Submitted:
10 February 2026
Posted:
11 February 2026
You are already at the latest version
Abstract
An alkene-tethered enaminone was synthesized in four steps from bromoacetic acid and 3,3-dimethylallyl alcohol. The enaminone was fully characterized, including UV-Vis spectra. TBADT-catalyzed HAT of the alkene-tethered enaminone initiated a fragmentation that yielded the literature-known phenylacetone-derived enaminone.
Keywords:
alkene
; enaminone
; hydrogen atom transfer (HAT)
; fragmentation
; tetrabutylammonium decatungstate (TBADT)
; phenylacetone
1. Introduction
Enaminones, which are masked 1,3-dicarbonyl compounds, are valuable starting materials for synthesizing diverse heterocyclic systems and are known for their robustness, scalability, and reproducibility in synthesis [1,2,3,4,5,6]. They can be prepared on a large scale from inexpensive, commercially available chemicals. Among the various synthetic approaches to enaminones [7], synthesis from compounds containing acidic methylene or methyl groups using amide acetals is notable for its simplicity [1,2]. Recently, enaminones have found diverse applications as starting materials in photocatalyzed transformations [8,9,10], including the photochemical [2+2] aza-de Mayo reaction [11].
Photocatalyzed HAT transformations have become a key area in photocatalysis, employing carbonyl-containing organic catalysts, organometallic catalysts, and nitrogen-centered photocatalysts [12,13,14,15,16]. Extending this methodology to asymmetric synthesis further enhanced its value in organic chemistry [17,18]. In this study, an alkene-tethered enaminone, 4-(dimethylamino)-1-((3-methylbut-2-en-1-yl)oxy)-3-phenylbut-3-en-2-one, was synthesized in four steps from 3,3-dimethylallyl alcohol and 2-bromoacetic acid to investigate an intramolecular HAT-catalyzed cyclization, which ultimately resulted in HAT-initiated fragmentation.
2. Results and Discussion
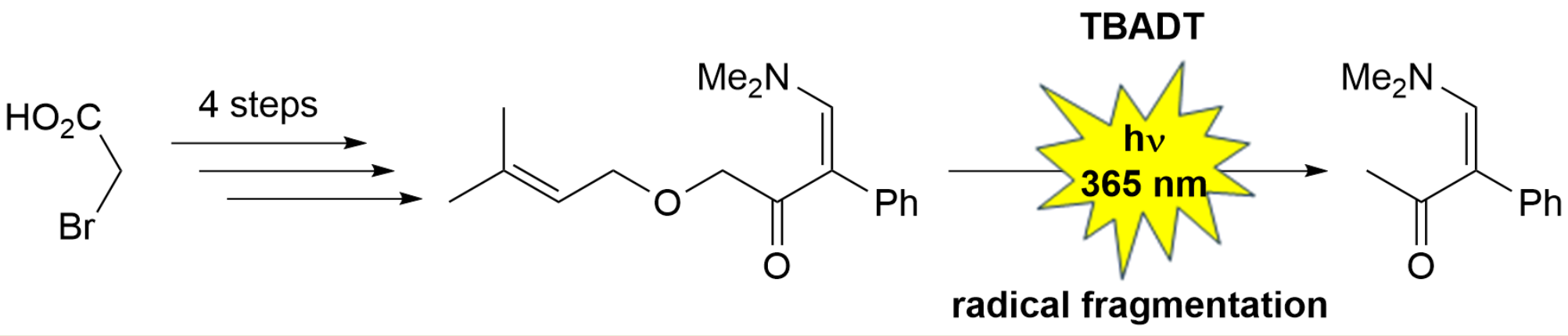
