
Submitted:
06 February 2026
Posted:
09 February 2026
You are already at the latest version
Abstract
Colorectal cancer(CRC) is among the leading causes of cancer-related deaths world-wide, but current chemotherapy strategies are insufficient, necessitating the identifica-tion of novel small molecules that can disrupt multiple survival pathways simultane-ously. In this study, the antiproliferative effects of benzimidazole-palladium complex compounds were evaluated in 4T1, MDA-MB-231, DLD-1, and HT-29 cancer cell lines using MTT assays. The autophagy and apoptosis-inducing capacities of the compounds, which showed significant cytotoxicity in CRC cells, were analyzed in detail using im-munofluorescence and flow cytometry methods. Furthermore, functional assays such as migration, adhesion, and colony formation were further investigated in CRC cells. The benzimidazole derivative drug candidate (3a) significantly reduced cell viability in HT-29 and DLD-1 cells. Mechanistic investigations using immunofluorescence and Annexin V/PI for LC3 and mTOR revealed that 3a triggered a dual response of au-tophagy and apoptosis. In CRC cells, compound 3a significantly impaired metastatic potential by inhibiting migration, colony formation, and cell adhesion. Gene expression analysis also showed that this compound suppressed the AKT/mTOR/STAT3 signaling axis, thereby triggering an autophagic response that compromised cell adhesion and accelerated apoptotic cell death. These findings suggest that compound 3a is a prom-ising anticancer candidate and warrants further mechanistic investigation.
Keywords:
autophagy
; colon cancer
; benzimidazole
; apoptosis
1. Introduction
Colorectal cancer (CRC), the third most common cancer worldwide, is a global health problem resulting in approximately 1.9 million new cases and 930,000 deaths according to 2020 data [1]. Current demographic projections predict this burden will reach 3.2 million new cases by 2040 [1]. While current therapeutic approaches primarily target actively proliferating cells, the development of chemoresistance and systemic toxicity necessitate the development of more specific and effective synthetic agents [2].
Benzimidazole, a key pharmacophore with an aromatic heterocyclic core, finds widespread use in drug research [3]. This framework has served as the basis for numerous synthetic derivatives exhibiting a variety of biological activities, including antibacterial, antiviral, antifungal, and antiproliferative effects [4]. Benzimidazole derivatives, in particular, have attracted attention as target-based chemical frameworks in cancer treatment and have been studied by researchers in various cancer cell lines [5,6]. Benzimidazole derivatives exhibit potent anticancer effects by inhibiting cancer cell proliferation, inducing apoptosis, and modulating critical cellular signaling pathways [7,8]. These molecules possess multifaceted mechanisms for cell cycle arrest through DNA binding, disruption of microtubule dynamics, and specific enzyme inhibition [9]. In addition to these comprehensive effects of benzimidazole derivatives, the inhibitory roles of benzimidazolium salts, particularly on the STAT3 signaling pathway, have been demonstrated [10].
Autophagy is a bidirectional mechanism in CRC that plays both suppressive and supportive roles in tumor development by regulating stress adaptation [11]. In CRC, the autophagy process is a central survival mechanism tightly controlled by the interaction between STAT3 and mTOR signaling pathways. The fact that STAT3 inhibition triggers autophagy by suppressing mTOR activity makes this axis a critical molecular target in terms of tumor progression and treatment response [12]. The lethal autophagy mechanism, as it is called, can transform this process into a death mechanism when cellular stress exceeds a certain threshold [13]. This phenomenon triggers pro-apoptotic signals from proteins such as Beclin-1 and Atg5, cleaved by caspases, leading the cell into an irreversible process of destruction. Consequently, the synergistic interaction between this initially protective mechanism and apoptosis presents a strategic therapeutic target that overcomes the chemoresistance shields of cancer cells and drives them into programmed cell death [14].
Further molecular and mechanistic studies supporting the anticancer effects of benzimidazolium salts are of great importance in determining the place of these compounds in the rational drug development process. This work begins with benzimidazoline salts synthesized by Akkoç, which were initially evaluated for efficacy by MTT-based cytotoxicity experiments [15]. Our preliminary experimental observations revealed that these compounds not only reduce cell viability but also cause significant changes in cell morphology. In particular, the formation of autophagosome-like vacuolar structures and a significant decrease in the adhesive properties of cells after drug application were observed. This series, exhibiting low IC₅₀ values and morphologically diverse effects, suggests that mechanisms beyond classical apoptotic cell death may be at play. Therefore, a more detailed evaluation of these compounds in different cancer cell types and elucidation of their mechanisms of action via apoptosis-autophagy-related molecular pathways constitute the main objective of the present study.
2. Materials and Methods
2.1. Synthesis of Benzimidazole-Based Compounds
The 2-hydroxyethyl substituted benzimidazolium salts evaluated in this study and the palladium complexes derived from these salts were successfully synthesized in a previous study by our research group (Scheme 1) [15]. The synthesis processes, structural characterizations, and fundamental physical properties of these compounds are reported in detail in the relevant study. The relevant publication comprehensively presents the structural characterization (¹H NMR, ¹³C NMR, FT-IR, elemental analysis, etc.), physical properties, and fundamental chemical data of the synthesized benzimidazolium salts and palladium complexes.
In our previous study, the antiproliferative activities of 2-hydroxyethyl substituted benzimidazolium salts, which are 1-(2-hydroxyethyl)-3-(4-methylbenzyl)-1H-benzo[d]imidazol-3-ium bromide (3a), 1-(2-hydroxyethyl)-3-(2,3,4,5,6-pentamethylbenzyl)-1H-benzo[d]imidazol-3-ium bromide (3b), and their palladium complexes, which are dibromo-[1-(2-hydroxyethyl)-3-(4-methylbenzyl)benzimidazol-2-ylidene]-N-(3-chloropyridine) palladium (II) complex (4a), dibromo-[1-(2-hydroxyethyl)-3-(2,3,4,5,6-pentamethylbenzyl)benzimidazol-2-ylidene]-N-(3-chloropyridine) palladium (II) complex (4b), were evaluated in four different cancer cell lines (HeLa, MDA-MB-231, HepG2, and DLD-1) using the MTT assay method. The results obtained revealed that the organic benzimidazolium salts possess significantly higher cytotoxic potential compared to their metal complexes. Specifically, compound 3b exhibited measurable cytotoxic activity with an IC₅₀ value of 80.64 ± 1.25 µM in the HeLa cell line, while the palladium complexes (4a and 4b) did not show any antiproliferative effect in the same cell line. Similarly, compounds 3a and 3b showed moderate cytotoxic activity in HepG2 cells, while the metal complexes were also found to be ineffective against this cell line.
The results obtained in MDA-MB-231 and DLD-1 cell lines more clearly demonstrated the potent and selective antiproliferative effects of organic compounds. In MDA-MB-231 breast cancer cells, compound 3b in particular showed the highest cytotoxic activity with an IC₅₀ value of 7.59 ± 0.68 µM and was found to be more effective than cisplatin, which was used as a positive control. In addition, compounds 3a and 3b were found to have a dose-dependent effect against breast cancer cells. In the DLD-1 CRC cell line, compound 3a showed a remarkable antiproliferative effect with an IC₅₀ value of 9.19 ± 1.51 µM, which was very close to the IC₅₀ value of cisplatin. In contrast, compounds 4a and 4b did not show cytotoxic activity in either cell line. These results indicate that the 2-hydroxyethyl substituted benzimidazolium salts synthesized in our previous study [15] are promising anticancer candidates, particularly MDA-MB-231 and DLD-1 cancer cells, and provide a strong basis for further biological studies. Therefore, mecanistic experiments were conducted in this study.
2.2. Cell Culture
Human breast cancer cell line MDA-MB-231, mouse breast cancer cell line 4T1, DLD-1 and HT-29 human CRC cell lines were obtained from the American Type Culture Collection (ATCC). MDA-MB-231, 4T1, DLD-1 and HT-29 cells were cultured in a single layer in RPMI 1640 or Dulbecco's Modified Eagle Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco), 4.5 g/L L-glutamine, 1 mM sodium pyruvate, 100 U/mL penicillin-streptomycin and 1% amphotericin B. Cultures were maintained at 37°C in a humidified incubator containing 5% CO₂.
