Submitted:
09 February 2026
Posted:
09 February 2026
You are already at the latest version
Abstract
Accurate determination of heavy metals in marine hydrate-associated muds is crucial for tracing methane seepage, yet it faces challenges from complex matrices. This study developed a matrix-matched microwave digestion ICP-OES method. By comparing XRF spectral profiles and statistical tests, the feasibility of using soil certified reference materials to simulate marine mud matrices was demonstrated, thereby optimizing digestion parameters (power/temperature) and ICP-OES spectral line selection. Method validation revealed detection limits of 0.0004 – 0.0105 mg/L for Cu, Zn, Pb, and Cr, with spike recoveries ranging from 95.5% – 103.7%. Accuracy was further verified using soil reference materials (GSS-4/5) and comparative t-tests with ICP-MS data. This efficient and reliable method provides a practical analytical tool for geochemical exploration of marine gas hydrates.
Keywords:
inductively coupled plasma optical emission spectrometer (ICP-OES)
; matrix effect
; marine gas hydrate mud
; microwave-enhanced digestion
; sediment analysis
1. Introduction
Natural gas hydrate (NGH) is a non-stoichiometric clathrate crystalline compound formed by the interaction of methane and water under high-pressure and low-temperature conditions. Recognized as one of the most promising new energy sources of the 21st century, NGH is regarded as a critical strategic resource for mitigating future energy shortages, garnering significant attention from scientists and governments worldwide [1,2]. NGH is predominantly found in deep-sea sediments and permafrost regions. Its exploration and extraction require the integration of multidisciplinary technologies, making it a prominent area of research in geological sciences. Traditional exploration methods rely on the identification of bottom-simulating reflectors (BSR) [3,4]. However, studies indicate that BSR does not always correlate with gas hydrate accumulation, necessitating complementary approaches such as geochemical exploration techniques. Among these, variations in trace element concentrations have been widely employed in hydrate exploration, identification, and related research.
Seafloor tectonic activity alters subsurface temperature and pressure conditions, leading to the dissociation of NGH and the subsequent release of significant methane volumes. The upward migration of methane triggers anaerobic oxidation of methane (AOM), a process coupled with the downward diffusion of sulfate [5,6]:
This reaction consumes a large amount of methane gas and ultimately produces carbonate and hydrogen sulfide [7]. The reaction formula is as follows:
The released H₂S accumulates within sediment pore spaces, while sulfate depletion establishes a strongly reducing environment. Variations in redox-sensitive elements (e.g., Cu, Zn, Pb, Cr) serve as effective geochemical proxies for methane hydrate seepage, supporting exploration efforts [8]. Consequently, trace element geochemistry in marine sediments provides critical insights for guiding NGH exploration. However, the compositional matrix of submarine mud samples (SMS) is highly complex, with numerous interfering factors. Thus, there is an urgent need to develop accurate analytical methods for quantifying trace elements in seafloor sediments. Given the challenges associated with marine sediment sampling, rapid and precise determination of inorganic element concentrations is essential to assess methane seepage intensity and its potential linkage to localized NGH dissociation. To address this, an efficient digestion method—such as wet acid digestion using inorganic acids/corrosive oxidizers—must be employed to fully decompose marine sediments and ensure complete dissolution of trace elements for analysis.
Microwave-assisted digestion operates through the interaction of samples with a high-frequency electromagnetic field [9]. Under microwave irradiation, the sample-acid mixture experiences rapid dielectric heating, where polar molecules continuously realign with the oscillating electromagnetic field. This molecular agitation generates intense internal friction, resulting in instantaneous temperature elevation and enhanced reaction kinetics. The sealed digestion system prevents volatile element loss while significantly accelerating sample decomposition [10]. This efficient technique has found extensive applications across diverse fields, including: carbon materials [11,12], catalyst analysis [13], sediment studies [14,15], energy resource characterization [16,17], food science [18,19], and pharmaceutical analysis [20,21].
When performing spectral analysis of metal elements in samples, matrix effects may significantly impact the accuracy of analyte quantification [22]. These interfering effects exhibit concentration-dependent behavior, varying with the abundance and relative concentration of matrix elements. However, the inherent complexity of SMS (Sample Matrix Similarity) spans the entire process from sample collection, processing to final analysis, often leading to extended analysis time. Traditional method development for precise testing and analysis typically requires substantial time investment. To overcome these challenges, we propose a method transfer and validation strategy based on matrix similarity. Previous studies have shown that multiple factors significantly influence XRF measurement accuracy, including sample stability, spectral interference, particle size distribution, moisture content, and measurement duration [23,24,25]. The proposed method handles samples while considering the aforementioned factors, and through XRF analysis and t-tests, it demonstrates that there is no significant difference in the overall effects between two matrices, which can serve as a preliminary screening criterion for selecting similar matrices. Finally, the applicability of this method to marine mud matrix is jointly validated through good spiked recovery rates of actual samples by ICP-OES, high accuracy verified using soil standard reference materials, and dedicated matrix interference experiments. The purpose of this method is to establish a robust analytical framework and significantly reduce the required testing time.
2. Materials and Methods
The analytical accuracy of our method was rigorously validated through the analysis of certified geochemical reference materials obtained from the Chinese National Standards Center, including: GBW07404 (GSS-4) - National standard soil reference material; GBW07405 (GSS-5) - National standard soil reference material; GSB07-3903-2021 - Certified soil quality control standard.
2.1. Reagents and Standards
Stock standard solutions (100 mg/L) for preparing mixed calibration standards were procured from the National Non-ferrous Metals and Electronic Materials Testing Center (China). Working standard solutions were prepared by serial dilution with 2% (v/v) HNO3, which was obtained by diluting ultrapure nitric acid (65%, Guangzhou Chemical Reagent, China) with ultrapure water (Millipore, USA). High-purity reagents were used throughout the study: Hydrogen peroxide (30% m/v); Hydrochloric acid (37% m/v); Hydrofluoric acid (40% m/v). All acids were Suprapur grade (Guangzhou Chemical Reagent, China). All laboratory ware, including plastic and glass volumetric vessels and funnels, underwent rigorous cleaning by soaking in 30% HNO3 for ≥24 h, followed by extensive rinsing with ultrapure water prior to use.
2.2. Instrumentation
Sample digestion was performed using a Multiwave PRO microwave digestion system (Anton Paar, Austria) equipped with eight high-pressure, pre-cleaned Teflon vessels. The system operates at maximum conditions of 80 bar pressure and 300 °C temperature. The digestion procedure was conducted as follows: 1) Precisely 0.20 g of dried sample was weighed into each acid-cleaned Teflon vessel; 2) 8 mL of inverse aqua regia (3:1 HNO₃:HCl) and 1 mL HF (40% m/v) were added to each vessel; 3) Microwave digestion was performed according to the optimized program (Table 1); 4) After cooling, samples were heated at 120°C to near-dryness (∼0.5 mL remaining) to ensure complete HF evaporation without requiring boric acid neutralization; 5) The digest was quantitatively transferred to a 25 mL volumetric flask and diluted to volume with deionized water (18.2 MΩ·cm); 6) Solutions were filtered through 0.45 µm Millipore membranes prior to analysis. Quality control measures included: Preparation of acid blanks for each sample batch; Use of pre-cleaned vessels to minimize contamination; Strict adherence to the digestion protocol for reproducibility.
