends on the responding cell. Another question is whether these functions are induced only in tumor cells or whether they are induced also in non-transformed cells in response to similar stimuli. Last but not least, one would like to know which is the reason as to why these functions are activated in tumor cells. At present, it is only possible to give some preliminary answers to these questions and formulate some hypotheses that await experimental verification.
One variable that may influence whether moonlighting functions are induced altogether, in subgroups or individually, is the duration and intensity of the stimulus that upregulates glycolysis. Thus, a relatively weak or short stimulus may induce one or a few of these functions, while a stronger stimulus or of longer duration may induce a larger number of functions. The occurrence of graded responses to stimuli of varying duration and intensity has already been demonstrated in other biological systems [117,118]. The other variable that may influence the spectrum of moonlighting functions is the responder cell. Cells of different tumor types or tumor stages may respond differently in response to the same stimuli. These are testable hypotheses that warrant experimental investigation in order to gain a more complete view of the breadth and significance of glycolytic moonlighting functions in tumorigenesis.
Concerning the question as to whether glycolysis-associated moonlighting functions occur also in normal, non-transformed cells, the answer is yes [119,120,121], although much less information is available than for tumor cells. Several examples of moonlighting functions occurring in non-transformed cells will be discussed in the following of this article. As we have discussed in the previous section, glycolysis-promoted epigenetic modifications can occur also in non-transformed cells [111]. Moreover, and most importantly, upregulated glycolysis can induce tumor-initiating events by promoting, among others, epigenetic changes; reviewed in [64]. On the other hand, the role of epigenetic modifications in facilitating neoplastic transformation and tumorigenesis is now firmly established [122,123,124]. Altogether, while no studies have been performed in order to investigate whether a stimulus that upregulates glycolysis induces inheritable epigenetic changes in non-transformed cells, the ensemble of the results that have been discussed here makes this a likely possibility that deserves investigation also because of its possible role in inducing tumor-initiating events. Thus, on a whole, these observations and considerations suggest that one of the moonlighting functions of glycolysis may contribute to neoplastic transformation in normal cells.
One of the most difficult questions to address is as to why moonlighting functions are induced in tumor cells but, as we have just discussed, also in non-transformed cells. Stated differently, why do glycolytic enzymes and metabolites, mediate functions that are totally unrelated to their role in glycolytic metabolism whether in transformed or non-transformed cells? In spite of the scarcity of data on this point, we can make some speculations that can be used as working hypotheses for future research. First of all, it is striking to note that most moonlighting functions promote the survival and expansion of the affected cell(s). This is the case for increased proliferation, promotion of DDR, antiapoptotic effects and reduction of drug accumulation. The induction of immunosuppressive effects also protects tumor cells, since they shield them from dangerous and potentially lethal immune responses. We have already proposed that glycolysis-associated drug resistance may represent a response of transformed or non-transformed cells to stimuli which, in principle, could undermine the integrity of the target cell(s) [28]. Herein, we propose that moonlighting functions, altogether, represent protective responses to potentially dangerous signals (Figure 4). Historically, the term danger signal was coined to define stimuli that induce innate immune responses that are initiated upon binding to pathogen-associated pattern-recognition receptors (PRR) [125,126]. Later it was found that also endogenous molecules, whether or not released from injured cells, are also able to interact with PRRs and non-PRRs in innate immune cells, thereby inducing sterile inflammatory responses aimed at repairing injured tissues [127]. Similarly, cell-autonomous or -nonautonomous stimuli induce upregulation of fermentative glycolysis and moonlighting functions. But which are the properties that endow these stimuli to be perceived as danger signals? We propose that this occurs when they exceed a given threshold of intensity. In this case, fermentative glycolysis is “overactivated” and one or more of the following changes occur. First, increased expression of a glycolytic enzyme until a critical concentration is achieved that allows it to interact with a non-canonical target. Second, induction of a PTM of a glycolytic enzyme that increases its affinity for a non-canonical target. Third, induction of a modification of the quaternary structure of a glycolytic enzyme allowing the enzyme to reach a non-canonical location (e.g., nucleus of mitochondria) where to perform its moonlighting function.
The crucial point concerning the model that we propose is that the affinity of the interaction between a given glycolytic enzyme for its substrate would be higher than the affinity of the interaction of the same enzyme (the ligand) with its non-canonical target. The latter interaction would become apparent only when the concentration and/or affinity of the ligand-target interaction exceed a given threshold in response to a dangerous or stressful stimulus.
But why are moonlighting functions associated with glycolytic metabolism? Possibly, upregulation of glycolysis and appearance of moonlighting functions occur in response to the same signals and this coincidence would have favored, energetically, the co-evolution of the two classes of functions, metabolic and moonlighting, within the same family of mediators.
It appears somehow paradoxical that a cellular response that is presumed to have protective effects towards the affected cell(s) supports tumorigenesis. This apparent paradox, however, is likely the result of the phenotype of the responding cells: in both non-transformed cells and transformed cells moonlighting functions have protective effects, but in tumor cells the responses to dangerous signals promotes survival thereby exacerbating the malignant properties.
So far, we have referred to moonlighting functions as the consequence of a supraphysiological increase of glycolysis in response stimuli that we have referred to as danger signals. It can be appreciated, however, that moonlighting functions encompass several cellular stress responses t (e.g., DDR, induction of autophagy etc.) [128,129]. This suggests that moonlighting functions induced by danger signals, represent an ensemble of cellular stress responses. The relationship and overlap between moonlighting functions and other cellular stress responses is a topic of obvious interest.
Still another question is whether the protein domains responsible for the moonlighting functions are different from those responsible for the catalytic activity. On the basis of what we have discussed so far, the answer is yes. In fact, we have referred to the glycolytic enzyme PKM2, which is catalytically most active in its tetrameric form but performs moonlighting functions upon shifting to a dimeric form and consequent exposure of a NLS which allows it to shuttle into the nucleus, the site of its non-canonical activities [76,77]. In addition, however, one would like to know the exact domains of a given protein (e.g., a glycolytic enzyme) that performs the moonlighting function(s) and where it maps compared to the catalytic domain. Unfortunately, very little information is available about this important point. The exception is GAPDH, the glycolytic enzyme that has been most deeply investigated as regards this aspect. In fact, the knowledge acquired so far suggests that the domains that mediate the moonlighting functions are not overlapping [130] with those that mediate the catalytic activity although a precise mapping of the different domains has, to our knowledge, not yet been reported. These observations suggest that also the moonlighting functions of other glycolytic enzymes may be mediated by protein domains different from those that are responsible for the catalytic activity.
5. Which Is the Most Important Output of Upregulated Glycolysis in Tumor Cells?
In a previous section we have summarized the most important outputs of upregulated glycolysis in tumor cells. A question that arises is as to which of these responses is the most crucial for tumor cell survival and expansion. In other words, is it possible to establish a hierarchy of relevance for tumorigenesis between the different outputs that have been discussed? This is not a question of mere academic interest, since an answer to it would allow to predict which of the outputs one would like to inhibit in order to achieve maximal antitumor efficacy without affecting the integrity of normal cells.
A partial answer to this question derives from many observations that blocking one energy-producing pathway leads to a compensatory upregulation of the other pathway. Thus, the inhibitory effect of metformin on OXPHOS has been shown to lead to a compensatory upregulation of glycolysis [131,132]. In fact, this upregulation is at the origin of the most dangerous side effect of metformin, lactic acidosis [133]. A similar upregulation of glycolysis has been observed in response to the other ETC complex I inhibitor IACS-010759 [134]. Vice versa, upregulation of OXPHOS has been observed in response to the glycolysis inhibitor 2-deoxy-D-glucose [135]. Similarly, human lymphoma and colorectal carcinoma cells exposed to the MCT1 inhibitor AZD3965 upregulated oxidative metabolism [136]. These are just a few examples of the many that have been reported in the literature. Of note, while compensatory upregulation of the alternative energy-producing pathway has been reported in most cases, there are also exceptions. Thus, inhibition of OXPHOS was found to successfully eradicate quiescent leukemic stem cells without any evidence of glycolysis upregulation aimed at supporting survival of the cells [137]. These and similar results suggest the possibility of eradicating some tumor cell populations through the use of inhibitors of only one energy-producing pathway while in most cases it appears necessary to use inhibitors of both pathways in order to achieve maximal eradication of metabolically heterogenous tumor cells [138,139].
