Submitted:
05 February 2026
Posted:
06 February 2026
You are already at the latest version
Abstract
The review presents the first attempt to build the virtual world of free-radical polymerization of vinyl monomers itself and in the presence of small additives of stable radicals in due course of extended virtual experiment. Chain-reaction essence of the chemical process; quantum-chemical concept of elementary reactions; spin theory of radicals; and digital-twin of their presentation lay the foundation of the world formed, which opens a new vision of the role of IT technologies for the virtualization of various chemical processes
Keywords:
virtual free-radical polymerization
; digital twins
; spin theory of radicals
; energy graphs of elementary reactions
; thermodynamic and kinetic descriptors
; polymerization passports
; vinyl monomers
; stable radicals
; fullerene C60
1. Introduction – the Metaverse and the Virtual Worlds of Science
“Over the entire year of 2020, the word ‘metaverse’ was searched less than 5,000 times in Yandex, but in November 2021, the Russian search engine recorded almost 76,000 such queries. The surge was associated with the announcement of the Meta project, launched on the Facebook platform. Many commentators labeled this event a ‘rebranding,’ devaluing its true meaning. In fact, the Greek word ‘meta’ symbolically launched a technological stage that has so far been little understood.” This is how the first section of V. Shabason and S. Malaikin’s book, “The Intelligent Metaverse: From Digital Apps to a New Habitat” [1] begins. It is echoed by the second book on this topic, M. Ball’s “The Metaverse: How It’s Changing Our World” [2]. Both books were published within a week of each other in July 2022 and immediately became bestsellers. The process of metaverse entering in human being’s life proved extremely rapid, and today, the bibliometrics of metaverse research is growing exponentially [3].
Both the Russian and American authors of the books mentioned above convincingly demonstrate that the advent of the metaverse is inevitable and that, unlike previous technological stages of community development, the metaverse is not just another stage of development, but rather a new meaning for the existence of human communities, which could be characterized by the word “hyperrealization” [1]. The metaverse is a space that creates opportunities for the latter. According to the authors, in the metaverse, the boundary between virtual and physical realities will disappear, and information will acquire an energetic equivalent. The metaverse does not yet exist. If it does appear, it will be singular. The authors believe [1,2] that its emergence is a matter of the next 25-30 years.
To understand how the above introduction relates to the stated topic, let us, along with the authors of the book [1], follow the chain of events preceding the establishment of the metaverse. Despite its absence, the metaverse has already been announced. However, what is commonly referred to today by this word is not yet a metaverse. The authors propose classifying Facebook, Microsoft, NVIDIA, Roblox, and so on as quasi-universes, a prelude to the emergence of a meta-universe. These are isolated islands of a new way of being, slowly rising from the receding maters of the old world. A new environment for the world’s existence, enabling a new way of community self-organization.
Quasi-universes are preceded by digital ecosystems that have become an everyday reality [1]. Perhaps everyone can name two or three large businesses that have decided to go beyond their usual boundaries to actively explore adjacent territories using digital technologies. Digital ecosystems are the result of the unification of digital products into larger entities. They represent a new way of life. Digital ecosystems have become a familiar part of the modern business landscape. There are many thousands of them. It is obvious that this process cannot but affect science.
An analysis of the history of scientific development convincingly leads us to the conclusion that, in this chain from the object of digital technology application to the metaverse, science can lay claim to a place as an ecosystem [4]. Indeed, both applied science, little distinguishable from real business, and pure science, anti-business in its goal-setting, are addressed to a large number of users and, throughout their development, have actively expanded beyond conventional boundaries and explored adjacent territories using digital technologies, from the simplest “accounting” to digital twins [5,6]. It is enough to look at structural chemistry and crystallography. Both of these branches of materials science are aimed at studying the atomic structure of real substances, and the first move beyond the realm of reality was marked by the recording of a chemical formula corresponding to the composition of the substances being studied. Actually, a C6H6 molecule or a corpuscle with the compound formula SiO2 do not exist. In reality, we are dealing with a colorless liquid with a pungent odor in the first case and admiring the sand on the seashore in the second. But today, materials scientists around the world know that when the above formulas are mentioned, they are referring to benzene and silica. Thus, chemical formulas became the first digital twins of substances. As analytical techniques improved, formulas were gradually replaced by tables containing specific digital information about the substance, including the periodic table numbers of the constituent atoms, the number of atoms of each type, and the geometry of the atoms’ arrangement in three-dimensional space. The digital twin matured and gradually transformed into vibrant, beautiful images, such as a truncated icosahedron representing the C60 fullerene molecule. Thus, a virtual world of visual images of corresponding digital twins was born. And today, a structural chemist or crystallographer, having received a set of digital data corresponding to the interaction of, for example, a beam of elementary particles with a substance of interest, plunges into a digital abyss from which there is no escape without digital twins of the object. At the dawn of structural chemistry and crystallography, no one could have imagined that chemical formulas and the still isolated digital twins built on them would mark the first steps of materials science into the metaverse.
The philosophical and analytical view of the metaverse by V. Shabason and S. Mileikin [1] receives strong support from the highly professional information technology practitioner M. Ball. As M. Ball writes, “the metaverse is a scalable and interoperable network of 3D virtual worlds, rendered in real time, that can be accessed synchronously and continuously by a virtually unlimited number of users with an individualized sense of presence and continuity of data such as identity, history, rights, objects, communications, and payments” [2]. And although Ball’s book takes us further into the problems of the technological implementation of the metaverse and the assessment of the costs it generates for humans on a commercial basis, Ball, like Shabason and Mileikin, speaks of virtual worlds that lie at the foundation of the metaverse and without which it is impossible. “Stylistically, virtual worlds,” continues M. Ball, “can accurately reproduce the real world (be so-called digital twins) or represent an artistically imagined version of reality.” Thus, while depicting the future of the world as the implementation of a 3D version of the Internet and information technologies as such (M. Ball) or as the habitat of homo informaticus – a new human species capable of solving problems inaccessible to homo sapiens (V. Shabason and S. MIleykin), all authors unanimously point to virtual worlds as the foundation of the coming transformation. In the hierarchy of stages of metaverse development, the creation of virtual worlds is assigned a place in ecosystems, placing them in the place of the main task. In light of this, the main direction that modern science will take becomes obvious – the creation of virtual worlds for each of its branches. And although such a formulation of the question today may seem unrealistic, it is impossible to cancel the advent of the metaverse and the objective laws of its development. Therefore, there is simply no other path for science.
One cannot but agree with the authors of the book [1] that the metaverse is a child already born and that homo informaticus already exists. The above example of structural chemistry and crystallography demonstrates that the development of these branches of science has consistently followed the goal of creating virtual worlds of the structure of matter. The development of new technical capabilities that facilitate the exploration of adjacent territories using digital technologies is accompanied by an increase in the complexity of the virtual worlds being created, and, therefore, gives rise to new scientific problems and new virtual worlds. The logic of the development of virtual worlds foresees dead-end situations [1], in which virtual worlds become unstable. Mandatory adherence to the objective laws of development will lead this system either to self-destruction or to transformation into previously uncharacteristic forms.
This review presents the first result of constructing a virtual world within a digital ecosystem—polymer chemistry. One of the computational platforms of quantum chemistry is used as the digital technology. The subject of this study is the free-radical polymerization of vinyl monomers.
2. Digital Twins as the Basis for Building A Virtual World
2.1. Changing the Concept of Modeling
Digital twins (DTs) burst into our lives in 2002, from the podium of the Society of Manufacturing Engineers conference, voiced by Michael Greaves [7]. Although the term itself had been proposed much earlier [8], it was Greaves who imbued it with the new meaning that DTs represent today. Greaves proposed DTs as a conceptual model underlying product lifecycle management. Eight years later, John Vickers implemented this concept [9], which has since evolved significantly [10]. Since then, the concept has spread widely across industry, healthcare, construction, business, education, public life, and other fields [6]. The book broadly presents the successes of the DT concept, achieved in various areas of human activity.
While nuanced, the general concept of digital data concerns the trinity of a physical object, a virtual/digital object, and the connections between them. These connections are provided by data flowing from the physical object to the digital/virtual object, and by information accessible from the digital/virtual object to the physical environment. This concept, while new to large, massive areas of human activity, has been widely used in academic research since the first computers became available. Known as simulations or modeling, the concept has enabled radical advances in academic research, particularly in the natural sciences.
Today, it is impossible to imagine modern physics, chemistry (or the same sciences, but with the prefixes bio- and geo-), and material science without modeling. The enormous leap in the development of computing software and tools over the past half-century has led to many previously primarily empirical sciences becoming virtual-empirical, and some, such as graphene science, are primarily virtual. Despite such rapid development, the relationship between a real object and its model, established as subordinate to the model, remained unchanged until recently. As it turns out, this circumstance significantly limits the further development of science in its predominantly virtual nature. And here is where the concept of digital domain, as formulated above, comes into play. Indeed, physical and virtual objects form the basis of both modeling and the concept of digital domain. The difference between these two approaches lies in the different meanings inherent in understanding the relationship between them. Using grammatical terms, this distinction can be thought of as the distinction between a complex sentence (main-subordinate) in the first case and a compound sentence (equal-equal) in the second. In every language, replacing one sentence with another changes the meaning of spoken language. The same is true in science. It is precisely this change in semantic speech, which the concept of digital twins brings to science, that transforms explanatory subordinate modeling into a free, legitimate virtual experiment. The first virtual experiments as legitimate participants in the research process took place and revealed their special and significant place in the technological sphere [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29]. However, the pioneer of the digital twin concept, Greaves, said in one of his interviews given in 2018 [7]: “It doesn’t necessarily have to be an all-or-nothing project. There is a wide range of information that I can collect and process using a double. Digital twins can also be used in very specific, very limited scenarios.” Such limited scenarios mark the beginning of legitimate virtual experiments in science [30,31,32,33,34,35,36,37,38,39,40,41,42]. But these first steps already showed that the contrast between the concept of a complex DT experiment and complex modeling reveals the high efficiency of the former for solving complex problems.
2.2. Computational Platform
The general algorithm for a virtual experiment, as applied to one of the limited scientific research scenarios, can be schematically represented as [6]
Digital Twins → Virtual Device → IT Product.
Diagram 1
Here, the digital twins are the digital models being studied, the virtual device is the carrier of the selected digital technology, and the IT product encompasses a wide range of computational results associated with the digital twin under various influences within the applied computing programs. The quality of the IT product largely depends on how broadly and deeply the selected digital twins encompass knowledge about the object under study and how adequately the virtual device matches the characteristics of that object. The severity of the first requirement can be significantly mitigated by a large set of corresponding digital twins. Digital twins are completely free from the statistical and random errors that accompany real experiments and are conducive to any modification. Given control of the virtual device, the digital twins lay the foundation for a free, independent virtual experiment concerning the process of interest.
Obviously, the choice of virtual device must be consistent with the set of selected digital twins and must ensure their reliable mathematical processing in order to obtain a reliable IT that answers the experimental questions. Regarding the latter, it represents one of the variants of Big Data (BD) sets, the comparative analysis of which presupposes the identification of regular trends in the behavior of the object of study. Naturally, the subject content of the selected DTs, virtual device, and BD set of the IT product depends on the object of study and the questions posed to it, which is well represented in experiments on virtual molecular dynamics [30,31,32,33], virtual graphenics [34,35,36], and virtual polymerization [37,38,39,40,41,42]. Below in this review, this issue will be considered in detail in relation to free-radical polymerization.
3. Fundamentals of A Virtual Free-Radical Polymerization Experiment
3.1. A General Concept of the Experiment
Computational polymer chemistry began as classical modeling over 60 years ago and is currently one of the most developed areas of chemical virtuality. Over the years, polymer modeling has come a long way, from the microscale representation of the problem to the intermediate, mesoscale, and macroscopic scales, which have served as the foundational models for quantum chemistry (QC) of polymers [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58], molecular dynamics [59], coarse-grained [60], and continuum concepts [53,60], respectively. While radically different in content, all concepts are nevertheless based on a common concept of polymers, which are directly modeled as a sequence of points in three-dimensional space. The aforementioned reviews and references provide a reliable overview of the current state of virtual polymer chemistry. At present, they need to be supplemented by the DT concept [12,16,26] and quantum computing [61].
Despite the significant contribution of computational chemistry to polymer science, few truly resolved issues remain. The high cost of calculations on models comparable to real systems, the limited capabilities of existing computational programs, and the lack of sophisticated software that simultaneously incorporates deterministic approaches such as thermodynamics, kinetics, and fluid mechanics undoubtedly constitute serious reasons for this fact. However, the exceptional complexity of a polymer object, the end product of multifaceted and multidimensional intermolecular interactions involving a large number of participants, renders the final material product the result of a choice among a vast number of possibilities. Modern traditional computational chemistry is unable to predict this choice. At best, the combination of all approaches, from micro to macro, allows one to explain the choices made either by Nature or by magical chemists, owing to their vast experience and intuition. However, as it turns out, not only the global concept of DTs, but even its partial modification within the framework of traditional quantum chemistry, allows to solve a number of problems that were previously inaccessible [34,35,36]. This circumstance prompted the author to turn to the QC of polymers from the perspective of the DT concept. It is quite logical that the first test of this new paradigm was made in favor of a question well studied experimentally and repeatedly considered virtually in traditional computational chemistry, namely, free-radical polymerization (FRP) of vinyl polymers.
This review presents the first description of a virtual world of free-radical polymerization, compiled on the basis of an extensive virtual experiment [37,38,39,40,41,42] consisting of individual experiments carried out under identical conditions. The overall design of the experiment is based on a number of fundamental concepts, including the following. The pilot concept is to represent the polymerization process as a chain reaction [62,63], including a set of elementary reactions. The latter are considered as independent and superpositional, which makes it possible to use all the accumulated experience of free-radical polymerization in considering individual chemical reactions [64,65,66]. Both the initial reactants and the final reaction products form a pool of DTs, which are the main objects of the virtual experiment. To transform the latter into digitalization tools, two types of descriptors are introduced: thermodynamic and kinetic, which are equally characteristic of all elementary reactions. The unification of descriptors allows for the introduction of polymerization passports, which are a “personal identification document” for each virtual reaction solution (VRS) and provide a potential comparison of all virtual characteristic features of the polymerization event being studied with empirical reality.
3.2. Dramatis Personae
The VRS of vinyl monomers [67,68] underlies the production of the vast majority of modern polymer products. Typically, the participants in this process are quite numerous, the main ones being monomers (), polymerization initiating free radicals (), monomer radicals (), continuously growing oligomer radicals (), and solvent. Together, these products define the actual reaction solution in which the reactions take place. All participants are linked by intermolecular interactions, leading to the formation of a large number of different intermolecular complexes. Thus, the interaction of a monomer with free radical leads to the formation of a monomer radical, whose interaction with the monomer underlies the growth of oligomer radicals, which constitute the polymer chain (polymerization propagation). The interaction of the latter with each other or with a monomer radicals, as well as with free radicals, determines the cessation of polymer chain growth. The interaction of terminated chains determines the formation of the bulk of the polymer material. The outcome of each of these elementary processes is greatly influenced by the interaction of their components with solvent molecules [69].
The real polymer product is determined by the simultaneous occurrence of all these events. Accordingly, a VRS as a DT container becomes the object of study in the virtual experiment. Since the DT concept works best when identifying trends, the virtual experiment considers a wide variety of VRS. A large amount of empirical data related to the FRC of vinyl monomers [67,68], as well as to their free-radical copolymerization (FRCP) with stable radicals such as and C60 fullerene [69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88], significantly facilitates the VRS modeling. As it turned out, real reaction solutions in all cases consist of a similar set of ingredients, including a solvent, a monomer, an initiator as a source of free radicals, and small additions of stable radicals. Obviously, each of the ingredients can vary significantly, further enhancing the variation with different concentrations. Therefore, the variation of possible VRS is large. This circumstance significantly complicates the selection of a limited number of compositions sufficient for the reliability of the identified trends, on the one hand, and the verification of these trends by comparison with experimental data, on the other. In light of this problem, when implementing a virtual experiment [37,38,39,40,41,42], attention was paid to a particular group of real experiments on the FRC of vinyl monomers and their FRCP with stable radicals [85,86,87]. In these studies, special attention was paid to the kinetics of the initial stages of the process, which was presented in terms of the time-dependent percentage consumption of the monomer . The experiments were carried out in the same solvents, at the same temperature, in the same reactor, that is, under virtually identical conditions, including monomers, initiators, and stable radicals, while the concentrations of these ingredients were strictly fixed. Based on these experiments, a set of VRSs was constructed [37,38,39,40,41,42] in which the monomer is one of three vinyls, namely, styrene, methyl methacrylate, or N-isopropylacrylamide (); the initiator is either the alkyl nitrile 2,2′-azobisisobutyronitrile () or benzoyl peroxide (), and the impurity stable radical is either or C60 fullerene, or both.
3.3. Elementary Reactions
The game-field in the experiment under discussion represents polymerization as a clearly traceable sequence of superposed elementary reactions. From a computational perspective, this view of polymerization is most favorable for the use of QC methods for its virtual analysis, reducing it to the consideration of individual elementary reactions [64,65,66]. The theory of elementary reactions and their QC analysis have existed for many decades [89,90,91]. The primary novelty of the DT concept concerns precisely this key point, since the large array of elementary reactions, which constitutes the experiment game-field, allows for clear distinctions between similar reactions conducted under identical conditions, with subsequent comparative analysis of their results, accompanied by the establishment of reliable trends. The “same conditions” status implies the use of the same QC tool, absolute zero temperature, and a vacuum environment.
