Submitted:
03 February 2026
Posted:
05 February 2026
You are already at the latest version
Abstract
This review presents the covalent chemistry of carbon within the spin-radical concept of electron interaction. Using the language of valence bond trimodality, the regions of classical spinless covalence and its spin counterpart are defined. Carbon is the only element exhibiting spin covalent chemistry. Classical covalent chemistry of carbon concerns molecular substances whose valence bond structure includes segregate or chained single sp3C-C bonds. Substances with double sp2C=C and triple sp1C≡C bonds are the subject of spin covalent chemistry of carbon. The mathematical apparatus of spin covalence forms the basis of algorithms governing the chemical modification of carbon substances, polymerization processes, and catalysis involving them, making it possible to supplement the empirical spin covalent chemistry of carbon with its virtual analog.
Keywords:
valence bond trimodality
; classical spinless covalent chemistry
; spin covalent chemistry
; spin-radical electron interaction
; virtual spin covalent chemistry
1. Introduction
The covalent realm of modern chemistry is populated
by pairs of bonded neutral atoms, representing one of the most deeply ingrained
and convenient graphical representations of interatomic interactions in matter.
The wide diversity of these pairs, possible for virtually any element in the
Mendeleev’s periodic table, is the fundamental basis of this representation's
perfection. Carbon stands out in this context—an atom that forms only covalent
bonds with atoms of other elements and possesses the unique ability to form
three types of covalent bonds with itself. The bond trimodality is so
widespread in nature that it allows us to distinguish a distinct, extensive
class of substances whose atoms are held together by purely carbon covalent bonds, realized as a three-mode set of sp3, sp2 and sp1bonds. The prefix spn used to
describe them denotes one of three possible configurations of the and electrons that provide the chemical bond between
two carbon atoms. The empirical discovery and theoretical justification of the
trimodality of bonds underly the covalent chemistry of pure
carbon compounds, which consist of both branched networks of and lone C2
atomic pairs. The first configuration is characteristic of solid-state pure spn
carbons and polymers, while the second applies to individual molecules, the
number of which is virtually countless.
This article discusses spn
carbons, both solid-state and molecular, whose remarkable properties allow us
to uncover the full depth of the chemical nature of this element. The article
is structured as follows. Section 2 briefly
presents the empirical picture of solid-state and molecular spn
carbon. The determining factors of the spin character of spn
carbon chemistry and the spin emergents of its covalent chemistry are presented
in Section
3. The spin chemistry of alkane, alkene, and alkyne bonds is
described in Sections
4, 5, and 6, respectively. Carbon catalysts are discussed in Section
7 in light on their spin covalence. Section
8 presents aposteriori reflections and concluding remarks.
2. Carbon in the Language of Covalent Bonds
The Earth's carbon world is extremely rich and
diverse. It comprises most of the planet's mineral kingdom and all living
matter. Thus, Nature, by bringing human beings into contact with carbon,
immediately places them in a world of big numbers, touching upon virtually
every aspect of the carbon environment: many species, many variations of each
species, countless cells, a vast number of atoms and molecules. From the moment
of contact with the carbon component of the world, humans have ceaselessly
searched for the patterns that govern it. The entire development of natural
science represents gradual steps toward understanding how this world unfolds.
Discovered first patterns, then replaced by others, carefully selected bits of
knowledge, and chemical, biological, physical, geological, and cosmogenic
understandings of the role and place of carbon in Nature has been gradually
developed. Today, the birth of a mathematical view of carbon as the foundation
of the carbon world of the metaverse is underway. The carbon atom and the
molecules it formed laid the foundation of these concepts. The atom as the
basic building block and the molecule as the fundamental configuration of
atoms, ensuring the existing reality of large numbers.
Molecular theory gave rise to the concept of
interatomic bonds, and from that moment on, the world of carbon took on new
colors. As it turned out, Nature endowed the carbon atom with the unique
ability to form stable bonds with virtually all chemical elements, particularly
highlighting its bonds with itself, noting their distinctive three-modal
nature: sp1, sp2, sp3. This trimodality concept proved not only a convenient mathematical device but
also touched upon the depths of the multi-dimensional nature of the carbon
world, greatly increasing the variability of its chemical, biological,
physical, geological, and cosmogenic manifestations, on the one hand, and
serving as a fundamental, common concept for all of these manifestations, on
the other. The differences in the properties of material carbon, determined by
the trimodality of its bonds, are also common to all manifestations, regardless
of whether they are observed in space or in a living organism on Earth.
This idea of the conceptual unity of the carbon
world underlies the discussion below. The subject of this discussion is digital
twins, which represent molecular configurations of carbon atoms corresponding
to solid forms of elemental carbon (crystals and amorphous forms) and a
selected set of real-life molecular carbon. The trimodality of covalent bonds and its influence on the properties of
digitized material objects is the main focus of this discussion. The patterns
established by this approach represent a common property of the entire carbon
world.
2.1. Crystalline Carbon
Solid carbon is a unique material with an internal
molecular structure, which can be viewed most favorably from the perspective of
sp1, sp2 and sp3trimodality.
This material carbon occupies a significant portion of the Earth's crust [1]
and is also present in small quantities in space. A large part of the material
solid carbon is produced industrially. Figure 1 shows the
currently known crystal structures of elemental carbon as one of the possible
sets of their digital twins. The well-known crystalline diamond with a cubic
structure (Figure
1a) opens the presentation. Each carbon atom is bonded to four
nearby (symmetrically located at the tetrahedron vertices) covalent sp3bonds. An ideal crystal can be imagined as a single
giant sp3 molecule [2]. Natural diamond
deposits, estimated at several million carats, are widespread across the
planet. Compared to this crystalline cubic carbon giant, lonsdaleite, or
hexagonal diamond, is an alien from outer space and is found primarily in
meteoritic debris [3,4]
(Figure
1b). As with diamond, its molecular chains of atoms are formed by
sp3 bonds.
In Figure 1c, the sp3
structure gives way to the sp2 structure of graphite, whose
triples of sp2bonds form a planar trigonal configuration [5].
Unlike the closely packed sp3 structures, graphite consists
of parallel layers formed by regular hexagons of carbon atoms linked to each
other by sp3bonds. As usually accepted, atoms of adjacent
layers are not covalently bound and the layers are coupled by Van der Waals
attraction. There are two modifications of graphite: α-graphite (hexagonal) and
β-graphite (rhombohedral), which differ in the layer arrangement. β-graphite is
not observed in pure form, as it is a metastable phase. However, in natural
graphite, the rhombohedral phase content can reach 30%. Graphite is widespread
in nature, with estimated reserves of 250-300 million tons.
As can be seen from the figure, diamond is a
three-dimensional structural carbon modification composed of sp3
atoms. Graphite is a two-dimensional layered allotropic carbon modification
formed by sp2-hybridized atoms. The third one-dimensional
linear allotropic modification, corresponding to the sp1
hybridization, could not be discovered for a long time. This substance was
first obtained artificially by Soviet chemists in the early 1960s and was named
"carbyne" [6].
According to the discoverers, the most difficult thing for them was to
determine what kind of bonds connected the carbon atoms in carbyne into a
chain. These could be alternating single and triple bonds of polyyne chains, only double bonds () of polyene chains, or both simultaneously. Over
time, it was possible to prove that there are no double bonds in the carbyne.
This substance, consisting of polyyne chains, was named α-carbyne. According to
microdiffraction data, single-crystal films of α-carbyne possess a hexagonal
lattice [7,8].
Figure
1d shows a carbyne spatial model. The numerals (l-5) denote
carbyne chains, the positions of which in a unit cell of the carbyne crystal in
a (0001) projection is shown in the inset. It can be seen that the carbyne
lattice is double-layered. The lower layer consists of closely packed carbyne
chains. The columns of the chains in the upper layer have vacancies presumably
due to impurity atoms such as Fe or K intercalated between the layers. These
atoms can provide saturation of the dangling bonds.
In 1968, carbyne-like carbon was found in
geological rocks formed in a meteorite crater [9]. To date,
approximately two tens of carbyne-like materials differing in structural
parameters have been artificially synthesized and revealed in nature [10–13].
Strong chemical activity and extreme instability at ambient conditions
characterize carbyne, an infinite sp1 - sp3
carbon chain. As a result, much less has been explored about carbyne as
compared to other carbon allotropes. Although end-capping groups can be used to
stabilize carbon chains, length limitations are still a barrier for production.
To overcome the difficulty, a method for the bulk production of long carbyne
linear chains protected by thin double-walled carbon nanotubes and composed of
more than 6000 atoms has been proposed [14].
Figure 1.
Digital twins of crystalline carbon allotropes. (a) Diamond. (b) Lonsdaleite. (c) Graphite. (d) Carbyne. (f) Y-diamond. (e) α (top) and β (bottom) Grahynes. (g) γ Grahyne. (i) Graphdiyne. Images taken from open sources.
Figure 1.
Digital twins of crystalline carbon allotropes. (a) Diamond. (b) Lonsdaleite. (c) Graphite. (d) Carbyne. (f) Y-diamond. (e) α (top) and β (bottom) Grahynes. (g) γ Grahyne. (i) Graphdiyne. Images taken from open sources.
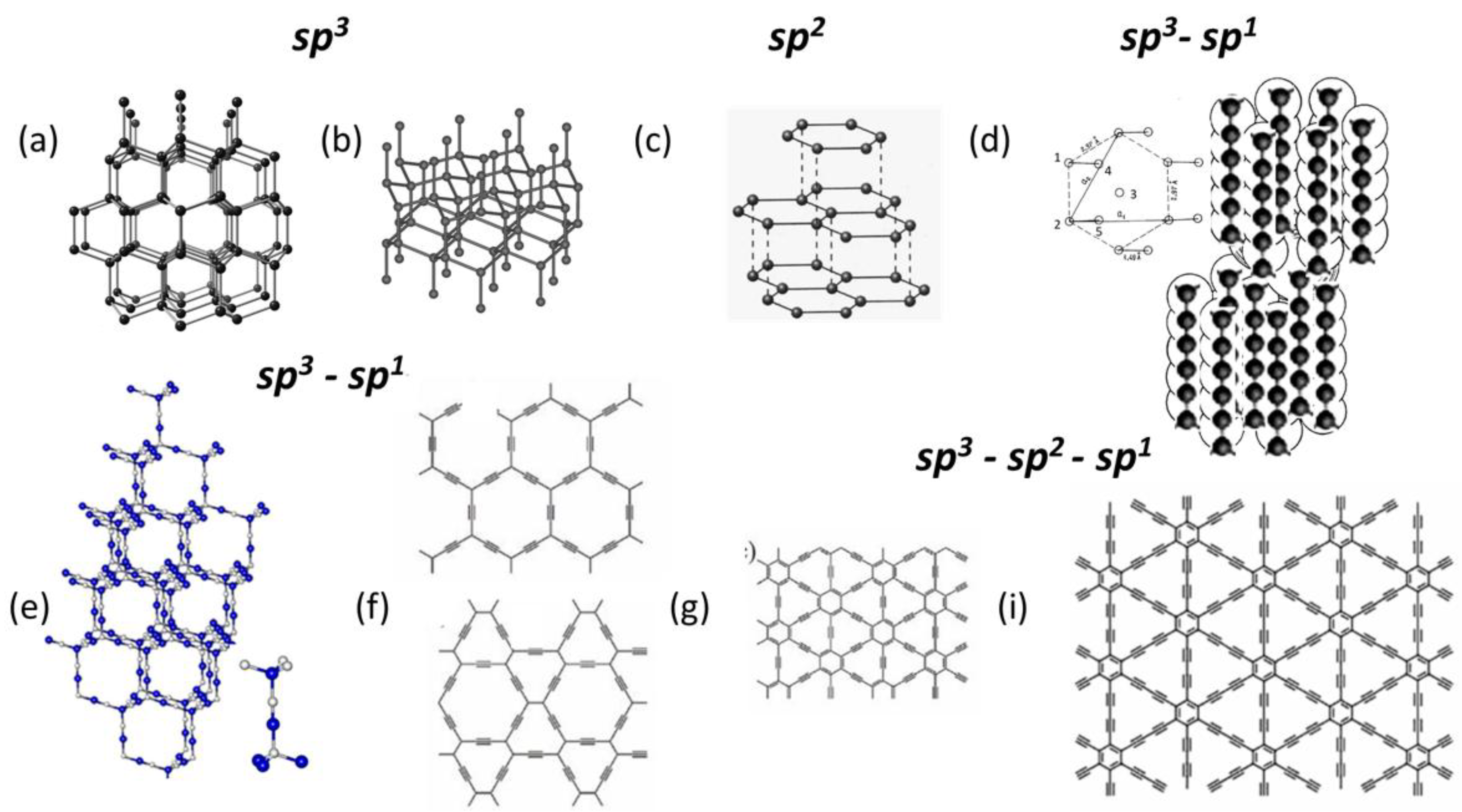
The existence of crystalline carbyne revealed a
very important property concerning the covalent bonds of carbon. It had to be
recognized that while stable polyyne configurations of carbon atoms linked
solely by sp1bonds are impossible, a stable configuration can
nevertheless be created by allowing these bonds to coexist with sp3and sp2 bonds. As it turned out, in addition to carbyne,
it is possible to construct regular carbon configurations involving a variety
of bond types. Thus, preliminary hints at the carbon sp3 – sp1
3D structures (which were called yne-diamonds or Y-diamonds) can be
found in early works [14–16],
where the insertion of acetylenic ) or diacetylenic linkages inside the sp3 skeleton
of diamond was assumed. Figure 1e presents the sp3–
sp1 configuration, demonstrating the structure of a virtual
Y-diamond crystal [17,18].
This mixed-bond configuration is predicted to be stable, so Y-diamond crystals
may be synthesized in the near future.
As can be seen from Figures 1a and 1e,
Y-diamond is the result of a structural transformation of diamond, in which all
sp3bonds are replaced by ternary sp3–
sp1– sp3 chains. A similar transformation, but
affecting all sp2bonds of the original two-dimensional sp2
carbon structures, leads to the formation of stable α and β graphynes [19],
the digital twins of which are shown in Figure 1f. It also
turned out that to obtain a stable planar regular structure, it is sufficient
to replace one third of the sp2bonds of graphite with acetylenic () or diacetylenic linkages [19–21]. Such a
transformation in the first case leads to the regular structure of γ graphyne
(see Figure
1g), and in the second - to graphdiyne (Figure
1i). Calculations showed the stability of structures of both
types, as well as other graph-n-ynes (n = 3, 4, 5, etc.). Numerous attempts to
synthesize this group of substances have been successful only in relation to
graphdiyne, the crystalline films of which have become the subject of careful
study [22–26].
A brief look at carbon crystallography, presented
above, shows that sp3 and sp2 bond
configurations are favorable for the formation of stable, regular structures,
supporting the existence of millions of tons of diamond and graphite deposits
on Earth and the existence of crystalline carbon in space. As for sp1
bonds, they cannot form regular structures on their own, so the latter are
formed only by combining sp1 bonds with sp3
and sp2 ones.
2.2. Amorphous Carbon
The trimodality of valence electron configuration
of carbon atoms forms the foundation of the unique three-mode amorphous state
of carbon solid. From the fundamentals of solid-state physics, sp3,
sp2, and sp1 amorphous carbons (s) are different species characterized by
conceptually different short-range orders, namely, groups of tetrahedrally
bonded sp3configured atoms, as well as
nanoscale-size-restricted sp2graphene domains, and sp3-
sp1 carbyne chains. Figure 2 provides a
basic overview of the current state of this carbon understanding.
As in the case of crystals, has an internal molecular structure, the
configuration of which depends on the type of bonds involved. Thus, tetrahedral () in Figure 2a [27,28]
represents the well-known configuration of atoms linked by sp3bonds. Another tetrahedral amorphous allotrope, -carbon in Figure 2b [29–31],
is a densely packed metastable phase formed by ultrafast quenching of carbon
melt in a super-undercooled state. After quenching, diamond tetrahedra are
randomly packed with >80% packing efficiency. Both s are not natural substances and constitute only a
small part of the vast amorphous wealth of carbon. The central part of the
latter is occupied by the sp2 . Recent purposeful studies [32–37]
have completed the gradually nascent change in the view of the solid as a
well-known, familiar physicochemical subject, the beginning of which was laid
back in 1941 [38],
transferring it to the rank of high-tech material of the modern nanotechnology.
We are talking about material known to humankind since the first decomposed
bonfire, which left behind black soot and charcoal. Today, this applies to
billions (anthracite coal) - millions (shungite carbon) - thousands
(anthraxolite and blackcarbon coatings, which are present everywhere and
accompanying almost all minerals in nature) tons of only discovered natural
deposits and hundred-million tons of synthetic black carbon produced
industrially. All this black carbon wealth is sp2, or a monoatomic solids without long-range order,
the atoms of which form sp2configured valence bonds with each other. It has a
unique common basis, namely, nano-micro-scale molecular compositions of
hexagonal honeycomb structures of carbon atoms (graphene domains) framed by
various heteroatom necklaces composed of oxygen, hydrogen, nitrogen, sulfur,
halogen and so forth. These necklaced graphene molecules are basic structural
units (BSUs), varying in size and shape, as well as differing in the necklace
chemical composition depending on the history of origin and/or method of production
of the black carbon, and form the first level structure of the bodies. The
discovered and experimentally confirmed graphene nature of this black gold has
led to a revolutionary revision of the theory, modeling and interpretation of
the experiments related to this class of solids [37].
Figure 2.
Digital twins of amorphous allotropes of carbon. (a) Tetrahedral amorphous carbon . (b) -carbon. (c) sp2 graphene-carbon.
(d) sp3- sp1 carbyne-carbon. Images taken
from open sources.
Figure 2.
Digital twins of amorphous allotropes of carbon. (a) Tetrahedral amorphous carbon . (b) -carbon. (c) sp2 graphene-carbon.
(d) sp3- sp1 carbyne-carbon. Images taken
from open sources.
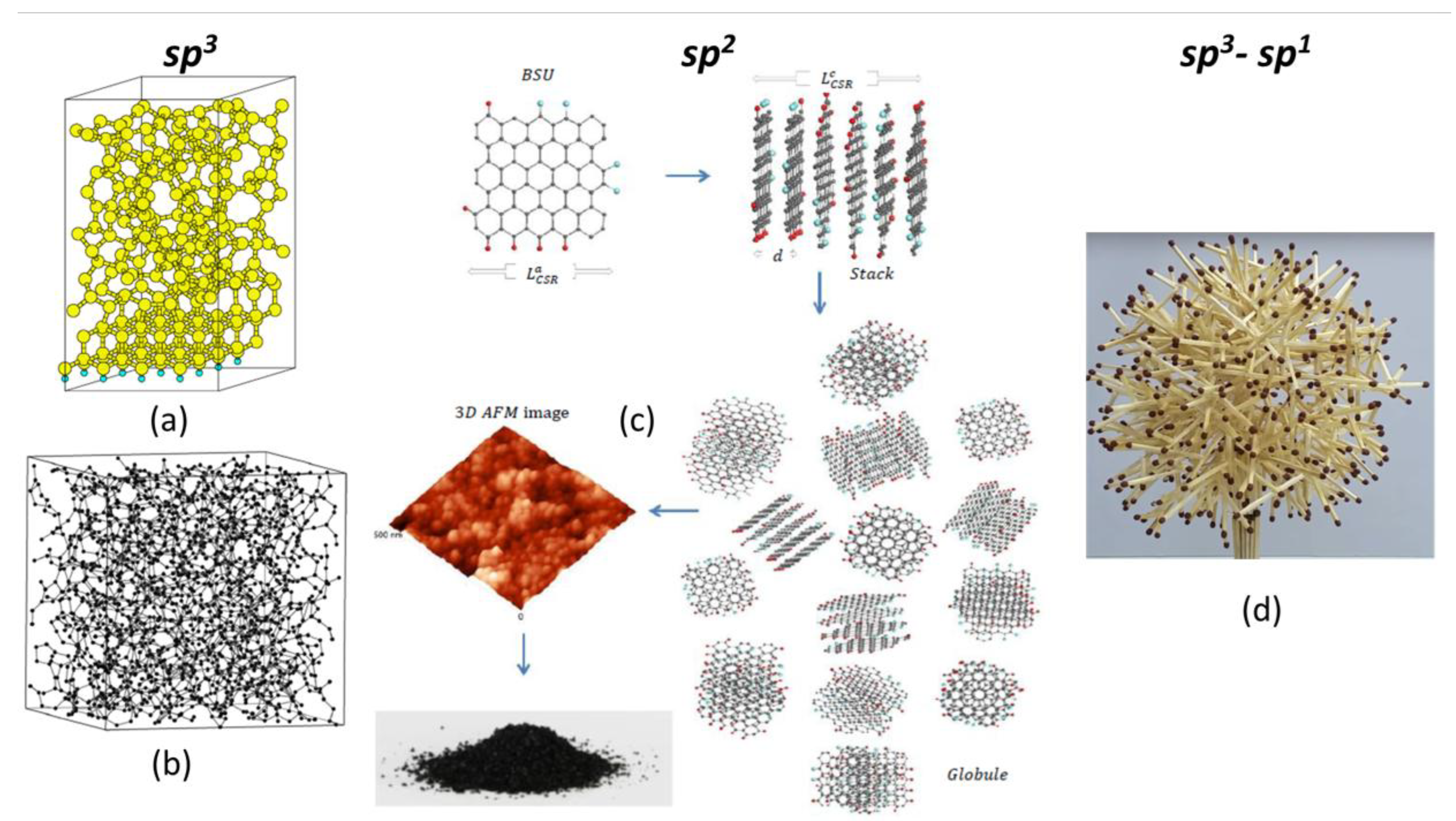
The second level structure pf the sp2s is provided with nano-thick BSUs stacks,
which are confidently recorded by X-ray and neutron diffraction structural
studies of sp2 s of all types [33]. The third-level
structure of these amorphics reliably follows from the porous structure
evidently observed experimentally [39,40]. It is
constructed from the BSUs stacks but the final compositions depend on the
stacks’ lateral dimension. When the latter is at the first nanometer level, the
composition presents globules of ~10 nm in size, which corresponds to pores,
size of which is first nanometers as well. Further aggregation of globules
leads to the formation of micro-nanosize agglomerates with pores of
tens-to-hundreds nm. Such a structure is typical to natural s such as shungite carbon, anthraxolite, anthracite
as well as black carbon coating of diamonds [41], mixed
carbon-silica spherical ‘sweets’ [42], black carbon in
meteorites [43,44]
and none of the exclusions has been known so far. Figure
2c schematically presents the evolution of this type of amorphic
structure from a single BSU to macroscopic powder. In contrast to natural
bodies, synthetic sp2 aCs are characterized by a large
dispersion of BSUs ranging in size from units to tens and/or over hundreds of
nanometers. At the low-limit end of the dispersion, the amorphic structure is
similar to that of natural species described above. At the high-limit end, the
BSU size does not prevent BSUs from packing in nanosize-thick stacks, while the
latter are extended and further packed into a paper-like structure.