2.3. MTT Cell Viability Test
The cytotoxic effect of compounds 3a, 3b, 4a and 4b on MDA-MB-231, 4T1, DLL-1, and HT-29 cells was evaluated using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] test. Cells were seeded into 96-well plates at a cell density of 6×10³ per well and incubated overnight. They were then treated with various concentrations of the compound (1–160 µM) for 48 hours. After treatment, 10 µL of MTT (5 mg/ml) solution was added to each well and incubated at 37 °C for 4 hours, and 50 µL of DMSO was added, and absorbance was measured at 570 nm using a microplate reader. Cell viability was expressed as a percentage compared to an untreated control group, which was considered to be 100%.
2.4. Immunofluorescence (IF) Staining
HT-29 and DLD-1 colorectal cancer cells were seeded onto sterile glass coverslips placed in 6-well culture plates and subsequently subjected to 3a treatment under the specified experimental conditions. Upon completion of the incubation period, cells were fixed in 4% paraformaldehyde for 15 min at room temperature and rinsed thoroughly with phosphate-buffered saline (PBS). Cellular permeabilization was achieved using Triton X-100 for 10 min at room temperature, followed by blocking of non-specific antibody binding with 5% bovine serum albumin (BSA). Immunostaining was performed by incubating the cells overnight at 4 °C in a humidified chamber with primary antibodies targeting LC3 (bs-8878R, Bioss) and mTOR (bs-1992R, Bioss), each diluted 1:200 in blocking buffer. After extensive washing with PBS, cells were incubated with a FITC-conjugated secondary antibody for 1 h at room temperature. Nuclear counterstaining and mounting were carried out using a DAPI-containing mounting medium (Sigma). Fluorescence images were acquired using an Olympus BX53 fluorescence microscope equipped with a DP74 camera system. Quantitative image analysis was conducted using ImageJ software. Mean fluorescence intensity values were determined from 30 cells per condition, selected from randomly chosen microscopic fields
2.5. Annexin V Apoptotic Analysis
To evaluate the apoptotic effects of compound 3a, DLD-1 and HT-29 cells were treated for 48 hours and then stained with Annexin V-APC and Propidium Iodide (PI). After staining, they were analyzed by flow cytometry to distinguish between viable and apoptotic cell populations. Apoptotic percentages were measured for each group and compared with controls.
2.6. Cell Migration Experiment
A wound healing experiment was performed to evaluate the effects of compound 3a on cell migration. Details of the method are given in our previous study [16]. Briefly, HT-29 CRC cells were seeded into 6-well plates at a density of 10*105 cells per well. After the cells reached approximately 70-80% density, at the end of 48 hours of incubation, a wound was created on the culture surface using a sterile 100 µL pipette tip. Wound widths were visualized at 0 and 48 hours and then percentages were determined using ImageJ software.
2.7. Cell Adhesion Experiment
HT-29 CRC cells were treated with compound 3a for 24 hours. Control group cells received no treatment. Then, the cell adhesion experiment was performed according to the literature [17]. Microplate wells were coated with 1% Poly-D-lysine and blocked with heat-denatured bovine serum albumin (BSA) to prevent non-specific binding. After fixing the cells with methanol, they were stained with 0.1% crystal violet, and the amount of bound cells was visualized by removing excess dye. In the final step, the dye bound to the cells was dissolved in 10% acetic acid and analyzed spectrophotometrically at a wavelength of 570 nm in a microplate reader.
2.8. Gene Expression Analysis
HT-29 cells were incubated with compound 3a at an IC50 (3.7 µM) concentration for 48 hours, followed by total RNA isolation using Trizol reagent (Invitrogen, USA) according to the manufacturer's instructions. cDNA synthesis was performed from the isolated RNA samples via reverse transcriptase enzyme; then, the expression levels of apoptosis-related (BCL-2, BAX, and Caspase-3), autophagy-related genes (Mtor and BECN1), adhesion-related (VIM and ITGβ1), AKT, and Stat3 genes were analyzed using quantitative real-time PCR (qPCR). Gene expression data were normalized relative to the beta-actin gene used as an internal control, and relative mRNA changes were calculated using the 2-∆∆Ct method. For statistical comparisons, the control group expression level was fixed at 1, and fold changes of 3a application on target genes were determined relative to this reference value.
2.9. Statistical Analysis
Statistical analyses were performed using GraphPad Prism software. Student t-test was applied for pairs of samples. For data with more than two samples, one-way ANOVA followed by Tukey's multiple comparison test was applied, IC50 values and confidence intervals were obtained from nonlinear regression analyses. A p-value < 0.05 was considered statistically significant.
3. Results
3.1. 2-hydroxyethyl Substituted Benzimidazolium Salts Demonstrates Significant Anti-Cancer Efficacy
In the previous study, the cytotoxicity of compounds 3a-b and 4a-b was tested in DLD-1 and MDA-MB-231 cells [15]. In this study, cytotoxicity tests were performed in four different cell lines, including 4T1 mouse breast cancer cells and HT-29 human CRC cells. In all four cell lines, the lowest IC50 values were observed in compound 3a(Table 1). The cytotoxic potential of compound 3a was evaluated against HT-29 and DLD-1 colorectal cancer cell lines using increasing concentrations (1-160µM). Our results demonstrated that 3a significantly suppressed cell proliferation in a dose-dependent manner in both models(Figure 1). Notably, HT-29 cells exhibited a more sensitive profile with an IC50 value of 3.7 µM, while DLD-1 cells showed a slightly higher resistance with an IC50 of 7.2 µM(Figure 1). These findings indicate that compound 3a possesses strong antiproliferative activity at low micromolar concentrations, establishing a robust foundation for further mechanistic investigations into its pro-apoptotic and autophagic effects.
3.2. Coordinated Regulation of LC3 and mTOR by 3a Induces Cytoprotective Autophagy in CRC Cells
Given the context-dependent role of autophagy in colorectal cancer progression and therapy response, immunofluorescence analyses of LC3 and mTOR were performed to evaluate whether 3a treatment modulates key regulators of autophagy-associated signaling. In HT-29 cells, 3a exposure resulted in a pronounced increase in LC3 fluorescence intensity compared to control cells, an effect that was statistically significant (p<0.0001; Figure 2). A similar enhancement of LC3 signal intensity was consistently observed in DLD-1 cells following 3a treatment, indicating a reproducible response across both colorectal cancer cell lines. Beyond quantitative changes, LC3 staining in 3a-treated cells exhibited a marked alteration in subcellular distribution. Specifically, LC3 displayed a prominent punctate cytoplasmic pattern characterized by dot-like aggregates dispersed throughout the cytoplasm, in contrast to the predominantly diffuse and low-intensity staining observed under control conditions (Figure 2)
In parallel, immunofluorescence analysis of mTOR revealed an opposite response to 3a treatment. In both HT-29 and DLD-1 cells, mTOR fluorescence intensity was significantly reduced following 3a exposure when compared to control cells (p<0.0001; Figure 3). This decrease in mTOR signal was consistently detected across independent experiments, suggesting a robust treatment-associated modulation of mTOR-related signaling.
Taken together, the observed increase in punctate LC3 accumulation accompanied by a concurrent reduction in mTOR expression indicates a coordinated cellular response to 3a treatment in colorectal cancer cells.
3.3. Compound 3a Triggers Apoptosis in CRC
To assess whether the growth inhibitory effects of compound 3a occur via programmed cell death, Annexin V-FITC/PI flow cytometry was used. Treatment at IC50 concentration for 48 hours resulted in a significant shift from a viable cell population to an apoptotic population in both colorectal cancer cell lines(Figure 4). This confirmed that the cytotoxicity of 3a is similarly driven by the activation of a cell death program.
3.4. Treatment of CRC Cells with 3a Suppressed Migration, Colonization, and Adhesion Properties
To evaluate the effect of compound 3a on the metastatic and tumor-forming properties of colorectal cancer cells, we performed a series of functional tests focusing on migration, adhesion, and colony formation. The wound healing test showed that control HT-29 cells almost completely shut down within 48 hours, while treatment with 3a significantly inhibited lateral migration, leaving a significant gap and disrupting the cellular metastasis mechanism(Figure 5A). This anti-metastatic potential was further supported by a marked decrease in the percentage of adhering cells, suggesting that 3a destabilizes cell-matrix interactions essential for survival and spread(Figure 5B). Furthermore, the colony formation test showed that 3a significantly suppressed the ability of single cancer cells to proliferate into independent colonies, thus severely impairing the long-term tumor-forming and survival potential of the HT-29 cell line(Figure 5C). Collectively, these findings demonstrate that compound 3a effectively attenuates the aggressive phenotype of CRC by inhibiting key processes such as colonization, adhesion, and migration.
