
Submitted:
03 February 2026
Posted:
04 February 2026
You are already at the latest version
Abstract
Preeclampsia is a double-hit vascular disorder centred on the VEGF-HO-1-CSE axis. First, excess placental soluble Flt-1 (sFlt-1) neutralises vascular endothelial growth factor (VEGF) and placental growth factor (PlGF), producing an angiogenic deficit that drives endothelial dysfunction, hypertension, proteinuria and end-organ injury. Second, failure of endogenous vascular brakes, heme oxygenase-1 (HO-1/CO) and cystathionine-γ-lyase (CSE)/hydrogen sulfide (H2S), amplifies anti-angiogenic factor release (sFlt-1, soluble endoglin) and oxidative-inflammatory stress, lowering the threshold at which VEGF loss precipitates severe disease. We synthesise human, animal and translational data that (i) establish placental sFlt-1 source and release, (ii) demonstrate human mechanistic causality via sFlt-1 removal, (iii) show prospective clinical validation that sFlt-1 rises and free PlGF falls before disease onset, and (iv) identify HO-1 and CSE/H2S as protective pathways that restrain anti-angiogenic drive. Finally, we summarise preclinical evidence that the oral H2S-donor MZe786 restores the HO-1/CSE axis, lowers sFlt-1, improves maternal haemodynamics and fetal outcomes across complementary pregnancy models, and outline the role of sFlt-1/PlGF-based triage (PROGNOSIS) in clinical decision-making. This reactive tool, while valuable, cannot sub-stratify among positive cases. Framing severe preeclampsia as a double-hit vascular disorder provides a biologically grounded framework that can inform risk stratification strategies like M-PREG®, a clinical decision support system, and prevention strategies, pairing early risk stratification with mechanism-informed interventions.
Keywords:
preeclampsia
; angiogenic imbalance
; soluble Flt-1 (sFlt-1)
; soluble endoglin (sEng)
; heme oxygenase-1 (HO-1)
; biliverdin reductase (BVR)
; hydrogen sulfide (H2S)
; cystathionine γ-lyase (CSE)
; VEGF
; PlGF
1. Epidemiology and Clinical Burden
Preeclampsia complicates 5–8% of pregnancies worldwide and remains a leading cause of maternal and perinatal morbidity and mortality. The burden is not evenly shared. In low- and middle-income countries, maternal death rates are many times higher than in wealthy nations due to limited access to antenatal and emergency care. Significant disparities persist even in high-resource settings; for example, in the United States, cause-specific mortality from preeclampsia/eclampsia is approximately five-fold higher among non-Hispanic Black women compared to non-Hispanic White women [1].
The clinical and human cost is staggering. Globally, preeclampsia is estimated to claim the lives of 76,000 mothers and 500,000 babies annually, and it leaves many more survivors with lasting cardiovascular, renal, and neurological sequelae [2]. The only definitive cure remains delivery of the placenta and baby, often prematurely, which creates serious challenges for newborn care.
Preeclampsia is characterised by new-onset hypertension after 20 weeks’ gestation with proteinuria and/or maternal organ dysfunction and is frequently associated with fetal growth restriction. If not recognised and treated, it can progress rapidly to eclampsia, a life-threatening convulsive state marked by seizures, stroke, and death.
This clinical syndrome, whose pathogenesis eluded medicine for decades, underscores an urgent, unmet need. While timely diagnosis and delivery mitigate mortality in high-resource settings, the disease itself remains unprevented. In low- and middle-income countries, where these safeguards are often absent, preeclampsia persists as a leading cause of maternal and perinatal death. This global burden, combined with the syndrome’s potential for rapid onset even in previously low-risk pregnancies, highlights the critical necessity for reliable early risk stratification and mechanism-informed prevention strategies.
2. The Angiogenic Imbalance Model: From Hypothesis to Validation (Hit 1)
2.1. Placental VEGF Signalling
A conceptual shift in the mid-1990s proposed that preeclampsia arose not from primary hypertension, but from a loss of vascular endothelial growth factor (VEGF) bioactivity within the maternal vasculature. This was grounded in foundational studies demonstrating that the human placenta expresses VEGF, its receptor VEGFR-1 (Flt-1), and placental growth factor (PlGF), establishing it as a tissue capable of autocrine and paracrine angiogenic signalling [3,4,5,6].
In 1997, Ahmed formalised this into an explicit “VEGF loss” hypothesis, proposing that preeclampsia results from the neutralisation of VEGF, plausibly by a soluble form of its receptor, sFlt-1 [7]. Shortly thereafter, Clark et al. identified soluble Flt-1 (sFlt-1) as the VEGF-binding protein released by the human placenta into the maternal circulation, providing the key molecular candidate predicted by the hypothesis [8].
Importantly, these early functional studies demonstrated that disease severity reflected the relative balance between anti-angiogenic and pro-angiogenic signals, rather than absolute concentrations alone (Figure 1). In 2001, Ahmed’s group showed that increasing placental sFlt-1 release, driven by hypoxia and VEGF itself, impaired endothelial migration, with the most severe angiogenic inhibition observed in preeclampsia compared with fetal growth restriction. Expressed as a PlGF/sFlt-1 relationship, lower values were associated with worse biological outcomes, anticipating later ratio-based approaches [9]. The key milestones preceding and supporting this hypothesis are summarised in Table 1.