A fairly complete list of elementary reactions associated with the events under study is presented in Table 1. The nominations presented in the table concern both the reactions and their final products. As can be seen, the number of the latter is quite large, so the digitalization of the chemical process under consideration appears quite convincing. A common characteristic of the reactions listed in the table is their radical nature. Reactions (1) and (2), combining a free radical with the monomer and its oligomers , clearly control the monomer’s FRP. The former is the cornerstone of the entire polymerization process, determining its feasibility. Selecting the most successful participants in this reaction empirically, the researchers settled on the free radicals and , obtained during the thermal decomposition of the initiators and , respectively. Stable radicals, such as TEMPO, proved unsuitable for this role, since the reaction (3), similar to reaction (1), always ends with the formation of a normal non-radical intermolecular -monomer complex. Only fullerene C60 occupies a special place among stable radicals. The reaction (4) is analogous to the reaction (3), however, the resulting monofullerenyl , unlike , is also a stable radical. The first wave of researchers who, being confident in the radical nature of fullerene, introduced it into polymerization, made repeated attempts to detect the reaction (5), accompanied by the formation of a monomer radical and the subsequent growth of a polymer chain initiated and added to fullerene (reaction (6)). Although the existence of reaction (5) was assumed in a number of cases, no unambiguous conclusion was made, and this reaction, like reaction (6), was classified as unlikely. The virtual experiment discussed made it possible to revise the experimental data and reliably establish the presence of both reactions at a certain composition of the VRS [41].
- Table 1. Nomination of elementary reactions and/or digital twins related to the initial stage of free radical copolymerization of vinyl monomers with stable radicals.
1) , denote DTs related to vinyl monomers, the initiating free radical, the C60 fullerene, and another stable radical, respectively. The superscript black dot denotes the radical molecule.
In contrast to the unreactive monomer, the capture of its monomer radical , described by the reactions (7) and (8), is traditionally highly expected for both stable radicals. These reactions are of particular importance because they have the potential to completely stop the polymerization process. These are followed by the reactions (9) and (10), representing the capture of free radicals by stable ones. Both reactions obviously influence the polymerization of the monomer by reducing the number of initiating free radicals. The reaction (11) takes into account the interaction of stable radicals with each other, while the reactions (12) and (13) describe the capture of the monomer radical by stable ones. This set of elementary reactions turned out to be quite complete for considering the initial stage of both the FRP of vinyl monomers and their FRCP with stable radicals. The corresponding DTs of final products, together with the starting ingredients, form a large pool of DTs, quite sufficient for digitalization of the chemical process under consideration.
3.4. Virtual Device
All quantum chemists working with the computational polymer chemistry of vinyl monomers recognized the radical nature of its participants, and discussions about the choice of appropriate software tools have been at the forefront throughout the long-term development of computational polymer chemistry (see reviews [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60] and references therein). The specifics of the QC description of stable radicals and open-shell electron systems have been repeatedly raised in relation to virtual polymer science [92,93,94,95]. However, current circumstances are such that the proportion of calculations using configuration interaction (CI) methods, even at the first level of complexity, in virtual polymer science is comparatively small. Justifying the long timeframes and high financial costs of such calculations, practicing quantum chemists have chosen various versions of the DFT technique, including unrestricted versions, as their primary calculation method. Unfortunately, both DFT and UDFT algorithms do not work in systems with open electron shells (see a detailed discussion of the problem in [96,97]). Accordingly, the numerous articles regularly appearing devoted to virtual DFT-based FRP do not advance us towards truly significant results in this area of science. In the virtual experiment [37,38,39,40,41,42], this aspect of FRP was considered self-consistently for the first time. The spin theory of radicals [98] served as the basis for the applied digital technology. The virtual device was a package of computational programs for the electronic properties of open-shell molecules, implementing a quantum chemical calculation based on the two-determinant (unrestricted) Hartree-Fock approximation (UHF). Each elementary reaction was considered through the prism of the spin density algorithm for step-by-step consideration of successive intermolecular chemical addition reactions [98], allowing for the confident determination of the target atoms of the reactants participating in the reaction. The competence of the applied QC covers several stages of radical polymerization, including the primary initiation of the process, the first steps of growth of a linear polymer chain and the act of terminating this growth.
The virtual device in the experiment was represented by the CLUSTER-Z1 software [99,100], which implements the AM1 version of the semi-empirical UHF [101]. The program has proven highly effective for open-shell electron systems such as fullerenes [102], graphene molecules [103], and stable radicals [104].
3.5. Descriptors of Virtual Polymerization
The main goal of the polymerization digitalization is to evaluate the properties of a chemical process numerically. To realize this desire, it is necessary to construct in silico models that establish a mathematical relationship between the structures of molecules and the properties under consideration. Molecular descriptors play a fundamental role in solving this problem, since they are formally the numerical representation of the molecular structure [105]. In the concept of a chain reaction, each polymerization process is the result of intense competition between bimolecular elementary reactions determined by their rates. Burdened with a large number of such reactions not only at the initial stage of the process, presented in Table 1, but also many others throughout its development, any outcome of this competition resembles a “pineapple-on-a-plantation” solution. The fate of a monomer (see Figure 1), caught in the field of interaction of numerous participants, can be figuratively represented as the state of a pineapple on a plantation, surrounded by sword-shaped leaves. Each leaf is one realized path of development in the corresponding direction. A large number of others are hidden within the individual scales of the fruit’s bark. Each step along this path is governed by the well-known energy graph shown in the figure. According to the graph, each elementary reaction occurs between a pair of reactants, the energy of intermolecular interaction between which, , follows a typical format as they approach each other in a chosen direction, , which represents the reaction coordinate. As can be seen from the figure, the graph relates the total energy of the reactants of the initial pair Y, , the energy of the final product of the interaction of the components of the pair X, ), and the transition state energy of the intermolecular complex under consideration, , which determines the height of the reaction’s energy barrier. Another important energy parameter is the enthalpy of reaction, , or the binding energy, =-. Both quantities are directly related to the atomic structure of the object and can rightfully be considered molecular descriptors. In the experiment conducted [37,38,39,40,41,42], plays this role of a virtual thermodynamic descriptor.
The execution of each of the swords surrounding the pineapple in Figure 1 and the opening of the windows on its cortex are controlled by the kinetics of the corresponding reactions, with the fastest of them emerging as the winner. Thus, reaction rate constants are unambiguous virtual descriptors of chemical reactions. The standard description of the rate constant is usually given by the Arrhenius relation [89,90,91]
Here is a complex entropy-frequency factor, and represents the activation energy, which is either the dissociation energy of product , , or the association energy of molecular pair , . The main difficulty in estimating the value of lies in the very complex nature of the factor [106]. Its definition touches upon complex problems of the rotational-vibrational dynamics of polyatomic molecules, complicated by a large number of both vibrational and rotational degrees of freedom, as well as anharmonicity of motion. However, for similar elementary reactions, it is expected that varies little [64,65,66], so that the activation energy becomes decisive in their comparative analysis. Its value can be determined by constructing graphs, or barrier profiles, corresponding either to the association of components of molecular pairs , or to the dissociation of the final product . An alternative approach to obtaining the activation energy is provided by the approximate Evans-Polanyi-Semenov (EPS) equation, which linearly relates the energy to the reaction enthalpy or the binding energy [48,49,55]
Here, and α are statistical parameters that are approximately constant for a given set of reactants but different for different sets. The activation energies defined in this manner act as virtual kinetic descriptors in the experiment.
3.6. Transition State
The barrier profiles required to determine were calculated for the dissociative reaction, starting with the energy at the equilibrium covalent bond length between the reaction participants and continuing until this bond is broken, following its regular elongation. As an example of one of the many calculations performed for the experiment [37,38,39,40,41,42], Figure 2 shows the barrier profiles corresponding to the initiation reaction of the FRP (1) of styrene () and () monomers stimulated by the free radical [37]. The insets in the figure represent the equilibrium structures of the corresponding products in the ground and transition states. The reaction coordinate in both cases links the carbon atom of the vinyl group of the monomers with the carbon atom of the free radical
As can be seen from the figure, the values are quite close in both cases. This means that the dissociation kinetics of both radical monomers should be similar. In contrast, differs significantly, showing a more than twofold decrease in the case of compared to . This feature is obviously related to the difference in the values of these substances and is of particular interest, indicating that the initiation of free radicals is kinetically more favorable for vinyl monomers with a set of sp2 bonds than for monomers with a single such bond. This conclusion correlates well with experimental data indicating the same trend, regularly observed in relation to the kinetic rate constants of FRP initiation reactions [67,68].
A careful analysis of the transition states in both cases revealed a certain general pattern in the behavior of the latter, despite the radically different kinetics. Firstly, the reaction coordinate , corresponding to the maximum of the barrier profile, is noteworthy. In both cases, the values are located near 2.1 Å. The latter is close to the value 2.11 Å, which determines the onset of sp3bond cleavage in ethane [107]. It is known that the value of varies quite noticeably, reacting to changes in the atomic configuration around the bond [103] and providing a very pronounced dispersion, which amounted to ~ 0.10 Å for the participants in the experiment [37]. This fact forced us to look closely at the intermolecular contacts in the monomer radicals , which in the case of vinyl monomers are represented by the connecting intermolecular sp3 bond and the adjacent intramolecular sp2bond of the vinyl units of the monomers. As it turned out, the lengths of these bonds are closely interrelated. In Figure 3, the red line depicts the dissociation of the sp3 bond, which begins at the point (1.535;1.471), indicating that the intermolecular contact in the radical monomer is a combination of a standard-length sp3 bond and an elongated sp2 bond of the vinyl group. Due to the elongation the latter is radicalized [107], thereby providing unpaired electrons with a total number of = 1.925 e, which leads to the formation of the target atom C3 with = 0.67 e. The value of is consistent with the general law that governs the gradual radicalization of sp2 bonds shown in the inset of the figure. Similar to sp3 bonds, the onset of sp2 bond radicalization corresponds to ≅1.395 Å for ethylene, the value of which also disperses, responding to the change in the bond environment. For vinyl monomers, the dispersion value is estimated to be ~0.02 Å. The dispersion zones for the values for both bonds are represented by vertical and horizontal light gray stripes, corresponding to the sp3 and sp2 bonds, respectively.
The dissociation of is completed at the point (4.203;1.342) (not shown), passing through the transition state at the point (2.129;1.387). As can be seen from the figure, is located in the region where the lengths of both bonds are close to their . Judging by the values at this point and referring to the inset, it can be concluded that the transition state corresponds to the onset of radicalization of both bonds. In this case, the sp2 bond is only at the very beginning of its long path to cleavage, whereas the onset of radicalization of the sp3 bond signifies the beginning of its cleavage. Therefore, the energy peak designated as indicates the reaction coordinate corresponding to the onset of cleavage of the sp3 bond during dissociation of the monomer radical. The sharp threshold transition, visible in the energy graphs around , is provided by the spin density of the object and marks either the exit of the spin density from zero values (dissociation) or the zeroing of the spin density upon the formation of a standard single bond (association). It has been suggested that it is precisely the spin-driven nature of the transition state that makes it so universally significant for any chemical reaction [106]. Clearly, the transition state described above, based on a single sp3 bond, is also the simplest, which becomes significantly more complex in the case of reaction coordinates represented by double or triple bonds.
The above statement that the coordinate of the transition state is determined by the bond providing the intermolecular contact in the reaction product was confirmed in the calculations of numerous barrier profiles performed during the experiment [37,38,39,40,41,42]. Figure 4 summarizes the revealed trends [37,40]. The and profiles describe the dissociation of the methyl methacrylate dimer radical and the capture of its monomer radical by the stable radical, respectively. The connecting bonds of the intermolecular contacts in this case are sp3 and sp3 bonds. The lower panel of the figure shows the dissociation curves of these bonds in ethane and ethylene glycol, respectively. It is clearly evident from the figure that the change in the position of in the profiles under consideration follows the trend in the position of of the corresponding bonds. While the position of , corresponding to the sp3 bond in the profile, remains virtually unchanged when the monomer is changed, it changes dramatically when the monomer is replaced by fullerene C60 in the profile, thereby demonstrating the influence of the topochemistry of monomer F on its reaction with the initiating free radical.
3.7. Digital Polymerization Passports
The results of the virtual experiment discussed [37,38,39,40,41,42] were presented in a matrix-table format. This format allowed for the generation of a universal “digital polymerization passport” (DPP) for the studied VRS. The availability, high efficiency, and relative simplicity of the applied QC computational method allow for the generation of such a passport for any VRS related to the FRP and/or FRCP of vinyl monomers. The passports themselves, on the one hand, are strictly specific to each of the VRS ingredients. On the other hand, they represent an informative source of knowledge about the process under study. The first such document were issued for the FRP of methyl methacrylate and its FRCP with C60 fullerene and the stable radical [40]. Similar studies followed for styrene [41] and [42]. The DPPs presented in these experiments consist of two pages. The first page contains text information regarding the VRS under consideration, including the nomination of elementary reactions and the corresponding DTs, supplemented by the thermodynamic and kinetic descriptors and , respectively. The second page displays images of the equilibrium structures of the DTs under consideration. This form of the DPP proved to be quite convenient for testing digitalized predictions using available empirical data. The main results of the virtual experiment under discussion are presented below in this same form.
4. Virtual Free-Radical Polymerization of Vinyl Monomers
Of the large set of virtual free-radical polymerizations considered during the experiment, only those containing styrene (), methyl methacrylate (), and () as vinyl monomers were selected to present the main results. The choice of monomers was based on the number of vinyls or sp2 bonds in the monomer. For example, monomer contains 7 sp2 bonds, while monomers and contain only one such bond, represented by a vinyl group. It was shown that the spin density and energy characteristics of these two monomer types differ significantly. These differences were so big that they allowed us to present a fairly complete picture of the process under consideration. We begin our presentation of the obtained results with the simplest virtual free-radical polymerizations, containing only the above-mentioned monomers and the initiator. The description below concerns the initiation of virtual free-radical polymerization, polymer chain growth, and, to some extent, chain termination. The first two processes are described by the elementary reactions (1) and of Table 1. The chain termination is illustrated by the final products of the reactions , and + .
4.1. Initiation of Free-Radical Polymerization and Chain Propagation
A detailed description of the virtual reactions involving the three aforementioned monomers within the elementary reactions (1) and (2) and the resulting VPPs for the corresponding VRSs are contained in [37,40,41,42]. The bulk of the QC calculations concerned linear chains of oligomer radicals with the number of chain links and the total length of the ‘clothesline’ connecting the monomers in them, . In the experiment [37,38,39,40,41,42], the number of links reached 8, and the corresponding length was ~30 Å. This was made possible by the high efficiency of the semiempirical UHF approximation, which underlies the virtual device used.
Analysis of the obtained results revealed the following patterns.
Oligomeric radicals , starting with the monomer radical, are formed by successive addition of monomer units. The first RM⦁ unit is formed at the initiation stage of polymerization, while all others arise during the gradual propagation of the latter. All studied vinyl monomers form stable oligomer radicals characterized by significant . The spin density and energy parameters clearly differentiate the monomers by the number of sp2bonds involved, thereby dividing the latter into single-group and multi-group ones.
The structural composition of all -oligomer radicals follows the same formation algorithm and is built around a basic ‘clothesline’ consisting of intermolecular bonds, each of which is formed by the subsequent addition of a monomer through an sp3 bond, which, in turn, causes elongation of its remaining sp2 bond with the radical attacking target. The successive sp2→sp3 transformation of covalent bonds in the area of intermolecular junction, accompanied by the elongation of the clothesline, in combination with the resistance to the emerging steric hindrances, leads to a bizarre, non-linear contour of the rope.
The transition state, characterized by , is the central point that allows for the direct determination of . The obtained data presented with kinetic descriptors are summarized in Table 2, which concern fragments of the studied VRSs. The corresponding passport photographs related to these solutions are presented in Figure 5. were determined by constructing barrier profiles for two oligomer radicals, marked in bold in Table 2. The remaining values were obtained using the EPS relation (2).
The obtained profiles clearly distinguish between single-group and multi-group monomers, indicating different kinetics of the latter. Against this background of difference, the transition state reveals a certain common feature associated with the identical position of the profile maximum on the reaction coordinate axis, which is an intermolecular sp3bond of the intermolecular junction, coinciding with ≅2.1 Å in all the cases. Simultaneously with the critical behavior of this sp3 bond, the length of the adjacent intramolecular sp2bond of the vinyl group takes on a value of 1.387 Å, which coincides with , which marks the onset of radicalization of sp2 bonds upon elongation. The framing of the energy parameter by two critical values, related to covalent sp3 and sp2 bonds, simultaneously indicates the spin-determining nature of the transition state of vinyl radical oligomers.
4.2. Chain Break and Polymerization Termination
The trends discussed are related to the initiation and propagation stages of the polymer chain elongation process, while identifying similar trends for polymer chain braik events also requires the development of a specific algorithm. For the considered vinyl monomer chain termination processes without additional additives, this algorithm involves considering the simplest reactions: , + and + as well DTs , , and (see Table 2). Figure 6 shows several examples of DTs of this type. Formed by the combination of radicals, the new intermolecular complexes are neutral molecules with a closed-shell electron system, completely devoid of any radical properties, thus indicating that each of the listed elementary reactions actually halts both polymerization itself and polymer chain growth. The intermolecular complexes shown in the figure represent only a small fraction of the potential products in VRSs. The species are characterized by a significant spread of values, from -45 to -4 kcal/mol, and, correspondingly, values, ranging from 23 to 2 kcal/mol. A significant portion of the dispersion in these energies should be attributed to steric effects. However, there is no doubt that the path to victory in these complex processes is not straightforward, and therefore the development of a specialized algorithm to predict this path is required.