A PANalytical Axios max-petro XRF spectrometer was used for semi-quantitative, label-free analysis of boric acid hemming presses. Samples were analyzed under a pressure of 30 MPa using P10 gas (methane:argon, 1:9 mass ratio). The X-ray tube was operated at 60 kV and 60 mA, and the analytical crystals employed were LiF200, Ge111, PE002, and PX1. A PerkinElmer Optima 8000 ICP-OES spectrometer was used for the determination of Cu, Pb, Cr, and Zn in digested samples. Argon gas (99.999%) was used for plasma generation. ICP-OES calibrations were performed using a series of standards at concentrations of 0.1, 0.2, 0.5, 1, and 2 mg/L, prepared from 1000 ppm single-element standards. A quality control sample (0.5 mg/L) was analyzed periodically to monitor instrument performance and ensure accuracy throughout the analysis. Comparative analyses of Cu, Pb, Cr, and Zn were also performed using an Agilent 7900 ICP-MS. Argon gas (99.999%) was used for plasma generation. The ICP-MS was tuned using a solution containing 1 µg/mL each of Ce, Co, Li, Y, and Tl in 2% HNO3 (Agilent Technologies, China) Co., Ltd. The specific operating conditions for elemental determination by ICP-OES and ICP-MS are detailed in Table 1.
2.3. Sample Preparation and Data Treatment
The SMS used in the experiment were collected from the Shenhu Sea area of the South China Sea, at a seawater depth of 2713 meters, with the sample number ZSQD78. After collecting, the samples were dried in an oven at 105 °C and then finely ground in an agate mortar. All data were processed using IBM SPSS Statistics 26 software and Excel 2013.
3. Results and Discussion
In this study, we applied Matrix similarity-based methods using XRF analysis. The data obtained were subsequently processed using IBM SPSS Statistics 26 software chemometric t-test to determine whether there was a statistical difference between the unknown SMS and the QC samples. The analysis revealed no statistically significant difference between the two samples. Therefore, the known QC samples were used to investigate analytical methods, and these methods were then inferred for the unknown SMS by analogy.
3.1. Matrix Effect of XRF Elemental
The independent samples t-test, a cornerstone statistical method in exploration data analysis [26], provides a robust framework for comparing population means between two independent groups. In this investigation, we employed this parametric test to statistically determine whether significant differences exist in the total concentrations of copper (Cu), zinc (Zn), lead (Pb), and chromium (Cr) between certified soil reference materials (GSB07-3903-2021, GSS-4, GSS-5) and SMS. Independent sample t-test analysis can reveal the difference between two independent samples more clearly and concisely. The calculation process is as follows: the meaning and variance of two independent samples are calculated, and the combined variance is used as an estimate of the variance of the two totals when the variances of the two samples are equal [27]. If the variances are not equal, the respective variances are used. The results are then used to calculate the t-statistics and F-statistics, along with the corresponding probability P-values. These P-values are compared with the significance level (usually 0.05 or 0.01) to determine whether there is a statistically significant difference between the two overall means.
Independent samples t-tests were performed to compare the concentrations of copper, zinc, lead, and chromium in soil QC samples (GSB07-3903-2021, GSS-4, GSS-5) and SMS analyzed by boric acid pressed slice X-ray fluorescence (XRF) spectrometry. The results of these analyses are detailed in Figure 1a, where bar graphs illustrate the outcome of the independent samples t-tests. A significance level of α = 0.05 was used for all statistical tests. In each graph, the left-hand bar represents the result of Levene’s test for equality of variances, which assesses whether the variances of the two independent samples are equal. The right-hand bar represents the mean equivalence t-test; a p-value below 0.05 was considered statistically significant. As shown in Figure 1a, Levene’s test yielded a p-value of 0.009, which is less than 0.05, indicating that the soil quality control sample and the SMS had significantly unequal variances. The subsequent t-test for unequal variances produced a p-value of 0.165, exceeding 0.05, suggesting that there was no statistically significant difference in mean concentrations between the two sample types. Based on these findings, the use of known soil quality control samples is justified for subsequent methodological studies in the analytical methods chapter. Similarly, Figure 1b and Figure 1c present the t-test results for SMS compared to GSS-4 and GSS-5 samples, respectively. The p-value for the comparison of SMS and GSS-4 was 0.939, and the p-value for the comparison of SMS and GSS-5 was 0.366. Both p-values are greater than 0.05, indicating no statistically significant differences between the SMS and either GSS-4 or GSS-5. This validation supports the accuracy of the XRF methodology. For a more detailed description of the calculations, please refer to the Electronic Supplementary Material (ESM).
3.2. Optimization of Microwave Digestion Program
The digestion procedure was optimized by evaluating the recovery of Cu, Cr, Pb, and Zn in the soil quality control sample GSB07-3903-2021. These elements are known to be difficult to dissolve, particularly in marine sediment samples containing silicates. Therefore, a mixture of inverse aqua regia (3HNO3: 1HCl) and hydrofluoric acid (HF) was selected as the digestion solution. HF effectively breaks Si-O bonds, forming SiF4 and other fluorosilicic compounds, thereby promoting the dissolution of samples with high silicon content. A volume of 1 mL of HF was found to be optimal, as further increases did not significantly improve analyte recoveries. The effect of microwave digestion temperature on analyte recovery was investigated using temperatures of 150, 180, 210, and 230 °C. In these experiments, 0.2000 g of GSB07-3903-2021 was digested using a mixture of 6 mL HNO3, 2 mL HCl, and 1 mL HF. Six replicate measurements were performed for each digested sample, and the average recoveries were calculated. As shown in Figure 2, copper recovery remained relatively constant (around 110%) across the tested temperature range, indicating that microwave digestion temperature has minimal impact on copper determination. The unusually high recovery rate of zinc at 180°C may be due to accidental contamination or enhanced interference from coexisting elements at a specific temperature, which suggests that reagent blanks and digestion consistency need to be strictly controlled in actual analysis. The recovery rate of chromium dropped to 87.9% at 230°C, which is consistent with the phenomenon reported in the literature that Cr may volatilize in the form of CrO₂Cl₂ at high temperatures. Therefore, in order to simultaneously ensure the complete dissolution of Pb (needs >180°C) and avoid the loss of Cr (needs <230°C), it is reasonable to choose 210°C as the compromise optimization temperature.
In summary, a digestion temperature of 150 °C or higher is generally sufficient for the microwave digestion of copper and zinc. However, caution is advised due to zinc’s susceptibility to contamination. For lead, the digestion temperature must exceed 180 °C to ensure complete digestion. The optimal digestion temperature for chromium lies between 180 °C and 210 °C; temperatures that are too low result in incomplete digestion, while excessively high temperatures can cause volatilization and consequently, low results.
3.3. Selection of Analytical Wavelengths
Analytical wavelengths for Copper, Zinc, Chromium, and Lead were determined by screening all spectral lines of these elements in the periodic table (Table 2). To verify the accuracy of these wavelengths, the soil quality control sample GSB07-3903-2021 was used. A 0.2000 g aliquot of the control sample was weighed, and the accuracy of the analytical wavelengths was confirmed following the optimized sample pretreatment method. Six replicate analyses were performed, and the average value was calculated. The relative error results are presented in Table 2. Among the three copper spectral lines, the 324.752 nm line exhibited the smallest relative error, high sensitivity, and minimal interference. For zinc, the 213.857 nm line, out of three, showed a small relative error and low interference. Among the four chromium spectral lines, the 267.716 nm line demonstrated a small relative error and low interference. For lead, the 261.418 nm line displayed a negative absorption peak in the analytical spectrum, indicating ineffective lead detection and poor sensitivity. The 217.0 nm and 283.306 nm lines had relative errors exceeding 100%, suggesting significant interference. Therefore, only the 220.353 nm line, with its small error, was suitable for selection. The selected spectral lines were 324.752 nm (Cu), 213.857 nm (Zn), 267.716 nm (Cr), and 220.353 nm (Pb).