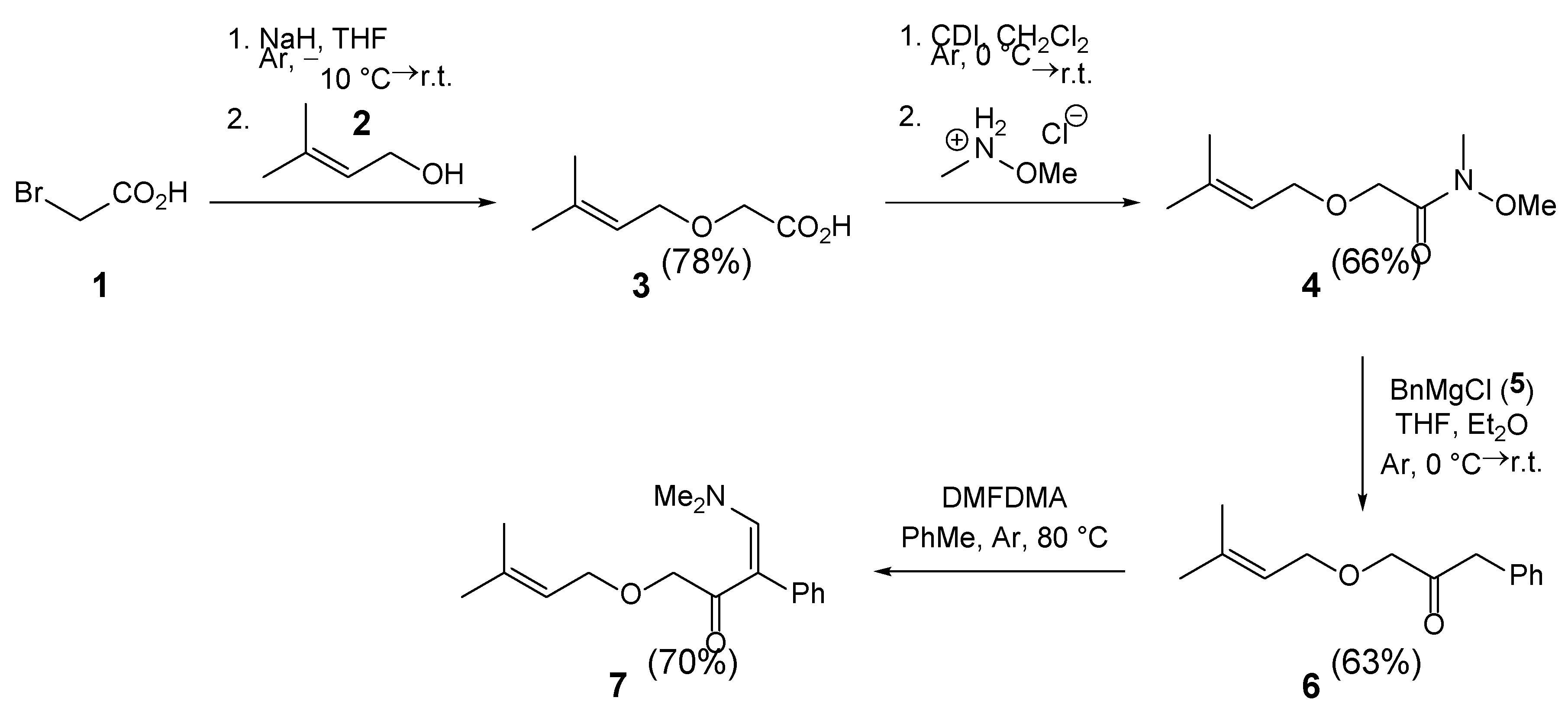
Enaminone 7 was synthesized in four steps from 2-bromoacetic acid (1) and 3,3-dimethylallyl alcohol (2) (Scheme 1). Following a literature procedure [19], 3,3-dimethylallyl alcohol (2) was alkylated with 2-bromoacetic acid (1) in the presence of excess sodium hydride to give acid 3 in 78% yield. Treatment of acid 3 with 1,1′-carbonyldiimidazole (CDI), followed by addition of N,O-dimethylhydroxylamine hydrochloride, afforded Weinreb amide 4 in 66% yield [20]. Subsequent reaction of amide 4 with excess benzylmagnesium chloride (5) gave ketone 6 in 63% yield. Although ketone 6 was isolated by column chromatography, it contained 45% benzyl alcohol [21] and 12% diphenylethane [22], as determined by proton and carbon NMR spectra. These impurities did not affect the subsequent enaminone formation with excess N,N-dimethylformamide dimethyl acetal (DMFDMA). The enaminone 7 was isolated in 70% yield by column chromatography (Scheme 1).
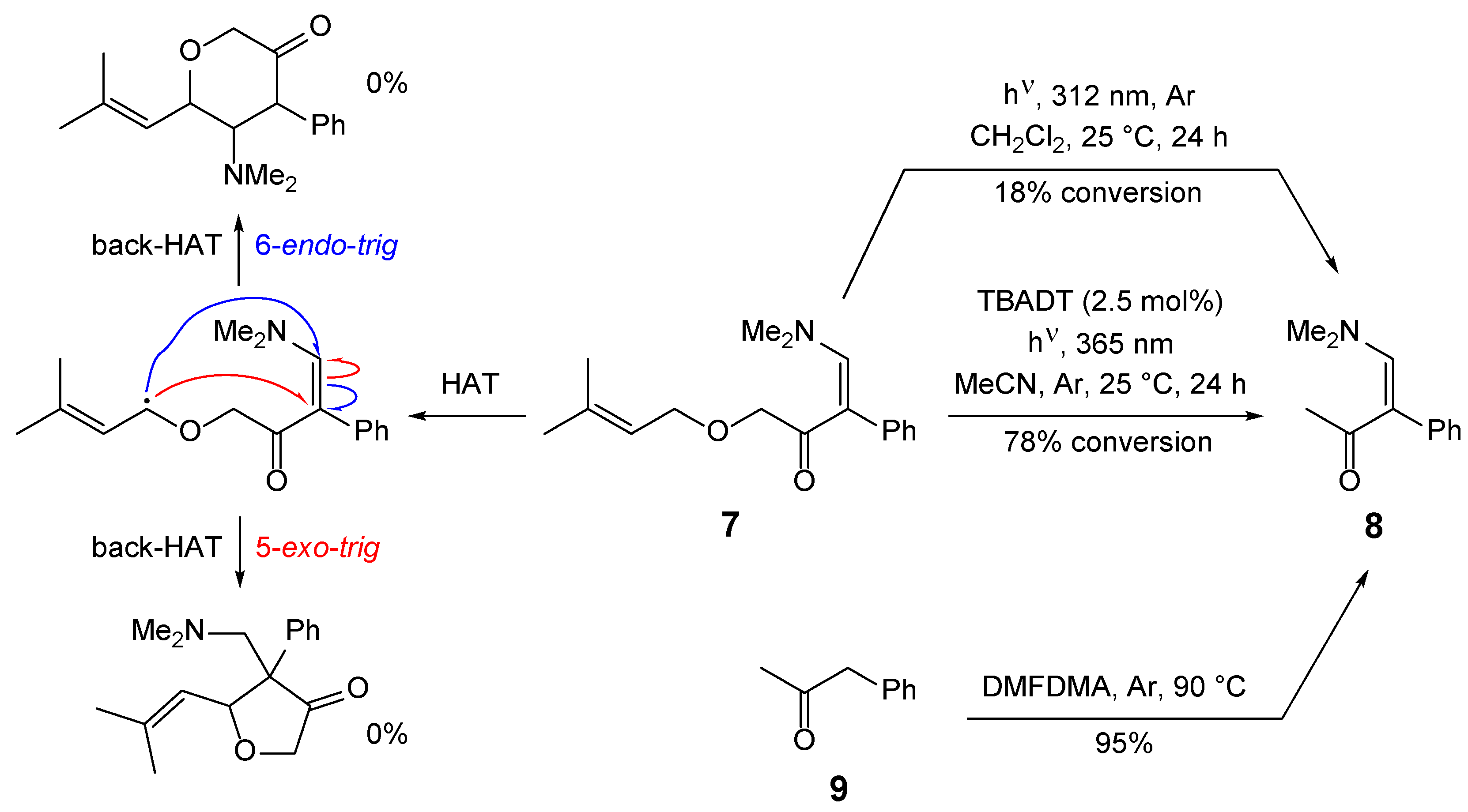
Next, the tetrabutylammonium decatungstate (TBADT) photocatalyzed HAT transformation of enaminone 7 was studied (Scheme 2). It was expected that 5- and/or 6-membered heterocycles would form via initial hydrogen atom abstraction at the allylic position adjacent to the oxygen atom, as that C–H bond has the lowest bond dissociation energy (BDE = 81.6 kcal/mol for allyl alcohol; 85.8 kcal/mol for benzyl methyl ether) [23]. Subsequent addition of the radical to the double bond, either in a 5-exo-trig or 6-endo-trig fashion followed by back-HAT, would yield the heterocyclic systems. However, irradiation of enaminone 7 at 365 nm with TBADT in anhydrous acetonitrile at room temperature for 24 hours resulted in 78% conversion to enaminone 8 [24]. Attempts to isolate enaminone 8 by column chromatography were unsuccessful. The identity of enaminone 8 was confirmed by independent synthesis from phenylacetone (9) and DMFDMA, which gave 8 in 95% yield