Hit 1 (Angiogenic Deficit): Excess placental sFlt-1 neutralizes VEGF and PlGF, driving maternal endothelial dysfunction. Hit 2 (Failed Brakes): Failure of the endogenous cytoprotective pathways HO-1/CO and CSE/H2S amplifies sFlt-1/sEng release and oxidative stress, lowering the threshold for clinical disease.
Left panel: Healthy pregnancy. Under normal conditions, the cystathionine γ-lyase/hydrogen sulfide (CSE/H2S) pathway modulates heme oxygenase-1 (HO-1) activity, maintaining high levels of specific microRNAs that bind to the 3′-UTR of FLT1 mRNA. This post-transcriptional regulation suppresses placental soluble Flt-1 (sFlt-1) release. Consequently, circulating levels of free vascular endothelial growth factor (VEGF) and placental growth factor (PlGF) remain high, supporting normal endothelial function, vascular homeostasis, and fetal-placental development. Hit 1 is absent. Right panel: Preeclampsia. When HO-1 and/or CSE/H2S pathways are compromised (Hit 2: failure of cytoprotective brakes), the microRNA-mediated brake on FLT1 mRNA is lifted. This leads to unchecked placental production and release of sFlt-1 into the maternal circulation (Hit 1: angiogenic deficit). Excess sFlt-1 neutralizes circulating VEGF and PlGF, resulting in decreased bioavailable pro-angiogenic factors. This imbalance drives maternal endothelial dysfunction, hypertension, proteinuria, systemic oxidative stress, and end-organ injury, culminating in the clinical syndrome of preeclampsia. Key to symbols: Green arrows indicate activation or production. Red arrows indicate inhibition or suppression or impaired signalling. The model integrates key evidence: human placental causality via sFlt-1 removal [10], prospective biomarker validation [11,12,13] and the therapeutic rationale for restoring protective brakes [14,15,16,17].
Systemic sFlt-1 in pregnant rats induces hypertension, proteinuria, and glomerular endotheliosis; reduced free VEGF/PlGF and endothelial dysfunction are reversible with VEGF/PlGF [18], demonstrating maternal causality in pregnant animals. This mechanistic link where elevated placental sFlt-1 impairs angiogenesis and sFlt-1 removal restores angiogenesis was first demonstrated directly in human placental tissue in 2004 using immunoprecipitation of sFlt-1 from preeclamptic placental conditioned media [10]. This experiment established human mechanistic causality in human tissue and provided the biological rationale for later translational approaches aimed at lowering circulating sFlt-1 in vivo [19,20].
2.2. Soluble Endoglin Strengthens the Anti-Angiogenic State
A second placenta derived anti-angiogenic factor, soluble endoglin (sEng), strengthened the angiogenic imbalance model by explaining why some women deteriorate rapidly with severe maternal end organ injury. Endoglin is a co receptor for TGF-β family signalling in the vasculature. Its soluble form antagonises pro-angiogenic and vasculoprotective signalling, compounding endothelial dysfunction [21]. Clinically, higher circulating sEng tracks severity and adverse outcomes, and biologically it acts synergistically with sFlt-1 to drive a more toxic anti-angiogenic state [22].
This matters mechanistically because sFlt-1 principally neutralises VEGF and PlGF, whereas sEng perturbs complementary endothelial signalling pathways. Together, they lower the threshold for vascular decompensation. The implication is simple: the most severe phenotype is not only a rise in sFlt-1, but a combined rise in anti-angiogenic drive in which sFlt-1 and sEng co-exist and amplify endothelial injury. This is the biological rationale for why restoring endogenous protective brakes, particularly HO-1 and CSE linked signalling, can be disease modifying rather than merely symptomatic.

Table 1.
Timeline of Key Discoveries and Translational Mapping.