The above presents the results of the first part of the virtual experiment [37,38,39,40,41,42], related to the FRP of vinyl monomers as such. Like its real-world counterpart, the virtual experiment proved to be very informative, providing researchers with a new set of concepts and characteristics that allow to develop a comprehensive understanding of the processes governing polarization. In assessing the reliability of the virtual results, it is necessary to turn to the experimental data. The latter, concerning the three studied VRSs, indicate that the FRP of the vinyl monomers under consideration occurs reliably over a wide range of changes in the concentrations of the participating ingredients, solvents, and temperature conditions. This is fully consistent with the obtained set of thermodynamic virtual descriptors , indicating the energetic stability of all the resulting monomer and oligomer radicals. The kinetic descriptors in Table 2 indicate that the time characteristics of polymerization initialization in the VRSs under consideration are largely similar, so that differences could be expected only in small details. However, this difference in the actual kinetics of the process in different VRSs has so far remained undetected. For their part, the data presented in the table acquire the status of reference data that confirm the polymerization of vinyl monomers. Based on these data, one can observe changes in the polymerization kinetics caused by the introduction of additives into the VRSs under consideration.
5. Virtual Free-Radical Copolymerization of Vinyl Monomers with Stable Radicals
5.1. Brief Review of Empirical Observations
Naturally, both real RSs and VRSs with their associated free-radical polymerization are not indifferent to the addition of stable radicals. Two of the latter are the most popular among practicing researchers: monotarget and polytarget C60 fullerene [69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87]. In the first case, the entire radical power is concentrated on a single oxygen atom of the molecule, while in the second, the radical activity is distributed across all carbon atoms. As expected, these stable radicals participate differently in the FRP of vinyl monomers, so the associated free-radical copolymerization (FRCP) of these monomers proceeds in varied ways. Within the framework of the chain reaction concept, this difference is determined by the diversity of elementary reactions involved in the process. As can be seen from Table 1, the participation of stimulates reactions (3), (8), and (11), while the presence of fullerene C60 causes reactions (4)-(7), (9), and (12). Experimental observations fully justify these expectations and provide rich material for detailed study. This is best demonstrated in studies of the kinetics of the initial stage of the FRCP of vinyl monomers in the presence of these two radicals, presented in the form of several graphs in Figure 7.
The figure accumulates empirical data presented as time-dependent total conversion of the monomer, which reflects well the kinetics of the initial stage of the FRP of vinyl monomers and their FRCP with fullerene C60 and upon initiation by free radicals and [86,87]. The panorama represents a set of experiments carried out under the same conditions with respect to temperature, solvent, monomer content, and chemical content of free and stable radicals. Graphs 1 in all panels represent the reference FRP graphs of the corresponding monomer. Figures 7a–7c demonstrate the effect of small additions of C60 on the reference FRP of methyl methacrylate (), styrene, and . As can be seen, the effect varies for different monomers, ranging from a simple decrease in the slope of reference graph 1 for MMA to a delay in the onset of the FRP of styrene and NIPA due to the appearance of low-intensity fractions in their graphs. Until recently, these fractions, also observed in other cases, were designated uniformly, referring to them as induction periods. Figure 7d and Figure 7e show the effect of the combined action of C60 and on the FRP of MMA and styrene. As can be seen from the figures, the presence of manifests itself identically in both cases, providing for the appearance of fairly long induction periods. The effect of additional C60 is completely analogous to that observed in the absence of (Figure 7d and Figure 7e), but is delayed relative to the onset of the FRP of the corresponding monomer on the duration of the induction period provided by the presence of . Analysis of the data presented in Figure 7a, Figure 7b and Figure 7d, and 7e reveals that stable and C60 radicals act on the polarizable medium in a superpositional manner. Figure 7e demonstrates that the mode of action of C60 on the monomer’s FRP depends not only on the monomer but also on the type of initiating free radical participating in the reaction. Thus, when is replaced by , the FRP of styrene in the presence of C60 is accompanied by a significant rearrangement of the low-intensity portion of the full graph, which precisely indicates a change in the process kinetics. The effects discussed are clearly expressed and intense enough to stimulate a persistent desire to understand their intricacies from the perspective of the spin theory of radicals in general and fullerenes in particular.
5.2. A Brief Excursion Into the Spin Theory of Fullerenes
5.2.1. the Origins of the Radicality of Fullerene C60
The digital twin of buckminsterfullerene C60 with continuous symmetry (99.9% ) [109] is a closed framework structure of sp2 covalent bonds, each of which is dynamically unstable. Being a conventional covalent bond connecting two atoms until its length exceeds the critical value = 1.395 Å (see Figure 8a) [108], above this value the bond transforms into a covalent bond between gradually radicalizing atoms. The bond elongation is accompanied by a violation of spin symmetry, since the α and β spins are in different electron orbitals [110,111,112,113,114]. This feature generates a non-zero spin density matrix, the real and imaginary parts of which represent the spin density and spin current density responsible for spin emergents [97], among which the total, , and partial, with respect to each atom, , numbers of effectively unpaired electrons occupy a special place [98]. Both quantities represent quantitative measures of atomic radicalization, thereby determining the molecular chemical susceptibility (MCS) of the molecule as a whole and the atomic chemical susceptibility (ACS) of individual atoms, respectively. Both quantities are amenable to computational and experimental verification, which has been performed repeatedly [115].
Figure 8 summarizes the above and shows that when atomic radicalization is taken into account, the long and short bonds of the molecule are significantly dispersed (panel b) and the bond length compositions of both groups change noticeably (panel c). The presence of a limited number of bond length groups is closely related to the specific distribution of spin density over the atoms of the molecule, the value of which in terms of is shown in Figure 8d, and its multicolor image is presented in the inset. As can be seen from this figure, the C60 DT is regioselective towards intermolecular interactions when exposed to the field of chemical attacks. The interaction is stronger, the higher the of the atom being attacked, as a result of which the ACS plotting is not only a spin portrait of the fullerene, but also the best descriptor underlying the algorithm of its virtual derivatization [107]. Figure 8 also demonstrates a profound conceptual difference between the C60 fullerene DT from the point of view of the spin-symmetric both RHF and DFT approaches, in contrast to the spin-asymmetric UHF.
5.2.2. Spin Density Algorithm for A Virtual Addition Reaction
The first application of spin theory to one of the fundamental events in molecular chemistry—molecule derivatization—reveals the following unique feature of C60, concerning the inevitable sp2→sp3 transformation of the attacked atoms of the molecule. This transformation affects not only all covalent bonds of the atom in question but also those of other atoms in its immediate vicinity. Resisting the imposed structural deformation and minimizing its impact, which could lead to the opening of the closed structure of the molecule, the entire covalent bond network is set in motion, relaxing all its atoms into a new equilibrium position. Figure 9 shows one example of such an event. The two-dimensional graph shown in the figure does not fully reflect the structural change in the valence bonds of the molecule, but concerns only their structural-passport part. The C60 atom at number 33 is attacked by a free radical, , which is the most frequently expected event in the FRP of vinyl monomers upon the addition of C60 to VRS (see [87] and references therein). The attack leads to the formation of monofullerenyl , shown in the figure. As a result, the number of valence bonds of atom 33 increases to 4, and their type changes from sp2 to sp3, which increases the length of all bonds. However, only the last bond, clearly shown in the figure, connects the fullerene to the radical. As for other three bonds, they remain in the system of sp2 bonds of other atoms, thus becoming bonds of mixed type. This peculiarity manifests itself in the fact that at the terminal atoms of the bonds (atoms 24, 32, and 34), the spin density does not disappear, as at atom 33, but takes on different values, which is expressed in values of 0.20, 0.36, and 0.55 e in fullerenyl instead of 0.20, 0.27, and 0.27 e in the original fullerene. Each atom of the trio under consideration is the terminal of two more bonds, each different, so the values represent the cumulative effect of a complex structural-spin rearrangement of the molecule caused by the addition of one addend through one sp3 bond. Thus, each act of derivatization of the molecule is accompanied by a strong collective perturbation, which explains the exceptional lability of the structural, electronic, and spin systems of the molecule, which is a unique characteristic of fullerene C60.
The purple spheres of varying sizes in the image mark the first five atoms at the top of the ACS Z→A list of values from 0.55 to 0.29 e. These atoms form a set of targets for subsequent attacks, located at distances of one, two, three, and four bonds from atom A. Which of these atoms participates in the next derivatization step depends on the addend structure due to the mandatory requirement to avoid steric hindrance when binding to the fullerene core. Each addend is an individual case, thereby completing the algorithmic selection of the top of the ACS list with a distance of a number of bonds from the previous binding site. Thus, in the case of the second binding of the radical to the fullerenyl , this act occurs at the fifth target atom, located at a distance of four bonds from atom A. The remaining four atoms in this case are “chemically dormant.” The third location of on the C60 core will occur on an atom from the top ACS list of the fullerenyl at a distance of four bonds from the second anchor, and further derivatization will proceed similarly. Apparently, the maximum number of radicals bound to the C60 core is 12. Such spin density algorithm was used in the virtual experiment [37,38,39,40,41,42] in all cases to determine the position of the target atom of the reaction under consideration.
5.3. Chain Reaction of Polymerization in View of Spin Theory
Let us consider the set of experimental data presented in Figure 7 in terms of the elementary reactions and their final products, presented in Table 1. According to the table. reactions (1) and (2), which combine a free radical with a monomer to form the monomer-radical and the oligomer-radical , control the FRCP of the monomers (graphs 1 in Figures 7a-7d). Reaction (3) culminates in the formation of a neutral product , whose detection in the final product is unlikely due to the low concentration of the radical. While the formation of a single product is possible in the case of , two similar products are possible in the case of fullerene C60, described by a doublet of reactions (4) and (5). This doublet reflects one of the unique features of fullerene, concerning the configuration of intermolecular junctions between it and the vinyl monomer. This junction is formed by two sp2bonds, one of which belongs to the fullerene, and the other is the vinyl group of the monomer. Accordingly, the compound can be either two-bond or one-bond coupled. While the first configuration leads to the formation of a [2x2] cycloadduct, a stable radical , similar to the initial C60, the second leads to the formation of a fullerene-initiated monomer radical , similar to and capable of further polymerizing vinyl. The equilibrium structures of these two DTs, related to methyl methacrylate, in Figure 10 are supplemented with ACS plottings revealing the radical properties of the fullerenyls under discussion, represented by the number of effectively unpaired electrons . The picture is common to all representatives of vinyls. The ACS of both DTs are plotted against a background representing the ACS distribution of the parent C60 fullerene. As can be seen from the figure, both fullerenyls retain the polytarget type of radicalization. The appearance of new targets with increased radicality in the fullerene core (see the most prominent atoms 35, 22, and 11 in Figure 10a and additional atoms 37 and 35 in Figure 10b) is an expected consequence of the reconstruction of the sp2 bond system of the fullerene caused by the formation of fullerenyls, discussed earlier in Section 5.2.2. The purple balls mark the carbon atoms with the three highest values. The bright scarlet balls mark the target atoms of the adduct, located at a distance of three bonds from the atoms of the first anchoring. The appearance of a new fullerenyl target , atom 62, linked to the vinyl bond of the monomer (see Figure 10b), with a predominant value of = 0.97e, demonstrates the undoubted readiness of the latter to continue association with other monomer molecules, similar to that observed in the case of standard FRP [38]. Accordingly, reaction (2) describes the growth of the polymer chain initiated by a free radical, and the reaction (5) describes the polymerization of the monomer initially grafted to the fullerene. Although the existence of reaction (5) was previously suggested in a number of cases, no confident conclusion about its existence was made, and this reaction, like reaction (3), was classified as unlikely. The first evidence of fullerene-initiated styrene polymerization was obtained precisely during the virtual experiment under discussion [41].
In contrast to the unreactive monomer, the capture of its monomer radical , described by reactions FRM (7) and (8), is traditionally expected in both cases with a high probability. These reactions should be highlighted since they have the potential to completely stop and/or delay monomer polymerization, causing an induction period in the graph. Reactions (9) and (10), describing the capture of the free radical by stable ones, also affect monomer polymerization by decreasing the number of initiating free radicals and thereby lowering the slope of the graphs. Reaction (12) and (13) are similar to reactions (9) and (10), but related to the monomer radical . The listed reactions are in qualitative agreement with all the characteristic changes presented by the experimental data. A quantitative description of their kinetics constituted the main objective of the virtual experiment [37,38,39,40,41,42].
Table 3 summarizes identical fragments of the text pages of the VPPs related to various VRSs. The content of elementary reactions is limited to the FRP of the monomers, as well as their FRCP with fullerene C60. Figure 11 presents fragments of the image pages of these data sheets related to the elementary reactions occurring in the considered VRSs. A more complete content of the issued VRSs can be found in [38]. In the composition of both the table and the figure, the data follow the order suggested by the presentation of the empirical data in Figure 7.
The collection of DTs shown in Figure 11 is composed as follows. The first row presents monomers and monomer radicals , grouped as pairs for methyl methacrylate (), styrene (), and N-isopropylacrylamide (). The three columns thus formed include pairs of monofullerenyls and (second row) and single monoaggregates (third row). All DTs are associated with elementary reactions where the alkyl nitrile acts as a free radical . The fourth row includes three DTs associated with the reactions , and , which are related to the FRCP of styrene with fullerene C60 in the case where the role of the initiating free radical is given to benzoyl peroxide . The full list of DTs considered during the project can be found in [38].
5.4. Virtual Free-Radical Copolymerization of Vinyl Monomers with C60 Fullerene
1. The set of elementary reactions introduced into the FRP by C60 fullerene is common to all VRSs of vinyl monomers and consists primarily of four members: , , ( and , initiated by the free radicals or , respectively), and and , as in the previous case). The considered fullerenyls , , and are monoadducts synthesized virtually in accordance with the C60 spin theory, with the corresponding addends attached to the same atom of the starting fullerene in all cases. The general structure of the fullerenyls and the dominant role of the fullerene in them suggest a similar type of associated reactions, which makes it possible to use the kinetic descriptors listed in Table 3 for a comparative assessment of the rates of the corresponding reactions.
2. According to the kinetic descriptors of methyl methacrylate VRSA, the reactions associated with fullerenes form the following series with respect to the rate constants:
The reaction of free radical capture by fullerene heads this series and has every reason to be the first. Without affecting the totality of monomers, this reaction does not interfere with the FRP of the latter, but it does reduce the current concentration of initiating free radicals, which should result in a decrease in the overall monomer conversion rate during its polymerization. This very response of the graph to the presence of fullerene in VRSA is observed experimentally (see Figure 7a). As expected, the conversion rate depends on the fullerene concentration and decreases with increasing concentration, which, as can be seen from the figure, is indeed the case experimentally.
3. The series ordering the sequence of reactions associated with fullerene in the styrene VRSA is fundamentally different from that discussed above and looks as follows:
giving a clear preference to the formation of the monomer radical . This reaction already affects the totality of the main monomer and can compete with the reaction (1). The final result of the competition depends on the ratio of the rate constants and . The ratio of the corresponding kinetic descriptors given in Table 3 indicates the comparability of both rates. However, additional considerations presented in [38] and concerning the factors A in equation (1) allowed to conclude that
So, polymerization in VRSA begins with the free-radical polymerization of styrene, initiated by fullerene and occurring on it. Until this reaction is complete, the fullerene-free styrene FRP does not begin. Thus, when all the fullerene content in the first stage of polymerization is exhausted, it is the turn of the standard styrene FRP , initiated by the free radical . Therefore, the graph of the total conversion should consist of two sections associated with the styrene FRP stimulated first by fullerene and then by . Since under normal conditions the [C60] content is three orders of magnitude lower than [], the first initial section of the graph should be very low in amplitude, which is observed in reality (see Figure 7b). It is clear that the duration of this stage increases with increasing [C60], which is fully confirmed by the data presented in the figure. It is noteworthy that the slope of the main linear portion of the graph is preserved, remaining the same as in the reference reactor without the addition of fullerene.
As can be seen from Table 3, replacing with dramatically changes the situation. Series (3) in the case of VRSB takes the form
and the reaction (7) of absorption of the monomer radical by fullerene comes to the fore. Naturally, this reaction has a direct influence on the polymerization of styrene, intervening between the two main reactions (1) and (2). According to the table and a detailed discussion of factor A [38], the sequence of reactions follows the ordering
so that the resulting monomer radical is captured by the fullerene, which prevents the monomer’s VRP, thereby ensuring the presence of a zero-amplitude conversion graph and revealing a classical induction period until the [C60] content is completely depleted. The FRP of styrene, starting at this point, is completely identical to the reference FRP. It is precisely this type of conversion graph that is presented in Figure 7f.
- 4. the Fullerene-Associated Reaction Sequence in Vrsa Forms A Series
The reaction (7), being barrier-free, is absolutely dominant, so that each of the newly formed monomer radicals is destined for immediate capture. The long induction period, which determines the complete depletion of [C60], becomes a distinctive feature of the conversion graph of VRSA , which is observed experimentally (see Figure 7c).