3.3. Calibration
For all concentration measurements, a five-point calibration procedure was implemented. Multi-element calibration standards were used, encompassing all four target elements. Calibration curves were generated by plotting the instrument response signal intensity for each element as a function of concentration and fitting a linear regression model. The method detection limit (MDL) was rigorously determined by performing ten replicate measurements of sample blanks and calculating three times the standard deviation (s) of the resulting blank intensity values (DL = 3 s/b, where b is the slope of the calibration curve). The limit of quantification (LOQ) was similarly calculated as ten times the standard deviation (s) of the ten blank intensity values (QL = 10s/b, where b is the slope of the calibration curve). A summary of these results is provided in Table 3.
3.4. Spiking Recovery Test
SMS A, of unknown composition, was collected from the Shenhu Sea area of the South China Sea. To assess method accuracy, a spike-and-recovery study was performed. A multi-element standard solution was added to the sample to achieve a 50% spike, resulting in a final concentration of 2 mg/L. The spiked samples were then digested using the established microwave digestion procedure, and the resulting solutions were analyzed to determine element recoveries. To ensure comprehensive quality control, the multi-element standard was added at the very beginning of sample preparation, before the digestion step. This approach allows for the detection of any potential analyte loss during the entire analytical process. The results of the spike-and-recovery study are summarized in Table 4. The observed recoveries for the target elements in SMS A fell within the range of 95.5% to 103.7%, demonstrating satisfactory method accuracy and reliability.
3.5. Matrix Element Influence
The XRF data indicate that the concentrations of iron, aluminum, magnesium, and manganese in soil matrix elements are high. According to HJ781-2016, copper (Cu) and lead (Pb) elements are interfered with by iron and aluminum, chromium (Cr) elements are interfered with by manganese and magnesium, and zinc (Zn) is interfered with by iron and magnesium. To assess potential spectral interferences, the effects of aluminum and iron on copper and lead spectral lines were investigated. Similarly, the effects of magnesium and manganese on chromium spectral lines, and magnesium and iron on zinc spectral lines, were examined. In each case, the concentration of the analyte (Cu, Pb, Cr, or Zn) was held constant at 0.1 mg/L, while the interfering elements (Al, Fe, Mg, Mn) were varied across concentrations of 10, 20, 50, and 100 mg/L. All samples were analyzed in triplicate, and the average results were used for subsequent calculations of relative errors. The results of these interference studies are presented in Figure 3 and Figure 4.
The degree of interference on copper spectral lines varies depending on the interfering element. The 222.778 nm and 224.700 nm lines were less affected by aluminum, while the 327.393 nm and 324.752 nm lines were less affected by iron. For example, at an Al/Cu mass ratio of 500, the relative error for the 222.778 nm Cu line was 1%, and for the 224.700 nm line, it was 3%. In contrast, at an Fe/Cu mass ratio of 500, the relative errors for the 222.778 nm and 224.700 nm Cu lines were 78% and 69%, respectively, while the errors for the 327.393 nm and 324.752 nm lines were 8% and 2%, respectively (Figure 3a and Figure 3b). The 220.353 nm lead spectral line exhibited the least interference from both aluminum and iron compared to other lead lines. For instance, at an Al/Pb mass ratio of 500, the relative error for the 220.353 nm line was 2%, while the errors for other lead lines exceeded 21%. Similarly, at an Fe/Pb mass ratio of 500, the relative error for the 220.353 nm line was 6%, while the errors for other lead lines were significantly higher (Figure 3c and Figure 3d). The 267.716 nm chromium spectral line was the least affected by interference from magnesium and manganese compared to other chromium lines. For example, at a Mg/Cr mass ratio of 200, the relative error for the 267.716 nm line was 1%, while the errors for other chromium lines ranged from 3% to 20%. At a Mn/Cr mass ratio of 100, the relative error for the 267.716 nm line was 1%, while the errors for other chromium lines ranged from 3% to 56% (Figure 4a and Figure 4b). Magnesium and iron exhibited varying degrees of interference on zinc spectral lines. The 202.548 nm and 334.501 nm lines were significantly affected by magnesium concentration, while the 202.548 nm and 206.200 nm lines were less affected by iron concentration. For example, at a Mg/Zn mass ratio of 500, the relative errors for the 202.548 nm and 334.501 nm Zn lines were 21% and 15%, respectively, while the errors for other zinc lines were less than 2%. In contrast, at a Fe/Zn mass ratio of 500, the relative errors for the 202.548 nm and 206.200 nm Zn lines were 2% and 0.98%, respectively. These lower errors suggest higher accuracy for these two lines under the tested conditions (Figure 4c and Figure 4d).
The interference errors of different matrix elements on the target element spectral line analysis vary. Therefore, it is recommended to select quality control samples with similar matrices to choose the test spectral line, thereby reducing errors in the testing process.
3.6. Sample Accuracy
To validate the accuracy and precision of the method, six aliquots of 0.1 g (±0.0001 g) of soil composition analysis standard materials GSS-4 and GSS-5 were weighed. Sample pretreatment and analysis were performed according to the described method, using the instrument parameters established in this study. Each aliquot was measured six times, and the average value and relative standard deviation (RSD) were calculated (Table 5). The measured values for each element were consistent with the certified reference values, with RSDs ranging from 0.67% to 2.3% (n = 6) and recoveries ranging from 97.5% to 108.6%. These results demonstrate that the analytical method exhibits high precision and accuracy and is suitable for the analysis and quantification of SMS.
3.7. Comparison Test
To verify the absence of significant interference during sample detection and to ensure the accuracy of the analytical data, Sediment Material Samples (SMS) were pre-treated using the optimized digestion method. The concentrations of lead, chromium, copper, and zinc were then determined by both ICP-MS and the ICP-OES method developed in this study. The data obtained from the two methods were compared using independent samples t-tests, and the results are summarized in Table 6 and Figure 5.
As shown in Table 6, the calculated t-test values were all less than the critical value (t0.05,5=2.571). This indicates, with 95% confidence, that there are no statistically significant differences between the results obtained using ICP-MS and the ICP-OES method. Figure 5 presents the results of the independent samples t-test analysis. For copper, Levene’s statistic was 0.818 (p>0.05), indicating equal variances between the two methods. The corresponding p-value for the t-test was 0.074 (p>0.05), confirming that there was no statistically significant difference in copper concentrations between the ICP-MS and ICP-OES methods. Similarly, for zinc, Levene’s statistic was 0.003 (p<0.05), indicating unequal variances. However, the p-value for the t-test was 0.922 (p>0.05), demonstrating no statistically significant difference in zinc concentrations between the two methods. For chromium, Levene’s statistic was 0.001 (p<0.05), indicating unequal variances, but the t-test p-value was 0.165 (p>0.05), again showing no significant difference. For lead, Levene’s statistic was 0.898 (p>0.05), indicating equal variances, and the t-test p-value was 0.132 (p>0.05), confirming no significant difference.