In contrast to sp3 and sp2s, information on sp1 s is currently practically absent, which indicates
great difficulties due to both the existence of carbyne chains of limited
length and the complexity of their mutual packing. We touched on this problem
above when describing the crystallization of carbyne chains. Since the end
atoms of the chains are very reactive, chemical approaches tend to stabilize
the chains by end groups. Different end groups have been used, ranging from
hydrogen or metal atoms to larger complexes. With larger end groups, chains of
up to 44 atoms have been synthesized [45]. It is assumed
that the extended end groups prevent cross-linking of the chains by keeping
them at a distance. Calculations show that both regular and irregular packing
of carbyne units is possible. The successful implementation of the former is
described in the previous section. Irregular, or amorphous, packing can be
imagined as a child's 'hedgehog made of matches', shown in Figure
2d. And only the future will tell how close our idea is to
reality.
Concluding the above comparative analysis of the
structures of solid carbon allotropes, it should be recognized that the
trimodality of the covalent bonds between carbon atoms is the primary reason
for their wide diversity. It is easy to imagine that similar vivid images would
accompany the description of any characteristic property of carbon molecules.
This, in turn, allows us to say that the type of covalent bond for each carbon
atom is a determining factor in its subsequent fate and behavior within a team
of surrounding atoms. This means that the analysis of these bonds becomes the
cornerstone of the entire carbon universe, from its chemistry to its biology
and cosmology. This fundamental problem will be further explored below within
the framework of the spin theory of the carbon atom
covalence.
3. Lengths of Bonds as a Governing Factor of the Carbon Covalent Chemistry
3.1. Spin-Radical Concept of the Carbon Atoms’ Covalence
Stretching and breaking of chemical bonds, leading
to open-shell character of molecular electronic systems, present key points of
the spin-radical concept of the covalence. The main statement declares that
open-shell character of both covalent carbon solids and molecules is the main
mechanism responsible for peculiar properties of the species at microscopic
level. Basically, the open-shell character is provided with either initially
unpaired odd electrons, which do not participate in the formation of covalent
bonds, or effectively unpaired electrons that are withdrawn from the covalent
bonding, in whole or in part, due to stretching of interatomic distance. In
both cases spacing between the electrons must exceed a critical value characteristic for partners of the covalent bond
under consideration.
Theoretically, the bond concept has come a long way
of development alongside the electron theory of chemical matter, and its
development is still ongoing. Particular epochs are associated with the valence
bond theory [46],
molecular orbital theory [47], and density functional theory [48].
A comprehensive collection of reviews exhibiting the modern concepts of the
chemical bonding is presented in two-part edited collections [49,50].
These theoretical approaches have laid the foundation of quantum chemistry
aimed at obtaining equilibrium multi-atomic configurations. However, a direct
solution of Schroedinger’s equation does not point to the bond within a
particular pair of atoms. Computationally, the bond justification consists in
finding evidence in space related to the electron density distribution in the
frame of either Bader’s atom-in-molecules theory [51] or some of its
developments (see Refs. [52,53] and references therein).
Empirically, in the majority of cases, the bond between two atoms is justified
by comparing the interatomic distance with one of standard bond lengths
accumulated on the basis of numerous structural data. In view of this interrelation,
on practice, the chemical bond is mainly associated with this structural
identificatory, with respect to which one can speak about ‘bond forming’, ‘bond
stretching’, or ‘bond breaking’. Speaking about the length of a covalent bond,
one usually addresses the data tabulated in numerous tables and presented in
numerous handbooks (see, for example, Refs. [54,55]). As seen
from the data, bond lengths for the same pair of atoms in various molecules are
rather consistent which makes it possible to speak about standard values
related to particular pairs of atoms. A standard length of 1.09Å is
attributed to the C-H pair while the lengths of 1.54 Å, 1.34 Å, and 1.20Å are
related to single, double and triple bonds, respectively. Complicated as a whole, the
set of the available data on bond lengths and bond energies provides a
comprehensive view on the equilibrium state of molecules and solids. On the
background of this self-consistency, the detection of extremely long bonds,
such as single sp3 bonds of 1.647 Å, 1.659 Å, and 1.704 Å instead of
1.54 Å [56]
and sp3 bonds of 1.54 Å [57] and 1.622 Å [58]
instead of 1.43 Å not only looks as a chemical curiosity but raises the
question of the bounds of covalent bonding. Two other questions are closely
related to the matter: 1) to which extent a chemical bond can be stretched and
2) on which length its breaking occurs. Empirically, this usually concerns
subjectively made estimations of critical values of a possible elongation of
bonds that broadly varied. Thus, the width of the region of admissible values
of bond’s lengths is significantly varied in different computer programs
aimed at molecular structure imaging. As for a bond rupture, this problem is
the most uncertain and the rupture is considered as a final result of a
continuous stretching only.
The problem of theoretical justification of the
chemical bond stretching and breaking concerns the criteria according to which
the considered bond is still alive or ceases to exist. Until now, two
approaches have been usually exploited. The first, based on the
atom-in-molecules theory [51], concerns the bond critical point
within the electron density distribution over an atomic composition, evidence
of which is considered as a proof of the bond existence. However, as shown [58],
the criterion, computationally realized, is not reliable in the case of weak
coupling due to which it cannot be used to fix the bond breaking. The second
approach overcomes the difficulty addressing directly to the correlation of
electrons involved in the bond [59] addressing the
entanglement among any pair of orbitals. The obtained results showed that
electron correlation is indeed the main determinant of stretching and breaking
of chemical bonds and the quantitative measure of the correlation may serve as
criteria for the fixation of the above processes.
Unrestricted two-determinant Hartree-Fock (UHF)
formalism is quite suitable for a quantitative description of electron
correlation thus providing four criteria able to characterize the extent of the
event [60]:
(i) misalignment of the energy of RHF () and UHF () solutions, where ; (ii) spin expressed contamination via
misalignment of squared spin ; here , is the UHF squared spin while presents the exact value; (iii) appearance
of effectively unpaired electrons of total number: (iv) molecular magnetism governed by
exchange integral . Moreover, the HF level of the theory is quite
appropriate for understanding the basic aspects of bonding [61].
3.2. Spin Emergents of Carbon Covalent Bonds
Besides UHF emergents, , , and , completely describing the open-shell nature of
the molecular configuration under consideration, dependent on the interatomic
distance graphs and their singularities present a perfect
benchmark for a quantitative description of stretching and breaking of chemical
bonds [62].
Bonds formed by two carbon atoms are the richest in content, and its general
representation in the form covers a set of traditionally matched single sp3, double sp2, and triple sp1 bonds. Throughout the article, the values of the
above parameters were obtained using the CLUSTER-Z1 software [63,64]
implementing the AM1 version of the semi-empirical unrestricted two-determinant
Hartree-Fock (UHF) approach [65]. The program showed itself highly
efficient concerning open-shell electronic systems such as fullerenes [66–68],
graphene molecules [69],
and stable radicals [70].
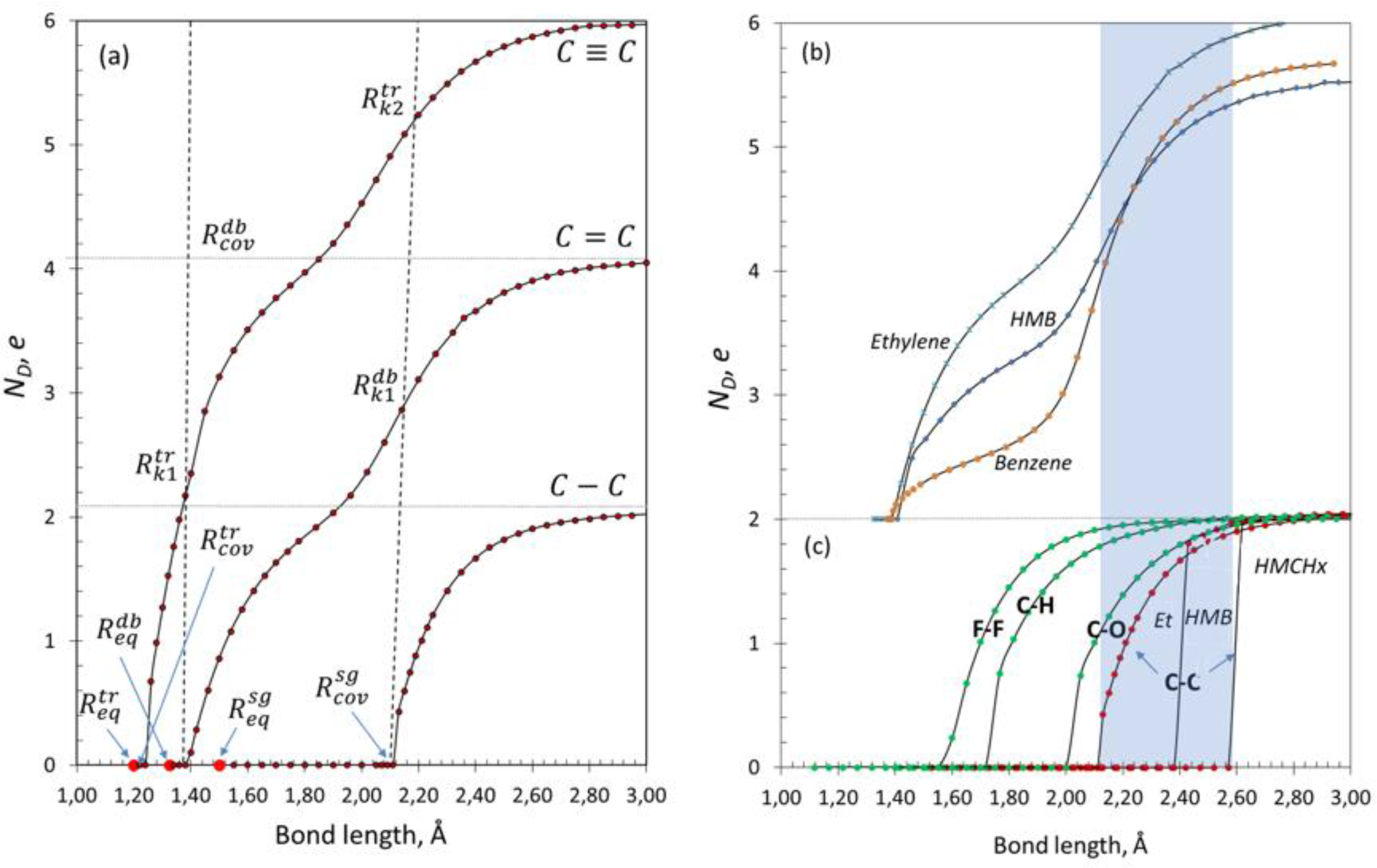
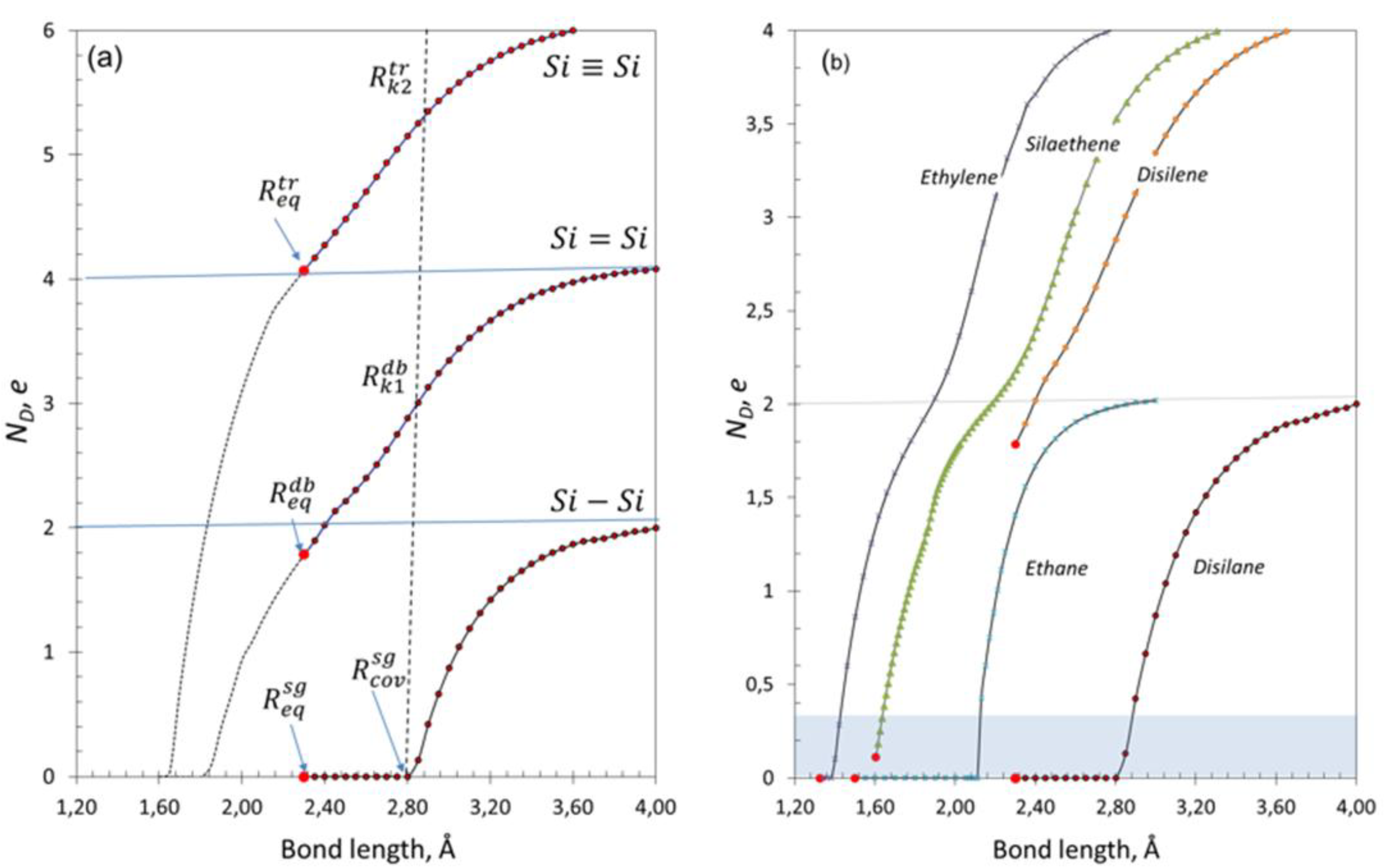
The graphs in Fig. 3a present a general view on the bond
family on an example of the gradual virtual dissociation of ethane (H3CC-CCH3),
ethylene (H2CC=CCH2), and propyne (H3CCCCH) molecules [62,69]. As seen in
the figure, all the studied graphs are of -like shape but significantly different. Thus, the
single-bond graph is of one-step -shape while for double and triple bonds -like curves are evidently of two- and three-step,
respectively. The number of steps evidently corresponds to the number of
electron pairs involved in the relevant bond. Each of the graphs starts by a
horizontal line corresponding to . Marked with large red dots, corresponds to the
equilibrium length of the bonds while the right-hand bound indicates at which
interatomic distance the covalent bonding is violated thus pointing to the
largest classical covalent bond length , from which the bond stops to be classical and
becomes to work as spin-dependent. This region can be characterized by both
absolute and relative width and and distinguishes areas where the bonds correspond
to the close-shell character of the relevant molecule electronic states.
Superscripts , and differentiate members of the family.
When reaching , each of the three graphs undergoes a jump that indicates the
beginning of the bond radicalization when stretching. The radicalization
gradually proceeds while the interatomic distance increases, although quite
differently for the three bonds. Thus, the radicalization of the sp3 bond of ethane, started at , is fully completed at ≤ 3Å and two single radicals are formed.
Radicalization of the sp2 bond of ethylene starts at Å and is saturated at the same as for the single bond where a pair of two-fold
radicals is formed. However, on the way to a completed radicalization a clearly
seen kink on the graph occurs. The kink critical point corresponds
to approaching 2 e and, exhibited by
differentiating, is located at = 2.12Å that is well consistent with of the single bond. Therefore, the bond
radicalization occurs in two steps, first of which is completed for a pair of π electrons by reaching ≈2 e
while the second should be attributed to the separation of the remaining σ electrons until ≈ 4 e
is reached.
graph of the sp1 bond of propyne, preserving a general -like pattern, shows a two-kink behavior. As seen
in Figure
3a, the bond radicalization starts at Å and the first kink is located in the region of ≈2
e at 1.40 Å that is consistent with 1.395 Å of the double bond of ethylene. In the
region of ≈4 e,
the second kink is observed, whose critical point at 2.10 Å is consistent with of the single bond of ethane. A pair of three-fold
radicals at Å completes the bond breaking. Therefore, a gradual
stretching of the sp1 bond of propyne can be presented as a consequent
completed radicalization of two pairs of π
electrons first and then terminated by the radicalization of σ electrons followed with the total bond
breaking.
Data presented in Fig. 3a allow speaking about a new aspect of chemical bonds concerning their radicalization. It should be remained that the radicalization is just a ‘chemical’ manifestation of the correlation of bond-involved valence electrons. From this viewpoint, single, double, and triple bonds are drastically different. Thus, the single bond is radicalized in the vicinity of its breaking. Derivation of the graph reliably highlights as a clear singularity thus allowing its attribution to the fixation of the bond breaking. In the case of double bond, determines the parting of π bond while , which coincides with , fixes the bond breaking. Similarly, on the graph marks the separation of π electrons of the first pair while and manifest the parting of π electrons of the second pair and the
remained σ electrons, respectively,
just completing the bond breaking. Therefore, according to the observed
consistency of of ethylene and of propyne with of ethane, all the three values can be attributed
to the interatomic distance at which any of the bonds of the discussed set can be considered as broken.
Figure 3.
graphs of carbon covalent bonds. (a) bond of ethane; bond of ethylene; bond of propyne. Dotted vertical lines mark
positions of and . Large red points mark positions in all the cases. (b) bonds of ethylene, benzene. and hexamethylbenzene
(HMB). (c) bonds of ethane, cyclohexane (CHx), and
hexamethylcyclohexane (HMCHx). Light blue band marks dispersion of the depending on the bond structure-elemental
surrounding. UHF AM1 calculations.
Figure 3.
graphs of carbon covalent bonds. (a) bond of ethane; bond of ethylene; bond of propyne. Dotted vertical lines mark
positions of and . Large red points mark positions in all the cases. (b) bonds of ethylene, benzene. and hexamethylbenzene
(HMB). (c) bonds of ethane, cyclohexane (CHx), and
hexamethylcyclohexane (HMCHx). Light blue band marks dispersion of the depending on the bond structure-elemental
surrounding. UHF AM1 calculations.
Fixation of the bond breaking allows introducing
such characteristic quantities as the absolute and relative width of the
radicalization region and , respectively, that in the case of double and
triple bonds of ethylene and propyne are of the form
for sp2 bond of ethylene; and
for sp1 bond of propyne. The corresponding sets of ,, , , data are listed in Table 1. As seen from
the table, while decreases when going from single to triple bond, inversely increases. The feature is the main
reason for a drastic difference in the chemical activity of the bonds (material
bodies and molecules) of different multiplicity.
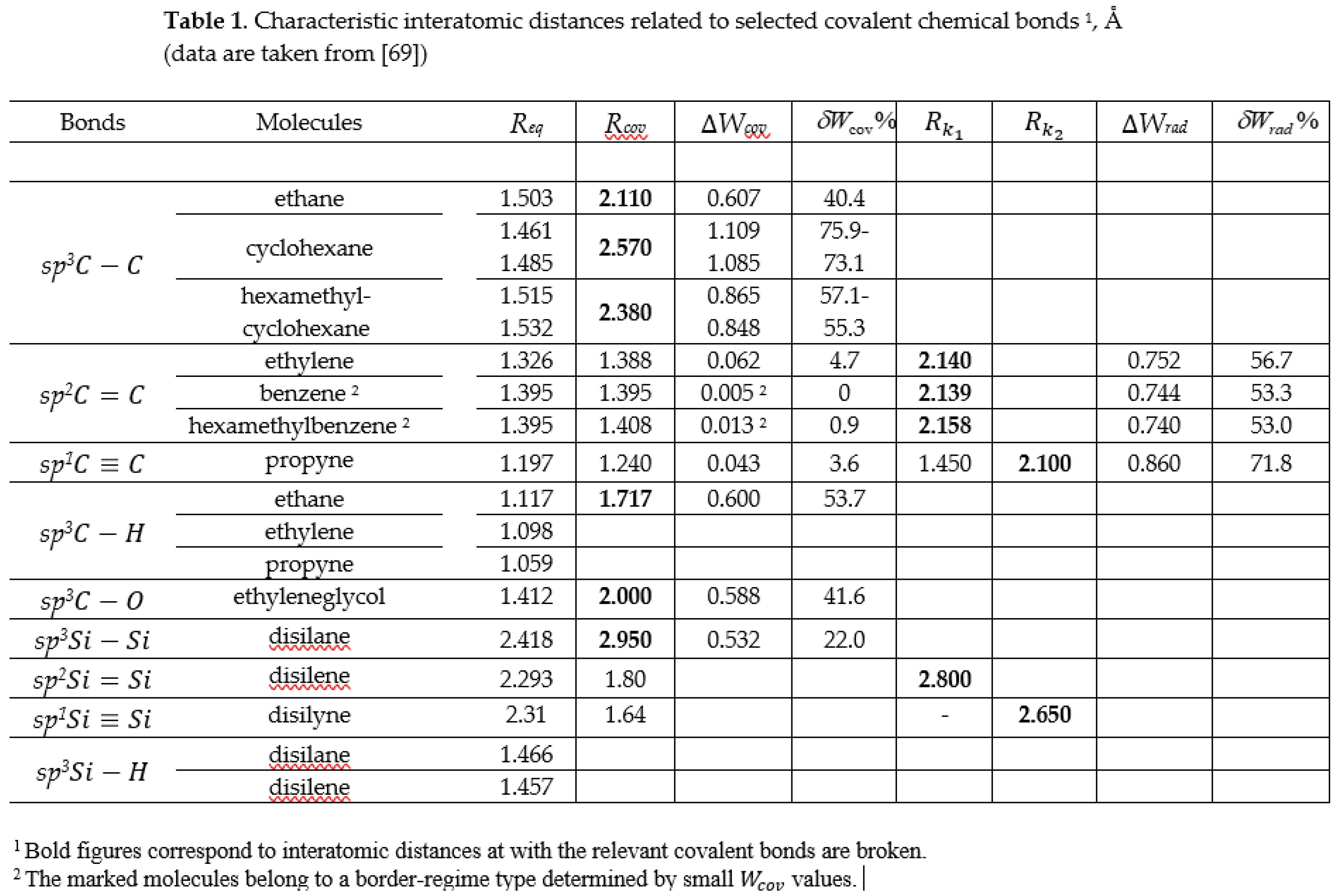
From the above it follows that any chemical bond
should be described by a set of characteristics, only one of which, namely, the
equilibrium length of chemical bond can be standardized. However, empirical data show
that is characterized with a significant dispersion
indicating the dependence of the quantity on surrounding atoms. From this
viewpoint, the data presented for the considered three molecules may change
when going to other atomic composition. Actually, data presented in Figure
3c for single sp3 bonds show that the absolute values of (see Table 1) are
different while the qualitative character of the relevant graphs is conserved. Particularly, it should be
noted that in polyatomic molecules the radicalization and breaking of these
bonds become more abrupt thus significantly narrowing the smoothing of the
region of their radicalization.