5. An important result of the virtual analysis [38] is the fact that, in contrast to the wide variety of responses to the presence of fullerene C60 in the VRS of vinyl monomers, the single-target stable radical behaves much more modestly. The diversity of the above-discussed VRSs is characterized by a single reaction, (8), which describes the capture of monomer radicals . As shown above, this reaction is characterized by a clearly defined conversion graph with zero amplitude, attributed to the induction period. The obtained predictions are fully confirmed by the experiments presented in Figure 7d and Figure 7e. Since, as was established during the virtual experiment, and C60 radicals do not interact, their action in the experimental reaction solutions is completely superpositional, as clearly seen in these same figures.
5.4. Virtual Star-Branched Polymers of C60 Fullerene
The discussion presented in the previous section concerned the initial stage of chemical transformations occurring in the reaction solution. The impressively diverse phenomena induced by the presence of C60 fullerene are not limited to these, even after the words about “complete utilization of the [C60] content by the end of reactions (4), (6), and (8)” were mentioned. The fact is that these three fundamental reactions of the initial stage generally culminate in the formation of -polyadducts , and , rather than monofullerenyls , , and . The radical activity of the fullerenyl polyadducts gradually decreases with increasing This does not occur immediately, but rather through a series of successive stages. Thus, in the case of the simplest addends, which are atomic hydrogen and fluorine, the adducts cease to be radicals when reaches 36 for hydrogen [116] and 48 for fluorine [117]. Naturally, with a complex addend structure, the result of derivatization is much more modest, since the number of attachment sites for such addends on the carbon core of the molecule is limited by the condition of preventing steric hindrance.
Analysis of numerous experimental data on the VRCP of vinyl monomers with C60 fullerene shows that the radical activity of the initial monofullerenyls is also not quenched, as a result of which the latter participate in the subsequent formation of the polymer mass of the studied monomer as stable radicals. It is often said that fullerene is incorporated into the polymer chains of the final product, becoming the center of star-shaped branching. From the standpoint of the spin theory of fullerenes, this assumption appears to be entirely justified. Indeed, while remaining radicals, polyfullerenyls actively interact with other radicals present in the reaction solution, the majority of which during massive polymerization are represented by oligomers of the base monomer , forming complex compositions of the type . The diversity of compositions is enormous, which obstructs a reliable prediction. Clearly, progress in this direction can only be expected after the development of general algorithms for the polyderivatization of fullerene C60, supplemented by a thorough kinetic analysis, which, in turn, requires a thorough elaboration of both experimental data and theoretical concepts. The main problem concerns the determination of the numbers and , as well as the identification of the locations of the target atoms of the fullerene core, which enable anchor couplings of and with the core. The difficulties encountered in this approach are clearly visible in Figure 12. The figure shows the formation of three simple stars, subjected to the ACS algorithm and based on the knowledge of the VRSs discussed in the previous sections, while maintaining the individuality of the FRCP processes in all cases [39].
The star C60- in Figure 12a accumulates a symbiosis of two main reactions occurring in this reaction solution: the capture of initiating radicals by fullerene and the simultaneous implementation of the VRP. Following Table 1, the result of the first reaction is the formation of a set of fullerenyls (4), while the formation of oligomer radicals (2) is a natural consequence of the VRP [40]. When constructing the star, the choice of numbers and was arbitrarily limited to 3 and 5, respectively. The design of fullerenyls begins with the formation of monofullerenyl , the principle of which is shown in Figure 9. As can be seen from the figure, the capture of the initiating free radical by the fullerene is accompanied by a reconstruction of both the sp2bond set and the spin density on the fullerene atoms. Pale lilac spheres of varying sizes represent the top five atoms from the Z→A atomic list of fullerenyl with values ranging from 0.55 to 0.29 e. The first four atoms form a set of targets quite suitable for subsequent addition, but, being one, two, and three bonds away from atom A, they are inaccessible to the radical addition due to steric hindrance, thus forming a four-atom set of “chemically dormant” atoms. Only the fifth, bright lilac atom, four bonds away from atom A, is ready to accept the addition of the second radical . This addition complements the previous four-atom set of dormant atoms in fullerenyl with another four-atom set of dormant atoms with values from 0.47 to 0.30 e, leaving only the ninth bright purple atom, located four bonds away from atom B, to accept the third radical. The set of dormant atoms of fullerenyl increases to 12 with values from 0.55 to 0.28 e, and the 13th atom with = 0.27, located four bonds away from atom C, is ready to serve as a target for the next addition. The addendum of this subsequent addition is the oligomer-radical , which has a single active carbon atom, the reactivity of which is ensured by the value of = 0.98 e. The addition of the first oligomer-radical to the fullerenyl provides the formation of the first branch of the star C60-. The bright lilac atom of this complex fullerenyl indicates the site of addition of the second oligomer-radical or the formation of the second branch of the star.
In the case of the star C60- in Figure 12b, the main reaction of the initial period is the capture of the monomer radical by the fullerene and the formation of the fullerenyl . Similar to the previous case, this addition is accompanied by the formation of a tetraatomic dormant set of carbon atoms with values from 0.55 to 0.29 e, while the fifth atom with = 0.29e is the target for the next addition. The addition addend in this case is the oligomer radical . A similar analysis of the fullerenyl reveals the target atom of the star C60-, which is ready for the next addition of either or .
The formation of the styrene star C60-, shown in Figure 12.c, proceeds similarly to the two previous cases. The first action in this reaction solution is the polymerization of styrene grafted onto the fullerene, accompanied by the formation of the fullerenyl . Although the composition is bulkier than and , the response of the fullerene spin density to the addition of the oligomer radical is virtually identical to the previous cases. Again, four of the five -highest carbon atoms are dormant, while the fifth accepts the addition of the oligomer radical . The latter, in turn, reveals five new target atoms, one of which is the target either for the repolymerization of styrene on fullerene or for the addition of a second oligomer radical.
6. Conclusive Remarks
A new vision of the role of digital technologies for the virtualization of various processes as an integral part of the trinity of a physical object, a virtual/digital object, and the connections between them emerged relatively recently [9] and was first and most confidently embodied in the materials industry [6,7,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29]. Apart from these achievements, the natural development of the digital component of materials science, as it turns out, also took place under the auspices of this trinity [30,31,32,33,34,35,36,37,38,39,40,41,42], beginning with the first introduction of the concept of digital twins [30,31,32], developing the concept of digital twins as a new modeling alternative capable of solving complex problems in materials science inaccessible to previous concepts [33,34,35,36], and finally, culminating in the trinity of a physical object, a digital object, and the connections between them in polymer chemistry [37,38,39,40,41,42]. The final project no longer focused on developing a new methodology for virtualizing the problem, but instead utilized the previously developed [30,31,32,33,34,35,36] concept of digital twins to solve a specific chemical problem. The results of the first virtual experiment of this type, presented in the previous sections, do not require repetition, but allow for several important conclusions.
First, the design of this experiment, its execution, and the analysis of the results convincingly represent the realization of the discussed trinity. Understanding the essence of the connection between the physical and digital objects allowed us to optimize the parameters of the digital twin concept most suited to solving the problem. In turn, feedback between the digital and physical objects allowed us to use the obtained results of the virtual experiment to establish new internal connections related to the physical object. The virtual device used naturally limited the range of common points of contact between the physical and digital objects. However, the origins of this limitation are clear, opening the possibility of expanding the scope of contact between the objects by changing the virtual device.
The second point brings us back to the title of this review. Undoubtedly, the virtual experiment yielded a vast number of results, previously uncharacteristic of traditional modeling. While this number still falls far short of Big Data, the dialectical transition of quantity to quality has nonetheless occurred, as the final trinity of information flowing from a digital object to a physical environment has made it possible to answer virtually all of the latter’s pressing questions. It is appropriate to recall now the remark of Greaves, a pioneer of the digital twin concept, regarding its applicability to limited scenarios [7]. Science, for its part, is a limited ecosystem on the path to a metaverse [4], and therefore all its projects can be considered limited scenarios. This allows us to conclude that the virtual experiment itself and the results obtained in its process represent the first significant contribution to the virtual world of polymer chemistry.
After this review was completed, the author became aware of the multidisciplinary, extensive international project CompSafeNano [118]. The project aims to develop and implement computational nanosafety and enhance the safety and innovation potential of nanomaterials by integrating advanced nanoinformatics, computational modeling, and predictive toxicology to enable the design of safer materials at the earliest stages of their development. It is expected that the number of similar projects at the intersection of information technology and scientific research will increase daily.
Acknowledgments
This paper has been supported by the RUDN University Strategic Academic Leadership Program.
References
- Shabason VE, Malaykin SN. Razumnaya metavselennaya. Ot tsifrovykh prilozheniy k novoy srede obitaniya. [Reasonable metaverse. From digital applications to a new environment]. Moscow, LitRes Publ., Juli 19 2022.
- Ball M. The Metaverse: And How It Will Revolutionize Everything. Liveright: New York, July 25, 2022.
- Li, H., Li, B. The state of metaverse research: a bibliometric visual analysis based on CiteSpace. J Big Data 11, 14 (2024). [CrossRef]
- Sheka EF. Metaverse and virtual worlds of science. Trends Comput Sci Inf Technol. 2024. 9, 103-105. [CrossRef]
- M. Liu, S. Fang, H. Dong, C. Xu. Review of digital twin about concepts, technologies, and industrial applications. J. Manufact. Syst. 58 (2021) 346–361. [CrossRef]
- Handbook of Digital Twins. Ed. Lv, Zh, CRC Press, Taylor and Francis, Boca Raton, 2024. [CrossRef]
- S. Waffenschmidt. Digital twin in the Industry 4.0: interview with a pioneer. t-systems.com›de/en/newsroom/best-practice/ issue 03, 2018.
- D. H. Gelernter (1991). Mirror Worlds: or the Day Software Puts the Universe in a Shoebox—How It Will Happen and What It Will Mean. Oxford; New York: Oxford University Press. ISBN 978-0195079067. OCLC 23868481.
- R. Piascik, et al., Technology Area 12: Materials, Structures, Mechanical Systems, and Manufacturing Road Map. 2010, NASA Office of Chief Technologist.
- Tao, F.; Cheng, J.; Qi, Q.; Zhang, M.; Zhang, H.; Sui, F. Digital Twin-Driven Product Design, Manufacturing and Service with Big Data. The International Journal of Advanced Manufacturing Technology 2018, 94 (9–12), 3563–3576. [CrossRef]
- Rasheed, A.; San, O.; Kvamsdal, T. Digital twins: Values, challenges and enablers from a modeling perspective. IEEE Access 2020. [CrossRef]
- Lazzari, S.; Lischewski, A.; Orlov, Y.; Deglmann, P.; Daiss, A.; Schreiner, E.; Vale, H. Toward a digital polymer reaction engineering. Adv. Chem. Eng. 2020, 56, 187-230.
- Zobel-Roos, S.; Schmidt, A.; Uhlenbrock, L.; Ditz, R.; Köster, D.; Strube, J. Digital Twins in Biomanufacturing; 2020; pp 181–262. [CrossRef]
- Gargalo, C. L.; de las Heras, S. C.; Jones, M. N.; Udugama, I.; Mansouri, S. S.; Krühne, U.; Gernaey, K. V. Towards the Development of Digital Twins for the Bio-Manufacturing Industry; 2020; pp 1–34. [CrossRef]
- Sinner, P.; Daume, S.; Herwig, C.; Kager, J. Usage of Digital Twins Along a Typical Process Development Cycle; 2020; pp 71–96. [CrossRef]
- Stanko, M.; Stommel, M. Digital Twin of the Polyurethane Rotational Moulding Process. In Advances in Polymer Processing 2020; Springer Berlin Heidelberg: Berlin, Heidelberg, 2020; pp 324–335. [CrossRef]
- Scheper, T.; Beutel, S.; McGuinness, N.; Heiden, S.; Oldiges, M.; Lammers, F.; Reardon, K. F. Digitalization and Bioprocessing: Promises and Challenges; 2020; pp 57–69. [CrossRef]
- Hürkamp, A.; Lorenz, R.; Ossowski, T.; Behrens, B.-A.; Dröder, K. Simulation-Based Digital Twin for the Manufacturing of Thermoplastic Composites. Procedia CIRP 2021, 100, 1–6. [CrossRef]
- Andronas, D.; Kokotinis, G.; Makris, S. On Modelling and Handling of Flexible Materials: A Review on Digital Twins and Planning Systems. Procedia CIRP 2021, 97, 447–452. [CrossRef]
- Qi, Q.; Tao, F.; Hu, T.; Anwer, N.; Liu, A.; Wei, Y.; Wang, L.; Nee, A. Y. C. Enabling Technologies and Tools for Digital Twin. J Manuf Syst 2021, 58, 3–21. [CrossRef]
- Lo, C. K.; Chen, C. H.; Zhong, R. Y. A Review of Digital Twin in Product Design and Development. Advanced Engineering Informatics 2021, 48, 101297. [CrossRef]
- Lebedev, I. V.; Ivanov, S. I.; Safarov, R. R.; Men’shutina, N. V. Digital Twins for the Porous Structures of Aerogels with the Use of the Cellular Automation Approach and Bezier Curves. Theoretical Foundations of Chemical Engineering 2023, 57 (4), 482– 488. [CrossRef]
- Wang, Y.; Tao, F.; Zuo, Y.; Zhang, M.; Qi, Q. Digital-Twin-Enhanced Quality Prediction for the Composite Materials. Engineering 2023, 22, 23–33. [CrossRef]
- Fernández-León, J.; Keramati, K.; Baumela, L.; González, C. A Digital Twin for Smart Manufacturing of Structural Composites by Liquid Moulding. The International Journal of Advanced Manufacturing Technology 2024, 130 (9–10), 4679–4697. [CrossRef]
- Isoko, K.; Cordiner, J. L.; Kis, Z.; Moghadam, P. Z. Bioprocessing 4.0: A Pragmatic Review and Future Perspectives. Digital Discovery 2024, 3 (9), 1662–1681. [CrossRef]
- Alajmi, A. Twinning the Future: Implementing Digital Twin Technology in the Optimisation of Fibre-Reinforced Polymers. MATEC Web of Conferences 2024, 401, 11005. [CrossRef]
- Jaboviste, K.; Sadoulet-Reboul, E.; Teloli, R. O.; Chevallier, G. Characterizing the Nonlinear Behavior of Viscoelastic Materials: A Bayesian Approach Combining Oberst Beam Experiments and Digital-Twin Simulations. Mech Syst Signal Process 2024, 208, 110978. [CrossRef]
- CEN and CENELEC. CEN and CENELEC Workshop Agreement: Methodology for the Data-Driven Management of Production; 50751:2024; 2024. https://www.cencenelec.eu/media/CEN-CENELEC/CWAs/RI/2024/cwa50751_2024.pdf (accessed 2024-12-19).
- Mitchell, A.; Wei, X,; Sun, R.; Yamamura, K.; Ye, L.; Corney, J.; Yu, N. Digital manufacturing for advanced manufacturing technologies: A review of successful implementation for plasma processing digital twin framework development. Available on 26 September 2024 at SSRN: https://ssrn.com/abstract=4846321 or. [CrossRef]
- E.F. Sheka, V.A. Popova. Virtual vibrational spectrometer for sp2 carbon clusters. 2. Fullerene C60 and its isomers. FNCN, 2021, 29, No. 12, 974-981. [CrossRef]
- E.F. Sheka and N. A. Popova. Virtual vibrational spectrometer for sp2 carbon clusters and dimers of fullerene C60. FNCN, 2022, 30, 777-786. [CrossRef]
- E.F. Sheka. Virtual vibrational spectrometry of stable radicals—necklaced graphene molecules. Nanomaterials 2022, 12(4), 597. [CrossRef]
- E.F.Sheka, N.A. Popova. Virtual vibrational analytics of reduced graphene oxide. Int. Journ. Mol. Sci. 2022, 23, 6978. [CrossRef]
- E.F.Sheka. Digital Twins solve the mystery of Raman spectra of parental and reduced graphene oxides. Nanomaterials, 2022, 12, 4209. [CrossRef]
- Е.Ф. Шека. Цифрoвые двoйники в технoлoгии графена. РЭНСИТ: Радиoэлектрoника. Нанoсистемы. Инфoрмациoнные технoлoгии, 2023, 15(1):59-80. [CrossRef]
- E.F. Sheka. Digital Twins in graphene technology. Chapter 47 of Handbook of Digital Twins, Ed. Lv, Zh, CRC Press, Taylor and Francis, Boca Raton, 2024, 19 pages. [CrossRef]
- E.F. Sheka. Virtual free-radical polymerization of vinyl monomers in view of digital twins. Polymers, 2023, 15, 2999. Published. [CrossRef]
- E.F. Sheka. Digitalization of free-radical polymerization. arXiv:2312.14163 [physics.chem-ph], 2023. [CrossRef]
- E.F. Sheka. The triumph of the spin chemistry of fullerene C60 in the light of its free radical copolymerization with vinyl monomers. Int. Journ. Mol. Sci. 2024, 25, 1317. [CrossRef]
- E.F. Sheka. Digital twins’ kinetics of virtual free-radical copolymerization of vinyl monomers with stable radicals. 1. Methyl methacrylate. FNCN, 2024. [CrossRef]
- E.F. Sheka. Digital twins’ kinetics of virtual free-radical copolymerization of vinyl monomers with stable radicals. 2. Styrol. arXiv:2311.02752 [cond-mat.mtrl-sci]. [CrossRef]
- E.F. Sheka. Digital twins’ kinetics of virtual free-radical copolymerization of vinyl monomers with stable radicals. 3. NIPA. arXiv:2311.06866 [cond-mat.mtrl-sci]. [CrossRef]
- Ladik, J., André, JM., Seel, M. (eds) Quantum Chemistry of Polymers — Solid State Aspects. NATO ASI Series, vol 123. Springer, Dordrecht, 1984.