These results provide strong evidence that the established ICP-OES analytical methods are accurate and reliable for the determination of these elements in SMS.
4. Conclusion
In summary, through t-test validation, we have demonstrated that soil QC samples can be effectively used to expedite method development for SMS analysis. The optimized ICP-OES method exhibits several advantageous characteristics, including low detection limits (0.0004–0.0105 mg/L) and high recoveries (95.5–103.7%), making it a robust and reliable tool for marine gas hydrate exploration.
The established analytical method is characterized by its short analysis time, low detection limits, good repeatability, and high sensitivity. It can be readily applied to the analysis of copper, zinc, lead, and chromium in marine sediment samples collected in the field, providing critical data to support the determination of natural gas hydrate presence at seafloor locations. This approach significantly reduces the time and resources required for developing analytical methods for novel marine sediment samples.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
All authors contributed to the study conception and design. Shujia Wang: Methodology, Formal analysis, Data curation, and Writing—original draft preparation. Peishan Li: Methodology, and Data curation. Jinan Guan: Conceptualization, Validation, Writing—original draft preparation, Writing—review and editing, Supervision, and Funding acquisition. Jingsheng Lu: Validation, and Data curation. Pibo Su: Methodology, Data curation, Formal analysis, Conceptualization, and Funding acquisition.
Data Availability Statement
The data supporting this article has been included in the article, as well as the electronic supplementary information.
Acknowledgments
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (U25A20799), and Hainan Province Key Research and Development Project (ZDYF2024GXJS002).
Competing Interests
The authors have no competing interests to declare that are relevant to the content of this article.
References
- Boswell, R.; Shipp, C.; Reichel, T.; Shelander, D.; Saeki, T.; Frye, M.; Shedd, W.; Collett, T.S.; Mcconnell, D.R. Prospecting for marine gas hydrate resources. J. Interpretation 2015, 4, SA13–SA24. [Google Scholar] [CrossRef]
- Guan, J.; Cong, X.; Archer, D.E.; Wan, L.; Liang, D. Spatio-temporal evolution of stratigraphic-diffusive methane hydrate reservoirs since the Pliocene along Shenhu continental slope, northern South China sea. J. Mar. Pet. Geol 2021, 125, 104864. [Google Scholar] [CrossRef]
- Rajput, S.; Müller, T.M.; Clennell, M.B.; Rao, P.P.; Thakur, N.K. Constraints on seismic reflections and mode conversions at bottom simulating reflectors associated with gas hydrates. J. J Petrol Sci Eng 2012, 88-89, 48–60. [Google Scholar] [CrossRef]
- Tong, S.; Wang, J.; Li, L.; Zhang, H.; Wu, Z.; Shao, Y. Study of the Sensitive Properties of Marine Gas Hydrate Based on the Prestack Elastic Inversion. J. J Ocean U China 2019, 18, 1086–1092. [Google Scholar] [CrossRef]
- Hu, Y.; Feng, D.; Liang, Q.; Xia, Z.; Chen, L.; Chen, D. Impact of anaerobic oxidation of methane on the geochemical cycle of redox-sensitive elements at cold-seep sites of the northern South China Sea. J. Deep-sea Res Pt II 2015, 122, 84–94. [Google Scholar] [CrossRef]
- Chen, F.; Hu, Y.; Feng, D.; Zhang, X.; Cheng, S.; Cao, J.; Lu, H.; Chen, D. Evidence of intense methane seepages from molybdenum enrichments in gas hydrate-bearing sediments of the northern South China Sea. J. Chem. Geol 2016, 443, 173–181. [Google Scholar] [CrossRef]
- Reeburgh, W.S. Oceanic methane biogeochemistry. J. Chemical Reviews 2007, 107, 486–513. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.N.; Chen, F.; Hu, Y.; Guo, Q.J.; Cao, J.; Chen, H.; Zhou, J.H.; Jiang, X.X.; Zhu, J. Methane seepage patterns during the middle Pleistocene inferred from molybdenum enrichments of seep carbonates in the South China Sea. J. Ore Geol. Rev 2020, 125. [Google Scholar] [CrossRef]
- Li, Y.; Campos, L.C.; Hu, Y. Microwave pretreatment of wastewater sludge technology—a scientometric-based review. J. Environ Sci Pollut R 2024, 31, 26432–26451. [Google Scholar] [CrossRef]
- Bizzi, C.A.; Pedrotti, M.F.; Silva, J.S.; Barin, J.S.; Nóbrega, J.A.; Flores, E.M.M. Microwave-assisted digestion methods: towards greener approaches for plasma-based analytical techniques. J. J. Anal. At. Spectrom 2017, 32, 1448–1466. [Google Scholar] [CrossRef]
- Patole, S.P.; Simoes, F.; Yapici, T.F.; Warsama, B.H.; Anjum, D.H.; Costa, P. An evaluation of microwave-assisted fusion and microwave-assisted acid digestion methods for determining elemental impurities in carbon nanostructures using inductively coupled plasma optical emission spectrometry. J. Talanta 2016, 148, 94–100. [Google Scholar] [CrossRef]