3.5. Interference with the AKT/mTOR/STAT3 Signaling Axis Using a Benzimidazole Derivative Compound 3a Restricts the Metastatic and Proliferative Potential of Human CRC Cells
Gene expression analyses performed in HT-29 cells confirmed that the molecular mechanism underlying the antitumor effect of molecule 3a is directly related to the induction of apoptosis and the suppression of survival pathways(Figure 6A,B). Examination of RT-qPCR data revealed a significant increase in pro-apoptotic BAX and the key executive caspase Caspase-3 mRNA levels, accompanied by a significant decrease in anti-apoptotic BCL2 expression, indicating that the compound strongly activates the mitochondrial apoptosis pathway(Figure 6A). At the cellular signaling level, the simultaneous decrease in the expression of AKT, Stat3, and Mtor genes, which have high oncogenic potential, demonstrates that 3a centrally targets the proliferation and survival networks of cancer cells. Furthermore, the coexistence of the increased trend in the autophagy marker BECN1 levels and mTor inhibition suggests that a cellular stress response is triggered; Downregulation of the ITGβ1 and VIM genes, associated with metastasis and cell adhesion, demonstrates that the molecule limits tumor invasion and the cell's capacity to adhere to the environment at the molecular level(Figure 6A).
4. Discussion
CRC, has become a global health crisis, presents a difficult barrier to overcome in current treatment protocols, particularly due to chemoresistance mechanisms developing in metastatic processes [18]. Despite the widespread application of conventional cytotoxic agents, the ability of cancer cells to evade apoptosis through genomic instability and disrupt DNA repair mechanisms significantly limits the clinical efficacy of existing regimens. To overcome these treatment bottlenecks and control resistant cell populations, the discovery of innovative synthetic compounds that simultaneously target cellular survival and adhesion dynamics is an indispensable priority for modern oncology research [18,19]. Benzimidazole salts stand out for their superior solubility and stability, which are critical for overcoming the bioavailability hurdles of neutral compounds. Their unique ionic profile enables more precise interaction with cellular targets, effectively dismantling oncogenic signaling to drive resistant cancer cells toward programmed death [5,20].
Benzimidazole derivative drugs have been proven to have a highly effective anticancer effect in many cancers, particularly colon cancer [5,6,7,21]. Our previous study showed that organic benzimidazolium salts (3a and 3b) have a much higher cytotoxic potential compared to metal complexes (4a and 4b) and exhibit antiproliferative activity comparable to cisplatin, particularly in DLD-1 and MDA-MB-231 cell lines15. In this study, the effects of these four compounds on cell proliferation were investigated in aggressive mouse breast cancer cells (4T1), aggressive human breast cancer cells (MDA-MB231), and aggressive HT-29 and DLD-1 cells. Similar to the previous study, 3a was the most effective compound in colon cancer, while 3a and 3b showed similar levels of anticancer activity in breast cancer. 4a and 4b did not show any anticancer activity, as in the previous study. In this case, the properties of benzimidazole suggest that the 2-hydroxyethyl substituent interacts more effectively with biological macromolecules in its free form, but complexation with palladium significantly reduces its anticancer activity by masking or halting the molecule's active pharmacological sites.
Autophagy functions as a protective mechanism in the early stages of cancer, preventing tumor formation by maintaining genomic stability, while in advanced stages, its over-induction becomes a strategic therapeutic weapon, triggering programmed cell death by creating a metabolic impasse in cancer cells [22]. LC3, a specific marker of autophagosome formation, is widely used in autophagy measurements [23]. One of the striking findings of this study is that 3a significantly induces autophagy. Immunofluorescence analyses show an increase in LC3 signaling and suppression of mTOR activity, indicating that 3a activates the autophagic flow. Autophagy can play both tumor-suppressing and tumor-promoting roles in CRC, depending on the context. Particularly in advanced cancers, autophagy can support cell survival under metabolic stress, while excessive or dysregulated autophagy can lead to cell death [24]. In this study, the observation of autophagy induction along with apoptosis suggests that 3a drives cells not towards protective autophagy, but towards a process described as "lethal autophagy." Numerous protein molecules involved in tumor pathogenesis have been found to influence tumor formation by regulating autophagy and apoptosis; most of these are via the autophagy protein LC3, beclin-1, the apoptosis protein family Bcl-2, BAX, or the interaction between the two [25]. Autophagy and apoptosis are in constant interaction via common molecular bridges such as the BH3 domain and p53 to maintain cellular homeostasis and make decisions about survival or death under stressful conditions [26]. Apoptosis analyses showed that compound 3a caused significant apoptotic cell death in both HT-29 and DLD-1 cells. This finding, confirmed by Annexin V/PI assays, is also supported by gene expression results. The decrease in anti-apoptotic BCL-2 expression, accompanied by an increase in pro-apoptotic BAX and caspase-3 expression, suggests that 3a activates the intrinsic apoptotic pathway. Given that apoptotic mechanisms are frequently suppressed in CRC, contributing to chemotherapy resistance, the ability of 3a to restore apoptotic balance is of therapeutic importance. It is thought that compound 3a causes cell apoptosis by triggering lethal autophagy.
The AKT/mTOR/STAT3 signaling pathways are key regulators of proliferation, survival, migration, and chemotherapy resistance in CRC cells [27]. Gene expression analyses revealed that compound 3a significantly suppressed AKT, mTOR, and STAT3 expression. The literature reports that constitutive activation of STAT3 is strongly associated with CRC progression, poor prognosis, and metastasis. STAT3 can also suppress autophagy by activating the mTOR pathway [28]. Therefore, the inhibition of the STAT3 and mTOR axis by compound 3a is mechanistically consistent with the activation of autophagy and the attenuation of cellular survival signals.
Functional analyses demonstrate that molecular findings directly reflect cellular behavior. In wound-healing, cell adhesion, and colony formation experiments, 3a was found to significantly inhibit the migration capacity, surface adhesion ability, and clonal growth of HT-29 cells. These findings are consistent with the decrease in Vimentin (VIM) and integrin β1 (ITGB1) expression observed in gene expression analyses. Integrins and EMT-related proteins play a critical role in CRC metastasis, and suppression of these proteins significantly reduces the invasive potential of tumor cells [29,30].
The literature increasingly features investigations on the anticancer potential of benzimidazole derivatives, however most of these molecules lack rigorous mechanistic evaluations [8]. This study is important because it shows that 3a not only suppresses proliferation but also reverses the autophagy-apoptosis balance against the tumor by targeting the AKT/mTOR/STAT3 axis. Especially in a heterogeneous and treatment-resistant tumor type like CRC, such multi-target agents hold promise for next-generation therapeutic strategies.
5. Conclusions
Although this study has some limitations, the findings clearly demonstrate that compound 3a exhibits a multifaceted anticancer effect in colon cancer cells. While the study was limited to in vitro models, the simultaneous induction of autophagy and apoptosis by 3a, and the association of these processes with the suppression of AKT/mTOR/STAT3 signaling pathways, provides strong clues about the compound's mechanism of action. Furthermore, the effective inhibition of migration, colony formation, and cell adhesion suggests that 3a can also target cellular characteristics associated with metastasis.
In conclusion, the benzimidazole derivative compound 3a emerges as a promising anticancer candidate that suppresses proliferation, triggers autophagy-mediated apoptotic cell death, and reduces metastatic potential in colon cancer cells. These findings strongly support the need for further evaluation of 3a's therapeutic potential through advanced mechanistic approaches and in vivo models.
Author Contributions
Conceptualization, M.Ç..; methodology, M.Ç. and E.A.; software, M.Ç. and E.A.; validation, M.Ç., E.A., S.A., H.Ö., H.A. and S.A.; formal analysis, M.Ç. and E.A.; investigation, M.Ç. and E.A.; resources, M.Ç.; data curation, M.Ç. and E.A.; writing—original draft preparation, M.Ç., E.A. and S.A.; writing—review and editing, M.Ç. and E.A.; visualization, M.Ç. and E.A.; supervision, M.Ç. ; project administration, M.Ç.; funding acquisition, M.Ç. . All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Research Fund of the Yüzüncü Yıl University. Project Number: TYD-2021-9343.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data supporting the reported results can be found by contacting the corresponding author.