| Year | Discovery | Explanation | References |
|---|---|---|---|
| 1993 | VEGF mRNA expression & localisation in human placenta, | VEGF mRNA localised to first trimester villous trophoblast, term extravillous trophoblast, fetal Hofbauer cells and maternal decidual cells, supporting placental and decidual VEGF expression during pregnancy. | [3] |
| 1994 | Discovery of VEGF receptor-1 (Flt-1) in placental trophoblast. | Molecular evidence that placental trophoblast expresses VEGF receptor 1 (Flt-1), indicating capacity for VEGF responsive signalling within placental tissue. | [4] |
| 1995 | VEGF–Flt-1 co-localisation in human placenta. | Co-localisation of VEGF protein and Flt-1 receptor in trophoblast, decidua and Hofbauer cells, supporting autocrine and paracrine VEGF signalling within the placenta. | [5] |
| 1996 | PlGF localisation in human placenta. | PlGF mRNA and protein localised in term human placenta with methodological detail and images, establishing PlGF as a placental ligand relevant to angiogenic balance. | [6] |
| 1997 | Loss of VEGF activity hypothesis in preeclampsia (Ahmed). | Proposed that preeclampsia arises from loss of VEGF bioactivity, plausibly mediated by endogenous soluble Flt-1. First explicit mechanistic hypothesis centred on VEGF antagonism. | [7] |
| 1998 | Human placenta produces and releases soluble VEGF receptor-1 (sFlt-1). | sFlt-1 mRNA demonstrated in trophoblast, villous explants release sFlt-1, and maternal serum contains a VEGF binding protein consistent with sFlt-1, establishing placental source and release into the maternal circulation. | [8] |
| 2000 | Heme Oxygenase-1 (HO-1) protective pathway in human placenta (first direct evidence). | HO-1 induction attenuates TNF-α-mediated cytotoxicity in human placental villous explants and causes carbon monoxide-dependent vasorelaxation; HO-1 protein is reduced in preeclamptic placenta, supporting HO-1 as an endogenous placental cytoprotective pathway in pregnancy. | [23] |
| 2001 | Functional imbalance (PlGF to sFlt-1 relationship). | Early evidence that lower PlGF to sFlt-1 relationship associates with worse biological outcomes, anticipating later ratio-based approaches. | [9] |
| 2003 | In vivo maternal causality (pregnant rats). | Systemic sFlt-1 in pregnant rats induces hypertension, proteinuria and glomerular endotheliosis; reduced free VEGF and PlGF and endothelial dysfunction are reversible with VEGF and PlGF. | [18] |
| 2004 | Human mechanistic causality. | Selective removal of sFlt-1 from preeclamptic placental conditioned medium restores angiogenesis ex vivo, providing direct human tissue evidence for sFlt-1 mediated angiogenic suppression. Direct human tissue proof of causality. | [10] |
| 2004 | Early clinical validation. | Rising sFlt-1 and falling free PlGF precede clinical preeclampsia, track severity, and fall postpartum, providing clinical evidence consistent with the angiogenic imbalance mechanism. Provided first large clinical evidence for the angiogenic imbalance theory. | [11,12,13] |
| 2006 | Soluble endoglin (sEng) enters the preeclampsia model. | Circulating sEng is elevated before preeclampsia and, together with sFlt-1, intensifies endothelial dysfunction; co administration of sEng and sFlt-1 produces a more severe preeclampsia like phenotype in pregnant animals, supporting synergistic anti-angiogenic drive. sFlt-1 plus sEng is a “high-toxicity” combination. | [21,22] |
| 2007 | HO-1/CO protective brake. | Up-regulation of HO-1 reduces sFlt-1 and sEng release; HO-1 deficiency increases both, identifying HO-1 as an endogenous pathway restraining anti-angiogenic factor release. | [14] |
| 2013 | CSE/ (H2S) protective brake. | CSE is reduced in preeclampsia; CSE inhibition increases sFlt-1 and sEng, whereas an H2S donor reduces anti-angiogenic factors and improves fetal growth in mice. | [15] |
| 2016 | Clinical triage adoption (PROGNOSIS). | Multicentre validation of the sFlt-1 to PlGF ratio; a cut-off ≤ 38 reliably rules out preeclampsia within 1 week in women with suspected disease. | [24] |
| 2019 | Real world effectiveness and implementation: PARROT trial. | Subsequent analyses extended rule out to up to 4 weeks and informed retesting strategies. A randomised trial of PlGF based testing in UK maternity units showed shorter time to diagnosis and reduced severe maternal adverse outcomes. | [25,26] |
| 2020-2021 | Oral small molecule therapy development (MZe786). | In refined mouse RUPP models, MZe786 lowers sFlt-1, reduces MAP and oxidative stress, and improves fetal outcomes. In an HO-1 compromised, high sFlt-1 pregnancy model, MZe786 reduces circulating sFlt-1 and sEng, whereas aspirin does not, with improved maternal and fetal outcomes across complementary models. | [16,17] |
| 2025-2026 | Translational/Developmental. | M-PREG is a clinical decision support system for risk stratification in suspected preeclampsia. In parallel, GMP grade manufacturing and formulation of MZe786 have been completed, and IND submission is in preparation. |
2.3. Heme Oxygenase-1: A Master Cytoprotective Brake
The heme oxygenase–biliverdin reductase (HO-1/BVR) system is a critical endogenous cytoprotective and signaling axis in vascular tissues. HO-1 degrades heme to biliverdin, carbon monoxide (CO), and iron. Biliverdin is subsequently reduced to bilirubin by biliverdin reductase (BVR). The products of this pathway confer protection via antioxidant effects (through a bilirubin↔biliverdin redox cycle) and vasodilatory signalling (via CO) [27,28].
In pregnancy, this axis is indispensable. In 2000, Ahmed et al. provided the first direct evidence of its role in the human placenta, demonstrating that HO-1 induction protects against cytokine-mediated cytotoxicity and mediates vasorelaxation, and that its expression is reduced in preeclampsia [23]. This established HO-1 as a vital guardian of placental and vascular health.
Within the double-hit framework (Figure 1), HO-1/BVR constitutes a major maternal cytoprotective “brake” (Hit 2). Its failure amplifies anti-angiogenic factor release and oxidative-inflammatory stress, lowering the threshold at which the angiogenic deficit (Hit 1) becomes clinically catastrophic. Subsequent studies showed that HO-1 suppresses the release of both sFlt-1 and soluble endoglin, confirming its central role as a protective brake [14].