- J. Ladik, Quantum Theory of Polymers as Solids. Plenum Press, New York, 1988.
- C. Tosy (ed). Proceedings of the First Donegani Scientific Workshop on Strategies for Computer Chemistry. October 12–13, 1987. Kluwer Academic Publishing: New York, Boston, Dordrecht, London, Moscow, 1989.
- Quantum Chemistry Aided Design of Organic Polymers: An Introduction to the Quantum Chemistry of Polymers and its Applications. Eds. J.-M. Andre, J. Delhalle, J.-L. Bredas, World Sci. Publ.: Singapoure, 1991.
- Computational Modeling of Polymers. Ed. J. Bicerano. CRC Press: Boca Raton, 1992.
- H. Fischer, L. Radom. Factors controlling the addition of carbon-centered radicals to alkenes. An experimental and theoretical perspective. Angew. Chem. Int. Ed. 2001, 40, 1340 – 1371.
- Heuts, J.P.A. Theory of radical reactions. In Handbook of Radical Polymerization, Eds. K. Matyjaszewski, T.P. Davis. John Wiley and Sons: Hoboken, 2002, pp. 1-76.
- Theory and Applications of Computational Chemistry: The First Forty Years. Eds. C. Dykstra, G. Frenking, K. Kim, G. Scuseria. Elsevier B.V.: Amsterdam, 2005.
- J.-M. Andre. Computational quantum chemistry on polymer chains: aspects of the last half century. In Theory and Applications of Computational Chemistry: The First Forty Years. Eds. C. Dykstra, G. Frenking, K. Kim, G. Scuseria. Elsevier B.V.: Amsterdam, 2005, pp. 1011-1045.
- M. L. Coote. Quantum-chemical modeling of free-radical polymerization. Macromol. Theory Simul. 2009, 18, 388–400.
- D. Gigmes, D. Bertin, C. Lefay, Y. Guillaneuf. Kinetic modeling of nitroxide-mediated polymerization: Conditions for living and controlled polymerization. Macromol. Theory Simul. 2009, 18, 402–419. [CrossRef]
- P. Deglmann, I. Mueller, F. Becker, A. Schaefer, K.-D. Hungenberg, H. Weiß. Prediction of propagation rate coefficients in free radical solution polymerization based on accurate quantum chemical methods: Vinylic and related monomers, including acrylates and acrylic acid. Macromol. React. Eng. 2009, 3, 496–515. [CrossRef]
- M. Dossi, G. Storti, D. Moscatelli. Initiation kinetics in free-radical polymerization: Prediction of thermodynamic and kinetic parameters based on ab initio calculations. Macromol. Theory Simul. 2010, 19, 170–178. [CrossRef]
- E. Mavroudakis, D. Cuccato, D. Moscatelli. On the use of quantum chemistry for the determination of propagation, copolymerization, and secondary reaction kinetics in free radical polymerization. Polymers 2015, 7, 1789-1819. [CrossRef]
- F. Ruipérez. Application of quantum chemical methods in polymer chemistry. Int. Rev. Phys. Chem. 2019, 38, 343-403. [CrossRef]
- Y. Hayashi, S. Kawauchi. Development of a quantum chemical descriptor expressing aromatic/quinoidal character for designing narrow-bandgap π-conjugated polymers. Polym. Chem., 2019, 10, 5584–5593. [CrossRef]
- J. Gay. Molecular Dynamics Studies of Polymer Systems. Department of Chemical Engineering at Worcester Polytechnic Institute: Worcester, 2012.
- T. E. Gartne, A. Jayaraman, Modeling and simulations of polymers: A roadmap. Macromol. 2019, 52, 755−786.
- C. Micheletti, P. Hauke, P. Faccioli. Polymer physics by quantum computing, Phys.Rev. Letts 2021, 127, 080501. [CrossRef]
- H.W. Starkweather, H.W.; Taylor, G.B. The kinetics of the polymerization of vinyl acetate. JACS 1930, 52, 4708-4714.
- Semenov, N.N. Tsepnyie Reakcii (Chain Reactions) Goschimizdat: Moskva, 1934 (in Russian).
- А.Bagdasar’yan, Kh.S., Teoriya radikal’noi polimerizatsii (Free_Radical Polymerization Theory), Moscow: Nauka, 1966. (in Russian).
- Gol’dfein, M.D.; Kozhevnikov, N.V.; Trubnikov, A.V. Kinetika i mekhanizm regulirovaniya protsessov obrazovaniya polimerov (Kinetics and Control of Polymerization Processes), Saratov: Saratov. Gos. Univ., 1989.
- Pross, A. Theoretical and Physical Principles of Organic Reactivity, Wiley, New York, 1995.
- Handbook of Radical Polymerization, Eds. K. Matyjaszewski, T. P. Davis. John Wiley & Sons, Inc. 2002.
- Handbook of Vinyl Polymers 2: Radical Polymerization, Process, and Technology, New Edition. Eds. M. Mishra, Y. Yagci. CRC Press: Boca Raton, 2019.
- Loy, D.A.; Assink, R.A. Synthesis of a C60-p-xylylene copolymer. J. Am. Chem. Soc. 1992, 114, 3977 – 3978.
- Bunker, C.E.; Lawson, G.E.; Sun, Y.P. Fullerene-styrene random copolymers. novel optical properties. Macromolecules 1995, 28, 3744 – 3746. [CrossRef]
- Cao, T.; Webber, S.E. Free-radical copolymerization of fullerenes with styrene. Macromolecules 1995, 28, 3741 – 3743. [CrossRef]
- Cao, T.; Webber, S.E. Free radical copolymerization of styrene and C60. Macromolecules 1996, 29, 3826 – 3830. [CrossRef]
- Camp, A. G.; Lary, A.; Ford, W. T. Free-radical copolymerization of methyl methacrylate and styrene with C60. Macromolecules 1995. 28, 7959–7961. [CrossRef]
- Sun, Y.P.; Lawson, G.E.; Bunker, C.E.; Johnson, R.A.; Ma, B.; Farmer, C.; Riggs, J.E.; Kitaygorodskiy, A. Preparation and characterization of fullerene−styrene copolymers. Macromolecules 1996, 29, 8441 – 8448. [CrossRef]
- Cao, T.; Webber, S.E. Radical copolymerization of styrene and C60. Macromolecules 1995, 28, 3741–3743.
- Geckeler, KE; Arsalani, N. Synthesis and properties of hydrophilic polymers. 4. Preparation and characterization of poly (oxyethylene) telechelics with different aromatic termini. J. Macromol. Sci., Part A: Pure and Appl. Chem. 1996, 33, 1165-1179. [CrossRef]
- Steward, D.; Imrie, C. T. Role of C60 in the free radical polymerisation of styrene. Chem. Commun. 1996, 13, 1383-1384.
- Chen, Y.; Lin, K.C. Radical polymerization of styrene in the presence of C60. J. Polym. Sci. A, Polym. Chem. 1999, 37, 2969 – 2975.
- Ford, W. T.; Graham, T. D.; Mourey, T. H. Incorporation of C60 into poly(methyl methacrylate) and polystyrene by radical chain polymerization produces branched structures. Macromolecules 1997, 30, 6422–6429. [CrossRef]
- Ford, W. T.; Nishioka, T.; McCleskey, S. C.; Mourey, T. H.; Kahol, P. Structure and radical mechanism of formation of copolymers of C60 with styrene and with methyl methacrylate. Macromolecules, 2000, 33, 2413–2423. [CrossRef]
- Nayak, P.L.; Yang, K.; Dhal, P.K.; Alva, S.; Kumar, J.; Tripathy, S.K. Polyelec-trolyte-containing fullerene I: synthesis and characterization of the copolymers of 4-vinylbenzoic acid with C60. Chem. Mater. 1998, 10, 2058 – 2066. [CrossRef]
- Jiang, G.; Zheng, Q. Synthesis and application of new fullerene derivative. J. Appl. Polym. Sci. 2005, 97, 2182 – 2185. [CrossRef]
- Kurmaz, S.V.; Pyryaev, A.N.; Obraztsova, N.A. Effect of fullerene on the radical homo_ and copolymerization of n_vinylpyrrolidone and (di)methacrylates. Polym. Sci., Ser. B, 2011, 53, 497–504. [CrossRef]
- Atovmyan, E.G.; Grishchuk, A.A.; Estrina, G.A.; Estrin, Ya.I. Formation of star-like water-soluble polymeric structures in the process of radical polymerization of N-isopropyl acrylamide in the presence of C60. Russ. Chem. Bull., Int. Ed. 2016, 65, 2082—2088. [CrossRef]
- Atovmyan, E. G. On the relationship between the fullerene reactivity and degree of substitution. Russ. Chem. Bull., Int. Ed. 2017, 66, 567-570. [CrossRef]
- Yumagulova, R. Kh.; Kuznetsov, S. I.; Diniakhmetova, D. R.; Frizen, A. K.; Kraikin, V. A.; Kolesov. S. V. On the initial stage of the free-radical polymerizations of styrene and methyl methacryate in the presense of fullerene C60. Kinetics and Catalysis 2016, 57, 380–387.
- Yumagulova, R. Kh.; Kolesov, S.V. Specific features of reactions between fullerene C60 and radicals stabilized by conjugation in the process of radical polymerization. Bullet, Bashkir University 2020, 25. 47-51. [CrossRef]
- Martin, N.; Giacalone, F. (Eds.) Fullerene Polymers. Synthesis, Properties and Applications. Wiley-VCH: Weinheim, 2009.
- Denisov, E. T. Constanty skorosni gomoliticheskikh zhidkofaznykh reakciy (Rate constants of homolytic liquid-phase reactions) Moskwa: Nauka. 1971.
- Denisov, E. T.; Sarkisov, O. M.; Likhtenshtein, G. I. Chemical Kinetics: Fundamentals and Recent Developments, Elsevier, Amsterdam, 2003.
- E.T. Denisov, I.B. Afanas’ev. Oxidation and Antioxidants in Organic Chemistry and Biology. Boca Raton, Florida: CRC Press. Taylor and Francis Group, 2005.
- H. Zipse. Radical stability—a theoretical perspective. Top Curr. Chem. 2006, 263: 163–189. [CrossRef]
- A.I. Krylov. The quantum chemistry of open-shell species. In Reviews in Computational Chemistry. Eds, A. L. Parrill, K. B. Lipkowitz. Vol. 30, First Edition. John Wiley & Sons, Inc.: Hoboken, 2017, pp. 151-224.
- Y. Huang, E. Egap. Open-shell organic semiconductors: an emerging class of materials with novel properties. Polym. Journ., 2018, 50, 603–614. doi.org/10.1038/s41428-018-0070-6.
- Z.X. Chen, Y. Li, F. Huang. Persistent and stable organic radicals: Design, synthesis, and applications. Chem 2021, 7, 288–332. [CrossRef]
- G. Kaplan. Symmetry properties of the electron density and following from it limits on the KS-DFT applications. Mol. Phys. 2018, 116, 658-665. [CrossRef]
- Sheka E F, Popova N A, Popova V A. Physics and chemistry of graphene. Emergentness, magnetism, mechanophysics and mechanochemistry Phys. Usp. 2018, 61, 645-691.
- E.F. Sheka, Chemical susceptibility of fullerenes in view of Hartree–Fock approach. Int. J. Quant. Chem. 2007, 107, 2803–2816. [CrossRef]
- Zayets, V. A. CLUSTER-Z1: Quantum-Chemical Software for Calculations in the s,p-Basis. Inst. Surf. Chem. Nat. Ac. Sci. of Ukraine: Kiev, 1990; (in Russian).
- Berzigiyarov, P.K.; Zayets, V.A.; Ginzburg, I.Ya.; et al. NANOPACK: Parallel codes for semiempirical quantum chemical calculations of large systems in the sp- and spd-basis. Int. J. Quantum Chem. 2002, 88, 449-462.
- Dewar M.J.S.; Zoebisch, E.G.; Healey, E.F.; Stewart, J.J.P. AM1: A new general-purpose quantum mechanical molecular model. J.Amer.Chem.Soc. 1985, 107, 3902-3909.
- Sheka, E.F. Fullerenes. Nanochemistry, Nanomagnetism, Nanomedicine, Nanophotonics. CRC Press, Taylor and Francis Group, Boca Raton, 2011.
- Sheka, E.F. Spin Chemical Physics of Graphene; Pan Stanford: Singapore, 2018.
- Sheka, E. F. Virtual vibrational spectrometry of stable radicals—necklaced graphene molecules. Nanomat. 2022, 12, 597.
- Mauri, A.; Consonni, V.; Todeschini, R. (2017). Molecular Descriptors. In: Leszczynski, J., Kaczmarek-Kedziera, A., Puzyn, T., G. Papadopoulos, M., Reis, H., K. Shukla, M. (eds) Handbook of Computational Chemistry. Springer, Cham.
- E. T. Denisov, O. M. Sarkisov, G. I. Likhtenshtein, Chemical Kinetics: Fundamentals and Recent Developments, Elsevier, Amsterdam 2003. 2015, 70, 111-161.
- Е.Ф. Шека. Спинoвая химия фуллерена. Издательствo РУДН, Мoсква, 2025.
- E.F.Sheka Stretching and breaking of chemical bonds, correlation of electrons, and radical properties of covalent species, Adv. Quant. Chem.
- Sheka, E.F.; Razbirin, B. S.; Nelson, D. K. Continuous symmetry of C60 fullerene and its derivatives. J. Phys. Chem. A 2011, 115, 3480-3490. [CrossRef]
- Löwdin, P-O. Quantum theory of many-particle systems. III. Extension of the Hartree-Fock scheme to include degenerate systems and correlation effects. Phys. Rev.1955, 97, 1509-1520. [CrossRef]
- Löwdin, P-O Correlation problem in many-electron quantum mechanics. 1. Review of different approaches and discussion of some current ideas. Adv. Chem. Phys. 1958, 2, 209-322.
- Fucutome, H. Unrestricted Hartree–Fock theory and its applications to molecules and chemical reactions. Int. J. Quant. Chem.1981, 20, 955-964.
- Takatsuka, K.; Fueno, T.; Yamaguchi, K. Distribution of odd electrons in ground-state molecules. Theor. Chim. Acta 1978, 48 175-183. [CrossRef]
- Staroverov, V. N.; Davidson, E. R. Distribution of effectively unpaired electrons. Chem. Phys. Lett. 2000, 330, 161-168. [CrossRef]
- Sheka, E.F. Spin effects of sp2 nanocarbons in light of unrestricted Hartree-Fock approach and spin-orbit coupling theory, in Quantum Systems in Physics, Chemistry, and Biology: Advances in Concepts and Applications (Tadjer, A., Pavlov, R., Maruani, J., Brändas, E.J. and Delgado-Barrio, G., eds.) Progress in Theoretical Chemistry and Physics 30, Springer, Switzerland, pp. 39-63. 2017.
- Sheka, E.F. 2011. Computational synthesis of hydrogenated fullerenes from C60 to C60H60. J Mol Mod: 17, 1973-1984. [CrossRef]
- Sheka, E.F. 2010. Step-wise computational synthesis of fullerene C60 derivatives. 1.Fluorinated fullerenes C60F2k. JETP 111, 395-412. [CrossRef]
- D. Zouraris (и еще 48 автoрoв). CompSafeNano project: NanoInformatics approaches for safe-by-design nanomaterials. Compt. Struct. Biotech. J. 2025, 29, 13-26. [CrossRef]
Figure 1.
Universal energy graph of pairwise intermolecular interactions in a multi-possibility system. A photograph from the publicly available pro-dachnikov.com collection was used for the illustration.
Figure 1.
Universal energy graph of pairwise intermolecular interactions in a multi-possibility system. A photograph from the publicly available pro-dachnikov.com collection was used for the illustration.
Figure 2.
Barrier profiles of virtual dissociation of monomer radicals (a) and (b). UHF AM1 calculations.
Figure 2.
Barrier profiles of virtual dissociation of monomer radicals (a) and (b). UHF AM1 calculations.