Acknowledgments
The authors express their gratitude to the staff of the GENKOK central laboratory at Erciyes University for their support. During the preparation of this article, the authors used Gemini (version 3.0) and Quilbot software for language editing and graphical abstract creation. The authors have reviewed and edited the output and are fully responsible for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global Burden of Colorectal Cancer in 2020 and 2040: Incidence and Mortality Estimates from GLOBOCAN. Gut 2023, 72(2), 338–344. [Google Scholar] [CrossRef]
- Islam, M.R.; Akash, S.; Rahman, M.M.; Nowrin, F.T.; Akter, T.; Shohag, S.; Rauf, A.; Aljohani, A.S.M.; Simal-Gandara, J. Colon Cancer and Colorectal Cancer: Prevention and Treatment by Potential Natural Products. Chem. Biol. Interact. 2022, 368, 110170. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Zulfiqar, T.; Jabeen, H.; Gohar, N.; Javed, T.; Munawar, Z.; Hassan, M.J.; Rasheed, M.M.; Amir, E. Benzimidazole Derivatives in Drug Design: Structure-Activity Relationships and Therapeutic Potential: A Review. Journal of Pharma and Biomedics 2024, 2(2), 71–80. [Google Scholar]
- Alheety, N.F.; Awad, S.A.; Alheety, M.A.; Darwesh, M.Y.; Abbas, J.A.; Besbes, R. Benzimidazole Derivatives: A Review of Advances in Synthesis, Biological Potential, Computational Modelling, and Specialized Material Functions. Chemistry (Easton). 2026, 8(1). [Google Scholar] [CrossRef]
- Akar, S.; Cakir, M.; Ozkol, H.; Akkoc, S.; Ozdem, B. A Benzimidazolium Salt Induces Apoptosis and Arrests Cells at Sub-G1 Phase in Epithelial Ovarian Cancer Cells. Mol. Biol. Rep. 2024, 51(1), 66. [Google Scholar] [CrossRef]
- Bitgen, N.; Cakir, M.; Akkoc, S.; Donmez-Altuntas, H. A Newly Synthesized Benzimidazolium Salt: 1-(2-Cyanobenzyl)-3-(4-Vinylbenzyl)-1H-Benzo [D] Imidazol-3-ium Chloride as a Potential Anticancer Agent for Colon Cancer Treatment, In Vitro Study. J. Biochem. Mol. Toxicol. 2025, 39(1), e70112. [Google Scholar] [CrossRef]
- Çakır, M.; Akar, S.; Çakır, Ş.; Akkoç, S.; Ozkol, H.; Tülüce, Y. Anticancer Effect of 1-(Anthracen-10-Ylmethyl)-3-(2-Cyanobenzyl)-1H-Benzo [d] Imidazol-3-Ium Chloride in 2D and 3D Cell Culture Models in Breast Cancer. Eastern Journal of Medicine 2024, 29(2), 244–251. [Google Scholar] [CrossRef]
- Mavvaji, M.; Akkoc, S. Recent Advances in the Anticancer Applications of Benzimidazole Derivatives. ChemistrySelect 2023, 8(35), e202302561. [Google Scholar] [CrossRef]
- Ren, L.; Li, W.; Zheng, X.; Liu, J.; Yang, Y.; Li, S.; Zhang, S.; Fu, W.; Xiao, B.; Wang, J. Benzimidazoles Induce Concurrent Apoptosis and Pyroptosis of Human Glioblastoma Cells via Arresting Cell Cycle. Acta Pharmacol. Sin. 2022, 43(1), 194–208. [Google Scholar] [CrossRef]
- Li, S.-S.; Chen, J.-J.; Zhang, M.-M.; Wang, W.-X.; Zhang, W.-Y.; Ma, C. Design, Synthesis, and Biological Evaluation of Novel Benzimidazole Derivatives as Anti-Cervical Cancer Agents through PI3K/Akt/MTOR Pathway and Tubulin Inhibition. Eur. J. Med. Chem. 2024, 271, 116425. [Google Scholar] [CrossRef]
- Li, Z.; Si, W.; Jin, W.; Yuan, Z.; Chen, Y.; Fu, L. Targeting Autophagy in Colorectal Cancer: An Update on Pharmacological Small-Molecule Compounds. Drug Discov. Today 2022, 27(8), 2373–2385. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, J.; Mao, Q.-F.; Wu, J.; Wang, Y. The Interaction between Autophagy and JAK/STAT3 Signaling Pathway in Tumors. Front. Genet. 2022, 13, 880359. [Google Scholar] [CrossRef]
- Kriel, J.; Loos, B. The Good, the Bad and the Autophagosome: Exploring Unanswered Questions of Autophagy-Dependent Cell Death. Cell Death Differ. 2019, 26(4), 640–652. [Google Scholar] [CrossRef]
- Xie, Q.; Liu, Y.; Li, X. The Interaction Mechanism between Autophagy and Apoptosis in Colon Cancer. Transl. Oncol. 2020, 13(12), 100871. [Google Scholar] [CrossRef] [PubMed]
- Akkoc, S. Antiproliferative Activities of 2-Hydroxyethyl Substituted Benzimidazolium Salts and Their Palladium Complexes against Human Cancerous Cell Lines. Synth. Commun. 2019, 49(21), 2903–2914. [Google Scholar] [CrossRef]
- Cakir, M.; Kuzu, B. Novel Imidazopyridine–Oxadiazole β-Tubulin Inhibitors Suppress Breast Cancer Migration and Induce Caspase-3-Mediated Apoptosis. ACS Omega 2026. [Google Scholar] [CrossRef]
- Pijuan, J.; Barceló, C.; Moreno, D.F.; Maiques, O.; Sisó, P.; Marti, R.M.; Macià, A.; Panosa, A. In Vitro Cell Migration, Invasion, and Adhesion Assays: From Cell Imaging to Data Analysis. Front. Cell Dev. Biol. 2019, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Karuniawati, H.; Jairoun, A.A.; Urbi, Z.; Ooi, D.J.; John, A.; Lim, Y.C.; Kibria, K.M.K.; Mohiuddin, A.K.M.; Ming, L.C. Colorectal Cancer: A Review of Carcinogenesis, Global Epidemiology, Current Challenges, Risk Factors, Preventive and Treatment Strategies. Cancers (Basel). 2022, 14(7), 1732. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhang, J.; Yan, T.; Miao, M.; Cao, Y.; Cao, Y.; Zhang, L.; Li, L. Novel Strategies to Reverse Chemoresistance in Colorectal Cancer. Cancer Med. 2023, 12(10), 11073–11096. [Google Scholar] [CrossRef]
- Satija, G.; Sharma, B.; Madan, A.; Iqubal, A.; Shaquiquzzaman, M.; Akhter, M.; Parvez, S.; Khan, M.A.; Alam, M.M. Benzimidazole Based Derivatives as Anticancer Agents: Structure Activity Relationship Analysis for Various Targets. J. Heterocycl. Chem. 2022, 59(1), 22–66. [Google Scholar] [CrossRef]
- Petersen, J.S.S.M.; Baird, S.K. Treatment of Breast and Colon Cancer Cell Lines with Anti-Helmintic Benzimidazoles Mebendazole or Albendazole Results in Selective Apoptotic Cell Death. J. Cancer Res. Clin. Oncol. 2021, 147(10), 2945–2953. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Shukla, N.; Singh, S.S.; Kushwaha, S.; Shrivastava, R. Mechanism of Interaction between Autophagy and Apoptosis in Cancer. Apoptosis 2021, 26(9), 512–533. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and Autophagy-Related Pathways in Cancer. Nat. Rev. Mol. Cell Biol. 2023, 24(8), 560–575. [Google Scholar] [CrossRef]
- Manzoor, S.; Muhammad, J.S.; Maghazachi, A.A.; Hamid, Q. Autophagy: A Versatile Player in the Progression of Colorectal Cancer and Drug Resistance. Front. Oncol. 2022, 12, 924290. [Google Scholar] [CrossRef]
- Prerna, K.; Dubey, V.K. Beclin1-Mediated Interplay between Autophagy and Apoptosis: New Understanding. Int. J. Biol. Macromol. 2022, 204, 258–273. [Google Scholar] [CrossRef]
- Xi, H.; Wang, S.; Wang, B.; Hong, X.; Liu, X.; Li, M.; Shen, R.; Dong, Q. The Role of Interaction between Autophagy and Apoptosis in Tumorigenesis. Oncol. Rep. 2022, 48(6), 208. [Google Scholar] [CrossRef]
- Li, J.-K.; Sun, H.-T.; Jiang, X.-L.; Chen, Y.-F.; Zhang, Z.; Wang, Y.; Chen, W.-Q.; Zhang, Z.; Sze, S.C.W.; Zhu, P.-L. Polyphyllin II Induces Protective Autophagy and Apoptosis via Inhibiting PI3K/AKT/MTOR and STAT3 Signaling in Colorectal Cancer Cells. Int. J. Mol. Sci. 2022, 23(19), 11890. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Li, M.Y.; Wang, Z.; Zuo, H.X.; Wang, J.Y.; Xing, Y.; Jin, C.; Xu, G.; Piao, L.; Piao, H. Convallatoxin Promotes Apoptosis and Inhibits Proliferation and Angiogenesis through Crosstalk between JAK2/STAT3 (T705) and MTOR/STAT3 (S727) Signaling Pathways in Colorectal Cancer. Phytomedicine 2020, 68, 153172. [Google Scholar] [CrossRef]
- Vlahakis, A.; Debnath, J. The Interconnections between Autophagy and Integrin-Mediated Cell Adhesion. J. Mol. Biol. 2017, 429(4), 515–530. [Google Scholar] [CrossRef]
- Gundamaraju, R.; Lu, W.; Paul, M.K.; Jha, N.K.; Gupta, P.K.; Ojha, S.; Chattopadhyay, I.; Rao, P.V.; Ghavami, S. Autophagy and EMT in Cancer and Metastasis: Who Controls Whom? Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2022, 1868(9), 166431. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Synthesis of compounds 2a-b, 3a-b and 4a-b.