The crucial point to consider is that several metabolic pathways branch out of glycolytic and oxidative metabolism and several metabolites of both pathways serve as precursors for biomass production. Thus, the pentose phosphate pathway (PPP) and glycogen synthesis pathway branch out of glycolysis, while glycerol, which is used for the synthesis of triglycerides and phospholipids, is produced from the glycolytic metabolite glyceraldehyde 3-phosphate. On the other hand, intermediates of the TCA cycle can serve as precursors of fatty acids and cholesterol which, in turn, can be used for the synthesis of steroid hormones, bile salts and vitamin D. They can also serve as precursors of many non-essential amino acids some of which, in turn, can serve as precursors of purines and pyrimidines. Porphyrins derive the majority of their carbon atoms also from TCA cycle intermediates. Eventually, oxalacetate is a precursor of glucose in the gluconeogenesis pathway. As can be immediately appreciated, glycolytic and oxidative metabolism supply intermediates for essentially non-overlapping pathways. There are a few exceptions to this rule. Thus, many amino acids can be generated from precursors of oxidative and glycolytic metabolism but in most, although not all cases (e.g., isoleucine), they differ between the two pathways. Another exception is reduced nicotinamide adenine dinucleotide phosphate, which can be generated during the oxidative phase of the PPP, but also by malic enzymes, by the TCA cycle enzymes isocitrate dehydrogenase 1 and 2, and by one-carbon metabolism [140]. Still another exception is ATP itself, albeit it is produced, as already discussed, with greatly different efficiencies by the two pathways. The reasons as to why glycolytic ATP production by glycolysis may be preferred over that of oxidative metabolism in spite of the lower efficiency have been discussed in a previous section. On a whole, this knowledge suggests the possibility that ATP production is the most important metabolic output for tumorigenesis. On the other hand, other effects like the moonlighting functions or the generation of intermediates for biomass production, may play a crucial role depending on the tumor type or tumor stage.
6. Conclusions
In this article we have briefly summarized current knowledge on the role fermentative glycolysis in tumorigenesis and, in the following, we have raised several unanswered questions on this issue. Thus, we have discussed evidence suggesting that different degrees of glycolysis upregulation may give rise to different responses. We have also raised the problem as to how fermentative glycolysis is maintained in aerobic conditions once the initiating stimulus has vanished and have suggested that this may be related to inheritable epigenetic changes that accompany upregulation of glycolysis. The reason as to why upregulation of glycolysis is accompanied by non-canonical, moonlighting functions of glycolytic enzymes and metabolites is also an unresolved problem. As regards this aspect, we have suggested that upregulation of glycolysis and appearance of non-canonical functions may be responses to stimuli acting as danger/stress signals. These responses may be aimed at preserving the integrity and promoting the proliferation of the affected cell which, in case of tumor cells, would imply an increased tumorigenicity and malignancy. Eventually, we have also asked which is the output of fermentative glycolysis that is most important in promoting growth and dissemination. Evidence has been discussed which suggests that ATP production may be, indeed, the single most relevant effect in this regard and the one which may be the most desirable to inhibit from a therapeutic point of view. Many inhibitors of glycolysis and oxidative metabolism leading to decreased energy production have been described over years but, so far, clinical trials that have been performed with these inhibitors have failed. It appears likely that, in order to obtain clinically relevant results, these compounds need to be targeted to the TME and tumor cells, whether as drug conjugates or nanoparticulate formulations [141,142]. As to the possibility to target moonlighting functions for therapeutic purposes, it seems necessary to obtain more information about their role in different tumor types and tumor stages as well as biomarkers before designing molecules able to interfere with these effects.
Author Contributions
F.M. was the originator of this work. S.W. and M.C. contributed to the elaboration and writing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
M.C is supported by grant RYC2021–031003I funded by MICIU/AEI/10.13039/501100011033 and, by European Union NextGenerationEU/PRTR.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
Marco Cordani reports financial relationships with OCA Global and EQA Certificados that include consulting or advisory. The other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Abbreviations
ATP, adenosine triphosphate; Bcl2, B-cell lymphoma 2; CoA, coenzyme A; DDR, DNA damage response; ETC, electron transport chain; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GBM, glioblastoma multiforme; GPR, G-protein-coupled receptor; LDH, lactate dehydrogenase; MCT, monocarboxylate transporter; NAD+, oxidized nicotinamide adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; NLS, nuclear localization signal; OXPHOS, oxidative phosphorylation; PEP, phosphoenolpyruvate; PKM2, Pyruvate kinase isoform M2; PPP, pentose phosphate pathway; PRR, pattern-recognition receptor; ROS, reactive oxygen species; TCA, tricarboxylic acid; TF, transcription factor; Th, T helper; TME, tumor microenvironment.
References
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Rumio, C.; Bontempi, G.; Strippoli, R.; Marcucci, F. Oxidative and glycolytic metabolism: Their reciprocal regulation and dysregulation in cancer. Cells 2025, 14, 1177. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell. 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Cordani, M.; Michetti, F.; Zarrabi, A.; Zarepour, A.; Rumio, C.; Strippoli, R.; Marcucci, F. The role of glycolysis in tumorigenesis: From biological aspects to therapeutic opportunities. Neoplasia 2024, 58, 101076. [Google Scholar] [CrossRef]
- Padder, R.A.; Bhat, Z.I.; Ahmad, Z.; Singh, N.; Husain, M. DRP1 promotes BRAFV600E-driven tumor progression and metabolic reprogramming in colorectal cancer. Front. Oncol. 2021, 10, 592130. [Google Scholar] [CrossRef]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-driven glycolysis is a therapeutic target in glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Jin, Y.; Cai, Q.; Shenoy, A.K.; Lim, S.; Zhang, Y.; Charles, S.; Tarrash, M.; Fu, X.; Kamarajugadda, S.; Trevino, J.G.; et al. Src drives the Warburg effect and therapy resistance by inactivating pyruvate dehydrogenase through tyrosine-289 phosphorylation. Oncotarget 2016, 7, 25113–25124. [Google Scholar] [CrossRef]