with 1H NMR data identical to the literature data [24]. A photochemical [2+2] aza-de Mayo reaction conducted at room temperature by irradiating a diethyl ether solution of enaminone 7 at 312 nm for 24 hours did not yield the expected [2+2] cycloadducts, although 18% conversion to enaminone 8 was observed (Scheme 2).
The TBADT-photocatalyzed fragmentation of enaminone 7 to enaminone 8 can be explained by an initial hydrogen atom transfer (HAT) at the allylic position adjacent to the oxygen atom, followed by fragmentation to 3,3-dimethylacrolein (not observed in the reaction mixture) and an α-methylketone radical, which, after back-HAT, yields the observed enaminone 8. The formation of a strong C=O bond could be the driving force of the reaction (Scheme 3).
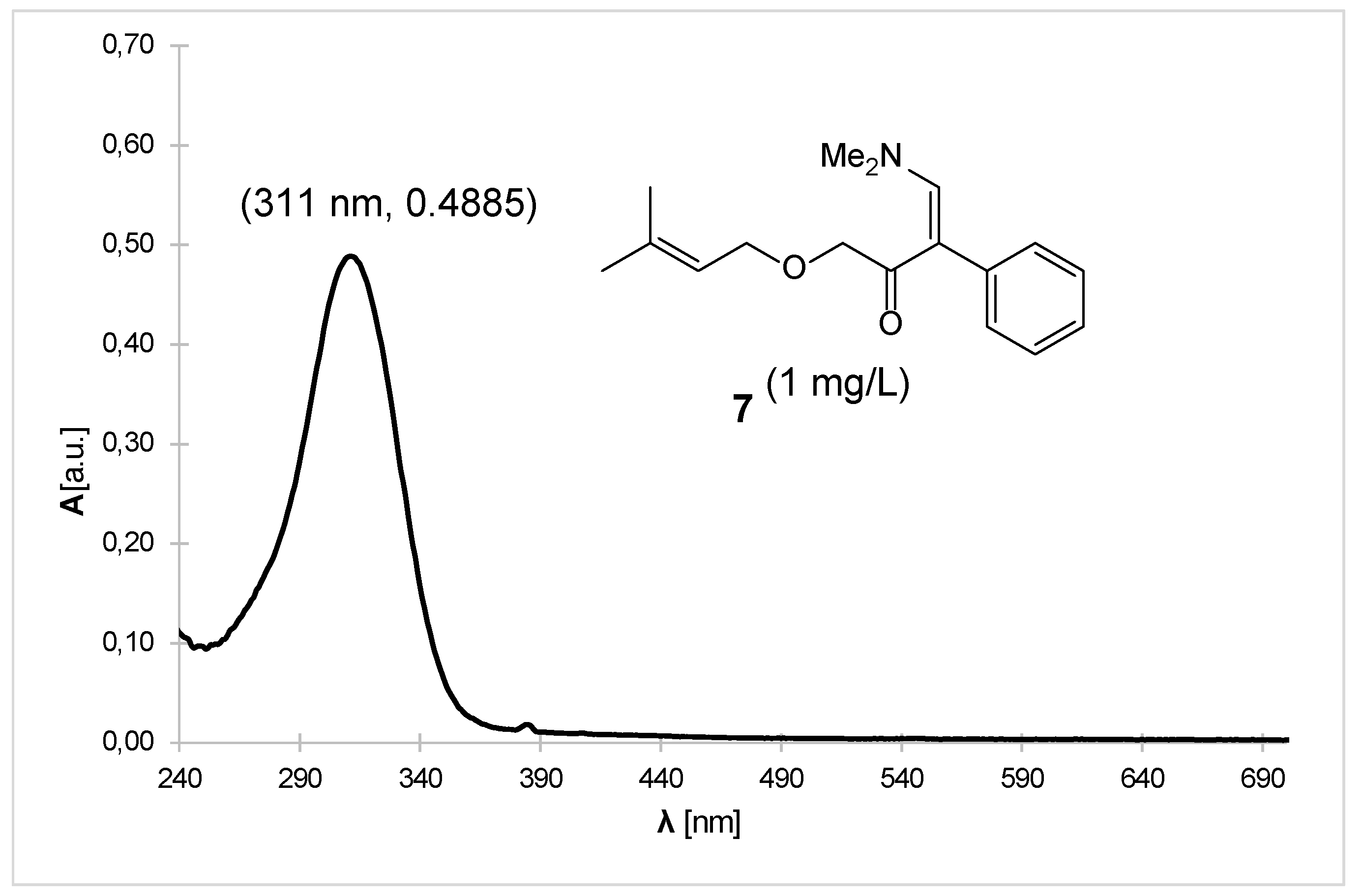
The structure of enaminone 7 was confirmed by 1H and 13C NMR, IR, high-resolution mass spectrometry, and UV-Vis spectra (Figure 1). Literature-known compounds 3 and 8 were confirmed by proton NMR spectra. Similarly, Weinreb amide 4 was characterized by 1H NMR, while ketone 6 was characterized by both proton and carbon NMR, as both were used in the synthesis of enaminone 7.
3. Materials and Methods
Solvents for extractions and chromatography were technical grade and distilled before use. Extracts were dried over technical grade anhydrous Na2SO4. The NMR spectra were recorded on a Bruker UltraShield 500 Plus spectrometer and a Bruker AVANCE NEO 600 MHz NMR spectrometer (Bruker, Billerica, Massachusetts, United States) at 500 and 600 MHz for 1H and at 126 and 150 MHz for 13C, respectively, using CDCl3 as solvent, with TMS as the internal standard. Mass spectra were recorded on an Agilent 6224 Accurate Mass TOF LC/MS (Agilent Technologies, Santa Clara, California, United States), and IR spectra were recorded on a Perkin-Elmer Spectrum BX FTIR spectrophotometer (PerkinElmer, Waltham, Massachusetts, United States). Photoinduced experiments were