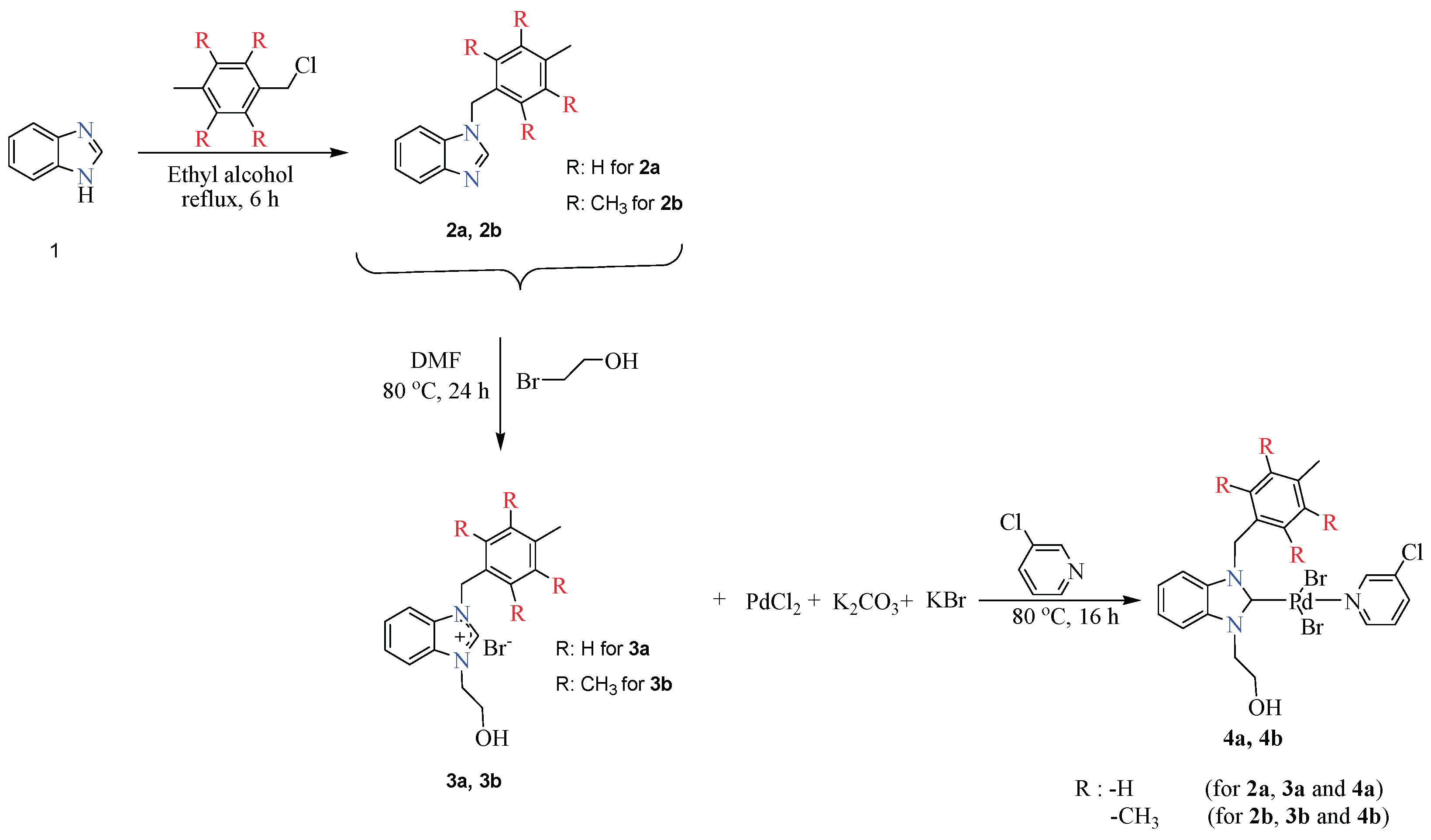
Figure 1.
Cytotoxic profile and dose-response curves of compound 3a in colorectal cancer cells. The bar charts and non-linear regression curves illustrate a significant, dose-dependent inhibition of cell proliferation in both HT-29 and DLD-1 cell lines after treatment. While compound 3a exhibited high potency across both lines, HT-29 cells showed greater sensitivity with an IC50 of 3.7 µM, compared to 7.2 µM in DLD-1 cells. Statistically significant differences compared to the control group are indicated as ns: non signifant and **** p < 0.0001.
Figure 1.
Cytotoxic profile and dose-response curves of compound 3a in colorectal cancer cells. The bar charts and non-linear regression curves illustrate a significant, dose-dependent inhibition of cell proliferation in both HT-29 and DLD-1 cell lines after treatment. While compound 3a exhibited high potency across both lines, HT-29 cells showed greater sensitivity with an IC50 of 3.7 µM, compared to 7.2 µM in DLD-1 cells. Statistically significant differences compared to the control group are indicated as ns: non signifant and **** p < 0.0001.