In the years that followed, independent studies confirmed and extended this model. Large prospective studies demonstrated that alterations in circulating PlGF and sFlt-1 precede clinical disease and can predict risk weeks before symptom onset [11,12]. Independent longitudinal studies further showed that circulating sFlt-1 rises weeks before the onset of clinical preeclampsia and correlates with disease severity [13].
Discoveries that reframe an entire field, offering a testable, mechanistic explanation for a major disease and guiding the path toward preventive are uncommon. The true significance of this conceptual shift lies not in priority, but in its impact on improving outcomes for mothers and babies worldwide.
2.4. Conceptual Evolution: From Protective Pathways to the Double-Hit Model
This framework, in which the failure of endogenous vascular protective systems determines disease severity, evolves from the accelerator-brake hypothesis first formally proposed to explain preeclampsia pathophysiology [29,30], it was posited that “the pathogenesis of preeclampsia is largely due to loss of HO activity,” resulting in excessive anti-angiogenic factor release [29]. This built upon the observation that the incidence of preeclampsia is reduced in smokers, a paradox linked to carbon monoxide (CO) from cigarette smoke suppressing placental sFlt-1 release. This provided an early epidemiological clue that augmenting the HO-1/CO ‘brake’ could modify disease risk.
This concept was refined using Bradford Hill criteria for causation. [31] It argued that the angiogenic imbalance model demonstrated strength, consistency, temporality, and biological plausibility, whereas other theories (e.g., defective spiral artery remodelling, systemic inflammation) appeared associative or consequential [32]. The subsequent identification of the CSE/H2S pathway as a second protective brake [15] completed the core circuitry of the endogenous cytoprotective system.
The synthesis presented here refines this into the testable ‘double-hit vascular disorder’ model. Hit 1 (angiogenic deficit) defines the necessary insult. Hit 2 (failed cytoprotective brakes) explains the variable penetrance and severity, providing a mechanistic basis for why only some pregnancies with placental stress progress to clinical disease. This model moves beyond association to provide a unified, causative framework that explains clinical heterogeneity, underpins biomarker utility, and directly informs therapeutic strategy.
3. Failure of Cytoprotective Brakes Amplifies Disease (Hit 2)
3.1. Cytoprotective Pathways: HO-1/CO and CSE/H2S
Identifying the trigger for preeclampsia was only part of the puzzle. The next question was simple: what natural systems normally keep the condition under control, and why do they fail?
Subsequent work identified a parallel cytoprotective system that also acts like a safety brake. The dysregulation of the cystathionine γ-lyase/hydrogen sulfide (CSE/H2S) pathway also contributes to maternal hypertension and placental abnormalities, establishing a second protective brake that fails in preeclampsia [15]. The pathways HO-1 and CSE keep those signals in balance and play a crucial role in pregnancy and preeclampsia. In healthy pregnancies, they stop things from spinning out of control. In preeclampsia, cytoprotective pathways fail, permitting unrestrained sFlt-1 release and VEGF activity is stripped away, and blood vessels take the hit (Figure 1).
3.2. HO-1 Promoter Activity and Genetic Vulnerability
Functional variation within the HMOX1 promoter refines the “protective brake” concept. HO-1 inducibility is strongly influenced by polymorphic elements within its promoter, most notably the length of a (GT)n microsatellite repeat, where longer repeat variants are associated with reduced transcriptional responsiveness and diminished HO-1 induction under stress [33,34].
In human pregnancy, long (GT)n promoter variants have been associated with susceptibility to late-onset and non-severe preeclampsia in the FINNPEC cohort, consistent with a model in which impaired HO-1 stress responsiveness lowers vascular resilience rather than acting as a primary trigger. This fits squarely within Hit-2 of the double-hit vascular disorder framework (Figure 1): once placental anti-angiogenic stress emerges, a weakened HO-1 brake permits greater endothelial injury and amplifies disease expression [33].
In population based mid trimester cohorts, participants of African ancestry show distinct baseline angiogenic set points with higher sFlt-1 and altered PlGF compared with other groups; a separation that can be approximated in some analyses as a ~20-30% shift in angiogenic profiles (for example, PlGF and/or sFlt-1/PlGF-derived risk metrics), depending on gestation and comparator, and which may help explain differences in susceptibility under equivalent placental stress. [35,36] In the context of our brake theory, ancestral enrichment of HMOX1 promoter variants linked to lower HO-1 inducibility provides a plausible biological substrate for reduced cytoprotective reserve, amplifying the impact of anti-angiogenic load once it emerges [33,37]. Together, these data support a mechanistic second hit in which constrained HO-1 inducibility and a shifted angiogenic equilibrium lower the threshold for maternal vascular decompensation when placental anti-angiogenic factors rise.