performed with a commercially available Penn PhD Photoreactor m2 (Penn Photon Devices, USA). Reactions were performed with stirring in borosilicate vials, which were placed approximately 1 cm from the light source. UV-a (365 nm) light was used at 100% intensity, unless specified differently. A photochemical reactor, Buckinghamshire model No. MLU/8, serial No. P/076 (λ = 312 nm) (Photochemical Reactors Ltd., Buckinghamshire, UK), was used for the photochemical reactions. Column chromatography was performed on silica gel (Silica gel 60, particle size 0.035–0.070 mm; Sigma-Aldrich, St. Louis, Missouri, United States). All commercially available chemicals were purchased from Sigma-Aldrich (St. Louis, Missouri, United States).
Synthesis of 2-((3-methylbut-2-en-1-yl)oxy)acetic Acid (3) [19]
To a suspension of NaH (60% dispersion in mineral oil, 32 mmol, 1.28 g) in anhydrous THF (20 mL) under argon, cooled to –10 °C, 2-bromoacetic acid (1) (97%, 10.6 mmol, 1.518 g) was added portionwise (3-5 portions), ensuring that hydrogen gas evolution ceased before each addition. After the addition of 2-bromoacetic acid, the reaction mixture was stirred at room temperature for 10 minutes, followed by slow addition of 3-methylbut-2-en-1-ol (2) (10.0 mmol, 1.016 mL). The reaction mixture was stirred at room temperature for 24 hours. Excess sodium hydride was carefully quenched with aqueous NaOH (1 M, 8 mL) and extracted with diethyl ether (20 mL). The organic phase was washed with aqueous NaOH (1 M, 4 × 40 mL), then discarded, and the combined aqueous phase was acidified with aqueous HCl (1 M) to pH 1 and extracted with ethyl acetate (3 × 20 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The crude acid 3 was used in the following transformation without further purification. Yield: 1.125 g (7.80 mmol, 78%) of yellowish oil. 1H-NMR (600 MHz, CDCl3): δ 1.70 (s, 3H), 1.77 (s, 3H), 4.11 (s, 2H), 4.11 (d, J=7.4 Hz, 2H), 5.33 – 5.39 (m, 1H), 10.10 (br s, 1H).