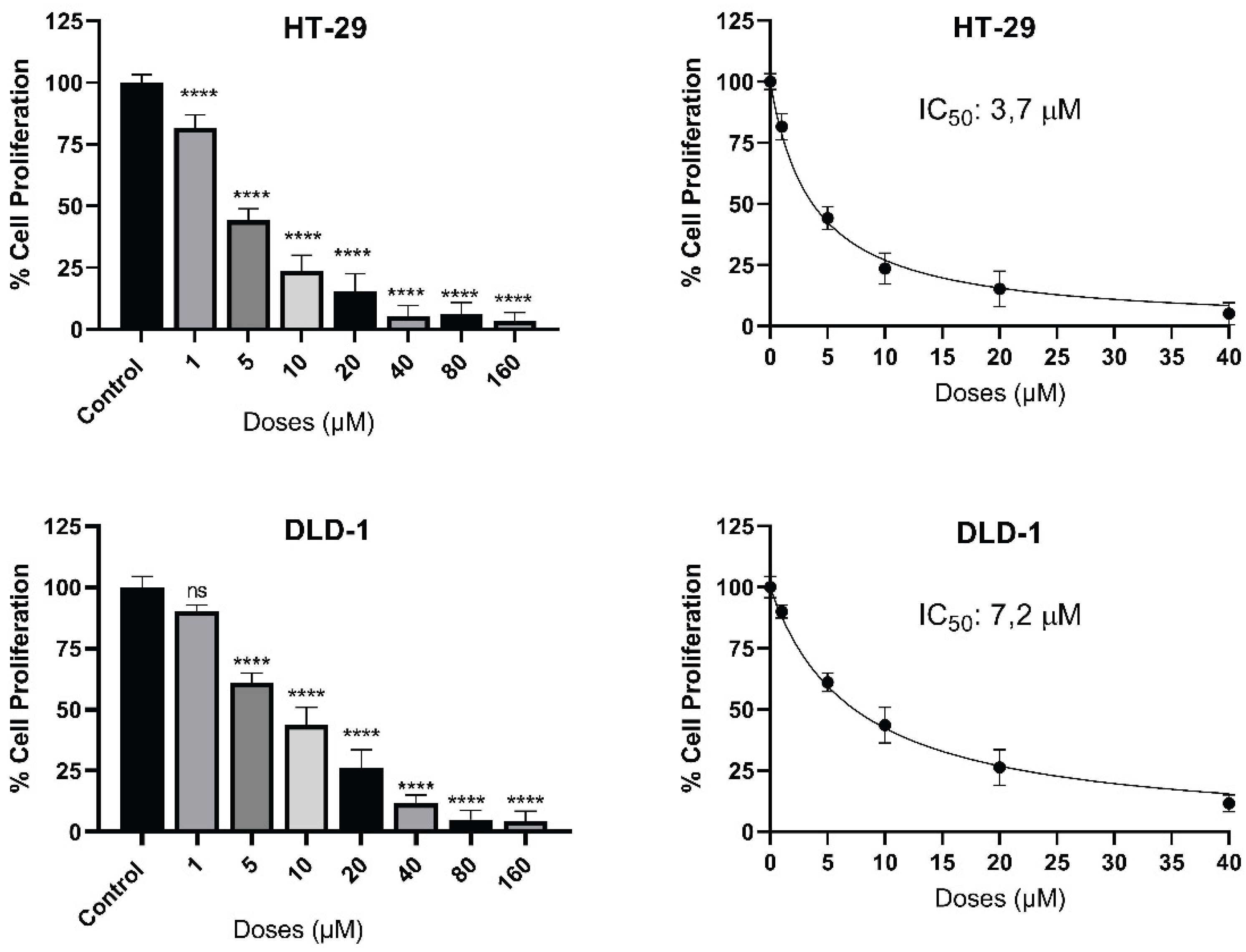
Figure 2.
3a treatment enhances LC3 expression and induces punctate LC3 accumulation in colorectal cancer cells. Representative immunofluorescence images of LC3 staining in HT-29 and DLD-1 colorectal cancer cells under control conditions and following 3a treatment. Low-magnification images and corresponding high-magnification (zoom) views illustrate LC3 localization patterns. Quantitative fluorescence analysis revealed a significant increase in LC3 mean fluorescence intensity in both HT-29 and DLD-1 cells upon 3a exposure compared to controls (****p < 0.0001). Notably, 3a-treated cells exhibited prominent punctate LC3-positive structures within the cytoplasm (indicated by arrows), in contrast to the predominantly diffuse LC3 distribution observed in control cells. Nuclei were counterstained with DAPI. Scale bar: 100 µm.
Figure 2.
3a treatment enhances LC3 expression and induces punctate LC3 accumulation in colorectal cancer cells. Representative immunofluorescence images of LC3 staining in HT-29 and DLD-1 colorectal cancer cells under control conditions and following 3a treatment. Low-magnification images and corresponding high-magnification (zoom) views illustrate LC3 localization patterns. Quantitative fluorescence analysis revealed a significant increase in LC3 mean fluorescence intensity in both HT-29 and DLD-1 cells upon 3a exposure compared to controls (****p < 0.0001). Notably, 3a-treated cells exhibited prominent punctate LC3-positive structures within the cytoplasm (indicated by arrows), in contrast to the predominantly diffuse LC3 distribution observed in control cells. Nuclei were counterstained with DAPI. Scale bar: 100 µm.
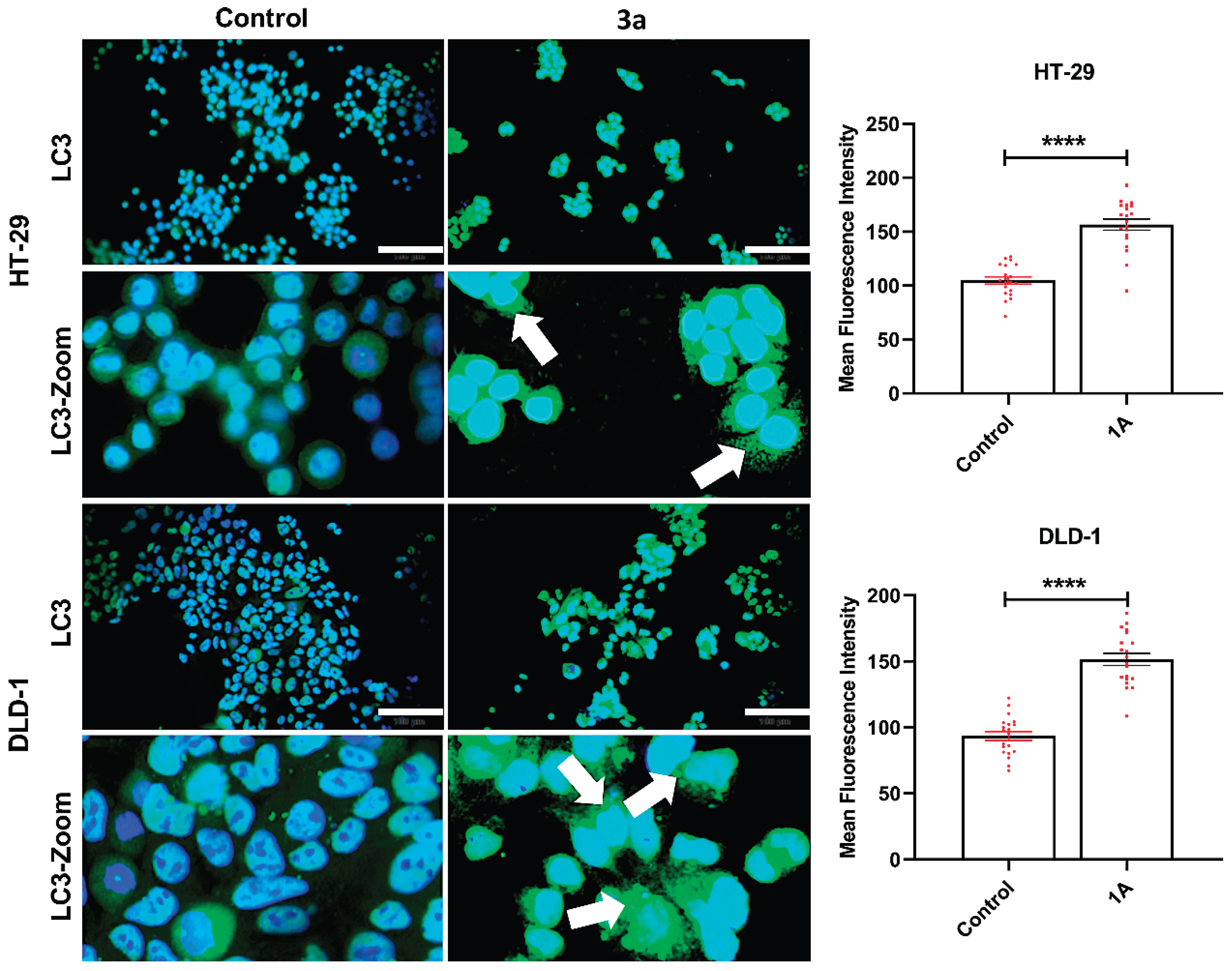
Figure 3.
3a treatment reduces mTOR fluorescence intensity in colorectal cancer cells. Representative immunofluorescence images depicting mTOR expression in HT-29 and DLD-1 cells under control conditions and following 3a treatment, shown at low magnification and corresponding zoomed-in views. Quantitative analysis demonstrated a significant reduction in mTOR mean fluorescence intensity in both colorectal cancer cell lines upon 3a treatment compared to control cells (****p < 0.0001). The decrease in mTOR signal intensity was consistently observed across independent experiments. Nuclei were counterstained with DAPI. Scale bar: 100 µm.