3.3. From Mechanism to Medicine: Translational Implications of Restoring the HO-1/CSE Axis
Crucially, this rationale supported development of therapies targeting these pathways. To strengthen the link to the model: Guided by the double-hit model, MZe786 was developed as an oral agent designed to restore HO-1/CSE pathway activity. Our recent preclinical work has demonstrated that MZe786 can prevent preeclampsia in pregnancies compromised by HO-1 deficiency and high sFlt-1, further validating the therapeutic strategy of restoring the protective brakes [16,17] In the refined Reduced Uterine Perfusion Pressure (RUPP) mouse model, MZe786 reduced circulating sFlt-1, improved maternal vascular function, and prevented hallmark features of preeclampsia [16]. In multiple preclinical models of preeclampsia, MZe786 reduced sFlt-1 levels, improved vascular function, and prevented the onset of disease-like symptoms. Its oral formulation is particularly important, as it makes the treatment practical to deliver in both high-income and low-resource settings. Clinical trials will be required to determine whether MZe786 can safely and effectively prevent preeclampsia in human pregnancy.
3.4. Validating the Axis: Recent Advances in Therapeutics and Subphenotyping
The predictive power of the double-hit model is demonstrated by its success in guiding therapeutic development and refining patient stratification.
3.5. Therapeutic Validation Targeting the Axis
The causal logic of the model dictates that interventions lowering sFlt-1 or boosting protective pathways should be beneficial. This has been validated on multiple fronts:
- Direct sFlt-1 Removal: Building on the foundational proof that sFlt-1 removal restores angiogenesis ex vivo [10] therapeutic dextran sulfate apheresis to lower circulating sFlt-1 in severe, early-onset preeclampsia has shown promise in prolonging gestation, providing direct in vivo human proof-of-concept [20].
- Restoring the Protective Brakes: The oral H2S-donor MZe786 represents a mechanism-informed approach targeting Hit 2. In complementary preclinical models, including a genetic model of HO-1 deficiency, MZe786 consistently reduces sFlt-1/sEng, improves maternal hemodynamics, and, critically, improves fetal growth [16]. This validates the core therapeutic strategy of brake restoration.
3.5. Subphenotyping and the Double-Hit Clinical Spectrum
The model explains the clinical continuum from late-onset to severe early-onset disease. Biomarker-based subphenotyping now aligns with this biology: a “placental” phenotype is characterized by profound angiogenic imbalance (severe Hit-1), often with fetal growth restriction, while a “maternal” phenotype may exhibit a more moderate imbalance but occur in individuals with underlying endothelial susceptibility (predisposition to Hit-2). This framework is operationalized in tools like M-PREG, which uses angiogenic markers in algorithms to stratify risk weeks before clinical onset.
3.6. Linking to Long-Term Cardiovascular Risk
The double-hit model also provides a biological lens through which to view the well-established link between preeclampsia and a woman’s lifelong risk of cardiovascular disease (CVD). A pregnancy complicated by preeclampsia may be the first major stress test revealing an individual’s inherent endothelial resilience and cytoprotective reserve (Hit-2 capacity). The same brake deficiency that amplifies placental anti-angiogenic stress may later contribute to endothelial dysfunction in response to metabolic or hypertensive challenges, explaining the shared pathophysiology.
Beyond direct sFlt-1 removal, a complementary strategy is the targeted inhibition of sFlt-1 production, for example using small interfering RNA (siRNA) technology. While this approach is mechanistically sound, its path to becoming a globally accessible preeclampsia therapy faces significant translational and practical hurdles (Table 2). In contrast, an orally active small molecule like MZe786, designed to restore the upstream protective HO-1/CSE axis, offers a more practical therapeutic profile. A direct comparison of the two modalities highlights the advantages of the mechanism-informed, oral brake-restoration strategy (Table 2).
4. From Biomarkers to Bedside Pathways: Short Term Triage and Real-World Implementation
Angiogenic biomarkers moved from mechanistic insight to clinical utility when they were shown to support time bounded decision making in women with suspected preeclampsia. The sFlt-1/PlGF ratio is now widely used as a triage aid to help clinicians rule out or rule in the likelihood of developing preeclampsia within a defined short horizon, supporting safer discharge, targeted surveillance, and earlier escalation when risk is high [24,26]. In practice, the value is not simply diagnostic accuracy, but the ability to reduce uncertainty in real time and align resources to those most likely to deteriorate.
Implementation studies in routine maternity care have further reinforced that PlGF based testing, when embedded into clinical pathways, can accelerate confirmation of diagnosis and support better maternal outcomes by shortening time to appropriate management decisions. Together, these advances established a pragmatic model: combine mechanistic biomarkers that reflect placental anti-angiogenic drive with a structured pathway that specifies what clinicians do next, rather than leaving results as isolated laboratory data. A recent preeclampsia landscape report highlights blood-based biomarkers, validated blood pressure devices, and task-sharing care models as key innovations for improving preeclampsia detection and management, while also identifying critical gaps in market access and health system readiness [38].
5. M-PREG: Clinical Decision Support for Risk Stratification
In parallel, M-PREG was developed as a clinical decision support system that integrates routinely available biomarker inputs (including sFlt-1 and PlGF) and other laboratory variables within a transparent computational framework to support risk stratification in women with suspected pre-eclampsia. The intended purpose is to help identify individuals at increased risk of deterioration earlier in the care pathway, supporting proportionate surveillance and escalation according to local protocols and clinical judgement.
This allows for close monitoring, early intervention, and, once MZe786 becomes available, the start of preventive therapy before irreversible damage occurs. Together, M-PREG and MZe786 are designed to work in synergy, one to predict, the other to protect.