- Yeung, C.; Gibson, A.E.; Issaq, S.H.; Oshima, N.; Baumgart, J.T.; Edessa, L.D.; Rai, G.; Urban, D.J.; Johnson, M.S.; Benavides, G.A.; et al. Targeting glycolysis through inhibition of lactate dehydrogenase impairs tumor growth in preclinical models of Ewing sarcoma. Cancer Res. 2019, 79, 5060–5073. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Wang, L.; Chen, S. Oncogenic virus-induced aerobic glycolysis and tumorigenesis. J. Cancer 2018, 9, 3699–3706. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef]
- Xie, H.; Hanai, J.; Ren, J.G.; Kats, L.; Burgess, K.; Bhargava, P.; Signoretti, S.; Billiard, J.; Duffy, K.J.; Grant, A.; et al. Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014, 19, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.O.; Li, C.-W.; Xia, W.; Lee, H.H.; Chang, S.S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR signaling enhances aerobic glycolysis in triple-negative breast cancer cells to promote tumor growth and immune escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef]
- Eriksson, M.; Ambroise, G.; Ouchida, A.T.; Lima Queiroz, A.; Smith, D.; Gimenez-Cassina, A.; Iwanicki, M.P.; Muller, P.A.; Norberg, E.; Vakifahmetoglu-Norberg, H. Effect of mutant p53 proteins on glycolysis and mitochondrial metabolism. Mol. Cell Biol. 2017, 37, e00328-17. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 2013, 123, 3664–3571. [Google Scholar] [CrossRef]
- Sullivan, W.J.; Mullen, P.J.; Schmid, E.W.; Flores, A.; Momcilovic, M.; Sharpley, M.S.; Jelinek, D.; Whiteley, A.E.; Maxwell, M.B.; Wilde, B.R.; et al. Extracellular matrix remodeling regulates glucose metabolism through TXNIP Destabilization. Cell. 2018, 175, 117–132.e21. [Google Scholar] [CrossRef]
- Viedma-Rodríguez, R.; Martínez-Hernández, M.G.; Martínez-Torres, D.I.; Baiza-Gutman, L.A. Epithelial Mesenchymal transition and progression of breast cancer promoted by diabetes mellitus in mice are associated with increased expression of glycolytic and proteolytic enzymes. Horm. Cancer 2020, 11, 170–181. [Google Scholar] [CrossRef]
- Damaghi, M.; West, J.; Robertson-Tessi, M.; Xu, L.; Ferrall-Fairbanks, M.C.; Stewart, P.A.; Persi, E.; Fridley, B.L.; Altrock, P.M.; Gatenby, R.A.; et al. The harsh microenvironment in early breast cancer selects for a Warburg phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2011342118. [Google Scholar] [CrossRef]
- Wang, J.; Yang, Y.; Shao, F.; Meng, Y.; Guo, D.; He, J.; Lu, Z. Acetate reprogrammes tumour metabolism and promotes PD-L1 expression and immune evasion by upregulating c-Myc. Nat Metab. 2024, 6, 914–932. [Google Scholar] [CrossRef]
- Abd-El-Raouf, R.; Ouf, S.A.; Gabr, M.M.; Zakaria, M.M.; El-Yasergy, K.F.; Ali-El-Dein, B. Escherichia coli foster bladder cancer cell line progression via epithelial mesenchymal transition, stemness and metabolic reprogramming. Sci. Rep. 2020, 10, 18024. [Google Scholar] [CrossRef]
- Yan, S.; Zhou, N.; Zhang, D.; Zhang, K.; Zheng, W.; Bao, Y.; Yang, W. PFKFB3 Inhibition attenuates oxaliplatin-induced autophagy and enhances its cytotoxicity in colon cancer cells. Int. J. Mol. Sci. 2019, 20, 5415. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.J.; Hopkins, G.L.; Rastogi, N.; Hodges, M.; Doyle, M.; Davies, S.; Hole, P.S.; Omidvar, N.; Darley, R.L.; Tonks, A. Reactive oxygen species drive proliferation in acute myeloid leukemia via the glycolytic regulator PFKFB3. Cancer Res. 2020, 80, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.T.; Cavallaro, F.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discov. 2020, 10, 608–625. [Google Scholar] [CrossRef] [PubMed]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell. 2021, 81, 691–707.e6. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic heterogeneity in human lung tumors. Cell. 2016, 164, 681–694. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C. Glycolysis-induced drug resistance in tumors - A response to danger signals? Neoplasia 2021, 23, 234–245. [Google Scholar] [CrossRef]
- Nowak, N.; Kulma, A.; Gutowicz, J. Up-regulation of Key Glycolysis Proteins in Cancer Development. Open Life Sci. 2018, 13, 569–581. [Google Scholar] [CrossRef]
- Coassolo, S.; Davidson, G.; Negroni, L.; Gambi, G.; Daujat, S.; Romier, C.; Davidson, I. Citrullination of pyruvate kinase M2 by PADI1 and PADI3 regulates glycolysis and cancer cell proliferation. Nat. Commun. 2021, 12, 1718. [Google Scholar] [CrossRef]
- Mojica-Benavides, M.; van Niekerk, D.D.; Mijalkov, M.; Snoep, J.L.; Mehlig, B.; Volpe, G.; Goksör, M.; Adiels, C.B. Intercellular communication induces glycolytic synchronization waves between individually oscillating cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2010075118. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Rumio, C. The role of oxidative metabolism in tumorigenesis and drug resistance. Discov. Med. 2024, 36, 1109–1126. [Google Scholar] [CrossRef]
- Jekabsons, M.B.; Merrell, M.; Skubiz, A.G.; Thornton, N.; Milasta, S.; Green, D.; Chen, T.; Wang, Y.H.; Avula, B.; Khan, I.A.; et al. Breast cancer cells that preferentially metastasize to lung or bone are more glycolytic, synthesize serine at greater rates, and consume less ATP and NADPH than parent MDA-MB-231 cells. Cancer Metab. 2023, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Velázquez, S.C.; Robledo-Cadena, D.X.; Hernández-Reséndiz, I.; Gallardo-Pérez, J.C.; Moreno-Sánchez, R.; Rodríguez-Enríquez, S. Energy metabolism drugs block triple negative breast metastatic cancer cell phenotype. Mol. Pharm. 2018, 15, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The power of mitochondrial metabolism to collaborate or replace fermentative glycolysis in cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef]
- Fogal, V.; Richardson, A.D.; Karmali, P.P.; Scheffler, I.E.; Smith, J.W.; Ruoslahti, E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell Biol. 2010, 30, 1303–1318. [Google Scholar] [CrossRef]
- Kumar, P.R.; Moore, J.A.; Bowles, K.M.; Rushworth, S.A.; Moncrieff, M.D. Mitochondrial oxidative phosphorylation in cutaneous melanoma. Br. J. Cancer 2021, 124, 115–123. [Google Scholar] [CrossRef]
- Rodrigues, T.; Ferraz, L.S. Therapeutic potential of targeting mitochondrial dynamics in cancer. Biochem. Pharmacol. 2020, 182, 114282. [Google Scholar] [CrossRef]
- Kukurugya, M.A.; Rosset, S.; Titov, D.V. The Warburg Effect is the result of faster ATP production by glycolysis than respiration. Proc. Natl. Acad. Sci. USA 2024, 121, e2409509121. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef]
- Fang, M.; Shen, Z.; Huang, S.; Zhao, L.; Chen, S.; Mak, T.W.; Wang, X. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 2010, 143, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, S.; Graziano, V.; Galavotti, S.; Henriquez, N.V.; Betts, J.; Saxena, J.; Minieri, V.; Deli, A.; Karlsson, A.; Martins, L.M.; Capasso, M.; et al. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Peng, Y.; Shi, L.; Wan, L.; Inuzuka, H.; Long, J.; Guo, J.; Zhang, J.; Yuan, M.; Zhang, S.; et al. Skp2 dictates cell cycle-dependent metabolic oscillation between glycolysis and TCA cycle. Cell Res. 2021, 31, 80–93. [Google Scholar] [CrossRef]