- Cruz, S.M.; Schmidt, L.; Dalla Nora, F.M.; Pedrotti, M.F.; Bizzi, C.A.; Barin, J.S.; Flores, E.M.M. Microwave-induced combustion method for the determination of trace and ultratrace element impurities in graphite samples by ICP-OES and ICP-MS. J. Microchem. J 2015, 123, 28–32. [Google Scholar] [CrossRef]
- Niemelä, M.; Pitkäaho, S.; Ojala, S.; Keiski, R.L.; Perämäki, P. Microwave-assisted aqua regia digestion for determining platinum, palladium, rhodium and lead in catalyst materials. J. Microchem. J 2012, 101, 75–79. [Google Scholar] [CrossRef]
- Chand, V.; Prasad, S. ICP-OES assessment of heavy metal contamination in tropical marine sediments: A comparative study of two digestion techniques. J. Microchem. J 2013, 111, 53–61. [Google Scholar] [CrossRef]
- Han, H.J.; Gysi, A.P.; Frey, B. An ICP-OES method for the precise and accurate quantification of rare earth elements in natural water: A comparative study from mine waste sites in New Mexico, USA. J. Spectrochimica Acta Part B 2026, 237, 107451. [Google Scholar] [CrossRef]
- Li, K.; Wu, X.; Chen, Z.; Luo, J.; Hou, X.; Jiang, X. A simple dilution method for the direct determination of trace nickel in crude oil with a miniaturized electrothermal atomic absorption spectrometer. J. J. Anal. At. Spectrom 2020, 35, 2656–2662. [Google Scholar] [CrossRef]
- Balarama Krishna, M.V.; Chandrasekaran, K.; Chakravarthy, S.; Karunasagar, D. An integrated approach based on oxidative pyrolysis and microwave-assisted digestion for the multi-elemental analysis of coal samples by ICP-based techniques. J. Fuel 2015, 158, 770–778. [Google Scholar] [CrossRef]
- Teklu, A.; Hymete, A.; Gebreyesus, S.H.; Ashenef, A. Determination of the level of heavy metals and assessment of their safety in cow’s milk collected from Butajira and Meskan districts, south central Ethiopia. J. Environ Monit Assess 2022, 195, 206. [Google Scholar] [CrossRef]
- Smoliński, A.; Stempin, M.; Howaniec, N. Application and Validation of Analytical Software (SQX) for Semi-Quantitative Determination of the Main Chemical Composition of Solid, Bulk and Powder Fuel Samples by Wavelength Dispersive X-ray Fluorescence Technique. J. Energies 2022, 15, 7311. [Google Scholar] [CrossRef]
- Senger, C.M.; Dornelles, I.S.; Queiroz, J.M.; Mello, P.A.; Muller, E.I.; Muller, A.L.H. Greening microwave-assisted digestion method using hydrogen peroxide for determination of elemental impurities by ICP-OES in antihypertensive active pharmaceutical ingredients. J. J Pharmaceut Biomed 2024, 238, 115802. [Google Scholar] [CrossRef]
- Pinheiro, F.C.; Barros, A.I.; Nóbrega, J.A. Evaluation of dilute-and-shoot procedure for determination of inorganic impurities in liquid pharmaceutical samples by ICP OES. J. Microchem. J 2019, 146, 948–956. [Google Scholar] [CrossRef]
- Raposo, F.; Barceló, D. Challenges and strategies of matrix effects using chromatography-mass spectrometry: An overview from research versus regulatory viewpoints. J. Trends Anal. Chem 2021, 134, 116068. [Google Scholar] [CrossRef]
- Finkelshtein, A.L.; Smely, R.V.; Amosova, A.A.; Chubarov, V.M. Estimation of the Biogenic Silica Content in Lacustrine Bottom Silicate Sediments by X-Ray Diffraction (XRD) and X-Ray Fluorescence (XRF). J. Anal. Lett 2022, 55, 1119–1130. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; An, Z.; Wang, Q.; Sun, D.; Chen, Z. New Arctic Marine Sediment Certified Reference Material for Elemental Analysis. J. Anal. Lett 2019, 52, 401–410. [Google Scholar] [CrossRef]
- Endriss, F.; Kuptz, D.; Wissmann, D.; Hartmann, H.; Dietz, E.; Kappler, A.; Thorwarth, H. Evaluation and Optimization of an X-ray Fluorescence Analyzer for Rapid Analysis of Chemical Elements in Solid Biofuels. J. Energy Fuels 2024, 38, 16426–16440. [Google Scholar] [CrossRef]
- Pollak, M.; Cohen, J. A comparison of the independent-samples t-test and the paired-samples t-test when the observations are nonnegatively correlated pairs. J. J Stat Plan Infer 1981, 5, 133–146. [Google Scholar] [CrossRef]
- Kim, T.K. T test as a parametric statistic. J. Korean J Anesthesiol 2015, 68, 540–546. [Google Scholar] [CrossRef]
Figure 1.
Matrix effects analysis: (a) t-Test comparison of soil QC (GSB07-3903-2021) and SMS; (b) and (c): SMS vs. GSS-4/GSS-5.
Figure 1.
Matrix effects analysis: (a) t-Test comparison of soil QC (GSB07-3903-2021) and SMS; (b) and (c): SMS vs. GSS-4/GSS-5.
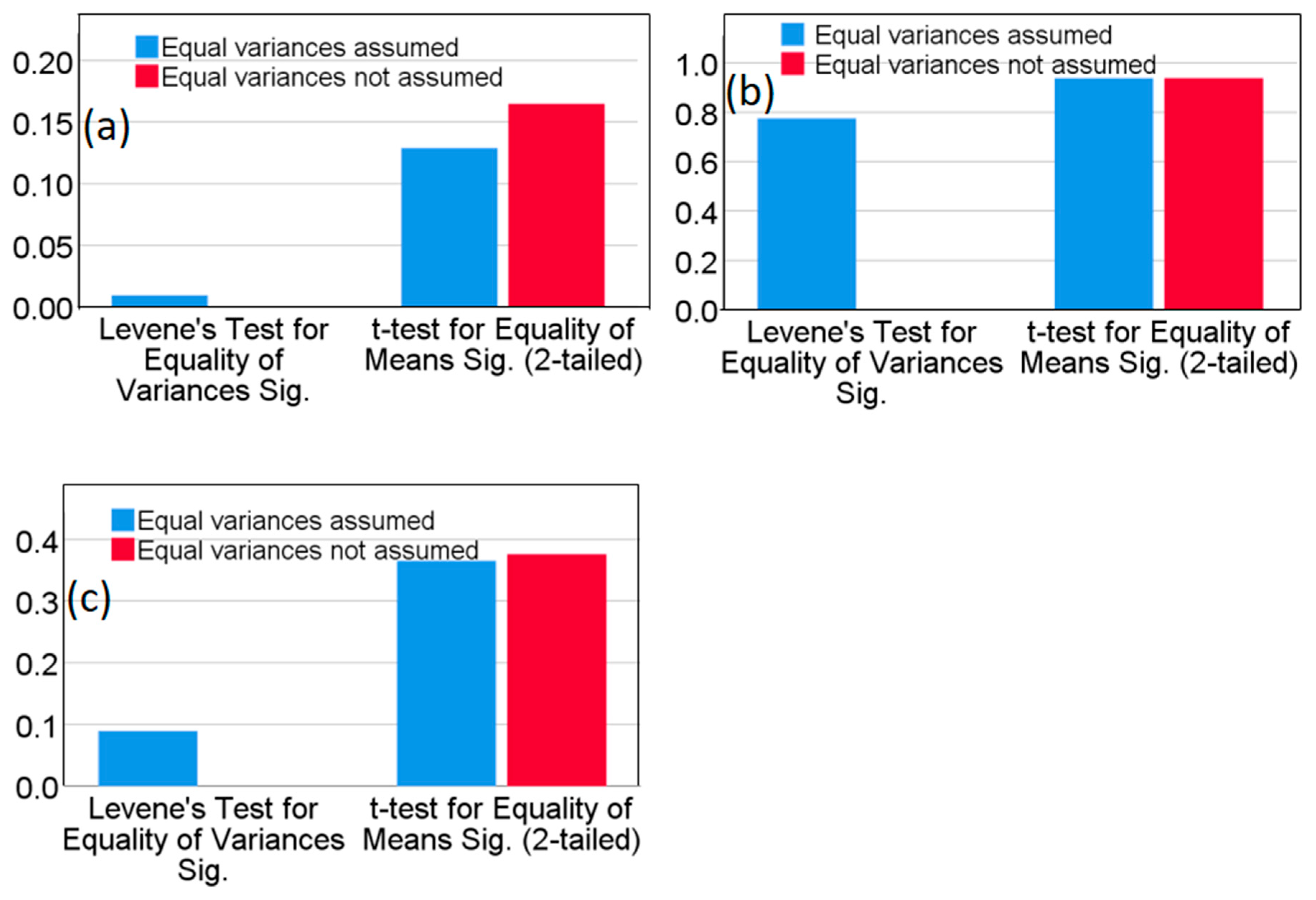
Figure 2.
The effect of different digestion temperatures on the recovery rates of Cu, Zn, Cr, and Pb (n=6).
Figure 2.
The effect of different digestion temperatures on the recovery rates of Cu, Zn, Cr, and Pb (n=6).