6. Towards Prediction and Prevention of Preeclampsia
This mechanistic understanding has informed translational development of two investigational approaches:
1. MZe786 is an orally active drug that stimulates the CSE and H2S pathway, restores angiogenic balance (a healthy balance for blood vessel growth factors), and protects maternal blood vessels. In preclinical studies, it has been shown to reduce sFlt-1, improve blood vessel function, and prevent the clinical features of preeclampsia in three separate animal models.
2. M-PREG is a digital diagnostic that has received the UKCA mark. It uses biomarkers such as sFlt-1 and PlGF to accurately predict which of the suspected preeclamptic pregnant cases are at high risk of developing preeclampsia. UKCA (UK Conformity Assessed) is the quality and safety mark required for medical devices in Great Britain, like the CE mark in the EU.
If validated in multicentre clinical trials, these approaches could enable integrated prediction and prevention strategies in preeclampsia research, transforming care for mothers and babies worldwide. While mechanistic synthesis is necessarily upstream of clinical adoption, such frameworks are essential for the development of decision-support tools, stratified trials, and ultimately guideline-level change.
7. Conclusions and Future Directions
The evidence reviewed here supports the double-hit vascular disorder model as a root cause of severe preeclampsia: an initiating placental anti-angiogenic state (Hit 1) that becomes clinically severe upon failure of endogenous vascular protective systems (Hit 2). This framework, built upon three decades of human, animal, and translational research and evolving from the earlier protective pathways’ hypothesis, moves beyond description to provide a biologically grounded, actionable roadmap.
It explains the success and limitations of biomarker-based triage (e.g., sFlt-1/PlGF ratio), informs the development of clinical decision-support tools like M-PREG for early risk stratification, and provides the mechanistic rationale for novel, mechanism-informed therapies like the oral H2S-donor MZe786 aimed at restoring the HO-1/CSE axis. Continued investigation through rigorous clinical trials is now essential to translate this unified understanding into strategies that can predict, prevent, and ultimately eliminate the global burden of preeclampsia. Furthermore, this model offers a novel perspective on the shared aetiology of pregnancy-specific vascular stress and lifelong cardiovascular risk, opening new avenues for postpartum monitoring and prevention.
Author Contributions
AA conceptualised and wrote the first draft of the manuscript. All authors were involved in the edits and approve the final version.
Funding
This study was supported by Innovate UK (Grant number 10066967) and MirZyme Therapeutics Limited. Funders had no role.
Use of Generative AI
ChatGPT (OpenAI) was used to assist with language and narrative editing; the authors verified all content and references.
Conflicts of Interest
AA is Founder and CEO of MirZyme Therapeutics. SKS is Chair of the Scientific Advisory Board of MirZyme Therapeutics. AA, SA and KW, are named inventors on patents related to diagnostics and therapeutics for preeclampsia. SKS declares no competing interests.
References
- MacDorman, M.F., et al., Racial and Ethnic Disparities in Maternal Mortality in the United States Using Enhanced Vital Records, 2016–2017. Am J Public Health 2021, 111(9), 1673–1681. [CrossRef] [PubMed]
- Grandi, S.M., et al., Cardiovascular Disease-Related Morbidity and Mortality in Women With a History of Pregnancy Complications. Circulation 2019, 139(8), 1069–1079. [CrossRef] [PubMed]
- Sharkey, A.M., et al., Expression of mRNA for vascular endothelial growth factor in human placenta. J Reprod Fertil 1993, 99(2), 609–15. [CrossRef] [PubMed]
- Charnock-Jones, D.S., et al., Vascular endothelial growth factor receptor localization and activation in human trophoblast and choriocarcinoma cells. Biol Reprod 1994, 51(3), 524–30. [CrossRef]
- Ahmed, A., et al., Colocalisation of vascular endothelial growth factor and its Flt-1 receptor in human placenta. Growth Factors 1995, 12(3), 235–43. [CrossRef]
- Khaliq, A., et al., Localisation of placenta growth factor (PIGF) in human term placenta. Growth Factors 1996, 13(3-4), 243–50, color plates I-II, pre bk cov.. [CrossRef]
- Ahmed, A. Heparin-binding angiogenic growth factors in pregnancy . Trophoblast Res 1997, 10, 215–258. [Google Scholar] [CrossRef]
- Clark, D.E., et al., A vascular endothelial growth factor antagonist is produced by the human placenta and released into the maternal circulation. Biol Reprod 1998, 59(6), 1540–8. [CrossRef]
- Ahmad, S.; Ahmed, A. Regulation of soluble VEGFR-1 by VEGF and oxygen and its elevation in pre-eclampsia and fetal growth restriction . Placenta 2001, 22(8-9), p. A.7. [Google Scholar]