Synthesis of N-methoxy-N-methyl-2-((3-methylbut-2-en-1-yl)oxy)acetamide (4) [20]
To a solution of 2-((3-methylbut-2-en-1-yl)oxy)acetic acid (3) (8.2 mmol, 1.182 g) in anhydrous CH2Cl2 (20 mL) under argon, cooled to –10 °C, 1,1′-carbonyldiimidazole (CDI) (97%, 9.84 mmol, 1.645) was added. The reaction mixture was stirred at –10 °C for 10 minutes, then at room temperature for 60 minutes, followed by the addition of N,O-dimethylhydroxylamine hydrochloride (98%, 12.3 mmol, 1.224 g). The reaction mixture was stirred at room temperature for 24 hours. The mixture was poured into aqueous HCl (0.5 M, 100 mL) and extracted with EtOAc (5 × 20 mL). The combined organic phase was washed with saturated aqueous Na2CO3 (3 × 20 mL) and saturated aqueous NaCl (3 × 20 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The crude amide 4 was used in the following transformation without further purification. Yield: 1.013 g (5.412 mmol, 66%) of yellowish oil. 1H-NMR (600 MHz, CDCl3): δ 1.70 (s, 3H), 1.76 (s, 3H), 3.20 (s, 3H), 3.69 (s, 3H), 4.11 (d, J=7.1 Hz, 2H), 4.25 (s, 2H), 5.40 (tt, J=1.4, 7.0 Hz, 1H).
Synthesis of 1-((3-methylbut-2-en-1-yl)oxy)-3-phenylpropan-2-one (6)
To a solution of N-methoxy-N-methyl-2-((3-methylbut-2-en-1-yl)oxy)acetamide (4) (5.40 mmol, 1.011 g) in anhydrous THF (20 mL) under argon, cooled to –10 °C, benzylmagnesium chloride (5) (1 M in Et2O, 10.8 mmol, 10.8 mL) was added. The reaction mixture was stirred at –10 °C for 10 minutes, then at room temperature for 24 hours. The reaction mixture was quenched with aqueous HCl (0.5 M, 30 mL) and extracted with EtOAc (3 × 20 mL). The combined organic phase was washed with saturated aqueous NaHCO3 (3 × 10 mL) and saturated aqueous NaCl (3 × 10 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The residue was purified by column chromatography (Silica gel 60; EtOAc/petroleum ether = 1:10). The fractions containing product 6 were combined, and the volatiles were evaporated in vacuo. The isolated ketone 6 contained 45% benzyl alcohol [21] (singlet at 4.69 ppm) and 12% 1,2-diphenylethane (singlet at 2.92 ppm) [22]. Ketone 6 was used in the following transformation without further purification. Yield: 743 mg (3.402 mmol, 63%) of yellowish oil. 1H-NMR (600 MHz, CDCl3): δ 1.64 (s, 3H), 1.74 (s, 3H), 3.76 (s, 2H), 4.00 (d, J=7.1 Hz, 2H), 4.06 (s, 2H), 5.29 – 5.35 (m, 1H), 7.18 – 7.39 (m, 5H). 13C-NMR (151 MHz, CDCl3): δ 18.12, 25.92, 46.34, 67.78, 74.25, 120.21, 127.09, 128.81, 129.61, 133.67, 138.52, 206.65.
Synthesis of 4-(dimethylamino)-1-((3-methylbut-2-en-1-yl)oxy)-3-phenylbut-3-en-2-one (7)
To a solution of 1-((3-methylbut-2-en-1-yl)oxy)-3-phenylpropan-2-one (6) (2.40 mmol, 524 mg) in anhydrous toluene (5 mL) under argon at room temperature, N,N-dimethylformamide dimethyl acetal (DMFDMA) (94%, 7.2 mmol, 1.018 mL) was added. The reaction mixture was stirred at 80 °C for 2 hours. Volatiles were evaporated in vacuo. The residue was purified by column chromatography (Silica gel 60; 1. EtOAc:petroleum ether = 1:10 to elute nonpolar impurities, 2. EtOAc to elute enaminone 7). Fractions containing pure product 7 were combined, and volatiles were evaporated in vacuo. Yield: 459 mg (1.68 mmol, 70%) of yellowish oil. EI-HRMS: m/z = 274.1806 (MH+); C17H24NO2 requires: m/z = 274.1802 (MH+); νmax 2912, 1661, 1561, 1495, 1428, 1385, 1263, 1200, 1069, 1027, 948, 841, 784, 718, 701, 628 cm-1. 1H-NMR (600 MHz, CDCl3): δ 1.59 (s, 3H), 1.69 (s, 3H), 2.69 (br s, 6H), 3.93 (s, 2H), 3.94 (d, J=7.1 Hz, 2H), 5.27 – 5.33 (m, 1H), 7.17 – 7.23 (m, 2H), 7.24 – 7.34 (m, 3H), 7.68 (s, 1H). 13C-NMR (151 MHz, CDCl3): δ 18.02, 25.81, 43.13, 67.28, 73.18, 108.68, 121.05, 127.05, 128.04, 132.40, 136.80, 137.16, 148.94, 194.10.