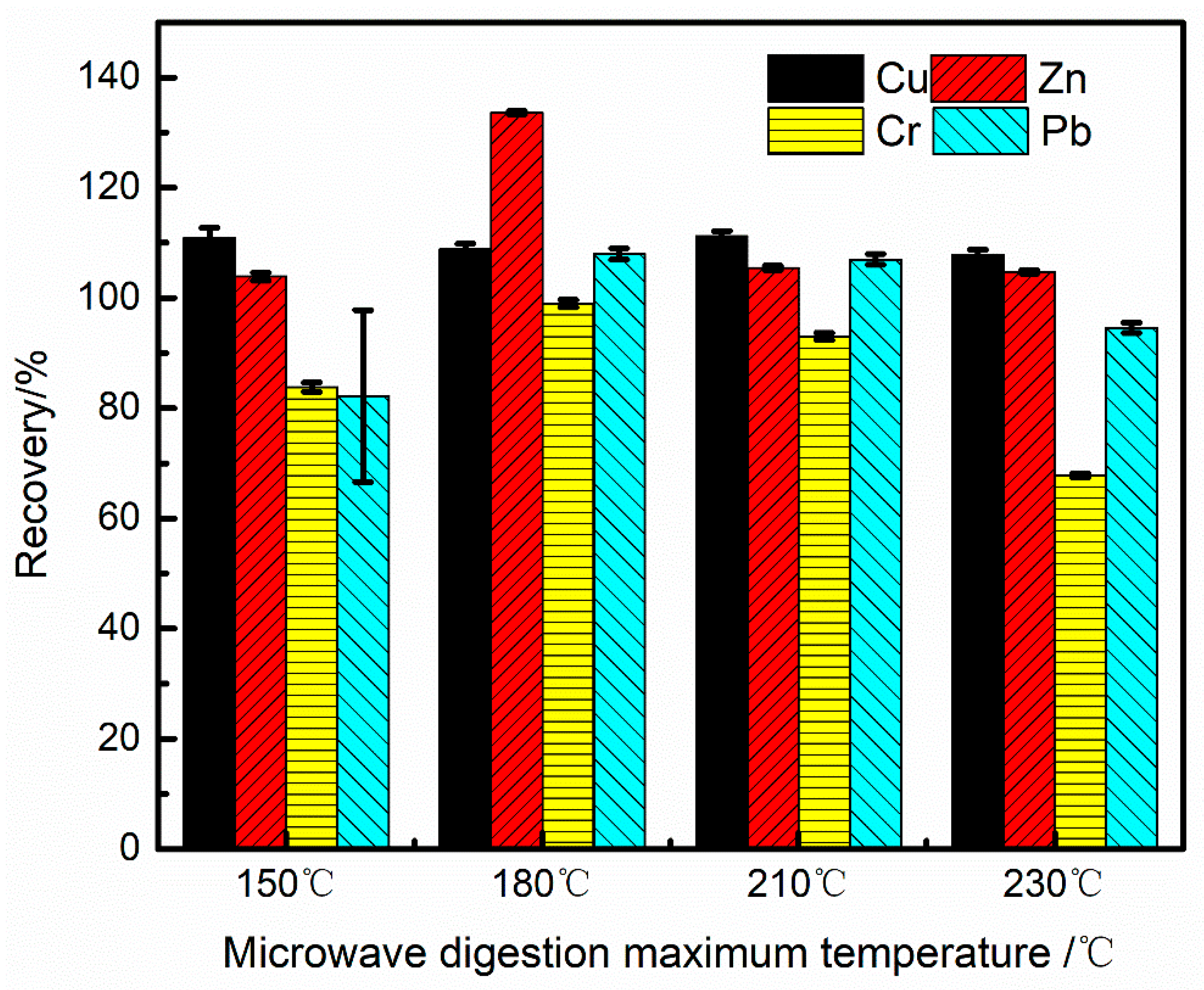
Figure 3.
Matrix element influence: (a) Effect of different Al concentrations on the interference of Cu spectral lines; (b) Effect of different Fe concentrations on the interference of Cu spectral lines; (c) Effect of different Al concentrations on the interference of Pb spectral lines; (d) Effect of different Fe concentrations on the interference of Pb spectral lines.
Figure 3.
Matrix element influence: (a) Effect of different Al concentrations on the interference of Cu spectral lines; (b) Effect of different Fe concentrations on the interference of Cu spectral lines; (c) Effect of different Al concentrations on the interference of Pb spectral lines; (d) Effect of different Fe concentrations on the interference of Pb spectral lines.
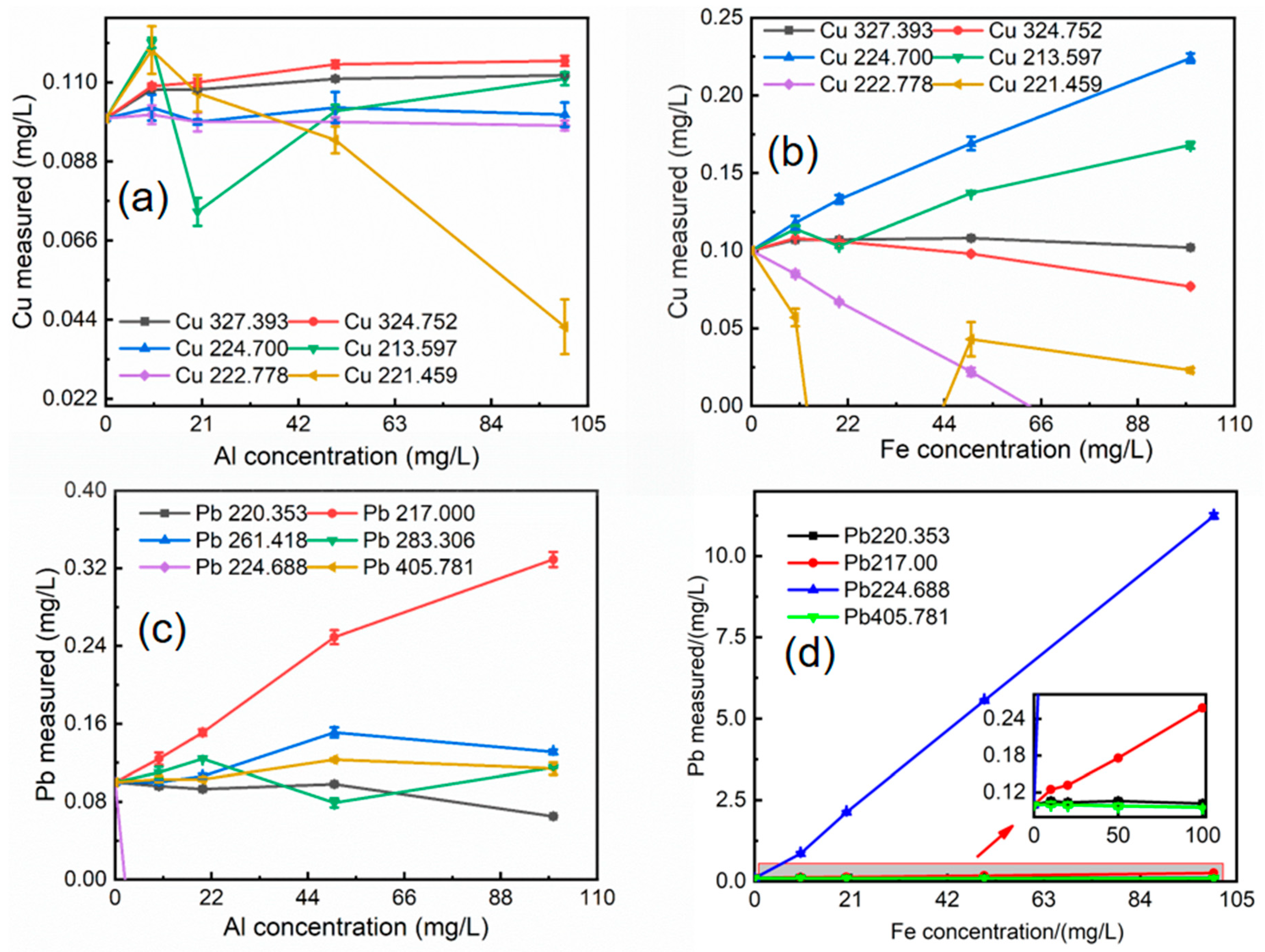
Figure 4.