- Munyon, W.H.; Merchant, D.J. The relation between glucose utilization, lactic acid production and utilization and the growth cycle of L strain fibroblasts. Exp. Cell Res. 1959, 17, 490–498. [Google Scholar] [CrossRef]
- Wang, T.; Marquardt, C.; Foker, J. Aerobic glycolysis during lymphocyte proliferation. Nature 1976, 261, 702–705. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell. 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Ghergurovich, J.M.; Lang, J.D.; Levin, M.K.; Briones, N.; Facista, S.J.; Mueller, C.; Cowan, A.J.; McBride, M.J.; Rodriguez, E.S.R.; Killian, A.; et al. Local production of lactate, ribose phosphate, and amino acids within human triple-negative breast cancer. Med. 2021, 2, 736–754. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Guo, Le Zhan Yanxiang; J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.X.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Signal Transduct. Target. Ther. 2022, 7, 305. [Google Scholar] [CrossRef]
- Zhang, L.; Xin, C.; Wang, S.; Zhuo, S.; Zhu, J.; Li, Z.; Liu, Y.; Yang, L.; Chen, Y. Lactate transported by MCT1 plays an active role in promoting mitochondrial biogenesis and enhancing TCA flux in skeletal muscle. Sci. Adv. 2024, 10, eadn4508. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Ng, C.P.; Jones, O.; Fung, T.S.; Ryu, K.W.; Li, D.; Thompson, C.B. Lactate activates the mitochondrial electron transport chain independently of its metabolism. Mol. Cell. 2023, 83, 3904–3920.e7. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Zhang, J.; Zhou, Q.; He, X.; Zheng, Z.; Wei, Y.; Zhou, K.; Lin, Y.; Yu, H.; Zhang, H.; et al. Hypoxia induces mitochondrial protein lactylation to limit oxidative phosphorylation. Cell Res. 2024, 34, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Lamonte, G.; Tang, X.; Chen, J.L.; Wu, J.; Ding, C.K.; Keenan, M.M.; Sangokoya, C.; Kung, H.N.; Ilkayeva, O.; et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013, 1, 23. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Tumour acidosis: from the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Corbet, C.; Pinto, A.; Martherus, R.; Santiago de Jesus, J.P.; Polet, F.; Feron, O. Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef]
- Feng, Y.; Wu, L. mTOR up-regulation of PFKFB3 is essential for acute myeloid leukemia cell survival. Biochem. Biophys. Res. Commun. 2017, 483, 897–903. [Google Scholar] [CrossRef]
- Yalcin, A.; Solakoglu, T.H.; Ozcan, S.C.; Guzel, S.; Peker, S.; Celikler, S.; Balaban, B.D.; Sevinc, E.; Gurpinar, Y.; Chesney, J.A. 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase-3 is required for transforming growth factor b1-enhanced invasion of Panc1 cells in vitro. Biochem. Biophys. Res. Commun. 2017, 484, 687–693. [Google Scholar] [CrossRef]
- Meng, Y.M.; Jiang, X.; Zhao, X.; Meng, Q.; Wu, S.; Chen, Y.; Kong, X.; Qiu, X.; Su, L.; Huang, C.; et al. Hexokinase 2-driven glycolysis in pericytes activates their contractility leading to tumor blood vessel abnormalities. Nat. Commun. 2021, 12, 6011. [Google Scholar] [CrossRef]
- Wang, F.; Li, L.; Zhang, Z. Platelet isoform of phosphofructokinase promotes aerobic glycolysis and the progression of non-small cell lung cancer. Mol. Med. Rep. 2021, 23, 1. [Google Scholar] [CrossRef]
- Guo, D.; Meng, Y.; Zhao, G.; Wu, Q.; Lu, Z. Moonlighting functions of glucose metabolic enzymes and metabolites in cancer. Nat. Rev. Cancer 2025, 25, 426–446. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C. Tumor cell glycolysis – At the crossroad of epithelial–mesenchymal transition and autophagy. Cells 2022, 11, 1041. [Google Scholar] [CrossRef]
- Xu, D.; Shao, F.; Bian, X.; Meng, Y.; Liang, T.; Lu, Z. The evolving landscape of noncanonical functions of metabolic enzymes in cancer and other pathologies. Cell Metab. 2021, 33, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Rumio, C. On the role of glycolysis in early tumorigenesis — Permissive and executioner effects. Cells 2023, 12, 1124. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, S.; Liao, D.; Zhang, Q.; Chen, C.; Yang, X.; Jiang, D.; Pang, J. Overexpression of PFKFB3 promotes cell glycolysis and proliferation in renal cell carcinoma. BMC Cancer 2022, 22, 83. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Ma, Y.; Cao, L.; Zhan, S.; Xu, Y.; Fu, F.; Liu, C.; Zhang, G.; Wang, Z.; Wang, R.; et al. B7-H3 promotes aerobic glycolysis and chemoresistance in colorectal cancer cells by regulating HK2. Cell Death Dis. 2019, 10, 308. [Google Scholar] [CrossRef]
- Ye, F.; Chen, Y.; Xia, L.; Lian, J.; Yang, S. Aldolase A overexpression is associated with poor prognosis and promotes tumor progression by the epithelial-mesenchymal transition in colon cancer. Biochem. Biophys. Res. Commun. 2018, 497, 639–645. [Google Scholar] [CrossRef]
- Jin, L.; Chun, J.; Pan, C.; Alesi, G.N.; Li, D.; Magliocca, K.R.; Kang, Y.; Chen, Z.G.; Shin, D.M.; et al. Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. Oncogene 2017, 36, 3797–3806. [Google Scholar] [CrossRef]
- Yang, T.; Ren, C.; Qiao, P.; Han, X.; Wang, L.; Lv, S.; Sun, Y.; Liu, Z.; Du, Y.; Yu, Z. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene 2018, 37, 5997–6009. [Google Scholar] [CrossRef]
- Zheng, Y.; Wu, C.; Yang, J.; Zhao, Y.; Jia, H.; Xue, M.; Xu, D.; Yang, F.; Fu, D.; Wang, C.; et al. Insulin-like growth factor 1-induced enolase 2 deacetylation by HDAC3 promotes metastasis of pancreatic cancer. Signal Transduct. Target. Ther. 2020, 5, 53. [Google Scholar] [CrossRef]
- Li, F.L.; Liu, J.P.; Bao, R.X.; Yan, G.; Feng, X.; Xu, Y.P.; Sun, Y.P.; Yan, W.; Ling, Z.Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fu, S.Q.; Yuan, X.; Yu, F.; Ji, Q.; Tang, H.W.; Li, R.K.; Huang, S.; Huang, P.Q.; Qin, W.T.; et al. A GAPDH serotonylation system couples CD8+ T cell glycolytic metabolism to antitumor immunity. Mol. Cell. 2024, 84, 760–775.e7. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Ju, H.; Fan, J.; Shi, X.; Cheng, Y.; Cang, X.; Zheng, Z.; Duan, X.; Yi, W. O-GlcNAcylation of PGK1 coordinates glycolysis and TCA cycle to promote tumor growth. Nat. Commun. 2020, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Arif, A.; Willard, B.; Smith, J.D.; Stuehr, D.J.; Hazen, S.L.; Fox, P.L. Protection of extraribosomal RPL13a by GAPDH and dysregulation by S-Nitrosylation. Mol. Cell. 2012, 47, 656–663. [Google Scholar] [CrossRef]
- Nakajima, H.; Itakura, M.; Kubo, T.; Kaneshige, A.; Harada, N.; Izawa, T.; Azuma, Y.T.; Kuwamura, M.; Yamaji, R.; Takeuchi, T. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) aggregation causes mitochondrial dysfunction during oxidative stress-induced cell death. J. Biol. Chem. 2017, 292, 4727–4742. [Google Scholar] [CrossRef]
- Lin, M.; Huang, L.; Huang, J.; Yu, J.; Yang, X.; Yang, J. Modulation of PKM2 inhibits follicular helper T cell differentiation and ameliorates inflammation in lupus-prone mice. J. Autoimmun. 2024, 145, 103198. [Google Scholar] [CrossRef]