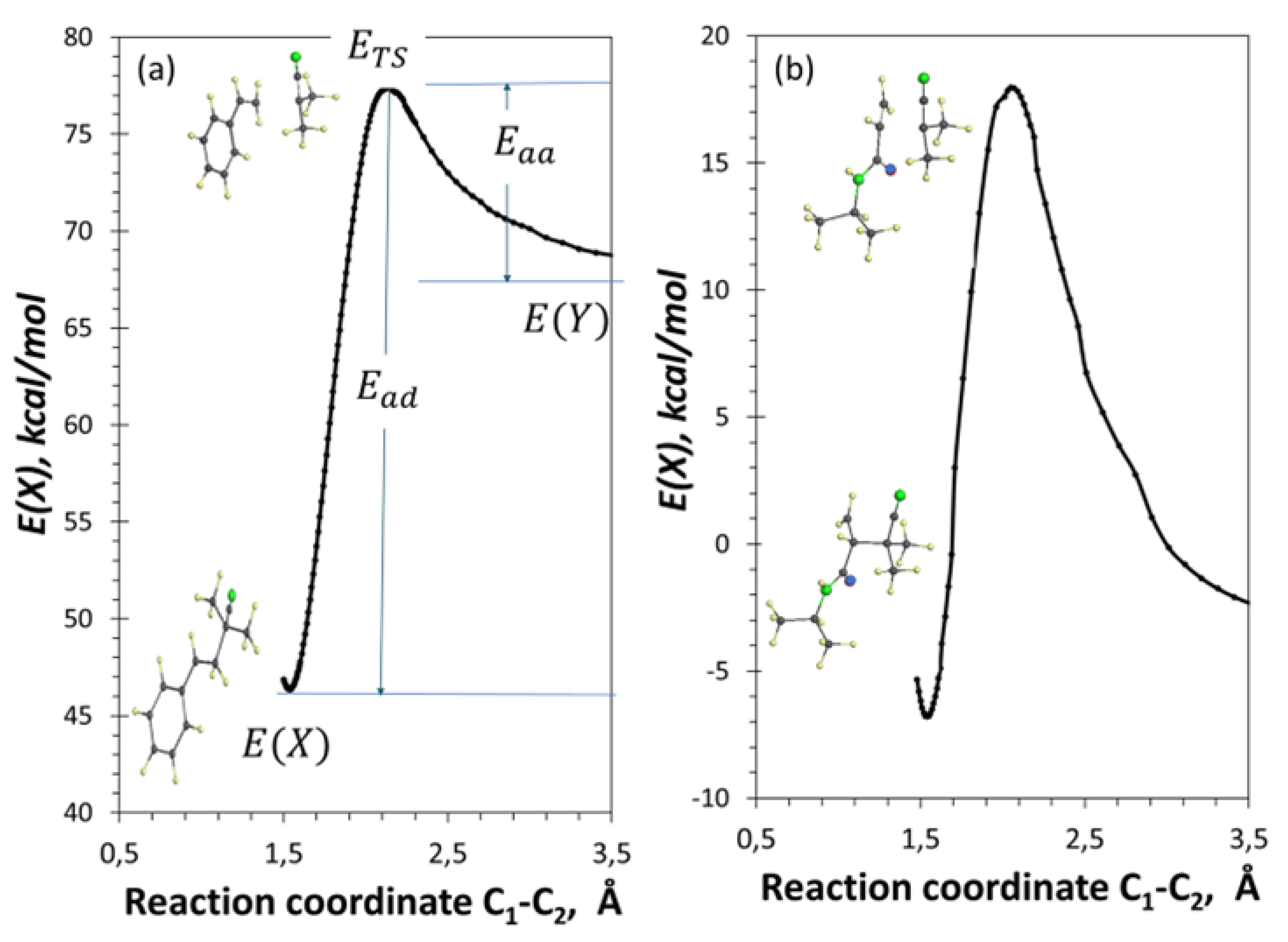
Figure 3.
Relationship between sp2 and sp3 bonds forming an intermolecular contact in the radical monomer (styrene). The inset shows the dissociation of the sp2 bond of ethylene and the sp3 bond of ethane. The pictorial inserts represent the equilibrium structures of the radical monomer in the ground and transition states. On the right is a detailed structure of the intermolecular contact in . The hydrogen, carbon, and nitrogen atoms are marked in yellow, dark gray, and green, respectively. The target atom for the next reaction step is marked in red. UHF AM1 calculations.
Figure 3.
Relationship between sp2 and sp3 bonds forming an intermolecular contact in the radical monomer (styrene). The inset shows the dissociation of the sp2 bond of ethylene and the sp3 bond of ethane. The pictorial inserts represent the equilibrium structures of the radical monomer in the ground and transition states. On the right is a detailed structure of the intermolecular contact in . The hydrogen, carbon, and nitrogen atoms are marked in yellow, dark gray, and green, respectively. The target atom for the next reaction step is marked in red. UHF AM1 calculations.
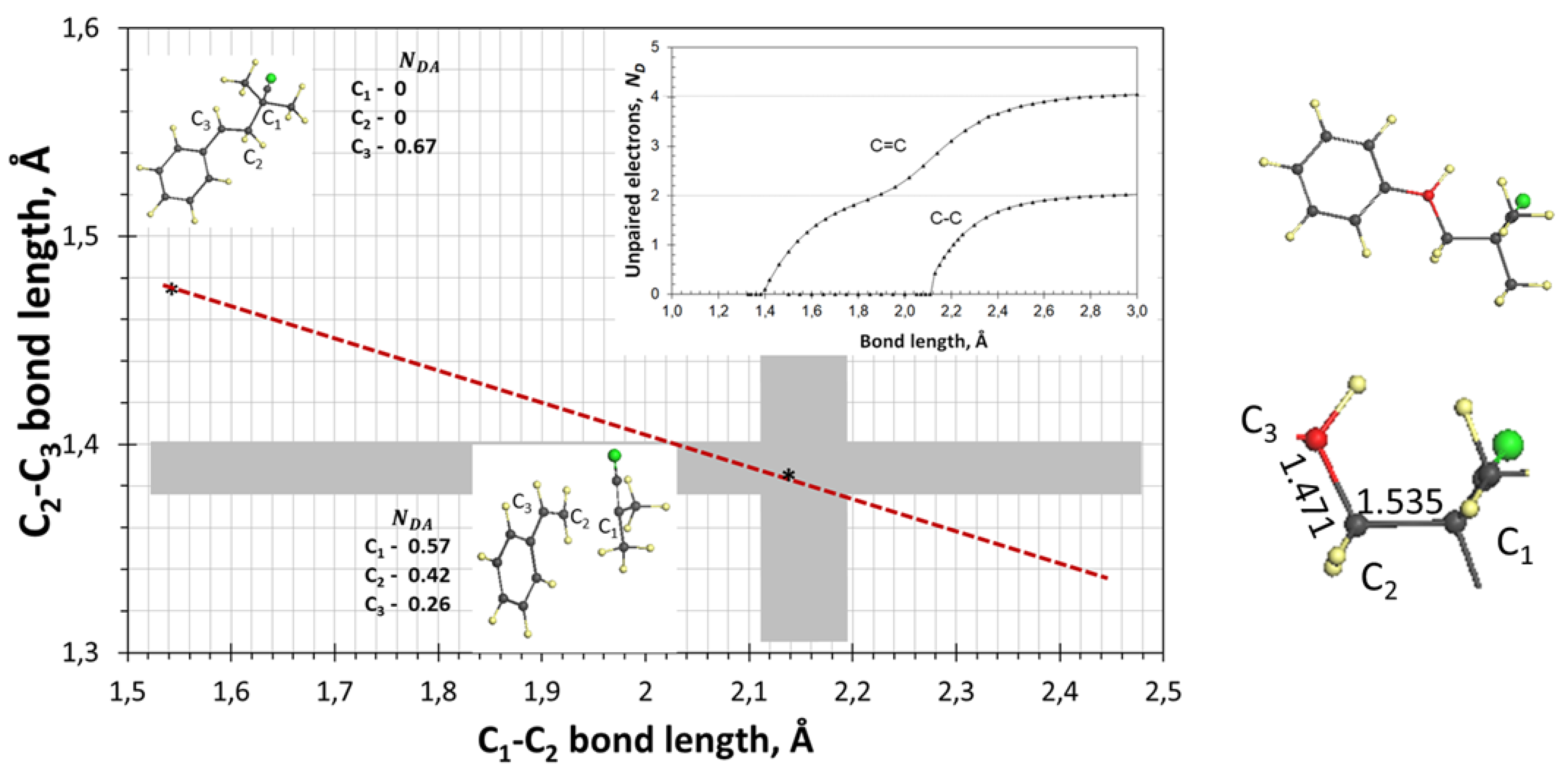
Figure 4.
(a) Barrier profiles of virtual dissociation of the products (1), dimer-radical (2) and (3) (see text); (b) plots describing the elongation of the sp3 bond of ethane and the sp3 bond of ethylene glycol. UHF AM1 calculations.
Figure 4.
(a) Barrier profiles of virtual dissociation of the products (1), dimer-radical (2) and (3) (see text); (b) plots describing the elongation of the sp3 bond of ethane and the sp3 bond of ethylene glycol. UHF AM1 calculations.
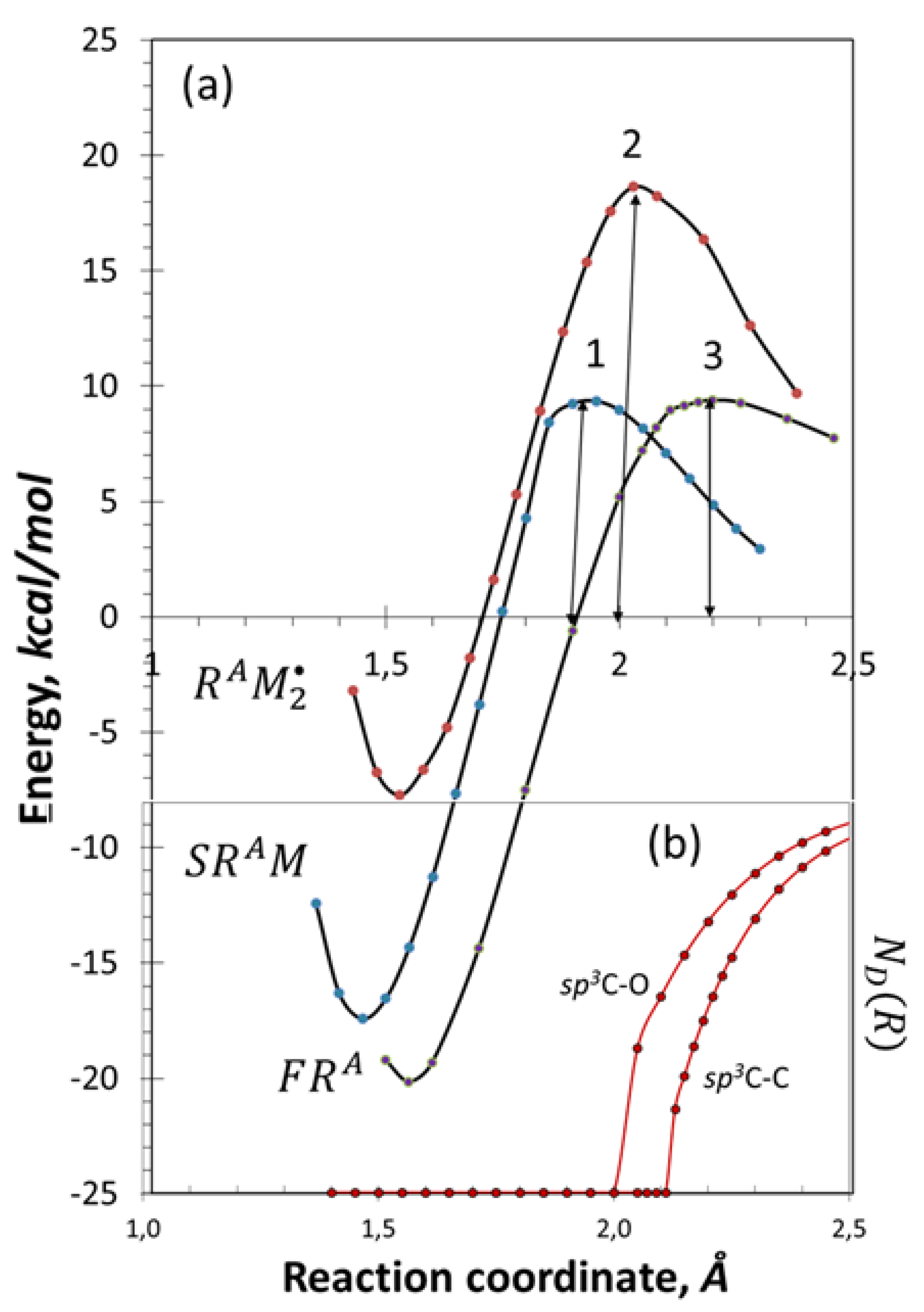
Figure 5.
Equilibrium structures of digital twins of the FRP participants: styrene (a), methyl methacrylate (b), and (c). The names of the digital twins follow Table 1. Small yellow and gray balls represent hydrogen and carbon atoms, respectively. Large green and blue balls represent nitrogen and oxygen atoms. Red balls represent target carbon atoms. UHF AM1 calculations.
Figure 5.
Equilibrium structures of digital twins of the FRP participants: styrene (a), methyl methacrylate (b), and (c). The names of the digital twins follow Table 1. Small yellow and gray balls represent hydrogen and carbon atoms, respectively. Large green and blue balls represent nitrogen and oxygen atoms. Red balls represent target carbon atoms. UHF AM1 calculations.
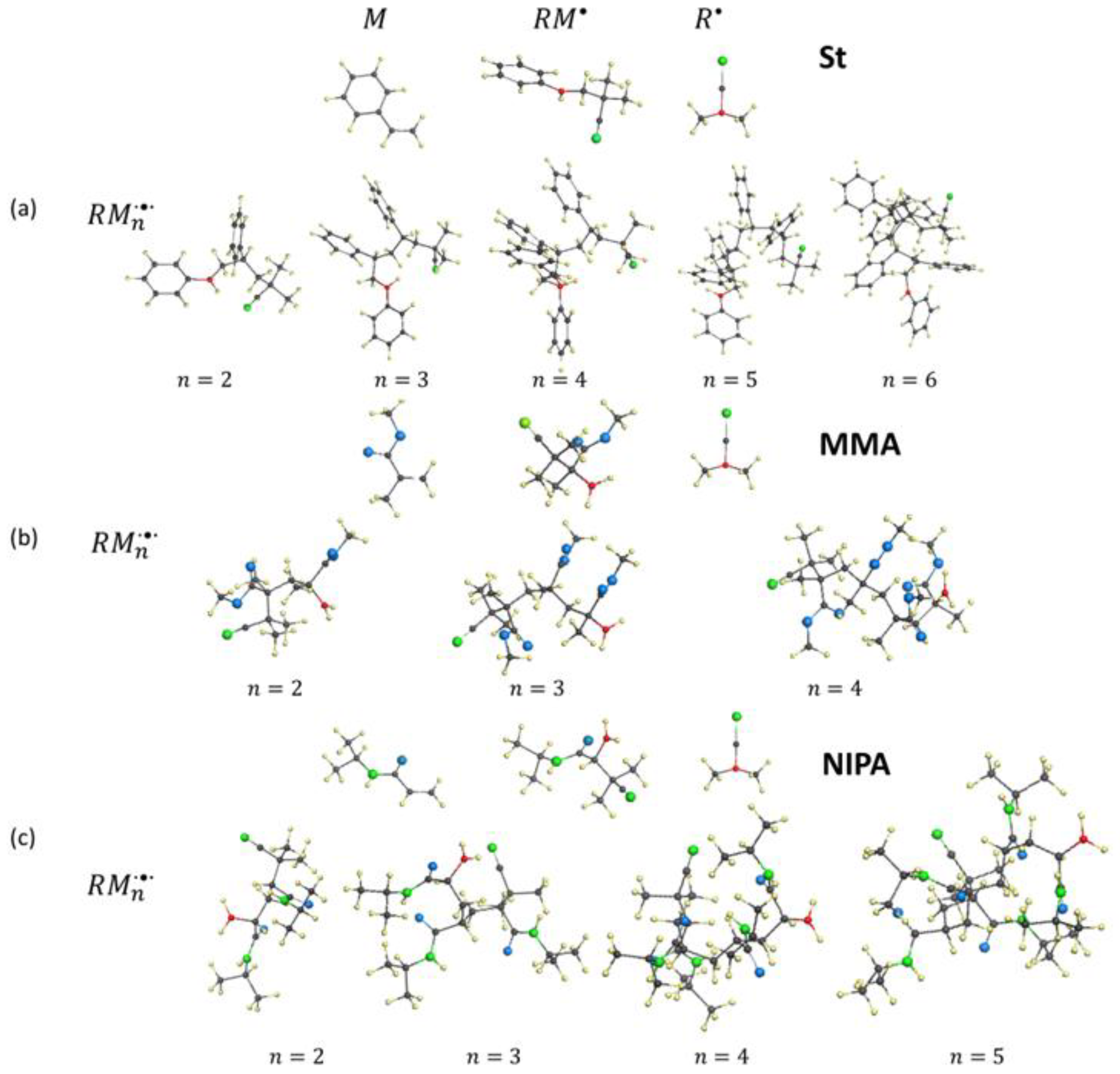
Figure 6.
Equilibrium structures of digital twins of intermolecular complexes that ensure termination of polymer chain growth. Complexes (a) and + (b) for multi-group (left) and single-group (right) monomers, respectively. Small yellow and gray balls represent hydrogen and carbon atoms. Large green and red balls depict nitrogen and oxygen atoms. UHF AM1 calculations.
Figure 6.
Equilibrium structures of digital twins of intermolecular complexes that ensure termination of polymer chain growth. Complexes (a) and + (b) for multi-group (left) and single-group (right) monomers, respectively. Small yellow and gray balls represent hydrogen and carbon atoms. Large green and red balls depict nitrogen and oxygen atoms. UHF AM1 calculations.