Matrix element influence: (a) Effect of different Mg concentrations on the interference of Cr spectral lines; (b) Effect of different Mn concentrations on the interference of Cr spectral lines; (c) Effect of different Mg concentrations on the interference of Zn spectral lines; (d) Effect of different Fe concentrations on the interference of Zn spectral lines.
Figure 4.
Matrix element influence: (a) Effect of different Mg concentrations on the interference of Cr spectral lines; (b) Effect of different Mn concentrations on the interference of Cr spectral lines; (c) Effect of different Mg concentrations on the interference of Zn spectral lines; (d) Effect of different Fe concentrations on the interference of Zn spectral lines.
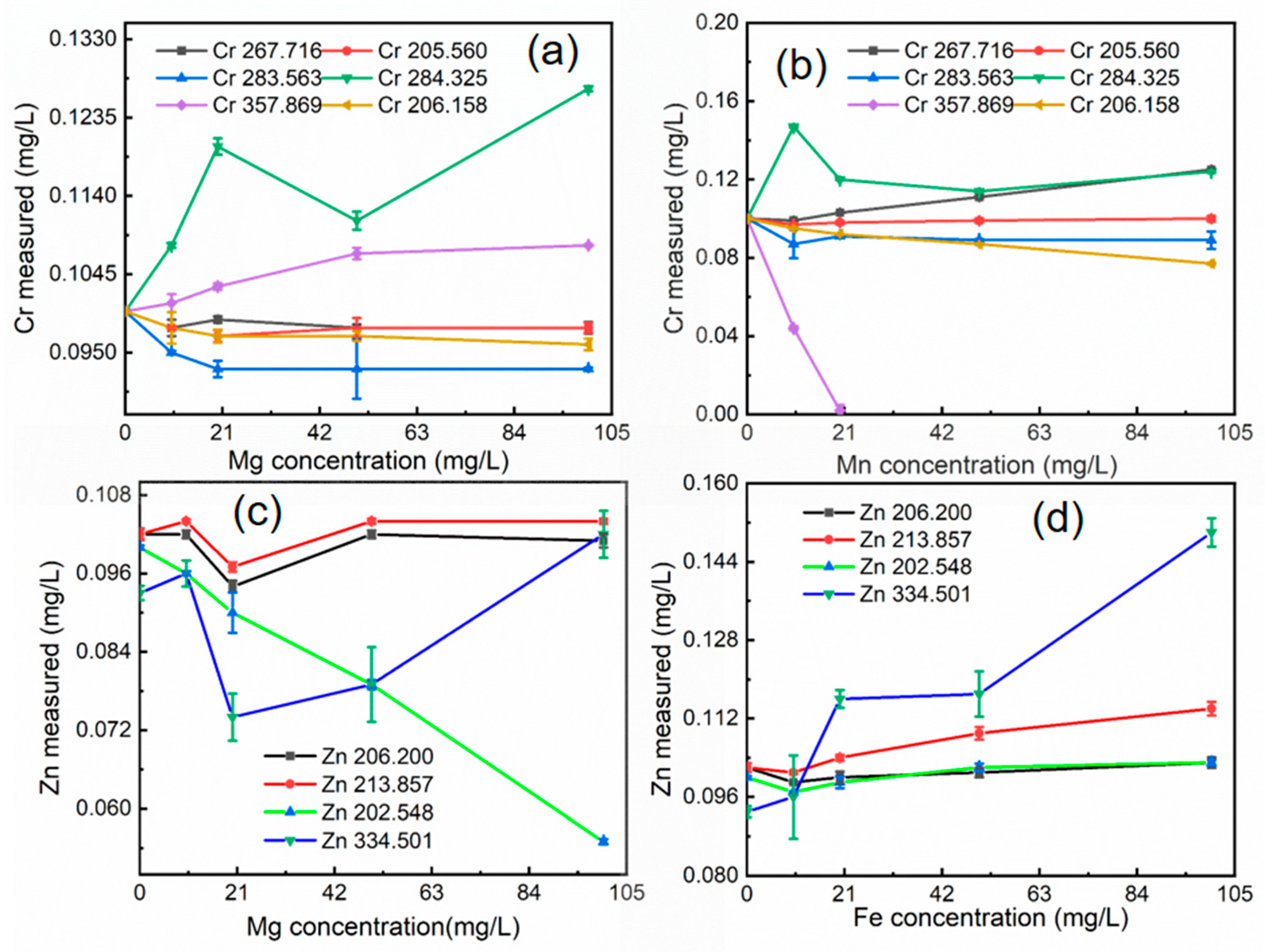
Figure 5.
Independent samples t-test data analysis results in P value.