Figure 3.
3a treatment reduces mTOR fluorescence intensity in colorectal cancer cells. Representative immunofluorescence images depicting mTOR expression in HT-29 and DLD-1 cells under control conditions and following 3a treatment, shown at low magnification and corresponding zoomed-in views. Quantitative analysis demonstrated a significant reduction in mTOR mean fluorescence intensity in both colorectal cancer cell lines upon 3a treatment compared to control cells (****p < 0.0001). The decrease in mTOR signal intensity was consistently observed across independent experiments. Nuclei were counterstained with DAPI. Scale bar: 100 µm.
Figure 4.
The effect of compound 3a on apoptosis induction colorectal cancer cells. HT-29 and DLD-1 colorectal cancer cells were treated with compound 3a at the respective IC50 concentrations for 48 hours. The percentage of apoptotic cells was determined by flow cytometry using Annexin V-FITC/PI double staining. Data are presented as mean ± SD of three independent experiments. Statistically significant differences compared to the control group are indicated as *p < 0.05 and **p < 0.01.
Figure 4.
The effect of compound 3a on apoptosis induction colorectal cancer cells. HT-29 and DLD-1 colorectal cancer cells were treated with compound 3a at the respective IC50 concentrations for 48 hours. The percentage of apoptotic cells was determined by flow cytometry using Annexin V-FITC/PI double staining. Data are presented as mean ± SD of three independent experiments. Statistically significant differences compared to the control group are indicated as *p < 0.05 and **p < 0.01.
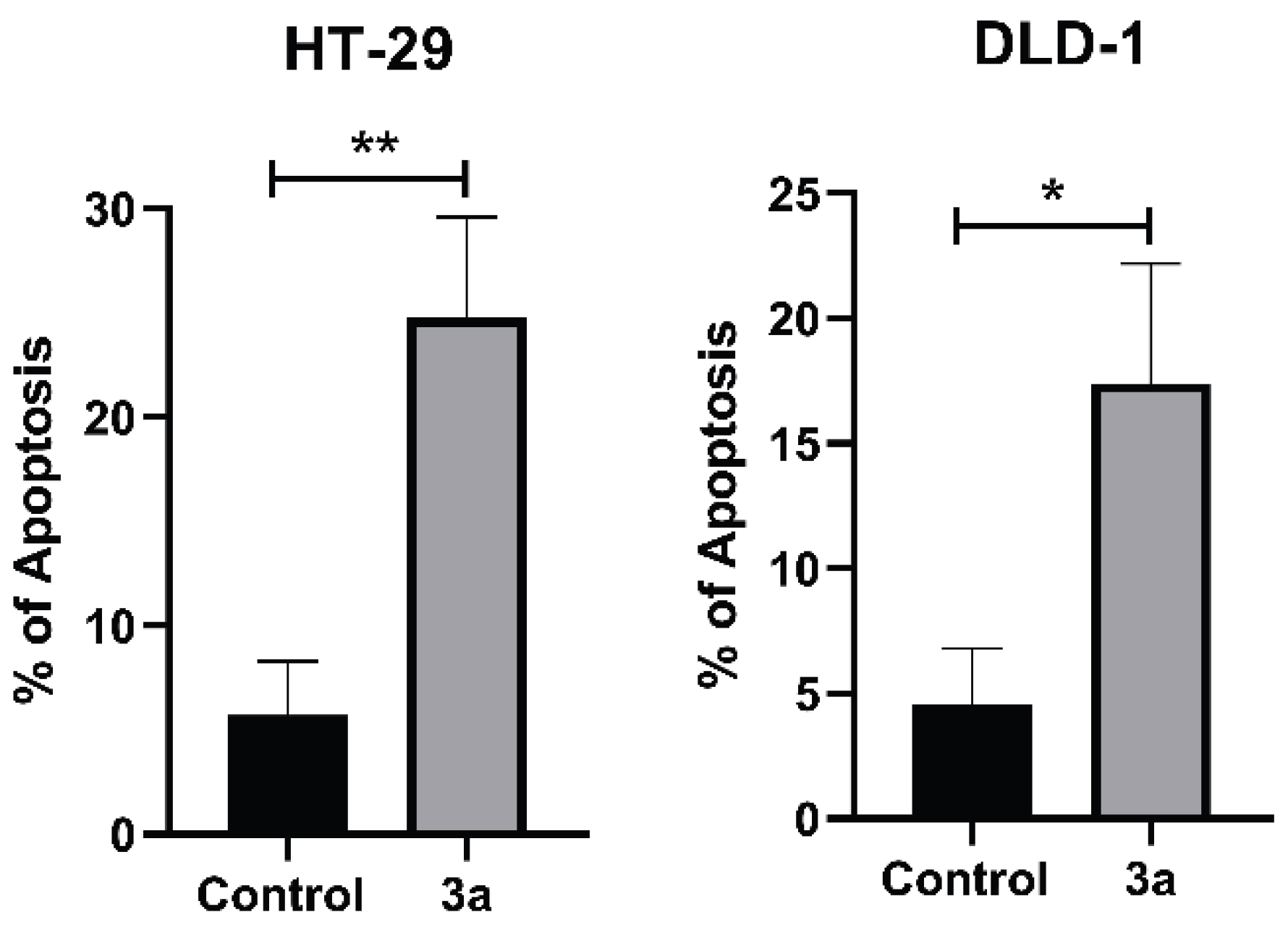
Figure 5.
Treatment of CRC cells with 3a decreased migratory, colonization, and adhesion characteristics. A) Comparative analysis of the inhibitory effects of compound 3a on the cell migration ability of HT-29 cells revealed that wound closure was not completed in the treatment group compared to the control group. This finding supports the strong potential anti-metastatic effect of 3a. In the study, wound widths were analyzed using Image J. B) A statistically significant decrease in the adhesion rate was observed between the control and treatment groups of HT-29 CRC cells treated with compound 3a. C)It is noteworthy that the treatment group showed a significant decrease in colony count in HT-29 cells compared to the control group. Data are presented as mean ± SD of three independent experiments. Statistically significant differences compared to the control group are indicated as ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
Figure 5.