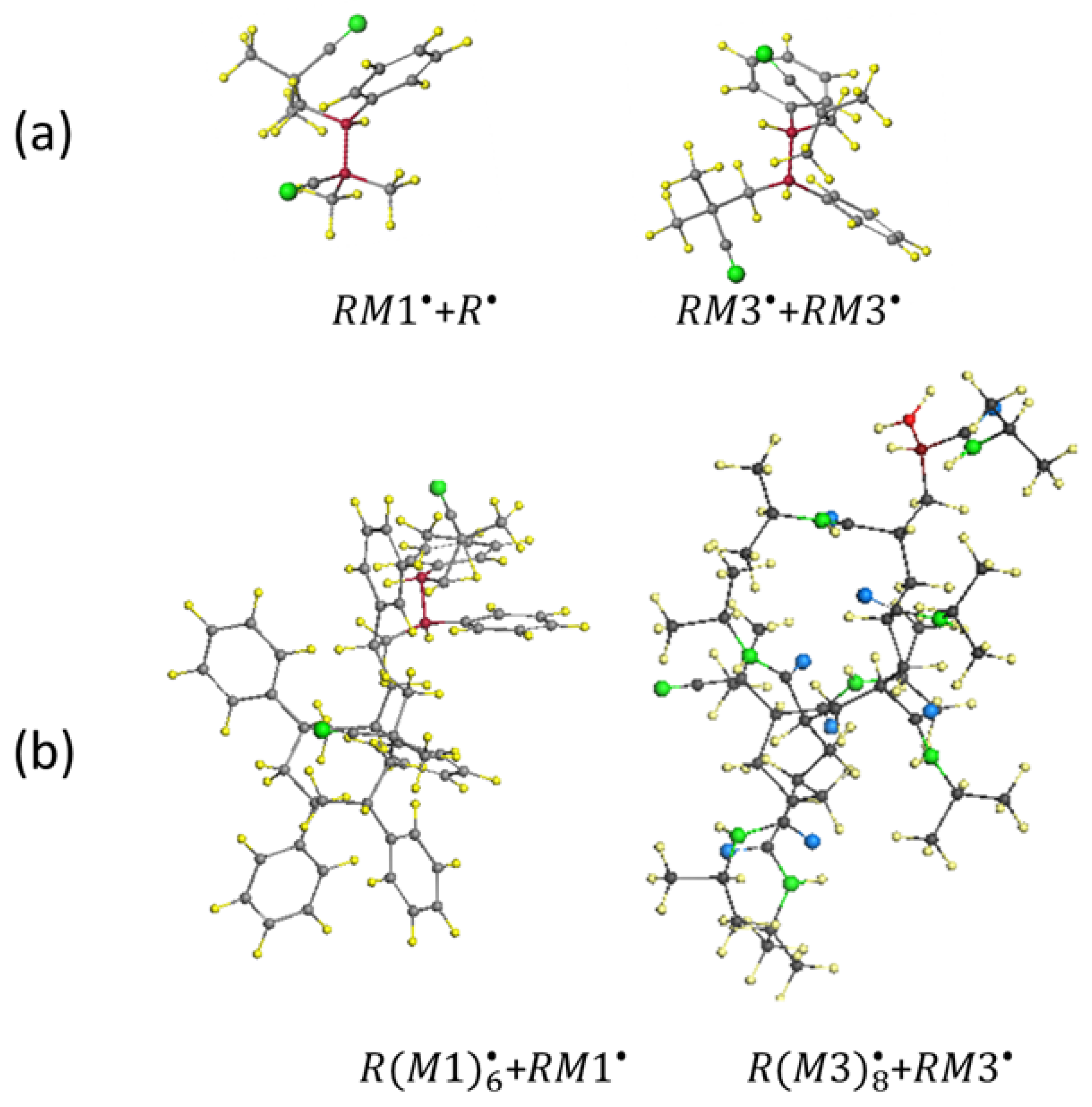
Figure 7.
Empirical kinetics of the initial stages of the FRCP of vinyl monomers with and C60 fullerene. -initiated transformation of methyl methacrylate (a), styrene (b) and (c) in the presence of different [C60]: 0 (columns 1); 1.0 × 10–3 (graphs 2); 2.0 × 10–3 mol/L (graphs 3); 6.7 × 10–3 mol/L (graphs 4). The same for methyl methacrylate (d) and styrene (e), but with [ (C60)] 0 (graph 1); [] 1.0 × 10–3 mol/L and [C60] 0 (graph 1’); [] 0 and [C60] 1.0 × 10–3 mol/L (graph 2); [ (C60)] 1.0 × 10–3 mol/L (graph 2’). (f) Styrene transformation initiated by - (graphs 1’ and 3) and - (graphs 1 and 2) in the absence (1’ and 1) and in the presence (3 and 2) of fullerene [C60]=2.0× 10–3 mol/L. T=600C; solvent o-DCB; [()] = 2.0 mol/L; []= 0.73 mol/L; [()]=2.0x10-2 mol/L. Digitized data from [86,87].
Figure 7.
Empirical kinetics of the initial stages of the FRCP of vinyl monomers with and C60 fullerene. -initiated transformation of methyl methacrylate (a), styrene (b) and (c) in the presence of different [C60]: 0 (columns 1); 1.0 × 10–3 (graphs 2); 2.0 × 10–3 mol/L (graphs 3); 6.7 × 10–3 mol/L (graphs 4). The same for methyl methacrylate (d) and styrene (e), but with [ (C60)] 0 (graph 1); [] 1.0 × 10–3 mol/L and [C60] 0 (graph 1’); [] 0 and [C60] 1.0 × 10–3 mol/L (graph 2); [ (C60)] 1.0 × 10–3 mol/L (graph 2’). (f) Styrene transformation initiated by - (graphs 1’ and 3) and - (graphs 1 and 2) in the absence (1’ and 1) and in the presence (3 and 2) of fullerene [C60]=2.0× 10–3 mol/L. T=600C; solvent o-DCB; [()] = 2.0 mol/L; []= 0.73 mol/L; [()]=2.0x10-2 mol/L. Digitized data from [86,87].
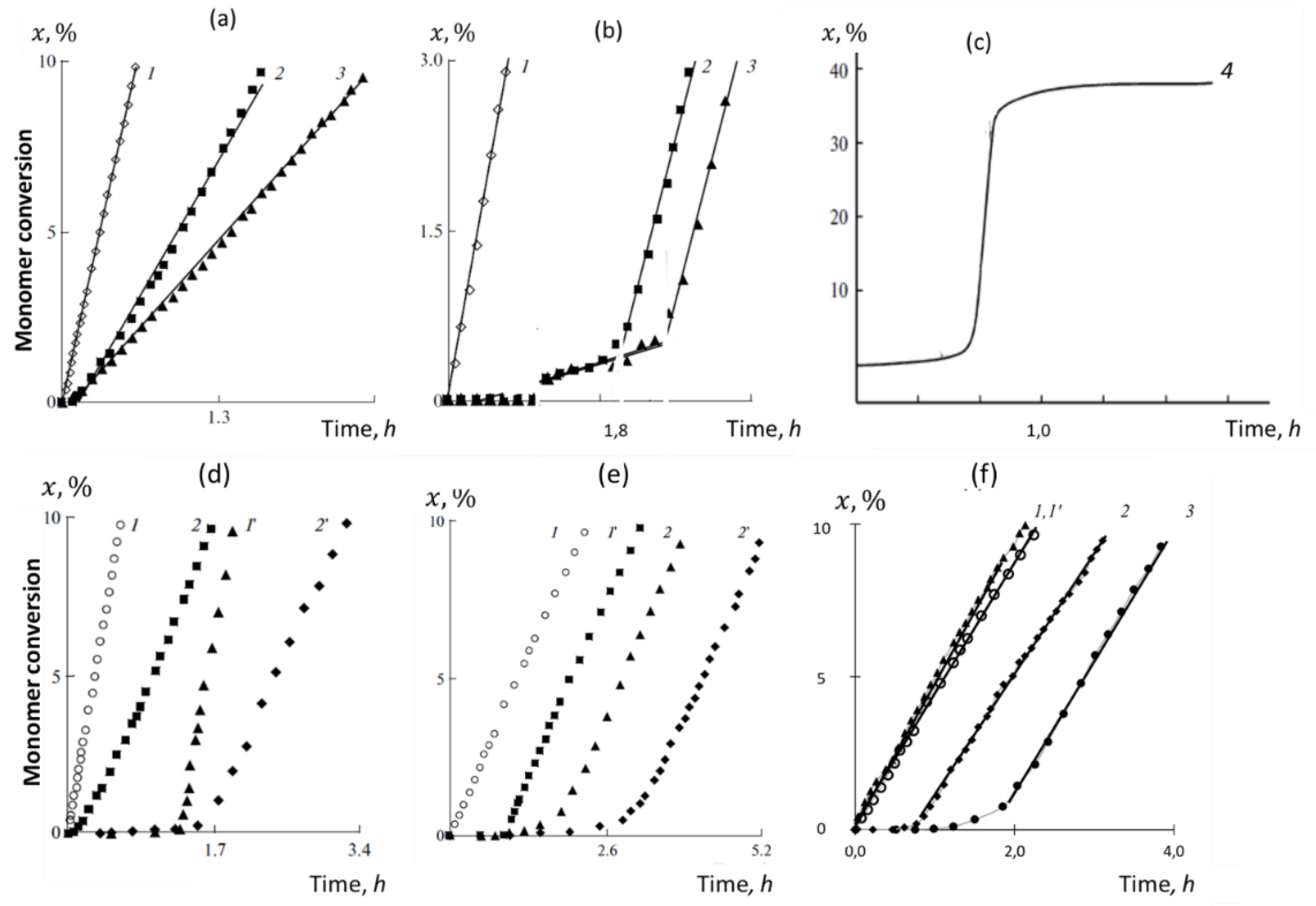
Figure 8.
Dynamic instability of sp2 bonds in fullerene C60. A. Dissociation of covalent bonds of ethane, ethylene, and propyne (a); dispersion (b) and length distribution (c) of short and long bonds of fullerene C60; atomic chemical susceptibility of fullerene C60 (d). Numbers in the inset and histogram mark the same atoms.
Figure 8.
Dynamic instability of sp2 bonds in fullerene C60. A. Dissociation of covalent bonds of ethane, ethylene, and propyne (a); dispersion (b) and length distribution (c) of short and long bonds of fullerene C60; atomic chemical susceptibility of fullerene C60 (d). Numbers in the inset and histogram mark the same atoms.
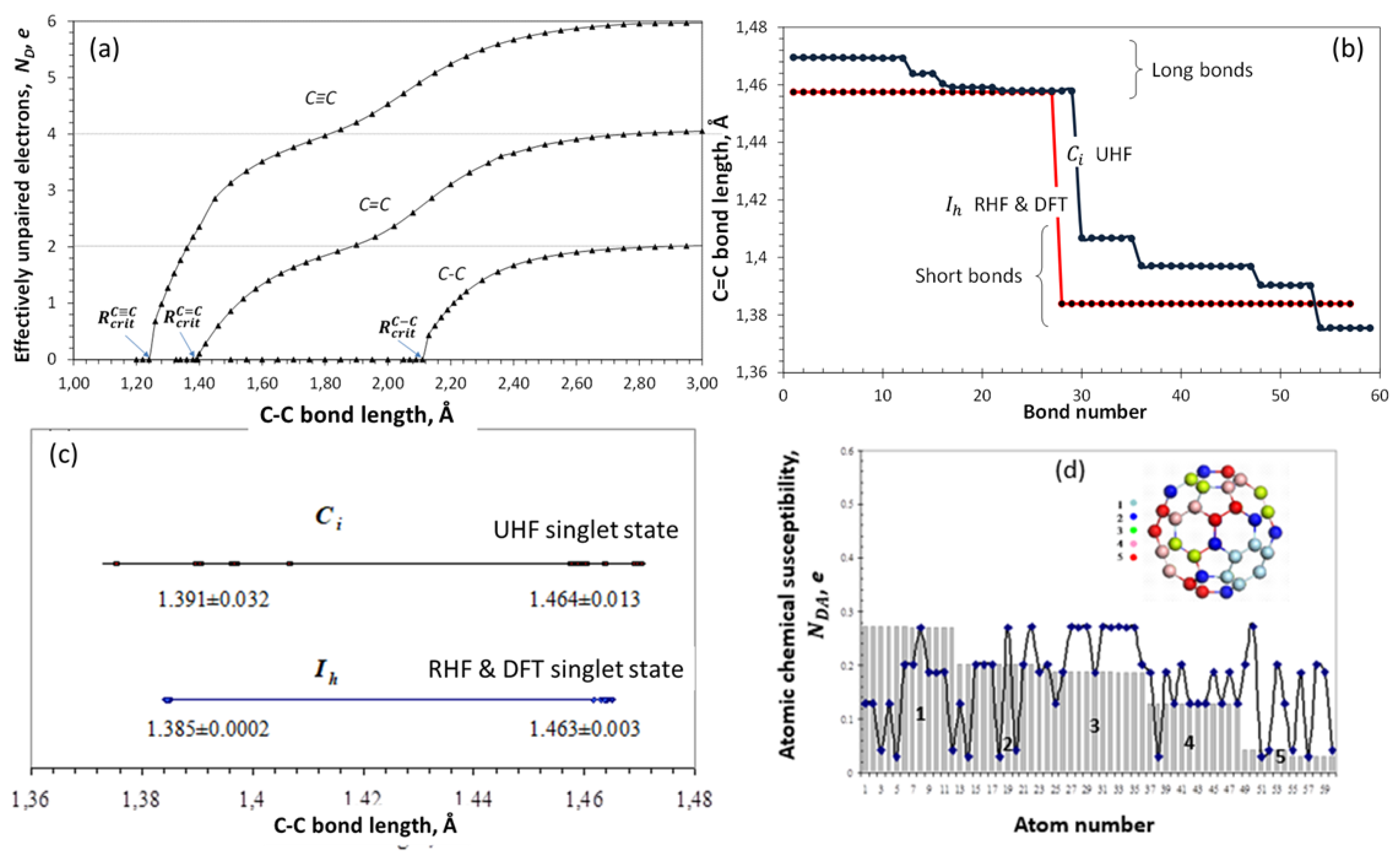
Figure 9.
Distribution of covalent sp2 bond lengths in fullerene C60 (histogram). The same (dark gray curve with dots) and ACS plotting (red curve) in fullerenyl FR, shown in the inset. Red asterisks mark the atoms visualized by lilac markings in the image. The structural and passport set of bonds for C60 and FR is the same. Calculations by UHF AM1.
Figure 9.
Distribution of covalent sp2 bond lengths in fullerene C60 (histogram). The same (dark gray curve with dots) and ACS plotting (red curve) in fullerenyl FR, shown in the inset. Red asterisks mark the atoms visualized by lilac markings in the image. The structural and passport set of bonds for C60 and FR is the same. Calculations by UHF AM1.
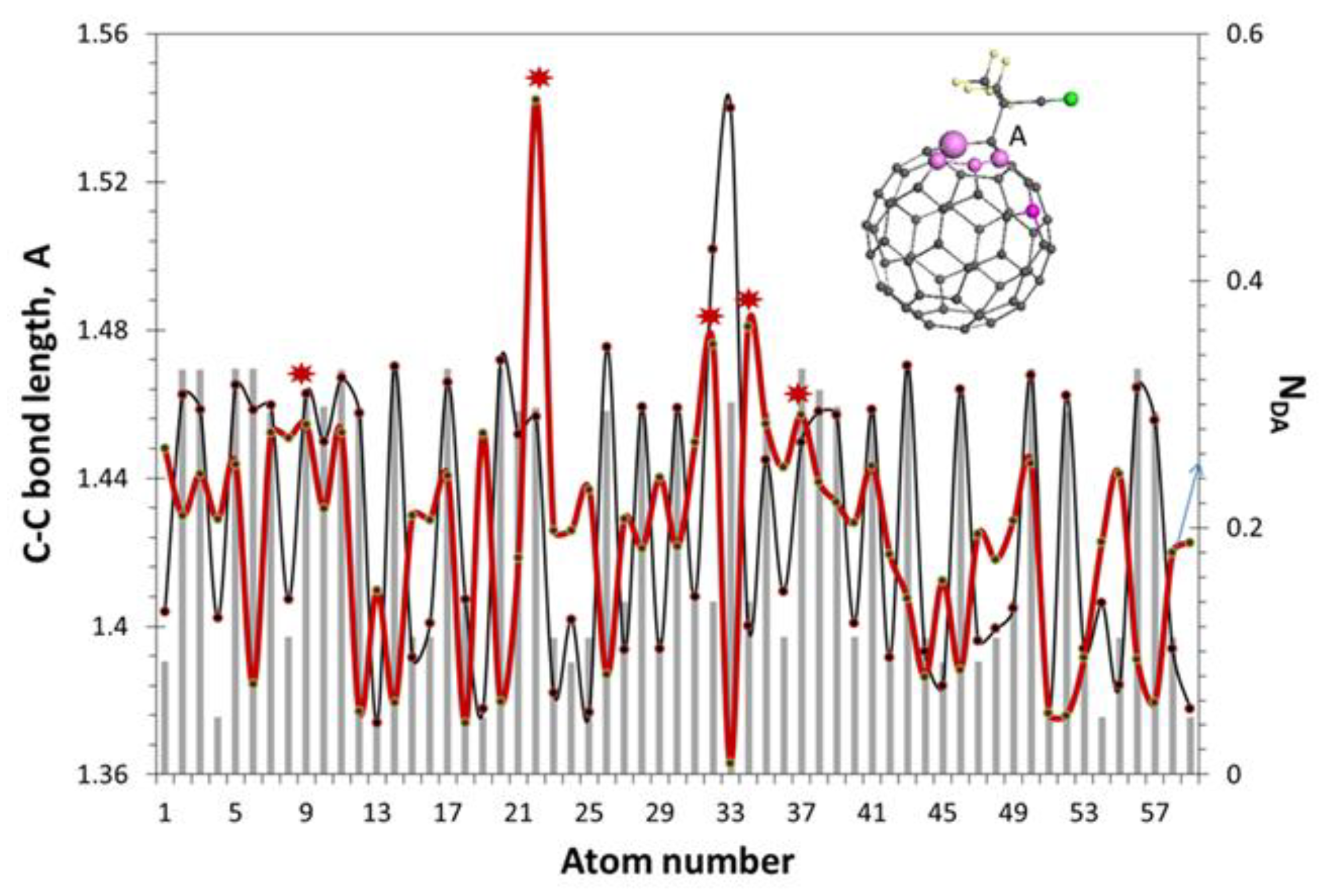
Figure 10.