Oxides and hydrides are the most popular species of
the carbon chemistry that is why sp3 and sp3 bonds deserve a particular attention. The relevant
graphs presented in Figure
3с are related to the dissociation of single bonds in ethylene glycol (-C-O) as well as bonds of ethane. parameters of the bond are listed in Table
1. As seen in the figure and follows from the table, the bonds
one-step behavior is similar to that of sp3 one. The elongation stage δWcov constitutes ~40-50%,
while the radicalization smoothing of is small enough. As in the case of sp3 bonds, one should expect a slight
difference in the characteristics of sp3 and sp3 bonds depending on the atomic surrounding.
Double sp2 bonds, similarly to the ethylene one, are
characteristic for a large family of alkenes. However, such bonds are more
often associated with benzene-based and other aromatic molecules. Their
stretching is of extreme significance for a large class of sp2 nanocarbons
[71].
Figure
3b presents a comparative view on the dissociation of a single sp2 bond of ethylene and one of the bonds of benzene
and hexamethylbenzene. The comparison reveals a common character of the
relevant graphs with some difference of values (see Table 1) as well as a
remarkable difference in the graphs’ shape. Nevertheless, all the graphs are
two-step -like with a kink located in the region of ~ 2e. The kink critical points are well
consistent with of the relevant single sp3 bonds.
Evidently, all the said above can be attributed to
species with sp1 bonds. The relevant critical point determines the onset of the transformation of
molecules involving triple bonds from closed-shell to open-shell ones.
3.2. Spin Emergents of Silicon Covalent Bonds
The current around-graphene science has represented
a new milestone of activity in the discussion of similarity-and/or-unlikeness
of different members of and higher tetrels (X=Si, Ge, Sn) families of covalent bonds, which
was a hot topic over a century [72]. Now this branch
is full of suggestions concerning new prototypes of graphene, foremost of which
are based on the equivalence of valence electrons of all tetrels atoms and
expected hexagon patterned one-atom-thick planar structures such as silicene,
germanene, and tinene-stanene. Covalent radii of tetrels make a series
0.76-0.73-0.69 Å, 1.11 Å, 1.20 Å, and 1.39 Å for carbon (sp3-sp2-sp1),
silicon, germanium, and tin, respectively [73]. To form a
reliable platform for a comparative analysis, the data presented below are
related to molecules of the common structure, namely, ditetralanes X2H6,
ditetrelenes X2H4, and
ditetrylynes X2H2 (C2H(CH3)
in the case of carbon) [74,75].
Figure
4a presents graphs related to complete sets of bonds while Figure 4b exhibits
the difference between carbon and silicon bonds [62,69,74,75]. As seen in the figure, the graphs of silicon covalent bonds
behave quite similar to those shown for carbon whilst shifted to longer
interatomic distances. The graph of disilane is one-step with clearly seen
points . The graph of disilene demonstrates only the
second part of the two-step radicalization, which is a reality for ethylene.
The equilibrium interatomic distance ≈2.3Å for
the latter greatly exceeds at 1.8 Å. Consequently, in contrast to covalently
saturated ethylene, equilibrium disilene is almost two-fold radical. When
proceeding with the bond elongation, the graph reveals kink positioned at , well consistent with . Thus, equilibrium disilene with separated π electrons, continues their dissociation
until parting the remained σ electrons
at . The three-step radicalized sp1 bond of propyne in Figure
3a is not reproduced as sp1 bond of disilyne in Figure
4a. The equilibrium distance ≈2.3 Å is
positioned much over thus presenting almost fourfold radical, which
means that π electrons of both pairs
are disunited. All parameters of the discussed silicon bonds are listed in Table
1.
Therefore, in contrast to stable carbonaceous
materials, possessing sp2 and sp1 bonds, siliceous ones with the shortest sp2 and sp1 bonds are radicals, chemical activity of which
drastically surpasses that of carbon species. The interaction of two odd
electrons, which are formed under transformation at any interatomic bond,
depends on the corresponding distance that is ~1.5 times larger for distances with respect to ones due to larger size of the atoms. At the same
time, the distance 1.4 Å is critical for these electrons to be
covalently coupled. Above the distance the electrons become effectively
unpaired, therewith the more the larger the distance. sp2 bond length of benzene just coincides with the
limit that provides a complete covalent bonding of the electrons, transforming
them into widely known π electrons.
Apparently, this is the way Nature has enabled benzene to play a particular
role for establishing and proving the aromaticity concept in the framework of
classical covalent chemistry as well as for introducing π electrons in organic chemistry.
Beside the discussed sp2 and sp2 bonds, one more double bond is
formed involving both carbon and silicon atoms. Figure
4b presents a comparative view on graphs related to the dissociation
of ethylene, silaethene, disilene as well as ethane and disilane molecules.
Comparing the graphs for sp2 and sp2 bonds, a drastic difference
becomes evident. In the case of ethylene, one can distinctly see that π
electrons govern the molecule continuous dissociation when interatomic distance
changes from 1.4 Å to 2.1 Å and thencomes the turn of σ
electrons until the dissociation is completed at 2.8 Å. At equilibrium the
molecule is closed-shell one with the sp2 bond of 1.326 Å in length.
Oppositely, π electrons are practically unobservable under disilene dissociation
since already in equilibrium they are almost fully transformed into a pair of
effectively unpaired electrons (=1.78 e). Therefore,
disilene has no closed-shell phase at all and in the equilibrium is open-shell
one. Essentially different is the situation with the sp2 bond of silaethene. As seen in the
figure, at = 1.605 Å the molecule is just at
the beginning of its open-shell status, radicalization of which constitutes = 0.153 e.
Figure 4.
graphs of silicon covalent bonds. (a) bond of disilane; bond of disilene; bond of disilyne. Dotted vertical line marks the
positions of . Large red points mark positions in all the cases. (b) , , and bonds of ethylene, disilene, and silaethene,
respectively, and , and bonds of ethane and disilane Light blue band
marks dispersion of the values characteristic for sp2
nanocarbons. UHF AM1 calculations.
Figure 4.
graphs of silicon covalent bonds. (a) bond of disilane; bond of disilene; bond of disilyne. Dotted vertical line marks the
positions of . Large red points mark positions in all the cases. (b) , , and bonds of ethylene, disilene, and silaethene,
respectively, and , and bonds of ethane and disilane Light blue band
marks dispersion of the values characteristic for sp2
nanocarbons. UHF AM1 calculations.
As can be seen from Figure 4, all
multiple covalent bonds involving silicon are radicalized, the sp2bond to the least extent and the sp1 bond to the greatest extent, and only the single sp3bond of disilane reliably binds neutral atoms. This
tendency of interatomic interaction is clearly visible in modern silicon
chemistry. Thus, individual disilanes and disilane components of more complex
structures constitute the main content of empirical silicon and organosilicon
chemistry. Disilenes turned out to be extremely chemically active, as a result
of which, from time to time, it is possible to stabilize them only in the form
of dimeric inclusions in other molecules [76,77]. In contrast,
silaethenes are successfully synthesized and analyzed [78,79].
Empirical evidence for the observation of silicene, a silicon analogue of
graphene, is a consequence of the erroneous classification of real sp3
structures of silicon on substrates as sp2-type structures
(see a detailed discussion of the issue in [74,75]). Individual
disilynes could not be synthesized, and the triple sp3 bond was only detected in the chain [80,81].
4. Spin Chemistry of Alkane Bonds
As follows from the analysis conducted in terms of
bond lengths, alkane bonds generated by classical covalent chemistry remain
faithful representatives of this chemistry, regardless of the causes and nature
of their origin. Single carbon sp3 bonds form the basic structural framework of
organic compounds. One example of their consolidated operation is the formation
of cord-like backbones of organic polymers. Figure 5 demonstrates
the action occurred in the course of virtual free-radical polymerization of
styrene [82–84].
Figure 5.
Digital twins present free radical oligomerization of styrene. Small yellow and gray balls mark hydrogen and common carbon atoms, respectively. Carbon targets are marked red. Larger green balls depict nitrogen atoms. UHF AM1 calculations.
Figure 5.
Digital twins present free radical oligomerization of styrene. Small yellow and gray balls mark hydrogen and common carbon atoms, respectively. Carbon targets are marked red. Larger green balls depict nitrogen atoms. UHF AM1 calculations.
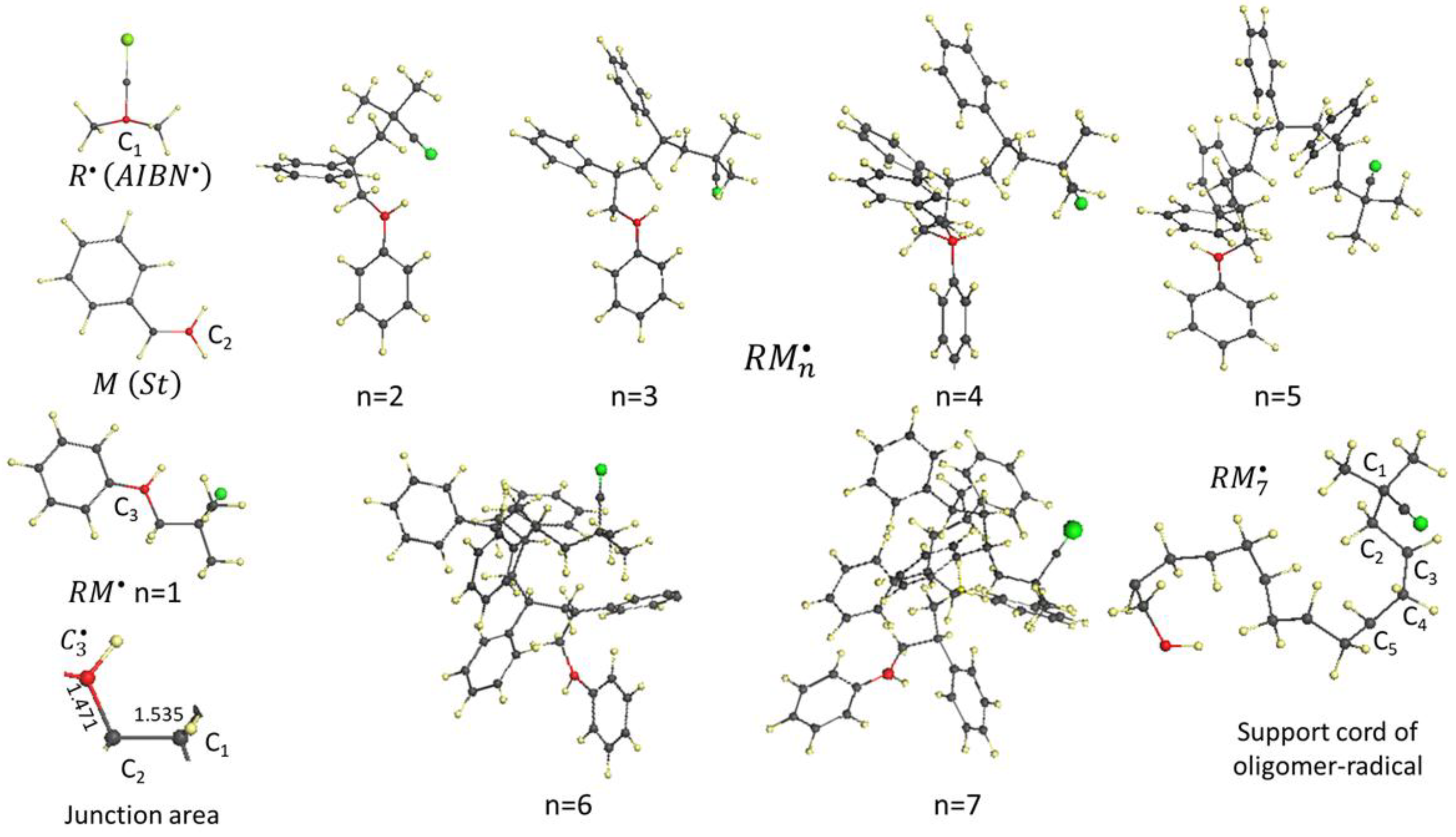
The reaction occurs in a virtual reaction solution
consisting of vinyl monomers M (styrene) and free radicals . The alkyl radical plays this role. The reaction begins with the formation of monomer radicals . The target atoms of these primary reactants are determined within the framework of the spin theory of radicals briefly described in the Sections 3.1 and 3.2. In the free radical, the target is the central carbon atom , whose odd electron appears as unpaired with a
partial electron number of = 0.807 e. In styrene, the target is the carbon
atom of the CH2 unit of the vinyl group with
= 0.106 e. The radical and monomer are bound by a
covalent bond between their target atoms, which leads to the formation of
monomer radical with an active target atom, which is the carbon
atom of the CH unit of the styrene vinyl group, the chemical activity of which is = 0.67 e. Intermolecular junction in the (see image in lower left corner of the figure) is configured by two covalent bonds and , the first of which is a standard alkane bond,
while the second retains the character of an alkene one due to the unpaired electron on the atom. The formation of a dimer radical (and each
subsequent member of the oligomer radical chain ) occurs through the formation of a single covalent
bond between the carbon atom of the CH component of the vinyl bond of the (or atom of the preceding oligomer radical) and the CH2 carbon atom of vinyl group of a new monomer, thus building a chain of alkane bonds that makes up the supporting cord of the oligomer molecule. The configuration of this cord in the oligomer radical is shown in the lower right corner of the figure. The cord consists of alternating CH2 and CH groups, the latter of which is suspended by a benzene ring. Each next-added monomer
Contributes two atoms of its vinyl group ( and in the case of the initial monomer-radical ) to this cord, forming an alkane bond with the preceding free radical and leaving the
alkene bond in a strained state. The addition of the next
monomer to with the pilot atom transforms the alkene bond into an alkane and completes the chain with two new bonds - an
alkane bond and an alkene bond . The pilot atom, , drives polymerization further, replacing the
alkene bonds of the oligomers with alkane bonds, leaving only one alkene bond
on the ever-elongating chain, marking the pilot carbon atom of the last monomer
added. The movement of the pilot alkene bond along the chain, leaving behind
only a chain of alkane bonds, is the essence of vinyl monomer polymerization.
As described above. the pilot alkene bond is
tightly interconnected with alkane bond that couples new monomer with the
current oligomer radical [82]. The feature is presented in Figure
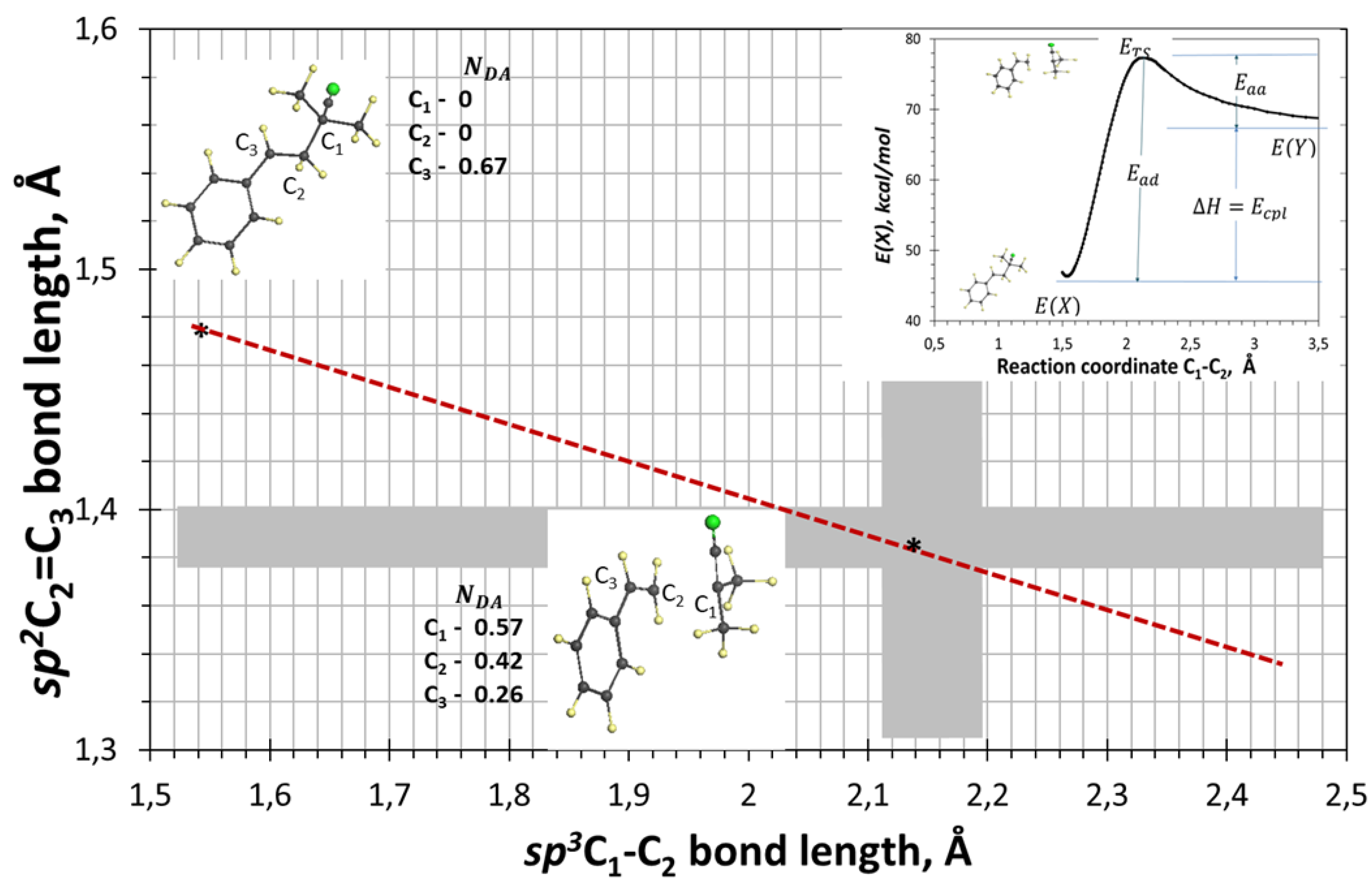
6 for the case of monomer radical . Red broken line depicts the elongation of the sp3 bond that connects free radical and monomer in the
course of the dissociation. The dissociation starts at the (x,y)
point (1,535; 1,471) indicating that the junction consists of a standard sp3bond and elongated sp2 one. Because of the elongation over , the sp2 bond is radicalized
thus providing generation of effectively unpaired electrons of the total number
= 1,925 e. The latter causes the generation
of the junction target atom C3 with = 0,67 e. The value is in agreement with a general law that
governs a gradual radicalization of the sp2 bonds presented in Figure 3a. When the
elongation of the sp3 bond proceeds, its length
approaches = 2.11 Å, while the sp2 bond
gradually shortens up to . Similar to the dispersion of sp3bonds lengths in the vicinity of , is dispersed responding to the changing the bond
surrounding. The dispersion zones for values for both bonds are presented with vertical
and horizontal light gray bands, related to the sp3 and sp2
bonds, respectively. The intersection of the bands determined the area of
transition state indicated on the energy graph presented in the inset of Figure
6. The graph describes the elementary reaction discussed above and reveals the transition state
occurring at the moment of cleavage of the sp3 bond that is the reaction coordinate under
consideration. This moment is the transition point for this bond from classical
to spin covalence. Molecular compositions of the transition state represent
open-shell electron systems, so their quantum-chemical analysis requires the
use of methods capable of correctly considering the spin-radical interaction of
electrons. Regarding the discussed sp3 bond, the presented
analysis allows us to conclude that every alkane covalent bond, classical
always and everywhere, becomes spin-covalent at the moment of its destruction
(or creation), which is always present in chemical reactions marking transition
state location in the space of reaction coordinates.
Figure 6.
The interconnection between the alkene sp2and alkane sp3bonds, composing the intermolecular junction of the
monomer radical . See the junction determination in Figure
5. Inset presents the energy graph describing the virtual dissociation of the . Light gray bands mark the dispersion of the (vertical) and (horizontal). Images are snap-shots corresponding to structures depicted with black points. UHF AM1 calculations.
Figure 6.
The interconnection between the alkene sp2and alkane sp3bonds, composing the intermolecular junction of the
monomer radical . See the junction determination in Figure
5. Inset presents the energy graph describing the virtual dissociation of the . Light gray bands mark the dispersion of the (vertical) and (horizontal). Images are snap-shots corresponding to structures depicted with black points. UHF AM1 calculations.
5. Spin Covalence of Alkene Bonds
Alkene bonds are present in a significant number of
covalent compounds. These can range from segregated bonds, such as those found
in vinyl monomers discussed above, to chains of bonds forming a closed sp2
framework, as in the case of polyaromatic hydrocarbons
(PAHs) and sp2 nanocarbons, including fullerenes, carbon
nanotubes, and graphene domains. The nature of the bonds determines all the
characteristic properties of these substances, and this could be discussed
endlessly. We will limit ourselves to those that relate to the unique spin
nature of these bonds, and will primarily use the quantities and , which determine the degree of bond
radicalization, as numerical characteristics.
5.1. Aromatic Hydrocarbons
A computational experiment performed for 14 n-PAHs
using a number of different CI approaches, such as UMP2, QCISD, as well as UHF
and UDFT [85],
showed that the species become open-shell molecules starting with naphthalene
(n = 2). The spin contamination values , indicating the open-shell nature of PAHs, are in
good agreement with each other for the first three methods, but remain zero and
indicate the closed-shell nature of these molecules in the UDFT case for all n.
Being based on wave functions, the UHF approximation is clearly well suited to
describing the effects of broken symmetry, while the DFT formalism is less
suited to considering the subtle features of the correlation of electrons with
different spins, as has been repeatedly noted in the literature [86–88].