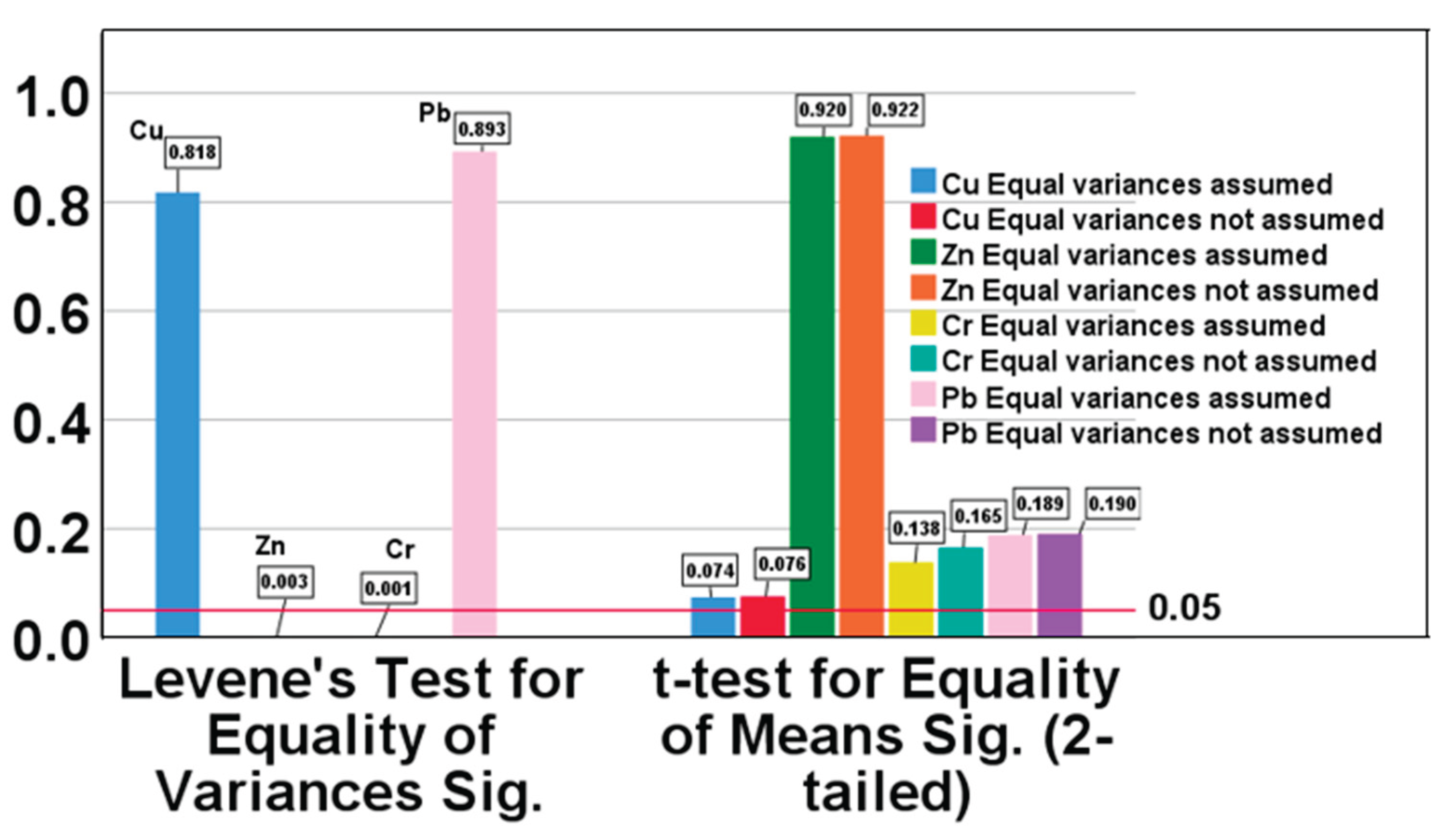

Table 1.
Instrumental Conditions for ICP-OES, ICP-MS And Microwave Digestion of Solid Samples.
| Operating conditions for ICP-OES | Operating conditions for ICP-MS | |||||
| RF power /W | 1300 | 1550 | ||||
| Plasma Ar gas flow /(L/min) | 10 | 14.5 | ||||
| Auxiliary Ar gas flow /(L/min) | 0.2 | 1.0 | ||||
| Nebulizer gas flow /(L/min) | 0.72 | 1.05 | ||||
| Pump feed volume /(mL/min) | 1.5 | 1.5 | ||||
| Observation distance | 15 | - | ||||
| Peak algorithm | Peak area | Intensity | ||||
| Number of points per peak | 7 | - | ||||
| Plasma observation | Axial | - | ||||
| Repetitions /times | 3 | 3 | ||||
| Internal standard element | - | Rh | ||||
| Collision Cell | - | He | ||||
| m/z per reading cycle | - | 63Cu, 66Zn, 208Pb, 52Cr | ||||
| Microwave Digestion Working Conditions | ||||||
| Steps | Power (W) | Hold time (min) | ||||
| 1 | 600 | 15 | ||||
| 2 | 600 | 5 | ||||
| 3 | 1200 | 10 | ||||
| 4 | 1200 | 30 | ||||
Table 2.
The Relative Error of the Analytical Spectrum of Copper, Zinc, Chromium, and Lead (n=6).
| Elements | Wavelength (nm) | Certified values (mean±SD) (mg·kg-1) | Measured Value (mg·kg-1) | Relative Error (%) |
| Cu | 327.393 | 36.4±5.1 | 27.5 | -24.4 |
| 324.752 | 39.2 | 7.7 | ||
| 224.7 | 68.5 | 88.2 | ||
| Zn | 206.2 | 110±12 | 97.5 | -11.3 |
| 213.857 | 106.0 | -3.7 | ||
| 202.548 | 97.1 | -11.7 | ||
| Cr | 205.56 | 77.1±8.6 | 62.0 | -19.6 |
| 283.563 | 99.4 | 28.9 | ||
| 284.325 | 43.4 | -43.7 | ||
| 267.716 | 69.6 | -9.7 | ||
| Pb | 220.353 | 35.2±5 | 30.5 | -13.3 |
| 217 | 134.1 | 281.0 | ||
| 261.418 | ND* | ND | ||
| 283.306 | 86.3 | 145.2 |
*ND indicates Not Detected.
Table 3.
Wavelength, Linear Equation, Correlation Coefficient, Detection Limit, and Quantitation Limit of the Working Curve.
Table 3.
Wavelength, Linear Equation, Correlation Coefficient, Detection Limit, and Quantitation Limit of the Working Curve.
| Element | Wavelength (nm) | Linear equation | r | DL /(mg·L-1) | QL /(mg·L-1) |
| Pb | 220.353 | Y=7579X+147.2 | 0.9999 | 0.0085 | 0.0338 |
| Cr | 267.716 | Y=202600X+4091.7 | 0.9999 | 0.0004 | 0.0017 |
| Cu | 324.752 | Y=671700X-6373.3 | 0.9999 | 0.0105 | 0.0419 |
| Zn | 213.857 | Y=88640X+4887.1 | 0.9997 | 0.0013 | 0.0054 |
Table 4.
Spiked Recovery Rates of SMS A (mean±SD) (n=3).
| Element |
Baseline level of A/(mg·L-1) |
Add amount /(mg·L-1) | Measurement of A/(mg·L-1) | Recovery/% |
| Pb | 0.172 | 2 | 1.037 | 95.5%±0.9 |
| Cr | 0.244 | 2 | 1.072 | 95.5%±0.2 |
| Cu | 1.832 | 2 | 1.987 | 103.7%±1.2 |
| Zn | 1.256 | 2 | 1.605 | 98.6%±0.5 |
Table 5.
Accuracy and Precision Test of Soil Quality Control Samples GSS-4 and GSS-5 (n=6).
| Sample | Element |
Mean /(mg·Kg-1) |
RSD/% | Certified /(mg·Kg-1) | Recovery/% |
| GSS-5 | Pb | 549±4 | 0.80 | 552±29 | 99.5 |
| Cr | 115±2 | 1.6 | 118±7 | 97.5 | |
| Cu | 149±2 | 1.1 | 144±6 | 103.5 | |
| Zn | 497±11 | 2.3 | 494±25 | 100.6 | |
| GSS-4 | Pb | 63±1 | 0.67 | 58±5 | 108.6 |
| Cr | 376±3 | 0.76 | 370±16 | 101.6 | |
| Cu | 40±1 | 1.59 | 40±3 | 100.0 | |
| Zn | 222±2 | 0.70 | 210±13 | 105.7 |
Table 6.
T-test Comparison of Analysis Results between This Method and ICP-MS Method (n=6).
| Sample | Element | ICP-OES /(mg·Kg-1) | ICP-MS /(mg·Kg-1) | t-value | Significance difference evaluation |
| A | Pb | 42.62±0.02 | 41.83±0.03 | 1.411 | No |
| Cr | 63.02±0.01 | 67.11±0.02 | 1.612 | No | |
| Cu | 432.60±0.02 | 440.75±0.01 | 1.993 | No | |
| Zn | 308.30±0.01 | 308.63±0.03 | 0.103 | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