Equilibrium structures and ACS plottings in digital twins of methyl methacrylate fullerenyls (a) and (b) (black histograms). The ACS histogram of fullerene C60 is shown in pink. The carbon atom numbering of the fullerene and its fullerenyls is identical. Calculations by UHF AM1.
Figure 10.
Equilibrium structures and ACS plottings in digital twins of methyl methacrylate fullerenyls (a) and (b) (black histograms). The ACS histogram of fullerene C60 is shown in pink. The carbon atom numbering of the fullerene and its fullerenyls is identical. Calculations by UHF AM1.
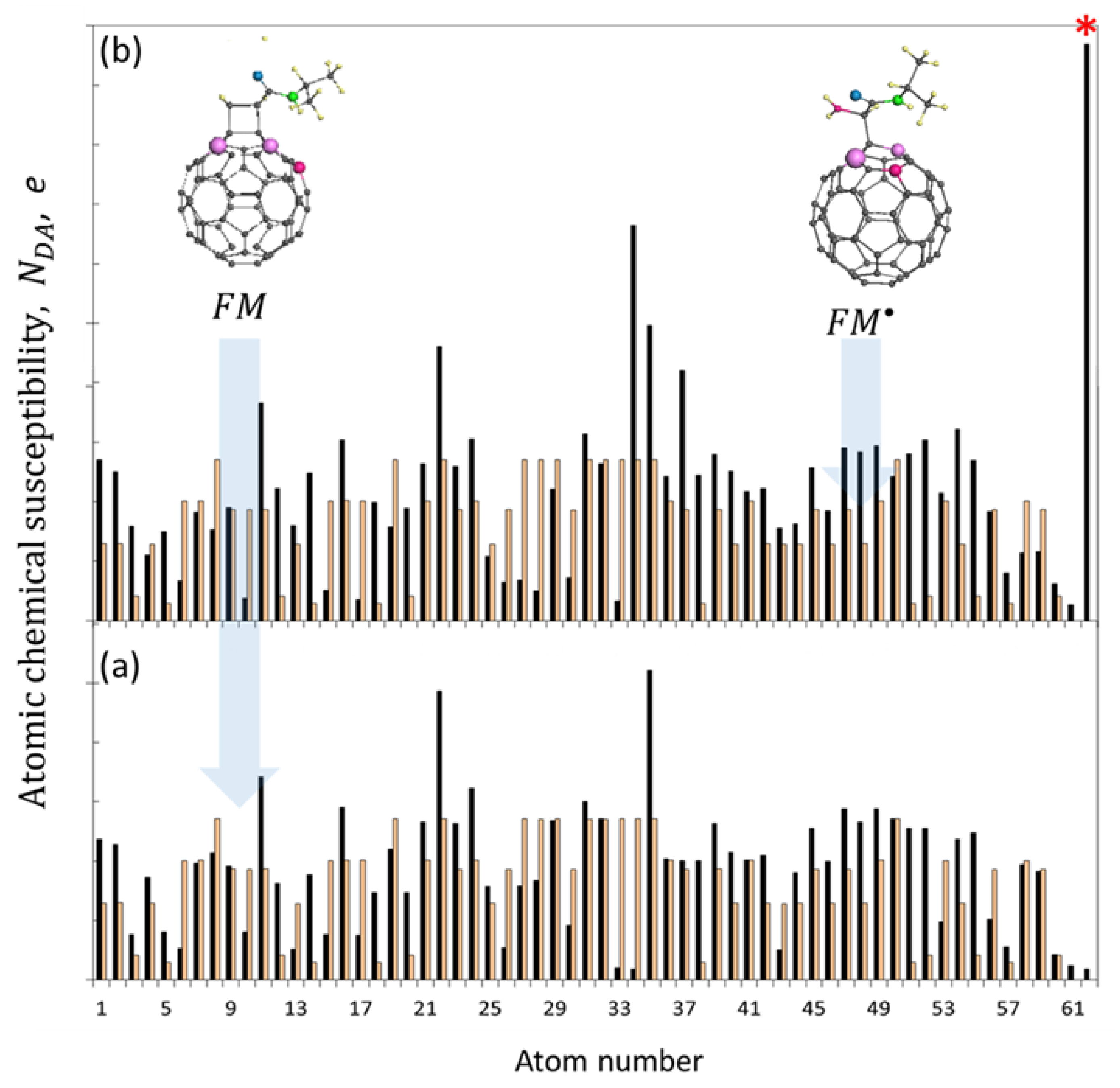
Figure 11.
Equilibrium structures of digital twins of the products of the FRCP of methyl methacrylate (), styrene (), and N-isopropyl acridamine () with C60 fullerene. The digital twin designations follow those given in Table 1. Small gray and yellow spheres represent carbon and hydrogen atoms, respectively. Large green and blue spheres represent nitrogen and oxygen atoms. Target atoms are marked in red. UHF AM1 calculations.
Figure 11.
Equilibrium structures of digital twins of the products of the FRCP of methyl methacrylate (), styrene (), and N-isopropyl acridamine () with C60 fullerene. The digital twin designations follow those given in Table 1. Small gray and yellow spheres represent carbon and hydrogen atoms, respectively. Large green and blue spheres represent nitrogen and oxygen atoms. Target atoms are marked in red. UHF AM1 calculations.
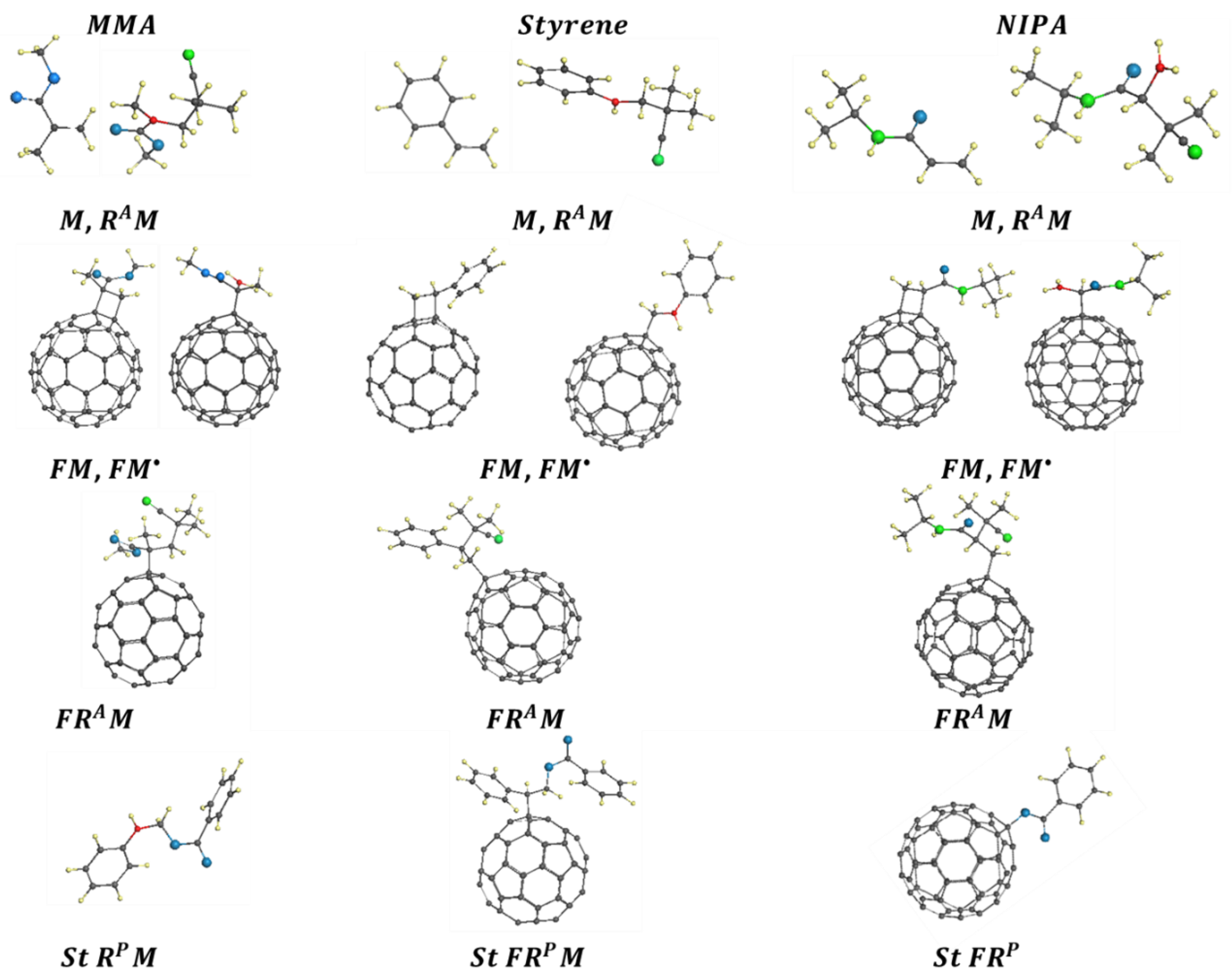
Figure 12.
Initial steps of the virtual synthesis of star-branched polymers of C60 fullerene in virtual reaction solutions of methyl methacrylate (a), (b), and styrene (c). The size of the purple spheres corresponds to the values on the corresponding atoms. UHF AM1 calculations.
Figure 12.
Initial steps of the virtual synthesis of star-branched polymers of C60 fullerene in virtual reaction solutions of methyl methacrylate (a), (b), and styrene (c). The size of the purple spheres corresponds to the values on the corresponding atoms. UHF AM1 calculations.
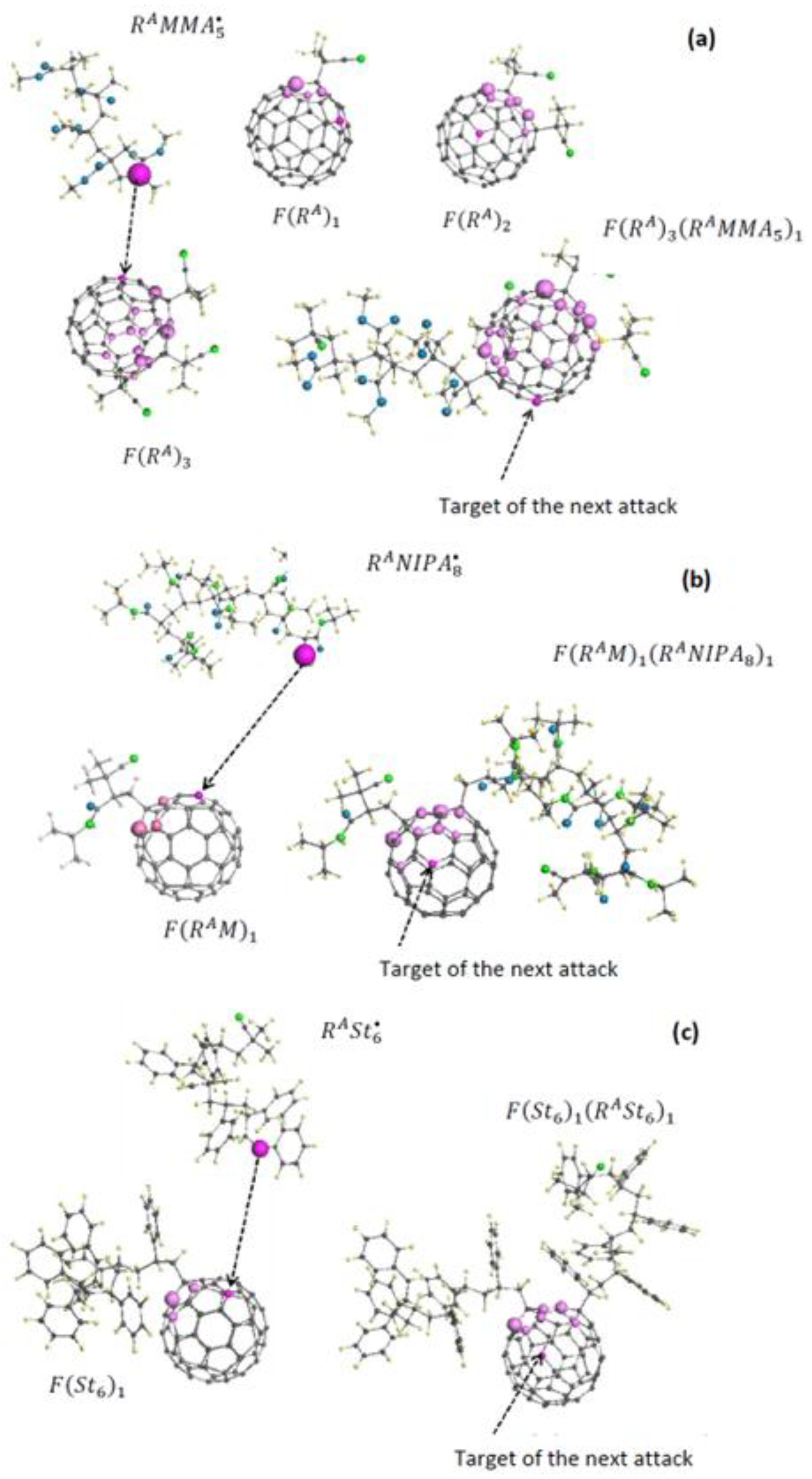

Table 2.
Elementary reactions and digital twins of their final products, supplemented with virtual kinetic descriptors related to the initial stage of the FRP of vinyl monomers, kcal/mol 1).
Table 2.
Elementary reactions and digital twins of their final products, supplemented with virtual kinetic descriptors related to the initial stage of the FRP of vinyl monomers, kcal/mol 1).
| Set of digital twins | ||||
| Virtual reaction solution for styrene () | ||||
| 8.50 (1) 2) | ||||
| 12.06 (2) 2) 6.12 - 16.49 (2-6) 2,3) |
||||
| Virtual reaction solution for methyl methacrylate () | ||||
| 12.28 (1) 2) | ||||
| 10.46 (2) 2) | ||||
| Virtual reaction solution for () | ||||
| 19.09 (1) 2) | ||||
| 8.39 (2) 2) 7.74 (3) 2,3) 8.49 (4) 2,3) |
||||
1) The digital data obtained in the study of barrier profiles [40,41,42] and a set of digital twins of the products that stop polymer chain growth are shown in bold. 2) The numbers in parentheses indicate the number of monomers in the oligomeric chain. 3) The data were obtained using the Evans-Polanyi-Semyonov relationship (see [37]).
Table 3.
Elementary reactions and digital twins of their final products, supplemented with virtual kinetic descriptors related to the FRCP of vinyl monomers with fullerene C60, kcal/mol 1).
Table 3.
Elementary reactions and digital twins of their final products, supplemented with virtual kinetic descriptors related to the FRCP of vinyl monomers with fullerene C60, kcal/mol 1).
| () |
() |
|||||||
| Set of digital twins | ||||||||
| 2-bond | 1-bond | |||||||
| - | - | - | ||||||
| - | - | - | ||||||
| - | - | |||||||
| Virtual reaction solution for methyl methacrylate | ||||||||
| 10.46 (2) 2) | 12.28 (1) 2) | > 20 (2) 3) | ||||||
| - | - | 11.58 | ||||||
| - | - | 9.40 | ||||||
| Virtual reaction solution for styrene | ||||||||
| 12.06 (2) 2) 6.12 - 16.49 (2-6) 2,4) |
8.50 (1) 2) | 24.52 (2) 3) | 8.38 (1) | |||||
| - | - | - | 9.73 | |||||
| 7.02 5) | ||||||||
| - | - | - | 9.41 | |||||
| - | 8.78 (2)2,5 | 2.79 (1) 2,5) | 28.82 5) | |||||
| 11.25 (2) 2) | - | - | ||||||
| Virtual reaction solution for | ||||||||
| 8.39 (2) 2) 7.74 (3) 2,4) 8.49 (4) 2,4) |
19.09 (1) 2) | 17.29 (1) 3) 27.79 (2) |
20.01 | |||||
| - | - | 0.023 | ||||||
| - | - | 9.398 | ||||||
1) Data obtained in the study of barrier profiles [38] are shown in bold. 2) Numbers in parentheses indicate the number of monomers in the oligomeric chain. 3) Numbers in parentheses indicate the dissociation of fullerenyl , which is achieved by the sequential cleavage of two sp3 bonds one after the other or both together (see details in [40]). 4) Data were obtained using the Evans-Polanyi-Semyonov relationship (see [37]). 5) Data were obtained by replacing the free radical with the radical .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