- Wang, H.; Fan, C.; Chen, X.; Zhou, W.; Guo, L.; Zhao, F.; Ye, S.; He, S.; Chen, Y. Pyruvate Kinase M2 Nuclear Translocation Regulate Ferroptosis-Associated Acute Lung Injury in Cytokine Storm. Inflammation 2024, 47, 1667–1684. [Google Scholar] [CrossRef]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–304. [Google Scholar] [CrossRef]
- Hosios, A.M.; Fiske, B.P.; Gui, D.Y.; Vander Heiden, M.G. Lack of Evidence for PKM2 Protein Kinase Activity. Mol. Cell. 2015, 59, 850–857. [Google Scholar] [CrossRef]
- Wang, X.; Liang, C.; Yao, X.; Yang, R.H.; Zhang, Z.S.; Liu, F.Y.; Li, W.Q.; Pei, S.H.; Ma, J.; Xie, S.Q.; et al. PKM2-induced the phosphorylation of histone H3 contributes to EGF-mediated PD-L1 transcription in HCC. Front. Pharmacol. 2020, 11, 577108. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, W.; Wang, Y.; Liu, J.; Hao, F.; Li, Y.; Tian, M.; Shu, H.; Dong, J.; Feng, Y.; et al. Pyruvate kinase M2 promotes the activation of dendritic cells by enhancing IL-12p35 expression. Cell Rep. 2020, 31, 107690. [Google Scholar] [CrossRef]
- Liu, P.; Sun, S.J.; Ai, Y.J.; Feng, X.; Zheng, Y.M.; Gao, Y.; Zhang, J.Y.; Zhang, L.; Sun, Y.P.; Xiong, Y.; et al. Elevated nuclear localization of glycolytic enzyme TPI1 promotes lung adenocarcinoma and enhances chemoresistance. Cell Death Dis. 2022, 13, 205. [Google Scholar] [CrossRef]
- Thomas, G.E.; Egan, G.; García-Prat, L.; Botham, A.; Voisin, V.; Patel, P.S.; Hoff, F.W.; Chin, J.; Nachmias, B.; Kaufmann, K.B.; et al. The metabolic enzyme hexokinase 2 localizes to the nucleus in AML and normal haematopoietic stem and progenitor cells to maintain stemness. Nat. Cell Biol. 2022, 24, 872–884. [Google Scholar] [CrossRef]
- Lv, L.; Xu, Y.P.; Zhao, D.; Li, F.L.; Wang, W.; Sasaki, N.; Jiang, Y.; Zhou, X.; Li, T.T.; Guan, K.L.; et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol. Cell. 2013, 52, 340–352. [Google Scholar] [CrossRef]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol. Cell. 2016, 61, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Cao, R.; Wang, X.; Zhang, Y.; Wang, P.; Gao, H.; Li, C.; Yang, F.; Zeng, R.; Wei, P.; et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 2017, 27, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Shegay, P.V.; Shatova, O.P.; Zabolotneva, A.A.; Shestopalov, A.V.; Kaprin, A.D. Moonlight functions of glycolytic enzymes in cancer. Front. Mol. Biosci. 2023, 10, 1076138. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, H.; Schnabel, L.; Meixensberger, J.; Gaunitz, F. Pyruvate attenuates the anti-neoplastic effect of carnosine independently from oxidative phosphorylation. Oncotarget 2016, 7, 85848–85860. [Google Scholar] [CrossRef]
- Wang, X.; Perez, E.; Liu, R.; Yan, L.J.; Mallet, R.T.; Yang, S.H. Pyruvate protects mitochondria from oxidative stress in human neuroblastoma SK-N-SH cells. Brain Res. 2007, 1132, 1–9. [Google Scholar] [CrossRef]
- Toshniwal, A.G.; Lam, G.; Bott, A.J.; Cluntun, A.A.; Skabelund, R.; Nam, H.J.; Wisidagama, D.R.; Thummel, C.S.; Rutter, J. The fate of pyruvate dictates cell growth by modulating cellular redox potential. Elife 2025, 13, RP103705. [Google Scholar] [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a metabolic checkpoint of anti-tumor T cell responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Hirota, M.; Sasaki, D.; Kalra, R.S.; Chien, H.C.; Tamai, M.; Sarkar, S.; Mi, Y.; Miyagi, M.; Seto, Y.; et al. Phosphoenolpyruvate regulates the Th17 transcriptional program and inhibits autoimmunity. Cell Rep. 2023, 42, 112205. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Torrini, C.; Nguyen, T.T.T.; Shu, C.; Mela, A.; Humala, N.; Mahajan, A.; Seeley, E.H.; Zhang, G.; Westhoff, M.A.; Karpel-Massler, G.; et al. Lactate is an epigenetic metabolite that drives survival in model systems of glioblastoma. Mol. Cell 2022, 82, 3061–3076.e6. [Google Scholar] [CrossRef]
- Zeng, S.; Hu, X. Lactic acidosis switches cancer cells from dependence on glycolysis to OXPHOS and renders them highly sensitive to OXPHOS inhibitors. Biochem. Biophys. Res. Commun. 2023, 671, 46–57. [Google Scholar] [CrossRef]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate transporters (SLC16): Function, regulation, and role in health and disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef]
- Certo, M.; Llibre, A.; Lee, W.; Mauro, C. Understanding lactate sensing and signalling. Trends Endocrinol. Metab. 2022, 33, 722–735. [Google Scholar] [CrossRef]
- Li, X.M.; Yang, Y.; Jiang, F.Q.; Hu, G.; Wan, S.; Yan, W.Y.; He, X.S.; Xiao, F.; Yang, X.M.; Guo, X.; et al. Histone lactylation inhibits RARg expression in macrophages to promote colorectal tumorigenesis through activation of TRAF6-IL-6-STAT3 signaling. Cell Rep. 2024, 43, 113688. [Google Scholar] [CrossRef]
- Sui, Y.; Shen, Z.; Wang, Z.; Feng, J.; Zhou, G. Lactylation in cancer: metabolic mechanism and therapeutic strategies. Cell Death Discov. 2025, 11, 68. [Google Scholar] [CrossRef]
- Zong, Z.; Ren, J.; Yang, B.; Zhang, L.; Zhou, F. Emerging roles of lysine lactyltransferases and lactylation. Nat. Cell Biol. 2025, 27, 563–574. [Google Scholar] [CrossRef]
- Zhou, J.; Gu, J.; Qian, Q.; Zhang, Y.; Huang, T.; Li, X.; Liu, Z.; Shao, Q.; Liang, Y.; Qiao, L.; et al. Lactate supports Treg function and immune balance via MGAT1 effects on N-glycosylation in the mitochondria. J. Clin. Invest. 2024, 134, e175897. [Google Scholar] [CrossRef]
- Qian, Y.; Galan-Cobo, A.; Guijarro, I.; Dang, M.; Molkentine, D.; Poteete, A.; Zhang, F.; Wang, Q.; Wang, J.; Parra, E.; et al. MCT4-dependent lactate secretion suppresses antitumor immunity in LKB1-deficient lung adenocarcinoma. Cancer Cell. 2023, 41, 1363–1380.e7. [Google Scholar] [CrossRef]
- Swietach, P.; Boedtkjer, E.; Pedersen, S.F. How protons pave the way to aggressive cancers. Nat. Rev. Cancer 2023, 23, 825–841. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Y.; Bozi, L.H.M.; Fischer, P.D.; Jedrychowski, M.P.; Xiao, H.; Wu, T.; Darabedian, N.; He, X.; Mills, E.L.; et al. Lactate regulates cell cycle by remodelling the anaphase promoting complex. Nature 2023, 616, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Monteith, A.J.; Ramsey, H.E.; Silver, A.J.; Brown, D.; Greenwood, D.; Smith, B.N.; Wise, A.D.; Liu, J.; Olmstead, S.D.; Watke, J.; et al. Lactate utilization enables metabolic escape to confer resistance to BET inhibition in acute myeloid leukemia. Cancer Res. 2024, 84, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhou, C.; Yu, L.; Hou, Z.; Liu, H.; Kong, L.; Xu, Y.; He, J.; Lan, J.; Ou, Q.; et al. Tumor-derived lactate promotes resistance to bevacizumab treatment by facilitating autophagy enhancer protein RUBCNL expression through histone H3 lysine 18 lactylation (H3K18la) in colorectal cancer. Autophagy 2024, 20, 114–130. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Chen, X.; Han, X.; Wang, Y.; Yu, D.; Li, Y.; Zhu, C.; Tong, Y.; Gao, S.; Wang, J.; et al.; 107 Lactate induces tumor progression via LAR motif-dependent Yin-Yang 1 degradation. Mol. Cancer Res. 2024, 22, 957–972. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Lin, R.; Huang, Y.; Wang, Z.; Xu, S.; Wang, L.; Chen, F.; Zhang, J.; Pan, K.; et al. Lactate drives epithelial-mesenchymal transition in diabetic kidney disease via the H3K14la/KLF5 pathway. Redox Biol. 2024, 75, 103246. [Google Scholar] [CrossRef]