Thus, for the already mentioned set of n-PAHs, Figure
7a presents the calculated values obtained using the UHF algorithms and the
density matrix renormalized group (DMRG) method [89]. As can be seen
from the figure, both data sets are virtually identical. Moreover, the linearly
increasing dependence of (n) on the number of benzenoid rings in the
molecules perfectly explains the difficulty of experimental synthesis of long
n-PAHs, among which pentacene (n = 5) is the last well-characterized polyacene.
As it turns out, higher polyacenes are indeed very reactive, as a result of
which PAHs with n = 6, 7, and 8 can exist either only in a modified form, when
additionally introduced protective groups inhibit the high chemical activity
inherent to these molecules, or in crystalline neutral matrices at very low temperatures.
Molecules with n>8 are not amenable to chemical synthesis at all. While
empirical confirmation of the correctness of UHF LDOS distributions in
open-shell molecules still faces significant
Difficulties, requiring significant refinement of
existing experimental methods [90], maps are reliably reproduced by scanning
open-shell molecules atom by atom in non-contact atomic force microscopy (AFM)
experiments. The greatest success has been achieved with microscopes with an
oxygen atom at the tip of the probe needle (see [91–95] and
references therein). Figure 7b shows AFM images of two
aromatic molecules, pentacene and olympicene, consisting of benzene rings. The
experiment was conducted with a gold needle with a CO molecule at its tip [91].
With the color scheme used in the experiment, brightly colored markings
indicate atoms of the molecule that interact weakly with the tip. Conversely,
the most shadowed atoms interact with it most strongly.
Figure 7.
(a) Total number of effectively unpaired electrons in polyacenes calculated by using DMRG (STO-3G)) [89]
and UHF AM1 formalism. No data scaling. (b) Atomic-resolved AFM image (top)
and the ACS maps (bottom) of pentacene (left) and olympicene
(right). (c) Digital twin (top), AFM images under the CuOx tip
(middle), and ACS maps (bottom) of
3,4,9,10-perylene-tetracarbonyl-dianhydride (left) and dicoronulene (right)
(see text). UHF AM1 calculations. (d) AFM images (top and bottom) and digital
twin (middle) of fullerene C60 [95].
Figure 7.
(a) Total number of effectively unpaired electrons in polyacenes calculated by using DMRG (STO-3G)) [89]
and UHF AM1 formalism. No data scaling. (b) Atomic-resolved AFM image (top)
and the ACS maps (bottom) of pentacene (left) and olympicene
(right). (c) Digital twin (top), AFM images under the CuOx tip
(middle), and ACS maps (bottom) of
3,4,9,10-perylene-tetracarbonyl-dianhydride (left) and dicoronulene (right)
(see text). UHF AM1 calculations. (d) AFM images (top and bottom) and digital
twin (middle) of fullerene C60 [95].
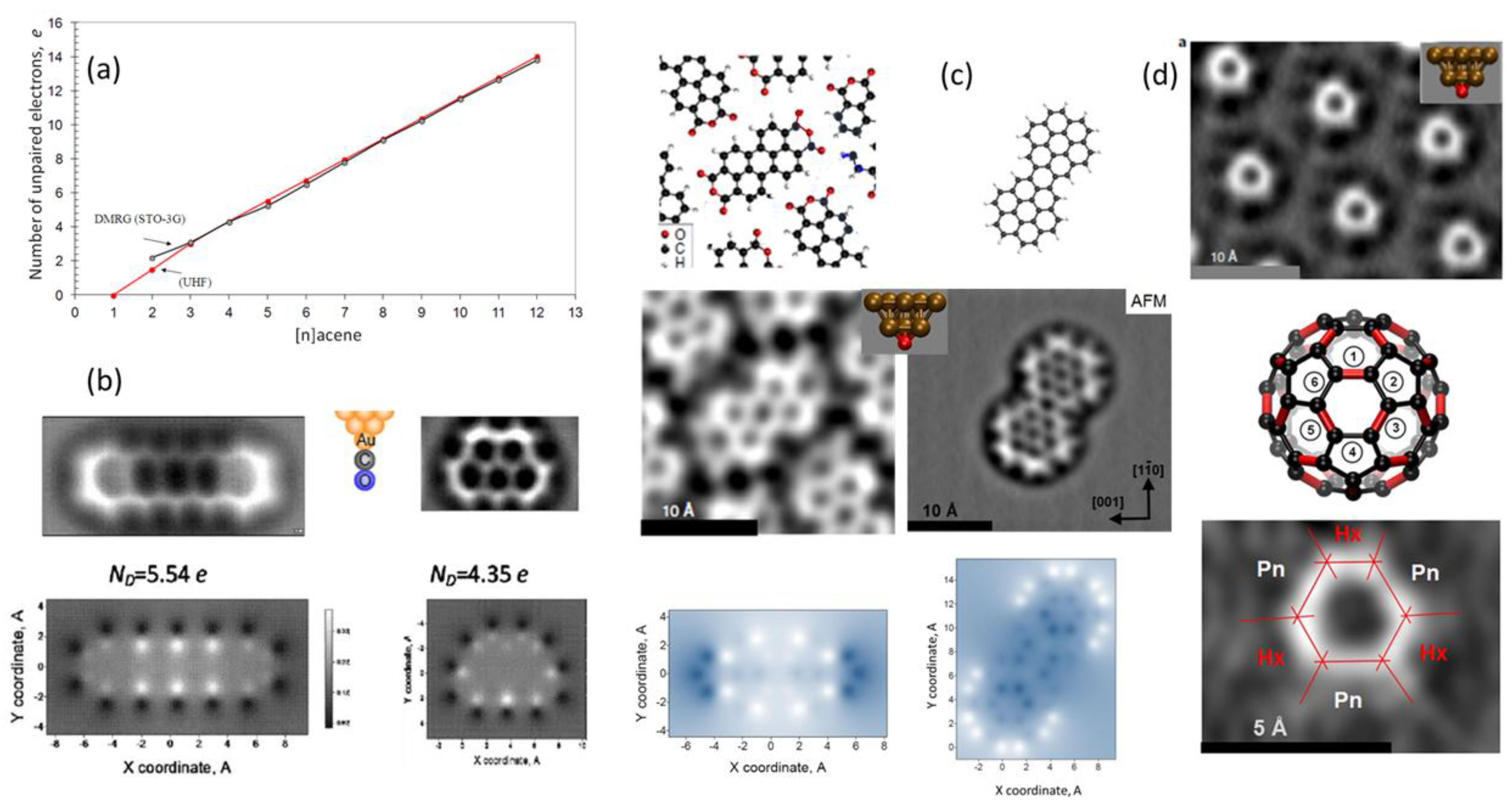
In conducting these and other AFM experiments with
an atomically sharpened tip, the authors aimed to obtain atomic-resolution
images of molecules. The fact that these images turned out to be uneven in
brightness was unexpected and was presented as experimental evidence of
differences in the interactions of the tip's terminal oxygen atom with the
atoms of the scanned molecule. The reason for this difference was not discussed
until the author of the current article proposed to associate this difference
with the ACS maps of the corresponding molecules. The maps of these molecules calculated by the author
are presented at the bottom of Figures 7b and 7c [71].
The color scheme of the virtual images in Figure 7b is inverted
with respect to the experimental one. A comparison of the empirical images and
virtual maps reveals that only carbon atoms are visible experimentally in AFM
experiments. As for the calculated values, they are fully confirmed experimentally,
correctly ranking the strong and weak interactions of the microscope tip with
the molecule under study when scanning their atoms. Thus, AFM with atomic
resolution is indeed a reliable method for reading ACS maps.
Figure 7c shows the
results using a different microscope tip configuration [95].
In this case, we are dealing with a copper tip delicately oxidized, so that an
oxygen atom is located at end of the CuOx tip. The figure presents
the results for 3,4,9,10-perylene-tetracarbonyl-dianhydride adsorbed on the
Ag(111) surface under Van der Waals attraction. The right side of the figure
presents similar results for the dicoronulene molecule, also adsorbed on the
Cu(110) surface. The brightness of carbon atoms in AFM is proportional to the
interaction strength of the oxygen atom of the tip with the atoms of the
molecule. The comparison of empirical images and virtual ACS maps confidently confirms the earlier conclusion
that atomic-resolution AFM is indeed a reliable method for reading ACS maps. Completing the AFM image in Figure
7d is a pinpoint image of C60 fullerene molecules
adsorbed on the Cu(111) surface and held there by Van der Waals forces [95].
The details of this image will be discussed in the next section.
5.2. Fullerenes
An analysis of the structural data for the molecule
[96]
showed that the diffraction pattern was consistent with the assumption that the
proper digital twin of the molecule is a truncated icosahedron with point group
symmetry, which is formed by two types of sp2bonds, one of which, separating two hexagons, has a
length of 1.398 Ǻ (short bonds), while the other, forming pentagons, has a
length of 1.455 Ǻ (long bonds). The same pattern was confirmed by a diffraction
study of C60 crystal, which again revealed two types of bonds
differing in length [97].
Based on the general concepts of bonds at that time, the former were associated
with double sp2carbon bonds, and the latter with single sp3bonds. Within the framework of this concept of the
molecular structure, experimental data related to neutron diffraction from C60
powder [98],
NMR data [99],
and repeated X-ray diffraction data for a C60 crystal [100]
were further explained. Numerous studies of STM images of C60
fullerene adsorbed on semiconductor surfaces [101,102] helped
confirm the icosahedral (nearly spherical) shape of the molecule. The molecule
is most fully represented using high-resolution non-contact AFM with an oxygen
atom at the tip of the probe (CuO needle) [95] in Figure
7d. As can be seen from the figure, the molecule is indeed a
truncated icosahedron with two types of interatomic bonds.
Numerous calculations of the molecule’ digital twin
confirmed its icosahedral shape, characterized by two sets of bonds, but
disagreed on the definition of symmetry. According to RHF and DFT calculations,
the molecule's symmetry is , while UHF shows . Recall that in the UHF formalism, the reduction
in symmetry concerns spin symmetry, but it affects subtle characteristics of
the geometric shape as well. In the case of the C60 fullerene, the
latter concerns the length dispersion of the sp2 bonds that form the molecule's framework. Figure
8a (top) shows the bonds distribution for two structures of C60
digital twins that correspond to the RHF and UHF solutions, while Figure
8a (bottom) details the distribution of these bond lengths in
each of the structures [66,67,71].
As can be seen from the figure, while the average bond lengths are virtually
identical for long bonds and differ slightly for short bonds, the corresponding
dispersions caused by the splitting of both types of bonds into four groups
differ by four and 16 times for long and short bonds, respectively. This large
difference in dispersion is an adequate response of the structure to a change
in the spin state. As can be seen from Figure 8a, all long sp2bonds of fullerene C60, and some of the
short ones, are located above the critical value = 1.395 Å, so the significant number of
effectively unpaired electrons , amounting to 9.84 e, seems quite natural.
The fragmentation of short and long bonds into
groups is expectedly reflected in their spin characteristics. As can be seen
from Figure
8b (top), the ACS graph over atoms of the molecule C60
also has a grouped appearance, clearly evident in the ordering of the values (see the black histogram
in Figure
8c (top)). A similar pattern is observed for effectively unpaired
electrons of fullerene C70 with a total number = 14.4 e. In the case of C60 and C70
molecules, both values constitute about 15-20% of the total number
of odd electrons. As for Si60 with = 63.52 e, not only all 60 odd π electrons but also
some σ electrons are unpaired, which evidences about a complete radicalization
of the molecule.
In terms of the spin-radical concept, the discussed
ACS graphs in Figures 8b and 8c
exhibit chemical activity of the molecules’ atoms. The formatted graph of fullerene C60
manifests five groups of 12 atoms each, which are characterized by the same value within the group. Each group atoms forms six
identical C2 pairs held by one of the sp2 bonds. The spin densities of the atoms in any pair
are equal in magnitude and opposite in sign, so that the spin density of the
molecule is zero [103].
According to the ACS graph, the C60 molecule consists of six
identical C10 compounds formed by five pairs corresponding to the
total number of groups. Distributing the atoms across six fragments according
to the map shown in the figure, we obtain a 6*C10 configuration
consisting of six identical naphthalene nuclei. Applying different colors to
atoms with different values, we obtain a multi-colored 'chemical portrait' of the C60 molecule in the singlet state in the UHF approximation [66,67,68,71,104] shown in Figure 8b (bottom). The same numbering concerns five groups indicated in Figure 8c (top) and five colors in Figure 8b (bottom). Accordingly, two hexagons formed by light blue atoms manifest the most active area of the molecule. Any first addition reaction with C60 starts with one of these atoms.
formatted ACS graph of the molecule C70 shown in Figure 8c (top) is much less contrasted in comparison with that of С60. Nevertheless, as previously, the graph points to a well-defined grouping. The D5h symmetry of the molecule in the UHF singlet state governs the molecule structure decomposition into three five-benzenoid fragments. Two 20-atom fragments A, one of which is shown in in the middle of the figure, are formed by conjugated benzenoids and look like a five-lobe flower each. Five benzenoids mutually coupled via a single sp2 bond form a 30-atom closed rarefied chain-bracelet B. The highest values are concentrated just in this area. It is clearly seen in Figure 8c (bottom), where a color chemical portrait of C70 is presented, but in terms of spin density that is fully consistent with values [68,69]. Any first addition reaction on C70 occurs on the chain-bracelet atom.
Figure 9 returns us to the comparison of carbon and silicon fullerenes. The theory of spin-radical interactions allows only the C60 fullerene to exist, thereby defining, in agreement with empirical data, the boundaries of the region of permissible per-one-bond values. This region is marked by the light-gray band in Figure 4b. The relatively small values determine the status of fullerene C60 as a stable radical. The corresponding to for the sp2 bond lies well above this region, once again confirming the impossibility of the Si60 fullerene existence. However, as Figure 4b shows, the value corresponding to of the sp2 bond falls within the region of permissible values. Thus, the spin-covalent chemistry of the sp2 bonds does not hinder formation of the (SiC)60 fullerene, which may allow it to be synthesized in the future.
5.3. Carbon Nanotubes
From the vast ocean of experimental and virtual data on carbon nanotubes (CNTs), we select only those related to the molecular structure of CNTs, which is based on chains of covalent bonds. Although diverse in their physical and chemical properties, CNTs are uniform from a molecular perspective. They all represent cylinders of varying diameters and lengths, formed by twisting a monatomic honeycomb web. The benzoid rings of the web are formed by sp2 bonds with lengths ranging from 1.39 to 1.45 Å, and their dispersion can vary. Most bonds are longer than the critical length , resulting in the electron system of the tubes containing a significant number of effectively unpaired electrons. Available virtual data show that the per-one- bond values fall within the region of permissible data, indicated in Figure 4b, evidencing a stable-radical character of the species.
The UHF approach was first applied to two fragments of (4,4) defect free and (4,4) 5–7 defect single-walled CNTs (SWCNTs) [105]. The obtained characteristic ACS graphs along the tube's atoms, manyfold supported in further studies [106], are shown in Figure 10. The graphs reliably monitor the tube atoms’ atomic chemical susceptibility as well as tight connection of the latter with the tube structure, thus highlighting the distribution of the sp2bond length excess over along the tube. The discussed graphs are related to CNT digital twins in the form of fragments of (4,4) SWCNT, equilibrium structures of whose digital twins are shown in the figure. One end of all fragments is capped, while the other is either open but hydrogen terminated (NT1) and empty (NT2), or capped (NT3). The total number of effectively unpaired electrons of the tubes is concentrated around 32 e. The distribution of these electrons over the tubes’ atoms forms characteristic ACS graphs that are shown in in the figure. As seen in Figure 10a, the tube can be divided into three regions. The first is related to the cap with adjacent atoms. The second concerns mainly the tube sidewall while the third refers to the open end terminated by hydrogen atoms. The biggest nonuniformity of the distribution is characteristic of the cap region. One should pay attention to the fact that the largest values belong to atoms that form the longest bonds. As for the sidewall region, the distribution is practically uniform, with the value scatter not bigger than 0.5%, which is consistent with a quite uniform distribution of the bond lengths as well. The graph in the end region is significantly affected by the hydrogenation. The curve with dots in the figure presents the free valence of the tube atoms that well coincides with the ACS expressed by . According to the latter, the tube cap is the most reactive part, while the tube sidewall is more passive, with ill-pronounced selectivity along the tube.
Removing hydrogen atoms at the tube’s open end, one obtains the graph of the NT2 fragment shown in Figure 10b. A tremendous contribution of the end atoms obviously dominates the graph. This is due to the fact that the sp2bonds are replaced in the region with the sp1ones. The transformation naturally results in increasing the total number of effectively unpaired electrons from 32.38 e to 39.59 e. The injection of additional effectively unpaired electrons disturbs the graph of the hydrogen-terminated tube (shown in the figure by bars) quite considerably. It is important to note that the changes occur not only in the vicinity of the open end, but also in the opposite cap end. Practically no changes occur along the tube sidewall, which seems to serve as a peculiar resonator for the electron conjugation. Addressing the chemical activity of the tube, dominant activity of the empty-end atoms is evident. Placing the cap at the open end of the tube (NT3 fragment) makes its ACS graph almost symmetrical (see Figure 10c).
Exhibited peculiarities of the discussed ACS graphs are generally common for all types of CNT and allow making general conclusions concerning addition reactions to be expected [106]. The space of chemical reactivity of any SWCNT coincides with its coordinate space, while remaining different for particular structure elements. Increasing the tube length leads to a broadening of zone 2 in its ACS graph without changing its amplitude. Increasing the tube diameter affects the graph' amplitudes if it is accompanied by a change in the dispersion of bond lengths within the tube. The apex and end atoms of the tubes are sensitive to the heteroatomic state of the surrounding environment. Local additions of short-length addends (involving individual atoms, simple radical, and so forth) to any SWCNT are the most favorable at open empty ends. Following these places in activity are end caps, defects in the tube sidewall, and the sidewall itself.
5.4. Graphene Domains
Graphene is currently one of the most actively studied objects of modern material science. Countless theoretical and experimental studies have already been performed, targeting electronic, magnetic, thermal, optical, structural, and vibrational properties. Low and homogeneous chemical reactivity of atoms throughout a graphene sheet is usually expected by the predominant majority of scientists dealing with graphene chemistry. However, the first UHF calculations [107] showed that this is not the case, since the equilibrium length of about half of sp2 bonds of graphene domains exceed the critical value ≅ 1.395 Ǻ. The domains are significantly radicalized, which is manifested with significant values of both and . Research-friendly rectangular digital twins of nanosize graphene domains (nanographenes) are nominated as (na,nz) NGrs following [108]. Here na and nz match the number of benzenoid units along the armchair and zigzag edges of the domains, respectively. Similar to CNTs, ACS graphs of graphene domains are quite typical, only slightly dependent on the domains size, while grows with the size.
Characteristic ACS graphs for (5,5) NGrs with bare and hydrogen-terminated edges, presented in Figure 11, demonstrates a rather significant variation of the quantity over atoms. As seen in the figure, the highest are characteristic of carbon atoms at the zigzag edges, while those of the armchair edges are similar to the values of atoms of the domain basal plane. The latter are comparable with those of fullerenes (ca. Figure 8) and SWCNT sidewalls (Figure 10). When hydrogen terminators are removed, values on both zigzag and armchair edges grow significantly, still conserving bigger values for zigzag ones. The obtained results alongside with many other known today [69,109,110] made allowance for the following conclusions concerning chemical reactivity of graphene materials. All reactions with graphene domain that is a spacious object are of topochemical nature. There is still no reliable algorithm for such reactions, although the main control element is unlikely to deviate in appearance from the well-tested form of an ACS graph. For now, we have to limit ourselves to this graph of ordinary chemistry when planning the course of successive reaction steps, as in the case of fullerenes and СNTs. Based on the graphs presented in Figure 11, it can be stated that any chemical addend will first be attached to the graphene zigzag edges, both hydrogen- and other heteroatom-terminated and empty. Chemical reactivity of basal-plane atoms depends on the edge termination and cannot be strictly correlated with activity of fullerenes and SWCNT sidewalls. The steadiness of stable radical status for the graphene domains is ensured by the state of edge atoms of the domain.
6. Spin Covalence of Alkyne Bonds
Comparing the ACS graphs of ethylene and propyine shown in Figure 3a, one can easily conclude that molecules with alkyne bonds should be more active. In practice, the situation is more complicated. Not many substances with alkyne bond chains are known and graphpolyines (GPYs), as related to modern graphene science, are of a particular interest. The species were object of numerous studies during last time (see comprehensive reviews [21,111,112,113] that discuss and summarize the state-of-the-art research of the issue. Introduced in Section 1.2, these allotropes are flat one-atom-thick carbon networks, which can be constructed by replacing some bonds in graphene by uniformly distributed polyacetylenic linkages (n–[–CC–]) forming GPYs, among which there are graphynes (GY, n=1), graphdiynes (GDY, n = 2), graphtriynes (GTY, n = 3), and so forth. GPYs are largely variable structures, a particular part of which is occupied by species consisting of networks that include C6 hexagons interconnected with polyacetylenic linkages. The main impression on promising interesting properties of GPYs as well as on their possible applications was provided virtually, while experimental evidences are rather scarce. However, almost all the computations were performed in closed-shell approximations without taking into account spin covalence of alkyne bonds. To compensate this drawback let look at some basic components of GPYs from the viewpoint of UHF formalism.
As seen in Figure 12a, diphenyl-n-acetylenes (n-DPHAs, phenyl terminated polyynes) consisting of two phenyls connected with a varying number of acetylenic linkages (ligaments below) are the basic GPY components. A set of n-DPHAs with n from 1 to 4 is shown on the left panels of the figure, while right panels display the relevant ACS maps that exhibit the presence of chemical reactivity of the molecules and disclose its distribution over the molecules’ atoms. Going from the top to bottom, one can see how the reactivity map changes in value and space when the acetylene linkage increases. The linkage of one acetylenic unit between benzene rings causes a considerable elongation of some of the ring bonds, thus promoting a significant radicalization of the rings as presented at the right-hand panel. The total number of effectively unpaired electrons = 1.22 e with fractional values on the ring carbon atoms from 0.10 e to 0.08 e. Similar small radicalization of = 0.06 e concerns the acetylenic unit. Inclusion of one more acetylenic unit between benzene rings promotes a remarkable elongation of both triple bonds, which, in turn, results in the enhancement of their radicalization just lifting both and values while characteristics of benzene rings remain practically unchanged. This trend is preserved with further increase in the number of triple bonds. As seen in the figure, in due course of growth of the triple bond number, the chemical reactivity of DPHAs is increasingly concentrated on the atoms of acetylenic units evidencing the growing elongation of the latter. This might be explained by the transformation of a quite rigid sole triple bond to a flexible chain of the bonds, which readily promotes the bond elongation, with quite regular spacing of 1.217–1.225 Å within the acetylenic units and of 1.327–1.332 Å between them.