- Ahmad, S.; Ahmed, A. Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia . Circ Res 2004, 95(9), 884–91. [Google Scholar] [CrossRef]
- Levine, R.J., et al., Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 2004, 350(7), 672–83. [CrossRef]
- Thadhani, R., et al., First trimester placental growth factor and soluble fms-like tyrosine kinase 1 and risk for preeclampsia. J Clin Endocrinol Metab 2004, 89(2), 770–5. [CrossRef] [PubMed]
- McKeeman, G.C., et al., Soluble vascular endothelial growth factor receptor-1 (sFlt-1) is increased throughout gestation in patients who have preeclampsia develop. Am J Obstet Gynecol 2004, 191(4), 1240–6. [CrossRef] [PubMed]
- Cudmore, M., et al., Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation 2007, 115(13), 1789–97. [CrossRef] [PubMed]
- Wang, K., et al., Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013, 127(25), 2514–22. [CrossRef]
- Saif, J., et al., Hydrogen sulfide releasing molecule MZe786 inhibits soluble Flt-1 and prevents preeclampsia in a refined RUPP mouse model. Redox Biol 2021, 38, 101814. [CrossRef]
- Rezai, H., et al., MZe786, a hydrogen sulfide-releasing aspirin prevents preeclampsia in heme oxygenase-1 haplodeficient pregnancy under high soluble flt-1 environment. Redox Biol 2021, 38, 101768. [CrossRef]
- Maynard, S.E., et al., Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. The Journal of clinical investigation 2003, 111(5), 649–58. [CrossRef]
- Thadhani, R., et al., Pilot study of extracorporeal removal of soluble fms-like tyrosine kinase 1 in preeclampsia. Circulation 2011, 124(8), 940–50. [CrossRef]
- Thadhani, R., et al., Removal of Soluble Fms-Like Tyrosine Kinase-1 by Dextran Sulfate Apheresis in Preeclampsia. J Am Soc Nephrol 2016, 27(3), 903–13. [CrossRef]
- Venkatesha, S., et al., Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 2006, 12(6), 642–9. [CrossRef] [PubMed]
- Levine, R.J., et al., Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 2006, 355(10), 992–1005. [CrossRef] [PubMed]
- Ahmed, A., et al., Induction of placental heme oxygenase-1 is protective against TNFalpha-induced cytotoxicity and promotes vessel relaxation. Mol Med 2000, 6(5), 391–409. [CrossRef]
- Zeisler, H., et al., Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N Engl J Med 2016, 374(1), 13–22. [CrossRef]
- Zeisler, H., et al., Soluble fms-like tyrosine kinase-1 to placental growth factor ratio: ruling out pre-eclampsia for up to 4 weeks and value of retesting. Ultrasound Obstet Gynecol 2019, 53(3), 367–375. [CrossRef]
- Duhig, K.E., et al., Placental growth factor testing to assess women with suspected pre-eclampsia: a multicentre, pragmatic, stepped-wedge cluster-randomised controlled trial. Lancet 2019, 393(10183), 1807–1818. [CrossRef]
- Baranano, D.E., et al., Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci U S A 2002, 99(25), 16093–8. [CrossRef]
- Lerner-Marmarosh, N., et al., Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc Natl Acad Sci U S A 2008, 105(19), 6870–5. [CrossRef]
- Ahmed, A. New insights into the etiology of preeclampsia: identification of key elusive factors for the vascular complications . Thromb Res 2011, 127 Suppl 3, S72–5. [Google Scholar] [CrossRef]
- Ahmed, A.; Ramma, W. Unravelling the theories of pre-eclampsia: are the protective pathways the new paradigm? . Br J Pharmacol 2015, 172(6), 1574–86. [Google Scholar] [CrossRef]
- Ramma, W.; Ahmed, A. Is inflammation the cause of pre-eclampsia? . Biochemical Society Transactions 2011, 39 Pt 6, 1619–1627. [Google Scholar] [CrossRef]
- Ahmed, A., H. Rezai, and S. Broadway-Stringer, Evidence-Based Revised View of the Pathophysiology of Preeclampsia. Adv Exp Med Biol 2017, 956, 355–374.
- Kaartokallio, T., et al., Microsatellite polymorphism in the heme oxygenase-1 promoter is associated with nonsevere and late-onset preeclampsia. Hypertension 2014, 64(1), 172–7. [CrossRef]
- Kaartokallio, T., et al., Fetal Microsatellite in the Heme Oxygenase 1 Promoter Is Associated With Severe and Early-Onset Preeclampsia. Hypertension 2018, 71(1), 95–102. [CrossRef]
- Wright, A., et al., Effect of race on the measurement of angiogenic factors for prediction and diagnosis of pre-eclampsia. BJOG 2023, 130(1), 78–87. [CrossRef]
- Yang, J., et al., Racial-ethnic differences in midtrimester maternal serum levels of angiogenic and antiangiogenic factors. Am J Obstet Gynecol 2016, 215(3), 359 e1–9. [CrossRef] [PubMed]
- Sun, T., et al., HMOX1 Genetic Polymorphisms Display Ancestral Diversity and May Be Linked to Hypertensive Disorders in Pregnancy. Reprod Sci 2022, 29(12), 3465–3476. [CrossRef]
- Unitaid. Commodity and service delivery innovations for detection and management of pre-eclampsia: Landscape report. Geneva: Unitaid (hosted by the World Health Organization); 2024.
Figure 1.
The double-hit vascular disorder model of preeclampsia.