Synthesis of 4-(dimethylamino)-3-phenylbut-3-en-2-one (8) [24]
To a solution of 4-(dimethylamino)-1-((3-methylbut-2-en-1-yl)oxy)-3-phenylbut-3-en-2-one (7) (0.11 mmol, 30.1 mg) in anhydrous MeCN (5 mL) under argon at room temperature, tetrabutylammonium decatungstate (TBADT) (2.75 μmol, 9.13 mg) was added. The reaction vessel was purged with argon until most of the solvent had evaporated and about 1 mL remained. The reaction mixture was irradiated in a commercially available Penn PhD Photoreactor m2 at 365 nm at 25 °C for 24 hours. During irradiation, the reaction mixture turned green; upon exposure to air, the color changed to brown. Volatiles were evaporated in vacuo (brown oil). Proton spectra recorded in CDCl3 indicated 78% conversion to enaminone 8. Attempted isolation of 8 by column chromatography (EtOAc) failed to separate the two enaminones; thus, 8 was obtained containing 28% of the starting enaminone 7. The identity of enaminone 8 was confirmed by independent synthesis from phenylacetone and DMFDMA (vide infra).
Synthesis of 4-(dimethylamino)-3-phenylbut-3-en-2-one (8) from phenylacetone (9) [24]
To a solution of phenylacetone (9) (10 mmol, 1.337 mL) under argon at room temperature, N,N-dimethylformamide dimethyl acetal (DMFDMA) (94%, 40 mmol, 5.653 mL) was added. The reaction mixture was stirred at 90 °C for 14 hours. Volatiles were evaporated in vacuo. The residue was purified by column chromatography (Silica gel 60; EtOAc). Fractions containing pure product 8 were combined, and volatiles were evaporated in vacuo. Yield: 1.798 g (9.50 mmol, 95%) of yellowish oil. 1H-NMR (500 MHz, CDCl3): δ 1.93 (s, 3H), 2.69 (br s, 6H), 7.17 – 7.22 (m, 2H), 7.23 – 7.28 (m, 1H), 7.29 – 7.34 (m, 2H), 7.61 (s, 1H).
4. Conclusions
An alkene-tethered enaminone 6 was synthesized in four steps from bromoacetic acid (1) and 3,3-dimethylallyl alcohol (2). The initial nucleophilic substitution yielded acid 3, which was converted to Weinreb amide 4. Treatment of amide 4 with benzylmagnesium chloride (5) gave ketone 6. Subsequent reaction with DMFDMA afforded enaminone 7. Enaminone 7 was fully characterized, including UV-Vis spectra. TBADT-catalyzed HAT of the alkene-tethered enaminone 7 initiated a fragmentation that produced the literature-known phenylacetone-derived enaminone 8.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. The following supporting information can be downloaded: copies of 1H- and 13C-NMR spectra; copies of 2D spectra; copies of HRMS reports; copies of IR spectra.
Author Contributions
Conceptualization, L.C., U.G., J.S. and B.Š.; methodology, L.C. and U.G.; software, L.C., A.B., U.G., J.S. and B.Š.; validation, L.C., N.P., A.B., U.G., J.S., F.P. and B.Š.; formal analysis, U.G., A.B. and L.C.; investigation, A.B., L.C. and U.G.; resources, L.C., U.G. and J.S.; data curation, L.C., N.P., A.B., U.G., J.S. and B.Š.; writing—original draft preparation, L.C., U.G., J.S. and B.Š.; writing—review and editing, L.C., N.P., U.G., J.S., F.P. and B.Š.; visualization, L.C., A.B., U.G., B.Š. and J.S.; supervision, U.G.; project administration, U.G. and J.S.; funding acquisition, U.G. and J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Slovenian Research and Innovation Agency (ARIS), research core funding No. P1-0179, and infrastructure program No. I0-0022.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Acknowledgments
We thank the EN-FIST Centre of Excellence, Dunajska 156, 1000 Ljubljana, Slovenia, for the use of their BX FTIR spectrophotometer.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Stanovnik, B.; Svete, J. Synthesis of Heterocycles from Alkyl 3-(Dimethylamino)propenoates and Related Enaminones. Chem. Rev. 2004, 104, 2433–2480. [Google Scholar] [CrossRef]
- Stanovnik, B. Enaminone, Enaminoesters, and Related Compounds in the Metal-Free Synthesis of Pyridines and Fused Pyridines. Eur. J. Org. Chem. 2019, 2019, 5120–5132. [Google Scholar] [CrossRef]
- Magoo, D.; Aggarwal, K.; Gupta, S.; Meena, K. Enamines and their variants as intermediates for synthesis of aza-heterocycles with applications in MCRs. Tetrahedron 2022, 103, 132545. [Google Scholar] [CrossRef]
- Huang, J.; Yu, F. Recent Advances in Organic Synthesis Based on N,N-Dimethyl Enaminones. Synthesis 2020, 53, 587–610. [Google Scholar] [CrossRef]
- Qu, Y.; Yang, F.; Han, Y.; Feng, F.; Wang, C. Recent Advances of Enaminones in Multi-component Reactions: A Brief Review. Eur. J. Org. Chem. 2025, 28, e202500167. [Google Scholar] [CrossRef]