Presented in Figure 12a exhibits a great increase in the chemical activity of polyynes when acetylenic chains becomes longer. Apparently, the finding explains the failure in the synthesis of linear ynes with n >11 [114]. Experimentally observed structures of polyynes with n from 2 to 11 manifested regularly distributed acetylenic units over the chain with characteristic interatomic spacing of 1.201–1.211 Å and 1.353–1.364 Å depending on the length and termination of the acetylenic chains. The data are in perfect agreement with those related to the DPHAs shown in the figure. It proved possible to overcome high chemical activity of long polyynes only placing them in a confining reactor [115]. The linear carbon chains were encapsulated in and protected by thin doublewalled carbon nanotubes. Exceptionally long and stable chains composed of more than 6,000 carbon atoms were obtained.
The considered DPHAs lay the foundation of various GPYs differing by the number of acetylene linkages between benzenoid rings. Thus, 1-DPHA, 2-DPHA, and 3-DPHA form the grounds of graphyne, graphdiyne, and graphtriyne, respectively. Independently of a concrete structure of the involved DPHA, the composition like a six-petaled flower lays the foundation of the structure of any GPY. Six-branched benzenoid hexagon C6 determines each of the flower centers, while another six hexagons terminate the flower petals. Each of these rings, one-branched previously, gradually becomes six-branched in due course of the GPY growth in plane, which results in a particular triangle pattering of the GPY body consisting of triangle-closed cycles. Three hexagons form the vertices of the triangle while ligaments lie along its sides. Basing on these structural grounds, it is easy to imagine a consequent formation of a regular extended structure of GDY. Thus, Figure 12 b presents one-triangle 2-DPH based compositions. The ACS map, or chemical portrait of the molecule at right-hand panel in the figure impressively exhibits changing in the 2-DPHA atomic reactivity caused by the triangle composition, which reflects a different changing in bond lengths.
A successive ranching of benzenoid hexagons of GDY recalls braiding Irish lace, whose main motive is selected by circle in Figure 12 c. The triangle fragment marked by red constitutes its main part related to the extended lace body. The knitting is obviously a multi-stage complex process, which is difficult to trace in all details. However, taking ACS maps as assistants, it is possible to disclose general trend and regularities for making the following conclusions. GDY presents a large cloth with a regular flower-like print where six-branched benzenoid hexagons play the role of the main floral motif while alkynic ligaments present thin twigs. The motive is a radical, but the status of its radicalization depends on the surrounding structure. The highest radicalization is related to that one surrounded by identical six-branched benzenoid hexagons. The motive main radicalization is concentrated on the hexagon, while each ligament is about half less reactive.
The GDY cloth as a whole is highly radicalized and, consequently, chemically reactive. The values of the motive atoms are similar to those that are characteristic for carbon atoms of fullerenes, nanotubes, and basal plane of graphene domains. Similar to the latter bodies, the per-one-alkyne bond fall inside the light gray band on Figure 12c, because of which GDY are stable radicals and can exist at ambient conditions, once inclined to a variety of chemical transformations. Cutting and saturation with defects will considerably enhance the body reactivity, which should be taken into account when discussing a possible controlling of electronic properties of GDY devices [113,116].
7. Carbon Catalysts in Light of Their Spin Covalence
The catalytic properties of carbon molecules are of interest to two rapidly developing branches of metal-free catalysis: organocatalysis [117,118,119,120] and carbocatalysis [121,122,123,124,125,126,127,128,129,130,131]. It should be noted that no bond-classified class of carbon molecules has attracted particular attention in organocatalysis: neither alkanes and alkenes, nor alkynes. In the literature, only a few references can be found to the involvement of alkenes [120] or alkynes [132]. Carbenes [133] have received somewhat more attention. Analysis of numerous manifestations of organocatalysis has revealed a clear correspondence between the catalytic eficiency of molecular catalysts and their acidic and alkaline properties. The release of catalysts from the metals has led to the only mechanism of catalysis associated with the presence of oxygen, sulfur, nitrogen, etc. heteroatoms.
In contrast to organocatalysis, carbocatalysis utilizes purely carbon catalysts, which mainly are representatives of sp2nanocarbons, including fullerenes, carbon nanotubes (CNTs), and graphene materials. The latter play a leading role in these reactions, while the use of fullerenes and CNTs is still scarce. Graphene carbocatalysis is divided into two large groups determined by the size of the catalysts used, namely, large-area and small-area ones. Catalysis in the first group is solid-state one, the features and control of which are determined by the surface properties of the reactants participating in the reaction (see [126,134,135,136,137,138] and references therein). The second group, combined with fullerenes and CNTs, is molecular carbocatalysis, a brief analysis of which, in light of the spin properties of their sp2 bonds, will be presented below. Interested primarily in the virtualization of this process, we begin our discussion by determining the structural dimensions of the catalysts and establishing the nature of their active sites. This primarily concerns graphene materials.
7.1. Graphene Carbocatalysis
The main types of molecular graphene structures are shown in Figure 13. Since sp2 graphene is chemically an atomic system with unsaturated valence bonds, the type of molecular structure is determined by the chemical state of two structurally sensitive regions of the graphene domain—its edge atoms with dangling bonds and basal plane atoms with unsaturated valences. The bare graphene domain is extremely chemically active and does not actually exist in practice. At ambient conditions, valence saturation of the domains occurs in two stages [109]. The first concerns the domain edge atoms, while maintaining the sp2 configuration of the carbon atoms in the basal plane. Such necklaced molecules are stable radicals [139] and are widely known as the basic building units (BSUs) of all types of sp2 amorphous carbon () [37]. Nanoscale synthetic reduced graphene oxide () also belongs to this group. The second stage of valence saturation affects the carbon atoms in the basal plane of the graphene domain, the sp2 configuration of which is replaced by the sp3 one [140]. At 100% valence saturation, the chemical activity of the molecule becomes zero. Graphene oxide () belongs to this type of molecule. does not exist in nature. It is synthesized by the harsh oxidation of highly fragmented graphite, so that the final product is a nanoscale powder, which is then used to obtain by chemical reduction.
The above-mentioned acid-base concept of the active cite of organocatalysts, proposed more than 30 years ago, were transferred to carbocatalysts (see details in review [141,142]). In the latter case, the concept has been somewhat corrected emphasizing a particular role of organic molecules due to many of them, possessing acidicity or basicity as well as redox activity, can catalyze reactions. The view on virtual carbocatalysis from the standpoint of the spin chemistry of graphene molecules was firstly proposed in [143] for catalysts, BSUs of which were presented with a conglomerate of graphene-oxynitrothiohydride stable radicals. The chemical activity of the BSUs atoms is reliably determined computationally, which allows mapping the distribution of active sites in these molecular catalysts. The presented maps reliably show the BSUs radicalization provided with carbon atoms only, the non-terminated edge part of which presents a set of active cites. Spin mapping of carbocatalysts active cites was suggested as the first step towards the spin carbocatalysis of the species.
Figure 14 presents a comparative view on the radical behavior of the studied s exemplified by ‘chemical portraits’ of the relevant digital twins. Each of the exhibited molecules heads a set of digital twins suggested for the studied amorphics [37]. The ACS map of the parent (5, 5) NGr domain accompanied with the formatted ACS graph is shown in Figure 14a. Bright spots on the map indicate cites of the most chemical activity. Quantitative presentation of this activity is shown by the ACS graph nearby. As seen in the figure, large values of 0.9÷1.3 e distinguish 22 edge atoms while values 0.3 e belong to basal-plane atoms.
This characteristic feature of bare graphene molecule is generally preserved for all necklaced graphene molecules as well while is evidently disturbed. As visible in Figure 14b and Figure 14c, and 14d, the inclusion of hydrogen and oxygen into the parent domain circumference expectedly inhibits the activity of the edge carbon atoms, directly involved in the new bonding. At the same time, the figures show enhancement of the activity of basal-plane atoms. The feature clearly demonstrates a peculiar collective character of chemical events occurring with graphene molecules [70]. The molecular chemical susceptibility of the parent domain C66 (33.49 e) considerably reduces in all the necklaced molecules and constitutes 23.65 e (C66O4H6), 20.49 e (C66O5H12), and 29.66 e (C66O4H3). As is visible in the figures, each decoration act causes remarkable changes in the ACS graphs thus revealing the bonds’ redistribution of the whole molecules. Despite the effect in each case is expectedly individual, the general pattern of the ACS graph is conserved. As for functional groups involving heteroatoms, a dominant role of which in carbocatalysis has been largely discussed until now, as seen in the figure, they completely lose their activity after attaching the carbon core and are not capable to further lead any chemical reaction while adjacent ‘empty’ carbon atoms are ready to play the role.
According to one of the most reputable experts in the field, “catalytic applications of carbon materials are as old as the discipline of physical chemistry, and probably even older” [144]. In fact, s materials such as activated carbons and carbon blacks have been used for ages in heterogeneous catalysis as either catalysts or catalyst supports. The first documentation of the issue was done about hundred years ago in a report about the aerobic oxidation of oxalic acid occurred on the surface of charcoal [145]. The latest achievements of this biocarbon catalyst can be found in reviews [121,146]. By the end of the nineties, research in the area of catalysis was a well-defined field of material science, whose further development is reflected in a large number of publications, thoroughly reviewed in a set of reviews and monographs [122,123,124,126,127,128,129,130,147,148].
In addition to serving as catalysts, small-area necklaced graphene BSUs of sp2 s provide a protective function similar to that of the large-area ones [137,138]. This is a reason of the carbon coating that accompanies great number of natural minerals. In the case of the dominant presence of carbon, say, of natural deposits of shungite, the carbon coating is transferred into encapsulating nanosize carbon sacks, whose skin is constructed from shungite’s BSUs while the interior is filled with ideally crystalline quartz [149,150,151]. Averaged size of the quartz particle constitutes (80 2) nm, which is well correlated with the size of a sphere, constructed with densely packed BSUs of shungite of 2.5 x 2) nm2 in size [37]. As occurred, sp2-carbon sacks were proposed for and realized in biotechnology as well [152], allowing to suspect that the presence of the nanosize graphene’s traces in the COVID vaccines [153,154] may be explained by the necessity to elongate the mRNA time of life encapsulating the latter in the carbon sacks.
7.2. Fullerene Carbocatalysis
The influence of the presence of fullerene, primarily of fullerene C60, on processes in reaction solutions is well known. It is enough to recall the effects it causes in the free-radical polymerization of vinyl monomers [68,84,155]. As for catalytic reactions involving fullerene, the author knows of only two: the oligomerization of terpenes [156] and the polymerization of styrene [83,84,155]. In the first case, the catalytic behavior of fullerene was suspected, while in the second, it has been convincingly proven. For a detailed description, we recall that catalysis has always been and remains the most difficult part of synthetic chemistry and is determined by a large number of thermodynamic and kinetic factors. In turn, the thermodynamics and kinetics of any elementary reaction between a pair of reactants are described by a characteristic graph [157,158], the main parameters of which are shown in the inset of Figure 6. The enthalpy of the reaction, or the coupling energy between its participants , determines whether the reaction will be energetically favorable. The height of the energy barrier determines the reaction rate . These two quantities are the main numerical descriptors of the reaction, and it is with them that the catalyst must work when the reaction is difficult or too slow. Obviously, the reaction is favored by , large in absolute value and negative in sign. If the thermodynamic descriptor of the reaction under consideration does not meet this requirement, the catalyst takes over the initial stage of the reaction, forming a pair with one of the participants with a good descriptor . A typical example is the free radical polymerization of vinyl monomers, in which free radicals act as effective catalysts of the first step of the reaction (see a detailed description in [82,84]). In turn, the height of the energy barrier in the case of participants from covalent chemistry depends on the energy gap , where and are the ionization potential and the electron affinity energy of the donor and acceptor participants in the reaction, respectively [159], so that a decrease in or an increase in is accompanied by a decrease in the value of and a corresponding increase in the kinetic descriptor . This circumstance is one of the main ones for metal catalysts characterized by low . A typical example is the kinetically hindered dimerization of fullerene C60 [160] and its monoderivatives [155], which becomes practically barrier-free in an electric field [160] or upon the addition of butyllithium to C60 [155].
A virtual quantum-chemical consideration of the thermodynamic and kinetic descriptors and of elementary reactions in a reaction solution containing styrene, the free radical , and fullerene C60 showed that the fastest reaction is the formation of the monomer-radical , in which fullerene C60 plays the role of the free radical, provided that the monomer is added to it through a single-bonded intermolecular contact [83,155] as shown in Figure 15a. As can be seen from the figure, confidently leads polymerization of styrene, the first members of which are shown in Figure 15b. Additionally, the formed oligomer-radical branches readily attach to the remaining free portions of the carbon frameworks of the fullerenyls generated during the reaction, forming star-shaped structures of the final polymerization products, represented in Figure 15c as an intermediate star . The resulting virtual picture is confirmed by a detailed analysis of the empirical data.
Following the path taken to establish the catalytic activity of fullerene, let us return to the oligomerization of α- and -pinenes in the presence of fullerene C60 and gaseous oxygen [156]. Thus, the reaction solution of interest contains a pinene, C60 fullerene, and degassed water. As follows from the experiment, pinene oligomerization is not observed; the monomer and fullerene retain the properties of the original molecules, indicating the absence of a productive interaction between them. The situation changes after passing a stream of oxygen through this solution. The transparent free-flowing water becomes viscous. Its color caused by fullerene [101] changes noticeably, indicating the formation of fullerosils. Fast atom bombardment mass spectrometric analysis reveals the presence of terpene’s dimers, trimers, and even traces of tetramers, which naturally explains the occurrence of the water viscosity, as well as fullerosils of the compositions C60O, C60O2, C60O3. Since separately pinene and fullerene dissolved in degassed water do not lead to similar changes, the conclusion follows that these changes are due to the interaction of the components of the pinene-fullerene-oxygen triad. What happens to this triad? To answer this question, it is necessary to consider virtually a set of elementary reactions, similar to what was done in the case of vinyls’ polymerization [82,84]. However, even without performing calculations, a certain prediction based on the experience of the styrene polymerization discussed above, can be made.
As follows from structure measurements [161], the particularly active sites of - and -pinene molecules are connected via sp2bond of their vinyl groups of 1.34 Å in length [161]. This value is much lower than , so that odd electrons of both carbon atoms are completely paired, as a result of which on both atoms is zero. Since the remaining carbon atoms are linked by alkane bonds and there are no other types of heteroatoms, the original pinene molecule is chemically inactive and its oligomerization, as in the case of benzene molecules [69, chapter 2], should be difficult. The same can be said about the oxidation of the molecule. In contrast, the oxidation of fullerene C60 and the formation of the aforementioned fullerosils are quite expected [67,68]. Moreover, each new added oxygen atom breaks one of the sp2 bond leaving the paired carbon atom with high value as a target for the next addition. If pinene is this addend, only one-bond connection between fullerene and one of its vinyl group carbons is possible which leads to generation of monomer radical analogous to that of styrene one discussed above. The resulting radical ensures the oligomerization of pinene on the fullerene body in the form of fullerosil C60Mn. The bulkiness of the terpene molecule and the resulting steric hindrances evidently prevent the formation of long oligomers. Room temperature of the solution leads to the splitting off the resulting oligomers into the solution. Virtualization of this process is entirely feasible, just like any other involving a monomer with an alkene bond, thus opening the way to virtual fullerene carbocatalysis.
8. Aposteriori Reflections and Conclusive Remarks
The bond type depicts the carbon family in which carbon-containing substance are born. Odd electrons, generated at either formation or rupture of the bonds, and spin-radical interaction between them are the main contributors to the treasure trove of special properties of these substances. This is how the credo of carbon covalent chemistry can be summarized today. Its first part reflects the trimodality of the bonds as a set of sp3, sp2, and sp1 ones, while the second refers to the peculiarities of the weak electron interaction, revealing its spin nature and transforming the classical covalent chemistry of carbon into spin covalent one. The degree of importance of the weak interaction varies for each of the three bond types, but in all cases is determined by the bonds’ length. This article represents the first review of existing results in carbon chemistry, considered from the perspective of the spin nature of bonds. Without rehashing the content of the article, we will formulate its main conclusions in the bond-length language
sp3bonds, known as single and alkane bonds. The life of these bonds occurs in the covalent regime, in the absence of odd electrons and the associated spin effects, which manifest themselves only at the moment of their rupture. Using the example of the ethane molecule, the life cycle of its lone sp3 bond lasts in the region of length = 0.607 Å that begins at = 1.503 Å, and ends when the bond length reaches = 2.110 Å, at which the bond starts to radicalize sharply and then goes to full rupture. As we can see, reveals a fairly wide range of possible bond elongation, while maintaining its spinless existence. After the bond rupture, two free radicals emerge with the summarized number of effectively unpaired electrons equal to 2. The equilibrium and critical within 2-3% retain the given values in almost all cases of observing these bonds in molecules not only carbonaceous, but also containing various heteroatoms. The values of are confirmed by numerous structural data. As for , spin covalent chemistry of carbon assigns it the place of the reaction coordinate localizing the transition state in reactions caused by either formation or rupture of this bond.
sp2bonds are known as double, alkene, and aromatic. The life cycle of the lone bond of ethylene within the limits of its length = 0.062 Å from = 1.326 Å to = 1.388 Å occurs in classical spinless regime, when odd electrons are tightly bound providing the electron contribution into the bond additionally to the one. When the bond length exceeds , the spin covalence begins to work stimulating at first radicalization and breaking the constituent of the bond reaching = 2 e by = 2.140 Å and accompanying further elongation of the bond by radicalization of its component up to complete bond rupture and growth of the number of effectively unpaired electrons to 4. The sp2 bond behavior above = 2.140 Å is similar to that of the sp3 one above = 2.110 Å.
is the main spin-marker of each sp2 bond. The value may be small and big, as well as, what is the most important, positive and negative. If it is positive, the bond is of classical spinless format and molecular compositions on their basis are spinless and non-radical. Therewith, the small marker value does not change the molecule behavior as it is in the case of benzene, for which 0.005 Å. However, already in the case of naphthalene, we encounter a situation where the carbon framework of the molecule is formed by bonds of two types: short and long, with positive for the former and negative for the latter. Thus, classical and spin covalence begin to compete, and the final result depends on the number of both bonds. The situation becomes sharp since ‘the safety interval’ is not very large making up only 10% of that one for alkane bonds, thus not presenting serious obstacles to the flow of alkene bonds from the group of short bonds to the group of long bonds and vice versa. In the PAH polyacene series, as the number of benzene rings increases, the number of long bonds increases faster than short ones, which correlates with the increasing radicality of the molecules, to the point where their synthesis at ambient conditions becomes impossible. We encounter a similar situation in the case of sp2 nanocarbons – fullerenes, carbon nanotubes and graphene domains. Clearly, the ratio of the numbers of negative (long bonds) and positive (or short bonds) determine the stable-radical status of these molecules. It appears that this ratio cannot be greater than one. Actually, close-to-one ratio is characteristic for stable PAHs and mentioned sp2 nanocarbons. However, more exact answer to this question requires further research.
The case of negative markers for all double bonds evidences highly reactive sp2 radical species. Molecular sp2 carbon of this type, neither virtual, nor real, has not been known. In contrast, it is typical case for virtual sp2 silicon. Even for lone sp2 bond of disilene, = -0.72 Å. This value is preserved with accepted accuracy in large molecules such as aromatic hydrosilicons similar to polyacenes and sp2 nanosilicons in form of fullerenes, tubes, and one-atom thick domains leaving no hope for the synthesis of the species in reality. However, there are still quite a few researchers who do not accept this verdict of spin covalent chemistry and continue to discuss, in particular, the properties of silicene as a real object analogous to graphene.
Carbon fullerenes, nanotubes, and necklaced graphene domains are stable radicals, properties of which are subordinated to spin theory of radicals. Thus, they are susceptible to chemical modification in various ways, in particular, to various derivatizations. For closed structures, such as frameworks of these substances, this property may lead to the return of spin-covalent sp2 bonds into classical spinless format. The fact is that any addition act with respect to a framework atom causes an inevitable sp2sp3 transformation of valence electrons hybridization of the latter. This rearrangement is accompanied with changing of spatial structure of the framework. Thus, the flat trigonal packing of C3 triad atoms connected with sp2 bonds is transformed into a three-dimensional tetrahedral packing of the C4 tetrahedron atoms connected with sp3 bonds. In the case of chemical modification of sp2 nanocarbons in full, this leads to the replacement of flat benzenoid basic units with their cyclohexanoid analogs. As a result, the chemical action is accompanied by physical restructuring, causing significant mechanical stress since the sp2sp3 rearrangement of the atomic system, requires an increase in space. Truncated icosahedron of C60 fullerene, cylindrical packings of carbon nanotubes, or graphene membrane domains, resist this stress which causes shortening some of remaining chemically unmodified sp2 bonds thus transforming their negative markers into positive ones, which was discussed above. This leads to removing the radicalization of the corresponding sp2 bonds, zeroing and stopping chemical reactions in which these bonds are involved. This effect, exceptionally important for modern fullerenics and grapheneics, was discovered during virtual fluorination [67,162] and hydrogenation [67,163] of fullerene C60, as well as hydrogenation of the graphene domain [69,164]. However, it was not taken into account for a long time until researchers encountered the paradoxical fact of a full identity of the characteristic Raman spectra of and presented in Figure 16. Virtual stepwise oxidation of the graphene domain [140] revealed the impossibility of its oxidation in full due to a significant shortening of approximately 20% of the sp2 C2 pairs in the basal plane of original domains to 1.35 Å, which is significantly less than the critical value . The percentage is fully sufficient to explain the presence of 20–10% unoxidized carbons in the sample array, which has been repeatedly established in practice (see reviews [165,166,167]. This finding of shortened sp2 bonds made it possible to explain the presence in the Raman spectrum of molecular of the G band of fully symmetrical vibrations of sp2 bonds next to the D band, which is caused by the vibrations of the same type but related to basic massive of sp3 bonds. In turn, the presence of the D band in the Raman spectrum of raised a similar question, while, the presence of G was is absolutely natural. The answer concerns the sp2sp3 transformation again. As shown [165], generally, rGO consists of multilayer stacks of BSUs presented with nanosize necklaced graphene domains [37]. Nanoscopic buckling of the latter forces some atoms of adjacent layers generate the fourth bond, which is supported with large enough ‘safety interval’ thus transforming the entire set of valence bonds of these atoms into sp3-type bonds abd ensuring D band emergence. The intensity and shape of this band allow to estimate the number of these domains in the BSU stack and the transverse size of the domains. The discussed characteristic doublet pattern of the Raman spectrum is widely used in practice as the main analytical evidence of the graphene nature of the material used, while not being able to separate the completely different sp3 and sp2 .
sp1bonds, known as triple or alkyne bonds. The life cycle of a sole bond in acetylene or propyne in spinless classical format is limited with very narrow region = 0.043 Å from = 1.240 Å to = 1.197 Å. The marker value indicates that behavior of sp2 and sp1 bonds is quite similar. Convincing confirmation of this statement can be seen in the ACS maps presented in Figure 12a for the border-regime sp2 bonds of benzene and sp1 bonds of carbyne chains. The lone sp1 bond cleavage starts at the point = 2.100 Å, practically coinciding with of ethane and showing that all three lone bonds break when their lengths reach the same value. The border-regime character of sp1 bonds naturally explains a comparative sparseness of population of the molecular world containing sp1 bonds. The bonds’ chain creation is evidently impossible. The situation is saved by the possibility of the existence of alternating polyyne chains . The presence of sp1 bonds suggests the spin-covalent nature of the doubly bonded links of these chains, however, the problem should be proven virtually.