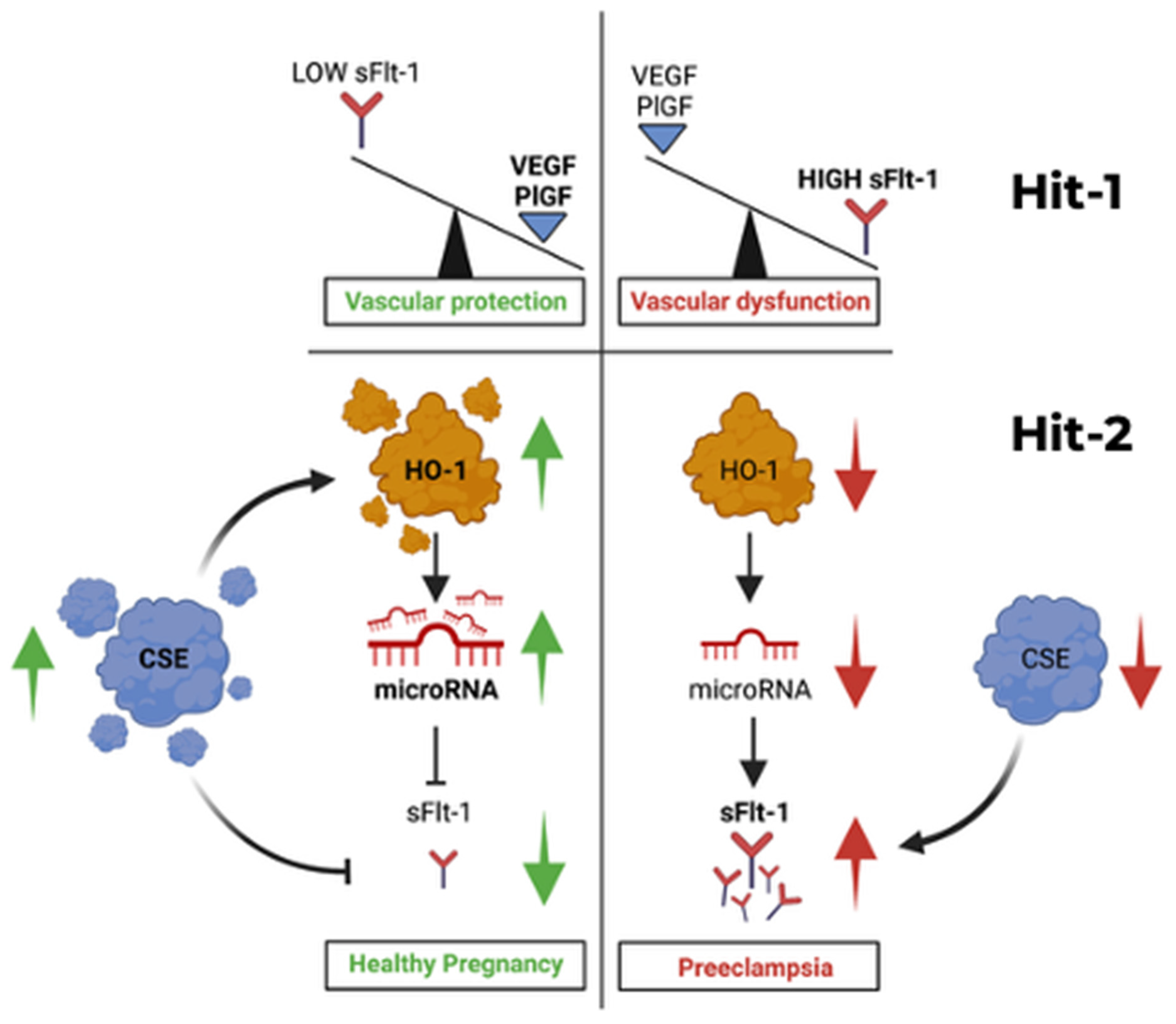
Table 2.
Comparative Analysis of Therapeutic Modalities for Preeclampsia: Oral Small Molecule vs. sFlt-1-targeted siRNA.
Table 2.
Comparative Analysis of Therapeutic Modalities for Preeclampsia: Oral Small Molecule vs. sFlt-1-targeted siRNA.
| Characteristic | Oral Small Molecule (MZe786) | sFlt-1-targeted siRNA |
|---|---|---|
| Route of Delivery | Oral pill (convenient, non-invasive) | Subcutaneous injection (invasive, requires clinical administration) |
| Patient Compliance | High (easy to use, home-based) | Low (pain, fear of injections, frequent dosing) |
| Manufacturing Cost | Low (simple chemical synthesis, scalable) | High (complex lipid nanoparticle (LNP) formulation, biologics production) |
| Storage & Distribution | Room temperature stable (robust supply chain) | Cold chain required (limits deployment, increases cost) |
| Bioavailability & Dosing | Sustained release possible; targets upstream pathway | Requires delivery vehicle (LNP); transient effect; repeated dosing needed |
| Safety & Immunogenicity | Low immune response (well-characterized small molecule) | Risk of immune system activation (e.g., complement activation, anti-drug antibodies) |
| Global Access | High potential (developing world access, primary care) | Limited (requires advanced healthcare infrastructure, high cost) |
| Regulatory Pathway | Well-understood (small molecule drug development) | Novel, with evolving regulatory challenges for oligonucleotides in pregnancy |
| Market & Health Equity | Global potential, aligns with WHO essential medicine goals | Likely limited to high-income markets, exacerbating health inequity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