- Romeo, E.; Caserta, C.A.; Rumio, C.; Marcucci, F. The vicious cross-talk between tumor cells with an EMT phenotype and cells of the immune system. Cells 2019, 8, 460. [Google Scholar] [CrossRef]
- Wang, S.; Huang, T.; Wu, Q.; Yuan, H.; Wu, X.; Yuan, F.; Duan, T.; Taori, S.; Zhao, Y.; Snyder, N.W.; et al. Lactate reprograms glioblastoma immunity through CBX3-regulated histone lactylation. J. Clin. Invest. 2024, 134, e176851. [Google Scholar] [CrossRef]
- Sajjanar, B.; Siengdee, P.; Trakooljul, N.; Liu, X.; Kalbe, C.; Wimmers, K.; Ponsuksili, S. Cross-talk between energy metabolism and epigenetics during temperature stress response in C2C12 myoblasts. Int. J. Hyperthermia. 2019, 36, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Luo, L.; Xu, Y.; Li, J.; Wu, Z.; Zhao, C.; Wen, J.; Jiang, P.; Zhu, H.; Wang, L.; et al. UHRF1-mediated epigenetic reprogramming regulates glycolysis to promote progression of B-cell acute lymphoblastic leukemia. Cell Death Dis. 2025, 16, 351. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Serra, P.; Marcilla, M.; Villanueva, A.; Ramos-Fernandez, A.; Palau, A.; Leal, L.; Wahi, J.E.; Setien-Baranda, F.; Szczesna, K.; Moutinho, C.; et al. A DERL3-associated defect in the degradation of SLC2A1 mediates the Warburg effect. Nat. Commun. 2014, 5, 3608. [Google Scholar] [CrossRef] [PubMed]
- Fitz-James, M.H.; Cavalli, G. Molecular mechanisms of transgenerational epigenetic inheritance. Nat. Rev. Genet. 2022, 23, 325–341. [Google Scholar] [CrossRef]
- Pan, R.Y.; He, L.; Zhang, J.; Liu, X.; Liao, Y.; Gao, J.; Liao, Y.; Yan, Y.; Li, Q.; Zhou, X.; et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022, 34, 634–648.e6. [Google Scholar] [CrossRef]
- Ziogas, A.; Novakovic, B.; Ventriglia, L.; Galang, N.; Tran, K.A.; Li, W.; Matzaraki, V.; van Unen, N.; Schlüter, T.; Ferreira, A.V.; et al. Long-term histone lactylation connects metabolic and epigenetic rewiring in innate immune memory. Cell 2025, 188, 2992–3012.e16. [Google Scholar] [CrossRef]
- Grossman, Z.; Singer, A. Tuning of activation thresholds explains flexibility in the selection and development of T cells in the thymus. Proc. Natl. Acad. Sci. USA 1996, 93, 14747–14752. [Google Scholar] [CrossRef]
- Tijs, C.; Konow, N.; Biewener, A.A. Effect of muscle stimulation intensity on the heterogeneous function of regions within an architecturally complex muscle. J. Appl. Physiol. 2021, 130, 941–951. [Google Scholar] [CrossRef]
- Bond, S.T.; Howlett, K.F.; Kowalski, G.M.; Mason, S.; Connor, T.; Cooper, A.; Streltsov, V.; Bruce, C.R.; Walder, K.R.; McGee, S.L. Lysine post-translational modification of glyceraldehyde-3-phosphate dehydrogenase regulates hepatic and systemic metabolism. FASEB J. 2017, 31, 2592–2602. [Google Scholar] [CrossRef]
- Brighenti, E.; Carnicelli, D.; Brigotti, M.; Fiume, L. The inhibition of lactate dehydrogenase A hinders the transcription of histone 2B gene independently from the block of aerobic glycolysis. Biochem. Biophys. Res. Commun. 2017, 485, 742–745. [Google Scholar] [CrossRef]
- Sirover, M.A. Subcellular dynamics of multifunctional protein regulation: Mechanisms of GAPDH intracellular translocation. J. Cell Biochem. 2012, 113, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Burdziak, C.; Alonso-Curbelo, D.; Walle, T.; Reyes, J.; Barriga, F.M.; Haviv, D.; Xie, Y.; Zhao, Z.; Zhao, C.J.; Chen, H.A.; et al. Epigenetic plasticity cooperates with cell-cell interactions to direct pancreatic tumorigenesis. Science 2023, 380, eadd5327. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xiao, B.; Hirukawa, A.; Smith, H.W.; Zuo, D.; Sanguin-Gendreau, V.; McCaffrey, L.; Nam, A.J.; Muller, W.J. Ezh2 promotes mammary tumor initiation through epigenetic regulation of the Wnt and mTORC1 signaling pathways. Proc. Natl. Acad. Sci. USA 2023, 120, e2303010120. [Google Scholar] [CrossRef] [PubMed]
- Heide, T.; Househam, J.; Cresswell, G.D.; Spiteri, I.; Lynn, C.; Mossner, M.; Kimberley, C.; Fernandez-Mateos, J.; Chen, B.; Zapata, L.; et al. The co-evolution of the genome and epigenome in colorectal cancer. Nature 2022, 611, 733–743. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP- sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C. How tumor cells choose between epithelial-mesenchymal transition and autophagy to resist stress—Therapeutic implications. Front. Pharmacol. 2018, 9, 714. [Google Scholar] [CrossRef]
- Sirover, M.A. Structural analysis of glyceraldehyde-3-phosphate dehydrogenase functional diversity. Int. J. Biochem. Cell Biol. 2014, 57, 20–26. [Google Scholar] [CrossRef]
- Dykens, J.A.; Jamieson, J.; Marroquin, L.; Nadanaciva, S.; Billis, P.A.; Will, Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol. 2008, 233, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Lee, M.; Park, M.; Kim, S.Y.; Shim, M.S.; Lee, C.Y.; Choi, D.H.; Cho, Y. Metformin and dichloroacetate suppress proliferation of liver cancer cells by inhibiting mTOR complex 1. Int. J. Mol. Sci. 2021, 22, 10027. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.; Fleming, G.A.; Chen, K.; Bicsak, T.A. Metformin-associated lactic acidosis: Current perspectives on causes and risk. Metabolism 2016, 6, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, F.; Bradbury, C.M.; Kaushal, A.; Li, L.; Spitz, D.R.; Aft, R.L.; Gius, D. 2- Deoxy-D-glucose-induced cytotoxicity and radiosensitization in tumor cells is mediated via disruptions in thiol metabolism. Cancer Res. 2003, 63, 3413–3417. [Google Scholar]
- Beloueche-Babari, M.; Wantuch, S.; Casals Galobart, T.; Koniordou, M.; Parkes, H.G.; Arunan, V.; Chung, Y.L.; Eykyn, T.R.; Smith, P.D.; Leach, M.O. MCT1 inhibitor AZD3965 increases mitochondrial metabolism, facilitating combination therapy and noninvasive magnetic resonance spectroscopy. Cancer Res. 2017, 77, 5913–5924. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Haugrud, A.B.; Zhuang, Y.; Coppock, J.D.; Miskimins, W.K. Dichloroacetate enhances apoptotic cell death via oxidative damage and attenuates lactate production in metformin-treated breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 539–550. [Google Scholar] [CrossRef]
- Li, B.; Li, X.; Ni, Z.; Zhang, Y.; Zeng, Y.; Huang, Y.; He, J.; Lyu, X.; Wu, Y.; Wang, Y.; et al. Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells. Oncotarget 2016, 7, 59458–59470. [Google Scholar] [CrossRef]
- Chandel, N.S. NADPH—The forgotten reducing equivalent. Cold Spring Harb. Perspect. Biol. 2021, 13, a040550. [Google Scholar] [CrossRef]
- Marcucci, F.; Caserta, C.A.; Romeo, E.; Rumio, C. Antibody-drug conjugates (ADC) against cancer stem-like cells (CSC) – is there still room for optimism? Front. Oncol. 2019, 9, 167. [Google Scholar] [CrossRef]
- Marcucci, F.; Lefoulon, F. Active targeting with particulate drug carriers in tumor therapy: Fundamentals and recent progresses. Drug Discov. Tod 2004, 9, 219–228. [Google Scholar] [CrossRef]
Figure 1.