The discussion, presented in this review in the language of valence bonds, is a discussion of the essence of covalent chemistry. This language was formed during the development and maturation of classical spinless covalent chemistry over a long historical period. Spin covalent chemistry has emerged from the vast experience of its predecessor and retains its language, imbuing it with new meanings and content. A significant advantage of the new scientific concept is the spin theory of electron-electron interactions, with particular emphasis on the interaction’s weakness under certain circumstances. This theory utilizes a well-developed mathematical apparatus, allowing for the construction of transparent algorithms describing the behavior of atoms in molecules and facilitating the widespread digitalization of individual chemical processes and the construction of their virtual worlds [168].
The review mentions the first results of virtual fullerenics, virtual graphenics, virtual polymerization of vinyl monomers, virtual graphene and fullerene carbocatalysis. But the main result of spin covalent chemistry is the obtained convincing proof of the uniqueness of the carbon atom as the only pretender on the king throne of covalent chemistry, while the closest contenders from the tetrel family - silicon, germanium, and tin - are deprived of this right according to the law of spin covalent chemistry due to the deprivation of their possibility of material embodiment. As for the spin covalent chemistry of carbon, it is taking its first steps, but even these still few successes open up broad horizons for a new vision of processes controlled by spin-radical electronic interactions, such as chemical modification or derivatization of any complexity, polymerization and catalysis of molecular compounds whose electronic properties have a spin aroma.
Acknowledgments
This paper has been supported by the RUDN University Strategic Academic Leadership Program.
References
- Allègre, CJ; Poirier, J-P; Humler, E; et al. The chemical composition of the Earth. Earth Planet Sci Lett. 1995, 134, 515–526. [Google Scholar] [CrossRef]
- Sladkov, A. M.; Kudryavtsev, Yu. P. Diamond, graphite, carbyne = allothropic forms of carbon. Priroda (Moscow) 1969, 5, 3. [Google Scholar]
- Bean, J. J.; Katiyar, N.K.; Forrest, R. M.; Zhou, X.; Goel, S. Resolving Lonsdaleite's decade-long controversy: Atomistic insights into a metastable diamond polymorph. Diam. Rel. Mat. 2025, 157, 112405. [Google Scholar] [CrossRef]
- Thapliyal, V.; Alabdulkarim, M. E.; Whelan, D. R.; Mainali, B.; Maxwell, J. L. A concise review of the Raman spectra of carbon allotropes. Diam. Rel. Mat. 2022, 127, 109180. [Google Scholar] [CrossRef]
- Ubbelohde, A.R. F. A. Lewis, Graphite and Its Crystal Compounds; Clarendon: Oxford, 1960. [Google Scholar]
- Kudryavtsev, Y.P. The discovery of carbyne. In Carbyne and Carbynoid Structures. Physics and Chemistry of Materials with Low-Dimensional Structures; Heimann, R.B., Evsyukov, S.E., Kavan, L., Eds.; Springer: Dordrecht, 1999; vol 21, pp. 1–6. [Google Scholar]
- Kudryavtsev, Y.P.; Evsyukov, S.E.; Guseva, M.B.; et al. Carbyne — the third allotropic form of carbon. Russ. Chem. Bull. 1993, 42, 399–413. [Google Scholar] [CrossRef]
- Kudryavtsev, Yu.P.; Evsyukov, S.E.; Babaev, V.G.; Gouseva, M.B. Oriented carbyne layers. Carbon 1992, 30, 213–221. [Google Scholar] [CrossRef]
- EL Goresy, A.; Donnay, G. A new allotropic form of carbon from the Ries Crater. Science 1968, 161, 363. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, A.G. Carbon: occurence of carbyne forms of carbon in natural graphite. Carbon 1979, 17, 21–24. [Google Scholar] [CrossRef]
- Bulychev, B. M.; Udod, I. A. Formation of ‘carbyne’ in the interaction of polyacetylene. Ross. Khim. Zh 1995, 39, 9–14. [Google Scholar]
- Heimann, R. B.; Evsyukov, S. E.; Kavan, L. (Eds.) Carbyne and Carbynoid Structures; Springer: Dordrecht, 1999. [Google Scholar]
- Heimann, R. B. The nature of carbyne – pros and cons. In Carbyne and Carbynoid Structures; Heinmann, R.B., Evsyukov, S.E., Kavan, L., Eds.; Springer: Dordrecht, 1999; pp. 1–15. [Google Scholar]
- A Hirsch, The era of carbon allotropes. Nat. Mater. 2010, 9, 868–871. [CrossRef]
- Jarowski, P.D.; Wodrich, M.D.; Wannere, C.S.; Schleyer, P.V.R.; Houk, K.N. How large is the conjugative stabilization of diynes? J. Am. Chem. Soc. 2004, 126, 15036–15037. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, L.; Altus, E.; Basch, H.; Hoz, S. Harder than diamond: determining the cross-sectional area and Young's modulus of molecular rods. Angew. Chem. Int. Ed. 2005, 44, 7432–7435. [Google Scholar] [CrossRef] [PubMed]
- Enyashin, A.N.; Ivanovskii, A.L. Structural, electronic, and elastic properties of Y-diamonds and their BN analogues. Diam. Rel. Mat. 2013, 38, 93–100. [Google Scholar] [CrossRef]
- Enyashin, A.N.; Ivanovskii, A.L. Structural and electronic properties of new 1D and 2D carbon allotropes with mixed sp1 - sp3 hybridization types. Chem. Phys. Lett. 2014, 609, 15–20. [Google Scholar] [CrossRef]
- Li, X.; Li, B-h.; He, Y-b.; Kang, F-y. A review of graphynes: Properties, applications and Synthesis. New Carbon Mat. 2020, 35, 619–629. [Google Scholar] [CrossRef]
- Baughman, A R. H.; Eckhardt, H. Structure-property predictions for new planar forms of carbon: Layered phases containing sp2 and sp atoms. J. Chern. Phys. 1997, 87, 6687. [Google Scholar] [CrossRef]
- Ivanovskii, L. Graphynes and graphdyynes. Progr. Solid State Chem. 2013, 41, 1–32. [Google Scholar] [CrossRef]
- Parvin, N.; Jin, Q.; Wei, Y.; Yu, R.; Zheng, B.; Huang, L. et.all. Few-layer graphdiyne nanosheets applied for multiplexed real-time DNA detection. Adv. Mater. 2017, 29, 1606755. [Google Scholar] [CrossRef]
- Li, G; Li, Y; Liu, H; et al. Architecture of graphdiyne nanoscale films. Chem. Communic 2010, 46, 3256–3258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J; Gao, X; Liu, R; et al. Synthesis of graphdiyne nanowalls using acetylenic coupling reaction. JACS 2015, 137, 7596–7599. [Google Scholar] [CrossRef]
- Wang, S S; Liu, H B; Kan, X N; et al. Superlyophilicity-facilitated synthesis reaction at the microscale: ordered graphdiyne stripe arrays. Small 2017, 13, 1602265. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, R; Sakamoto, R; Hoshiko, K; et al. Crystalline graphdiyne nanosheets produced at a gas/liquid or liquid/liquid interface. JACS 2017, 139, 3145–3152. [Google Scholar] [CrossRef] [PubMed]
- Caro, M. A.; Zoubkoff, R.; Lopez-Acevedo, O.; Laurila, T. Atomic and electronic structure of tetrahedral amorphous carbon surfaces from density functional theory: Properties and simulation strategies. Carbon 2014, 77, 1168–1182. [Google Scholar] [CrossRef]
- Palomäki, T.; Wester, N.; Caro, M. A.; Sainio, S.; Protopopova, V.; Koskinen, J.; Laurila, T. Electron transport determines the electrochemical properties of tetrahedral amorphous carbon (ta-C) thin films. Electrochim. Acta 2017, 225, 1–10. [Google Scholar] [CrossRef]
- Gupta, S.; Bhaumik, A.; Sachan, R.; et al. Structural evolution of Q-carbon and nanodiamonds. JOM 2018, 70, 450–455. [Google Scholar] [CrossRef]
- Narayana, J.; Bhaumika, A.; Guptaa, S.; Haquea, A.; Sachan, R. Progress in Q-carbon and related materials with extraordinary properties. Mater. Res. Lett. 2018, 6, 353–364. [Google Scholar] [CrossRef]
- Sachan, R.; Gupta, S.; Narayan, J. Nonequilibrium structural evolution of Q-carbon and interfaces. ACS Appl. Mater. Interf. 2020, 12, 1330–1338. [Google Scholar] [CrossRef]
- Sheka, E.F.; Hołderna-Natkaniec, K.; Natkaniec, I.; Krawczyk, J. X.; Golubev, Ye.A.; Rozhkova, N.N.; Kim, V.V.; Popova, N.A.; Popova, V.A. Computationally supported neutron scattering study of natural and synthetic amorphous carbons. J. Phys. Chem. C 2019, 123, 15841–15850. [Google Scholar] [CrossRef]
- Golubev, Ye. A.; Rozhkova, N. N.; Kabachkov, E. N.; Shul'ga, Y. M.; Natkaniec-Hołderna, K.; Natkaniec, I.; Antonets, I. V.; Makeev, B. A.; Popova, N. A.; Popova, V. A.; Sheka, E. F. sp2 Amorphous carbons in view of multianalytical consideration: normal, expeсted and new. J. Non-Cryst. Solids 2019, 524, 119608. [Google Scholar] [CrossRef]
- Sheka, E. F.; Natkaniec, I.; Ipatova, E. U.; Golubev, Ye. A.; Kabachkov, E. N.; Popova, V. A. Heteroatom necklaces of sp2 amorphous carbons. XPS supported INS and DRIFT spectroscopy. FNCN 2020, 28, 1010–1029. [Google Scholar]
- Sheka, E.F.; Golubev, Y. A.; Popova, N. A. Graphene domain signature of Raman spectra of sp2 amorphous carbons. Nanomat 2020, 10. [Google Scholar] [CrossRef]
- Sheka, E.F.; Golubev, Ye.A.; Popova, N.A. Amorphous state of sp2 solid carbons. FNCN 2021, 29, 107–113. [Google Scholar] [CrossRef]
- Sheka, E.F. A neoteric view of sp2 amorphous carbon. Nanomat. 2023, 13, 1648. [Google Scholar] [CrossRef]
- Warren, B.E. X-ray diffraction in random layer lattices. Phys. Rev. B 1941, 59, 639–643. [Google Scholar] [CrossRef]
- Avdeev, M. V.; Tropin, T. V.; Aksenov, V. L.; Rosta, L.; Garamus, V. M.; Rozhkova, N. N. Pore structures in shungites as revealed by small-angle neutron scattering. Carbon 2006, 44, 954–961. [Google Scholar] [CrossRef]
- Golubev, Ye.A.; Ulyashev, V.V.; Veligzhanin, A.A. Porosity and structural parameters of Karelian shungites according to the data of small-angle synchrotron radiation scattering and microscopy. Crystallgr. Rep. 2016, 61, 66–77. [Google Scholar] [CrossRef]
- Duan, X.; Ao, Z.; Zhang, H.; Saunders, M.; Sun, H.; Shao, Z.; Wang, S. Nanodiamonds in sp2 /sp3 configuration for radical to nonradical oxidation: Core-shell layer dependence. Appl. Cat. B: Envirom 2018, 222, 176–181. [Google Scholar] [CrossRef]
- Sadovnichii, R.V.; Rozhkov, S.S.; Rozhkova, N.N. The use of shungite processing products in nanotechnology: Geological and mineralogical justification. Smart Nanocomp 2016, 7, 111–119. [Google Scholar]
- Garvie, L.A.J.; Buseck, P.R. Carbonaceous materials in the acid residue from the Orgueil carbonaceous chondrite meteorite. Meteor. Planet. Sci. 2006, 41, 633–642. [Google Scholar] [CrossRef]
- Taskaev, S.; Skokov, K.; Khovaylo, V.; Donner, W.; Faske, T.; Dudorov, A.; Gorkavyi, N.; Muratov, D. S.; Savosteenko, G.; Dyakonov, A.; Baek, W.; Kuklin, A.; Avramov, P.; Gutfleisc, O. Exotic carbon microcrystals in meteoritic dust of the Chelyabinsk superbolide: Experimental investigations and theoretical scenarios of their formation. Eur. Phys. J. Plus 2022, 137, 562. [Google Scholar] [CrossRef]
- Ivanovskii, AL. Graphynes and graphdyines. Prog. Solid State Chem. 2013, 41, 1–19. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press, 1960. [Google Scholar]
- Locke, W. Introduction to Molecular Orbital Theory. 1997. retrieved May 18, 2005.
- March, N.H. Electron Density Theory of Atoms and Molecules; Academic Press, 1992. [Google Scholar]
- Frenking, G.; Shail, S. The Chemical Bond. Fundamental Aspects of Chemical Bonding; John Wiley and Sons, 2014. [Google Scholar]
- Frenking, G..; Shail, S. The Chemical Bond. Chemical Bonding Across the Periodic Table; John Wiley and Sons, 2014. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules - A Quantum Theory; Oxford University Press: London, 1990. [Google Scholar]
- Gazquez, J.L.; del Campo, J.M.; Trickey, S.B.; Alvarez-Mendez, R.J.; Vela, A. Analysis of generalized gradient approximation for exchange energy. In Concepts and Methods in Modern Theoretical Chemistry. Vol. 1. Electronic Structure and Reactivity; Ghosh, S.K., Chattaraj, P.K., Eds.; CRC Press, Taylor and Francis Group: Boca Raton, 2013; pp. 295–307. [Google Scholar]
- Scherer, W.; Fisher, A.; Eickerling, G. The experimental density perspectives of chemical bonding. In The Chemical Bond: Fundamental Aspects of Chemical Bonding; Frenking, G., Shaik, S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA, 2014; pp. 309–343. [Google Scholar]
- Sanderson, R.T. Chemical Bonds and Bond Energy; Academic Press: New York, London, 1976. [Google Scholar]
- Darwent, B. de B. National Standard Reference Data Series; National Bureau of Standards 31: Washington, DC, 1970. [Google Scholar]
- Schreiner, P.R.; Chernish, L.V.; Gunchenko, P.A.; Tikhonchuk, E.Yu.; Hausmann, H.; Serafin, M.; Schlecht, S.; Dahl, J.E.P.; Carlson, R.M.K.; Fokin, A.A. Overcoming lability of extremely long alkane carbon–carbon bonds through dispersion forces. Nature 2011, 477, 308–311. [Google Scholar] [CrossRef]
- Mascal, M.; Hafezi, N.; Meher, N.K.; Fettinger, J.C. Oxatriquinane and oxatriquinacene: Extraordinary oxonium ions. JACS 2008, 130, 13532–13533. [Google Scholar] [CrossRef]
- Lane, J.R.; Contreras-García, J.; Piquemal, J.-P.; Miller, B.J. Are bond critical points really critical for hydrogen bonding? J. Chem. Theory Comput. 2013, 9, 3263–3266. [Google Scholar] [CrossRef]
- Boguslawski, K.; Tecmer, P.; Legeza, O.; Reiher, M. Entanglement measures for single- and multireference correlation effects. J. Phys. Chem. Lett. 2012, 3, 3129–3135. [Google Scholar] [CrossRef] [PubMed]
- Sheka, E F; Popova, N A; Popova, V A. Physics and chemistry of graphene. Emergentness, magnetism, mechanophysics and mechanochemistry. Phys. Usp. 2018, 61, 645–691. [Google Scholar] [CrossRef]
- Mayer, I. Covalent bonding: the role of exchange effects. J. Phys. Chem. A 2014, 118, 2543–2546. [Google Scholar] [CrossRef] [PubMed]
- Sheka, E.F. Stretching and breaking of chemical bonds, correlation of electrons, and radical properties of covalent species. Adv. Quant. Chem. 2015, 70, 111–161. [Google Scholar]
- Zayets, V. A. CLUSTER-Z1: Quantum-Chemical Software for Calculations in the s,p-Basis; Inst. Surf. Chem. Nat. Ac. Sci. of Ukraine; Kiev, 1990. [Google Scholar]
- Berzigiyarov, P.K.; Zayets, V.A.; Ginzburg, I.Ya.; et al. NANOPACK: Parallel codes for semiempirical quantum chemical calculations of large systems in the sp- and spd-basis. Int. J. Quantum Chem. 2002, 88, 449–462. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healey, E.F.; Stewart, J.J.P. AM1: A new general- purpose quantum mechanical molecular model. JACS 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Sheka, E.F. Chemical susceptibility of fullerenes in view of Hartree–Fock approach. Int. J. Quant. Chem. 2007, 107, 2803–2816. [Google Scholar] [CrossRef]
- Sheka, E.F. Fullerenes. Nanochemistry, Nanomagnetism, Nanomedicine, Nanophotonics, CRC Press, Taylor and Francis Group: Boca Raton, 2011.
- Sheka, E.F. Spin Teory of Fullerene; RUDN Press: Moscow, 2025. [Google Scholar]
- Sheka, E.F. Spin Chemical Physics of Graphene; Pan Stanford: Singapore, 2018. [Google Scholar]
- Sheka, E. F. Virtual vibrational spectrometry of stable radicals—necklaced graphene molecules. Nanomat. 2022, 12, 597. [Google Scholar] [CrossRef]
- Sheka, E. F. Spin effects of sp2 nanocarbons in light of unrestricted Hartree-Fock approach and spin-orbit coupling theory. In Quantum Systems in Physics, Chemistry, and Biology: Advances in Concepts and Applications; Tadjer, A., Pavlov, R., Maruani, J., Brändas, E.J., Delgado-Barrio, G., Eds.; Progress in Theoretical Chemistry and Physics; Springer: Switzerland, 2017; Volume 30, pp. 39–63. [Google Scholar]
- Nagase, S. Structure and reactions of compounds containing hevier main group elements. In The Transition State. A Theoretical Approach; Fueno, T., Ed.; Kodansha Ltd: Tokyo, Japan; Gordon and Breach Sci Pubs.: Amsterdam, The Netherlands, 1999; pp. 140–157. [Google Scholar]
- Cordero, B.; Gґomez, V.; Platero-Prats, A.E.; Revґes, M.; Echeverrґıa, J.; Cremades, E.; Barragґan, F.; Alvarez, S. Covalent radii revisited. Dalton Trans 2008, 2832–2838.
- Sheka, E.F. Why sp2-like nanosilicons should not form: Insight from quantum chemistry. Int. J. Quant. Chem. 2013, 113, 612–618. [Google Scholar] [CrossRef]
- Sheka, E.F. Silicene is a material phantom. Nanosystems: Phys. Chem. Math. 2016, 7, 983–1001. [Google Scholar] [CrossRef]
- Gunbas, G.; Hafezi, N.; Sheppard, W.L.; Olmstead, M.M.; Stoyanova, L.V.; Tham, F.S.; Meyer, M.P.; Mascal, M. Extreme oxatriquinanes and a record C–O bond length. Nat. Chem. 2012, 4, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Iwamoto, T.; Kira, M. Fused tricyclic disilenes with highly strained Si-Si double bonds: Addition of a Si-Si single bond to a Si-Si double bond. Ang. Chem. Int. Ed. 2006, 45, 6371–6373. [Google Scholar] [CrossRef]
- Gusel’Nikov, L. E.; Flowers, M. C. The thermal decomposition of 1,1-dimethyl-1-silacyclobutane and some reactions of an unstable intermediate containing a silicon-carbon double bond. Chem. Commun. (London). 1967, 864–865. [Google Scholar]
- Brook, A.G.; Abdesaken, F.; Gutekunst, B.; Gutekunst, G.; Kallury, R.K. A solid silaethene: isolation and characterization. J. Chem. Soc. Chem. Comm. 1995, 24, 2473–2670. [Google Scholar] [CrossRef]
- Karni, M.; Apeloig, Y. The quest for a stable silyne, RSi≡CR_. The effect of bulky substituents. Sil. Chem. 2002, 1, 61–66. [Google Scholar]
- Sekiguchi, A.; Kinjo, R.; Ichinohe, M. A stable compound containing a silicon-silicon triple bond. Science 2004, 305, 1755–1757. [Google Scholar] [CrossRef]
- Sheka, E.F. Virtual Free-Radical Polymerization of Vinyl Monomers in View of Digital Twins. Polymers 2023, 15, 2999. [Google Scholar] [CrossRef]
- Sheka, E.F. Digital twins’ kinetics of virtual free-radical copolymerization of vinyl monomers with stable radicals. 2. Styrene. arXiv:2311.02752 [cond-mat.mtrl-sci], 2023.