- Chattopadhyay, A. K.; Hanessian, S. Cyclic enaminones. Part II: applications as versatile intermediates in alkaloid synthesis. Chem. Commun. 2015, 51, 16450–16467. [Google Scholar] [CrossRef]
- Govindh, B.; Diwakar, B. S.; Murthy, Y. L. N. A brief review on synthesis & applications of β-enamino carbonyl compounds. Org. Commun. 2012, 5, 105–119. [Google Scholar]
- Han, Y.; Zhou, L.; Wang, C.; Feng, S.; Ma, R.; Wan, J.-P. Recent advances in visible light-mediated chemical transformations of enaminones. Chin. Chem. Lett. 2024, 35, 108977. [Google Scholar] [CrossRef]
- Hu, W.; Diao, X.; Yuan, J.; Liang, W.; Yang, W.; Yang, L.; Ma, J.; Zhang, S. Photoredox-Catalyzed Tandem Cyclization of Enaminones with N-Sulfonylaminopyridinium Salts toward the Synthesis of 3-Sulfonaminated Chromones. J. Org. Chem. 2024, 89, 644–655. [Google Scholar] [CrossRef]
- Sun, J.; Wei, Y.; Wang, W.; Lin, W.; Wang, C.; Zhou, J.; Wang, G.; Li, J. Organic Photoredox Catalytic Sulfonylation of Enaminones to Access 3-Sulfonyl Chromones. Adv. Synth. Catal. 2025, 367, e202401396. [Google Scholar] [CrossRef]
- Salaverri, N.; Alemán, J.; Marzo, L. Harnessing the Power of the De Mayo Reaction: Unveiling a Photochemical and Photocatalytic Masked [2+2] Methodology for Organic Synthesis. Adv. Synth. Catal. 2024, 366, 156–167. [Google Scholar] [CrossRef]
- Funes-Ardoiz, I.; Garrido-Barros, P. Controlling selectivity of hydrogen atom transfer (HAT) in photoredox catalysis. Chem Catalysis 2024, 4, 100930. [Google Scholar] [CrossRef]
- Meger, F.S.; Murphy, J.A. Recent Advances in C–H Functionalisation through Indirect Hydrogen Atom Transfer. Molecules 2023, 28, 6127. [Google Scholar] [CrossRef]
- Capaldo, L.; Ravelli, D. Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2017, 2056–2071. [Google Scholar] [CrossRef]
- Capaldo, L.; Ravelli, D.; Fagnoni, M. Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration. Chem. Rev. 2022, 122, 1875–1924. [Google Scholar] [CrossRef]
- Zhang, J.; Zhuang, K.; Trevino, R.; Dhungana, B. R.; Sun, H.; Yin, S.; Li, Y.; Sanchez, J. A.; Xia, X.; Elerian, R.; Sun, Y.; Fremin, S. O.; Huang, C.; He, M.; Cheng, M.; Larionov, O. V.; Jin, S. Dispersion-Enhanced Nitrogen-Centered Photocatalysis of the Direct Hydrogen Atom Transfer. Angew. Chem. Int. Ed. 2026, 65, e22022. [Google Scholar] [CrossRef]
- Feng, A. G.; Pinto Pereira Junior, M. V.; Miller, S. J.; Knowles, R. R. Asymmetric Hydrogen Atom Transfer. ACS Catal. 2026, 16, 844–865. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, Y.; Zhang, Y.-Q. Where Enantioselection is Set: A Mechanistic Framework for Asymmetric Hydrogen-Atom Transfer. Angew. Chem. Int. Ed. 2026, e26135. [Google Scholar] [CrossRef]
- Yatsuzuka, K.; Kawasaki, M.; Shirai, R. Enantioselective [2,3]-Wittig Rearrangement of Carboxylic Acid Derived Enolates by Tetradentate Chiral Lithium Amide. Synlett 2023, 34, 1727–1731. [Google Scholar] [CrossRef]
- Xia, Z.; Hu, J.; Gao, Y.-Q.; Yao, Q.; Xie, W. Facile access to 2,2-disubstituted indolin-3-ones via a cascade Fischer indolization/Claisen rearrangement reaction. Chem. Commun. 2017, 53, 7485–7488. [Google Scholar] [CrossRef]
- Babij, N. R.; McCusker, E. O.; Whiteker, G. T.; Canturk, B.; Choy, N.; Creemer, L. C.; Amicis, C. V. D.; Hewlett, N. M.; Johnson, P. L.; Knobelsdorf, J. A.; Li, F.; Lorsbach, B. A.; Nugent, B. M.; Ryan, S. J.; Smith, M. R.; Yang, Q. NMR Chemical Shifts of Trace Impurities: Industrially Preferred Solvents Used in Process and Green Chemistry. Org. Process Res. Dev. 2016, 20, 661–667. [Google Scholar] [CrossRef]
- Yoo, B. I.; Kim, Y. J.; You, Y.; Yang, J. W.; Kim, S. W. Birch Reduction of Aromatic Compounds by Inorganic Electride [Ca2N]+•e– in an Alcoholic Solvent: An Analogue of Solvated Electrons. J. Org. Chem. 2018, 83, 13847–13853. [Google Scholar] [CrossRef]
- Luo, Y.-R. Comprehensive Handbook of Chemical Bond Energies, 1st ed.; CRC Press, 2007. [Google Scholar] [CrossRef]
- Kozmin, S. A.; Iwama, T.; Huang, Y.; Rawal, V. H. An Efficient Approach to Aspidosperma Alkaloids via [4 + 2] Cycloadditions of Aminosiloxydienes: Stereocontrolled Total Synthesis of (±)-Tabersonine. Gram-Scale Catalytic Asymmetric Syntheses of (+)-Tabersonine and (+)-16-Methoxytabersonine. Asymmetric Syntheses of (+)-Aspidospermidine and (−)-Quebrachamine. J. Am. Chem. Soc. 2002, 124, 4628–4641. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Four-step synthesis of enaminone 7.