Stimuli that upregulate fermentative glycolysis. Cell-autonomous or –nonautonomous stimuli can induce overexpression or PTM of glycolytic enzyme(s). These, in turn, promote overall upregulation of fermentative glycolysis with increased production of metabolites, in particular lactate, which can induce several biological effects. PTM, post-translational modification; ECM, extracellular matrix; ROS, reactive oxygen species, CK, cytokine.
Figure 1.
Stimuli that upregulate fermentative glycolysis. Cell-autonomous or –nonautonomous stimuli can induce overexpression or PTM of glycolytic enzyme(s). These, in turn, promote overall upregulation of fermentative glycolysis with increased production of metabolites, in particular lactate, which can induce several biological effects. PTM, post-translational modification; ECM, extracellular matrix; ROS, reactive oxygen species, CK, cytokine.
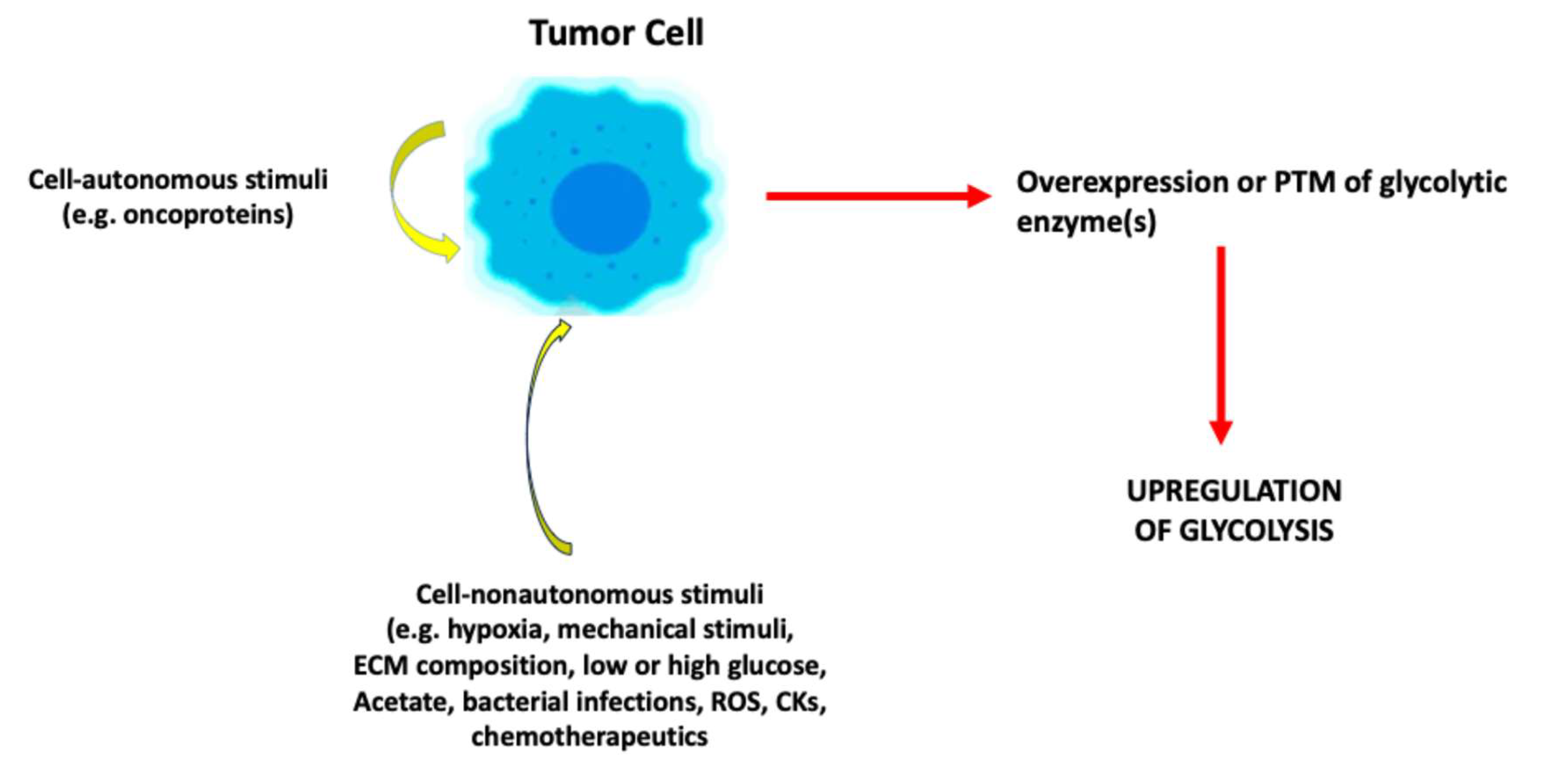
Figure 2.
Outputs of upregulated fermentative glycolysis in tumor cells. The figure depicts the outputs of fermentative glycolysis that has been induced/upregulated in response to cell-autonomous or –nonautonomous stimuli. ATP, adenosine triphosphate.
Figure 2.
Outputs of upregulated fermentative glycolysis in tumor cells. The figure depicts the outputs of fermentative glycolysis that has been induced/upregulated in response to cell-autonomous or –nonautonomous stimuli. ATP, adenosine triphosphate.
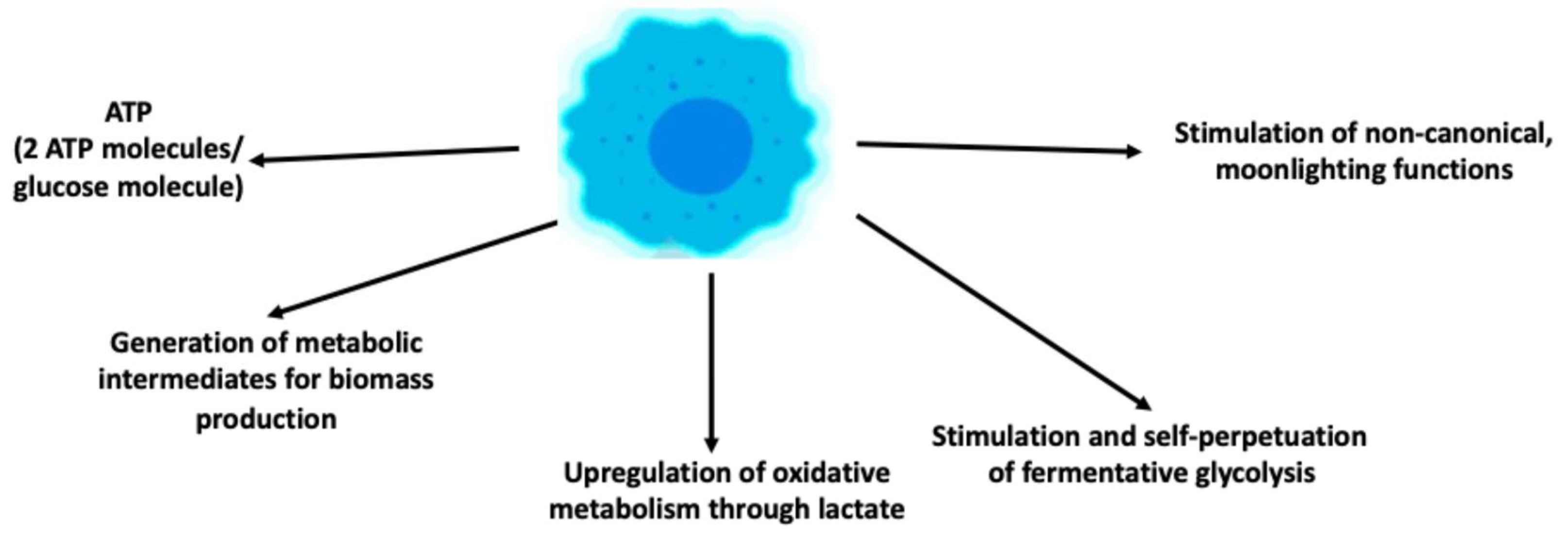
Figure 3.