- Sheka, E.F. Digitalization of free-radical polymerization. Adv. Quant. Chem. 2025, 92, 286–317. [Google Scholar]
- Blanquart, G. Effects of spin contamination on estimating bond dissociation energies of polycyclic aromatic hydrocarbons. J. Quant. Chem. 2015, 115, 796–801. [Google Scholar] [CrossRef]
- Kaplan, I. Problems in DFT with the total spin and degenerate states. Int. J. Quant. Chem. 2007, 107, 2595–2603. [Google Scholar] [CrossRef]
- Jacob, C.R.; Reiher, M. Spin in density-functional theory. Int. J.Quant. Chem. 2012, 112, 3661–3684. [Google Scholar] [CrossRef]
- Kaplan, I. Symmetry properties of the electron density and following from it limits on the KS-DFT applications. Mol. Phys. 2018, 116, 658–665. [Google Scholar] [CrossRef]
- Hachmann, J; Dorando, JJ; Avilés, M; Chan, GK-L. The radical character of the acenes: a density matrix renormalization group study. J. Chem. Phys. 2007, 127, 134309. [Google Scholar] [CrossRef]
- Spethmann, J.; Khanh, N. D.; Yoshimochi, H.; Takagi, R.; Hayami, S.; Motome, Y.; Wiesendanger, R.; Seki, S.; von Bergmann, K. SP-STM study of the multi-Q phases in GdRu2Si2. Phys. Rev. Mat. 2024, 8, 064404. [Google Scholar]
- Gross, L.; et al. Bond-order discrimination by atomic force microscopy. Science 2012, 337, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Mistry, A.; et al. The synthesis and STM/AFM imaging of ‘olympicene’ benzo[cd]pyrenes. Chem. Eur. J. 2014, 21, 2011–2018. [Google Scholar] [CrossRef]
- Pavliček, N.; et al. Synthesis and characterization of triangulene. Nature Nanotech 2017, 12, 308–311. [Google Scholar] [CrossRef]
- Van der Lit, J.; et al. Suppression of electron-vibron coupling in graphene nanoribbons contacted via a single atom. Nat. Commn 2013, 4, 2023. [Google Scholar] [CrossRef]
- Mönig, H.; Amirjalayer, S.; Timmer, A.; et al. Quantitative assessment of intermolecular interactions by atomic force microscopy imaging using copper oxide tips. Nature Nanotech. 2018, 13, 371–375. [Google Scholar] [CrossRef]
- Hedberg, K.; Hedberg, L.; Bethune, D. S. lengths in free molecules of buckminsterfullerene, C60, from gas-phase electron diffraction. Science 1991, 234, 410–412. [Google Scholar] [CrossRef]
- Liu, S.; Lu, Y.-J.; Kappes, M. M.; Ibers, J. A. The structure of the C60 molecule: X-ray crystal structure determination of a twin at 110 K. Science 1991, 254, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, F.; Damay, P.; Foukani, M.; Chieux, P.; Bellisent-Funnel, M.C.; Rassat, A.; Fabre, C. Precise determination of the molecular geometry in fullerene C60 powder: A study of the structure factor by neutron scattering in a large momentum-transfer range. Phys. Rev. B 1993, 48, 2748–2758. [Google Scholar] [CrossRef]
- Yanonni, C.S.; Bernier, P.P.; Bethune, D.S.; Meijer, G.; Salem, J.K. NMR determination of the bond lengths in C60. J. Am. Chem. Soc. 1991, 113, 3190–3192. [Google Scholar] [CrossRef]
- Slovokhotov, Yu.L.; Moskaleva, I.V.; Shil’nikov, V.I. Molecular and crystal structures of C-60 derivatives: CSD statistics and theoretical modeling. Mol. Mater. 1996, 8, 117–124. [Google Scholar]
- Li, Y. Z.; Chander, M.; Patrin, J. C.; Weaver, J. H.; Chibante, L. P. F.; Smalley, R. E. Adsorption of individual С60 molecules on Si(111). Phys. Rew. B 1992, 1, 13837–13840. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, P. J. Fullerene adsorption on semiconductor surfaces. Surf. Sci. Rep. 2010, 65, 175–227. [Google Scholar] [CrossRef]
- Sheka, E. F. ‘Chemical’ portrait of fullerenes. J. Struct. Chem. 2006, 47, 593–599. [Google Scholar] [CrossRef]
- Sheka, E.F.; Chernozatonskii, L.A. Bond length effect on odd electrons behavior in single-walled carbon nanotubes. J. Phys. Chem. C. 2007, 111, 10771–10779. [Google Scholar] [CrossRef]
- Sheka, E.F.; Chernozatonskii, L.A. Broken symmetry approach and chemical susceptibility of carbon nanotubes. Int. Journ. Quant. Chem. 2010, 110, 1466–1480. [Google Scholar] [CrossRef]
- Sheka, E.F.; Chernozatonskii, L. A. Broken spin symmetry approach to chemical reactivity and magnetism of graphenium species. J. Exp. Theor. Phys. 2010, 110, 121–132. [Google Scholar] [CrossRef]
- Gao, X.; Zhou, Z.; Zhao, Y.; Nagase, S.; Zhang, S. B.; Chen, Z. Comparative study of carbon and BN nanographenes: Ground electronic states and energy gap engineering. J. Phys. Chem. A 2008, 112, 12677–12682/. [Google Scholar] [CrossRef]
- Sheka, E.F. Molecular theory of graphene chemical modification. In Graphene Science Handbook: Mechanical and Chemical Properties, Vol.4; Aliofkhazraei, M., Ali, N., Miln, W.I., Ozkan, C. S., Mitura, S., Gervasoni, J., Eds.; CRC Press, Taylor and Francis Group: Boca Raton; pp. 312–338.
- Sheka, E F; Popova, N A; Popova, V A. Physics and chemistry of graphene. Emergentness, magnetism, mechanophysics and mechanochemistry. Phys. Usp. 2018, 61, 645–691. [Google Scholar] [CrossRef]
- Li, Y.; Xu, L.; Liu, H.; Li, Y. Graphdiyne and graphyne: From theoretical predictions to practical construction. Chem. Soc. Rev. 2014, 43, 2572–2586. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, G.; Yao, H.; Liu, L.; Jiang, L.; Zheng, Y. A simple tight-binding model for typical graphyne structures. New J. Phys. 2014, 14, 113007. [Google Scholar] [CrossRef]
- Peng, Q.; Dearden, A.K.; Crean, J.; Han, L.; Liu, S.; Wen, X.; De, S. New materials graphyne, graphdiyne, graphone, and graphane: Review of properties, synthesis, and application in nanotechnology. Nanotech. Sci. Appl. 2014, 7, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Chalifoux, W.A.; Tykwinski, R.R. Synthesis of polyynes to model the sp-carbon allotrope carbine. Nature Chem. 2010, 2, 967–971. [Google Scholar] [CrossRef]
- Shi, L.; Rohringer, P.; Suenaga, K. Confined linear carbon chains as a route to bulk carbine. Nature Mat. 2016, 15, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Wang, D.; Shua, Z. Electronic properties and charge carrier mobilities of graphynes and graphdiynes from first principles. WIREs Comput. Mol. Sci. 2015, 5, 215–227. [Google Scholar] [CrossRef]
- Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels-Alder reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature, 2008, 455|18 September 2008, 304-308. [CrossRef] [PubMed]
- Xiang, S-H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nature Comm. 2020, 11, 3786. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Ahn, B.K.; Lee, J.Y.; Kim, H.S.; Ha, D.-C. Organocatalysis for the asymmetric Michael addition of cycloketones and α, β-unsaturated nitroalkenes. Catalysts 2021, 11, 1004. [Google Scholar] [CrossRef]
- Lee, J.; Kim, K.H.; Kwon, E.E. Biochar as a catalyst. Renew. Sustain. Energy Rev. 2017, 77, 70–79. [Google Scholar] [CrossRef]
- Donet, J.-B.; Bansal, R.C.; Wang, M.-J. Carbon Black. Science and Technology; Marcel Dekker: New York, NY, USA; Basel, Switzerland, 1993. [Google Scholar]
- Francisco, R.R. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Radovich, L.R. Carbon materials as adsorbents in aqueous solutions. In Chemistry and Physics of Carbon; Marcel Dekker: New York, NY, USA, 2001; Volume 27, pp. 227–406. [Google Scholar]
- Toyoda, M.; Tsumura, T.; Tryba, B.; Mozia, S.; Janus, M.; Morawski, A.W.; Inagaki, M. Carbon materials in photocatalysis. In Chemistry and Physics of Carbon; Radovich, L.R., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2013; pp. 171–267. [Google Scholar]
- Machado, B.; Serp, P. Graphene-based materials for catalysis. Catal. Sci. Technol. 2012, 2, 54–75. [Google Scholar] [CrossRef]
- Serp, P.; Machado, B. (Eds.) Nanostructured Carbon Materials for Catalysis; Royal Society of Chemistry: Croydon, UK, 2015. [Google Scholar]
- Bandosz, T.J. Surface chemistry of carbon materials. Spectroscopic methods. In Carbon Materials for Catalysis; Serp, P., Figueiredo, J.L., Eds.; Wiley: Hoboken, NJ, USA, 2009; pp. 45–92. [Google Scholar]
- Bandosz, T.J. Nanoporous carbons: Looking beyond their perception as adsorbents, catalyst supports and supercapacitors. Chem. Rec. 2016, 16, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Matos, I.; Bernardo, M.; Fonseca, I. Porous Carbon: A versatile material for catalysis. Catal. Today 2017, 285, 194–203. [Google Scholar] [CrossRef]
- Yam, K.M.; Guo, N.; Jiang, Z.; Li, S.; Zhang, C. Graphene-based heterogeneous catalysis: Role of graphene. Catalysts 2020, 10, 53. [Google Scholar] [CrossRef]
- Salvio, R.; Molitermo, M.; Bella, M. Alkynes in organocatalysis. Asian J. Org. Chem. 2014, 3, 340–351. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–406. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, G.; Zhang, F. Multiple roles of graphene in heterogeneous catalysis. Chem. Soc. Rev. 2015, 44, 3023–3035. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Yam, K.M.; Zhang, C. Substrate engineering of graphene reactivity: Towards high-performance graphene-based catalysts. npj 2D Mater. Appl. 2018, 2, 1. [Google Scholar] [CrossRef]
- Deng, D.; Novoselov, K.S.; Fu, Q.; Zheng, N.; Tian, Z.; Bao, X. Catalysis with two-dimensional materials and their heterostructures. Nat. Nanotech 2016, 11, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Fu, Q.; Zhang, Y.-Y.; Weng, X.; Li, H.; Chen, M.; Jin, L.; Dong, A.; Mu, R.; Jiang, P.; et al. Graphene cover-promoted metal-catalyzed reactions. Proc. Natl. Acad. Sci. USA 2014, 111, 17023–17028. [Google Scholar] [CrossRef] [PubMed]
- Sutter, P.; Sadowski, J.T.; Sutter, E.A. Chemistry under cover: Tuning metal–graphene interaction by Reactive Intercalation. J. Am. Chem. Soc. 2010, 132, 8175–8179. [Google Scholar] [CrossRef]
- Sheka, E.F. Virtual vibrational spectrometry of stable radicals—necklaced graphene molecules. Nanomat. 2022, 12, 597. [Google Scholar] [CrossRef] [PubMed]
- Sheka, E.F. Digital Twins solve the mystery of Raman spectra of parental and reduced graphene oxides. Nanomat. 2022, 12, 4209. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.C.; Donnet, J.-B. Surface groups on carbon blacks. In Carbon Black. Science and Technology; Donet, J.-B., Bansal, R.C., Wang, M.-J., Eds.; Marcel Dekker: New York, NY, USA; Basel, Switzerland, 1993; pp. 152–181. [Google Scholar]
- Navalon, S.; Dhakshinamoorthy, A.; Alvaro, M.; Antonietti, M.; García, H. Active sites on graphene-based materials as metal-free catalysts. Chem. Soc. Rev. 2017, 46, 4501–4529. [Google Scholar] [CrossRef] [PubMed]
- Sheka, E.F. Graphene oxyhydride catalysts in view of spin radical chemistry. Materials 2020, 13, 565. [Google Scholar] [CrossRef] [PubMed]
- Radovich, L.R. Physicochemical properties of carbon materials: A brief overview. In Carbon Materials for Catalysis; Serp, P., Figueiredo, J. L., Eds.; Wiley, Hoboken, 2009; pp. 1–44. [Google Scholar]
- Rideal, K.; Wright, W.M. Low temperature oxidation at charcoal surfaces. Part I. The behaviour of charcoal in the absence of promoters. J. Chem. Soc. Trans. 1925, 127, 1347–1357. [Google Scholar] [CrossRef]
- Lonkar, S.P.; Abdala, A.A. Applications of graphene in catalysis. J. Thermodyn. Catal 2014, 5, 1–6. [Google Scholar]
- Chung, C; Kim, YK; Shin, D; Ryoo, SR; Hong, BH; et al. Biomedical applications of graphene and graphene oxide. Acc Chem Res 2013, 46, 2211–2224. [Google Scholar] [CrossRef]
- Rozhkov, S.; Goryunov, A.; Rozhkova, N. Molecular serum albumin unmask nanobio properties of molecular graphenes in shungite carbon nanoparticles. Int. J. Mol. Sci. 2024, 25, 2465. [Google Scholar] [CrossRef] [PubMed]
- Sadovnichii, R.V.; Rozhkova, N.N. The minerals associations of carbon-rich shungite rocks of the Maksovo deposit (the onega structure). Trans. KarRC RAS 2014, 1, 148–158. [Google Scholar]
- Sadovnichii, R.V.; Mikhaylina, A.A.; Rozhkova, N.N.; Inina, I.S. The Morphological and structural features of quartz of shungite rocks from Maksovo deposit. Trans. KarRC RAS 2016, 2, 73–88. [Google Scholar]
- Sadovnichii, R.V.; Rozhkov, S.S.; Rozhkova, N.N. The use of shungite processing products in nanotechnology: geological and mineralogical justification. Smart Nanocomp 2016, 7, 111–120. [Google Scholar]
- Chen, Y.; Guo, F.; Jachak, A.; Kim, S. P.; Datta, D.; Liu, J.; Kulaots, I.; Vaslet, C.; Jang, H. D.; Huang, J.; et al. Aerosol Synthesis of Cargo-Filled Graphene Nanosacks. Nano Lett. 2012, 12, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Campra, P. Graphene oxide detection in aqueous suspension. Observational study in optical and electron microscopy. June 28, 2021. Available online: https://www.docdroid.net/rNgtxyh/microscopia-devial-.
- Yong, R.O. Scanning and transmission microscopy reveals graphene oxide in CoV-19 vaccines. Acta Sci. Med. Sci. 2022, 6, 98–111. [Google Scholar]
- Sheka, E.F. The triumph of the spin chemistry of fullerene C60 in the light of its free radical copolymerization with vinyl monomers. Int. J. Mol. Sci. 2024, 25, 1317. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Lawandy, N.M.; Suuberg, E.M. Fullerene-catalyzed oxidation of organic solvents. En. Fuels 1994, 8, 810–811. [Google Scholar] [CrossRef]
- Bagdasar’yan, K.S. Teoriya Radikal’noi Polimerizatsii (Free_Radical Polymerization Theory); Nauka: Moscow, Russia, 1966. [Google Scholar]
- Pross, A. Theoretical and Physical Principles of Organic Reactivity; Wiley: New York, NY, USA, 1995. [Google Scholar]
- Sheka, E.F. Intermolecular interaction in C60-based donor acceptor complexes. Int. J. Quant. Chem. 2004, 100, 388–406. [Google Scholar] [CrossRef]
- Sheka, E.F. Donor-acceptor origin of fullerene C60 dimerization. Int. J. Quant. Chem. 2007, 107, 2361–2371. [Google Scholar] [CrossRef]
- Neeman, E.M.; Ramón, J.; Moreno, A.; Huet, T.R. The gas phase structure of -pinene, a main biogenic volatile organic compound. J. Chem. Phys. 2017, 147, 214305. [Google Scholar] [CrossRef] [PubMed]
- Sheka, E.F. Step-wise computational synthesis of fullerene C60 derivatives. Fluorinated fullerenes C60F2k. J. Exp. Theor. Phys. 2010, 111, 397–414. [Google Scholar] [CrossRef]
- Sheka, E.F. Computational synthesis of hydrogenated fullerenes from C60 to C60H60. J. Mol. Mod. 2011, 17, 1973–1984. [Google Scholar] [CrossRef]
- Sheka, E.F.; Popova, N.A. Odd-electron molecular theory of the graphene hydrogenation. J. Mol. Model. 2012, 18, 3751–3768. [Google Scholar] [CrossRef] [PubMed]
- D. R. Dreyer, S. Park, C. W. Bielawski, R. S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [CrossRef] [PubMed]
- Chen, D.; Feng, H.; Li, J. Graphene oxide: Preparation, functionalization, and electrochemical applications. Chem. Rev. 2012, 112, 6027−6053. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Bongiorno, A. Origin of the Chemical and kinetic stability of graphene oxide. Sci. Rep. 2013, 3, 2484. [Google Scholar] [CrossRef] [PubMed]
- Sheka, E.F.; Golubev, Y.A.; Popova, N.A. Graphene domain signature of Raman spectra of sp2 amorphous carbons. Nanomat. 2020, 10. [Google Scholar] [CrossRef]
- Sheka, E.F. Metaverse and virtual worlds of science. TCSIT 2024, 9, 103–105. [Google Scholar]
Figure 8.
sp2 Fullerenes in light of the spin-radical concept. (a) Space structure in terms of covalent bonds. (b) and (c) Effectively unpaired electrons of C60 and C70, respectively. UHF AM1 calculations..
Figure 8.
sp2 Fullerenes in light of the spin-radical concept. (a) Space structure in terms of covalent bonds. (b) and (c) Effectively unpaired electrons of C60 and C70, respectively. UHF AM1 calculations..
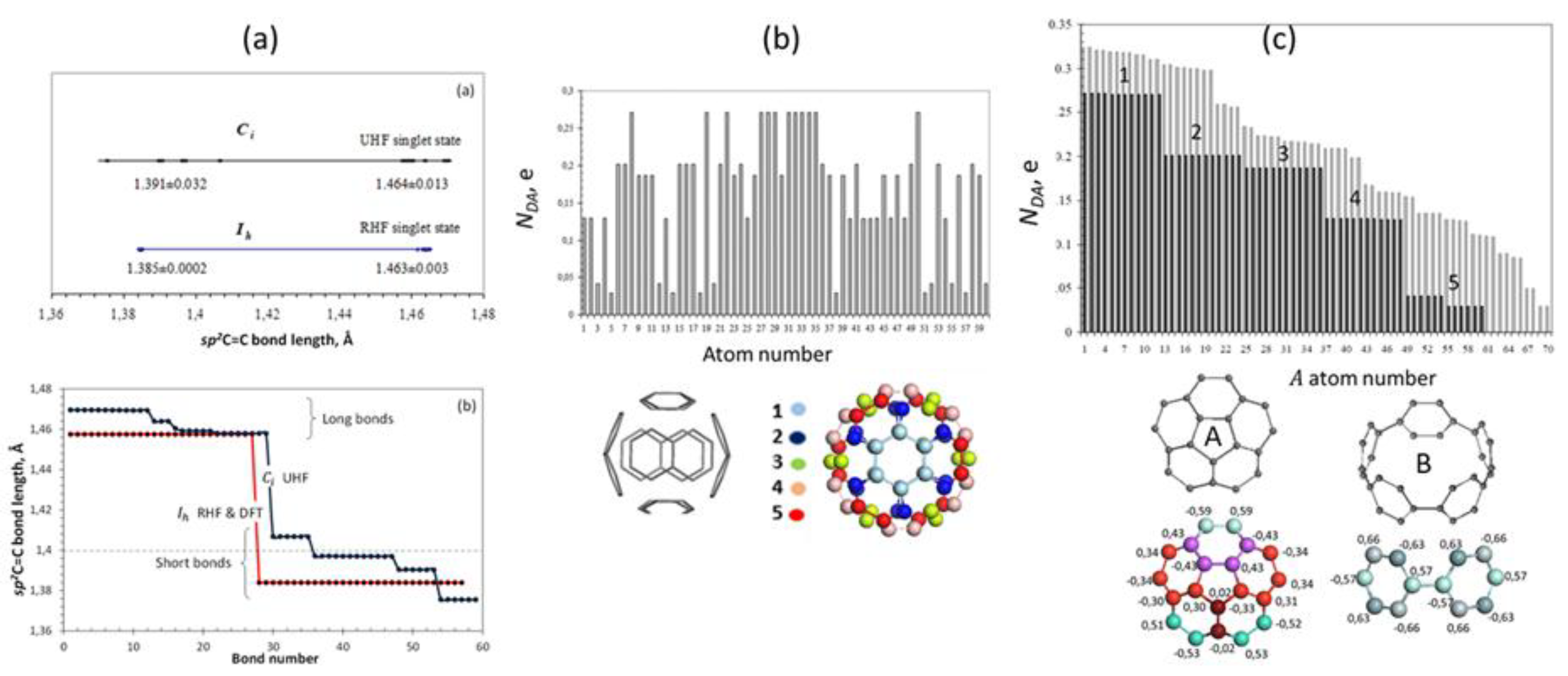
Figure 9.
Digital twins of carbon-silicon fullerenes. UHF AM1 calculations.
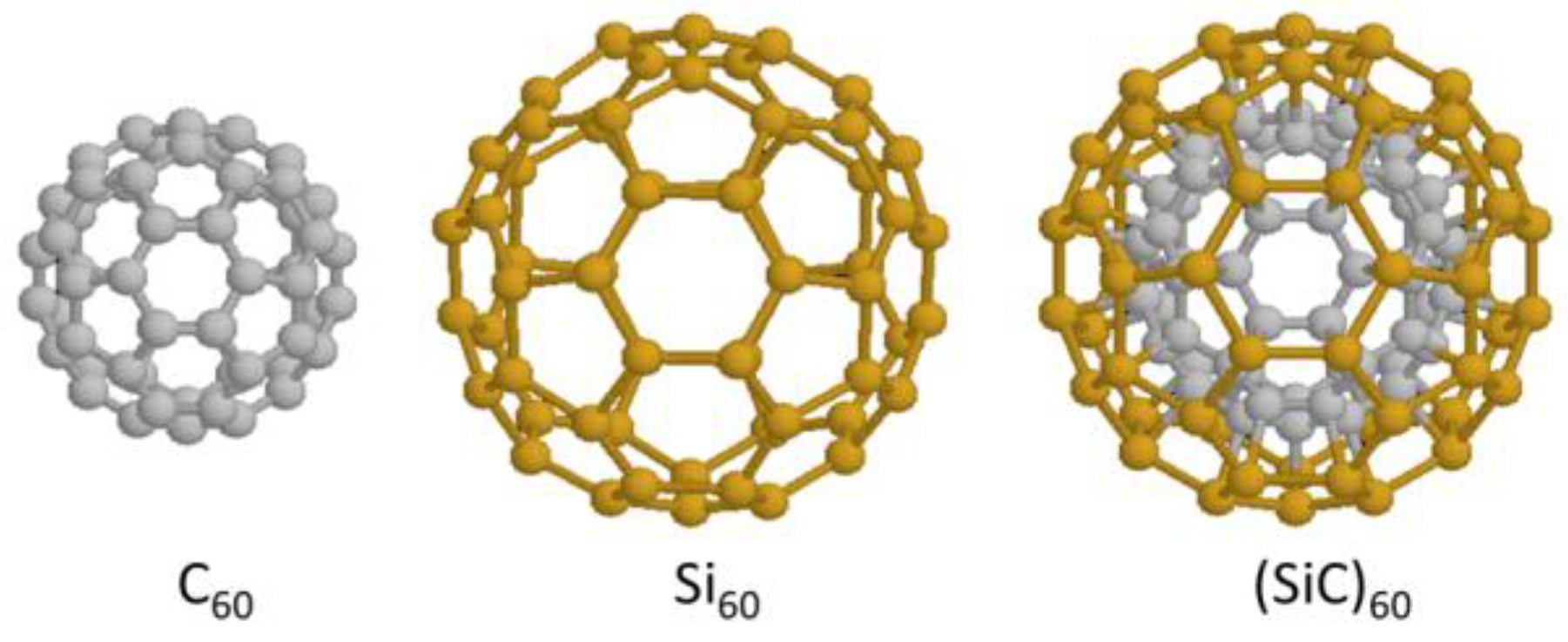
Figure 10.