Treatment of CRC cells with 3a decreased migratory, colonization, and adhesion characteristics. A) Comparative analysis of the inhibitory effects of compound 3a on the cell migration ability of HT-29 cells revealed that wound closure was not completed in the treatment group compared to the control group. This finding supports the strong potential anti-metastatic effect of 3a. In the study, wound widths were analyzed using Image J. B) A statistically significant decrease in the adhesion rate was observed between the control and treatment groups of HT-29 CRC cells treated with compound 3a. C)It is noteworthy that the treatment group showed a significant decrease in colony count in HT-29 cells compared to the control group. Data are presented as mean ± SD of three independent experiments. Statistically significant differences compared to the control group are indicated as ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
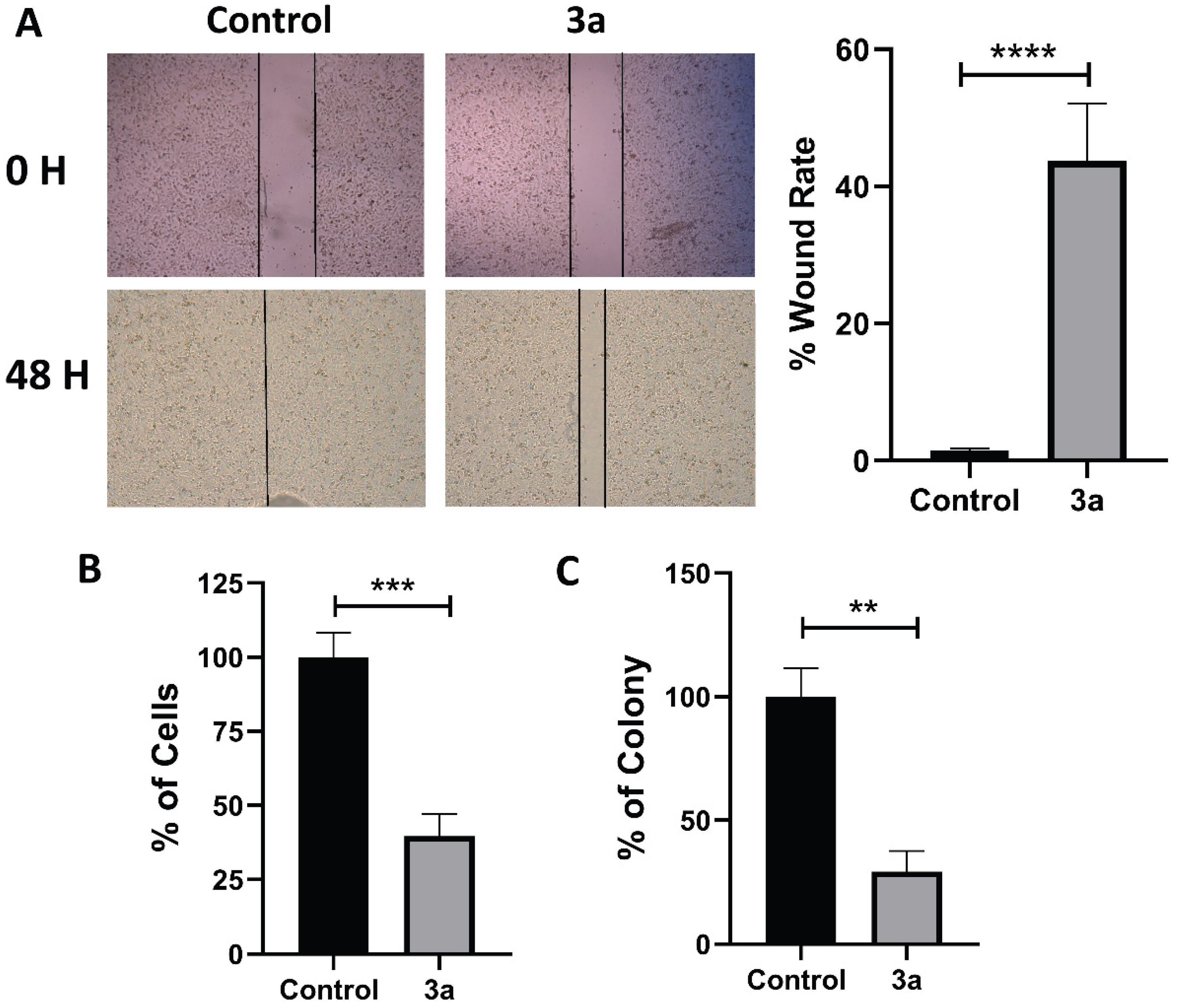
Figure 6.
Gene expression profile of HT-29 cells treated with compound 3a. A) mRNA expression levels of key genes involved in apoptosis (BAX, BCL2, Caspase-3), autophagy (mtor, BECN1), survival signaling (AKT, Stat3), and adhesion (VIM, ITGβ1) were analyzed by RT-qPCR after 48 hours of treatment with compound 3a. GAPDH was used as the housekeeping gene. Fold change values are shown with the control group fixed at 1. Significant downregulations were observed in the AKT/mTOR/STAT3 axis and adhesion markers, while pro-apoptotic markers were significantly upregulated.B) Schematic representation of altered gene expression in HT-29. Drawn by artificial intelligence based on information from the literature, then revised and checked before being added.
Figure 6.
Gene expression profile of HT-29 cells treated with compound 3a. A) mRNA expression levels of key genes involved in apoptosis (BAX, BCL2, Caspase-3), autophagy (mtor, BECN1), survival signaling (AKT, Stat3), and adhesion (VIM, ITGβ1) were analyzed by RT-qPCR after 48 hours of treatment with compound 3a. GAPDH was used as the housekeeping gene. Fold change values are shown with the control group fixed at 1. Significant downregulations were observed in the AKT/mTOR/STAT3 axis and adhesion markers, while pro-apoptotic markers were significantly upregulated.B) Schematic representation of altered gene expression in HT-29. Drawn by artificial intelligence based on information from the literature, then revised and checked before being added.
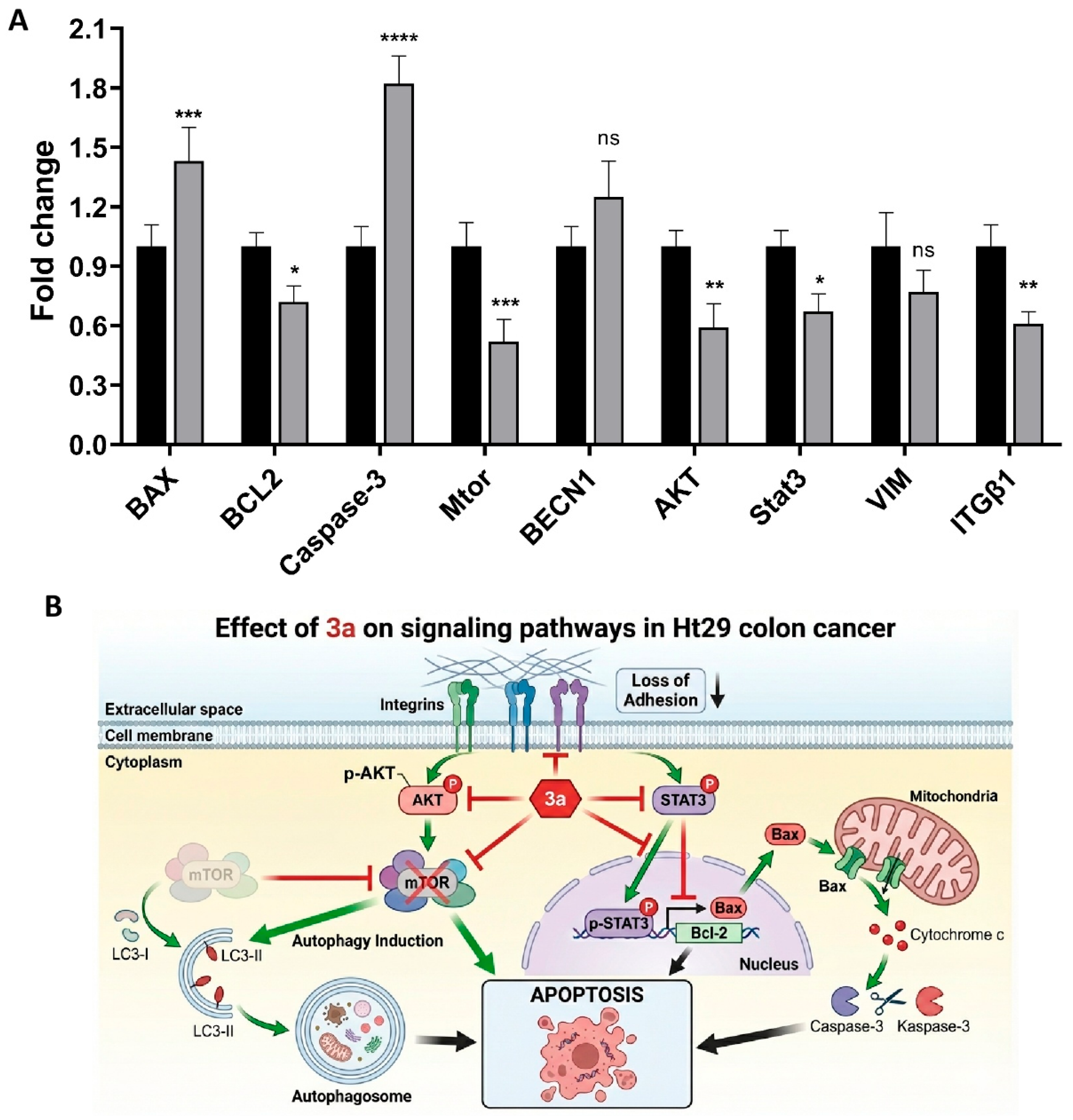

Table 1.
IC50 values of compounds at 72 hours in 4 different cancer cell(Breast and Colon) types.
| IC50 (µM) | ||||
|---|---|---|---|---|
| MDA-MB-231 | 4T1 | HT-29 | DLD-1 | |
| 3a | 8,2 | 6,4 | 3,7 | 7,2 |
| 3b | 8,7 | 10,2 | 28,6 | 43,4 |
| 4a | 100< | 100< | 100< | 100< |
| 4b | 100< | 100< | 100< | 100< |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