Mechanisms underlying moonlighting functions of glycolytic enzymes or metabolites. Moonlighting functions may become detectable following overexpression of glycolytic enzymes which may induce overall upregulation of fermentative glycolysis and to increased levels of glycolytic metabolites (e.g., lactate); PTM of glycolytic enzymes; alteration of the quaternary structure of glycolytic enzymes leading to their aggregation or to deoligomerisation (e.g., tetramer-dimer/monomer conversion of PKM2). PKM2, pyruvate kinase isoform M2; PTM, posttranslational modification.
Figure 3.
Mechanisms underlying moonlighting functions of glycolytic enzymes or metabolites. Moonlighting functions may become detectable following overexpression of glycolytic enzymes which may induce overall upregulation of fermentative glycolysis and to increased levels of glycolytic metabolites (e.g., lactate); PTM of glycolytic enzymes; alteration of the quaternary structure of glycolytic enzymes leading to their aggregation or to deoligomerisation (e.g., tetramer-dimer/monomer conversion of PKM2). PKM2, pyruvate kinase isoform M2; PTM, posttranslational modification.
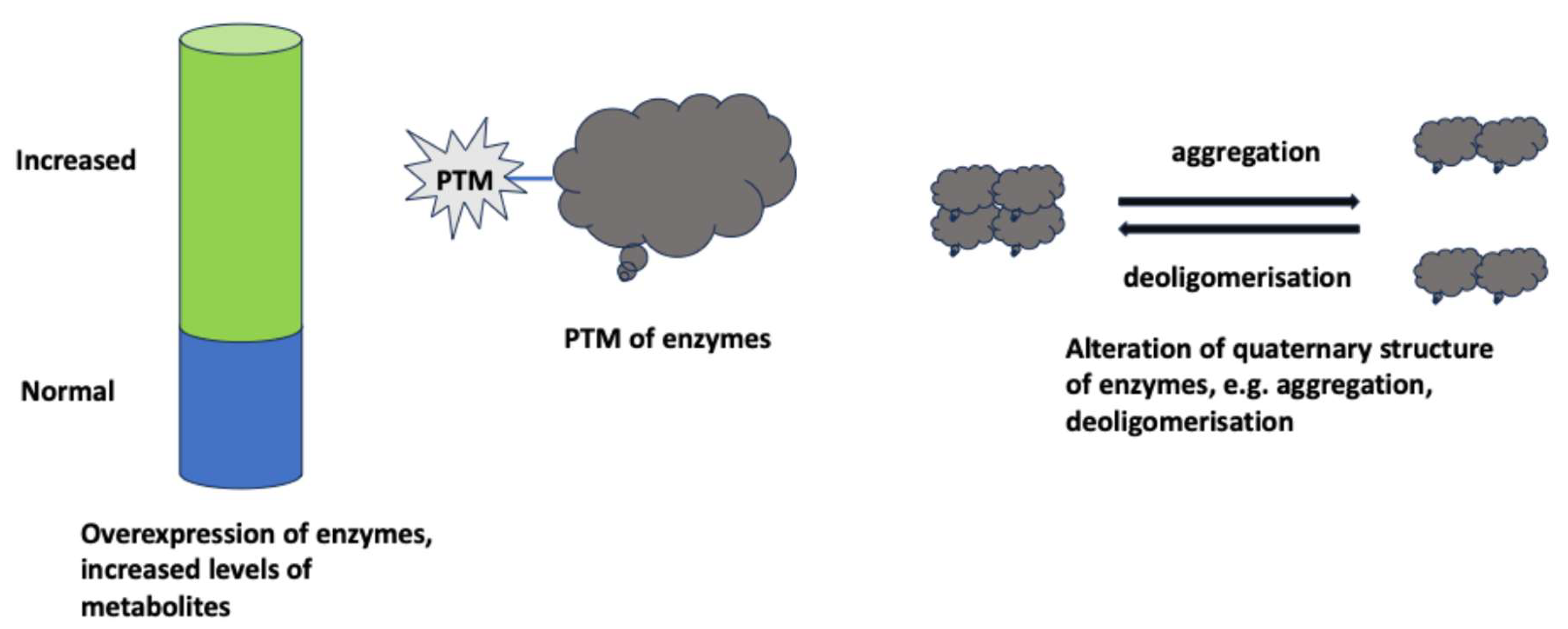
Figure 4.
Stimuli inducing upregulation of glycolysis. Some stimuli, referred to as physiological, induce a limited increase of fermentative glycolysis which responds to the metabolic demands of the cell. Other stimuli, referred to as supraphysiological, whether cell-autonomous or –nonautonomous, are perceived as danger signals by the affected cell and lead to strong upregulation of fermentative glycolysis with the appearance of moonlighting functions.
Figure 4.
Stimuli inducing upregulation of glycolysis. Some stimuli, referred to as physiological, induce a limited increase of fermentative glycolysis which responds to the metabolic demands of the cell. Other stimuli, referred to as supraphysiological, whether cell-autonomous or –nonautonomous, are perceived as danger signals by the affected cell and lead to strong upregulation of fermentative glycolysis with the appearance of moonlighting functions.
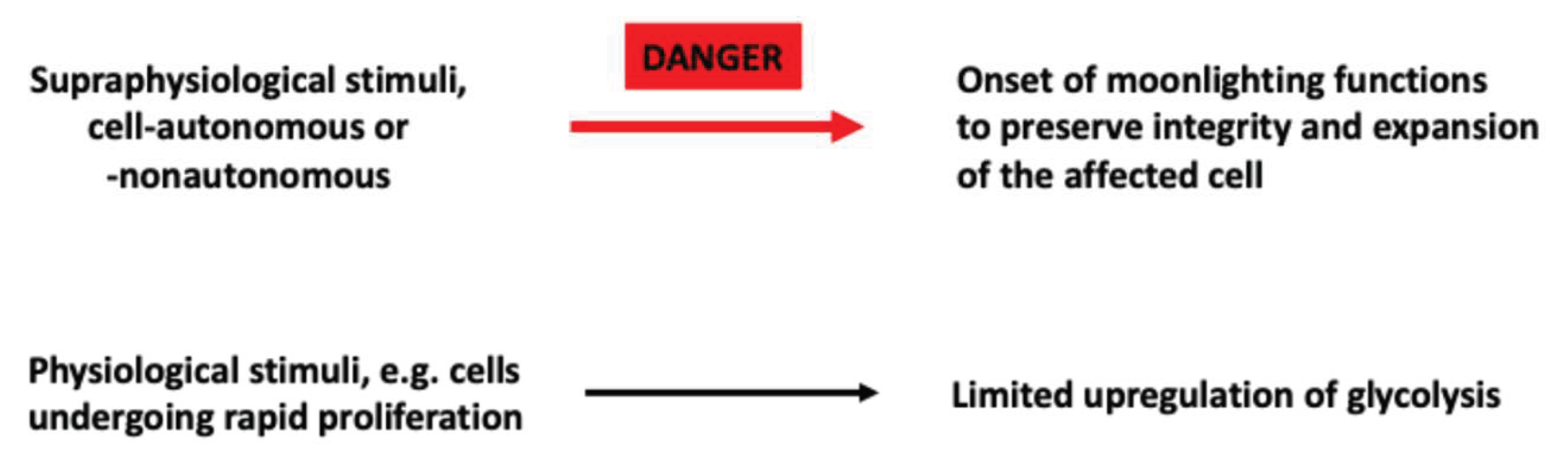
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