Digital twins of fragments of the (4,4) SWCNT and their ACS graphs. (a), (b), and (c) are related to NT1, NT2, and NT3 species, respectively. UHF AM1 calculations.
Figure 10.
Digital twins of fragments of the (4,4) SWCNT and their ACS graphs. (a), (b), and (c) are related to NT1, NT2, and NT3 species, respectively. UHF AM1 calculations.
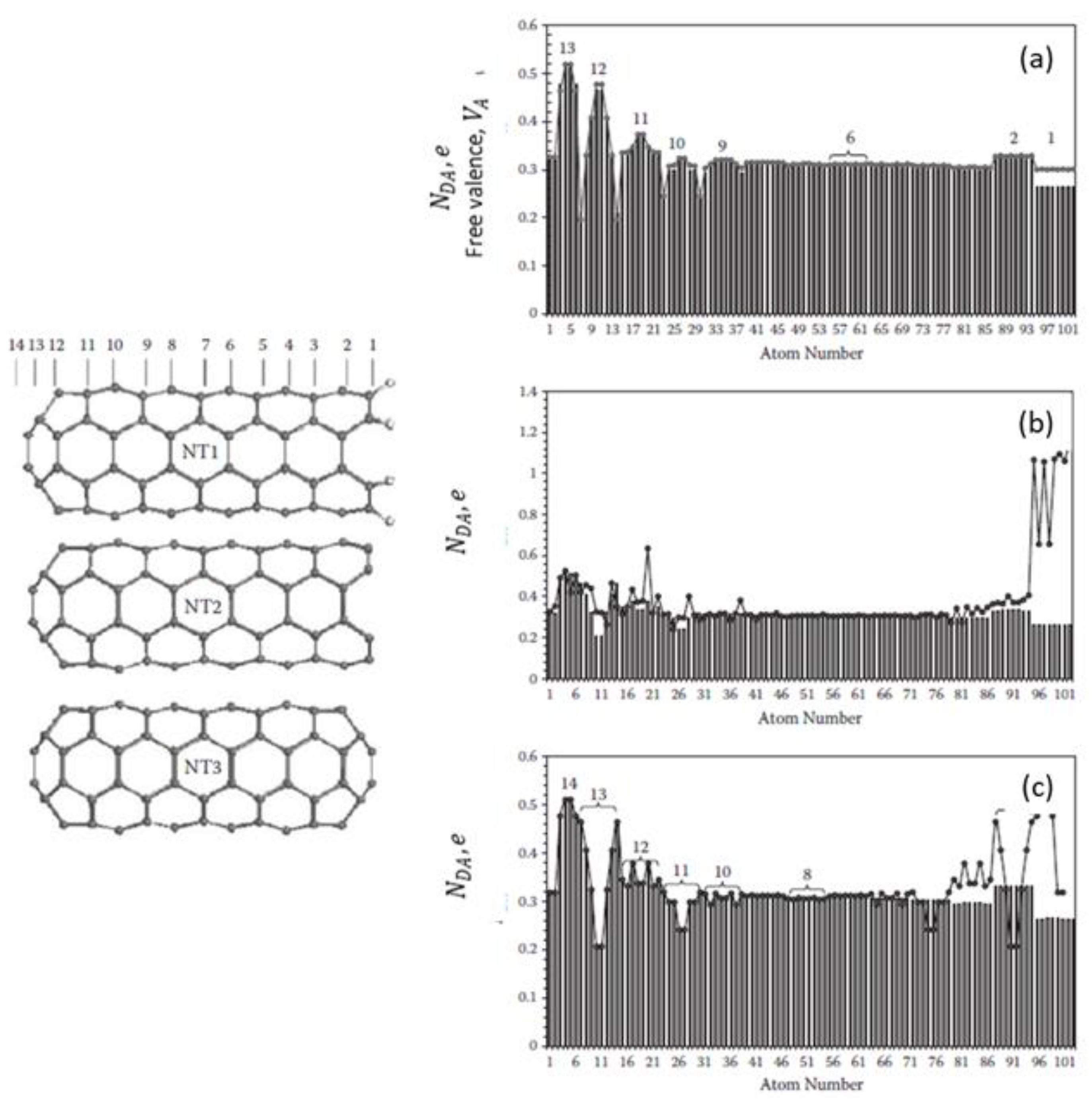
Figure 11.
Digital twins of bare and hydrogen-terminated (5,5) NGrs and their ACS graphs. UHF AM1 calculations.
Figure 11.
Digital twins of bare and hydrogen-terminated (5,5) NGrs and their ACS graphs. UHF AM1 calculations.
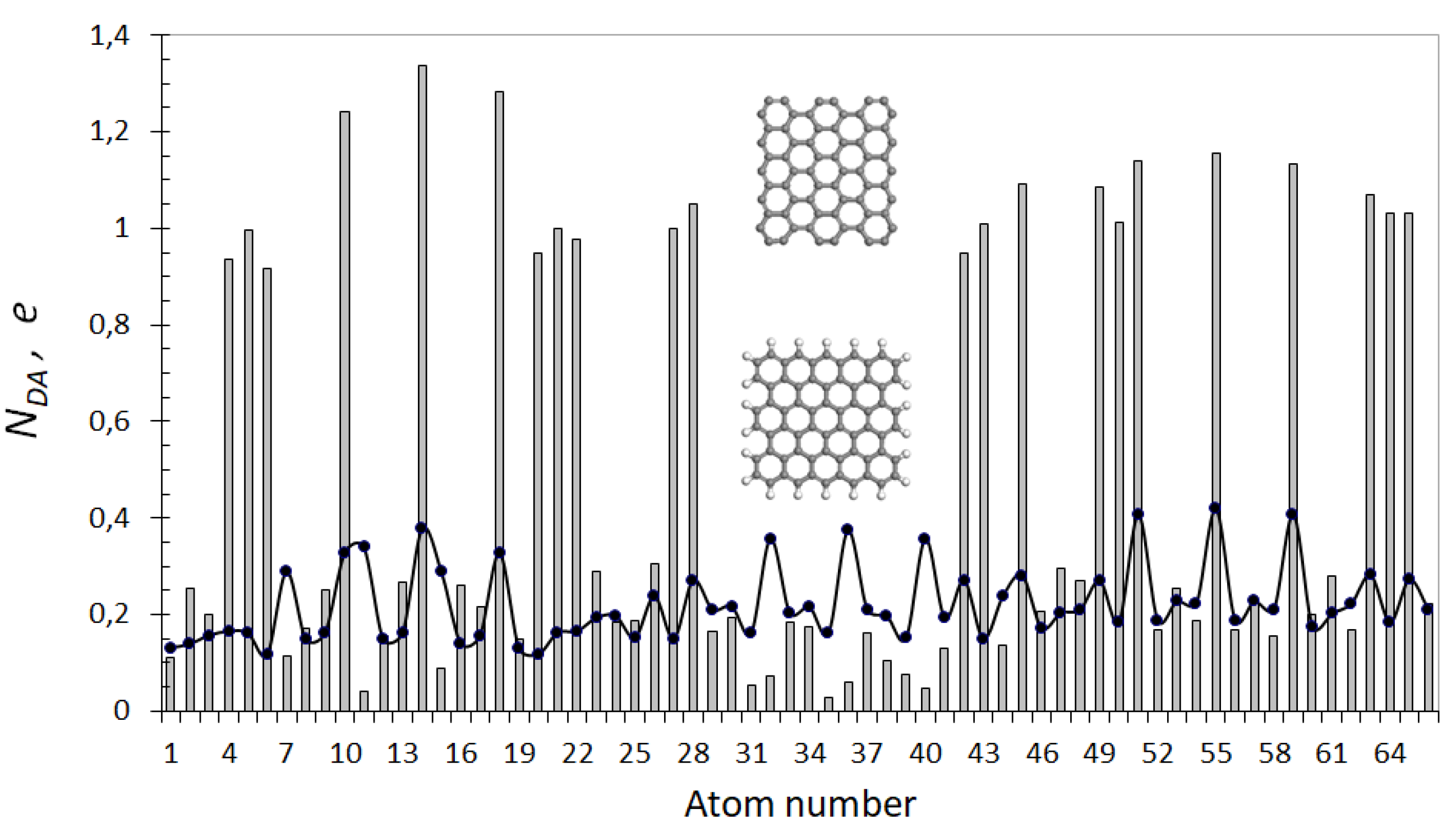
Figure 12.
Equilibrium structures (left) and ACS image maps (right) of graphpoyynes digital twins. (a) n-diphenylacetylens; n=1, 2, 3, 4. The maximum value of the map intensity scale varies from 0.10 e for 1-DPHA and 2-DPHA to 0.16 e for 3-DPHA and 0.30 e for 4-DPHA. (b) One-triangle 2-DPHA-based compositions with two-branched hexagons C6. (c) ‘Irish lace’ GDY pattern. The maximum value of the map intensity scale is 0.23 e in (b) and 0.30 e in (c). Figures present summarized values related to hexagons C6 and ligaments. Gray and red balls mark carbon and hydrogen atoms, respectively. UHF AM1 calculations.
Figure 12.
Equilibrium structures (left) and ACS image maps (right) of graphpoyynes digital twins. (a) n-diphenylacetylens; n=1, 2, 3, 4. The maximum value of the map intensity scale varies from 0.10 e for 1-DPHA and 2-DPHA to 0.16 e for 3-DPHA and 0.30 e for 4-DPHA. (b) One-triangle 2-DPHA-based compositions with two-branched hexagons C6. (c) ‘Irish lace’ GDY pattern. The maximum value of the map intensity scale is 0.23 e in (b) and 0.30 e in (c). Figures present summarized values related to hexagons C6 and ligaments. Gray and red balls mark carbon and hydrogen atoms, respectively. UHF AM1 calculations.
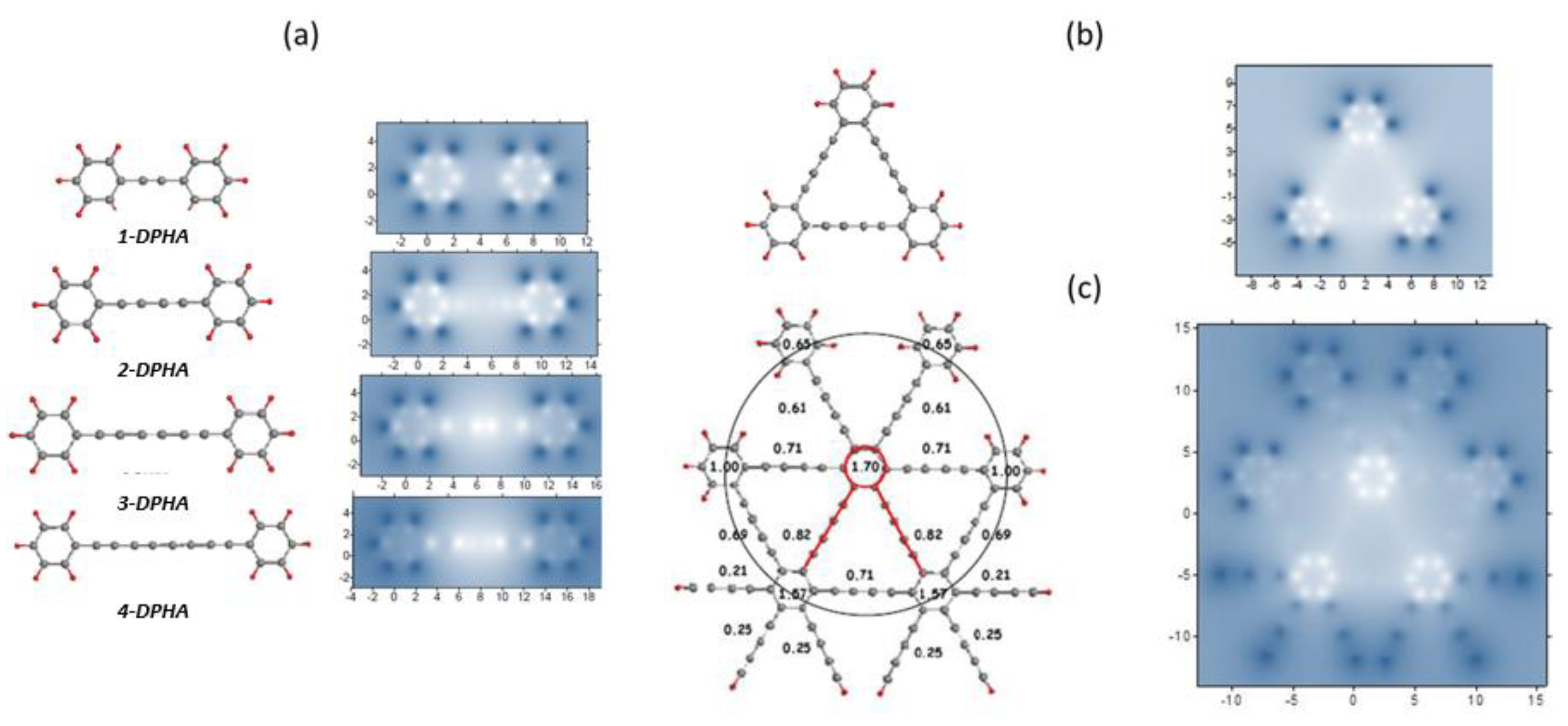
Figure 13.
Digital twins of typical graphene catalysts.
Figure 14.
Spin character of digital twins of graphene catalysts. (a) Parent graphene domain C66. Graphene oxyhydrides C66O4H6, C66O5H12, and C66O4H3 from right to left on (b) and (c) and from top to bottom on (d), respectively.
Figure 14.
Spin character of digital twins of graphene catalysts. (a) Parent graphene domain C66. Graphene oxyhydrides C66O4H6, C66O5H12, and C66O4H3 from right to left on (b) and (c) and from top to bottom on (d), respectively.
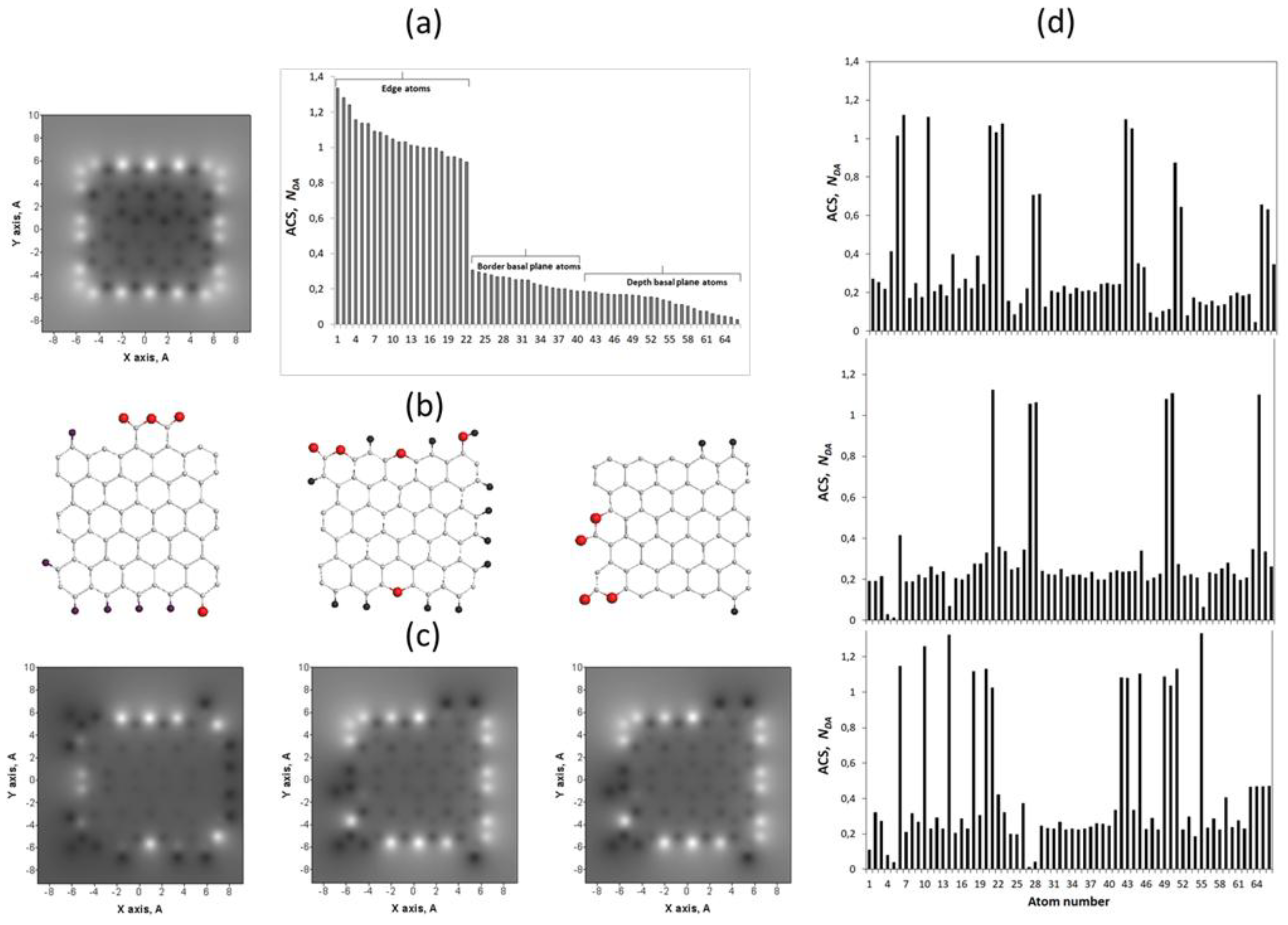
Figure 15.
Digital twins of the virtual polymerization of styrene catalyzed with fullerene C60. (a) Reaction participants. (b) oligomers for n from 1 till 7. (c) Two-branch element from a star-branch polymer product. UHF AM1 calculations.
Figure 15.
Digital twins of the virtual polymerization of styrene catalyzed with fullerene C60. (a) Reaction participants. (b) oligomers for n from 1 till 7. (c) Two-branch element from a star-branch polymer product. UHF AM1 calculations.
Figure 16.
Raman spectra of graphene oxide and reduced graphene jxide.
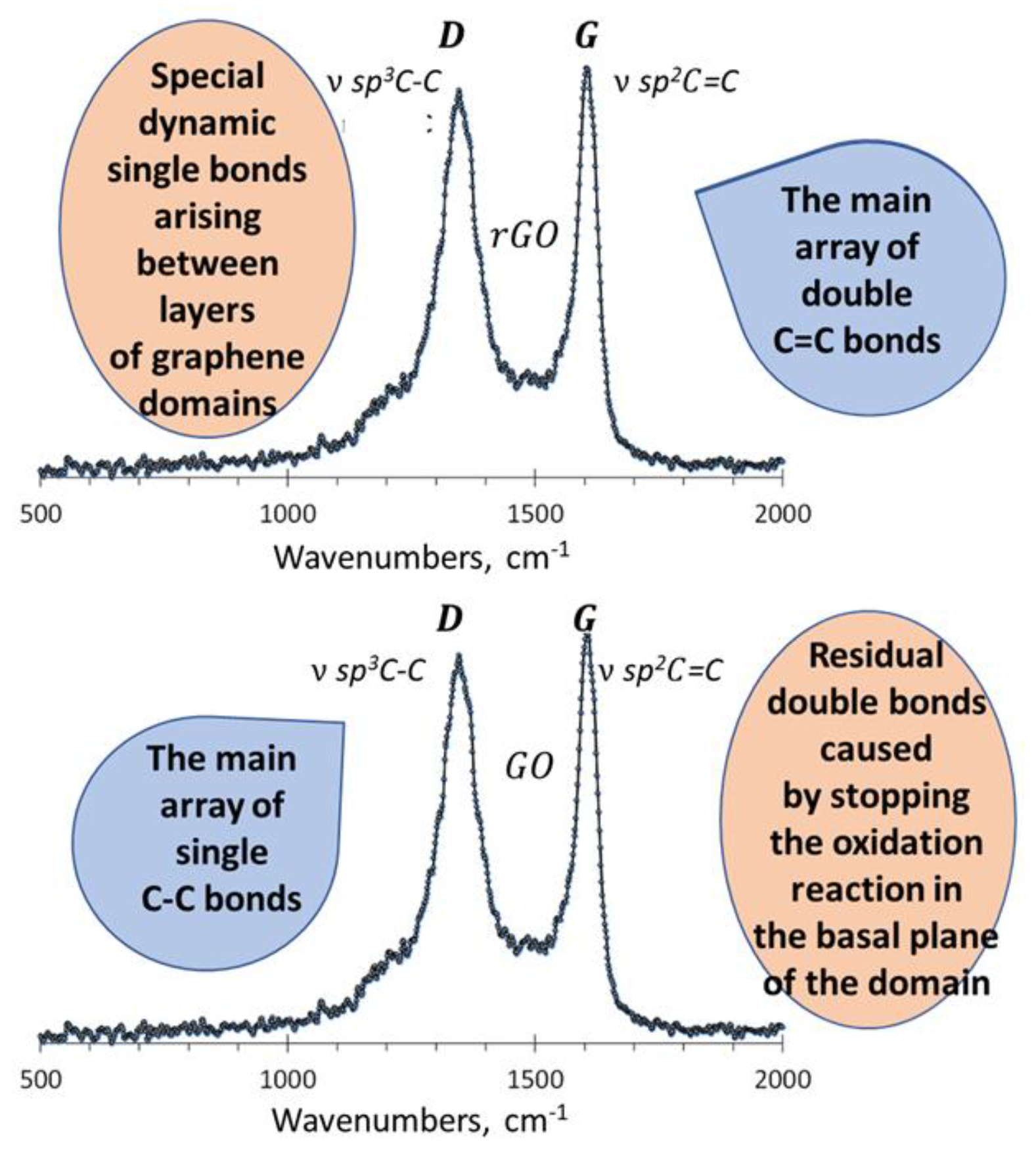
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