Scheme 2.
Photocatalyzed transformations of enaminone 7.
Scheme 3.
Proposed mechanism for the photocatalyzed fragmentation of enaminone 7.
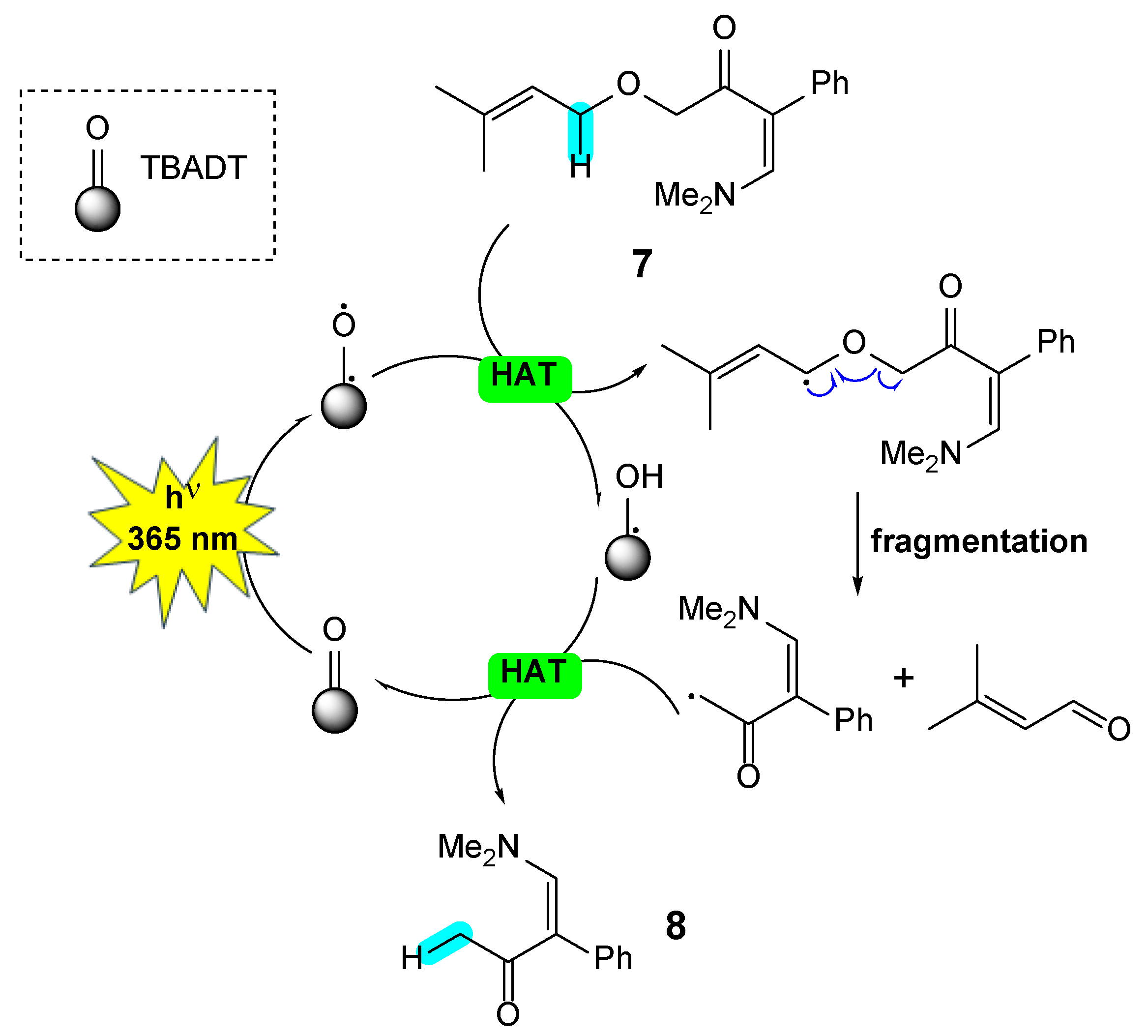
Figure 1.
UV-Vis spectrum of enaminone 7 in dichloromethane.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



